

Abstract
Fibrillar aggregates of the α-synuclein (αS) protein are the hallmark of Parkinson’s Disease and related neurodegenerative disorders. Characterization of the effects of mutations and post-translational modifications (PTMs) on the αS aggregation rate can provide insight into the mechanism of fibril formation, which remains elusive in spite of intense study. A comprehensive collection (375 examples) of mutant and PTM aggregation rate data measured using the fluorescent probe thioflavin T is presented, as well as a summary of the effects of fluorescent labeling on αS aggregation (20 examples). A curated set of 131 single mutant de novo aggregation experiments are normalized to wild type controls and analyzed in terms of structural data for the monomer and fibrillar forms of αS. These tabulated data serve as a resource to the community to help in interpretation of aggregation experiments and to potentially be used as inputs for computational models of aggregation.
Keywords: Parkinson’s Disease, neurodegenerative disorder, amyloid, fibril, thioflavin T
Introduction
Synucleinopathies are a family of age-related neurodegenerative diseases including Parkinson’s Disease (PD), Parkinson’s Disease with Dementia (PDD), Multiple System Atrophy (MSA), and Dementia with Lewy Bodies (DLB) that are characterized by inclusions of the abundant neuronal protein α-synuclein (αS). PD is the second most common neurodegenerative disease after Alzheimer’s Disease, affecting an estimated 0.3% of the world’s population with [variable pathologies of synucleinopathies1] and accounting for about 15% of all dementia cases [2]. Clinically, PD has an average age of onset of 55 years and is distinguished by impaired motor functions such as resting tremors, rigidity, and bradykinesia as well as non-motor impairments such as sleep disorders, hallucinations, dementia, autonomic dysfunction, and mood disorders [1–3]. Pathologically, PD diagnostic criteria include a loss of dopaminergic neurons in the substantia nigra and intracellular inclusions of aggregated αS in Lewy Bodies and Lewy Neurites shown in Figure 1 [4–6].
Figure 1.
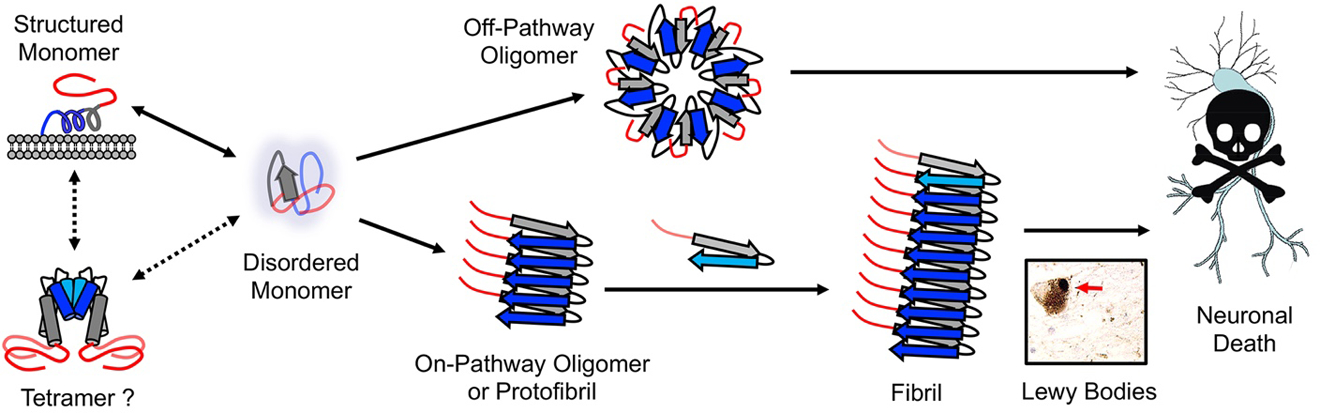
Conformational states and aggregation pathways of αS. N-terminal, NAC, and C-terminal regions colored as shown in Figure 2.
αS is 140 amino acids in length and is typically characterized in terms of three regions. The N-terminal region consists of residues 1 to 60 with 7 imperfect 11 amino acid repeats, each including a conserved KTKEGV motif (Figure 2). This amphipathic region drives αS’s interactions with membranes [7]. Directly adjacent to this region is the central non-amyloid-β component (NAC) section, comprising resides 61 to 95, and is a largely hydrophobic region thought to be necessary for fibril formation. Finally, the C-terminal domain includes residues 96 to 140. It is acidic and remains disordered under all reported conditions [7, 8]. While the tertiary structure of wild-type (WT) αS is not agreed upon (Figure 1), the more widely accepted hypothesis is that αS exists as an intrinsically disordered monomer [9]. Upon binding to a membrane, αS’s N-terminus and NAC region adopt a helical conformation while the C-terminus remains unstructured [10]. Depending on the curvature of the membrane, the helical region may also adopt a set of antiparallel helices [11, 12]. The other major hypothesis argues that αS exists as a partially ordered tetramer in solution that is aggregation-resistant. Disruptions of these non-toxic tetramers release monomers which are prone to misfolding and aggregation [13, 14].
Figure 2.
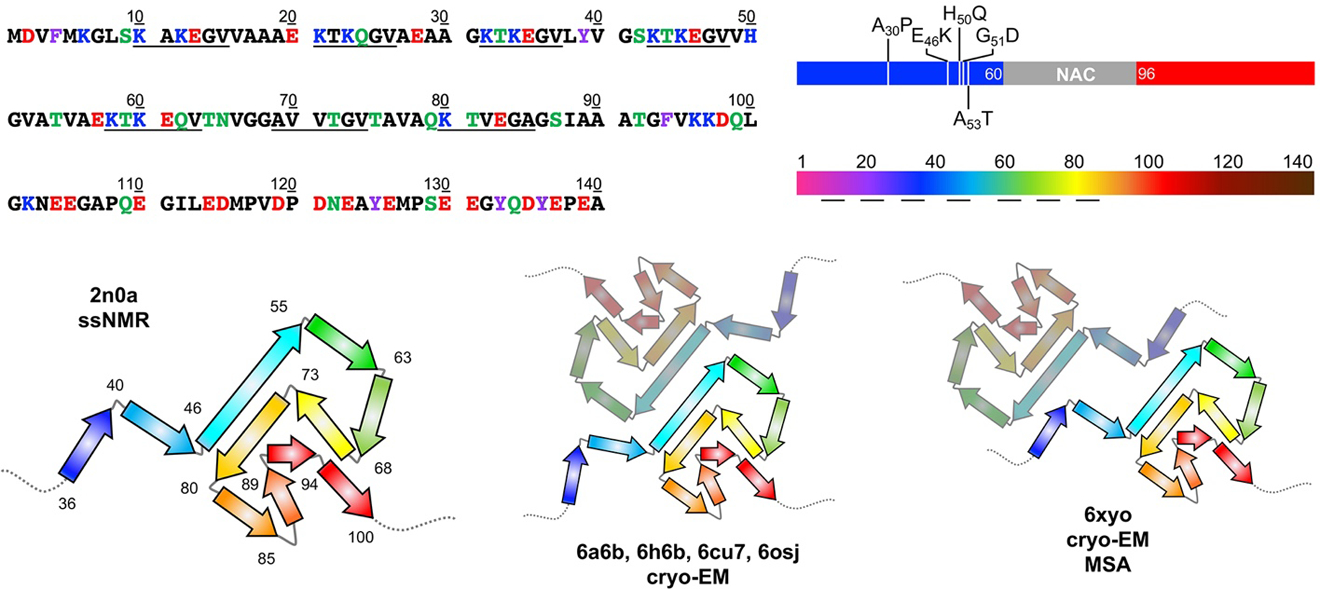
Top Left: αS sequence with amino acids colored by properties: hydrophobic (black), polar (green), aromatic (purple), acidic (red), or basic (blue). Imperfect repeats are underlined. Top Right: αS structure segments colored blue (N-terminal), grey (NAC), and red (C-terminal) as in Figures 1 and 3, with familial mutants indicated. Rainbow-colored by sequence number as in structural images below and Figures 4, 6, 7, and 8. Bottom: 2n0a ssNMR structures of αS fibrils viewed down the helical axis. The core region of residues 36–100 is shown in cartoon form with the residues at turns between β-stands noted. Cryo-EM structures of fibrils (6a6b, 6h6b, 6cu7, 6osj) showing a similar fold with a common protofibril packing motif observed in structures from several independent studies. A similar fold is also observed in fibrils from MSA patients (6xyo) with a different protofibril arrangement. PDB IDs are noted.
While αS’s native function is not fully understood, it is primarily localized to the presynaptic termini of neurons [15]. Interactions have been identified with partners including lipid membranes, synaptic vesicles, SNARE protein complexes, proteins involved in dopamine homeostasis, proteins involved in calcium regulation, and more [16]. These observations have spawned proposals of αS functions such as suppressing apoptosis, modulating calcium levels via calmodulin, helping to assemble SNARE complexes and regulate the release of neurotransmitters, antioxidation, neuronal differentiation, dopamine biosynthesis, and maintaining polyunsaturated fatty acid levels [7].
Aggregation
In PD pathology, monomeric αS aggregates into oligomers (Figure 1). These oligomers can continue to add monomeric αS and become fibrils which are the major components of Lewy Bodies and Lewy Neurites [6, 17]. The toxicity associated with the formation of Lewy Bodies is thought to result from αS aggregation and interactions between these aggregates and organelles such as mitochondria, autophagosomes, and endo/lysosomes [18]. Much like prions, fibrils play a role in disease progression because of their ability to fragment and form seeds which can recruit unstructured monomeric αS to form new oligomers and fibrils [19]. Seeding also has the ability to trigger aggregation events in nearby neurons, causing the progressive loss of neuronal function seen in PD pathology [20–23]. While mature fibrils are the form of αS primarily found in Lewy Bodies [24], oligomers also exhibit toxicity [25]. The relative toxicity of these structures and their exact role in disease progression is still unclear due to the wide range of aggregates that are reductively referred to as oligomers and fibrils despite having different structures and molecular weights [17, 26, 27]. The use of different in vitro cell and animal models as well as varied aggregation conditions further complicates the comparison of αS studies [28–30]. αS can also form oligomers that do not lead to standard fibril formation [17, 31].
The αS aggregation pathway is generally divided into a series of microscopic steps termed primary nucleation, elongation, and secondary nucleation. Primary nucleation involves monomeric αS assembly into small oligomers termed nuclei. This step largely consists of monomeric disordered αS undergoing a conformational change to become a partially folded intermediate capable of aggregation [32]. After nucleation follows elongation, where nuclei act as templates and additional monomers add to their ends to form longer fibrils. Secondary nucleation refers to the formation of nuclei from already existing aggregates termed seeds. Secondary nucleation is thought to occur at specific locations on the surface of fibrils [33, 34] or through fibril fragmentation [35]. For fibrils produced in vitro, sonication can be used to break fibrils into smaller seeds so that secondary nucleation and elongation can be studied separately from primary nucleation [36].
αS aggregation can be studied using fibrils formed in vitro. Generally, these fibrils are formed from recombinantly expressed human αS that has been agitated in solution [37], although fibrils can also be formed under quiescent conditions over a longer period of time [38]. These fibrils have been shown to be similar to those derived from PD patients through a variety of biochemical and low resolution structural methods, but no high resolution structure of PD patient fibrils yet exists for comparison [39, 40]. Fibril formation and kinetics can be measured using a number of dyes and reporters, the most common being Thioflavin T (ThT, Figure 3). ThT is a fluorescent dye whose absorption and emission spectra shift upon binding to the cross-β structures of amyloid fibrils [41, 42]. There are two groups of ThT aggregation assays. The first consists of removing aliquots of aggregating αS at specific time points, adding ThT, and then monitoring fluorescence. The second method involves aggregating αS in the continued presence of ThT using a fluorescence plate reader [43]. While there are other techniques used to measure aggregation, such as sedimentation or chromophore reporters Congo Red and K114, this review focuses on ThT because it is the most widely used.
Figure 3.
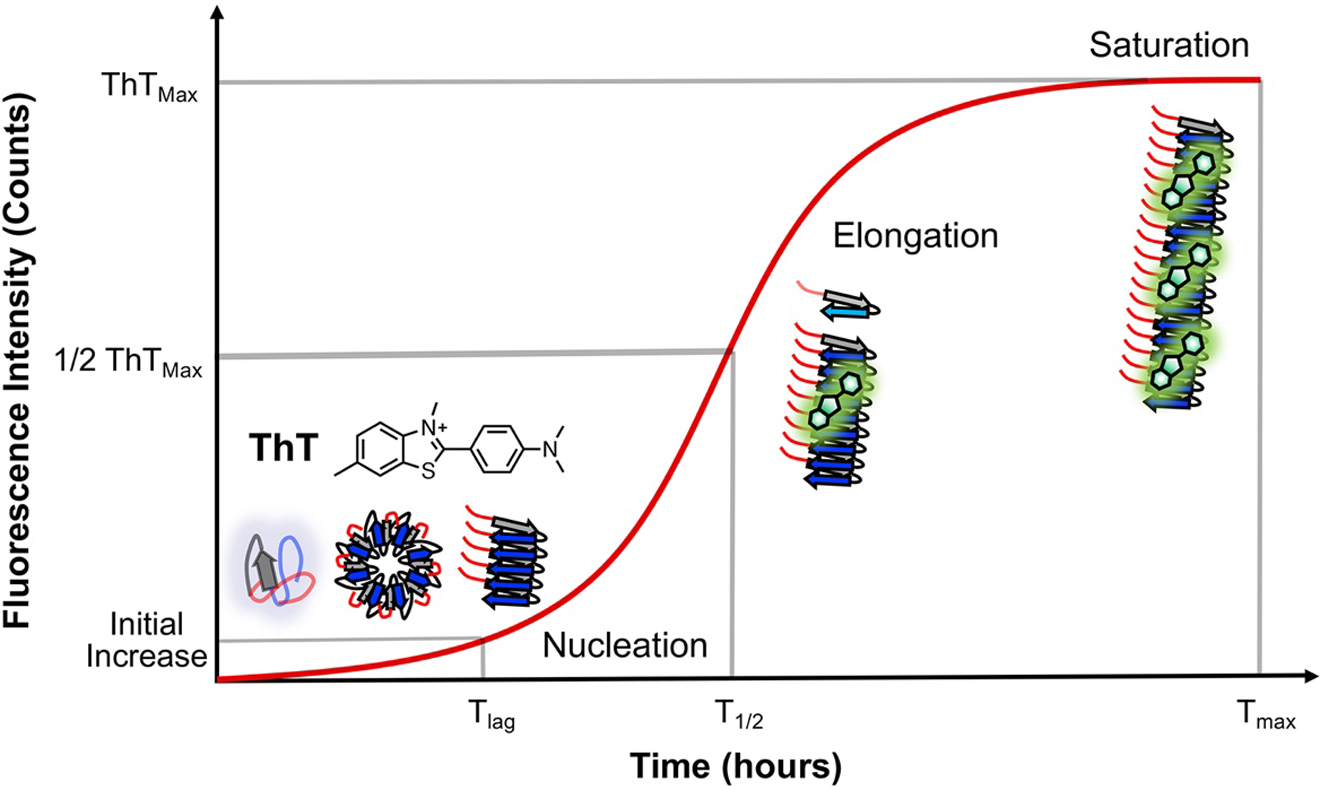
Use of ThT to monitor aggregation. ThT emission changes are shown with major αS conformational states populated at each stage. The timepoints of half maximal ThT fluorescence (T1/2) are used to determine the effects of mutations and PTMs on αS aggregation.
Aggregation progress, as reported by ThT fluorescence, is generally sigmoidal and can be described in terms of three phases (Figure 3) [3, 44]. The first phase, the lag phase, is characterized by a lag time, Tlag, which represents the amount of time required to detect an initial change in fluorescence. The second phase, or elongation phase, exhibits exponential growth due to addition of monomer to fibril ends as well as fragmentation of fibrils to increase the number of fibril seeds onto which monomeric αS can be added. T1/2, or the time to reach half of the maximum fluorescence, occurs during the middle of the elongation phase. The final, or saturated, phase occurs when monomer is depleted such that net growth of fibrils is very slow or non-existent. Tmax is the time to reach maximum fluorescence.
While these three phases may seem separate, this is largely a result of the low sensitivity and lack of specificity of ThT fluorescence. Primary and secondary nucleation occur during all phases, with differences between phases resulting from the relative rates of primary and secondary nucleation [45]. The overall rate of fibrillization, as measured by fitting the sigmoidal ThT curve, is a function of the initial concentration of both monomer and seeds. Seeds are able to accelerate the rate of fibril formation by skipping the primary nucleation event and shortening Tlag [36]. Modifications and interactions of αS that impact aggregation rate as studied by ThT fluorescence include mutations, post-translational modifications (PTMs), small molecules, membranes, surfaces, salt, pH, temperature, agitation, and other peptides and proteins [45]. It is important to note that the conditions under which aggregation experiments are performed can significantly impact the observed effects. Therefore, one must always conduct an experiment with the WT protein under identical conditions for comparison. Our analysis of the effects of aggregation conditions (see Supporting Information) among experiments on the same mutant indicates that while the magnitude of a mutational effect may vary somewhat, it is generally robust to changes in conditions when normalized to a corresponding WT aggregation.
While ThT is very useful for monitoring aggregation, it is worth discussing some of the drawbacks to these assays. ThT has been shown to bind preferentially to certain fibril polymorphs, even if the polymorphs have similar β-sheet content [46]. This may happen because the distribution of ThT binding sites may be different for fibrils with different structure. In terms of interpreting ThT data, overall fluorescence cannot be taken as an indicator of fibril concentration [47, 48]. Additionally, when ThT is incubated with monomeric αS as in plate reader assays, it may impact the resulting fibril structure. ThT may bind to early nuclei or αS monomer and stabilize structures that are aggregation-prone. By stabilizing certain intermediates and structures, ThT may preferentially promote certain fibril pathways over others [49]. ThT is also known to increase the rate of fibril formation. This may occur because of the stabilization of intermediates or because of ThT’s interactions with the C-terminal domain which interrupt intermolecular interactions that protect αS from aggregating [43, 49]. While not discussed in this review, ThT is often used to monitor the impact of inhibitors on the rate of αS. ThT and certain types of inhibitors may compete for binding sites or inhibitors may quench ThT fluorescence, making ThT an unreliable reporter of certain inhibitors [43].
A few modifications to ThT assays have been suggested to increase the reproducibility and to mitigate some of these shortcomings. ThT’s impact on αS fibril structure and aggregation rate is dependent on the concentration of ThT. At concentrations below 30 μM, these effects are diminished [49]. Choosing the timepoint assay as opposed to the plate reader assay also reduces ThT’s impact on aggregation as ThT is not able to interfere with monomeric αS or nuclei during aggregation [43]. Additionally, primary nucleation is a stochastic process, meaning that observed lag times are subject to some variation [37]. Bypassing or reducing the need for primary nucleation through seeded aggregation, the addition of SDS, and agitation of samples produces more reproducible results [37, 50]. Small beads have also been added to plate reader assays to accelerate aggregation by inducing fibril breakage and secondary nucleation [50] or by increasing the size of the hydrophobic interface [51]. In spite of the noted concerns with ThT assays and the alternatives, the ubiquity of data collected under relatively consistent conditions still makes it the assay of choice for a comprehensive review such as this.
Fibril Structure and Morphology
αS is able to form multiple “strains” of fibrils with different morphologies, toxicities, and seeding capacities. The strains hypothesis is important in understanding the difference between synucleinopathies, as fibrils derived from patients with different synucleinopathies have unique structural and biochemical characteristics [52, 53]. Oligomers formed under different conditions were shown to have distinct morphologies, impacts on intracellular calcium levels, seeding abilities, and membrane permeabilities [26]. Bousset et al. found that different in vitro aggregation conditions led to two distinct strains, termed “fibrils” and “ribbons,” that had different structures and toxicities [46]. In rat models, injection of ribbons or fibrils induced different histopathological and behavioral phenotypes [54]. In these studies, distinct strains have been able to recruit WT monomer to form fibrils of the same strain. However, new evidence seems to show that solution or cellular conditions of fibril formation also have an impact on fibril morphology. MSA inclusions, called glial cytoplasmic inclusions (GCIs), are located in oligodendrocytes. Lewy Bodies, which are located in neurons, were injected into oligodendrocytes in mice. The resulting inclusions were immunohistochemically characterized as GCIs rather than Lewy Bodies, indicating that strains may not always propagate under different cellular conditions [53]. Under solution conditions different from a seed’s creation, the seed may be unstable, making it unable to trigger secondary nucleation [55].
Solid state NMR (ssNMR) and cryo-electron microscopy (cryo-EM) have proven useful in understanding the structures of αS fibrils (Figure 2). The first fibril structure was solved using ssNMR (2n0a), and established the Greek Key motif (formed by residues 36–100) that has been observed in many of the subsequent structures [56]. Using cryo-EM, research groups have solved the structure of full-length WT [57, 58], C-terminally truncated WT [59], N-terminally acetylated WT [60], G51D [61], H50Q [62], acetylated A53T [63], and both acetylated and non-acetylated E46K αS (Figure 8) [64, 65]. All of the cryo-EM structures feature two protofilaments comprised largely of β-strands with a steric zipper at the interface between the protofilaments. While these structures differ slightly in helical rise and twist, the biggest differences are the interfaces between the two protofilaments, electrostatic interactions among sidechains, and the orientation of the β-sheets. A detailed analysis of these structures is found in the Discussion section.
Figure 8.
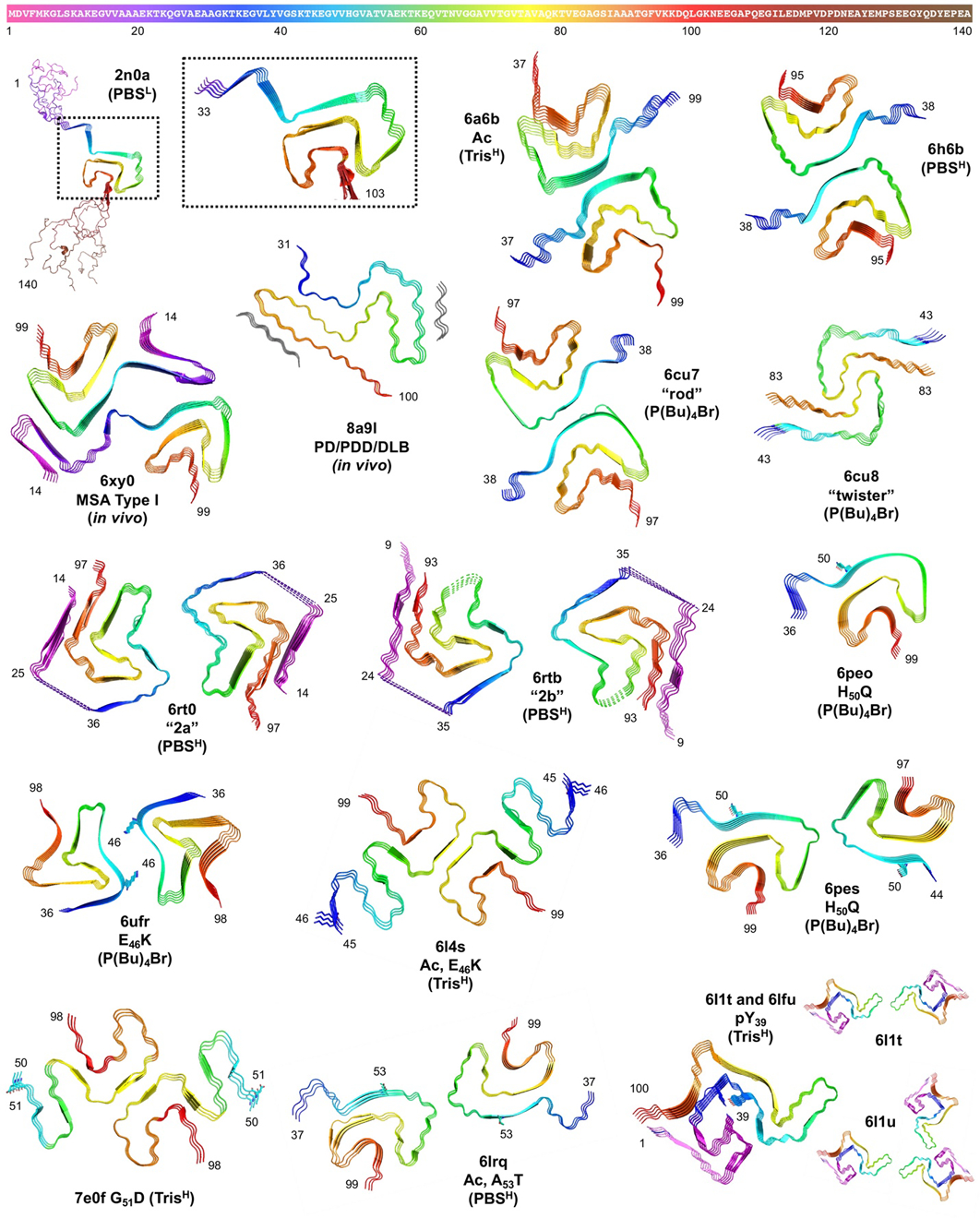
Top: αS sequence colored as in structures below and Figures 2, 4, 6, and 7. Bottom: Fibril structures from ssNMR (2n0A) and cryo-EM (all other PDB IDs) with αS modifications, aggregation conditions (L = low salt, H = high salt), and key residues labeled and shown as sticks. 6l1t and 6l1u share a common fold with two- or three-stranded fibril packing.
Modifications
The SNCA gene that encodes αS is subject to a number of mutations that are linked to inherited forms of PD such as A30P, A53T, A53E, H50Q, G51D, and A53V [66–73]. These mutants have different average ages of onset than sporadic PD, with A30P, E46K, A53T, A53E, A53V, and G51D leading to early-onset PD and H50Q leading to late-onset PD [73, 74]. Additionally, familial mutants have different biophysical properties like aggregation rate [75, 76], lipid binding [77], and fibril structure as compared to WT αS [63, 65, 71, 78–80]. Importantly, these mutant strains have shown variation in their ability to cross-seed, or recruit monomers of different isoforms to form fibrils [81, 82]. Although these inherited forms of PD account for less than 10% of total cases of PD, these mutations have been instrumental in understanding the aggregation pathway of αS and the importance of specific residues to the normal function of αS. In addition to familial mutations in SNCA, two sporadic mutations, A18T and A29S, have been discovered and characterized as having different biophysical properties than WT αS [83, 84].
αS also undergoes a number of PTMs which have an impact on toxicity and aggregation rate (Figure 4). The most common PTM in patient derived Lewy Bodies is phosphorylation, specifically at Ser129. Phosphorylation at Ser129 (pS129) is viewed as a hallmark of Lewy Bodies, occurring in more than 90% of Lewy Bodies as compared to around 4% of αS under physiological conditions [85–87]. N-terminal acetylation is also a common feature of mammalian αS [86]. Other PTMs that are less prevalent include ubiquitination of lysine [88], nitration of tyrosine [89], the addition of small ubiquitin-like modifiers (SUMOylation) to lysine [90], N-acetylglucosamine modification (O-GlcNAcylation) of serine and threonine [91, 92], glycation of lysine [93], adenylation of serine, threonine, and tyrosine [94], methionine oxidation [95], modification by the lipid peroxidation product 4-hydroxy-2-nonenal (HNE) [96], glutamate arginylation [97], and truncation by proteolysis [4, 86].
Figure 4.
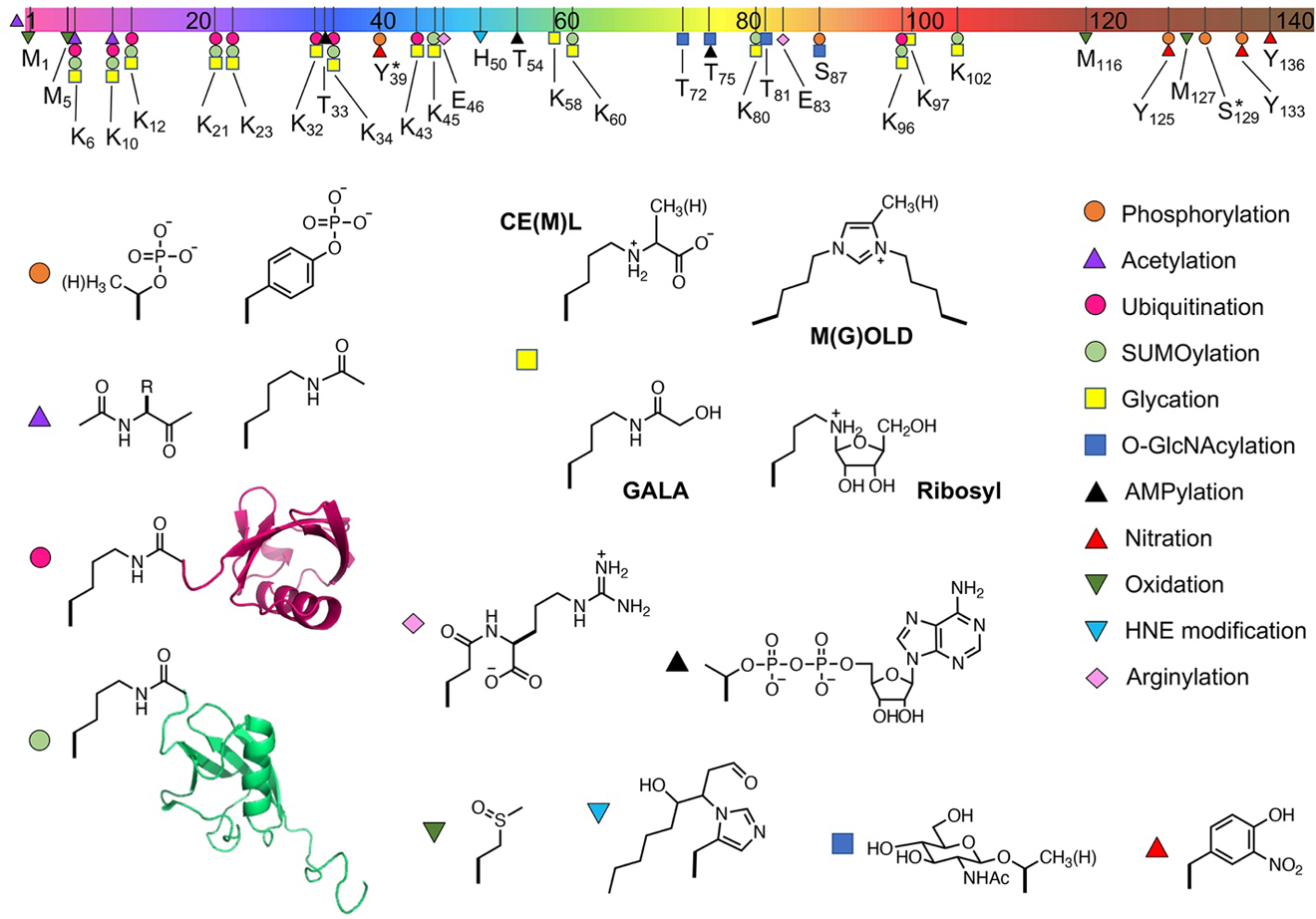
PTMs on αS with characterized effects on aggregation. Top: αS sequence with segments colored as in Figure 2 and locations of PTMs noted. No PTMs occur simultaneously at a given site, but multiple PTMs can occur at different sites in the same αS monomer. Bottom: Structures of PTMs. All PTMs studied to date have either had no effect on aggregation or slowed aggregation when present in 100% of αS monomers. Effects on aggregation rates are summarized in Table 2. Some PTMs exert different effects when present at lower percentages (e.g., pY39) mixed with WT, indicated by a *. CML: N(ε)-(carboxymethyl)lysine; CEL: N(ε)-(carboxyethyl)lysine; GOLD: glyoxal-lysine dimer; MOLD: methylglyoxal-lysine dimer; GALA: N6-(glycoloyl)lysine
Generally, in vitro studies use bacterially overexpressed αS. However, protein produced with this method lacks PTMs, even N-terminal acetylation unless acetyltransferase NatB is co-expressed [98]. Naturally occurring PTMs are installed through a variety of processes and may involve small molecules or enzymes. PTMs with the potential to occur at multiple residues are ideally studied using αS generated using native chemical ligation (NCL) so that the PTM occurs only at specific sites (Figure 5). These methods have been used to study nitration [99], phosphorylation [100–102], ubiquitination [103–105], SUMOylation [106, 107], O-GlcNAcylation [108], arginylation,[109] and N-terminal acetylation [110]. Given that these procedures can be synthetically intensive, limiting the amounts of protein available and in vivo applications, PTMs have also been studied by nonspecific modification at multiple residues or by using mutant “mimics” that best represent the physico-chemical properties of a PTM from among the natural amino acids. For example, a serine, threonine or tyrosine phosphorylation mimic might be substitution with a negatively charged amino acid like Asp or Glu [111]. While these substitutions are imperfect, they provide some insight into the role of PTMs on αS aggregation.
Figure 5.
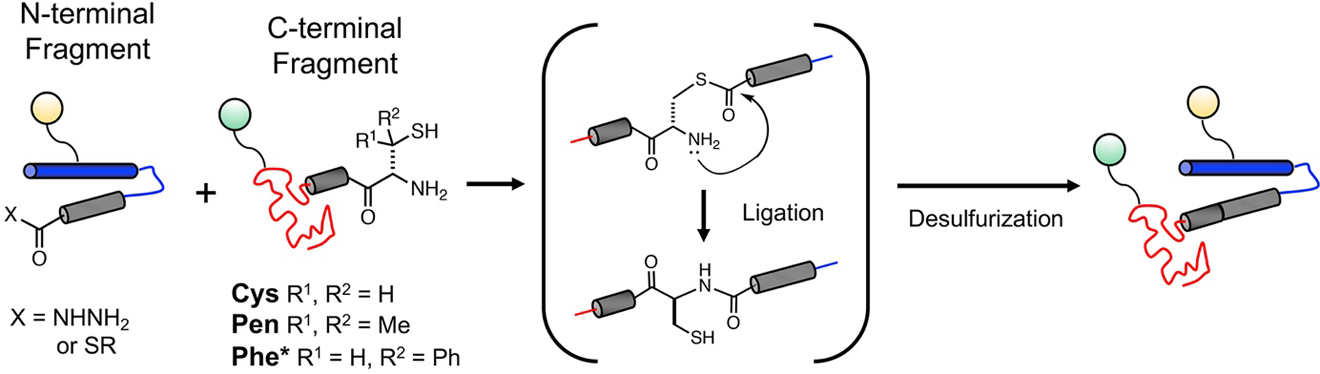
Synthesis of αS containing PTMs through NCL. N- and C-terminal fragments can be generated with PTMs at specific sites through solid phase peptide synthesis or protein expression and enzymatic modification. After ligation, Cys or Cys analogs can be desulfurized to make the ligation “traceless.” PTMs are indicated by colored spheres.
Summary of Modification Data
The following sections present characterization of the effects of mutations and PTMs on αS aggregation rates as measured by ThT fluorescence and, in a few cases, Congo Red absorbance. The mutational data are summarized in Table 1, the PTM data are summarized in Table 2, and all data are provided in a spreadsheet format in Supplementary Information. Aggregation rates for mutants, T1/2(Mut), mutants are scored by qualitatively comparing the relative rate, T1/2(Mut), to the corresponding WT control, T1/2(WT), as follows:
Table 1.
Summary of mutation effects on αS aggregation
| Position | Mutant | Rate | Aggregation Conditions | Ref. |
|---|---|---|---|---|
| Val3 | V3W | 0 | 20 mM Gly-NaOH, 0.01% NaN3, pH 7.4, 300 μM, 50 rpm | [118] |
| Met5 | M5L | −1 | 20 mM Tris-HCl, 100 μM NaCl, pH 7.5, 35 μM αS, 600 rpm | [154] |
| Lys10 | K10Q | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Lys12 | K12E | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu13 | E13K | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| Val15 | V15E | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val16 | V16P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala17 | A17T | 1 | 20 mM sodium phosphate buffer, 0.1% NaN3, pH 7.4, 300 μM αS, 300 rpm | [84] |
| Ala18 | A18P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| A18T | 1 | 20 mM sodium phosphate buffer, 0.1% NaN3, pH 7.4, 300 μM αS, 300 rpm | [84] | |
| Ala19 | A19T | 1 | 20 mM sodium phosphate buffer, 0.1% NaN3, pH 7.4, 300 μM αS, 300 rpm | [84] |
| Lys21 | K21Q | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Thr22 | T22P | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Lys23 | K23E | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Gln24 | Q24K | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| Q24P | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Val26 | V26P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala27 | A27P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala29 | A29P | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| A29S | 1 | 20 mM sodium phosphate buffer, 0.1% NaN3, pH 7.4, 300 μM αS, 300 rpm | [84] | |
| Ala30 | A30Pa | −1 | PBS, pH 7.4, 250 μM αS, “slight agitation” | [25] |
| Lys32 | K32Q | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu35 | E35K | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| E35Q | 1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Gly36 | G36N | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val37 | V37E | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| V37P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Leu38 | L38P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Tyr39 | Y39A | −2 | 100 mM phosphate buffer, 100 mM NaCl, pH 7.4, 72 μM αS, 600 rpm | [161] |
| Y39E | −2 | 31 mM Tris, 100 mM NaCl, pH 7.4, 100 μM αS, 1300 rpm | [102] | |
| Val40 | V40P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Lys45 | K45E | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| K45R | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Glu46 | E46K | 1 | PBS, pH 7.4, 250 μM αS, “slight agitation” | [25] |
| Gly47 | G47Q | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val48 | V48P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val49 | V49P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| His50 | H50A | 1 | PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [144] |
| H50D | 1 | PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [144] | |
| H50Q | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] | |
| H50R | −2 | PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [144] | |
| Gly51 | G51D | −1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| Val52 | V52P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala53 | A53E | −1 | 10 mM sodium phosphate, 0.02% NaN3, pH 7, 60 μM αS, 600 rpm | [132] |
| A53K | −1 | 20 mM Glycine-NaOH buffer, 0.01% NaN3, pH 7.4, 200 μM αS, 50 rpm | [124] | |
| A53T | 1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| A53V | 1 | 20 mM Glycine-NaOH buffer, 0.01% NaN3, pH 7.4, 300 μM αS, 20 rpm | [76] | |
| Ala56 | A56P | −1 | 50 mM sodium phosphate, 100 mM NaCl, 0.01% NaN3, pH 7.4, 100 μM αS, 200 rpm | [131] |
| Glu57 | E57K | 1 | PBS, pH 7.4, 250 μM αS, agitation | [25] |
| Thr59 | T59P | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Lys60 | K60Q | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu61 | E61K | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| Val63 | V63E | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| V63G | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| V63P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Thr64 | T64P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Asn65 | N65P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val66 | V66P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| V66R | −2 | PBS, 0.05% NaN3, pH 7, 210 μM αS, shaking | [150] | |
| V66S | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Gly67 | G67P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Gly68 | G68A | 1 | PBS, 0.05% NaN3, pH 7, 210 μM αS, shaking | [150] |
| G68E | −2 | PBS, 0.05% NaN3, pH 7, 210 μM αS, shaking | [150] | |
| G68P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| G68R | −2 | PBS, 0.05% NaN3, pH 7, 210 μM αS, shaking | [150] | |
| Ala69 | A69P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val70 | V70E | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| V70G | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| V70P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| V70T | −1 | 10 mM Tris-HCl, 0.02% NaN3, pH 7.4, 140 μM αS, shaking | [122] | |
| Val71 | V71W | 0 | 20 mM Gly-NaOH, 0.01% NaN3, pH 7.4, 300 μM, 50 rpm | [118] |
| Thr72 | T72A | 1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| T72E | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| T72K | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| T72P | −2 | 100 mM sodium acetate buffer, pH 7.4, 350 μM αS, 1050 rpm | [137] | |
| Gly73 | G73P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val74 | V74E | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| V74G | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| V74R | −2 | PBS, 0.05% NaN3, pH 7, 210 μM αS, shaking | [150] | |
| Thr75 | T75A | −1 | PBS, 140 μM αS, shaking, glass bead | [152] |
| T75K | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| T75P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Ala76 | A76G | 0 | 100 mM sodium acetate, pH 7.4, 350 μM, shaking | [138] |
| A76P | −1 | 50 mM sodium phosphate buffer, 100 mM NaCl, 0.01% NaN3, pH 7.4, 100 μM αS, 200 rpm | [131] | |
| A76V | −1 | 100 mM sodium acetate, pH 7.4, 350 μM, shaking | [138] | |
| Ala78 | A78T | −1 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Lys80 | K80Q | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Thr81 | T81P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val82 | V82K | 0 | 10 mM Tris-HCl, 0.02% NaN3, pH 7.4, 140 μM αS, shaking | [151] |
| Glu83 | E83K | 1 | PBS, pH 7.3, 70 μM αS, 500 rpm | [140] |
| E83P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] | |
| Ala85 | A85P | −1 | 100 mM sodium acetate, pH 7.4, 350 μM, shaking | [138] |
| Ser87 | S87A | 0 | 10 mM Tris, pH 7.4, 100 μM αS, shaking | [164] |
| S87D | −2 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [187] | |
| S87E | −2 | 10 mM Tris, pH 7.4, 100 μM αS, shaking | [164] | |
| S87K | 1 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [187] | |
| S87W | 1 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [187] | |
| Ala89 | A89P | −2 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala90 | A90P | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Gly93 | G93S | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Val95 | V95S | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Asn103 | N103K | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu104 | E104A | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu105 | E105A | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Pro108 | P108A | 1 | 20 mM HEPES, 150 mM NaCl, 0.02% NaN3, pH 7.4, 70 μM αS, 270 rpm | [159] |
| Asp115 | D115A | 0 | 20 mM Tris, pH 7.2, 10 μM αS, 300 rpm | [117] |
| Met116 | M116L | −1 | 20 mM Tris-HCl, 100 μM NaCl, pH 7.5, 35 μM αS, 600 rpm | [154] |
| Pro117 | P117A | 1 | 20 mM HEPES, 150 mM NaCl, 0.02% NaN3, pH 7.4, 70 μM αS, 270 rpm | [159] |
| Asp119 | D119A | 0 | 20 mM Tris, pH 7.2, 10 μM αS, 300 rpm | [117] |
| Pro120 | P120A | 1 | 20 mM HEPES, 150 mM NaCl, 0.02% NaN3, pH 7.4, 70 μM αS, 270 rpm | [159] |
| Asp121 | D121A | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Glu123 | E123A | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Ala124 | A124W | 0 | 20 mM Gly-NaOH, 0.01% NaN3, pH 7.4, 300 μM, 50 rpm | [118] |
| Tyr125 | Y125A | −1 | 100 mM phosphate buffer, 100 mM NaCl, pH 7.4, 72 μM αS, 600 rpm | [161] |
| Y125E | −2 | 20 mM Tris-HCl, pH 7.4, 50 μM αS, 270 rpm | [162] | |
| Y125F | −2 | 20 mM Tris-HCl, pH 7.4, 50 μM αS, 270 rpm | [162] | |
| Y125W | −1 | 30 mM Tris, 100 mM NaCl, pH 7.2, 70 μM αS, 600 rpm | [160] | |
| Met127 | M127L | −1 | 20 mM Tris-HCl, 100 μM NaCl, pH 7.5, 35 μM αS, 600 rpm | [154] |
| Ser129 | S129A | 0 | 20 mM Tris-HCl, pH 7.4, 50 μM αS, 270 rpm | [162] |
| S129D | 0 | 20 mM Tris-HCl, pH 7.4, 50 μM αS, 270 rpm | [162] | |
| S129E | 0 | 20 mM Bis-Tris propane, 100 mM LiCl, pH 7.4, 100 μM αS, shaking | [111] | |
| Glu130 | E130A | 0 | PBS, pH 7.4, 150 μM αS, 150 rpm | [129] |
| Tyr133 | Y133A | −2 | 100 mM phosphate buffer, 100 mM NaCl, pH 7.4, 72 μM αS, 600 rpm | [161] |
| Tyr136 | Y136A | −1 | 100 mM phosphate buffer, 100 mM NaCl, pH 7.4, 72 μM αS, 600 rpm | [161] |
| Y136E | −2 | 25 mM Tris-HCl, 1 mol NaCl, pH 7.5, 70 μM αS, 170 rpm | [156] | |
| Y136F | −1 | 25 mM Tris-HCl, 1 mol NaCl, pH 7.5, 70 μM αS, 170 rpm | [156] | |
| Y136L | −2 | 25 mM Tris-HCl, 1 mol NaCl, pH 7.5, 70 μM αS, 170 rpm | [156] | |
| Y136S | −2 | 25 mM Tris-HCl, 1 mol NaCl, pH 7.5, 70 μM αS, 170 rpm | [156] | |
| Y136W | 0 | 25 mM Tris-HCl, 1 mol NaCl, pH 7.5, 70 μM αS, 170 rpm | [156] | |
| Ala140 | A140C | 1 | 6 mM phosphate, 9 mM NaN3, 0.1 mM EDTA, 1 mM DTT, pH 7.4, 70 μM αS, 1000 rpm | [157] |
| A140W | 0 | 20 mM Gly-NaOH, 0.01% NaN3, pH 7.4, 300 μM, 50 rpm | [118] |
There is significant disagreement in the literature on the effect of the A30P mutation on aggregation.
Table 2.
Summary of PTM effects on αS aggregation
| Position | Mutant | Rate | Aggregation Conditions | Ref. |
|---|---|---|---|---|
| N-terminus | Ac-αSa | 0 | PBS, 137 mM NaCl, pH 7.4, 140 μM αS, 600 rpm | [168] |
| Lys6 | K6Ub | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys10 | K10Ub | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys12 | K12Ub | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys21 | K21Ub | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys23 | K23Ub | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys32 | K32Ub | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Lys34 | K34Ub | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Tyr39 | nY39 | −1 | 50 mM Tris, 150 mM NaCl, pH 7.5, 10 μM αS, 1000 rpm | [99] |
| pY39 | −2 | 20 mM Tris, 100 mM NaCl, pH 7.4, 100 μM αS, 1300 rpm | [102] | |
| Lys46 | K46Ub | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] |
| Thr72 | gT72 | −2 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [189] |
| Thr75 | gT75 | −2 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [189] |
| Thr81 | gT81 | −2 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [189] |
| Ser87 | gS87 | −2 | 10 mM phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [187] |
| pS87, S129Ab | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [164] | |
| Lys96 | K96 SUMO1 | −2 | 10 mM sodium phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [178] |
| K96 SUMO3 | −1 | 10 mM sodium phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [178] | |
| K96Ub | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [103] | |
| Lys102 | K102 SUMO1 | −2 | 10 mM sodium phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [178] |
| K102 SUMO3 | −2 | 10 mM sodium phosphate, 0.05% NaN3, pH 7.4, 50 μM αS, 1000 rpm | [178] | |
| Tyr125 | nY125 | −2 | 50 mM Tris, 150 mM NaCl, pH 7.5, 10 μM αS, 1000 rpm | [99] |
| pY125 | −2 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [162] | |
| Ser129 | pS129 | −1 | 10 mM PBS, 0.05% NaN3, pH 7.4, 100 μM αS, shaking | [162] |
There is significant disagreement in the literature on the effect of N-terminal acetylation on aggregation.
While all other mutations and PTMs are presented as single site modifications, pS87 is included with the accompanying S129A mutation since those were the only data available. It is scored as −1 using the S129A aggregation rate as a pseudo-WT control to isolate the effect of Ser87 phosphorylation.
+1 for T1/2(Mut) < 0.5 T1/2(WT)
0 for 0.5 T1/2(WT) ≤ T1/2(Mut) ≤ 2 T1/2(WT)
−1 for 2 T1/2(WT) < T1/2(Mut) < 4 T1/2(WT)
−2 for T1/2(Mut) > 2 T1/2(WT) or very shallow, non-sigmoidal aggregation curve
The mutation T1/2 data are also summarized graphically in Figure 6, and PTM and labeling data are presented in Figure 4 and Figure 7, respectively. For cases in which multiple studies reported aggregation rates, the most representative data (based on inclusion in a larger study with other mutants for benchmarking, a more common buffer system, consistency with other reports, etc) are included in Table 1 and Table 2, but all studies are included in Supplementary Information. For the familial mutations, aggregation rates have been measured by many independent studies, which are compared in the discussion below, but only one value is included in Table 1 for clarity. Studies in which multiple mutations or PTMs are introduced simultaneously are discussed, but only data for single site modifications are included in the figures or in Table 1 or Table 2. Finally, cases in which the modified αS was studied as a mixture with WT αS, which has been done for mutants,[112] PTMs,[102] and fluorescent labels,[113] have been omitted from Table 1 and Table 2, so all of the rate data are directly comparable, describing the effect of a single site modification present in 100% of the αS molecules.
Figure 6.
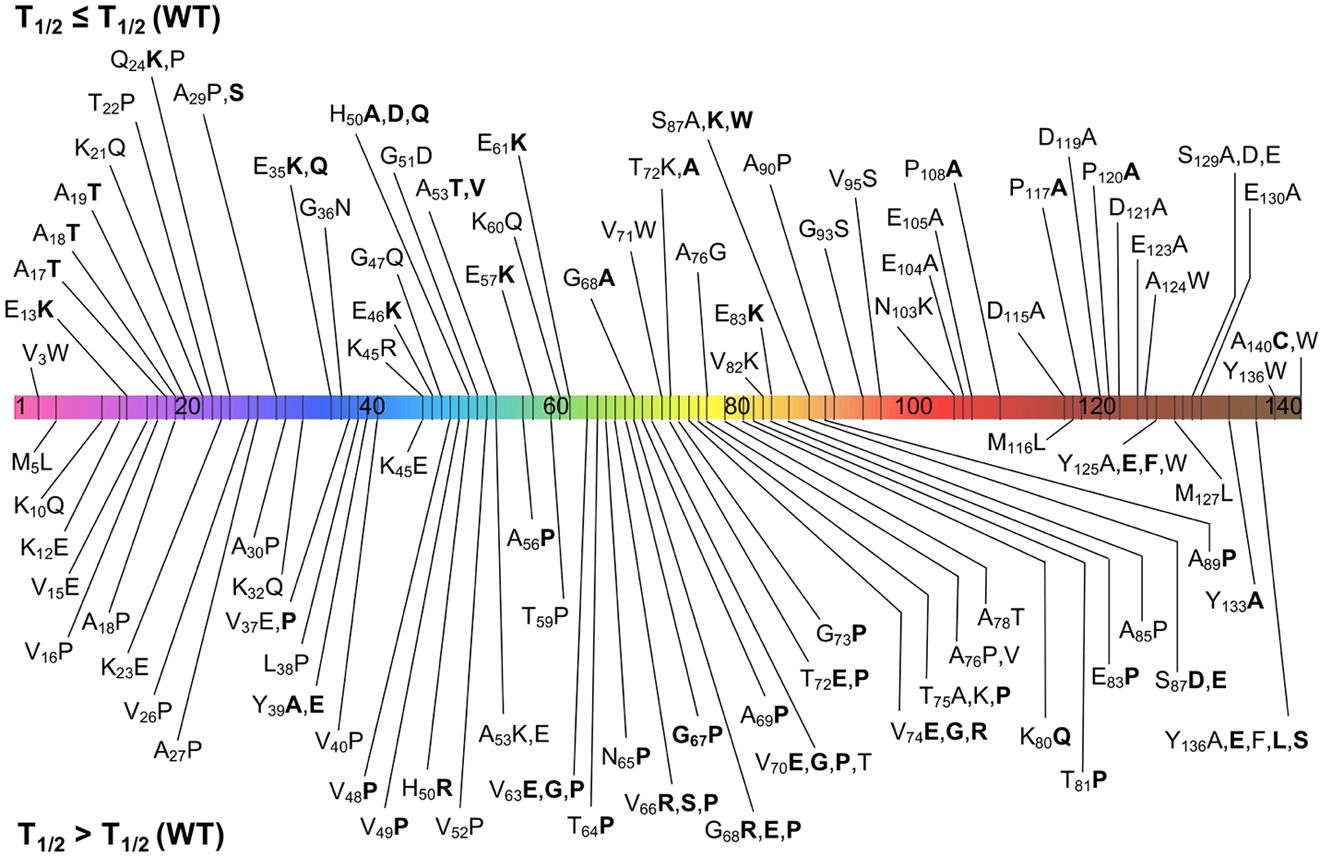
Mutations known to affect αS aggregation. αS sequence with segments colored as in Figure 2 and locations of mutations noted. Mutations above the sequence are either neutral (0 in Table 1, normal type) or accelerate aggregation (+1 in Table 1, bolded). Mutations below the sequence slow the sequence moderately (−1 in Table 1, normal type) or severely (−2 in Table 1, bolded).
Figure 7.
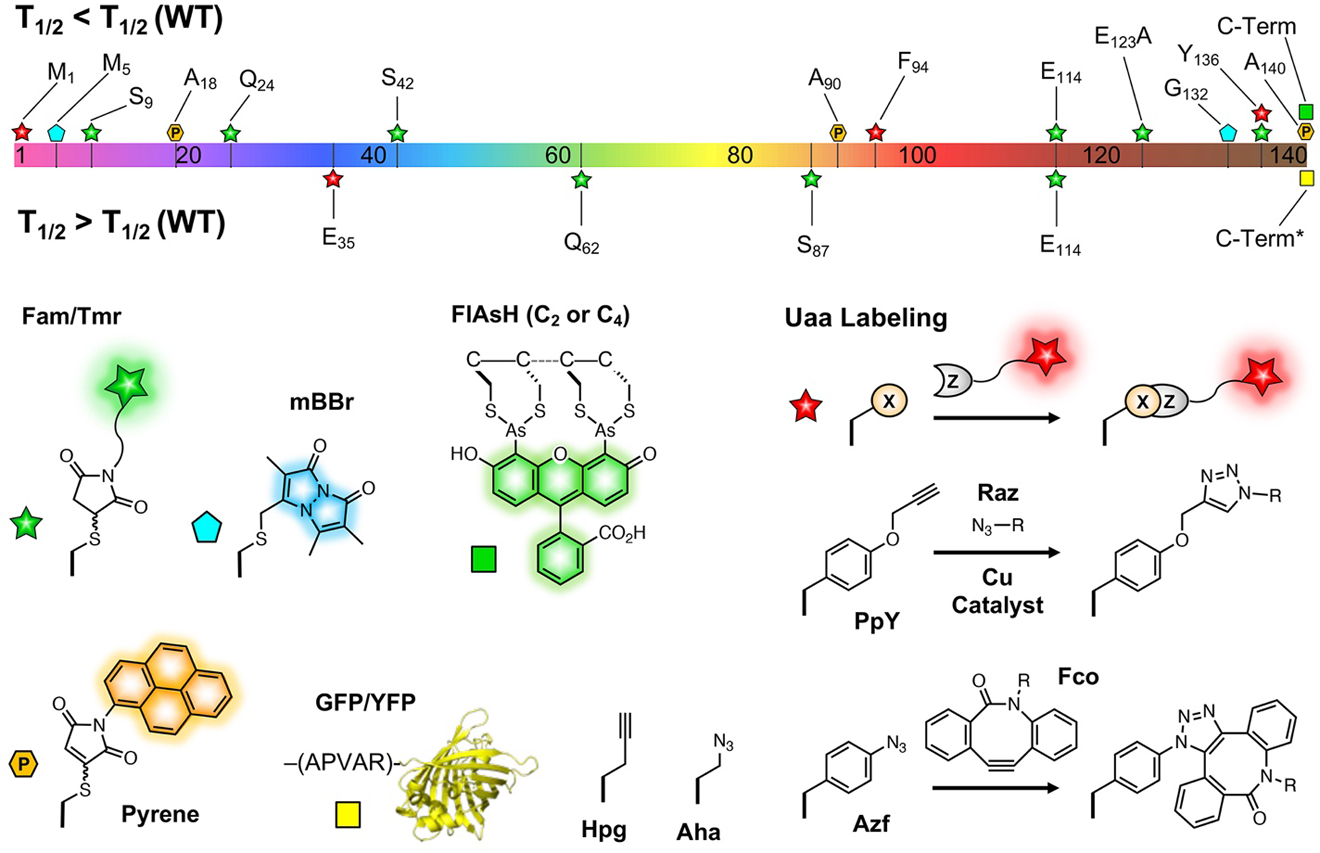
Spectroscopic labels with characterized effects on αS aggregation. Top: αS sequence with segments colored as in Figure 2 and locations of modifications noted. Modifications above the sequence are neutral. Modifications below the sequence slow aggregation moderately. *C-terminal GFP was reported to slow aggregation, C-terminal YFP with APVAR linker was reported to accelerate aggregation.
Familial and Sporadic Mutations
The A53T mutation was discovered in 1997 by Polymeropoulos et al. in a mostly Italian cohort and three unrelated Greek families. This mutation was the first familial mutation to be linked to PD [66]. Just a year later, the A30P mutation was discovered in a population in Germany [67]. The discovery and characterization of the first two familial mutants paved the way for numerous studies using mutant forms of αS as a way to understand PD pathology. Since then, five new mutations, E46K, H50Q, G51D, A53E, and A53V, have been discovered. Each of these mutations causes forms of PD that differ in their age of onset, clinical features, and neuropathological features [114]. Despite extensive characterization, the impact of these mutations on the aggregation pathway of αS is not well understood. Potential reasons for altered aggregation rates include changes in charge, hydrophobicity, long-range interactions between different areas of αS, and propensity to form secondary structures.
A53T
Since A53T was the first familial mutant to be discovered, it is also the most well-studied. In addition to in vitro assays, A53T is the most widely used mutation in mouse models of PD. The first two biophysical characterizations of A53T by Conway et al. and Narhi et al. confirmed that fibrils made in vitro from mutant αS were similar to those derived from patients. They also found that A53T αS aggregated faster than WT αS and was able to accelerate the rate of fibril formation of WT αS. Conway et al. determined that the increased toxicity of A53T was related to the increased rate of oligomer formation, emphasizing the importance of studying the rate of aggregation as one way of understanding the aggregation pathway [38, 82]. Other studies have since confirmed that A53T mutation increases the rate of fibril formation [81, 115–118].
Since its initial characterization, many groups have studied A53T using its rate of aggregation, with nearly every group agreeing that A53T increases aggregation rate relative to WT by ThT fluorescence [25, 48, 72, 76, 81, 82, 84, 94, 114, 117–130]. Two studies do state that A53T aggregates at the same rate as WT αS. Karpinar et al.’s work also points to a much lower maximum fluorescence intensity for A53T which is not consistent with other results. They propose that the gel-like behavior of aggregated A53T leads to interference with ThT binding, leading to suboptimal aggregation curve data [131]. Hoyer et al. found that their no salt preparation aggregated at the same rate as WT, however, with salt, they found that A53T aggregates faster than WT [125]. A53E and A53V are also familial mutations, discovered in 2014 and 2018, respectively. As these are newly discovered mutants, the amount of data on these mutants is limited. However, based on current research, A53V aggregates faster than WT [76], and A53E mutants aggregate slower than WT [76, 117, 124, 130, 132], with the exception of one study where A53E was shown to aggregate at about the same rate as WT [114]. The changes in aggregation rate in these three mutations can be partially explained by the introduction of amino acids with different properties. A53V’s increased hydrophobicity is thought to contribute to its oligomerization. For A53E, the substitution of a glutamate, a negatively charged amino acid, may impact the intermolecular interactions needed to stabilize β-sheets, disfavoring aggregation [76]. For A53T, the threonine is thought to introduce hydrogen bonds that stabilize the partially folded intermediate [76, 124]. To further study the importance of Ala53’s physico-chemical properties to aggregation, Ghosh et al. also created an A53K mutant which has a positive charge 76. Like A53E, this mutant delayed aggregation relative to WT, suggesting that any charged group at position 53 may inhibit fibril formation [124].
A30P
A30P is the mutant with the least agreement regarding its impact on aggregation rate. Different laboratories have found that A30P aggregates faster [48, 130, 131], at the same rate [79, 126], or slower than WT [25, 82, 114, 117–121, 124, 125, 127, 128, 133, 134]. These results are particularly striking because many of these studies also measured the aggregation rate of A53T, where the overwhelming majority of them concluded that A53T forms fibrils faster than WT, implying that their methods are consistent. One potential explanation is that A30P’s aggregation pathway is unique. A30P was shown to have different kinetic stages of aggregation as compared to WT [48]. While the other familial mutants also had different kinetic steps, it is possible that A30P’s aggregation intermediates are not amenable to ThT aggregation assays. Additionally, A30P is thought to favor a species of oligomer that takes longer to seed fibril formation as compared to WT [127]. Because ThT binds specifically to fibrils and not oligomers [135], ThT may miss this stage in A30P’s aggregation. The findings that A30P aggregates faster than WT are somewhat surprising considering that proline has a very low β-sheet propensity due to its rigid structure, although whether this region adopts a β-sheet in fibrils is debated [125]. Indeed, proline substitutions at other residues have been shown to slow or nearly prevent aggregation [122, 131, 136–138]. Ultimately, A30P was assigned a rate of −1 in Table 1 and Figure 6 since the majority of studies found it to be a moderately retarding mutation.
E46K
E46K aggregates faster than WT αS [25, 48, 117, 118, 121, 123, 126, 129, 130, 134, 139, 140]. Since this region is thought to have long-range contacts with the C-terminal domain, the increased aggregation rate as compared to WT could be indicative of a disruption to the long-range contacts in different areas of αS [134, 140, 141]. However, Rospigliosi et al. found that the E46K mutation enhances interactions with the negatively charged C-terminal region. Although such interactions have typically been viewed as stabilizing the monomer, they proposed that the increase in net charge drives faster aggregation [142]. Another possible explanation for the increased aggregation rate of glutamic acid mutants is that glutamic acid has a low β-sheet propensity, potentially reducing the probability of aggregation of WT as compared to this family of mutants [140]. In contrast, three groups found that E46K aggregates slower than WT [64, 65, 114]. Boyer et al. hypothesize that the E46K mutant lacks the E46-K80 salt bridge found in WT αS fibrils (Figure 2), leading to a different, slower misfolding pathway. Their slower aggregation rate may also be explained by the use of tetrabutylphosphonium bromide as their aggregating buffer, which is unique among the set of experiments reviewed here [65]. Zhao et al. similarly found that N-terminally acetylated E46K αS aggregated slower than WT. Similar to Boyer et al., their cryo-EM structure (Figure 2) points to altered electrostatic interactions that lead to a different fibril folding pathway [64].
Glu46 is located in the fourth imperfect repeat of αS (Figure 2). Glutamic acid is located at similar positions in the other repeats with the exceptions of repeat five which has a glutamine and repeat six which is poorly conserved. The repeats have been shown to protect against fibril formation [143]. In order to investigate the role of this position and the imperfect repeats more broadly on the rate of αS aggregation, multiple groups have mutated these glutamic acid residues (and the lone glutamine residue). Harada and colleagues looked at this position in every imperfect repeat. They created mutants E13K, E35K, E46K, E61K, and E83K, as well as Q24K. All of these mutants also aggregated faster than WT [140].
H50Q
The H50Q familial mutation has been shown to increase the rate of aggregation [75, 114, 117, 123, 128, 130, 141, 144]. There are two exceptions: Khalaf et al.’s preparation at 45 μM [128], and Rutherford et al.’s preparation at 5 mg/ml (340 μM) [75], which aggregated at about the same rate as WT. However, these data points appear to be anomalous since both groups did their aggregation assays at multiple concentrations, and for each of their assays done with lower concentrations of αS, H50Q aggregated faster than WT. His50 is the only histidine present in the protein and is known to associate with copper [145], meaning that mutations to His50 may be toxic because of changes in binding to copper or other metals. This residue is also the site of HNE modification [146]. Because of His50’s known interactions and modifications, multiple groups have attempted to characterize it by creating non-familial mutations. Ghosh et al. created an H50A mutation that aggregated faster than WT much like the H50Q mutation [123]. Xiang and colleagues studied mutant H50R which suppressed aggregation [147]. Chi et al. characterized mutations H50A and H50R in addition to a new mutant, H50D, designed to test the impact of negative charge at His50. The H50A and H50D mutations both accelerated fibril formation relative to WT but were slower than H50Q. Given that WT αS has a positively charged histidine at residue 50 and that arginine is positively charged, it seems as though positively charged residues at position 50 have the ability to suppress aggregation [144].
G51D
The impact of the G51D mutation on aggregation is not entirely clear, with various reports showing that the rate of aggregation is faster [114], the same [72, 80], and slower than WT [75, 117, 130, 141, 148]. Both Ranjan et al. and Stephens et al., who reported that G51D aggregates slower, found that the mutation strengthened long-range interactions between the αS N- and C-termini [117, 141]. In contrast, Fares et al., who also reported slower aggregation, did not find a perturbation in long-range contacts [148]. Another explanation for the slower aggregation of G51D may be related to the structure of its aggregates and their affinity for ThT. Fares et al. found that early aggregates are ThT-negative but readily sediment, indicating that these oligomers may be off-pathway [148]. Hayakawa et al. and Ruf et al. also suggest that ThT may not bind to early G51D oligomers [80, 114]. One group found that G51D aggregates faster than WT. They explain this result by G51D’s reduced affinity for lipid vesicles, which would increase the concentration of unbound αS, leading to faster aggregation [114]. However, Fares et al. also found that G51D had reduced lipid binding but that G51D still aggregated slower than WT [148]. The different reported rates of aggregation of G51D are likely due to differences in aggregation conditions. However, given that results with slower rates were obtained with a range of concentrations, multiple pH values, and also with Lys114 and sedimentation as aggregation reporters, G51D clearly aggregates slower than WT under most conditions.
Sporadic Mutations
In addition to the familial mutations, two mutations that may be associated with sporadic PD, A18T and A29S, were discovered. Each of these mutations were found in a single PD patient out of a cohort of 629 patients and were absent in healthy patients. Because of the small sample size, it is not clear if these mutations are related to PD pathology. However, these two mutations are the first two potentially sporadic mutations identified, underscoring the importance of point mutations in PD pathology [83]. These mutants are also interesting because of their location. They occur within the imperfect repeats and fall on the same face of the α-helix when αS undergoes a conformational change upon lipid binding. Additionally, these mutants are different than the familial mutants (except for A30P) in that they are located in the first α-helical segment of αS. Both A18T and A29S mutants increased the rate of αS aggregation [84, 130]. To better understand this region, two additional mutations, A17T and A19T, were generated. Both of these mutants aggregated faster than WT but slower than the neighboring A18T, suggesting that Ala18 has more of an influence on αS aggregation. In addition to these two mutants, an A18P mutant was found to aggregate faster than WT and at a comparable rate to the A18T mutation [84],128.
Systematic Mutational Studies
Despite only differing from WT by a single residue, the familial mutations have a significant impact on αS’s aggregation and toxicity. As a result, many groups have made other mutants of αS to study the effects on aggregation, the most notable being Koo and colleagues’ series of systematic mutations at positions throughout αS [129]. These mutants are essential to understanding the role of charge, hydrophobicity, and secondary structure in the different domains of αS. The charge of each region of αS plays a role in aggregation rate (Figure 2). The N-terminal region has a net charge of +4 while the NAC region, which is largely hydrophobic, has a charge of −1. The C-terminal region has many Asp and Glu residues, resulting in a charge of −12 [149].
N-Terminus
The impact of charge appears to be most pronounced in the N-terminal domain. Mutations E13K, Q24K, E35Q, E35K, E46A, and E57K which increase the positive charge of the N-terminal domain also increased the rate of fibril formation [25, 129, 140]. However, mutation A53K slowed aggregation [124, 128]. Similarly, mutations K10Q, K12E, V15E, K23E, K32Q, V37E, and K45E which decreased the positive charge also decreased the rate of aggregation. Outliers include K21Q and K60Q which aggregate at around the same rate as WT [129]. Mutations at residue 50 also did not agree with these trends, as H50D and H50A aggregated faster [123, 144] and H50R aggregated slower [144]. Since residue 50 is the site of a familial mutation, metal interactions, and a PTM, factors more complex than charge alone may impact the rate of aggregation of its mutants. Mutations which introduce polar residues have varying effects. As noted above, A17T, A18T, A19T, and A29S increase the rate of aggregation [84], while mutations G36N and G47Q have no impact on the rate of aggregation [129]. Mutation K45R, which maintains the positive charge [129], and mutation V3W, which maintains the hydrophobicity of Val, do not impact the rate of aggregation [118].
NAC Region
In the NAC region, hydrophobicity is thought to play a role in fibril formation. Mutations V63E, V66R, G68E, G68R, V70E, T72E, V74R, and T75K, which swapped hydrophobic residues for charged residues, all decreased the rate of aggregation [129, 150]. In addition to the impact of charge on aggregation rate, the backbone flexibility changes for each of these mutants, which may also play a role in aggregation rate. Mutations T72K [129] and V82K [151] did not significantly alter the rate of fibril formation. V66S, V70T, A78T, and V95S, mutants that increase polarity, decreased the rate of aggregation [122, 129] while G93S does not significantly alter the rate of aggregation [129]. The results for T72A, which decreases polarity and β-branching, were mixed, with Koo et al. finding that T72A was the fastest aggregating mutant in the NAC region in their series [129] (and Kochen et al. were in agreement [152]), while Marrotta et al. found that T72A αS barely aggregates [108]. Kochen et al. also studied T75A and fond that it aggregates more slowly, framing a hypothesis for threonine mutation effects in terms of “cavities” that regulate fibril growth [152]. Differences in fibril preparation and aggregation conditions likely account for this discrepancy. NAC mutations which retain hydrophobicity have different effects on aggregation, with G68A aggregating faster than WT [150], V63G, V70G, and V74G aggregating slower than WT [129], and A76G, A76V, and V71W aggregating at the same rate as WT [118, 138]. These different effects may result from changes in backbone flexibility. A few groups made Glu to Lys mutations in the NAC region, finding that the E61K mutation aggregated at the same rate [129] or faster [140] than WT, and that the E83K mutation aggregated faster than WT [140]. Faster aggregation may occur because a Glu to Lys change disrupts long range contacts, destabilizing monomeric αS and promoting aggregation [140]. Mutant K80Q, which decreases the positive charge, inhibits fibril formation almost completely [129]. The electrostatic trends presented here are imperfect and serve as a reminder that the influence of point mutations on aggregation cannot be easily predicted based on the amino acid change and the region.
C-Terminus
The charge of the C-terminus has been extensively studied using C-terminally truncated mutants with different net charges, where increased positive charge is directly proportional to aggregation rate [153]. Hokenson et al. created a series of single, double, and triple Met to Leu substitutions at Met5, Met116, and Met127. All of these mutants aggregated slower than WT, with each additional leucine mutation decreasing the rate of aggregation even more [154]. Mutations N103K, E105A, D115A, D119A, D121A, E123A, and E130A, which decreased the overall negative charge of the C-terminal domain, did not have a significant impact on the rate of fibril formation [117, 129]. Afitska et al. made a series of mutants with an increasing number of Lys or Asp residues in the C-terminus. They showed that the more positively charged mutants aggregated faster only in low salt media, suggesting that larger overall changes in charge are needed for detectable differences in aggregation rate [155]. Izawa et al. also reduced net negative charge by mutating five or six negatively charged amino acids in the C-terminal domain to Asn. Both αS variants aggregated faster than WT, with the mutant with six substitutions aggregating faster than the mutant with five substitutions [156]. Mutations A124W, A140W, and A140C which do not change the charge of the C-terminal domain do not impact the rate of fibril formation [118, 157]. Mutation Y136C aggregates slightly slower than WT [158].
Pro Scanning
The family of variants exhibiting the most consistent effects are proline mutations. As mentioned above, proline has low β-sheet propensity. These mutants are important because the partially folded intermediate in αS’s aggregation pathway is known to have β-sheet character. Disrupting these β-sheets with proline mutations should lead to longer aggregation times [131]. Indeed, nearly every Pro mutant decreases the rate of aggregation. This effect is most prominent in the NAC region. The NAC region comprises the fibril core and is the major site of β-sheet formation during aggregation, making it particularly susceptible to Pro mutants. Many Pro mutations in the NAC region severely reduce the rate of fibril formation, and several inhibit fibril formation almost entirely [129, 137]. Outliers include A76P [131], A85P [138], and A90P [129] which aggregate just slightly slower than WT. Proline mutants in the N-terminal domain have a less dramatic effect, with some mutants aggregating slower or at the same rate as WT. Two slow aggregating mutants, A56P [131, 136] and A70P [122, 129], have inconsistent results. Both aggregate slower than WT, although different studies have come to different conclusions about the extent to which the rate is reduced as compared to WT. Mutant A18P also has inconsistent results, with Koo et al. finding that it aggregates slower than WT [129] and Kumar et al. finding that it aggregates faster than WT [84]. These discrepancies likely arise from differences in aggregation conditions.
There are five native prolines in the C-terminal domain. Meuvis et al. made three single mutations, P108A, P117A, and P120A to study the impact of these Pro residues on aggregation and structure. These three mutants aggregated faster than WT. They also made some mutants with multiple Pro to Ala substitutions, the most interesting being a mutant where all five Pro were converted to Ala. This mutant had significantly more α-helical structure than WT, suggesting that C-terminal prolines are important for maintaining its disordered structure. In addition, this mutant had an extended C-terminus which reduced the shielding of the hydrophobic NAC region, allowing it to aggregate more easily [159].
Aromatic Mutations
The final group of mutants is the tyrosine mutants. Tyr residues are important because Tyr39 and Tyr125 undergo phosphorylation, and all Tyr residues can undergo nitration and participate in the formation of di-tyrosine crosslinks. There are long-range intermolecular interactions between the C-terminal region and the central region of αS [160]. While these interactions are thought to be largely electrostatic in nature, hydrophobic interactions may also play a role. To study long-range hydrophobic interactions, Ulrih et al. mutated Tyr residues to Ala residues. Complete inhibition of fibril formation was shown in the triple mutant Y125A/Y133A/Y136A. Additionally, Y39A and Y133A inhibited fibril formation entirely, while Y125A and Y136A reduced the rate of fibril formation as compared to WT. Ulrih et al. explain these results by proposing that Tyr125, Tyr 133, and Tyr136 form a hydrophobic cluster which interacts with Tyr39, with the strongest contacts occurring between Tyr39 and Tyr133 [161]. Another potential explanation is that Ala, which is less bulky than Tyr, has a lower β-sheet propensity, potentially inhibiting the formation of β-sheets in αS’s aggregation pathway. In contrast, Izawa et al. found that Y125A, Y133A, and Y125A/Y133A αS aggregated faster than WT whereas Y136A and double and triple mutants containing Y136A aggregated slower than WT. Since Y136A seemed to be the cause of slowed aggregation, they made a series of mutations at Y136. Y136W aggregated around the same rate as WT and Y136F only slightly slower than WT, suggesting that aromaticity at this position matters. Y136E, Y136S, and Y136L all aggregated slower than WT [156]. Again, the differences between these conclusions about the rate of aggregation of the single Tyr mutants can likely be explained by differences in salt concentration. Ulrih used 100 mM NaCl while Izawa used 1 M NaCl which is a tenfold increase and well above physiological salt concentrations. Additional mutations were made at position 125, with Y125F and Y125E aggregating much slower than WT [162] and Y125W aggregating slightly slower than WT [160].
Post-Translational Modifications
Ser. Thr, and Tyr Phosphorylation
Phosphorylation of αS is a hallmark of PD pathology, as 90% of Lewy Bodies have phosphorylated αS, while around 4% of αS in healthy brains is phosphorylated. αS can be phosphorylated at Ser87, Ser129, Tyr125, Tyr133, and Tyr136 (where phosphorylation of the residue is denoted as in pS129 for phosphoserine 129) [163]. A number of kinases have been shown phosphorylate αS in vitro, with reasonable specificity. Casein Kinase I (CKI) phosphorylates Ser87 and Ser129, Casein Kinase II (CKII) phosphorylates Ser129, the G-protein coupled receptor kinases (GRK 1,2,5 and 6) phosphorylate Ser129, Leucine Rich Repeat Kinase 2 (LRRK2) phosphorylates Ser129, and the Polo-like Kinases (PLKs) phosphorylate Ser129 [164]. The impacts of phosphorylation on aggregation rate depend on which residue is phosphorylated, the method of phosphorylation, and the fraction of the αS population that is phosphorylated.
Generally, pS129 is thought to reduce the rate of fibril formation [117, 153]. Schreurs and coworkers found that mutants S129A, and S129D, as well as pS129 that was phosphorylated by PLK-2 all aggregated at about the same rate as WT [162]. However, PLK-2 only phosphorylated a fraction of the total αS (46% by MALDI-TOF/TOF analysis and 35% by LC-ESI-MS/MS analysis). In at least some cases, it has been observed that different ratios of phosphorylated αS to WT αS can have different impacts on aggregation [162]. Paleologou and coworkers also found that S129E aggregates at the same rate as WT, however, S129A aggregated much faster. They also used CK1 to phosphorylate Ser129. Unlike PLK-2, phosphorylation by CK1 decreased the rate of aggregation. To address concerns about nonspecific phosphorylation, they used CK1 to phosphorylate an S87A αS mutant. Much like WT αS phosphorylated by CK1, this construct inhibited fibril formation. Since these constructs were heterogenous mixtures of phosphorylated and WT structures, they purified S87A/pS129 and tested the aggregation of mixtures with different ratios of S87A/pS129 to WT. At 5% of S87A/pS129, there were no differences in fibril formation, but at 20%, fibril formation was inhibited [111]. Thus, in spite of the discrepancy in the findings of the Schreurs and Paleologou studies, which may be attributable to the levels of phosphorylation, both agree that disruption in aggregation rates by this PTM is due to changes in long-range contacts.
Both groups applied similar methods to study phosphorylation at other sites. Paleologou and coworkers examined the effects of phosphorylation on Ser87. They found that S87A aggregates like WT while phosphomimic S87E does not aggregate [111, 164]. They also examined enzyme-mediated phosphorylation by using CK1 to phosphorylate S129A mutants to ensure that they obtained singly phosphorylated constructs at Ser87. This species inhibited fibril formation. In agreement with the above studies, Ser mutants seem to be reliable models for understanding the aggregation rates of Ser phosphorylated αS [164]. In studies of Tyr phosphorylation, Schreurs and coworkers found that Y125E and Y125F αS both aggregated slower than WT. They then used Fyn kinase to phosphorylate Tyr125 and found that unlike the mutants, pY125 does not alter the rate of aggregation, indicating that the glutamate phosphomimic is not as useful a model for Tyr sites as it is for Ser sites. However, Fyn also led to some phosphorylation at Tyr133 and Tyr136 which may explain some of the differences in aggregation rate between the phosphomimics and the in vitro phosphorylated constructs [162].
Generally, differences between the phosphomimics and in vitro phosphorylated αS can be explained by charge and steric effects. Asp and Glu have a charge of −1 while phosphate groups have a charge of −2. Additionally, while Asp and Glu may share some structural features with pSer, they are not able to replicate the structure and properties of pTyr very well. To overcome the limitations of phosphomimics, several groups have used semisynthetic methods to install phosphate groups at specific residues. Lashuel and coworkers used a semisynthetic strategy to create pY125 αS, finding that phosphorylation at this residue does not have a large impact on aggregation rate [100]. They also synthesized pY39, finding that aggregation of pY39 αS is significantly slower than WT [101]. Petersson and Rhoades used a chemoenzymatic synthesis to create pY39 αS and compared aggregation at different percentages of phosphorylated protein rather than with homogenous pY39. Interestingly, they found that the rate of aggregation increased between 1–5% pY39 but began to decrease above 10% pY39. They found a similar phenomenon with the Y39E phosphomimic, however, the turning point for acceleration was between 10–25% [102]. Their follow up study using NMR[165] and a cryo-EM structure obtained by Liu and coworkers[166] indicate that this difference comes from a change to the fibril fold rather than changes in monomer structure.
N-Terminal and Lys Acetylation
N-terminal acetylation (Ac-αS) is a ubiquitous eukaryotic co-translational modification that is installed on αS by N-terminal acetylase NatB [98, 167]. Bacterially overexpressed αS is not N-terminally acetylated. In order to study N-terminally acetylated αS, the protein is either expressed with NatB or synthesized using NCL [110]. Multiple groups have found that N-terminal acetylation has local effects on structure, leading to increased helicity of the first 9 [110, 168], 10 [110], or 12 residues [98]. Because of this change in helicity in the N-terminal domain, the impact of N-terminally acetylated αS on aggregation is likely different when studied in the presence of lipids or in vivo as compared to the in vitro studies discussed in this section. The helicity induced by N-terminal acetylation may also impact the structure of other areas in the N-terminal domain, specifically regions with familial mutations A30P, E46K, and A53T. Despite a change in charge in the N-terminal domain, N-terminal acetylation does not seem to interfere with long-range contacts with the C-terminal domain [98]. However, altered residues in the N-terminal domain may be important because they abolish copper binding at Met1 and Asp2 [169].
N-terminal acetylation is generally thought to decrease the rate of aggregation of αS [168–172], however some studies also found that N-terminal acetylation does not have an impact on aggregation rate [98, 110]. Possible reasons for the decrease in aggregation rate may include the stabilization of the N-terminal region of the protein, or subtly different electrostatic and hydrophobic regions [168]. N-terminally acetylated αS also has a lower β-sheet content which may make it less prone to aggregation [170]. On the other hand, N-terminally acetylated αS may aggregate at the same rate as WT because the effects of N-terminal acetylation are mostly local and do not seem to impact the NAC domain [98]. These results are somewhat complicated by the fact that two groups found that N-terminally acetylated fibrils resulted in lower maximum ThT fluorescence than WT even when the percentage of aggregated material was similar by sedimentation, indicating that N-terminal acetylation may lead to structurally unique aggregates [110, 170].
Lys residues in αS can also be acetylated. De Olivera et al. determined that αS was acetylated at Lys6 and Lys10. In order to study acetylation at these residues, they made two mutants, K6R/K10R and K6Q/K10Q. The Arg mutants, which represent αS that cannot be acetylated, aggregate faster than WT whereas the Gln mutants, which are mimics of acetylated αS, aggregate slower than WT [172]. However, more precise studies involving isolated acetylation of Lys6 and Lys10 by semisynthetic methods should be carried out in the future to confirm the impact of acetylation at these residues. The recent cryo-EM study of fibrils isolated from MSA patients included reports of Lys acetylation at several sites throughout the protein [173], but additional biochemical characterization of these PTMs has yet been reported.
Lys Ubiquitination and SUMOylation
Ubiquitination and SUMOylation are PTMs that involve attaching small proteins to Lys residues via an isopeptide linkage. In vivo, these linkages are conferred using three ubiquitin ligases. These ligases are not site-specific, so in order to study the impact of ubiquitination or SUMOylation on a specific residue, groups have turned to semisynthetic methods [107, 174].
Ubiquitination generally targets proteins for degradation, and serves as a potential way to clear excess or aggregated αS [87]. αS has been shown to undergo ubiquitination at 9 out of its 15 Lys residues; αS is generally only monoubiquitinated which could be sufficient as a small protein to lead to degradation by a purified proteasome, to a varied extent depending on the modification site [175, 176], but not necessarily in the presence of deubiquitinases, in which case de-ubiquination likely occurs fast enough to prevent degradation [105]. On the other hand, di- and tetraubiquinated αS were degraded more efficiently, as αS remains ubiquinated for longer times, allowing for the proteasome activity. Interestingly, Lys6, Lys10, and Lys12 can be ubiquitinated after fibril formation [103]. Lashuel and Brik first synthesized mono-ubiquitinated αS using NCL. They found that ubiquitination at K6 (K6Ub) inhibits fibril formation in vitro, possibly because ubiquitination in the N-terminal domain stabilizes the monomeric form of αS, preventing oligomerization [104]. Since NCL is synthetically intensive, groups have made ubiquitinated analogues involving different linkers between αS and ubiquitin. In Meier et al., Pratt and coworkers mutated Lys residues that are known to be ubiquitinated to Cys residues and then formed a disulfide linkage between a ubiquitin attached to a linker with a free Cys and αS. Ubiquitination at K6Ub, K10Ub, K12Ub, K21Ub and K23Ub showed moderate inhibition of aggregation, while K32Ub, K34Ub, K43Ub, and K96Ub displayed no fibril formation. Complete inhibition of aggregation occurs when αS is ubiquitinated on residues thought to be in the center of the fibril core (22–36 to 90–98, depending on analytical method), potentially pointing to steric effects of ubiquitination. In terms of N-terminal ubiquitination, possible causes for slowed aggregation may be shielding positively charged Lys residues from interacting with the C-terminal domain [103]. Lewis et al. also used a synthetic linker, this time one made from Bis-thio-acetone (BTA). Unlike proteins with disulfide linkers, proteins with BTA linkers can be used under reducing conditions. Ubiquitinated analogues at Lys23, Lys43, and Lys96 completely inhibited aggregation, while some small, off-pathway fibrils were formed for Lys6. In the Pratt study noted above, Lys23 ubiquitination caused slower aggregation but did not completely inhibit fibril formation, indicating that different linkers have different effects on fibril formation [107].
There are three related SUMO proteins, SUMO1 and the nearly identical SUMO2 and SUMO3. In general, SUMOylation alters sub-cellular protein localization and solubility [177]. Although αS can undergo SUMOylation at 11 out of the 15 Lys residues, most SUMOylation occurs at K96 and K102 [106]. Krumova and colleagues expressed SUMOylated αS by overexpressing the required E1 and E2 ligases, as well as SUMO1 with αS. They tested the impact of SUMOylation at different concentrations when mixed with WT αS, as only a small fraction of αS is SUMOylated in vivo. At 100% and 50% SUMOylated αS, SUMO completely inhibited fibril formation. At even 10% of SUMOylated αS, SUMO delayed fibril formation significantly. Much like ubiquitination, SUMOylation is thought to sterically inhibit fibril formation, or shield positively charged Lys residues. Additionally, SUMOylation is known to increase solubility, potentially preventing aggregation by keeping αS in solution [106]. Abeywardana and Pratt used a disulfide linkage similar to the one used in Meier et al. to site-specifically introduce SUMO modifications. They tested SUMOylation at Lys96 and Lys102 with SUMO1 and SUMO3 (K96SUMO1, K102SUMO1, K96SUMO3, K102SUMO3) finding that while all modifications decreased the rate of fibril formation, modification at Lys102 and with SUMO1 had the largest effect [178]. Lewis et al. also used their BTA linker to attach SUMO3 at Lys96 and Lys102, finding that SUMOylation at Lys96 does not impact the rate of aggregation and that SUMOylation at Lys102 inhibits fibril formation. The differences in the effects among these residues is surprising when one considers that Lys96 is typically found in the folded region of fibrils (Figure 8) while Lys102 is not, reflecting the complexity of PTM effects on aggregation[107].
Lys Glycation
αS’s Lys residues can also undergo glycation. During glucose metabolism or ribose metabolism, several byproducts which are nonreducing sugars condense with Lys residues and then undergo a series of reactions. The result is an irreversible modification called an advanced glycation end-product (AGE) [179]. In the context of αS, methylglyoxal (MGO), glyoxal (GO), pyruvic acid, and D-ribose can serve as glycating agents. Lys reacts with α-dicarbonyl compounds MGO, GO or pyruvic acid to form either carboxyalkylated lysine, an imidazolium cross-link or amide modifications, all of which typically occur under oxidative conditions[180]. Carboxyalkylated lysine products include N(ε)-(carboxyethyl)lysine (CEL, from MGO or pyruvic acid) and N(ε)-(carboxymethyl)lysine (CML, from GO). Formation of carboxylic acid CML is generally favored over generation of the isomeric amide, N6-(glycoloyl)lysine (GALA, from GO). Imidazolium cross-links, such as MGO-lysine dimer (MOLD, from MGO) and GO-lysine dimer (GOLD, from GO), are commonly found in vivo[181, 182]. An intermediate is shared between the imidazolium cross-link, GOLD, and an amide cross-link, N6 –(2-((5-amino-5-carboxypentyl)amino)-2-oxoethyl)lysine (GOLA, from GO). In the presence of excess GO, formation of GOLD is favored over GOLA. Lys also directly condensates with glucose or ribose to form a Schiff base, which rearranges into a stable ketoamine, Amadori product – oxidative cleavage of this also leads to the formation of carboxyalkylated lysine products or imidazolium cross-links. Glycation is thought to reduce membrane binding, inhibit the clearance of αS, and promote the accumulation of oligomeric species [93]. Glycation can occur on any of the 15 Lys residues in α-Syn. All of these studies were done by incubating αS with glycating agents, meaning that the site-specific effects of glycation are not yet understood.
αS glycation with MGO promotes the oligomerization of off-pathway aggregates [93, 183, 184]. Because of MGO-modified α-Syn’s propensity to form off-pathway oligomers, the rate of aggregation is not well characterized by ThT, with reports showing faster [184], the same rate [93], or inhibited [183] aggregation. MGO glycated αS is thought to primarily impact Lys sidechains in the N-terminal domain, extending them and decreasing the conformational flexibility and also neutralizing the positive charge at the N-terminus. This lack of flexibility is thought to be responsible for inhibited fibril formation [93, 184, 185]. Similar to the MGO effects, Lee et al. found that GO glycation decreases conformational flexibility and causes the formation of off-pathway oligomers. The rate of aggregation seems to be faster than WT αS, but off-pathway oligomers may not be amenable to characterization by ThT [184]. Mariño et al. formed N(ε)-(carboxyethyl)lysine (CEL) modified αS by incubating WT αS with pyruvic acid and NaBH3CN, a reducing agent. CEL-modified αS did not aggregate [185]. It should be noted that CEL modifications can also occur as a result of MGO modification with subsequent oxidation. Ribosylation can occur at all 15 Lys residues however it favors C-terminal Lys residues. Ribosylation is similar to other glycation modifications in that it also creates off-pathway oligomers. These oligomers have a similar rate of formation to that of WT fibrils [186].
Ser/Thr O-GlcNAcylation
O-GlcNAcylation modification of αS includes the addition of N-acetyl-glucosamine units to residues Thr33, Thr44, Thr54, Thr59, Thr64, Thr72, Thr75, Thr81, and Ser87 [187]. O-GlcNAcylation universally reduces the rate of aggregation. Zhang et al. were able to co-express αS with a shortened version of the enzyme O-GlcNAc transferase in order to generate non-site specifically O-GlcNacylated αS (g-αS). g-αS inhibits fibril formation and instead forms small oligomers [188]. Pratt and coworkers have used native chemical ligation to understand the site-specific impacts of O-GlcNAcylation on αS aggregation, specifically at residues Thr72, Thr75, Thr81, and Ser87 [108, 187, 189]. O-GlcNAcylated Thr72 (gT72, where g indicates O-GlcNAcylation) αS has been shown to greatly reduce the rate of aggregation. As little as 10% gT72 was able to delay aggregation kinetics, potentially because gT72 is unable to form oligomers. This is supported by the fact that after sedimentation, most of the O-GlcNAcylated αS remained in solution [108]. gS87 also reduced the rate of aggregation, but to a lesser extent than gT72 as gS87 was able to form shorter fibrils and smaller structures. Interestingly, at a lower concentration, gS87 behaved more like gT72. It is possible that gT72’s equilibrium favors a monomeric form more than gS87, and that gS87’s propensity for its monomeric form is overcome at higher concentrations, leading to somewhat increased oligomerization [187].
Levine et al. studied gT72, gT75, gT81, and gS87, finding that all four modifications lead to reduced aggregation. They found that gT81 was the most inhibitory, forming very few small amorphous aggregates, followed by gT75 which also formed amorphous aggregates and gT72 which formed short, broken fibrils, and then gS87 which formed some fibrils that are morphologically different than WT [189]. Levine et al. also studied a gT72/gT75/gT81 triple mutant which completely inhibited monomeric aggregation and aggregation seeded by WT seeds, suggesting that the triple mutant is incompatible with adopting a conformation that would allow it to be added to the ends of fibrils. They also found that an A53T/gT72/gT75/gT81 mutant did not aggregate, showing that O-GlcNAcylation could suppress an aggregation-prone familial mutant [189].
Since Ser87 can be phosphorylated and undergo O-GlyNAcylation, Lewis et al. created mutants S87E, S87D, S87A, S87K, and S87W to further characterize the impact of charge and steric hinderance on Ser87. S87E, a phosphomimic, inhibited aggregation. S87D, which has the same charge as S87E, and S87A, which represents a loss-of-function mutation for either O-GlcNAcylation or phosphorylation, both aggregated slower than WT. S87W, which mimics the steric bulk of O-GlyNAcylation but is hydrophobic, and S87K, which has the opposite charge of phosphorylation and the S87D/E mutants, both aggregated faster than WT. These results indicate that inhibition of aggregation by O-GlyNAcylation and phosphorylation at Ser87 is driven by hydrophilicity and negative charge rather than just steric hinderance [187].
To understand how O-GlcNAcylation impacts aggregation, Galesic et al. made a number of monosaccharide modifications at Thr72. They chose β-O-GalNAc and β-O-glucose and α-O-mannose in addition to the traditional N-Acetyl-Glucosamine responsible for O-GlcNAcylation because this group of monosaccharides are structurally similar. They found that modification at Thr72 by all of the monosaccharides resulted in slower aggregation, with modification by α-O-mannose reduced the rate of aggregation more than modification by β-O-GalNAc and β-O-glucose. Given the differences in the impact of aggregation of these modifications despite the similarities in their structures, it is unlikely that purely steric effects are responsible for the slower aggregation [190].
Thr AMPylation
AMPylation, or adenylylation, consists of adding adenosine monophosphate (AMP) to proteins. Huntingtin yeast interacting protein (HYPE) is a protein localized in the ER and is implicated in cellular stress responses like the unfolded protein response [94]. A mutant form of HYPE is able to directly AMPylate αS at Thr33, Thr54, and Thr75. αS that was incubated with constitutively active HYPE showed similar rates of fibril formation, but much lower amounts of overall fibrils in the plateau phase, indicating that AMPylation impacts elongation rather than nucleation [94]. However, more studies still need to be done to understand the site-specific impact of AMPylation. Additionally, since AMPylation and O-GlyNAcylation occur on some of the same residues, physiological changes are likely to impact which PTM is more prevalent in a population of αS molecules.
Tyr Nitration
αS is also known to undergo modifications due to oxidative and nitrative stress, including the nitration of Tyr residues, di-Tyr oligomer formation, methionine sulfidation, and lipid peroxidation, resulting in the addition of HNE to Lys and His residues. αS has four Tyr residues at positions 39, 125, 133, and 136, all of which can undergo nitration. Nitration of αS occurs as a result of ONOO− degrading into •NO2 and then reacting with tyrosine at either of the two carbons ortho to the hydroxyl group to form 3-nitrotyrosine (nY). Additionally, under oxidizing conditions, Tyr residues are able to form either intramolecular or intermolecular di-tyrosine (DiY) linkages. Unfortunately, nitrated αS formed by incubation with oxidizing agents often forms a heterogenous mixture of nitrated products including monomers (both nitrated and intramolecularly crosslinked), dimers, and high molecular weight oligomers. The formation of many nitrated species, alongside the variety of methods used to carry out nitration, has made it difficult to achieve clear consensus on nitration’s impact on aggregation rate.
Nitrating agents like tetranitromethane (TNM), NaNO2, or ONOO− can be used to nitrate αS in vitro and creates a heterogenous mixture of nitrated and DiY products [191]. Yamin et al. and Uversky et al. found that incubation with TNM leads to largely octameric species. Additionally, they found that even at concentrations as low as 1:10 TNM-nitrated to WT α-Syn, nitration was able to delay aggregation at physiological pH [192, 193]. Similarly, Xiang et al. and Burai et al. found that TNM-nitrated αS showed little aggregation by ThT [99, 194]. The lack of fibril formation may occur because nitrated αS and WT αS are able to form stable heterooligomers, effectively decreasing the concentration of WT protein that is able to aggregate into fibrils [192, 194]. In contrast, using NaNO2 as a nitrating agent, Liu et al. found that αS aggregates at about the same rate as WT αS but forms amorphous oligomers instead of fibrils which may be challenging to characterize by ThT [195]. This indicates that the choice of nitrating agent may also play a role in the rate of aggregation of in vitro nitrated αS aggregation.
In order to get homogenous nitration, Long et al. developed a light-controlled method for tyrosine nitration. This method depends on a dinitroimidazole reagent and cleanly converts all four tyrosine residues to nY residues in around 10 minutes, resulting in homogenously nitrated populations with no DiY oligomers. Their tetra-nitrated αS does not aggregate. Co-incubation of WT αS with this species shows concentration-dependent reduction in the rate of aggregation [196]. While this strategy is able to create consistently nitrated α-Syn, one limitation is that it nitrates all four tyrosine residues which does not allow for the investigation of the site-specific effects of nitration.
Nitration at specific residues has been accomplished by mutating other Tyr residues to Phe residues, however, that may alter some of the properties of αS. To overcome this issue, Burai et al. used expressed protein ligation coupled with a novel desulfurization strategy that does not reduce the nitro groups to site-specifically incorporate nY39 and nY125. Both modifications delayed aggregation, with Tyr125 showing slower aggregation than Tyr39. However, the levels of soluble protein after the aggregation assay were about the same as WT, indicating that the ThT assay may not accurately reflect protein aggregation due to fluorescence quenching by nY residues[99].
Met Oxidation
Met residues are also susceptible to oxidative stress, forming methionine sulfoxide (MetO) residues. αS has four Met residues, at positions 1, 5, 116, and 127. Unlike many of the other PTMs discussed, Met oxidation is known to be reversed, by methionine sulfoxide reductases. Met can also be irreversibly oxidized to form a methionine sulfone, however, this is rare [197]. Under near physiological conditions, Met oxidation is thought to essentially inhibit fibril formation, instead promoting oligomer formation.
When incubated with H2O2, all four Met residues are oxidized, and at physiological pH, MetO-αS does not aggregate [95, 160, 194]. Additionally, MetO-αS is able to inhibit fibril formation of WT αS in a dose-dependent manner, with 25% MetO-αS leading to a reduced rate of fibrillation [95]. This may be because MetO is more polar than Met, causing a reduction in aggregation-promoting hydrophobicity [95], or because MetO allows αS to adopt a conformation that strengthens auto-inhibitory long-range contacts [160].
UV irradiation can also oxidize αS. In Carmo-Gonçalves et al.’s preparation, they found that only Met5 was oxidized by mass spectrometry and NMR [198]. Despite having fewer Met residues oxidized, this preparation was found to be more inhibitory than H2O2-oxidized αS, as just 5% 5-MetO-αS was able to slow fibril formation. UV irradiated αS also formed a small population of dimers and trimers which may be DiY products which could impact the observed rate of aggregation of the 5-MetO-αS [198]. Interestingly, oligomers formed from H2O2-induced MetO-αS were found to be non-toxic to primary rat neurons [160] whereas those induced by UV irradiation were found to be toxic to primary mouse neurons [198]. If these oligomers are indeed toxic, then Met oxidation may not be protective. Since the mechanism of UV-induced Met oxidation is not clear, more research is needed to understand if this preparation is relevant to in vivo oxidation.
Hokenson et al. investigated the impact of site-specific Met oxidation, finding that Met oxidation also reduces the rate of fibril formation. They made single, double, triple, and quadruple Met-to-Leu mutants and then oxidized the proteins using H2O2. They found that the number of Met residues was proportional to the rate of aggregation, with single mutants containing three Met residues aggregating the fastest, followed by double, triple and finally quadruple mutants. Like the other studies using H2O2, they found that all Met residues had about the same impact on fibril formation, as all of the single mutants had roughly the same lag times [154]. However, certain conditions such as acidic pH [95, 160] or incubation with Ti3+, Al3+, Zn2+, and Pb2+ been shown to induce fibril formation in MetO-αS [154, 199]. Collectively, these studies show that Met oxidation generally reduces the rate of fibril formation, with increasing concentrations of MetO-αS or an increasing number of oxidized Met residues leading to greater inhibition of fibril formation.
His Lipid Adducts
Oxidative stress can also cause lipid peroxidation. This leads to the production of reactive aldehydes such as HNE and 4-oxy-2-noneal (ONE) that are able to react with Lys and His residues in αS through either Michael addition or Schiff base formation [200]. Generally, one, two, or three HNE molecules are added, with His50 being the most reactive position for HNE addition [96, 147, 194]. Both ONE and HNE modification result in the inhibition of fibril formation and the promotion of oligomers that are not detectable by ThT [96, 194, 200, 201].
Glu Arginylation
Arginylation occurs when the arginyltransferase ATE1 transfers Arg from tRNA to protein N-termini or Glu and Asp sidechains. Arginylation occurs on αS at residues Glu46 and Glu83 and is thought to have a neuroprotective effect [97]. Pan et al. used semisyntheic methods to create αS that was arginylated at Glu46 and/or Glu83. They found that arginylation at Glu83, but not Glu46, slows the rate of aggregation. Interestingly, arginylation at both Glu83 and Glu46 decreases the rate of aggregation even further [109].
Labeling Modifications
ThT is useful for tracking the kinetics of aggregation as defined by primary and secondary nucleation steps. However, there are certain aspects of aggregation that ThT is not able to detect that may be relevant to the aggregation pathway of αS, such as early species like folding intermediates and oligomers as well as the role of each domain of αS in fibril formation.[202] Site-specifically labeled αS can be used in Förster resonance energy transfer (FRET) [203–205] and fluorescence polarization (FP) [206, 207] experiments to overcome some of these challenges while also serving as a reliable reporter of aggregation kinetics. In addition to its use in kinetics assays, site-specifically labeled αS has applications in Raman Spectroscopy [208], fluorescence correlation spectroscopy (FCS) [209, 210], and cell-based imaging [211–214]. In the context of this review, site-specific labeling can be considered another type of “mutation” that can have an impact on the aggregation rate. Since site-specific labeling is often used as a reporter of aggregation kinetics, it is important to select residues and labels that do not perturb the aggregation of αS. This collection of information on label perturbation shown in Figure 7 will be generally valuable to those wishing to introduce labels to αS without perturbing its aggregation. Note: We will include exclusively sidechain modifications in this discussion. While there have been some studies of αS involving backbone modification, such as the Petersson laboratory’s usage of thioamides [215–218], we do not feel that those effects are directly comparable to the larger body of mutation data.
Different laboratories have applied a variety of labeling techniques to αS. By far the most common is Cys labeling. Since αS lacks naturally occurring Cys residues, by using site-directed mutagenesis, groups are able to incorporate a Cys which can then be covalently linked to a reporter. Thirunavukkuarasu et al. attached a pyrene probe to mutants A18C, A90C, and A140C in order to observe oligomerization through excimer formation. In spite of the addition of the bulky, hydrophobic pyrene group, all three pyrene-labeled constructs aggregate at about the same rate as WT [219]. Many groups also use this technique to create fluorescent probes for fluorescence polarization assays, as FP is able to report on local dynamics, oligomers, and fibril formation. Marvian et al. labeled M5C and G132C αS with monobromobimane (mBBr) to study the N- and C-terminal domains of αS independently. By ThT, the aggregation rate of labeled protein at either position was similar to WT [220]. Haney et al. describe a collection of Cys mutants at residues 9, 24, 42, 62, 87, 114, 123, and 136 with fluorescein maleimide (Fam) for use in FP assays. By Congo Red absorbance, Fam labels at positions 9, 24, 42, 123, and 136 have similar aggregation kinetics to WT while positions 62, 87, and 114 showed slightly slower kinetics [206, 221]. Cys labeling is also used for the introduction of FRET pairs. Engelborghs et al. created singly and doubly labeled αS, with the donor Alexa 488 carboxylic acid succinimidyl ester at the N-terminus and the acceptor tetra-methyl rhodamine at A140C. While the protein labeled with the acceptor only aggregated at a similar rate to WT, the construct labeled with the donor and the double labeled construct aggregated slightly faster. The authors attributed this to pH changes to avoid labeling Lys residues when labeling the N-terminal domain, but it may be that the label itself is perturbing. They also created a version of αS labeled at A140C with Alexa-488 C5 maleimide for fluorescence correlation spectroscopy which aggregated at the same rate as WT [222].
Since αS also lacks Trp residues, the incorporation of Trp, which is intrinsically fluorescent, has been used to monitor fibril formation. Generally, Trp residues replace one of αS’s aromatic residues to reduce potential perturbation. Dusa et al. used a Y39W mutant to track the motion of the N-terminal domain during fibril formation, however, the kinetics of this mutant as measured by ThT were slower than WT, indicating that this mutation may not be an accurate reporter of fibril formation [223]. Kaylor et al. made a Y125W/Y133F/Y136F construct so that Tyr39 and Trp125 could be used as a FRET pair to detect earlier aggregation species like oligomers, with the other Tyr residues mutated to Phe so that there was only one donor. By ThT, this mutant showed slightly faster nucleation based on the lag time but had roughly the same rate of elongation, potentially because of a difference in hydrophobicity [224].
Unnatural amino acids (Uaas) can also be incorporated into αS and can provide a variety of functionalities. Uaas may be intrinsically fluorescent or they can serve as handles for the addition of probes using strategies like click chemistry reactions. Uaas can also serve as probes for other types of spectroscopy. Flynn et al. incorporated homopropargylglycine (Hpg) at Met positions 1, 5, 116, and 127 using native cellular machinery. Hpg has a terminal alkyne that can be used as a probe for Raman spectroscopy to characterize aggregation kinetics and secondary structural changes during aggregation. Their full-length construct aggregated at about the same rate as WT, however, their M115 truncated version aggregated faster than WT [208]. Since truncated αS generally aggregates faster than WT αS, it’s possible that this difference can be attributed to the truncation of the C-terminus rather than perturbation as a result of Hpg. Haney et al. used evolved aminoacyl-tRNA synthetase/tRNA pairs to site-specifically incorporate Uaas that served as handles for fluorescent probes. They incorporated azidophenylalanine (AzF) at site 94, which they then labeled with fluorescein-dibenzyocyclooctyne (Fco) and fluorescein-alkyne (Fak) and propargyltyrosine (Ppy) at sites 39 and 94, which were labeled with tetramethylrhodamine-azide (Raz). Using Congo Red absorbance, they showed that a 5% concentration of PpyRaz94-αS is non-perturbing when aggregated with WT. Haney et al. used this method along with a cysteine mutant strategy to orthogonally label αS at residues 9 and 94. A 5% addition of CysFam9/PpyRaz94-αS to WT aggregated at similar rates to pure WT protein. In addition to using translation machinery to incorporate UAAs, they also used E. Coli aminoacyl transferase, an enzyme that transfers Leu, Phe, Met, or hydrophobic UAAs to proteins with an N-terminal Lys or Arg. Using this method, they created an αS variant that spanned residues 2–140 and had a D2K mutation for labeling at the N-terminal Lys. Here, Lys2 was labeled with Azf, Ppy, or azidohomoalanine (Aha), which can then be modified with Fco or Rco [221].
Groups have also used linkers at the N- and C-terminus of αS to probe its aggregation using fluorescent groups, including fluorescent proteins which are larger than αS itself. van Ham et al. used an Ala-Pro-Val-Ala-Thr linker to attach a yellow fluorescent protein (YFP) to the C-terminal domain for use in fluorescence polarization experiments. YFP-labeled αS aggregates slightly faster than WT [225]. However, this seems to be specifically attributable to the linker as a C-terminal GFP fusion aggregated dramatically slower than WT [226]. Kotzbauer et al. attached a six amino acid bi-cysteine tag at the N-terminus (C2-αS). The association of two or more αS molecules is reported by fluorescein arsenical hairpin binder (FlAsH). C2-αS and WT αS aggregate at the same rate, and aggregation kinetics as followed by the FlAsH assay are very similar to those reported by ThT [227].
Discussion
The aggregation of αS is central to the progression of the synucleinopathies. Various studies have shown that αS fibrils, oligomers, and the process of Lewy Body formation [17, 18] are toxic to cells. Therefore, inhibiting aggregation or degrading toxic aggregates may be useful in treating the synucleinopathies [228]. Mutations and modifications at single residues, whether familial mutations, designed mutations, PTMs, or labels and probes, can have significant impacts on αS’s aggregation as measured by ThT and other reporters of fibril formation. While some of these mutations may produce on-pathway oligomeric species, recent ssNMR and cryo-EM structures of fibrils formed from modified αS show that single mutations or modifications can lead to fibrils that are structurally different than WT αS [62–65]. Taken together, the impact of these mutations, modifications, and labels on αS’s rate of aggregation speak to the sensitivity of αS to small perturbations in its environment and physico-chemical properties.
Perhaps the largest issue with in vitro aggregation is that aggregates created under different solution conditions have different properties. While αS is able to form aggregates under a wide range of conditions, only specific conditions yield aggregates which are able to seed further aggregation [28]. Hoyer et al. aggregated WT αS under different pH and salt conditions and found that low pH or high salt aggregations resulted in amorphous, off-pathway aggregates that formed quickly and had low seeding abilities [30]. De Olivera et al. compared the aggregation of the familial mutants A30P, E46K, and A53T at different salt concentrations. For the no salt condition, each mutant showed a unique kinetic profile with multiple inflection points that likely represent distinct microscopic processes like primary nucleation, elongation, or secondary nucleation. At high salt, however, each mutant’s kinetics appear to be sigmoidal and the rate of aggregation for all mutants is increased [48].
The observed pH effects may stem from a reduction in charge in the C-terminal domain. At low pH, the C-terminal region, which is usually negatively charged, becomes hydrophobic, leading to the collapse of the C-terminal domain. Similarly, the addition of salt shields N- and C-terminal Coulombic interactions [30]. The connection between increased charge and faster aggregation is not entirely understood. Wu et al. propose that while the contacts between the N- and C-terminal domains remain intact, at low pH there are more contacts between the NAC and C-terminal domain whereas at high pH there are more contacts between the NAC and the N-terminal domain. This change, alongside the different charge distribution at low pH, may lead to increased aggregation [149]. McClendon et al. agree that the interactions between the N- and C-terminal domain are preserved, however also argues that at low and high pH values, the NAC domain interacts with both the N- and C-terminal domains. In this case, hydrophobicity, not the release of long-range contacts is not the source of increased aggregation [229].
While it is possible that mutations and PTMs exert effects on more subtle intermediate phases of aggregation, a simple analysis might assume that an accelerating modification acts by either destabilizing the monomer state or stabilizing the fibril state. Structural data, in the form of solution phase NMR studies of the disordered monomer ensembles, as well as ssNMR and cryo-EM studies of the of the fibrils, can provide some insight into the effects of buffer conditions, mutation, and aggregation rates.
A common and sensitive technique to analyze solution phase NMR is by chemical shift perturbation (CSP) analysis of 1H-15N heteronuclear single-quantum correlation (HSQC) spectroscopy data which provides residue-level information on the backbone environment. For the more common familial mutations, A30P, E46K, H50Q, G51D, and A53T, experiments in a variety of conditions agree that nearby residues are structurally perturbed [134, 141, 148, 230–233]. However, these studies do not agree on the extent of distal changes which may also be linked to buffer conditions or trace contaminants. For example, at pH 6.0, Ranjan et al. noted C-terminal perturbations for all common mutants including A53E, but at pH 7.4, the long-range effects disappeared or were greatly diminished with the exception of E46K [141]. These effects were thought to destabilize the monomer, promote NAC exposure, and lead to increased aggregation. Bhattacharyya et al. noticed similar effects for E46K and A30P, however, the spectra are not entirely similar [134]. In general, it is hard to decouple mutational effects from differences in pH and buffer when comparing these studies, so we have restricted analysis to comparisons within publications using consistent preparations. In addition to monomer effects, fibrils may be more stabilized as a result of mutation, and ssNMR provides insight into how mutations may contribute to increased aggregation. Data from Heise et al. on the A53T mutation show that fibrils may have a β-sheet region extended by a few residues, thereby increasing overall stability [234]. Lemkau et al. also noted small changes in structure due to the A53T mutation but noticed large differences in the resonances assigned to E46K indicating a difference in fibril fold which was later confirmed via cryo-EM [235].
Cryo-EM has been used to solve fibril structures at near-atomic resolution. However, the interpretation of these structures is complicated by the influence of different fibril preparations. Four fibril preparations produced roughly the same structure. Stahlberg’s 2018 structure was produced from C-terminally truncated αS1–121 (Figure 8, 6h6b) [59], Li and Liu solved the structure of N-terminally acetylated αS [60] (Figure 8, 6a6b), which was independently corroborated by Lee and coworkers (6osj), who also obtained structures of N-terminally and C-terminally truncated fibrils (6osl and 6osm) [236], and Eisenberg and Jiang solved the structure of the “rod” polymorph (Figure 8, 6cu7) [57]. All three of these structures have a β-arch and the interface between the two protofibrils is created through hydrophobic packing [58]. In addition, the fibril interface is formed by residues His50-Glu57, encompassing familial mutant residues His50, Gly51, and Ala53. In these structures, Glu46 forms a salt bridge [57, 59, 60]. Eisenberg and Jiang also solved the structure of the “twister” polymorph (Figure 8, 6cu8). This structure is similar in that it has a β-arch and hydrophobic packing; however, it has an interface consisting of residues Val66-Ala78. Additionally, the pre-NAC region where familial mutants are located is at the peripheral region of the fibril core, indicating that these mutations may not have a strong impact on the twister fibril structure [57]. In 2019, Stahlberg’s group solved two more structures, termed “2a” (Figure 8, 6rto) and “2b” (Figure 8, 6rtb), of WT αS. These differ from the other structures because they have an additional N-terminal β-strand, their β-arch runs in the reverse direction, and the interface is connected through electrostatic interactions involving Glu46. The difference between 2a and 2b is that 2a has an interface consisting of a salt bridge between residues Lys45-Glu57 whereas 2b includes a salt bridge between residues Lys45-Glu46 [58]. These differences highlight the fact that changes in fibril preparation can dramatically impact morphology.
In addition to structures of WT αS, several laboratories have solved the structures of fibrils created from αS with familial mutations. Two groups have solved structures of E46K fibrils. Eisenberg and Jiang’s structure (Figure 8, 6ufr) resembles the WT 2a overall structure, but lacks the Greek key motif of some of the WT structures and has a solvent-filled interface spanning residues 45–57 [65]. Li and Liu solved the structure of acetylated E46K fibrils (Figure 8, 6l4s), showing that these fibrils have a unique serpentine fold [64]. This fold is thought to occur because of a smaller fibril core region and rearrangement of β-strands in the C-terminal region of the fibril core. Structural differences are thought to be a result of the loss of the Glu46-Lys80 salt bridge [64, 65]. Eisenberg and Jiang solved the structure of H50Q fibrils, finding a “narrow” fibril consisting of one protofilament (Figure 8, 6peo) and a wide fibril consisting of two protofilaments (Figure 8, 6pes). Protofilament A has an ordered β-arch whereas protofilament B does not, likely because it lacks an interaction between Lys45 and Gln50 that is present in protofilament A. The wide fibril’s protofilament interface consists of residues 58–61 which is much smaller than interfaces found in other structures [62]. Liu solved the structure of G51D fibrils (Figure 8, 7eof) [61]. The fibril core is made up of residues 50–98 and the topology is unique in that it consists of six beta strands in a serpentine fold. This structure is most similar to Liu and Li’s E46K structure (Figure 8, 6l4s) except that a β-hairpin in the E46K structure leads to a longer resolvable region in the N-terminus [61]. Liu also solved the structure of acetylated A53T fibrils (Figure 8, 6lrq) which folds into a Greek key-like motif. These fibrils have a core composed of residues 37–99 which is similar to the acetylated WT structure (Figure 8, 6a6b) and the wide H50Q structure (Figure 8, 6pes). However, the interface between the protofilaments involves fewer residues than the acetylated WT structure and the overall morphology differs from the H50Q structure [63]. Among the PTMs, only pY39 fibrils have been structurally characterized, where Liu and coworkers reported structures of fibrils composed of a common fold with either two (Figure 8, 6l1t) and or three (Figure 8, 6l1u) protofibrils [166]. The fold that they observed is different than any other structure, where the effects of pY39 modification can be clearly seen to enable new salt bridges with Lys21 and Lys34 that cause the N-terminus to fold inward.
In analyzing the collection of αS fibril structures, one can see that modifications like mutations and PTMs impact the structure of αS fibrils, but that the clear mechanistic explanation identified for the pY39 fibrils is exceptional. Since the majority of familial mutations occur at the protofibril interface formed by residues 50–60 in WT structures 6a6b, 6h6b, and 6cu7, it is not surprising that they tend to destabilize that interface resulting in changes in morphology. The new fibril morphologies that they adopt appear to result more from this destabilization than from new favorable contacts that are adopted, since none of the mutations are found at the newly formed interfaces. However, seeming inconsistencies confound full mechanistic interpretation. For example, the two E46K structures (Figure 8, 6ufr and 6l4s) are different, and while they differ in buffer and N-terminal acetylation, neither of these aspects affect the WT fibril polymorphs (Figure 8, 6a6b, 6h6b, 6cu7). Thus, it seems that some interplay of protein modifications and the effects of the medium dictate fibril morphology. The fact that multiple polymorphs such as 2a and 2b (Figure 8, 6rto, 6rtb) or rod and twister (Figure 8, 6cu7, 6cu8) can be obtained under roughly the same conditions underscores this issue. Moreover, comparison of the many experiments performed for each of the familial mutations (see Supporting Information spreadsheet) shows that changes in buffer, salt, temperature, αS concentration, and shaking speed can alter the effect of a mutation, even when normalized to a WT experiment performed under the same conditions.
When one considers the impact of aggregation conditions on morphology, one must question which, if any, of these structures most resembles fibrils in patients, where aggregation conditions are clearly different. Scheres and Goedert have used cryo-EM to solve the structure of MSA patient-derived GCI fibrils (Figure 8, 6xyo as well as other polymorphs) and, very recently, PD, PDD, and DLB patient-derived Lewy Body fibrils (Figure 8, 8a9l). The in vitro fibril fold that MSA fibrils most closely resemble is the 2n0a ssNMR structure, but this structure is monomeric, whereas the MSA fibrils consist of two protofilaments, The two types of MSA filaments each contain two similar, but non-identical protofilaments, with non-proteinaceous molecules at their interface [173]. The PD/PDD/DLB fibril fold bears some similarity to the various in vitro fibril folds, but does not closely match any of them [237]. Like the MSA fibrils, non-proteinaceous density is observed. While these structures are tremendously significant in terms of their physiological relevance, they are of WT αS and the PTMs are not well-defined, so it is difficult to say whether the different polymorph is the result of PTMs, the non-proteinaceous co-aggregates, or other aspects of the intracellular milieu, which has been shown to affect αS fibril formation [53]. Scheres and coworkers have shown that using MSA patient material is insufficient to template fibrils of similar morphology using recombinant WT αS, implying that one or more of these other components is necessary [238]. Thus, in spite of the availability of fibril structures, there are still complexities to the aggregation mechanism that are not well understood. The comprehensive view of positional effects on aggregation that we present here can be used to address these complexities.
The mutational data presented in Figure 6 and Table 1 can also be analyzed to identify trends that affect aggregation without relying on structural models. We have categorized each mutation by the properties of the amino acid change, which in some cases may include multiple categories (e.g. A53T as nβ→β for non-β-branched to β-branched and nP→P for nonpolar to polar), and averaged the +1, 0, −1, −2 scoring for each mutation category for the entirety of αS and within the N-terminal, NAC, and C-terminal domains. Those mutation categories that had the strongest effects for the entire protein are depicted in Figure 9 along with control categories (control categories involve mutation within the same category; for example, nP Cntrl includes nonpolar to nonpolar mutations such as Ala-to-Leu) to aid in interpreting the significance of effects. The category definitions are found in the caption to Figure 9. In addition to these control categories, anti-correlation of the inverse mutation category (i.e. nβ→β favors aggregation while β→nβ disfavors aggregation) lends further support for the importance of a particular type of mutation. In some cases, these effects are apparent for the entire protein. In other cases, their impact differs depending on the region of αS, as shown in Figure 10.
Figure 9.
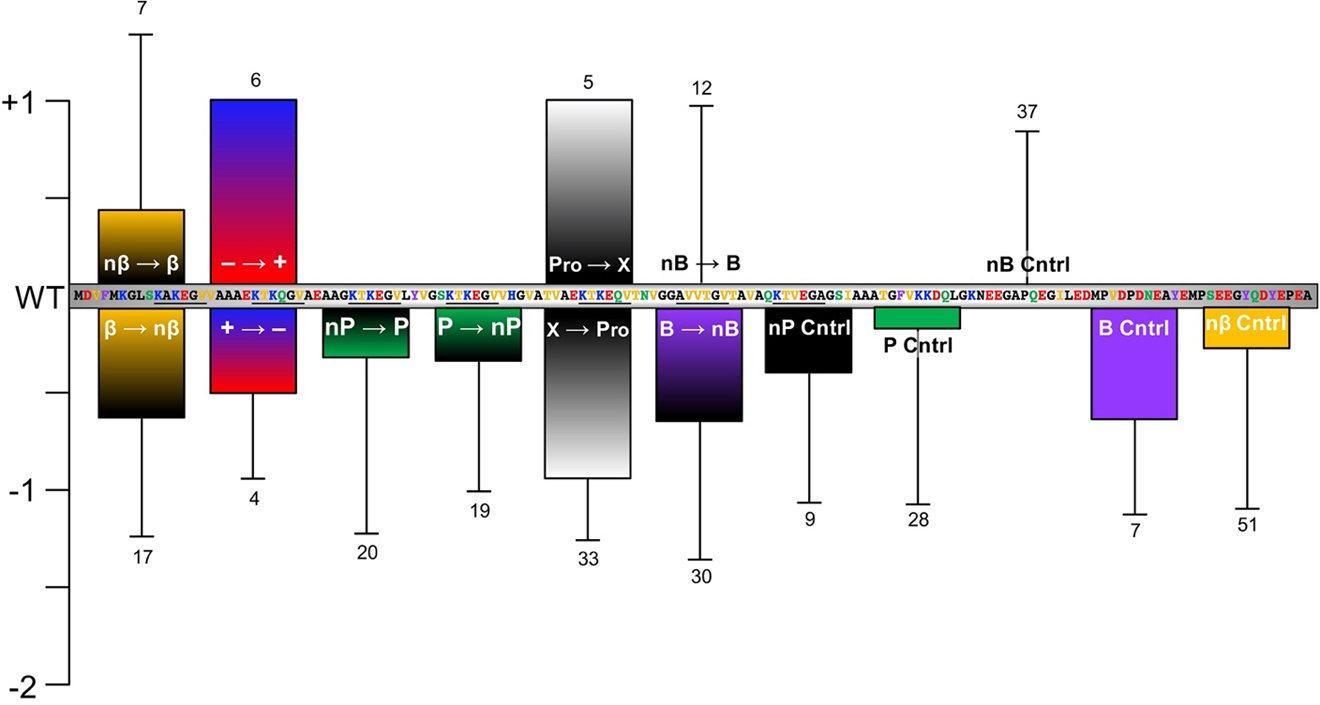
Categorized Mutation Effects Averaged Over Entire αS Protein. Sequence shown colored according to amino acid type: non-polar, Gly, and Pro (black), polar (green), negatively charged (red), positively charged (blue), aromatic (purple), β-branched (yellow). Imperfect repeat sequences are underlined. Bars depict average aggregation scores for each category, unidirectional error bars represent standard deviation, number of examples given above or below bar. Mutation definitions: nβ→β) non-β-branched to β-branched; β→nβ) β-branched to non-β-branched; – → +) negatively charged to positively charged; + → –) positively charged to negatively charged; nP→P) non-polar to polar; P→nP) polar to non-polar; Pro→X) proline to any other amino acid; X→Pro) any amino acid to proline; nB→B) non-bulky to bulky; B→nB) bulky to non-bulky; Cntrl) control groups represent mutation to the same type of amino acid. Category definitions: β) Ile, Thr, Val; nβ) all others except Gly and Pro; +) Arg, His, Lys; –) Asp, Glu; nP) Ala, Ile, Leu, Met, Phe, Trp, Val; P) all others except Gly and Pro; X) all others except Pro; B) His, Ile, Leu, Phe, Thr, Trp, Tyr, Val; nB) all others except Pro.
Figure 10.
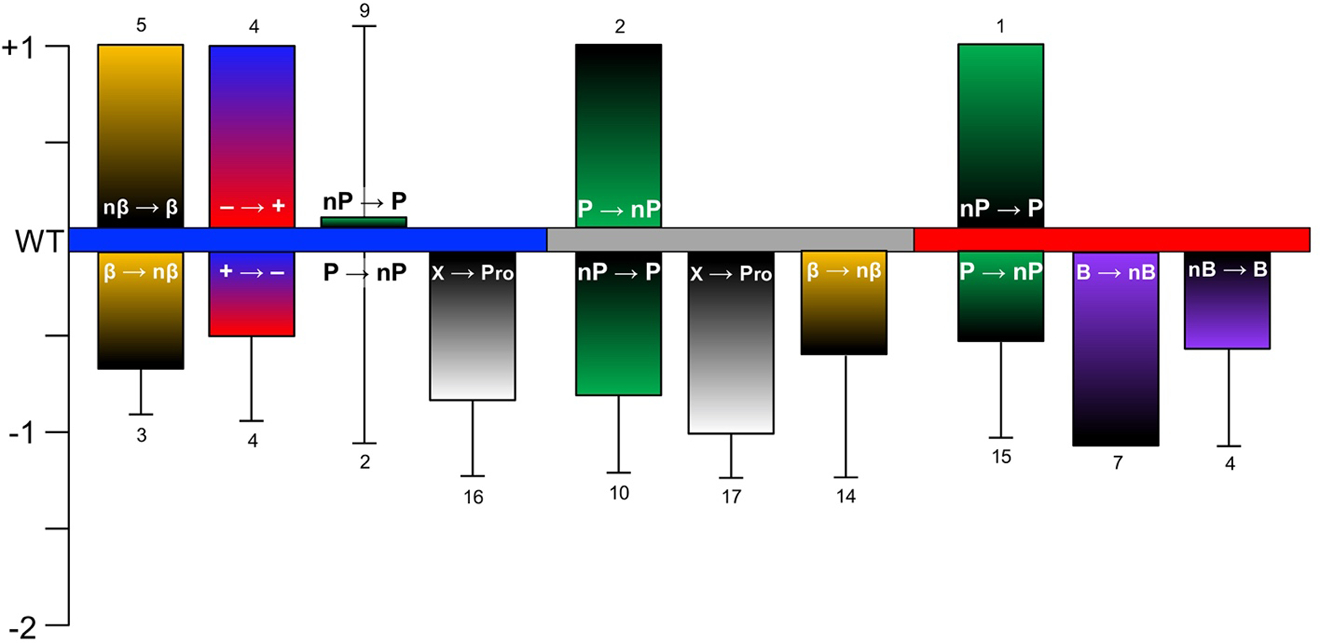
Categorized Mutation Effects Averaged Over αS Regions. N-Terminal, NAC, and C-terminal regions of αS indicated by blue, grey, and red respectively. Bars depict average aggregation scores for each category, unidirectional error bars represent standard deviation, number of examples given above or below bar. Mutation definitions: nβ→β) non-β-branched to β-branched; β→nβ) β-branched to non-β-branched; – → +) negatively charged to positively charged; + → –) positively charged to negatively charged; nP→P) non-polar to polar; P→nP) polar to non-polar; Pro→X) proline to any other amino acid; X→Pro) any amino acid to proline; nB→B) non-bulky to bulky; B→nB) bulky to non-bulky; Cntrl) control groups represent mutation to the same type of amino acid. Category definitions: β) Ile, Thr, Val; nβ) all others except Gly and Pro; +) Arg, His, Lys; –) Asp, Glu; nP) Ala, Ile, Leu, Met, Phe, Trp, Val; P) all others except Gly and Pro; X) all others except Pro; B) His, Ile, Leu, Phe, Thr, Trp, Tyr, Val; nB) all others except Pro.
In examining the mutational trends for the whole protein, three effects stand out. Firstly, one can see that β-branched amino acids clearly favor aggregation, where nβ→β has an average aggregation score of +0.43 and β→nβ has an average score of −0.56, while the nβ control category has a near-WT score of −0.21. This is true for the protein as a whole as well as within the N-terminus and NAC domains. Secondly, conversion of negatively charged residues to positively charged residues significantly favors aggregation (– → +, +1.00) and the converse type of mutation disfavors aggregation (+ → –, −0.50). These data derive primarily from the N-terminus (8 of 10 examples), so comparison across regions is not meaningful. Thirdly, mutation of any amino acid to proline slows aggregation (X→Pro, −0.88) and mutation of proline to other amino acids speeds aggregation (Pro→X, +1.00). This effect is significant for all three regions of αS, although the examples in the C-terminus are limited to 5 Pro→X cases. These are the only categories that appear to be meaningful when scores are averaged over the whole protein. For example, although conversion of bulky to non-bulky residues slows aggregation (B→nB, −0.63), the converse has no effect (nB→B, 0.00) and the bulky control category (B Cntrl, −0.57) has a similar effect, indicating a general sensitivity of those residues to mutation. Indeed, when one examines these effects across the three regions, taking the prevalence of different amino acid types into account (see SI Tables S3–S6), one can see that the bulk effects are predominantly captured by the β-branched effects in the N-terminus and NAC, and that the C-terminal cases show that mutations of the native aromatic amino acids slow aggregation (Figure 10).
Analysis of the mutational effects with respect to the three regions of αS provides greater insight into categories that are inconsequential when analyzed over the entire protein, such as polarity. Changes in polarity in the N-terminus have no impact on aggregation. Increased polarity in the NAC slows aggregation, while increased polarity in the C-terminus speeds aggregation. Thus, one can see that polarity is indeed quite important when viewed in terms of the protein regions and one can also see why these effects negate each other when averaged over the whole protein. Charge swapping effects are very clear in the N-terminus, but the data are too limited in the NAC or C-terminus to draw any conclusions. Likewise, although the influence of β-branched amino acids favoring aggregation is clear in the N-terminus and NAC, there are no C-terminal examples in our data set. The fact that aggregation is slowed by changes to introduce or to remove bulk in the C-terminus, involving mutations of the native aromatic amino acids as well as introduction of non-native aromatic amino acids, implies that this region is sensitive to steric effects that may alter the flexibility of the highly dynamic portion of αS.
Given the clear correlations of some of these quantified effects, we wished to determine whether we could predict whether a mutation would lead to faster, near-WT, or slower aggregation by separating the data into three clusters using an unsupervised machine learning KMeans partitioning algorithm. Once the data were clustered, we projected the data into two dimensions using Principal Component Analysis (PCA) for visualization. As one can see in Figure 11, there is clear separation between the three clusters, but enrichment for the aggregation effects in each cluster is limited. For example, while Cluster 3 (black) includes no examples of accelerated aggregation and a small fraction of near-WT aggregation, Clusters 1 (red) and 2 (blue) have mixtures of all three types and very mild enrichment for near-WT and acceleration, respectively. Indeed, Cluster 2 is enriched for acceleration in the sense that 76% of accelerated cases are included in that cluster, but the cluster is also composed of significant portions of near-WT and slowed aggregation cases. Thus, one can see that using machine learning holds some promise for prediction of aggregation effects, but a more sophisticated approach will be required, presumably using supervised learning.
Figure 11.
Two Dimensional Principal Component Analysis (PCA) of K-means Clustered Aggregation Data. Circle, triangular, and square markers represent mutations which slow aggregation rate, have no effect (near-WT), or accelerate aggregation rate, respectively. The color of each marker represents the cluster assignment from K-means where red is Cluster 1, blue is Cluster 2, and black is Cluster 3. Since mutants may have exactly the same bit vector representation, (e.g., two Val-to-Pro mutations in the N-terminus with aggregation score −1) many data points overlap in PCA space. Marker size is used to indicate the number of data points at that position in PCA space.
Outlook
ThT is useful in understanding the effects of mutations and PTMs on the aggregation kinetics of αS because the assays are easy to implement across many laboratories to obtain consistent data that can be compared, as we have done here. In spite of the issues with ThT that we have discussed, the convenience of a small molecule probe that can be added without the need to label the protein or use specialized techniques still makes it the method of choice for high throughput studies of aggregation effectors. The resulting data can be interpreted in terms of a variety of more sophisticated biophysical, structural, and cell biological assays. We hope that the collection of data that we present here will be valuable to the αS field, where we have attempted to present a consistent categorization of the effects mutation, PTMs, and labeling on aggregation, accounting for the overlaying influence of the experimental conditions.
We also anticipate that the database we have created will be useful to computational modeling. Computational methods are increasingly being used to study protein folding or protein interactions and are particularly useful for intrinsically disordered proteins like αS. For example, machine learning has recently been used to screen many mutants to determine which structures are responsible for particular activities of αS [239]. Monte Carlo methods have been used to rapidly predict αS structural ensembles,[240] and can potentially sample a large number of mutations and modifications to predict structures that will behave differently than WT. While modeling alone may not be sufficiently predictive, experimental data such as the lag times presented in this review can be incorporated into machine learning protocols to better understand the propensity of each residue to cause aggregation. Instead of mutating every residue and testing proteins in vitro, machine learning can produce predictions that inform experimental studies, allowing researchers to focus on mutations or modifications that are thought to have a large impact on aggregation.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (NIH R01-NS103873 to E.J.P). S.X.P. thanks the Vagelos Molecular Life Science Scholars program for support. M.S. thanks the Nakajima Foundation for scholarship funding. R.M.P. thanks the NIH for funding through the Chemistry Biology Interface Training Program (T32 GM133398). S.G.G. thanks the National Science Foundation for funding through the Graduate Research Fellowship Program (DGE-1845298).
References
- [1].Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nature Reviews Disease Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- [2].Meade RM, Fairlie DP, Mason JM. Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Molecular Neurodegeneration. 2019;14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK. α-synuclein aggregation and its modulation. International Journal of Biological Macromolecules. 2017;100:37–54. [DOI] [PubMed] [Google Scholar]
- [4].Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. American Journal of Pathology. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- [5].Goedert M Alpha-synuclein and neurodegenerative diseases. Nature Reviews Neuroscience. 2001;2:492–501. [DOI] [PubMed] [Google Scholar]
- [6].Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–40. [DOI] [PubMed] [Google Scholar]
- [7].Emamzadeh FN. Alpha-synuclein structure, functions, and interactions. J Res Med Sci. 2016;21:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Uversky VN, Fink AL. Amino acid determinants of α-synuclein aggregation: putting together pieces of the puzzle. FEBS Letters. 2002;522:9–13. [DOI] [PubMed] [Google Scholar]
- [9].Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–603. [DOI] [PubMed] [Google Scholar]
- [11].Trexler AJ, Rhoades E. α-Synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry. 2009;48:2304–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Middleton ER, Rhoades E. Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophysical Journal. 2010;99:2279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proceedings of the National Academy of Sciences. 2011;108:17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Iwai A, Masliah E Fau - Yoshimoto M, Yoshimoto M Fau - Ge N, Ge N Fau - Flanagan L, Flanagan L Fau - de Silva HA, de Silva Ha Fau - Kittel A, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. [DOI] [PubMed]
- [16].Benskey MJ, Perez RG, Manfredsson FP. The contribution of alpha synuclein to neuronal survival and function – Implications for Parkinson’s disease. Journal of Neurochemistry. 2016;137:331–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Alam P, Bousset L, Melki R, Otzen DE. α-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. Journal of Neurochemistry. 2019;150:522–34. [DOI] [PubMed] [Google Scholar]
- [18].Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, et al. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 2020;117:4971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. α-Synuclein fibrillogenesis is nucleation-dependent: Implications for the pathogenesis of Parkinson’s disease. Journal of Biological Chemistry. 1999;274:19509–12. [DOI] [PubMed] [Google Scholar]
- [20].Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. Journal of Neurochemistry. 2009;111:192–203. [DOI] [PubMed] [Google Scholar]
- [21].Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Volpicelli-Daley LA, Luk KC, Lee VMY. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nature Protocols. 2014;9:2135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nature Medicine. 2008;14:504–6. [DOI] [PubMed] [Google Scholar]
- [24].Mehra S, Sahay S, Maji SK. alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. BBA-Proteins Proteomics. 2019;1867:890–908. [DOI] [PubMed] [Google Scholar]
- [25].Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guerrero-Ferreira R, Kovacik L, Ni D, Stahlberg H. New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Current Opinion in Neurobiology. 2020;61:89–95. [DOI] [PubMed] [Google Scholar]
- [28].Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TPJ, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Peelaerts W, Baekelandt V. ?-Synuclein strains and the variable pathologies of synucleinopathies. Journal of Neurochemistry. 2016;139:256–74. [DOI] [PubMed] [Google Scholar]
- [30].Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V. Dependence of α-Synuclein Aggregate Morphology on Solution Conditions. Journal of Molecular Biology. 2002;322:383–93. [DOI] [PubMed] [Google Scholar]
- [31].Fagerqvist T, Näsström T, Ihse E, Lindström V, Sahlin C, Fangmark Tucker SM, et al. Off-pathway α-synuclein oligomers seem to alter α-synuclein turnover in a cell model but lack seeding capability in vivo. Amyloid. 2013;20:233–44. [DOI] [PubMed] [Google Scholar]
- [32].Uversky VN, Li J Fau - Fink AL, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. [DOI] [PubMed]
- [33].Scheidt T, Łapińska U, Kumita JR, Whiten DR, Klenerman D, Wilson MR, et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Science Advances. 2019;5:eaau3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, et al. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Quarterly Reviews of Biophysics. 2017;50. [DOI] [PubMed] [Google Scholar]
- [35].Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, et al. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science. 2009;326:1533. [DOI] [PubMed] [Google Scholar]
- [36].Shvadchak VV, Claessens MMAE, Subramaniam V. Fibril Breaking Accelerates α-Synuclein Fibrillization. The Journal of Physical Chemistry B. 2015;119:1912–8. [DOI] [PubMed] [Google Scholar]
- [37].Giehm L, Lorenzen N, Otzen DE. Assays for α-synuclein aggregation. Methods. 2011;53:295–305. [DOI] [PubMed] [Google Scholar]
- [38].Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nature Medicine. 1998;4:1318–20. [DOI] [PubMed] [Google Scholar]
- [39].Conway KA, Harper JD, Lansbury PT Jr. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–63. [DOI] [PubMed] [Google Scholar]
- [40].Giasson BI, Uryu K, Trojanowski JQ, Lee VMY. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. Journal of Biological Chemistry. 1999;274:7619–22. [DOI] [PubMed] [Google Scholar]
- [41].Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–9. [DOI] [PubMed] [Google Scholar]
- [42].LeVine H, 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Coelho-Cerqueira E, Pinheiro AS, Follmer C. Pitfalls associated with the use of Thioflavin-T to monitor anti-fibrillogenic activity. Bioorg Med Chem Lett. 2014;24:3194–8. [DOI] [PubMed] [Google Scholar]
- [44].Sulatskaya AI, Rodina NP, Sulatsky MI, Povarova OI, Antifeeva IA, Kuznetsova IM, et al. Investigation of α-synuclein amyloid fibrils using the fluorescent probe thioflavin T. International Journal of Molecular Sciences. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Arosio P, Knowles TPJ, Linse S. On the lag phase in amyloid fibril formation. Physical Chemistry Chemical Physics. 2015;17:7606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nature Communications. 2013;4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sidhu A, Vaneyck J, Blum C, Segers-Nolten I, Subramaniam V. Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in -synuclein fibrils. Amyloid-J Protein Fold Disord. 2018;25:189–96. [DOI] [PubMed] [Google Scholar]
- [48].de Oliveira GAP, Silva JL. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun Biol. 2019;2:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kumar H, Singh J, Kumari P, Udgaonkar JB. Modulation of the extent of structural heterogeneity in -synuclein fibrils by the small molecule thioflavin T. Journal of Biological Chemistry. 2017;292:16891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Giehm L, Otzen DE. Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal Biochem. 2010;400:270–81. [DOI] [PubMed] [Google Scholar]
- [51].Pronchik J, He X, Giurleo JT, Talaga DS. In vitro formation of amyloid from α-synuclein is dominated by reactions at hydrophobic interfaces. J Am Chem Soc. 2010;132:9797–803. [DOI] [PubMed] [Google Scholar]
- [52].Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;578:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018;557:558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4. [DOI] [PubMed] [Google Scholar]
- [55].Peduzzo A, Linse S, Buell AK. The Properties of α-Synuclein Secondary Nuclei Are Dominated by the Solution Conditions Rather than the Seed Fibril Strain. ACS Chemical Neuroscience. 2020;11:909–18. [DOI] [PubMed] [Google Scholar]
- [56].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23:409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nature Communications. 2018;9:3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Guerrero-Ferreira R, Taylor NMI, Arteni A-A, Kumari P, Mona D, Ringler P, et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife. 2019;8:e48907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Guerrero-Ferreira R, Taylor NMI, Mona D, Ringler P, Lauer ME, Riek R, et al. Cryo-EM structure of alpha-synuclein fibrils. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, et al. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Research. 2018;28:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sun Y, Long H, Xia W, Wang K, Zhang X, Sun B, et al. The hereditary mutation G51D unlocks a distinct fibril strain transmissible to wild-type α-synuclein. Nature Communications. 2021;12:6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, et al. Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat Struct Mol Biol. 2019;26:1044–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sun Y, Hou S, Zhao K, Long H, Liu Z, Gao J, et al. Cryo-EM structure of full-length α-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Research. 2020;30:360–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nature Communications. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Boyer DR, Li B, Sun C, Fan W, Zhou K, Hughes MP, et al. The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:3592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. [DOI] [PubMed] [Google Scholar]
- [67].Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nature Genetics. 1998;18:106–8. [DOI] [PubMed] [Google Scholar]
- [68].Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Annals of Neurology. 2004;55:164–73. [DOI] [PubMed] [Google Scholar]
- [69].Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Movement Disorders. 2013;28:811–3. [DOI] [PubMed] [Google Scholar]
- [70].Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel α-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiology of Aging. 2014;35:2180.e1-.e5. [DOI] [PubMed] [Google Scholar]
- [72].Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D alpha-Synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Annals of Neurology. 2013;73:459–71. [DOI] [PubMed] [Google Scholar]
- [73].Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol Aging. 2017;57:248.e7-.e12. [DOI] [PubMed] [Google Scholar]
- [74].Flagmeier P, Meisl G, Vendruscolo M, Knowles TPJ, Dobson CM, Buell AK, et al. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:10328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rutherford NJ, Moore BD, Golde TE, Giasson BI. Divergent effects of the H5OQ and G51D SNCA mutations on the aggregation of alpha-synuclein. Journal of Neurochemistry. 2014;131:859–67. [DOI] [PubMed] [Google Scholar]
- [76].Mohite GM, Kumar R, Panigrahi R, Navalkar A, Singh N, Datta D, et al. Comparison of Kinetics, Toxicity, Oligomer Formation, and Membrane Binding Capacity of alpha-Synuclein Familial Mutations at the A53 Site, Including the Newly Discovered A53V Mutation. Biochemistry. 2018;57:5183–7. [DOI] [PubMed] [Google Scholar]
- [77].Alza NP, Iglesias González PA, Conde MA, Uranga RM, Salvador GA. Lipids at the Crossroad of α-Synuclein Function and Dysfunction: Biological and Pathological Implications. Frontiers in Cellular Neuroscience. 2019;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Aguirre C, Ikenaka K, Mochizuki H. Two New Polymorphs in H50Q Determined by CryoEM Suggest a Mechanism That Explains Its Faster Kinetics In Vitro. Movement Disorders. 2020. [DOI] [PubMed] [Google Scholar]
- [79].Nielsen SB, Macchi F, Raccosta S, Langkilde AE, Giehm L, Kyrsting A, et al. Wildtype and A30P mutant alpha-synuclein form different fibril structures. PLoS One. 2013;8:e67713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hayakawa H, Nakatani R, Ikenaka K, Aguirre C, Choong CJ, Tsuda H, et al. Structurally distinct alpha-synuclein fibrils induce robust parkinsonian pathology. Movement Disorders. 2020;35:256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sidhu A, Segers-Nolten I, Subramaniam V. Conformational Compatibility Is Essential for Heterologous Aggregation of alpha-Synuclein. Acs Chemical Neuroscience. 2016;7:719–27. [DOI] [PubMed] [Google Scholar]
- [82].Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hoffman-Zacharska D, Koziorowski D, Ross OA, Milewski M, Poznański J, Jurek M, et al. Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism & Related Disorders. 2013;19:1057–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kumar S, Jangir DK, Kumar R, Kumari M, Bhavesh NS, Maiti TK. Role of Sporadic Parkinson Disease Associated Mutations A18T and A29S in Enhanced alpha-Synuclein Fibrillation and Cytotoxicity. Acs Chemical Neuroscience. 2018;9:230–40. [DOI] [PubMed] [Google Scholar]
- [85].Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology. 2002;4:160–4. [DOI] [PubMed] [Google Scholar]
- [86].Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. Journal of Biological Chemistry. 2006;281:29739–52. [DOI] [PubMed] [Google Scholar]
- [87].Chen H, Zhao YF, Chen YX, Li YM. Exploring the Roles of Post-Translational Modifications in the Pathogenesis of Parkinson’s Disease Using Synthetic and Semisynthetic Modified alpha-Synuclein. Acs Chemical Neuroscience. 2019;10:910–21. [DOI] [PubMed] [Google Scholar]
- [88].Nonaka T, Iwatsubo T, Hasegawa M. Ubiquitination of α-synuclein. Biochemistry. 2005;44:361–8. [DOI] [PubMed] [Google Scholar]
- [89].Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–9. [DOI] [PubMed] [Google Scholar]
- [90].Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 2006;281:9919–24. [DOI] [PubMed] [Google Scholar]
- [91].Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci U S A. 2012;109:7280–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wang Z, Park K, Comer F, Hsieh-Wilson LC, Saudek CD, Hart GW. Site-Specific GlcNAcylation of Human Erythrocyte Proteins: Potential Biomarker(s) for Diabetes. Diabetes. 2009;58:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Vicente Miranda H, Szego EM, Oliveira LMA, Breda C, Darendelioglu E, de Oliveira RM, et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain. 2017;140:1399–419. [DOI] [PubMed] [Google Scholar]
- [94].Sanyal A, Dutta S, Camara A, Chandran A, Koller A, Watson BG, et al. Alpha-Synuclein Is a Target of Fic-Mediated Adenylylation/AMPylation: Possible Implications for Parkinson’s Disease. Journal of Molecular Biology. 2019;431:2266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human α-synuclein in vitro. FEBS Letters. 2002;517:239–44. [DOI] [PubMed] [Google Scholar]
- [96].Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J Biol Chem. 2007;282:5862–70. [DOI] [PubMed] [Google Scholar]
- [97].Wang J, Han X, Leu NA, Sterling S, Kurosaka S, Fina M, et al. Protein arginylation targets alpha synuclein, facilitates normal brain health, and prevents neurodegeneration. Scientific Reports. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Maltsev AS, Ying J, Bax A. Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry. 2012;51:5004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Burai R, Ait-Bouziad N, Chiki A, Lashuel HA. Elucidating the Role of Site-Specific Nitration of alpha-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J Am Chem Soc. 2015;137:5041–52. [DOI] [PubMed] [Google Scholar]
- [100].Hejjaoui M, Butterfield S, Fauvet B, Vercruysse F, Cui J, Dikiy I, et al. Elucidating the Role of C-Terminal Post-Translational Modifications Using Protein Semisynthesis Strategies: alpha-Synuclein Phosphorylation at Tyrosine 125. J Am Chem Soc. 2012;134:5196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Dikiy I, Fauvet B, Jovičić A, Mahul-Mellier AL, Desobry C, El-Turk F, et al. Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by PD Mutations. ACS Chem Biol. 2016;11:2428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pan Y, Rhoades E, Petersson EJ. Chemoenzymatic Semisynthesis of Phosphorylated alpha-Synuclein Enables Identification of a Bidirectional Effect on Fibril Formation. ACS Chem Biol. 2020;15:640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Meier F, Abeywardana T, Dhall A, Marotta NP, Varkey J, Langen R, et al. Semisynthetic, Site-Specific Ubiquitin Modification of alpha-Synuclein Reveals Differential Effects on Aggregation. J Am Chem Soc. 2012;134:5468–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hejjaoui M, Haj-Yahya M, Kumar KSA, Brik A, Lashuel HA. Towards Elucidation of the Role of Ubiquitination in the Pathogenesis of Parkinson’s Disease with Semisynthetic Ubiquitinated alpha-Synuclein. Angew Chem-Int Edit. 2011;50:405–9. [DOI] [PubMed] [Google Scholar]
- [105].Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, Hejjaoui M, Bavikar SN, Karthikeyan SV, et al. Synthetic polyubiquitinated α-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proceedings of the National Academy of Sciences. 2013;110:17726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, et al. Sumoylation inhibits α-synuclein aggregation and toxicity. Journal of Cell Biology. 2011;194:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lewis YE, Abeywardana T, Lin YH, Galesic A, Pratt MR. Synthesis of a Bis-thio-acetone (BTA) Analogue of the Lysine Isopeptide Bond and its Application to Investigate the Effects of Ubiquitination and SUMOylation on alpha-Synuclein Aggregation and Toxicity. ACS Chem Biol. 2016;11:931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, et al. O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson’s disease. Nature Chemistry. 2015;7:913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pan B, Kamo N, Shimogawa M, Huang Y, Kashina A, Rhoades E, et al. Effects of Glutamate Arginylation on α-Synuclein: Studying an Unusual Post-Translational Modification through Semisynthesis. J Am Chem Soc. 2020;142:21786–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D, et al. Characterization of Semisynthetic and Naturally N-alpha-Acetylated alpha-Synuclein in Vitro and in Intact Cells IMPLICATIONS FOR AGGREGATION AND CELLULAR PROPERTIES OF alpha-SYNUCLEIN. Journal of Biological Chemistry. 2012;287:28243–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. Journal of Biological Chemistry. 2008;283:16895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Koo H-J, Choi MY, Im H. Aggregation-defective α-synuclein mutants inhibit the fibrillation of Parkinson’s disease-linked α-synuclein variants. Biochem Biophys Res Commun. 2009;386:165–9. [DOI] [PubMed] [Google Scholar]
- [113].Haney CM, Petersson EJ. Fluorescence spectroscopy reveals N-terminal order in fibrillar forms of α-synuclein. Chem Commun. 2018;54:833–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ruf VC, Nubling GS, Willikens S, Shi S, Schmidt F, Levin J, et al. Different Effects of alpha-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. Acs Chemical Neuroscience. 2019;10:1649–59. [DOI] [PubMed] [Google Scholar]
- [115].Brucale M, Sandal M, Di Maio S, Rampioni A, Tessari I, Tosatto L, et al. Pathogenic Mutations Shift the Equilibria of alpha-Synuclein Single Molecules towards Structured Conformers. ChemBioChem. 2009;10:176–83. [DOI] [PubMed] [Google Scholar]
- [116].Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, et al. The E46K mutation in α-synuclein increases amyloid fibril formation. Journal of Biological Chemistry. 2005;280:7800–7. [DOI] [PubMed] [Google Scholar]
- [117].Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, et al. Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nature Communications. 2020;11:2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Sahay S, Ghosh D, Dwivedi S, Anoop A, Mohite GM, Kombrabail M, et al. Familial Parkinson disease-associated mutations alter the site-specific microenvironment and dynamics of α-synuclein. J Biol Chem. 2015;290:7804–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40:11604–13. [DOI] [PubMed] [Google Scholar]
- [120].Li J, Uversky VN, Fink AL. Conformational behavior of human alpha-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology. 2002;23:553–67. [DOI] [PubMed] [Google Scholar]
- [121].Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Anthony Crowther R, et al. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Letters. 2004;576:363–8. [DOI] [PubMed] [Google Scholar]
- [122].Sode K, Usuzaka E, Kobayashi N, Ochial S. Engineered alpha-synuclein prevents wild type and familial Parkin variant fibril formation. Biochem Biophys Res Commun. 2005;335:432–6. [DOI] [PubMed] [Google Scholar]
- [123].Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, et al. The parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry. 2013;52:6925–7. [DOI] [PubMed] [Google Scholar]
- [124].Ghosh D, Sahay S, Ranjan P, Salot S, Mohite GM, Singh PK, et al. The newly discovered Parkinsons disease associated finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry. 2014;53:6419–21. [DOI] [PubMed] [Google Scholar]
- [125].Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43:16233–42. [DOI] [PubMed] [Google Scholar]
- [126].Sidhu A, Segers-Nolten I, Raussens V, Claessens MMAE, Subramaniam V. Distinct Mechanisms Determine α-Synuclein Fibril Morphology during Growth and Maturation. ACS Chemical Neuroscience. 2017;8:538–47. [DOI] [PubMed] [Google Scholar]
- [127].Tosatto L, Horrocks MH, Dear AJ, Knowles TPJ, Dalla Serra M, Cremades N, et al. Single-molecule FRET studies on alpha-synuclein oligomerization of Parkinson’s disease genetically related mutants. Scientific Reports. 2015;5:16696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, et al. The H50Q Mutation Enhances alpha-Synuclein Aggregation, Secretion, and Toxicity. Journal of Biological Chemistry. 2014;289:21856–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Koo HJ, Lee HJ, Im H. Sequence determinants regulating fibrillation of human alpha-synuclein. Biochem Biophys Res Commun. 2008;368:772–8. [DOI] [PubMed] [Google Scholar]
- [130].Guan Y, Zhao X, Liu F, Yan S, Wang Y, Du C, et al. Pathogenic Mutations Differentially Regulate Cell-to-Cell Transmission of α-Synuclein. Frontiers in Cellular Neuroscience. 2020;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Karpinar DP, Balija MBG, Kugler S, Opazo F, Rezaei-Ghaleh N, Wender N, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. Embo J. 2009;28:3256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Lazaro DF, Dias MC, Carija A, Navarro S, Madaleno CS, Tenreiro S, et al. The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol Commun. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lemkau LR, Comellas G, Kloepper KD, Woods WS, George JM, Rienstra CM. Mutant Protein A30P alpha-Synuclein Adopts Wild-type Fibril Structure, Despite Slower Fibrillation Kinetics. Journal of Biological Chemistry. 2012;287:11526–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Bhattacharyya D, Kumar R, Mehra S, Ghosh A, Maji SK, Bhunia A. Multitude NMR studies of α-synuclein familial mutants: probing their differential aggregation propensities. Chem Commun. 2018;54:3605–8. [DOI] [PubMed] [Google Scholar]
- [135].Stsiapura VI, Maskevich AA, Kuzmitsky VA, Uversky VN, Kuznetsova IM, Turoverov KK. Thioflavin T as a Molecular Rotor: Fluorescent Properties of Thioflavin T in Solvents with Different Viscosity. The Journal of Physical Chemistry B. 2008;112:15893–902. [DOI] [PubMed] [Google Scholar]
- [136].Zweckstetter M, Karpinar P, Griesinger C. Mutant alpha-synuclein, and methods using same. Google Patents; 2016. [Google Scholar]
- [137].Waxman EA, Giasson BI. A novel, high-efficiency cellular model of fibrillar alpha-synuclein inclusions and the examination of mutations that inhibit amyloid formation. Journal of Neurochemistry. 2010;113:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Waxman EA, Mazzulli JR, Giasson BI. Characterization of Hydrophobic Residue Requirements for alpha-Synuclein Fibrillization. Biochemistry. 2009;48:9427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, et al. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry. 2007;46:7107–18. [DOI] [PubMed] [Google Scholar]
- [140].Harada R, Kobayashi N, Kim J, Nakamura C, Han SW, Ikebukuro K, et al. The effect of amino acid substitution in the imperfect repeat sequences of alpha-synuclein on fibrillation. Biochim Biophys Acta-Mol Basis Dis. 2009;1792:998–1003. [DOI] [PubMed] [Google Scholar]
- [141].Ranjan P, Kumar A. Perturbation in Long-Range Contacts Modulates the Kinetics of Amyloid Formation in α-Synuclein Familial Mutants. ACS Chemical Neuroscience. 2017;8:2235–46. [DOI] [PubMed] [Google Scholar]
- [142].Rospigliosi CC, McClendon S, Schmid AW, Ramlall TF, Barré P, Lashuel HA, et al. E46K Parkinson’s-Linked Mutation Enhances C-Terminal-to-N-Terminal Contacts in α-Synuclein. Journal of Molecular Biology. 2009;388:1022–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kessler JC, Rochet JC, Lansbury PT, Jr. The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–8. [DOI] [PubMed] [Google Scholar]
- [144].Chi YC, Armstrong GS, Jones DN, Eisenmesser EZ, Liu CW. Residue histidine 50 plays a key role in protecting α-synuclein from aggregation at physiological pH. J Biol Chem. 2014;289:15474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Dudzik CG, Walter ED, Millhauser GL. Coordination Features and Affinity of the Cu2+ Site in the α-Synuclein Protein of Parkinson’s Disease. Biochemistry. 2011;50:1771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Bae E-J, Ho D-H, Park E, Jung JW, Cho K, Hong JH, et al. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of α-synuclein. Antioxid Redox Signal. 2013;18:770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Xiang W, Menges S, Schlachetzki JCM, Meixner H, Hoffmann AC, Schlotzer-Schrehardt U, et al. Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Molecular Neurodegeneration. 2015;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Fares M-B, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovičić A, Kiely A, et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Human Molecular Genetics. 2014;23:4491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J. Structural reorganization of α-synuclein at low pH observed by NMR and REMD simulations. J Mol Biol. 2009;391:784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Du HN, Tang L, Luo XY, Li HT, Hu J, Zhou JW, et al. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry. 2003;42:8870–8. [DOI] [PubMed] [Google Scholar]
- [151].Sode K, Ochiai S, Kobayashi N, Usuzaka E. Effect of reparation of repeat sequences in the human alpha-synuclein on fibrillation ability. Int J Biol Sci. 2007;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Nathan Kochen N, Vasandani V, Seaney D, Pandey AK, Walters MA, Braun AR, et al. Threonine Cavities Are Targetable Motifs That Control Alpha-Synuclein Fibril Growth. ACS Chemical Neuroscience. 2022;13:2646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Levitan K, Chereau D, Cohen SIA, Knowles TPJ, Dobson CM, Fink AL, et al. Conserved C-Terminal Charge Exerts a Profound Influence on the Aggregation Rate of alpha-Synuclein. Journal of Molecular Biology. 2011;411:329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Hokenson MJ, Uversky VN, Goers J, Yamin G, Munishkina LA, Fink AL. Role of Individual Methionines in the Fibrillation of Methionine-Oxidized α-Synuclein. Biochemistry. 2004;43:4621–33. [DOI] [PubMed] [Google Scholar]
- [155].Afitska K, Fucikova A, Shvadchak VV, Yushchenko DA. Modification of C Terminus Provides New Insights into the Mechanism of α-Synuclein Aggregation. Biophysical Journal. 2017;113:2182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Izawa Y, Tateno H, Kameda H, Hirakawa K, Hato K, Yagi H, et al. Role of C-terminal negative charges and tyrosine residues in fibril formation of alpha-synuclein. Brain Behav. 2012;2:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Mučibabić M, Apetri MM, Canters GW, Aartsma TJ. The effect of fluorescent labeling on α-synuclein fibril morphology. Biochimica et Biophysica Acta - Proteins and Proteomics. 2016;1864:1419–27. [DOI] [PubMed] [Google Scholar]
- [158].Barinova KV, Kuraysky ML, Arutyunyan AM, Serebryakova MV, Schmalhausen EV, Muronetz VI. Dimerization of Tyr136Cys alpha-synuclein prevents amyloid transformation of wild type alpha-synuclein. International Journal of Biological Macromolecules. 2017;96:35–43. [DOI] [PubMed] [Google Scholar]
- [159].Meuvis J, Gerard M, Desender L, Baekelandt V, Engelborghs Y. The Conformation and the Aggregation Kinetics of alpha-Synuclein Depend on the Proline Residues in Its C-Terminal Region. Biochemistry. 2010;49:9345–52. [DOI] [PubMed] [Google Scholar]
- [160].Zhou W, Long C, Reaney SH, Di Monte DA, Fink AL, Uversky VN. Methionine oxidation stabilizes non-toxic oligomers of α-synuclein through strengthening the auto-inhibitory intra-molecular long-range interactions. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2010;1802:322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ulrih NP, Barry CH, Fink AL. Impact of Tyr to Ala mutations on alpha-synuclein fibrillation and structural properties. Biochim Biophys Acta-Mol Basis Dis. 2008;1782:581–5. [DOI] [PubMed] [Google Scholar]
- [162].Schreurs S, Gerard M, Derua R, Waelkens E, Taymans J-M, Baekelandt V, et al. In vitro phosphorylation does not influence the aggregation kinetics of WT α-synuclein in contrast to its phosphorylation mutants. International journal of molecular sciences. 2014;15:1040–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Zhang JM, Li XP, Li JD. The Roles of Post-translational Modifications on alpha-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front Neurosci. 2019;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. Journal of Neuroscience. 2010;30:3184–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Pan B, Park JH, Ramlall T, Eliezer D, Rhoades E, Petersson EJ. Chemoenzymatic Semi-synthesis Enables Efficient Production of Isotopically Labeled α-Synuclein with Site-Specific Tyrosine Phosphorylation. ChemBioChem. 2021;22:1440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zhao K, Lim Y-J, Liu Z, Long H, Sun Y, Hu J-J, et al. Parkinson’s disease-related phosphorylation at Tyr39 rearranges alpha-synuclein amyloid fibril structure revealed by cry-EM. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:20305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Deng S, Pan B, Gottlieb L, Petersson EJ, Marmorstein R. Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. Elife. 2020;9:e57491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Science. 2012;21:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Moriarty GM, Minetti C, Remeta DP, Baum J. A Revised Picture of the Cu(II)-alpha-Synuclein Complex: The Role of N-Terminal Acetylation. Biochemistry. 2014;53:2815–7. [DOI] [PubMed] [Google Scholar]
- [170].Watson MD, Lee JC. N-Terminal Acetylation Affects alpha-Synuclein Fibril Polymorphism. Biochemistry. 2019;58:3630–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Bartels T, Kim NC, Luth ES, Selkoe DJ. N-Alpha-Acetylation of alpha-Synuclein Increases Its Helical Folding Propensity, GM1 Binding Specificity and Resistance to Aggregation. Plos One. 2014;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].de Oliveira RM, Vicente Miranda H, Francelle L, Pinho R, Szegö ÉM, Martinho R, et al. The mechanism of sirtuin 2–mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLOS Biology. 2017;15:e2000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Moon SP, Balana AT, Pratt MR. Consequences of post-translational modifications on amyloid proteins as revealed by protein semisynthesis. Current Opinion in Chemical Biology. 2021;64:76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, Hejjaoui M, et al. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Molecular Cell. 2012;48:87–97. [DOI] [PubMed] [Google Scholar]
- [176].Abeywardana T, Lin Yu H, Rott R, Engelender S, Pratt Matthew R. Site-Specific Differences in Proteasome-Dependent Degradation of Monoubiquitinated α-Synuclein. Chemistry & Biology. 2013;20:1207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Plotegher N, Bubacco L. Lysines, Achilles’ heel in alpha-synuclein conversion to a deadly neuronal endotoxin. Ageing Research Reviews. 2016;26:62–71. [DOI] [PubMed] [Google Scholar]
- [178].Abeywardana T, Pratt MR. Extent of Inhibition of alpha-Synuclein Aggregation in Vitro by SUMOylation Is Conjugation Site- and SUMO Isoform-Selective. Biochemistry. 2015;54:959–61. [DOI] [PubMed] [Google Scholar]
- [179].Vicente Miranda H, El-Agnaf OMA, Outeiro TF. Glycation in Parkinson’s disease and Alzheimer’s disease. Movement Disorders. 2016;31:782–90. [DOI] [PubMed] [Google Scholar]
- [180].Glomb MA, Pfahler C. Amides are novel protein modifications formed by physiological sugars. J Biol Chem. 2001;276:41638–47. [DOI] [PubMed] [Google Scholar]
- [181].Glomb MA, Pfahler C. Amides are novel protein modifications formed by physiological sugars. International Congress Series. 2002;1245:501–2. [DOI] [PubMed] [Google Scholar]
- [182].Degenhardt TP, Thorpe SR, Baynes JW. Chemical modification of proteins by methylglyoxal. Cell Mol Biol (Noisy-le-grand: ). 1998;44:1139–45. [PubMed] [Google Scholar]
- [183].Padmaraju V, Bhaskar Jj Fau - Prasada Rao UJS, Prasada Rao Uj Fau - Salimath PV, Salimath Pv Fau - Rao KS, Rao KS. Role of advanced glycation on aggregation and DNA binding properties of α-synuclein. [DOI] [PubMed]
- [184].Lee D, Park CW, Paik SR, Choi KY. The modification of α-synuclein by dicarbonyl compounds inhibits its fibril-forming process. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2009;1794:421–30. [DOI] [PubMed] [Google Scholar]
- [185].Mariño L, Ramis R, Casasnovas R, Ortega-Castro J, Vilanova B, Frau J, et al. Unravelling the effect of N(ε)-(carboxyethyl)lysine on the conformation, dynamics and aggregation propensity of α-synuclein. Chemical Science. 2020;11:3332–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Chen L, Wei Y, Wang X, He R. Ribosylation Rapidly Induces α-Synuclein to Form Highly Cytotoxic Molten Globules of Advanced Glycation End Products. PLOS ONE. 2010;5:e9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, et al. O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem Biol. 2017;12:1020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Zhang JM, Lei HZ, Chen YB, Ma YT, Jiang F, Tan JQ, et al. Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci Lett. 2017;655:90–4. [DOI] [PubMed] [Google Scholar]
- [189].Levine PM, Galesic A, Balana AT, Mahul-Mellier AL, Navarro MX, De Leon CA, et al. alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:1511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Galesic A, Rakshit A, Cutolo G, Pacheco RP, Balana AT, Moon SP, et al. Comparison of N-Acetyl-Glucosamine to Other Monosaccharides Reveals Structural Differences for the Inhibition of α-Synuclein Aggregation. ACS Chem Biol. 2021;16:14–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Wördehoff MM, Shaykhalishahi H, Groß L, Gremer L, Stoldt M, Buell AK, et al. Opposed Effects of Dityrosine Formation in Soluble and Aggregated α-Synuclein on Fibril Growth. Journal of Molecular Biology. 2017;429:3018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. Febs Letters. 2003;542:147–52. [DOI] [PubMed] [Google Scholar]
- [193].Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, et al. Effects of nitration on the structure and aggregation of alpha-synuclein. Mol Brain Res. 2005;134:84–102. [DOI] [PubMed] [Google Scholar]
- [194].Xiang W, Schlachetzki JCM, Helling S, Bussmann JC, Berlinghof M, Schaffer TE, et al. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci. 2013;54:71–83. [DOI] [PubMed] [Google Scholar]
- [195].Liu Y, Qiang M, Wei Y, He R. A novel molecular mechanism for nitrated α-synuclein-induced cell death. Journal of Molecular Cell Biology. 2011;3:239–49. [DOI] [PubMed] [Google Scholar]
- [196].Long T, Liu L, Tao Y, Zhang W, Quan J, Zheng J, et al. Light-Controlled Tyrosine Nitration of Proteins. Angewandte Chemie International Edition. 2021;60:13414–22. [DOI] [PubMed] [Google Scholar]
- [197].Chavarría C, Souza JM. Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Archives of Biochemistry and Biophysics. 2013;533:25–32. [DOI] [PubMed] [Google Scholar]
- [198].Carmo-Gonçalves P, Pinheiro AS, Romão L, Cortines J, Follmer C. UV-induced selective oxidation of Met5 to Met-sulfoxide leads to the formation of neurotoxic fibril-incompetent α-synuclein oligomers. Amyloid. 2014;21:163–74. [DOI] [PubMed] [Google Scholar]
- [199].Yamin G, Glaser CB, Uversky VN, Fink AL. Certain metals trigger fibrillation of methionine-oxidized alpha-synuclein. J Biol Chem. 2003;278:27630–5. [DOI] [PubMed] [Google Scholar]
- [200].Näsström T, Fagerqvist T, Barbu M, Karlsson M, Nikolajeff F, Kasrayan A, et al. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radical Biology and Medicine. 2011;50:428–37. [DOI] [PubMed] [Google Scholar]
- [201].Näsström T, Wahlberg T, Karlsson M, Nikolajeff F, Lannfelt L, Ingelsson M, et al. The lipid peroxidation metabolite 4-oxo-2-nonenal cross-links α-synuclein causing rapid formation of stable oligomers. Biochem Biophys Res Commun. 2009;378:872–6. [DOI] [PubMed] [Google Scholar]
- [202].Shimogawa M, Petersson EJ. New strategies for fluorescently labeling proteins in the study of amyloids. Current Opinion in Chemical Biology. 2021;64:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR. α-synuclein structures from fluorescence energy-transfer kinetics: Implications for the role of the protein in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Trexler AJ, Rhoades E. Single Molecule Characterization of α-Synuclein in Aggregation-Prone States. Biophysical Journal. 2010;99:3048–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Veldhuis G, Segers-Nolten I, Ferlemann E, Subramaniam V. Single-Molecule FRET Reveals Structural Heterogeneity of SDS-Bound α-Synuclein. ChemBioChem. 2009;10:436–9. [DOI] [PubMed] [Google Scholar]
- [206].Haney CM, Cleveland CL, Wissner RF, Owei L, Robustelli J, Daniels MJ, et al. Site-Specific Fluorescence Polarization for Studying the Disaggregation of α-Synuclein Fibrils by Small Molecules. Biochemistry. 2017;56:683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Luk KC, Hyde EG, Trojanowski JQ, Lee VMY. Sensitive Fluorescence Polarization Technique for Rapid Screening of α-Synuclein Oligomerization/Fibrillization Inhibitors. Biochemistry. 2007;46:12522–9. [DOI] [PubMed] [Google Scholar]
- [208].Flynn JD, Gimmen MY, Dean DN, Lacy SM, Lee JC. Terminal Alkynes as Raman Probes of α-Synuclein in Solution and in Cells. ChemBioChem. 2020;21:1582–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Nath S, Meuvis J, Hendrix J, Carl SA, Engelborghs Y. Early Aggregation Steps in alpha-Synuclein as Measured by FCS and FRET: Evidence for a Contagious Conformational Change. Biophysical Journal. 2010;98:1302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. 2006;90:4692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Roberti MJ, Bertoncini CW, Klement R, Jares-Erijman EA, Jovin TM. Fluorescence imaging of amyloid formation in living cells by a functional, tetracysteine-tagged α-synuclein. Nature Methods. 2007;4:345–51. [DOI] [PubMed] [Google Scholar]
- [212].Jun JV, Haney CM, Karpowicz RJ, Giannakoulias S, Lee VMY, Petersson EJ, et al. A “Clickable” Photoconvertible Small Fluorescent Molecule as a Minimalist Probe for Tracking Individual Biomolecule Complexes. J Am Chem Soc. 2019;141:1893–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Karpowicz RJ Jr., Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VMY. Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. Journal of Biological Chemistry. 2017;292:13482–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Marotta NP, Ara J, Uemura N, Lougee MG, Meymand ES, Zhang B, et al. Alpha-synuclein from patient Lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta Neuropathol Commun. 2021;9:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Wissner RF, Wagner AM, Warner JB, Petersson EJ. Efficient, Traceless Semi-Synthesis of α-Synuclein Labeled with a Fluoro-phore/Thioamide FRET Pair. Synlett. 2013;24:2454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Batjargal S, Huang Y, Wang YJ, Petersson EJ. Synthesis of thioester peptides for the incorporation of thioamides into proteins by native chemical ligation. J Pept Sci. 2014;20:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Batjargal S, Wang YJ, Goldberg JM, Wissner RF, Petersson EJ. Native chemical ligation of thioamide-containing peptides: Development and application to the synthesis of labeledα-synuclein for misfolding studies. J Am Chem Soc. 2012;134:9172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Wissner RF, Batjargal S, Fadzen CM, Petersson EJ. Labeling proteins with fluorophore/thioamide Förster resonant energy transfer pairs by combining unnatural amino acid mutagenesis and native chemical ligation. J Am Chem Soc. 2013;135:6529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Thirunavukkuarasu S, Jares-Erijman EA, Jovin TM. Multiparametric Fluorescence Detection of Early Stages in the Amyloid Protein Aggregation of Pyrene-labeled α-Synuclein. Journal of Molecular Biology. 2008;378:1064–73. [DOI] [PubMed] [Google Scholar]
- [220].Marvian AT, Aliakbari F, Mohammad-Beigi H, Ahmadi ZA, Mehrpooyan S, Lermyte F, et al. The status of the terminal regions of α-synuclein in different forms of aggregates during fibrillization. International Journal of Biological Macromolecules. 2020;155:543–50. [DOI] [PubMed] [Google Scholar]
- [221].Haney CM, Wissner RF, Warner JB, Wang YJ, Ferrie JJ, J. Covell D, et al. Comparison of strategies for non-perturbing labeling of α-synuclein to study amyloidogenesis. Organic & Biomolecular Chemistry. 2016;14:1584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Engelborghs Y The analysis of time resolved protein fluorescence in multi-tryptophan proteins. Spectrochimica Acta - Part A Molecular and Biomolecular Spectroscopy. 2001;57:2255–70. [DOI] [PubMed] [Google Scholar]
- [223].Dusa A, Kaylor J, Edridge S, Bodner N, Hong D-P, Fink AL. Characterization of Oligomers during α-Synuclein Aggregation Using Intrinsic Tryptophan Fluorescence. Biochemistry. 2006;45:2752–60. [DOI] [PubMed] [Google Scholar]
- [224].Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in α-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F α-synuclein. Journal of Molecular Biology. 2005;353:357–72. [DOI] [PubMed] [Google Scholar]
- [225].van Ham TJ, Esposito A, Kumita JR, Hsu S-TD, Kaminski Schierle GS, Kaminski CF, et al. Towards Multiparametric Fluorescent Imaging of Amyloid Formation: Studies of a YFP Model of α-Synuclein Aggregation. Journal of Molecular Biology. 2010;395:627–42. [DOI] [PubMed] [Google Scholar]
- [226].Afitska K, Fucikova A, Shvadchak VV, Yushchenko DA. Modification of C Terminus Provides New Insights into the Mechanism of α-Synuclein Aggregation. Biophysical Journal. 2017;113:2182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Dhavale DD, Tsai C, Bagchi DP, Engel LA, Sarezky J, Kotzbauer PT. A sensitive assay reveals structural requirements for α-synuclein fibril growth. Journal of Biological Chemistry. 2017;292:9034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Frontiers in Molecular Neuroscience. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].M S, R CC, E D. Charge Neutralization and Collapse of the C-terminal Tail of Alpha-Synuclein at Low pH. Protein science : a publication of the Protein Society. 2009;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Porcari R, Proukakis C, Waudby CA, Bolognesi B, Mangione PP, Paton JFS, et al. The H50Q Mutation Induces a 10-fold Decrease in the Solubility of α-Synuclein *. Journal of Biological Chemistry. 2015;290:2395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, et al. The Parkinson’s Disease-Associated H50Q Mutation Accelerates α-Synuclein Aggregation in Vitro. Biochemistry. 2013;52:6925–7. [DOI] [PubMed] [Google Scholar]
- [232].Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, et al. The Impact of the E46K Mutation on the Properties of α-Synuclein in Its Monomeric and Oligomeric States. Biochemistry. 2007;46:7107–18. [DOI] [PubMed] [Google Scholar]
- [233].Bertoncini CW, Fernandez CO, Griesinger C, Jovin TM, Zweckstetter M. Familial Mutants of α-Synuclein with Increased Neurotoxicity Have a Destabilized Conformation*. Journal of Biological Chemistry. 2005;280:30649–52. [DOI] [PubMed] [Google Scholar]
- [234].Heise H, Celej MS, Becker S, Riedel D, Pelah A, Kumar A, et al. Solid-State NMR Reveals Structural Differences between Fibrils of Wild-Type and Disease-Related A53T Mutant α-Synuclein. Journal of Molecular Biology. 2008;380:444–50. [DOI] [PubMed] [Google Scholar]
- [235].Lemkau LR, Comellas G, Lee SW, Rikardsen LK, Woods WS, George JM, et al. Site-Specific Perturbations of Alpha-Synuclein Fibril Structure by the Parkinson’s Disease Associated Mutations A53T and E46K. PLOS ONE. 2013;8:e49750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Ni X, McGlinchey RP, Jiang J, Lee JC. Structural Insights into α-Synuclein Fibril Polymorphism: Effects of Parkinson’s Disease-Related C-Terminal Truncations. Journal of Molecular Biology. 2019;431:3913–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Yang Y, Shi Y, Schweighauser M, Zhang X, Kotecha A, Murzin AG, et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Lövestam S, Schweighauser M, Matsubara T, Murayama S, Tomita T, Ando T, et al. Seeded assembly in vitro does not replicate the structures of α-synuclein filaments from multiple system atrophy. FEBS Open Bio. 2021;11:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Newberry RW, Leong JT, Chow ED, Kampmann M, DeGrado WF. Deep mutational scanning reveals the structural basis for α-synuclein activity. Nature Chemical Biology. 2020;16:653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Ferrie JJ, Petersson EJ. A Unified De Novo Approach for Predicting the Structures of Ordered and Disordered Proteins. The Journal of Physical Chemistry B. 2020;124:5538–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.