

Abstract
Drug resistance contributes to poor therapeutic response in urothelial carcinoma (UC). Metabolomic analysis suggested metabolic reprogramming in gemcitabine‐resistant urothelial carcinoma cells, whereby increased aerobic glycolysis and metabolic stimulation of the pentose phosphate pathway (PPP) promoted pyrimidine biosynthesis to increase the production of the gemcitabine competitor deoxycytidine triphosphate (dCTP) that diminishes its therapeutic effect. Furthermore, we observed that gain‐of‐function of isocitrate dehydrogenase 2 (IDH2) induced reductive glutamine metabolism to stabilize Hif‐1α expression and consequently stimulate aerobic glycolysis and PPP bypass in gemcitabine‐resistant UC cells. Interestingly, IDH2‐mediated metabolic reprogramming also caused cross resistance to CDDP, by elevating the antioxidant defense via increased NADPH and glutathione production. Downregulation or pharmacological suppression of IDH2 restored chemosensitivity. Since the expression of key metabolic enzymes, such as TIGAR, TKT, and CTPS1, were affected by IDH2‐mediated metabolic reprogramming and related to poor prognosis in patients, IDH2 might become a new therapeutic target for restoring chemosensitivity in chemo‐resistant urothelial carcinoma.
Keywords: chemoresistance, hypoxia‐inducible factor‐1α, isocitrate dehydrogenase 2, metabolomic reprogramming, urothelial carcinoma
Subject Categories: Cancer, Metabolism
Metabolic rewiring of bladder cancer cells contributes to chemoresistance via IDH2/ HIF‐1α‐mediated production of competing dCTP.
Introduction
The poor response to chemotherapies due to the development of resistance in tumors remains a significant clinical challenge and contributes to a poor overall patient prognosis. Metastatic urothelial carcinoma (UC) has one of the highest recurrence rates among carcinomas and is difficult to cure (Roupret et al, 2021; Witjes et al, 2021). The gold standard therapy is a combination of gemcitabine (GEM) and cisplatin (CDDP), but resistance to this treatment is acquired over time (von der Maase et al, 2000). Although pembrolizumab was introduced as a second‐line therapy for patients with resistance, the response rate is only 15–20% (Bellmunt et al, 2017), which is still far from the therapeutic goal; thus, more therapies are needed for drug‐resistant UC.
Numerous studies have focused mainly on the biological mechanisms of acquired CDDP resistance, putting the most emphasis on combating CDDP resistance since it was recognized as the key cytotoxic component (Van Allen et al, 2014). We previously investigated the stimulation of drug efflux and reinforcement of antioxidant defense in CDDP‐resistant (CR) UC cells (Shigeta et al, 2020), but the entire landscape of changes involved in acquiring chemoresistance has yet to be fully investigated. On the other hand, the mechanism of GEM resistance has also been underexplored; however, GEM is still applied as part of this first‐line chemotherapy strategy and is administered 3 times per cycle.
Antimetabolites such as GEM are deoxycytidine analogs that hamper DNA replication and therefore arrest tumor growth, but very small evidence has shown the acquisition of GEM resistance. The efficacy of several anticancer therapies is linked to tumor cell survival and therefore to their effect on metabolic alterations in tumor cells (DeBerardinis et al, 2008). In this respect, recent studies have elucidated that the metabolic network in cancer cells undergoes reprogramming during malignant transformation, cell proliferation, and drug resistance acquisition (Navarro et al, 2022). Dr. Warburg first proposed the metabolic phenotype of increased metabolism of glucose to lactate even under oxygen‐rich conditions, a phenomenon called “aerobic glycolysis” or the “Warburg effect” (Warburg, 1956; Vander Heiden et al, 2009). In addition to this well‐known adaptive survival strategy, the metabolic reprogramming of cancer cells extends far beyond glycolysis to glutamine metabolism and lipid metabolism (Sun & Denko, 2014). One novel study presented strong evidence that GEM resistance is facilitated by alterations in intracellular metabolic dynamics. The authors suggested that cells acquire GEM chemoresistance due the establishment of molecular competition, which results in a metabolic shift from the aerobic pathway to the anaerobic pathway (Shukla et al, 2017). In particular, the stabilization of hypoxia‐inducible factor‐1α (Hif‐1α), the master regulator of glucose metabolism, plays a critical role in triggering metabolic reprogramming in cancer cells under normoxic conditions to “mimic” the metabolome under hypoxic conditions (Semenza, 2003, 2010). Previous studies have also shown that higher expression of Hif‐1α is closely related to tumor cell chemoresistance, as it promotes antioxidant and detoxification activities (Semenza, 2013). Other studies indicated that glucose metabolism and reductive glutamine metabolism are strongly accelerated in cancer cells, especially after chemoresistance is acquired, in pancreatic cancer (Yokoi & Fidler, 2004). UC cells metabolize glucose at higher rates, since metastatic sites commonly show high signals from fluorodeoxyglucose positron emission tomography (Voskuilen et al, 2021). However, the precise fates of glucose and its downstream metabolites and their roles in chemoresistant UC cells have yet to be fully investigated.
Our aim is to clarify the intratumor cell metabolic alterations that facilitate chemoresistance in UC cells and to identify a new therapeutic target for enhancing the effectiveness of chemotherapy from a metabolic perspective.
Results
GR UC cell lines rely on aerobic glycolysis to maintain cell survival, and this phenotype is not observed in wild‐type or CR UC cells
To evaluate the metabolic basis in wild‐type (WT) cells and cisplatin resistant (CR)/gemcitabine resistant (GR) UC cells, we generated bladder cancer cell line (T24 and UMUC3) models with acquired chemoresistance. We cultured WT cells with increasing concentrations of CDDP and GEM (up to 3 μM over a period of ~ 12 and 18 months, respectively). The resistance status of T24CR and UMUC3CR cells was shown in our previous study (Shigeta et al, 2020), the resistance status of T24GR and UMUC3GR cells was determined by calculating the half maximal inhibitory concentration (IC50) of GEM using WST cytotoxicity assays (Fig 1A). A 5‐ to 10‐fold or greater IC50 was observed in GR cells compared with WT cells in both cell lines (Fig 1B).
Figure 1. Metabolic differences among WT, CR, and GR UC cells.
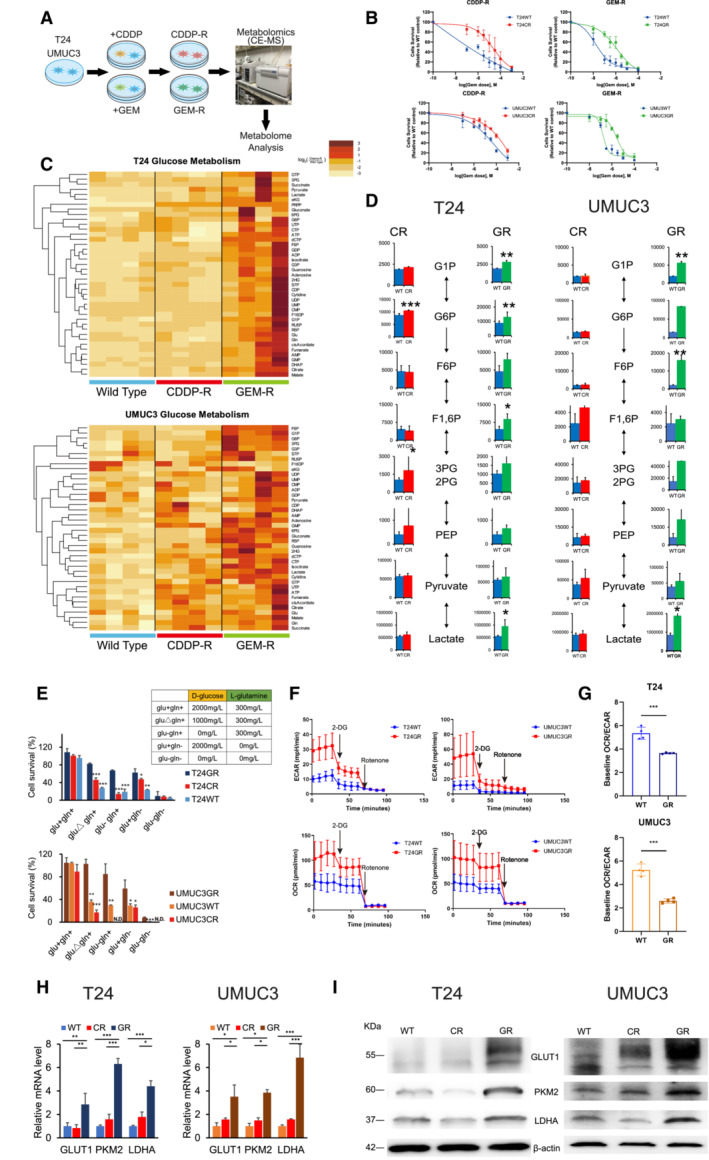
- T24 and UMUC3 cisplatin‐resistant (CR) and gemcitabine‐resistant (GR) urothelial cancer (UC) cells were generated by exposing the corresponding wild‐type (WT) cells to up to 3 μM cisplatin (CDDP) or gemcitabine (GEM) over 12 or 18 months, respectively. Brief schema showing the generation of CR and GR cell lines. Metabolites, including CR and GR, were analyzed by using CE‐MS metabolomics.
- Graphs show changes in cytotoxicity between WT and GR T24 and UMUC3 cells exposed to increasing concentrations of GEM, and WST assays were performed 48 h after treatment (n = 3, biological replicates). The data are shown as the mean values ± SDs.
- Hierarchical clustering of significantly regulated metabolites among T24 and UMUC3 cells, including WT, CR, and GR cells (n = 4, biological replicates).
- Major metabolites altered in the glycolytic pathway in CR cells and GR cells compared with WT cells. The data are shown as the mean values ± SDs (n = 4) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- Bar graph showing the survival of WT, CR, and GR cells in glucose‐ and glutamine‐replete (glu+, gln+), glutamine limitation (gluΔ, gln+), glucose deprivation (glu−, gln−), glutamine deprivation (glu+, gln−), or glucose and glutamine deprivation (glu−, gln−) medium. The table shows the glucose and glutamine concentrations in each medium. Significant differences were observed in GR cells compared with WT and/or CR cells after 48 h. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001. N.D., non‐detectable.
- WT and GR cells were seeded in 24‐well plates and exposed to 2‐DG and rotenone to measure baseline ECAR and baseline OCR. The data are shown as the mean values ± SDs (n = 3, biological replicates).
- Baseline OCR/ECAR ratios in WT and GR cells (upper: T24, lower: UMUC3). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. ***P < 0.001.
- Relative mRNA levels of GLUT1, PKM2, and LDHA, which are enzymes that regulate glycolysis. The data are shown as the mean values ± SEs (n = 3, biological replicates) and were analyzed by Student's t‐test, and the values are plotted relative to the expression levels in WT cells. *P < 0.05, **P < 0.01, ***P < 0.001.
- Western blot analysis of GLUT1, PKM2, LDHA, and β‐actin compared among WT, CR, and GR cells.
Source data are available online for this figure.
To investigate the metabolic alterations underlying chemoresistance, we performed capillary electrophoresis and mass spectrometry (CE‐MS) based metabolome analysis of T24 and UMUC3 cells followed by supervised hierarchical clustering. CE‐MS analysis indicated similar metabolic pools between WT and CR cells, while differential metabolic pools were observed in GR cells (Fig 1C). Specifically, the intermediate metabolites of glycolysis were especially increased in GR cells. We observed an increase in metabolites including glucose 1‐phosphate (G1P), glucose 6‐phosphate (G6P), fructose 6‐phosphate (F6P), and fructose 1,6‐phosphate (F1,6P). We also observed increased intracellular lactate acid levels in GR cells (Fig 1D).
To confirm glycolysis‐dependent metabolism in GR cells, we cultured cells under glucose limitation and deprivation conditions. We compared the survival of three types (WT, CR, and GR) of two cell lines after 72 h under these conditions (Fig 1E). For the glucose limitation medium, we used 1,000 mg/l D‐glucose, and for the glucose deprivation medium, we used 0 mg/l D‐glucose. As a result, we confirmed that GR cells of both cell lines showed tolerance under both glucose‐limited and under glucose‐deprived conditions, while CR and WT cells could not survive. We also evaluated the change in cell survival under glutamine deprivation conditions. We confirmed that GR cells still showed higher cell survival rates than CR/WT cells in glucose+/glutamine− (glu+gln−) medium. However, no parental or resistant cells could survive under both glucose and glutamine deprivation.
Furthermore, we conducted XF24 extracellular flux analysis to measure the baseline extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) and compare these rates between WT and GR cells. In comparison with the WT cell lines, GR cells demonstrated an increased ECAR (Fig 1F). As a result, GR cells showed a lower OCR/ECR ratio than WT cells (T24: OCR/ECR, 5.35 ± 1.98 in WT cells and 3.45 ± 0.75 in GR cells; UMUC3: OCR/ECR, 5.23 ± 2.69 in WT cells and 2.40 ± 1.47 in GR cells; Fig 1G). These results suggested that glycolysis is upregulated in GR cells compared with WT cells.
We further confirmed the expression levels of key enzymes that promote glycolysis metabolism, including glucose transporter 1 (GLUT1), pyruvate kinase M2 (PKM2), and lactate dehydrogenase A (LDHA), in all 3 cell lines. The mRNA levels of all three enzymes were significantly higher in GR cells than in WT or CR cells in both cell lines (e.g., GLUT1, PKM2, and LDHA mRNA levels in T24GR cells were 2.84 ± 0.95, 6.30 ± 0.47, and 4.47 ± 0.45 relative to that in T24WT cells, respectively; Fig 1H). Similar results were observed by confirming the higher protein expression of glycolytic enzymes in GR cells by Western blot (WB) analysis (Fig 1I).
From these results, we confirmed that compared with CR and WT cells, GR cells have different metabolic mechanisms for cell survival, suggesting the stimulation of glycolysis‐dependent metabolism in these cells.
Increased pyrimidine biosynthesis from pentose phosphate pathway (PPP) generates dCTP for acquiring molecular competition against GEM
CE‐MS‐based metabolomics profiling of GR cells revealed an increase in the levels of intermediate metabolites of the PPP (Fig 2A). We observed significant increases in the level of both oxidative and nonoxidative PPP metabolites (Fig 2B), which was similar to the results of previous studies in GR pancreatic cancer (Shukla et al, 2017). Since the PPP promotes nucleotide biosynthesis, we compared the amounts of metabolic end products of purine and pyrimidine synthesis between WT and GR cells. CE‐MS metabolomics demonstrated that the metabolic end products produced in pyrimidine synthesis were the most upregulated in GR cells, while no significant differences were observed among the metabolic end products in purine synthesis (Fig 2C and D). From RT–PCR analysis, we observed that GR cells had significantly increased expression of the glucose 6 phosphate dehydrogenase (G6PD), TP53‐inducible glycolysis and apoptosis regulator (TIGAR), and transketolase (TKT) genes, which are involved in promoting the metabolic shift of glucose flux from glycolysis to PPP (Fig 2E; Jiang et al, 2016; Tseng et al, 2018; Cheung et al, 2020). Furthermore, we observed a significant increase in cytidine triphosphate synthase 1 (CTPS1) expression at both the mRNA and protein expression levels in GR cells, which are suspected to be key enzymes that increase cytidine triphosphate, which are the essential components for DNA elongation (Fig 2F). Overall, the relative mRNA expression levels of pyrimidine metabolism enzymes were significantly higher in GR cells than in WT cells, while slight differences in the relative mRNA expression levels of purine metabolism enzymes were observed between WT and GR cells (Fig EV1A).
Figure 2. GR cells increases pyrimidine biosynthesis to generate dCTP.
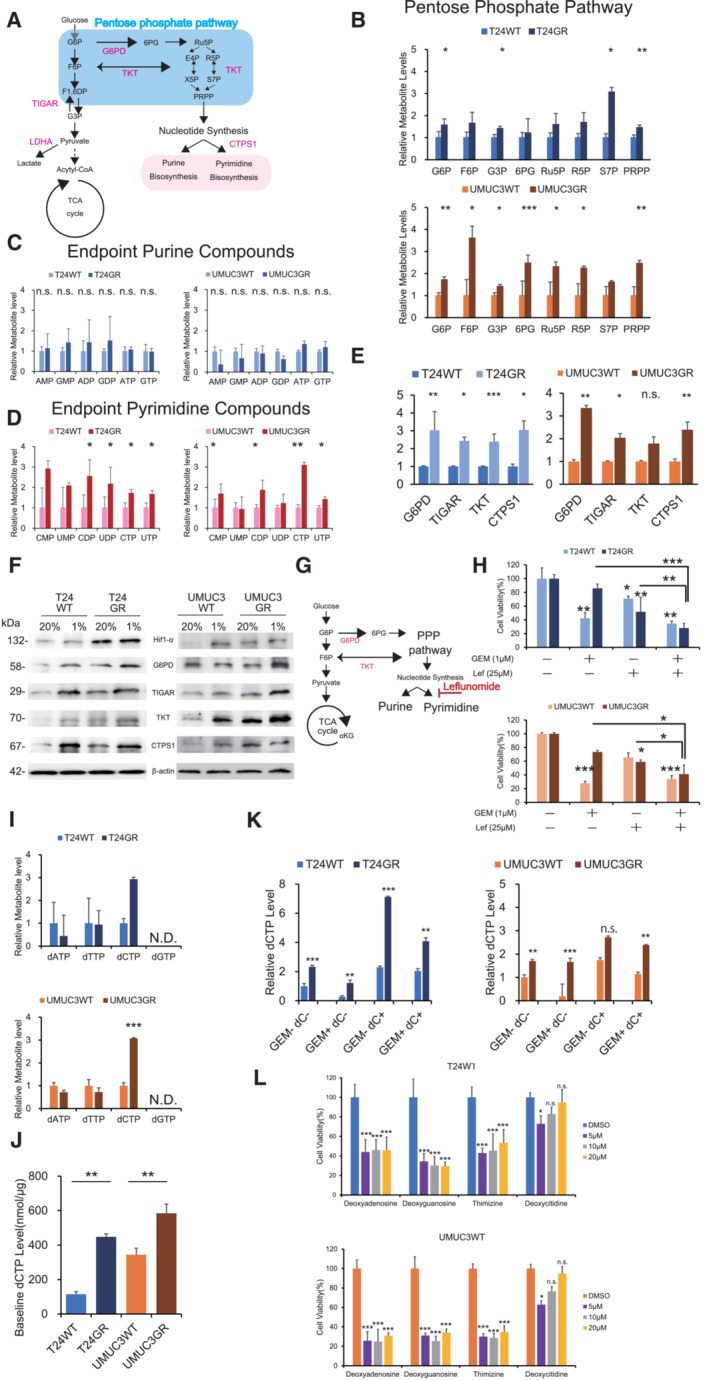
- Schematic of altered mechanisms in GR cells in glycolysis and the pentose phosphate pathway (PPP). Glucose is transferred from aerobic glycolysis to PPP. Glucose flux into the PPP results in increased nucleotide biosynthesis, including purine and pyrimidine biosynthesis.
- PPP metabolite levels in T24GR and UMUC3GR cells relative to WT cells based on targeted CE‐MS metabolomics The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- Levels of endpoint metabolites of the purine synthesis pathway in GR cells relative to those in WT cells as determined by CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. n.s., non‐significant
- Levels of endpoint metabolites of the pyrimidine synthesis pathway in GR cells relative to those in WT cells as determined by CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- Relative mRNA expression levels of G6PD, TIGAR, TKT, and CTPS1, which are the genes responsible for activating the PPP and pyrimidine biosynthesis. WT and GR cells are shown. The data are shown as the mean values ± SEs (n = 3, biological replicates). The expression levels were analyzed by Student's t‐test and plotted relative to expression levels in WT cells (left: T24 cells, right: UMUC3 cells). *P < 0.05, **P < 0.01, ***P < 0.001, n.s., non‐significant.
- Western blot analysis of Hif‐1α, G6PD, TIGAR, TKT, CTPS1, and β‐actin in WT and GR cells under normoxia and hypoxia (left: T24 cells, right: UMUC3 cells).
- Simple schematic of the glycolytic pathway and the PPP. Glucose flux into the PPP results in increased nucleotide biosynthesis, including purine and pyrimidine biosynthesis. Leflunomide inhibits pyrimidine biosynthesis.
- The viability of WT and GR cells was assessed by WST assays under treatment with gemcitabine (Gem), leflunomide, or Gem with leflunomide. The data are shown as the mean values ± SDs (n = 3, biological replicates). Comparisons were made with respect to the corresponding controls or the indicated groups, followed by analysis with Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- CE‐MS‐based metabolite detection for diphosphate nucleosides in WT and GR cells. The data are shown as the mean values ± SDs (n = 3, biological replicates). The levels in GR cells are presented relative to those in WT control cells and analyzed by Student's t‐test. ***P < 0.001. N.D., non‐detectable.
- Comparison of baseline dCTP levels in WT and GR cell lines determined by CE‐MS metabolomic analysis. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
- dCTP levels as determined by CE‐MS metabolomic analysis under treatment with 1 μM GEM and/or 10 μM deoxycytidine (dC) (left: T24 cells; right: UMUC3 cells). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by one‐way ANOVA with the Bonferroni test. **P ≤ 0.01, ***P ≤ 0.001. n.s., nonsignificant.
- Effect of deoxycytidine and other nucleosides (deoxyadenosine, deoxyguanosine, and thymidine) at various concentrations with 1 μM GEM in WT cells, as evaluated by WST assays 48 h post‐treatment (upper: T24 cells; lower: UMUC3 cells). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by one‐way ANOVA with the Bonferroni test. *P ≤ 0.05, ***P ≤ 0.001. n.s., non‐significant.
Source data are available online for this figure.
Figure EV1. Stimulation of PPP is responsible for chemoresistance in GR cells.
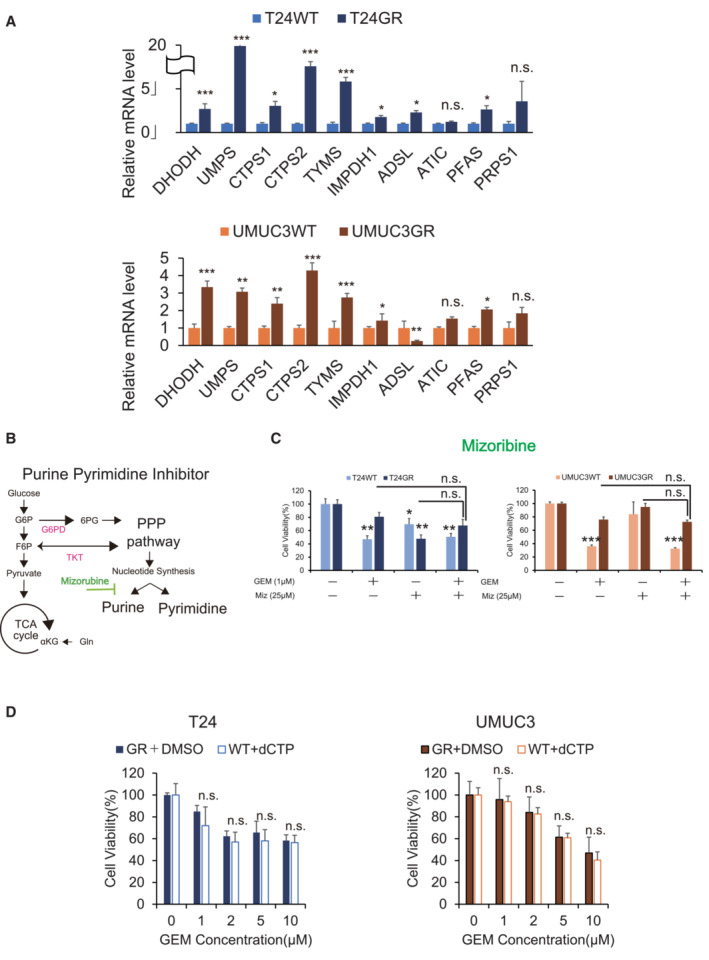
- Relative mRNA expression levels of DHODH, UMPS, CTPS1, CTPS2, TYMS, IMPDH1, ADSL, ATIC, PFAS, and PRPS1 in WT and GR cells (upper: T24, lower: UMUC3). The data are shown as the mean values ± SEs (n = 3, biological replicates) and were analyzed by Student's t‐test, and plotted relative to expression levels in WT cells. *P ≤ 0.05, ***P ≤ 0.001. n.s., non‐significant.
- Simple schematic of the glycolytic pathway and the pentose phosphate pathway (PPP). Glucose flux into the PPP results in increased nucleotide biosynthesis, including purine and pyrimidine biosynthesis. Mizoribine inhibits purine biosynthesis.
- The viability of WT and GR cells was evaluated by WST assays under treatment with gemcitabine (GEM), mizoribine, or GEM with mizoribine. The data are shown as the mean values ± SDs (n = 3, biological replicates). Comparisons were made with respect to the corresponding controls or the indicated groups, followed by Student's t‐test. **P < 0.01, ***P < 0.001. n.s., non‐significant.
- Bar graph showing the viability of cells exposed to various concentrations of GEM for 48 h and treated with DMSO (GR cells) or 10 μM dC (WT cells) (left: T24 cells; right: UMUC3 cells). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. n.s., non‐significant.
We further examined the cytotoxic effect of a pyrimidine synthesis inhibitor (leflunomide) with GEM in GR cells (Fig 2G). We examined the combination of 25 μM leflunomide with GEM and found a significant decrease in cell survival with the combination of 1 μM GEM (T24GR cell viability rate with 1 μM GEM vs. 1 μM GEM combined with 25 μM leflunomide: 86.1 ± 6.1% vs. 28.1 ± 6.8%, P < 0.001; Fig 2H). In contrast, compared with 1 μM GEM alone, the combination of a purine inhibitor (mizoribine, 25 μM) with 1 μM GEM did not show a significant effect on the cell viability rate (Fig EV1B and C). Similar results were obtained in UMUC3GR cells, and these results indicated that GEM resistance derives from an increase in pyrimidine biosynthesis.
As previous studies have shown, GEM is a deoxycytidine analog, and therefore deoxycytidine triphosphate (dCTP) synthesis directly affect GEM efficacy (Shukla et al, 2017). We suspected that pyrimidine biosynthesis through the PPP stimulation generates dCTP for acquiring molecular competition against GEM. CE‐MS analysis revealed that GR cells showed a significant increase in pyrimidine biosynthesis end product compounds (CTP and UTP), and we also found that dCTP was specifically increased in GR cells compared with dATP, dTTP, and dGTP (Fig 2I). We also measured the baseline dCTP levels in WT and GR cells (Fig 2J). The baseline dCTP levels were significantly higher in T24GR and UMUC3GR cells (447.65 nmol/μg ± 17.8 nmol/μg and 584.65 nmol/μg ± 53.2 nmol/μg, respectively) than in T24WT and UMUC3WT cells (114.59 nmol/μg ± 15.5 nmol/μg and 343.93 nmol/μg ± 37.3 nmol/μg, P = 0.001 and P = 0.004).
We also measured intracellular dCTP levels in WT and GR cells treated with deoxycytidine (dC) (10 μM) and GEM (1 μM) (Fig 2K). CE‐MS analysis revealed that dCTP levels remained high in GR cells and WT cells treated with GEM in combination with dC. Indeed, we confirmed that the cytotoxic efficacy of GEM in WT cells was reduced by the addition of 10 μM dC (Fig EV1D).
Finally, we compared the therapeutic response with 1 μM GEM observed on treatment with other nucleoside analogs, such as 2‐deoxyadenosine monohydrate, 2‐deoxyguanosine monohydrate, and thymidine, with that observed on treatment with dC in WT cells. WT cells treated with dC at concentrations of 10 μM or higher showed GEM resistance, while other nucleoside analogs did not show any molecular competition with GEM (Fig 2L). These results demonstrated that pyrimidine biosynthesis is increased in GR cells through stimulation of the PPP, which leads to accumulation of dCTP and may cause competitive inhibition of GEM activity.
Reductive carboxylation due to glutamine carbon flux preferentially produces 2‐HG to stabilize Hif‐1α in GR UC cells
We further compared the metabolism of the TCA cycle in WT and GR cells. From the entire metabolomic analysis, we found that the intermediate metabolites of former steps of the TCA cycle (citrate, cis‐Aconitate, and isocitrate) were increased but metabolites of later steps were unchanged or decreased in GR cells compared with WT cells. In specific, glutamine (gln) and glutamate (Glu) showed increase while α‐ketoglutarate (α‐KG) showed significant decrease in both GR cells compared with WT (Fig 3A). Since recent studies have proposed metabolic reprogramming in cancer cells, as indicated by citrate generation via the reductive carboxylation of glutamine (Mullen et al, 2011), we used [13C5‐]‐labeled glutamine to identify the pathways by which glutamine carbon contributes to citrate production in WT and GR cells (Fig 3B). WT and GR cells were cultured in medium containing [13C5‐] glutamine, and intracellular metabolites were extracted and analyzed by CE‐MS after 1 h of labeling. When comparing WT and GR cells, the cit + 5, and α‐KG + 5, pools constituted the majority of the enriched 13C5 glutamine in GR cells (Fig 3C). Cit + 5 derived from the reductive carboxylation of glutamine‐derived α‐KG + 5 to isocitrate+5, followed by the isomerization of isocitrate + 5 to cit + 5. Thus, Cit + 5 constituted a significantly smaller pool in WT cells than GR cells while cit + 4 to the total citrate pool was significantly lower in GR cells than in WT cells (Fig 3D). On the other hand, malate m + 4 constituted a significantly smaller pool in GR cells compared with WT. The m + 3 species, which can be derived from the cit + 5 generated by reductive carboxylation, showed significantly larger pool in GR cells than WT cells (Fig 3E).
Figure 3. Reductive carboxylation produces 2‐HG to stabilize Hif‐1α.
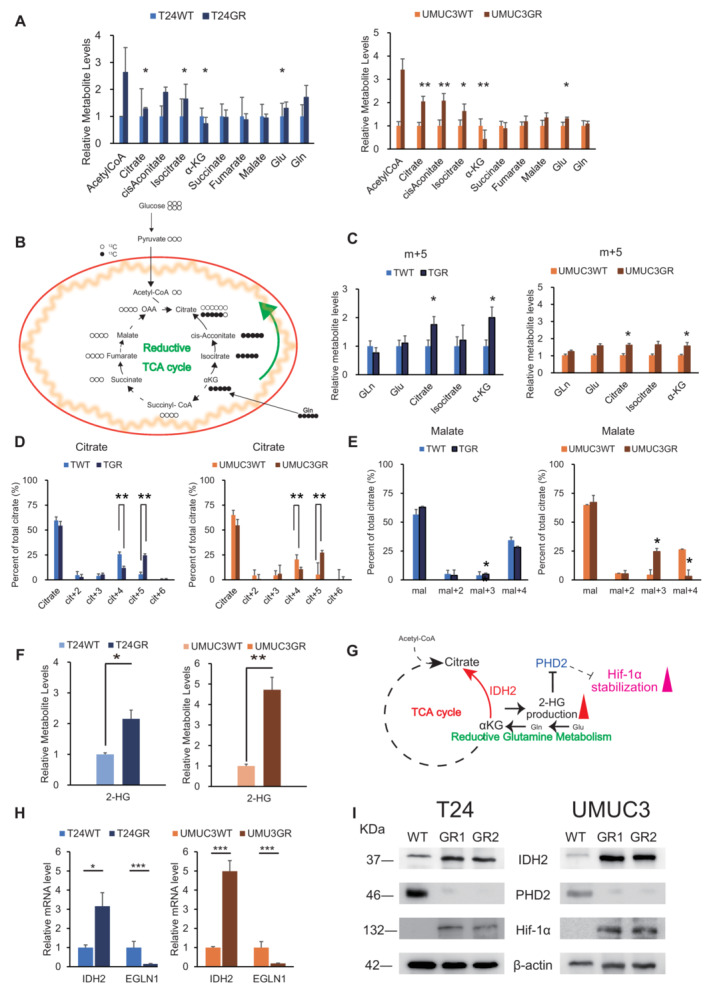
- Levels of intermediate metabolites of the tricarboxylic acid cycle (TCA) cycle in GR cells relative to those in WT cells based on targeted CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- Schematic describing the details of the reductive TCA cycle. Model depicting the pathways for citrate+4 (cit+4) and cit+5 production from [13C5‐] glutamine (glutamine+5). Glutamine+5 is catabolized to α‐ketogutarate+5, which can then contribute to citrate production by two divergent pathways. Normal metabolism in WT cells produces succinate+4, fumarate+4, malate+4, and oxaloacetate+4, which can condense with unlabeled acetyl‐CoA to produce citrate+4. In GR cells, altered mechanisms in the TCA cycle were observed in terms of high glutamate metabolism. The [13C5]‐labeled glutamate‐derived metabolite levels are indicated in black.
- The relative levels of metabolites contributing to citrate production through increased reductive carboxylation [glutamine (gln), glutamate (glu), α‐KG, and citrate] in T24GR cells versus WT cells. Cells were cultured for 24 h in 10 mM glutamine and then cultured for 1 h in medium supplemented with [13C5‐] glutamine. 13C enrichment in cellular citrate was quantitated by CE‐MS. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
- 13C enrichment in cellular citrate determined by CE‐MS and normalized to the total pool size for the relevant metabolite (left: T24GR, right: UMUC3GR) The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
- 13C enrichment in cellular malate determined by CE‐MS and normalized to the total pool size for the relevant metabolite (left: T24GR, right: UMUC3GR) The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
- Relative 2‐hydroxyglutarate (2‐HG) production level based on targeted CE‐MS metabolomics comparing among WT and GR cells (left panel: T24, right panel: UMUC3). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
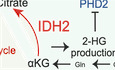
- Schematic describing the wild type (WT) IDH2 gain of function which mediated the reductive glutamine metabolism. 2‐HG accumulation driven by WT‐IDH2 suppresses EGLN1/PHD2 expressions to stabilize Hif‐1α expression.
- Relative mRNA expression levels of IDH2 and EGLN1 genes compared between WT and GR cells (left panel: T24, right panel: UMUC3). The expression levels were analyzed by Student's t‐test and plotted relative to expression levels in WT cells. The data are shown as the mean values ± SEs (n = 3, biological replicates). *P < 0.05, ***P < 0.001.
- Western blot analysis of IDH2, PHD2, and β‐actin in WT and GR cells (left panel: T24, right panel: UMUC3).
Source data are available online for this figure.
Recent work has found that reductive glutamine metabolism in the TCA cycle is catalyzed by isocitrate dehydrogenase (IDH2) expression in mitochondria (Wise et al, 2011). IDH2 is presumed to catalyze the reductive carboxylation of α‐KG to isocitrate and citrate for further lipid biosynthesis, or to generate 2‐ hydroxylate glutamate (2‐HG) for further energy production (Fan et al, 2013). We further performed whole‐exome sequencing (WES) on DNA from four cell lines (T24WT, T24GR, UMUC3WT, and UMUC3GR). Several genetic differences were observed between WT and GR cells, but no IDH1/2 mutations were found in the WES analysis in either GR cell line (Fig EV2).
Figure EV2. Whole‐exome sequencing analysis of DNA in UC cell lines.
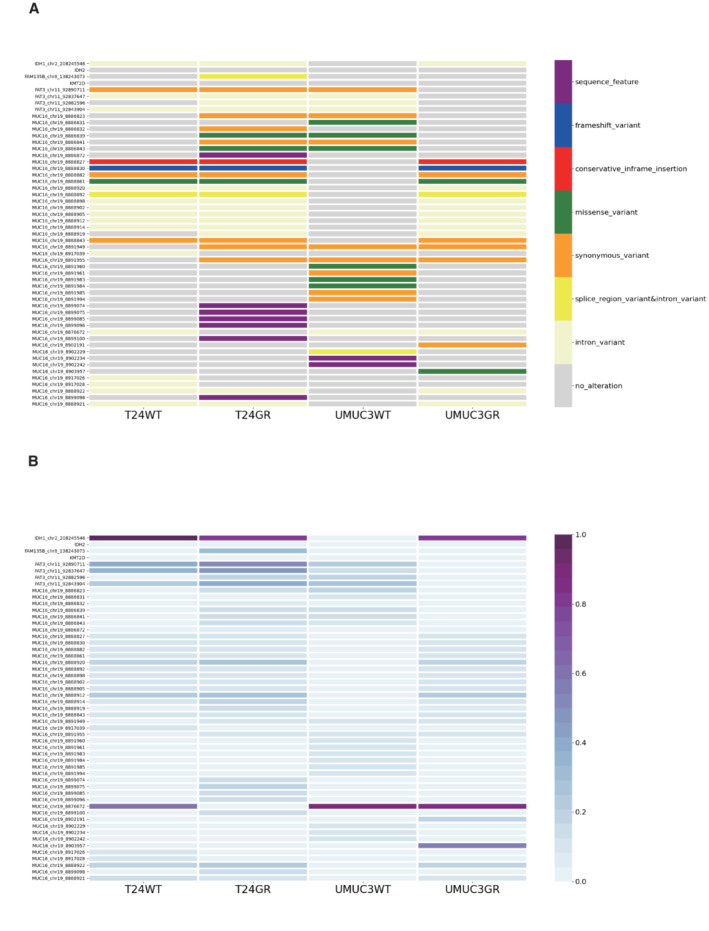
- Details of the whole‐exome sequencing analysis of DNA in T24WT, T24GR, UMUC3WT, and UMUC3GR cells. The vertical axis shows the genes and their genomic positions. The horizontal axis shows the tumor samples (T24WT, T24GR, UMUC3WT, and UMUC3GR).
- Details of the alteration rates of genomic positions in each gene. The alteration rates were calculated by the following equation: alteration depth/(alteration depth + reference depth).
Although our current study did not show IDH2 mutations, both GR cell lines showed significantly increased wild‐type IDH2 (WT IDH2) mRNA expression levels (T24GR: 3.16‐fold higher than WT, P = 0.045; UMUC3GR: 4.99‐fold higher than WT, P < 0.001) (Fig 3H). In addition, we measured the 2‐HG levels in both WT and GR cells. We found that 2‐HG production was appreciably increased in GR cells compared with WT cells in both cell lines.
Specifically, T24GR cells produced 2.15 times more 2‐HG than the corresponding WT cells (P = 0.027), while UMUC3GR cells produced 4.72 times more 2‐HG than the corresponding WT cells (P = 0.015) (Fig 3F).
From these results, we found that WT‐IDH2 gain of function would lead to increase 2‐HG production, which is consistent with the recent findings in cancer metabolism.
Many previous articles have shown that IDH2 gain of function promotes 2‐HG accumulation and stabilizes Hif‐1α expression by suppressing the activity of EGLN/PHD family members, which generally hydroxylate Hif‐1α and lead to its degradation under normoxic conditions (Fig 3G; Du & Hu, 2021). Therefore, we conducted RT–PCR and WB analyses to determine whether PHD2 expression is suppressed in GR cells. We confirmed that EGLN1 (the gene encoding PHD2) was significantly suppressed in GR cells (Fig 3H). WB analysis also showed a significant decrease in PHD2 protein expression (Fig 3I).
These results suggested that GR cells are dependent on reductive glutamine metabolism, which results from wild‐type (WT) IDH2 gain of function. IDH2 promotes reductive carboxylation and leads to 2‐HG accumulation, which results in Hif‐1α stabilization in GR cells.
Downregulation of HIF1A or IDH2 restores chemosensitivity in UC cells
To clarify the relationship between Hif‐1α expression and chemoresistance, we further conducted Hif‐1α knockdown by using HIF1A‐targeting small interfering RNAs (siRNAs) (siHIF1A#1: s6539 and siHiF1A#2: s6541) in both GR cell lines. The mRNA expression levels of glycolytic enzymes (GLUT1, G6PD, TIGAR, TKT, CTPS1, PKM2, and LDHA) decreased significantly along with HIF1A downregulation (Fig 4A). Similar results were obtained in the WB analysis, indicating that the expression of key metabolic enzymes (TIGAR, TKT, and CTPS1) involved in glycolysis and the PPP shift was significantly suppressed by Hif‐1α downregulation (Fig 4B). We confirmed that HIF1A‐downregulated GR cells (transfected with #siHIF1A#1 and #2) showed clear restoration of the cytotoxic effect with GEM treatment (Fig 4C). These results indicate that the key enzymes related to aerobic glycolysis and the PPP are regulated by Hif‐1α and are stabilized in GR cell lines under normoxic conditions.
Figure 4. Downregulation of HIF1A and IDH2.
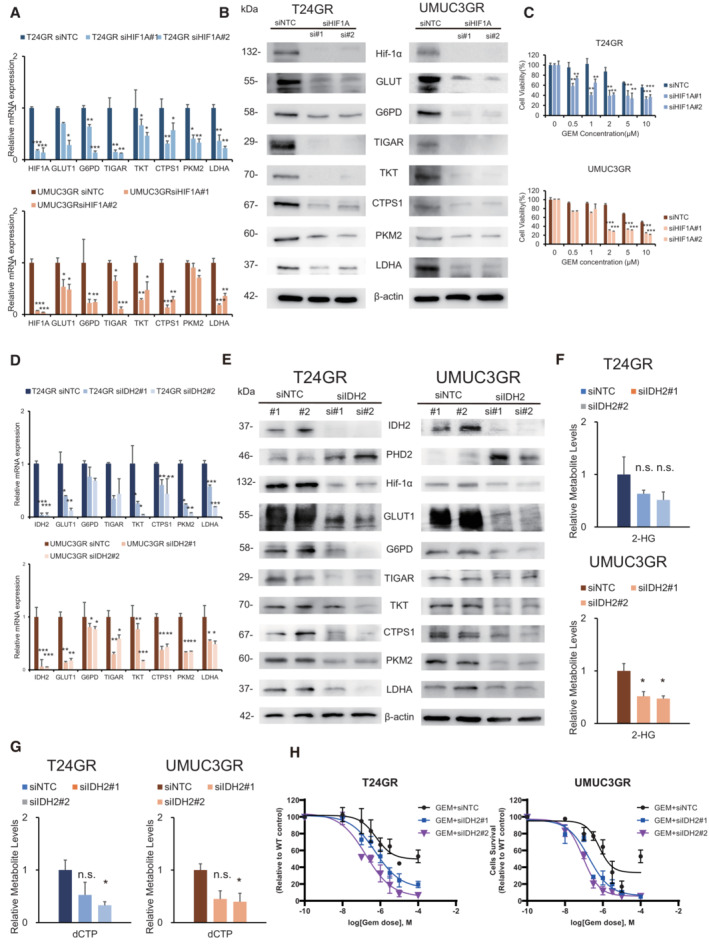
- Relative mRNA expression levels of HIF1A, GLUT1, G6PD, TIGAR, TKT, CTPS1, PKM2, and LDHA in cells transfected with siHIF1A#1 and #2 compared with cells transfected with siNTC (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SEs (n = 3, biological replicates) and were analyzed by Student's t‐test, and plotted relative to expression levels in GR cells transfected with siNTC. *P < 0.05, **P < 0.01, ***P < 0.001.
- Western blot analysis of Hif‐1α, GLUT1, G6PD, TIGAR, TKT, CTPS1, PKM2, LDHA, and β‐actin after transfection with NTC and IDH2‐targeting (siHIF1A#1 and #2) siRNAs (left: T24GR, right: UMUC3GR).
- Bar graph showing the viability of GR cells exposed to various concentrations of GEM for 48 h after transfection with NTC and HIF1A‐targeting (siHIF1A#1 and #2) siRNA (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01, ***P < 0.001.
- Relative mRNA expression levels of IDH2, GLUT1, G6PD, TIGAR, TKT, CTPS1, PKM2, and LDHA in cells transfected with siIDH2#1 and #2 compared with cells transfected with siNTC (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SEs (n = 3, biological replicates) and were analyzed by Student's t‐test, and plotted relative to expression levels in GR cells transfected with siNTC. *P < 0.05, **P < 0.01, ***P < 0.001.
- Western blot analysis of IDH2, PHD2, HIF‐1α, G6PD, GLUT1, TIGAR, TKT, CTPS1, PKM2, LDHA, and β‐actin after transfection with NTC and IDH2‐targeting (siIDH2#1 and #2) siRNA (left: T24GR, right: UMUC3GR).
- Relative 2‐HG production level based on targeted CE‐MS metabolomics in GR cells after transfection with NTC and IDH2‐targeting (siIDH2#1 and #2) siRNA (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05. n.s. = non‐significant.
- Relative dCTP level based on targeted CE‐MS metabolomics in GR cells after transfection with NTC and IDH2‐targeting (siIDH2#1 and #2) siRNA (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05. n.s., non‐significant.
- Graph showing the viability of GR cells exposed to various concentrations of GEM for 48 h after transfection with NTC and IDH2‐targeting (siIDH2#1 and #2) siRNA (left: T24GR, right: UMUC3GR) The data are shown as the mean values ± SDs (n = 3, biological replicates).
Source data are available online for this figure.
Next, to clarify the relationship between IDH2 expression and chemoresistance, T24GR cells and UMUC3GR cells were transfected with two siRNA sequences targeting IDH2 or with a nontargeting control (NTC) siRNA and incubated for 2 days. After siIDH2 transfection, the mRNA expression levels of IDH2 and other key metabolic enzymes, including Hif‐1α, was confirmed by RT–PCR (Fig 4D). The mRNA expression levels of key glycolytic and PPP enzymes decreased significantly with IDH2 downregulation. Similar results were obtained in the WB analysis, indicating that IDH2 protein expression was significantly suppressed, which then suppressed the expression of Hif‐1α and other key glycolytic and PPP enzymes (Fig 4E). Regarding 2‐HG production, significantly lower 2‐HG levels were observed in both GR cell lines transfected with siIDH2 (Fig 4F). Similarly, significantly lower dCTP levels were observed in GR cells transfected with siIDH2 (Fig 4G). The entire spectrum of metabolic changes in GR cells transfected with siIDH2 is shown in Fig EV3. When IDH2 was silenced, GEM sensitivity was restored in both T24GR and UMUC3GR cells (Fig 4H). These results indicated that the function of Hif‐1α is regulated by IDH2 expression, which in turn suggested that all the observed metabolic reprogramming results from WT IDH2‐dependent stabilization of Hif‐1α.
Figure EV3. IDH2 down regulation suppresses metabolic rewiring.
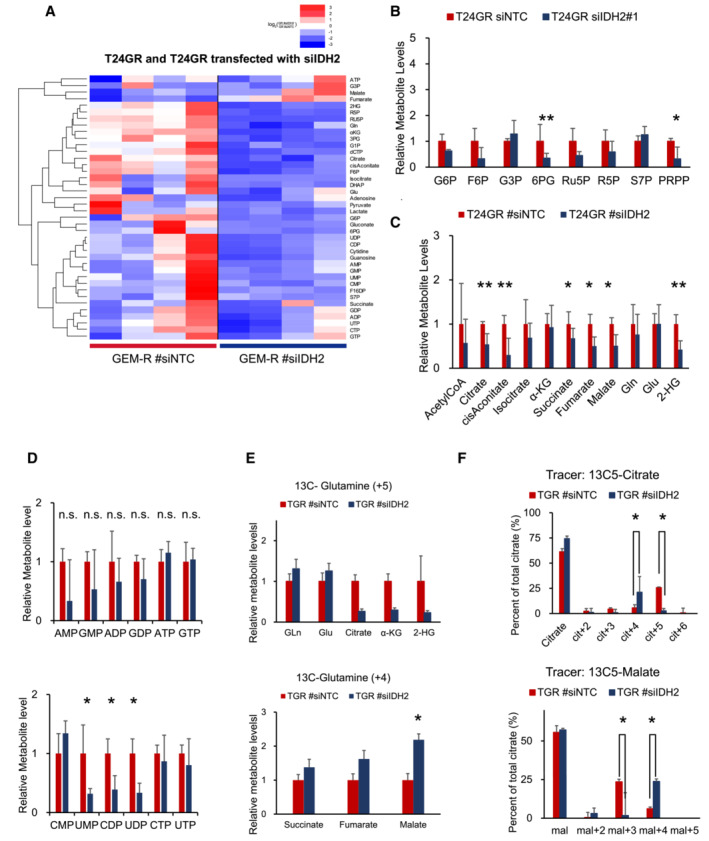
- The entire heat map showing intracellular levels of metabolites in T24GR cells transfected with NTC and IDH2. For each cell line, four independently derived cell lines are shown, and the levels were normalized to those in WT cells.
- PPP metabolite levels in T24GR cells transfected with siIDH2 compared with GR cells transfected with NTC based on targeted CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- The intermediate metabolite levels of the TCA cycle in T24GR cells transfected with NTC and siIDH2 based on targeted CE‐MS/MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- Levels of endpoint metabolites of the purine and pyrimidine synthesis pathway in GR cells relative to those in WT cells as determined by CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05. n.s., non‐significant.
- The levels of +5 metabolites and +4 metabolites, including early metabolites and late metabolites, in the TCA cycle compared between T24GR cells transfected with NTC and those transfected with siIDH2. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
- 13C enrichment in cellular citrate and malate between T24GR cells transfected with NTC and those transfected with siIDH2 as determined by CE‐MS; the levels were normalized to the total pool size of the relevant metabolite. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
IDH2 overexpression recapitulate metabolic rewiring
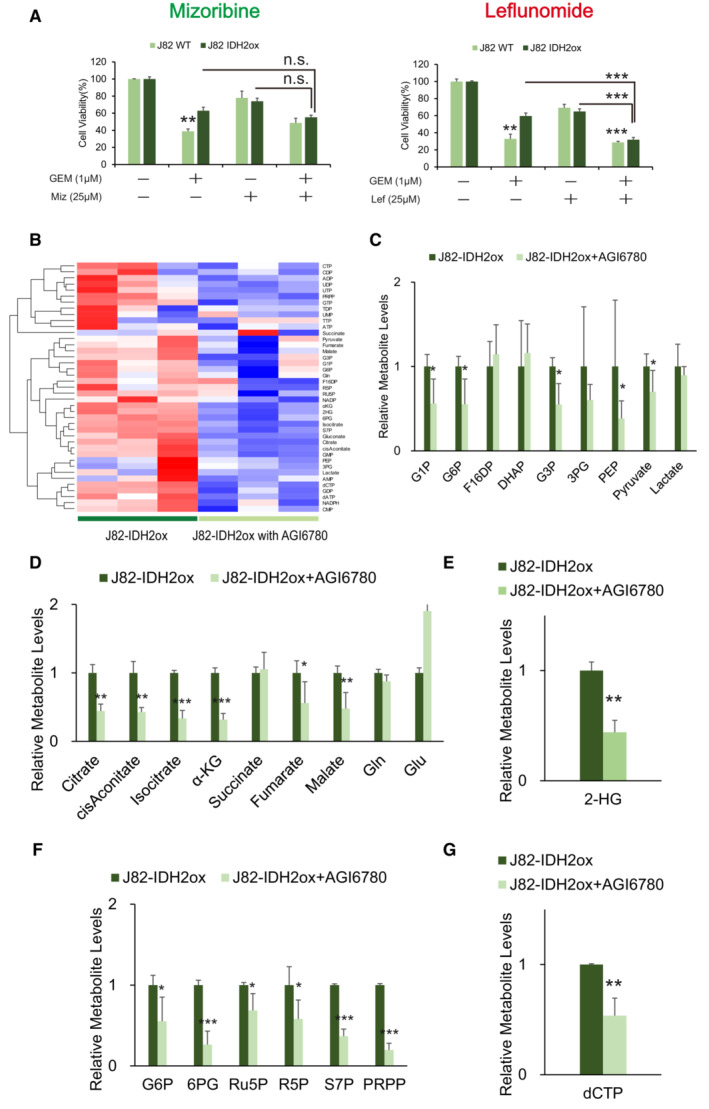
To clarify whether IDH2 overexpression induces metabolic reprogramming, we performed a gain‐of‐function experiment by overexpressing WT IDH2 in J82 cells, which exhibit low IDH2 expression. The results are shown in Fig 5. RT–PCR and WB analyses showed significant increases in IDH2 and Hif‐1α expression along with increased expression of key metabolic enzymes (Fig 5A and B). CE‐MS metabolome analysis revealed that WT IDH2‐overexpressing J82 (J82‐IDH2ox) cells showed significant increases in glutamine, α‐KG, isocitrate and citrate (Fig 5C and D). The labeling analysis also showed increased α‐KG (+5) and decreased malate (+4) levels, suggesting clear activity of reductive glutamine metabolism (Fig 5F and G). Furthermore, we confirmed a significant increase in 2‐HG production in J82‐IDH2ox cells compared with WT cells (Fig 5E). CE‐MS metabolome analysis also revealed that the metabolome of glycolysis and PPP intermediates was significantly enhanced in IDH2ox cells (Fig 5H and I). Finally, a significantly high level of dCTP was found in J82‐IDH2ox cells (Fig 5J); therefore, J82‐IDH2ox cells showed GEM resistance relative to WT cells (Fig 5K). The current experiment demonstrated that WT IDH2 overexpression recapitulates the reductive TCA cycle, stabilizing Hif‐1α expression via 2‐HG production and resulting in stimulation of aerobic glycolysis and PPP bypass.
Figure 5. IDH2 overexpression recapitulate metabolic rewiring.
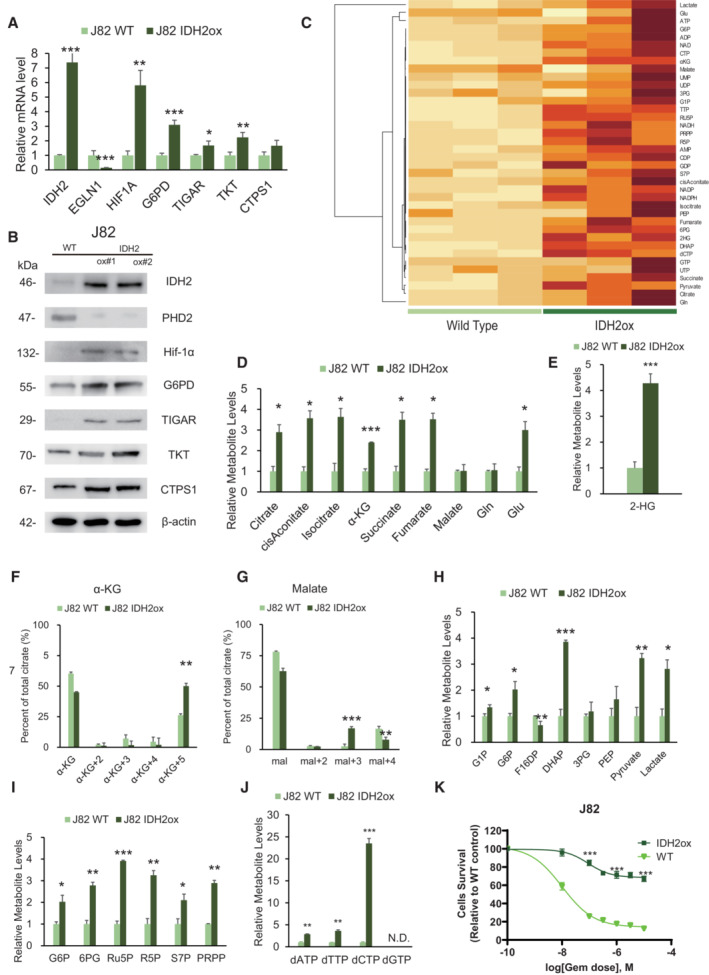
- Relative mRNA expression levels of IDH2, EGLN1, Hif‐1α, G6PD, TIGAR, TKT, and CTPS1 between J82 wild‐type cells (J82WT) and J82 cells with IDH2 overexpression (J82‐IDH2ox cells). The data are shown as the mean values ± SEs (n = 3, biological replicates). The expression levels were analyzed by Student's t‐test and are plotted relative to the expression levels in WT cells. *P < 0.05, **P < 0.01, ***P < 0.001.
- Western blot analysis of IDH2, PHD2, Hif‐1α, G6PD, TIGAR, TKT, CTPS1, and β‐actin in J82WT and J82‐IDH2ox cells.
- Hierarchical clustering of significantly regulated metabolites in J82WT and J82‐IDH2ox cells (n = 3, biological replicates each).
- Levels of intermediate metabolites of the tricarboxylic acid (TCA) cycle based on targeted CE‐MS/MS metabolomics in GR cells relative to WT cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, ***P < 0.001.
- Relative 2‐HG production level based on targeted CE‐MS metabolomics in J82‐IDH2ox cells compared with WT cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. ***P < 0.001.
- 13C enrichment in cellular α‐KG, as determined by CE‐MS and normalized to the total pool size of the corresponding metabolite. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
- 13C enrichment in cellular malate determined by CE‐MS and normalized to the total pool size of the corresponding metabolite. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01, ***P < 0.001.
- Major metabolites in the glycolytic pathway altered in J82‐IDH2ox cells compared with WT cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- PPP metabolite levels in J82‐IDH2ox cells compared with WT cells based on targeted CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- Relative dATP, dTTP, and dCTP level based on targeted CE‐MS metabolomics in J82‐IDH2ox cells compared with WT cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P ≤ 0.01, ***P ≤ 0.001. N.D., non‐detectable.
- Graph showing the viability of J82WT and J82‐IDH2ox cells exposed to various concentrations of GEM for 48 h. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. ***P < 0.001.
Source data are available online for this figure.
Increased intracellular NADPH levels enhance the antioxidant defense in GR cells, which leads to cross resistance to CDDP
In the course of our experiment, we found that GR cells showed significant resistance to CDDP. The results were confirmed by exposing T24GR and UMUC3GR cells to various concentrations of CDDP for 48 h. The IC50 value was more than 5.0‐fold higher in T24GR cells than in T24WT cells. Similarly, the IC50 value was 4.4‐fold higher in UMUC3GR cells than in UMUC3WT cells (Fig 6A). Similar results were obtained from the in vivo experiment (tumor volume in UMUC3GR‐implanted mice treated with GEM: 516.9 ± 57.9 mm3, tumor volume in UMUC3GR‐implanted mice treated with CDDP: 461.1 ± 120.9 mm3, tumor volume in UMUC3WT‐implanted mice treated with GEM: 210.1 ± 92.4 mm3, and tumor volume in UMUC3WT‐implanted mice treated with CDDP: 180.8 ± 82.6 mm3; Fig 6B and C). The Ki‐67 score was not significantly different in GR cells before and after chemotherapy administration, while it was significantly decreased in WT cells (Ki‐67 score: 74.7% vs. 57.7% vs.68.8% in GR cells, 74.7% vs. 36.0% vs. 16.3% in WT cells; Fig 6D).
Figure 6. Increased intracellular NADPH levels leads to cross resistance to CDDP.
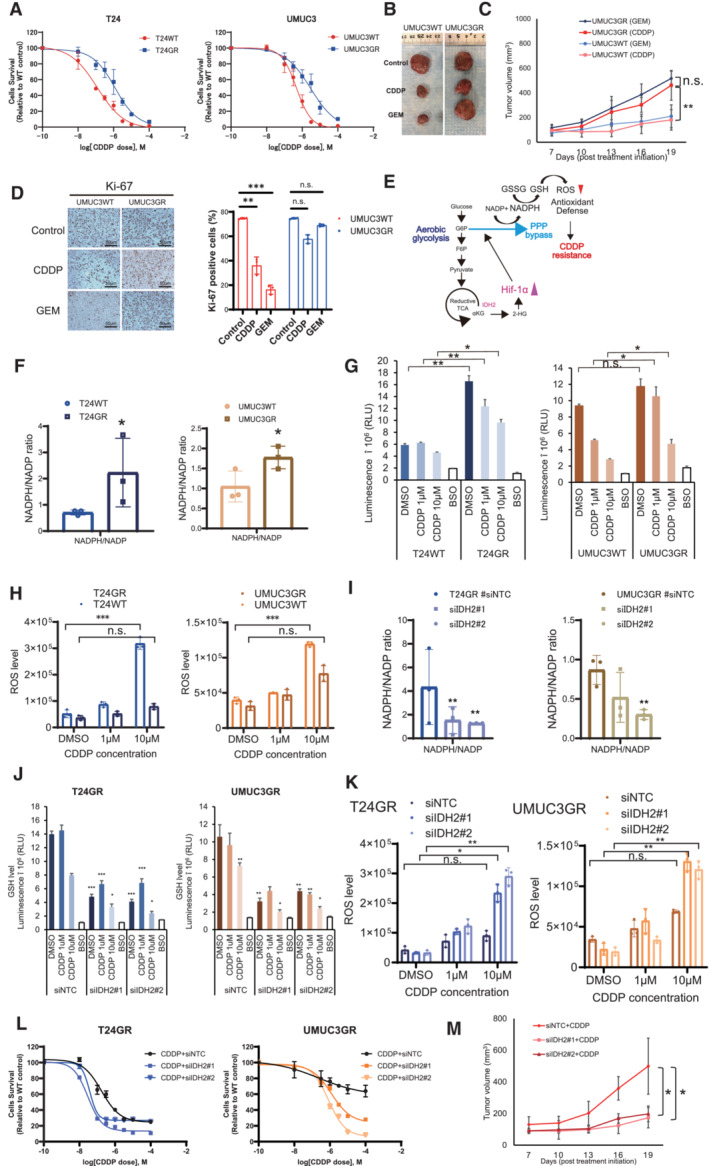
- Changes in cytotoxicity between WT and GR T24 and UMUC3 cells exposed to increasing concentrations of CDDP for 48 h. The data are shown as the mean values ± SDs (n = 3, biological replicates).
- Representative image showing the effect of CDDP in GR xenograft UC mouse models.
- Graph showing the cytotoxic effect of CDDP and GEM in WT and GR cells orthotopically implanted mouse models (n = 5 mice per group). UMUC3GR cells (2 × 106 cells) were implanted subcutaneously into the flanks of nude mice. After 1 weeks of implantation, the groups were treated with GEM (i.p. injection of 50 mg/kg weekly) or CDDP (intraperitoneal injection of 15 mg/kg on weekly). The mean growth of tumor volume (mm3) ± SD for each group is shown. Data were analyzed by Student's t‐test. **P < 0.01. n.s., non‐significant.
- Immunohistochemistry (IHC) staining of Ki‐67 in UMUC3WT and GR cells exposed to CDDP/GEM. Ki‐67 staining quantitation of UMUC3WT and UMUC3GR cells exposed to CDDP/GEM are shown as bar graph The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by one‐way ANOVA with the Bonferroni test. **P < 0.05, ***P < 0.001. n.s., non‐significant.
- Schematic of the mechanism by which GR UC cells acquire an antioxidant defense. Bypass of the oxidative PPP via aerobic glycolysis is suspected to be responsible for increased NADPH production. The increased NADPH leads to an increase in the GSH level. The increase in the GSH level decreases ROS generation and leads to the acquisition of cross‐resistance to CDDP.
- NADPH/NADP ratios in WT and GR cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
- Intracellular GSH levels in WT and GR cells after 24 h of exposure to the DMSO, CDDP (1 μM and 10 μM). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01. n.s., non‐significant.
- Intracellular ROS generation in WT and GR cells after 24 h of exposure to CDDP (1 and 10 μM). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. ***P < 0.001. n.s., non‐significant.
- NADPH/NADP ratios of GR cells transfected with NTC, IDH2 siRNA#1, and siRNA#2 (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
- Intracellular GSH levels in GR cells transfected with NTC, IDH2 siRNA#1, and siRNA#2 (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- ROS generation levels in GR cells transfected with NTC, IDH2 siRNA#1, and siRNA#2 (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01. n.s., non‐significant.
- Graph shows the viability of WT and GR cells exposed to various concentrations of GEM for 48 h after transfection with NTC, IDH2 siRNA#1, and siRNA#2 (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates).
- Treatment in vivo using orthotopically implanted mouse models (n = 5 mice per group). We implanted UMUC3GR cells (2 × 106 cells) transfected with #siNTC for 48 h, UMUC3GR cells (2 × 106 cells) transfected with siIDH2#1 and siIDH2#2 for 48 h, and then injected subcutaneously into the flank of each BALB/c‐nu/nu mice. One week after implantation, cells were treated with CDDP (i.p. injection of 15 mg/kg weekly). The mean tumor volume (mm3) ± SD for each group is shown. Data were analyzed by Student's t‐test. *P < 0.05.
Recent findings have revealed that increased production of NADPH contributes to reductive biosynthesis and elevates antioxidant defense (Kalyanaraman et al, 2018; Zhang et al, 2022). In particular, NADPH production mainly relies upon oxidative PPP (Chen et al, 2019). To verify this, we used a G6PD inhibitor (G6PD‐i), which is known as a major PPP inhibitor, to confirm whether NADPH production is dependent on the oxidative PPP. We found that CDDP sensitivity was restored in both GR cell lines by combined treatment with the G6PD inhibitor and CDDP (Fig EV4A and B). Moreover, we confirmed that the NADPH/NADP ratio and GSH level in GR cells were significantly decreased by G6PD‐i administration, which suggests that NADPH production results mostly from oxidative PPP stimulation (Fig EV4C–F). Thus, we hypothesized that the NADPH/NADP increase delivered through PPP stimulation may contribute to further GSH production and lowering reactive oxygen species (ROS) generation, which inhibits the therapeutic effect of CDDP by reinforcing antioxidant defense (Fig 6E).
Figure EV4. Suppression of oxidative PPP lowers NADPH level.
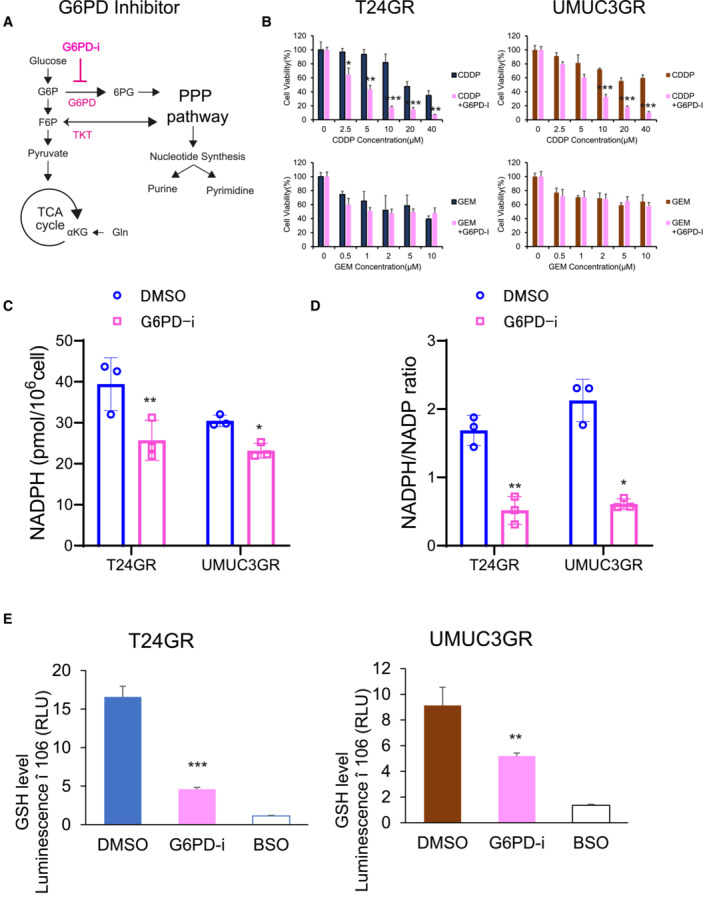
- Simple schematic of the glycolytic pathway and the pentose phosphate pathway (PPP). The G6PD inhibitor (G6PD‐i) inhibits the oxidative bypass pathway coupling glycolysis to the PPP.
- Bar graph showing the viability of GR cells exposed to various concentrations of CDDP and GEM for 48 h and treated with DMSO or G6PD‐i (10 μM) (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- Total NADPH levels in T24GR and UMUC3GR cells after 48 h of exposure to DMSO or G6PD‐i (10 μM). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- NADPH/NADP ratio in T24GR and UMUC3GR cells after 48 h of exposure to DMSO or G6PD‐i (10 μM). The data are shown as the mean values ± SDs (n = 3, biological replicates). Data were analyzed by Student's t‐test and are plotted relative to the levels in DMSO group. *P < 0.05, **P < 0.01.
- Intracellular GSH levels in GR cells after 48 h of exposure to DMSO or G6PD‐i (10 μM) (left: T24GR, right: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01, ***P < 0.001.
To verify this hypothesis, we measured NADPH/NADP ratio in WT and GR cells. As a result, NADPH/NADP ratio was significantly elevated in GR cells (Fig 6F). Moreover, high GSH levels were confirmed in GR cells exposed to CDDP, which suggests reductive biosynthesis in GR cells (Fig 6G). The ROS generation level was also significantly lower in GR cells than in WT cells, which suggests that the antioxidant defense is induced in GR cells treated with CDDP (Fig 6H). However, with the transfection‐mediated downregulation of IDH2, the NADPH/NADP ratio and GSH level were decreased in GR cells (Fig 6I and J). ROS generation was higher in siIDH2 GR cells (Fig 6K), which clearly suggests that IDH2 gain of function is related to the acquisition of cross‐resistance to CDDP in GR UC cells (Fig 6L). To confirm all the results of the in vitro experiments, we conducted an in vivo experiment. In a UMUC3GR cell‐derived mouse xenograft UC model, GR cell transfected with siNTC, GR cell transfected with siIDH2#1, and siIDH#2 were treated with CDDP, respectively. As a result, IDH2 knock down group restored chemosensitivity upon CDDP (tumor volume in siNTC transfected UMUC3GR‐implanted mice: 499.6 ± 177.3 mm3, tumor volume in siIDH2#1 transfected UMUC3GR‐implanted mice: 173.6 ± 49.4 mm3, and tumor volume in siIDH2#2 transfected UMUC3GR‐implanted mice: 198.2 ± 64.3 mm3, respectively; Fig 6M).
According to these results, GEM resistance also influences therapeutic sensitivity to CDDP due to stimulation of the oxidative PPP, which is a major source of NADPH production, leading to increased antioxidant defense against CDDP.
IDH2 inhibitor (AGI6780) restores chemoresistance in UC cells
To determine the pharmacological effect of an IDH2 inhibitor on GR cells, we used the IDH2 inhibitor AGI‐6780, a chemical compound known to potently inhibit both mutant and WT IDH2 (Chen et al, 2020). We evaluated its impact on cell viability and proliferation. We found that the IC50 values of AGI6780 were 6.4 μM in T24GR cells and 8.9 μM in UMUC3GR cells. Thus, we treated T24GR and UMUC3GR cells with 10 μM AGI6780 in combination with various concentrations of GEM to evaluate whether AGI6780 restores GEM sensitivity. As a result, a significant cytotoxic effect was observed in cells treated with the combination of GEM and AGI6780 compared with those treated with GEM alone (Fig 7A). Measurement of 2‐HG production revealed significantly lower 2‐HG levels observed in both types of GR cells treated with AGI6780 (0.632‐fold lower in T24GR cells and 0.518‐fold lower in UMUC3GR cells compared with vehicle control‐treated cells; Fig 7B). Similarly, significantly lower dCTP levels were observed in GR cells treated with AGI6780 (0.529‐fold lower in T24GR cells and 0.191‐fold lower in UMUC3GR cells compared with vehicle control‐treated cells; Fig 7C).
Figure 7. AGI6780 restores chemosensitivity in chemo‐resistant UC cells.
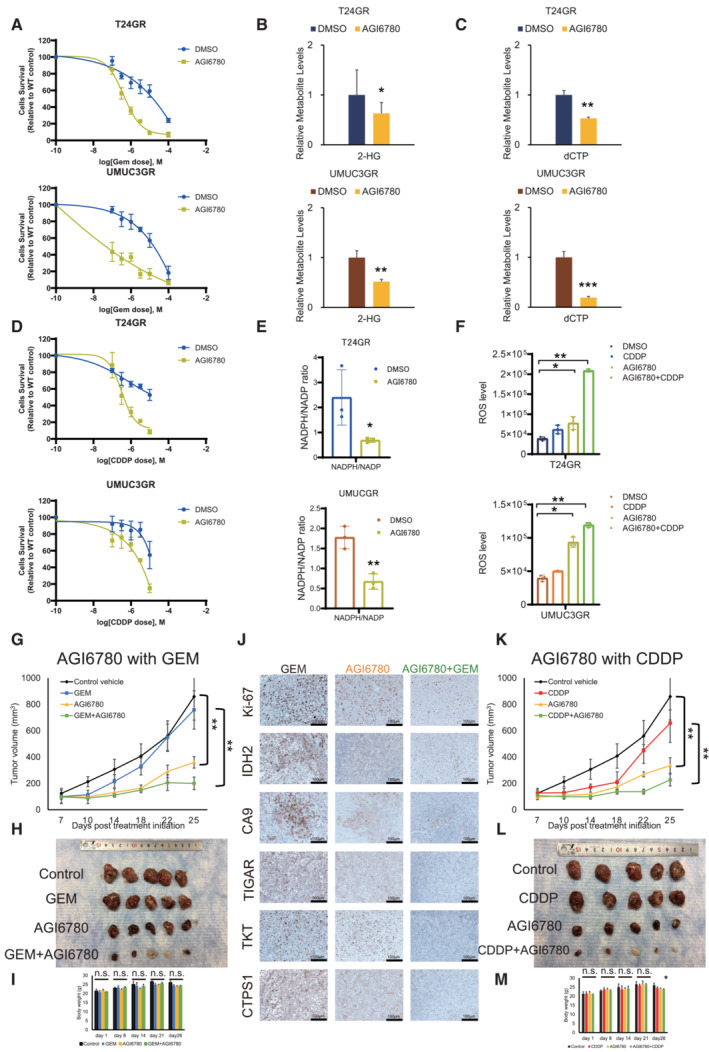
- Graph showing the viability of GR cells exposed to various concentrations of GEM for 48 h in combination with DMSO or 10 μM AGI6780 (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates).
- Relative 2‐HG production level based on targeted CE‐MS metabolomics in GR cells exposed to DMSO or 10 μM AGI6780 for 24 h (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01.
- Relative dCTP level based on targeted CE‐MS metabolomics in GR cells exposed to DMSO or 10 μM AGI6780 for 24 h (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01, ***P < 0.001.
- Graph showing the viability of GR cells exposed to various concentrations of CDDP for 48 h in combination with DMSO or 10 μM AGI6780 (upper: T24GR, lower: UMUC3GR). The data are shown as the mean values ± SDs (n = 3, biological replicates).
- NADPH/NADP ratio in GR cells exposed to DMSO or 10 μM AGI6780 for 24 h. The data are shown as the mean values ± SDs (n = 3, biological replicates). The data were analyzed by Student's t‐test (upper: T24GR, lower: UMUC3GR). *P < 0.05, **P < 0.01.
- Intracellular ROS generation in GR cells exposed to DMSO or 10 μM AGI6780 for 24 h. The data are shown as the mean values ± SDs (n = 3, biological replicates). The data were analyzed by Student's t‐test (upper: T24GR, lower: UMUC3GR). *P < 0.05, **P < 0.01.
- In vivo treatment of orthotopically implanted mouse models (n = 5 mice per group). UMUC3GR cells (2 × 106 cells) were implanted subcutaneously into the flanks of nude mice. One week after implantation, the groups were treated with GEM alone (i.p. injection of 50 mg/kg weekly), AGI6780 alone (i.p. injection of 50 mg/kg daily), or a combination of GEM and AGI6780 (i.p. injection of 50 mg/kg GEM weekly and i.p. injection of 50 mg/kg AGI6780 daily). The mean tumor volume (mm3) ± SD for each group is shown. Data were analyzed by Student's t‐test. **P < 0.01.
- Representative images of tumors upon necropsy.
- Body weights of mice with the indicated treatments. The data are shown as the mean values ± SDs (n = 5 mice per group), and were analyzed by Student's t‐test. n.s. = non‐significant.
- IHC staining for IDH2, Ki‐67, CAIX, TIGAR, TKT, and CTPS1 in groups with GEM alone, AGI6780 alone, and the combination of GEM and AGI6780.
- In vivo treatment of orthotopically implanted mice (n = 5 mice per group). UMUC3GR cells (2 × 106 cells) were implanted subcutaneously into the flanks of nude mice. One week after implantation, the groups were treated with CDDP alone (weekly i.p. injection of 15 mg/kg), AGI6780 alone (daily i.p. injection of 50 mg/kg), or a combination of GEM and AGI6780 (weekly i.p. injection of 15 mg/kg CDDP and daily i.p. injection of 50 mg/kg AGI6780). The tumor volume (mm3, mean ± SD) in for each group is shown. Data were analyzed by Student's t‐test. **P < 0.01.
- Representative images of tumors upon necropsy.
- Body weights of mice treated as indicated. The data are shown as the mean values ± SDs (n = 5 mice per group) and were analyzed by Student's t‐test. n.s. = non‐significant.
Source data are available online for this figure.
Regarding CDDP sensitivity, we further treated T24GR and UMUC3GR cells with 10 μM AGI6780 in combination with various concentrations of CDDP. Similar to the GEM experiment, AGI6780 combined with CDDP showed a significant cytotoxic effect on both GR cell lines (Fig 7D). Less NADPH/NADP production and higher ROS generation were observed in the AGI6780‐treated group (Fig 7E and F).
To verify the therapeutic effect of AGI6780 in vivo, we performed an animal study to evaluate the therapeutic effects of AGI6780 alone and in combination with GEM. On Day 25 after the start of treatment, the tumor volume (mean ± standard deviation [SD]) in mice treated with AGI6780 alone was 360.0 ± 43.9 mm3, which was significantly lower than that in mice treated with vehicle control (859.8 ± 178.9 mm3, P = 0.006). Furthermore, the tumor volume in mice treated with the combination of GEM and AGI6780 was 200.2 ± 47.8 mm3, which was significantly lower than that in mice treated with GEM alone (758.4 ± 145.8 mm3, P = 0.002; Fig 7G and H). There were no apparent toxic effects, such as a decrease in body weight, in mice in any treatment group (Fig 7I). IHC revealed that the expression of IDH2, CAIX (a surrogate marker of hypoxia, similar to Hif‐1α), TIGAR, TKT, and CTPS1 was decreased in the AGI6780 and GEM/AGI6780 combination treatment groups compared with the control group (Fig 7J). Likewise, similar results were obtained regarding AGI6780 in combination with CDDP in the murine UC xenograft model (the tumor volume [mean ± SD]) on Day 25 in mice treated with CDDP and AGI6780 was 226.4 ± 45.6 mm3, which was significantly lower than that in mice treated with CDDP alone (658.0 ± 100.1 mm3, P = 0.001; Fig 7K–M).
Furthermore, we also tested the impact of AGI221, another selective IDH2 inhibitor called enasidenib, in both GR cell lines. Similar to the above results, we found a significant cytotoxic effect with the combination of GEM and AGI221 in both in vitro and in vivo experiments (Appendix Fig S1).
High IDH2, CAIX, TIGAR, TKT, and CTPS1 levels in human UC patients are correlated with a poor prognosis
Based on the in vitro and in vivo experiments, we conducted IHC studies on tumor specimens from UC patients who underwent surgery. By staining all anaerobic enzymes, we found high expression of IDH2, CAIX, TIGAR, TKT, and CTPS1 in tumor specimens with advanced upper tract urothelial carcinoma (UTUC) patients (Fig 8A and B). Furthermore, we focused on 74 of the 214 (34.6%) UTUC patients who received adjuvant chemotherapy after surgical intervention. Overall, 34 (45.9%) showed recurrence after adjuvant chemotherapy and 30 (40.5%) died by cancer death. By dividing the cohort with chemo‐sensitive and chemo‐resistant UTUC, it revealed that CAIX, TIGAR, TKT, and CTPS1 expression were higher in the chemo‐resistant group than chemo‐sensitive group (Fig 8C). High IDH2 expression was observed in 37 (50.0%) of the 74 patients. The Kaplan–Meier method also revealed that patients who had high expression of CAIX, TIGAR, TKT, and CTPS1 showed significantly lower CSS than those with low expression (Fig 8D). In particular, the 5‐year CSS was 45.0% in high IDH2 expression group, which was significantly lower than 63.7% of low IDH2 expression group (P = 0.041). These results suggest that UTUC tumor specimens with high IDH2 expression may follow poor prognosis. The expressions of anaerobic enzymes may indicate the metabolic alterations in UC cells, revealing the potential for acquiring chemoresistance. To further verify the association between chemoresistance and IDH2 expression in advanced UC, we obtained another 32 muscle‐invasive bladder carcinoma (MIBC) tumor specimens from patients who underwent neoadjuvant GC therapy before radical cystectomy (RC) (Table 2). High IDH2 expression was observed in 14 (43.8%) of the 32 patients. Similar to the results for UTUC tumor specimens, CAIX, TIGAR, TKT, and CTPS1 showed higher rates of expression in the high IDH2 expression groups (Fig 8E–G). MIBC patients with high IDH2, TIGAR, TKT, and CTPS1 expression showed a poor prognosis after neoadjuvant GC therapy (Fig 8H).
Figure 8. High IDH2 expressed patients follow poor prognosis.
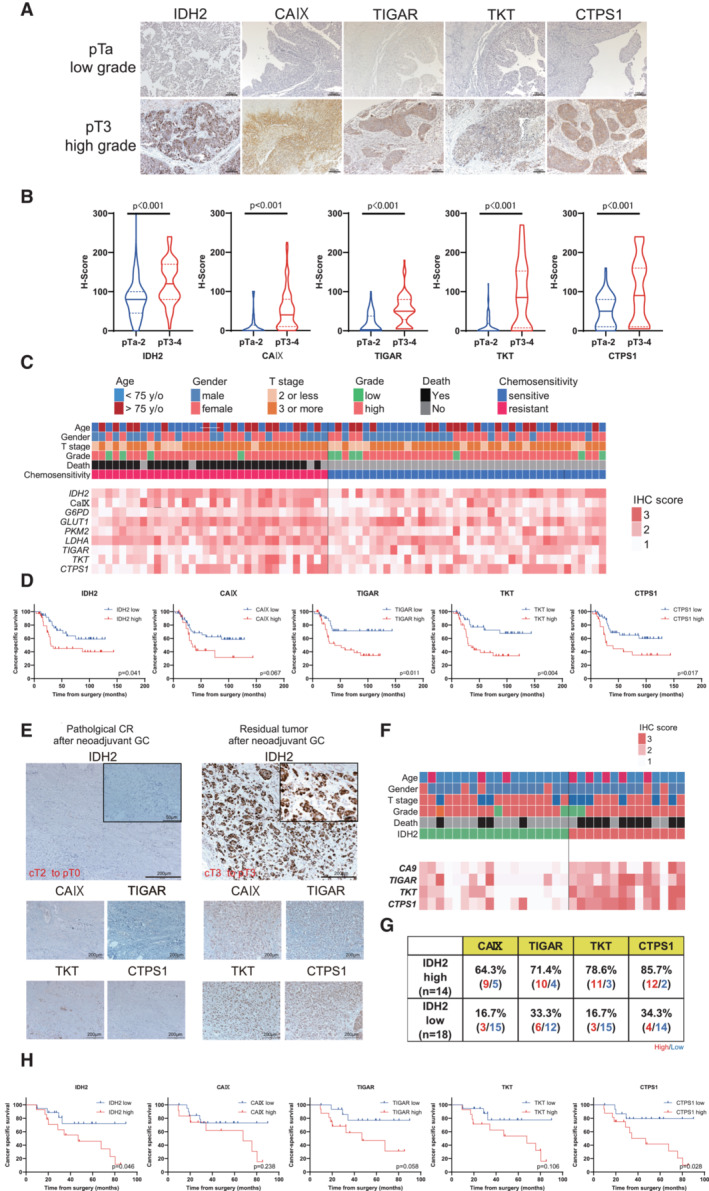
- Representative immunostaining of IDH2, CAIX (surrogate marker of Hif‐1α), TIGAR, TKT, and CTPS1 in surgical specimens from upper tract urothelial carcinoma (UTUC) patients. The upper panel represents the immunostaining of metabolic enzymes of early‐stage UTUC tumors, whereas the lower panel represents the images of invasive UTUC tumors. The power field scale bar = 100 μm.
- Comparison of histoscores (H‐scores) for each metabolic enzyme divided by early‐ or advanced‐stage tumors. The H‐score was calculated by applying the following formula: mean percentage × intensity (range, 0–300). Violin plots were divided by pTa‐2: 116 UC patients and pT3‐4: 98 UC patients. Data were analyzed by Student's t‐test.
- Heat map describing the IHC score in UTUC patients treated with GEM and CDDP (GC) adjuvant chemotherapy (n = 74). The heat map of metabolic enzymes in patients was classified by chemo‐sensitivity with information for age, sex, pT stage, grade, and death.
- Kaplan–Meier curves showing the cancer‐specific survival (CSS) of 74 UTUC patients treated with GC adjuvant chemotherapy. Metabolic enzymes were classified according to the results of receiver operating curve (ROC) analysis. Data were compared with the log‐rank test.
- Representative immunostaining of IDH2, CAIX, TIGAR, TKT, and CTPS1 in surgical specimens from muscle invasive urothelial carcinoma (MIBC) patients with neoadjuvant GC chemotherapy. Case 1 was a transurethral resection of bladder tumor (TURBT) specimen that induced a pathological complete response (pT0) in a radical cystectomy (RC) specimen. Case 2 was indicated as residual pT3 after neoadjuvant GC therapy. The power field scale bars = 200 and 50 μm.
- Heat map describing the IHC score in MIBC patients treated with GC chemotherapy (n = 32). The heat map of metabolic enzymes in patients classified by IDH2 expression with information for age, sex, pT stage, grade, and death.
- Details of high and low expression of CAIX, TIGAR, TKT, and CTPS1 classified with IDH2 expression.
- Kaplan–Meier method showing the CSS of 32 MIBC patients treated with GC chemotherapy. The cutoffs for metabolic enzyme levels were determined according to the results of ROC analysis. Data were compared with the log‐rank test.
Table 2.
Patient and tumor characteristics of MIBC patients who received neoadjuvant chemotherapy.
| Patient characteristics | MIBC patients with NAC | |
|---|---|---|
| Total n = 32, (%) | ||
| Follow‐up duration | Mean ± SD | 46.0 ± 9.35 |
| Time to death | Mean ± SD | 14.1 ± 4.6 |
| Time to meta | Mean ± SD | 33.4 ± 4.21 |
| Age | < 75 | 24 (75.0) |
| ≥ 75 | 8 (25.0) | |
| Sex | Male | 25 (78.1) |
| Female | 7 (21.9) | |
| ECOG‐PS | 0–1 | 29 (90.6) |
| 2 | 3 (9.4) | |
| Tumor histology | Pure UC | 27 (84.4) |
| Non‐pure UC | 5 (15.6) | |
| Pathological T stage | T0 | 11 (34.4) |
| 1 | 4 (12.5) | |
| 2 | 7 (21.9) | |
| 3 | 6 (18.7) | |
| 4 | 4 (12.5) | |
| Pathological N stage | 0 | 24 (75.0) |
| 1,2 | 8 (25.0) | |
| Tumor grade | Low | 2 (6.2) |
| High | 30 (93.8) | |
| LVI | Absent | 18 (56.2) |
| Present | 14 (43.8) | |
| Concomitant CIS | No | 29 (90.6) |
| Yes | 3 (9.4) | |
| Systemic adjuvant chemotherapy | No | 25 (78.1) |
| Yes | 7 (21.9) | |
| Tumor recurrence | No | 18 (56.2) |
| Yes | 14 (43.8) | |
| Cancer specific death | No | 19 (59.4) |
| Yes | 13 (40.6) |
CIS; carcinoma in situ; ECOG‐PS; Eastern Cooperative Oncology Group‐ Performance Status; LVI; lymphovascular invasion; MIBC; muscle invasive bladder cancer; NAC; neoadjuvant chemotherapy; SD; standard deviation; UC; urothelial carcinoma.
In summary, we found that GR UC cells exhibit reductive glutamine metabolism‐mediated aerobic glycolysis with PPP bypass for chemoresistance acquisition. For the establishment of complete metabolic reprogramming, WT IDH2 gain of function drives Hif‐1α stabilization via 2‐HG production. Stabilized Hif‐1α under normoxic conditions stimulates aerobic glycolysis and leads to increased production of dCTP for molecular competition with GEM through PPP stimulation. In the course of aerobic glycolysis to PPP bypass, the production of strong antioxidants such as NADPH is increased to reinforce the antioxidant defense against CDDP. Knockdown or pharmacological suppression (i.e., AGI6780 treatment) of IDH2 inhibits the complete metabolic reprogramming and the expression of aerobic glycolysis/PPP related components, which results in restoration of GEM/CDDP sensitivity (Fig 9).
Figure 9. Mechanism of metabolic reprogramming.
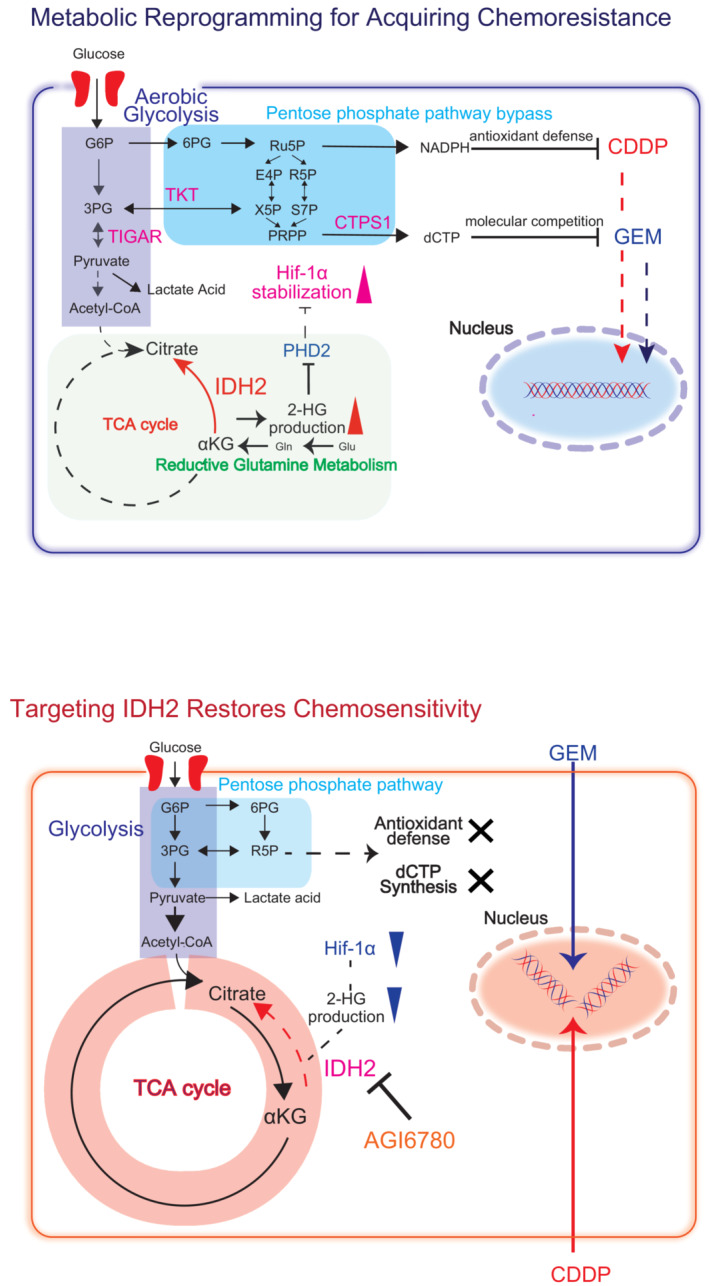
Graphical summary of the metabolic basis of GEM resistance in UC cells. GR cells demonstrate 2‐HG production by reductive glutamine metabolism stimulated by IDH2 gain of function. The increase of 2‐HG production suppresses PHD2 function and stabilizes Hif‐1α expression, which regulates the key metabolic enzymes TIGAR, TKT, and CTPS1, which are involved with aerobic glycolysis and PPP bypass. With the stimulation of aerobic glycolysis metabolism, it promotes the generation of dCTP for acquiring therapeutic competition against GEM. At the same time, oxidative PPP stimulation increases NADPH/NADP production and enhances antioxidant defense, which facilitates cross‐resistance to CDDP. Thus, inhibition of IDH2 suppress the entire metabolic reprogramming and therefore restore both GEM and CDDP sensitivity in UC cells.
Discussion
In this study, we presented an underlying mechanism by which WT IDH2 gain of function induces metabolic reprogramming in chemo resistant UC cells.
Altered metabolism is one of the important hallmarks of cancer (Hanahan & Weinberg, 2011). The association between intracellular metabolic reprogramming and the acquisition of drug resistance has been assessed in recent studies (Metallo et al, 2011; Semenza, 2013). Previous studies have revealed that intracellular Hif‐1α stabilization confers resistance to CDDP, oxaliplatin, and paclitaxel in various cancers (Ai et al, 2016; Rankin & Giaccia, 2016). One novel study revealed that GR pancreatic cancer cells showed a metabolic shift from aerobic metabolism to anaerobic metabolism, accompanied by increased glycolytic metabolism fed through the nonoxidative arm of the PPP, which resembles our present results (Shukla et al, 2017). However, it remained unknown about the relationships between metabolic reprogramming and chemoresistance in UC cells. From the metabolome analysis, we found a unique metabolic characteristic in GEM resistant UC cells that they appeared to alter the intracellular metabolome to generate molecules that can compete against GEM by increasing pyrimidine biosynthesis. Specifically, we revealed that WT IDH2 mediated glutamine dependent reductive carboxylation increasing 2‐HG production to stabilize Hif‐1α, which is an important feature for bridging the metabolomic stimulation to aerobic glycolysis and PPP bypass.
The concept of reductive glutamine metabolism was recently clarified based on stable isotope labeling studies in cancer cells exposed to hypoxia or with genetic deficiencies related to mitochondrial function (Mullen et al, 2011; Wise et al, 2011). These studies demonstrated metabolic reprogramming of the reductive flux of 13C‐labeled glutamine to citrate and acetyl‐CoA, presumably via IDH2‐catalyzed reductive carboxylation of α‐KG to isocitrate and citrate. This pathway for glutamine metabolism is called glutaminolysis and is suggested to provide substrates for the expansion of biomass during cancer cell proliferation. “Reverse” flux from α‐KG to citrate has often been observed in several cancers with IDH2 gene mutations, such as low‐grade glioma and AML (Cairns & Mak, 2013; Chan et al, 2015). Production of 2‐HG by mutant IDH2 is considered a key oncogenic mechanism, which led to the development of drugs specifically targeting mutant IDH2 (Xu et al, 2011). Interestingly, however, the presence of tumor cells harboring mutant IDH2 has often been associated with a better response to treatment than the presence of tumor cells harboring WT IDH2 (Molenaar et al, 2018). Indeed, IDH2 mutations have been associated with improved overall survival in AML patients (Xu et al, 2017).
In contrast, several recent articles have demonstrated that glutamine‐dependent reductive TCA metabolic flux is also observed after WT IDH2 gain of function, leading to cancer cell proliferation with a poor treatment response (Barnabas et al, 2021; Zeng et al, 2022). Although the function of WT IDH2 is still under investigation, many recent studies have compiled the potential mechanisms of WT IDH2, which involve aerobic glycolysis, glutamine‐dependent reductive carboxylation, and/or 2‐HG production (Li et al, 2018; Jezek, 2020; Shi et al, 2020). According to these articles, several differences have been observed in metabolic reprogramming mediated by WT IDH2 compared with that mediated by mutant IDH2, including upregulation of c‐MYC expression, a modest increase in 2‐HG, and poorer oncological outcomes. One novel study by Zeng et al (2022) recently confirmed that WT IDH2 gain of function in AML patients plays a critical role in activating reductive TCA metabolic flux and thus promotes the survival of acute myelocyte leukemia (AML) cells and leads to poor prognosis. These recent articles support our findings in indicating that metabolic reprogramming may also begin with WT IDH2 gain of function, resulting in stimulation of glycolysis and the PPP for chemoresistance acquisition. On the other hand, the exact mechanism by which WT IDH2 increases its expression with long‐term GEM exposure is still under investigation. One assumption is that aerobic glycolysis followed by PPP activity may trigger the entire metabolic hub to induce glutamine metabolism, which results in IDH2 overexpression (Barnabas et al, 2021). Indeed, our results showed that both glucose and glutamine metabolism were stimulated in GR cells, while WT cells relied on only glucose metabolism, suggesting that aerobic glycolysis may stimulate secondary glutamine metabolism as another energy source to drive the reductive TCA. Another assumption is that Hif‐1α or other glycolytic enzymes (e.g., TKT or CTPS1) might affect the expression of another key transcription factor (e.g., Nrf2) that regulates IDH2 (Rojo de la Vega et al, 2018; Dai et al, 2022). However, these possibilities need to be further investigated in future studies.
Another new insight in our study is our finding of a high NADPH/NADP ratio in GR cells, which suggested the potential mechanism of cross‐resistance to CDDP. Generally, NADPH is consumed in the presence of reductive TCA cycle activity, when citrate and acetyl‐CoA produced by reductive carboxylation are used for fatty acid synthesis (Mullen et al, 2014). Thus, we asked why the total NADPH level is increased in GR cells, since IDH2 consumes NADPH in the converting a‐KG to isocitrate. From the experiment of G6PD inhibitor treatment (Fig EV4A–E), we found that NADPH production largely relies upon the oxidative PPP and/or production through the PPP, and that the NADPH/NADP ratio therefore increases because of metabolic stimulation of aerobic glycolysis and PPP bypass. Since IDH2 downregulation significantly decreased the NADPH/NADP ratio in GR cells, our current data suggested that IDH2 inhibition would restore PPP activity to normal levels and would therefore no longer support NADPH production, consistent with the results of a recent study (Zhang et al, 2021). NADPH is metabolized to GSSG/GSH, which is also a strong reducing agent that promotes antioxidant and detoxification activities. This chain reaction of reduction activities, which diminishes cytotoxic effects on UC cells, explains the sensitivity to CDDP‐based chemotherapy. Thus, we consider that IDH2‐mediated metabolic reprogramming contributes to not only GEM resistance but also to CDDP resistance by increasing glucose flux into the PPP and, as a consequence, increases NADPH production.
Importantly, we showed that specific knockdown of WT IDH2 led to suppression of metabolic reprogramming (Fig EV5A–G) and re‐sensitization of chemoresistant UC cells to chemotherapeutic agents, indicating that this molecule could be a potential therapeutic target. Indeed, pharmacological inhibition of IDH2 by using AGI6780 showed significant cytotoxic effects when combined with GEM in GR cells both in vitro and in vivo (Wang et al, 2013; Liu et al, 2022). It also restored CDDP sensitivity to some degree by decreasing the NADPH/NADP ratio and increasing ROS generation. In addition, recent articles have confirmed that AGI6780 shows apparent cytotoxicity in AML, Epstein–Barr virus‐related head and neck squamous carcinoma, and chemoresistant chronic myeloid leukemia (Shi et al, 2020; Liu et al, 2022; Zeng et al, 2022). Collectively, these recent data suggest that WT IDH2 can become a “druggable” target and that it might be a feasible approach for the clinical treatment of chemo‐resistant UC.
Figure EV5. AGI6780 suppresses the entire metabolic reprogramming.
- The viability of J82WT and J82‐IDH2ox cells was assessed by WST assays under treatment with GEM, mizorubine, GEM with mizorubine, leflunomide, or GEM with leflunomide. The data are shown as the mean values ± SDs (n = 3, biological replicates). Comparisons were made with respect to the corresponding controls or the indicated groups, followed by analysis with Student's t‐test. **P ≤ 0.01, ***P ≤ 0.001. n.s., non‐significant.
- Complete heatmap showing the intracellular levels of metabolites in J82‐IDH2ox cells treated with AGI6780. For each cell line, data for three individual biological replicates are shown, and the levels were normalized to those in J82‐IDH2ox cells.
- Major metabolites in the glycolytic pathway altered in J82‐IDH2ox cells treated with AGI6780 compared with untreated J82‐IDH2ox cells. The data are shown as the mean values ± SDs (n = 3, biological replicates). The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05.
- Levels of intermediate metabolites of the tricarboxylic acid (TCA) cycle based on targeted CE‐MS metabolomics in J82‐IDH2ox cells treated with AGI6780 relative to untreated J82‐IDH2ox cells based on targeted CE‐MS metabolomics. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
- Relative 2‐HG production level based on targeted CE‐MS metabolomics in J82‐IDH2ox cells treated with AGI6780 compared with untreated J82‐IDH2ox cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
- PPP metabolite levels based on targeted CE‐MS metabolomics in J82‐IDH2ox cells treated with AGI6780 relative to untreated J82‐IDH2ox cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. *P < 0.05, ***P < 0.001.
- Relative dCTP level based on targeted CE‐MS metabolomics in J82‐IDH2ox cells treated with AGI6780 compared with untreated J82‐IDH2ox cells. The data are shown as the mean values ± SDs (n = 3, biological replicates) and were analyzed by Student's t‐test. **P < 0.01.
For the two decades, the first‐line chemotherapy regimen of GEM and CDDP has not been improved. Although high‐dose methotrexate, vinblastine sulfate, adriamycin, and cisplatin (MVAC), GEM combined with carboplatin, and GEM combined with paclitaxel have been introduced as alternative regimens for UC (Sternberg et al, 2001; Calabro et al, 2009; De Santis et al, 2012), no significant advances have resulted from such efforts. As the current study suggests the decreased sensitivity of GEM and CDDP to metabolic reprogramming in UC cells, clinicians need to consider that the combination of GEM and CDDP needs to be questioned, and other options should be considered to maximize the therapeutic effect for metastatic UC patients. Although several immune‐check point inhibitors (ICIs) have been emerged for 2nd‐line treatment for UC, only a small amount of patients receive benefit. Since the energetic interplay between tumor and immune cells confronts “metabolic competition” between tumor proliferation and immune activity, limiting nutrient availability in immune cells may hinder immune cell function (Afonso et al, 2020). Thus, the present study may suggest that the hallmarks of metabolic reprogramming in UC cells after the acquisition of chemoresistance may also be related to the treatment efficacy of immunotherapy (Scholtes et al, 2021). To confirm this hypothesis, however, further experiments are warranted.
To conclude, our study delineates the core metabolic alterations that mediate GEM resistance in UC cells by elucidating the entire landscape of the metabolic mechanism of chemoresistant UC cells. WT‐ IDH2 gain of function may play an important role for metabolic reprogramming in UC cells; therefore, targeting IDH2 may provide new insights for defeating chemoresistant UC cells.
Materials and Methods
Reagents and Tools table
| Reagent | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti‐IDH2 monoclonal antibody in mouse | Abcam | Cat#: ab55271; RRID: AB_943793 |
| Purified Mouse Anti‐Human Hif1‐α monoclonal antibody in mouse | Santa Cruz Biotechnologies | Cat#: sc‐13515; RRID: AB_627723 |
| Carbonic anhydrase IX monoclonal antibody in mouse | BioScience | Cat# DF13218; RRID: AB_2846237 |
| Anti‐GLUT1 (A‐4) monoclonal antibody in mouse | Santa Cruz Biotechnologies | Cat# sc‐377228; RRID: AB_2190936 |
| Anti‐TIGAR polyconal antibody in rabbit | Invitrogen | Cat# PA1‐41487; RRID: AB_2259272 |
| PKM2 (D78A4) XP Rabbit monocloncal antibody in rabbit | Cell Signaling Technology | Cat# 4053; RRID: AB_1904096 |
| LDHA‐Specific polyclonal antibody in rabbit | Proteintech | Cat# 199871‐1‐AP |
| Anti‐ G6PD imonocloncal antibody in mouse | Santa Cruz Biotechnologies | Cat# sc‐373886; RRID: AB_10918100 |
| Anti‐TKT polyclonal antibody produced in rabbit | Sigma‐Aldrich | Cat#HPA029481; RRID: AB_10558885 |
| Anti‐CTPS1 (416‐430) polyclonal antibody produced in rabbit | Sigma‐Aldrich | Cat# SAB1101072; RRID: AB_10606733 |
| Anti‐PHD2 polyclonal antibody in rabbit | Invitrogen | Cat# PA1‐16525; RRID: AB_568563 |
| Ki‐67, mouse monoclonal anibody | Dako | # Cat: M7240 |
| Anti‐β‐actin monoclonal antibody in mouse | Sigma‐Aldrich | #Cat: A5441 |
| Chemicals | ||
| Cisplatin | Nippon Kayaku | N/A |
| Gemcitabine | Nippon Kayaku | N/A |
| 2'‐Deoxycytidine | Sigma‐Aldrich | #Cat: D3897 |
| AGI6780 | Selleck Chemicals | #Cat: S7241 |
| AGI221 (Enadesinib) | Cayman Chemical | #Cat: 21277 |
| [13C5]‐glutaime | Millipore Sigma | #Cat: 605166 |
| G6PD‐inhibitor | Selleck Chemicals | #Cat: S9617 |
| Leflunomide | Selleck Chemicals | #Cat: S1247 |
| Mizoribine | Selleck Chemicals | #Cat: S1384 |
| BPTES | Selleck Chemicals | #Cat: S7753 |
| 2'‐Deoxyadenosine monohydrate | Selleck Chemicals | #Cat: S9351 |
| 2'‐Deoxyguanosine monohydrate | Selleck Chemicals | #Cat: S9007 |
| Thymidine | Selleck Chemicals | #Cat: S4803 |
| Galiiac acid | Selleck Chemicals | #Cat: S4603 |
| Digoxin | Selleck Chemicals | #Cat: S4290 |
| Buthionine Sulfoximine | Sigma‐Aldrich | #Cat: 415383 |
| Critical commercial assays | ||
| Blood & Cell Culture DNA extraction Kits | Qiagen | #Cat: 13323 |
| Cell Tox Green Cytotoxicity Assay | Promega | #Cat: G8741 |
| NADP/NADPH Quantitation Colorimetric Kit | Biovision | #Cat: K347‐100 |
| CellROX® Orange | Thermo Fischer Scientific | #Cat: C10443 |
| GSH‐Glo Glutathione | Promega | #Cat: V6911 |
| Dharmafect transfection Reagent 1 | Thermo Fischer Scientific | #Cat: T‐2001 |
| Xfect™ Transfection Reagent | Takara Bio | 631317Z1317N |
| pcDNA™3.1 (+) Mammalian Expression Vector | Invitrogen | V79020 |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent Technologies | 103344‐100 XF |
| Seahorse XFe24 FluxPak mini | Agilent Technologies | 102342‐100 |
| Seahorse XF RPMI assay medium pack | Agilent Technologies | 103681‐100 |
| Experimental models: cell lines | ||
| Human: T24 | American Type Culture Collection | #Cat: ATCC HTB‐4 |
| Human: UMUC3 | American Type Culture Collection | #Cat: ATCC CRL‐1749 |
| Human: J82 | American Type Culture Collection | #Cat: ATCC HTB‐1 |
| Experimental models: oraganisms/strains | ||
| BALB/cAJcl‐nu/nu | CLEA Japan, Inc. | #Cat: 6496 |
| Oligonucleotides | ||
| Taqman Probe hIDH2 | Applied Biosystems | Hs00953879_m1 |
| Taqman Probe hEGLN1 | Applied Biosystems | Hs00254392_m1 |
| Taqman Probe hHIF1A | Applied Biosystems | Hs00153153_m1 |
| Taqman Probe hGLUT1 | Applied Biosystems | Hs00892681_m1 |
| Taqman Probe hG6PD | Applied Biosystems | Hs00166169_m1 |
| Taqman Probe hCTPS1 | Applied Biosystems | Hs01041852_m1 |
| Taqman Probe hTIGAR | Applied Biosystems | Hs00608644_m1 |
| Taqman Probe hTKT | Applied Biosystems | Hs0115545_m1 |
| Taqman Probe hPKM2 | Applied Biosystems | Hs00987255_m1 |
| Taqman Probe hLDHA | Applied Biosystems | Hs03405707_g1 |
| Taqman Probe hDHODH | Applied Biosystems | Hs00361406_m1 |
| Taqman Probe hUMPS | Applied Biosystems | Hs00923517_m1 |
| Taqman Probe hCTPS2 | Applied Biosystems | Hs00219845_m1 |
| Taqman Probe hTYMS | Applied Biosystems | Hs00426586_m1 |
| Taqman Probe hIMPDH1 | Applied Biosystems | Hs04190080_gH |
| Taqman Probe hATIC | Applied Biosystems | Hs00269671_m1 |
| Taqman Probe hPFAS | Applied Biosystems | Hs00389822_m1 |
| Taqman Probe hATIC | Applied Biosystems | Hs00269671_m1 |
| Taqman Probe hPRPS1 | Applied Biosystems | Hs00751338_s1 |
| Taqman Probe hACTB | Applied Biosystems | Hs01060665_g1 |
| Recombinant DNA | ||
| siRNA‐hIDH2 target sequence #1 | Thermo Fischer Scientific | s227 |
| siRNA‐hIDH2 target sequence #2 | Thermo Fischer Scientific | 106706 |
| Silencer siRNA Non‐targeting control | Invitrogen | #Cat:AM4611 |
| siRNA‐hHIF1A target sequence #1 | Thermo Fischer Scientific | s6539 |
| siRNA‐hHIF1A target sequence #2 | Thermo Fischer Scientific | s6541 |
| Software and Algorithms | ||
| GraphPad Prism 8 software | Graph Pad | N/A |
| Statistical Package of Social Sciences software ver 24.0 | IBM | N/A |
Methods and Protocols
Cell lines
T24 UMUC3, and J82 urothelial carcinoma (UC) cells were obtained from the American Type Culture Collection (Rockville, MD, USA). Both cell lines were cultured in RPMI 1640 (containing 11.1 mM glucose and 2 mM L‐glutamine) supplemented with 10% fetal bovine serum and penicillin (100 mg/ml) and incubated at 37°C in a humidified chamber under 5% CO2 (normoxia). All cells were passaged every 5 days using 0.05% trypsin–EDTA in PBS when they reached a confluency of 75–80%. The cell lines were validated by short tandem repeat (STR) profiling (BEX CO. LTD, Japan). We generated CR cell lines (T24CR and UMUC3CR) by increasing the CDDP concentration to 3 μM over 12 months as previously shown (Shigeta et al, 2020). Likewise, GEM‐resistant (GR) UC cell lines (T24GR and UMUC3GR) were generated by increasing the GEM concentration to 3 μM over 18 months. These cells were passaged within 1 month of drug exposure to confirm the persistence of CDDP/GEM resistance.
CE‐MS metabolome analysis
T24 (WT, CR, GR) and UMUC3 (WT, CR, GR), and J82 (WT and IDH2ox) were washed with Dulbecco's PBS (D‐PBS) and cultured under specific conditions. The cells were washed in 10% mannitol (Wako) and then plunged into methanol that contained internal standards of methionine sulfone for cation exchange and 2‐morpholinoethanesulfonic acid for anion exchange (300 μM each). The cells were collected for analysis in a capillary electrophoresis system equipped with an Agilent 1100 series MSD mass spectrometer (capillary electrophoresis and mass spectrometry: CE‐MS) and an Agilent 1100 series isocratic high‐performance liquid chromatography pump with a G1603A adaptor kit and G1607A sprayer kit as described previously (Shintani et al, 2009; Tohyama et al, 2013, 2016).
WT, GR, and transfected cells (1 × 107 cells per 10 cm2 dish) were incubated with RPMI 1640 containing 13C5‐labeled glutamine (1 g/l) for 1 h, a sufficient period of time to identify 13C5‐glutamine conversion of the downstream intermediates belonging to the tricarboxylic acid (TCA) cycle. Following rinsing with PBS, the cells were washed with 5% mannitol and then frozen in liquid nitrogen. Metabolites were extracted from cells with a solvent composed of methanol, distilled water and chloroform. After centrifugation at 20,000 g at 4°C for 15 min, the supernatants were filtered through a 5‐kDa cutoff filter (Millipore). Metabolites including those that had at least one 13C atom in the filtrate were analyzed by using CE‐MS. Labeled metabolites containing at least one 13C atom in a molecule were collectively indicated as 13C‐labeled forms. All values were normalized to the cell number.
Cell survival assay under glucose and glutamine deprivation culture media
Modified RPMI 1640 medium (with 1 mM HEPES and without phenol red, sodium bicarbonate, D‐glucose, and L‐glutamine) was prepared by Agilent. Then, 1,000 mg/l D‐glucose and 300 mg/l L‐glutamine were added to prepare the glucose‐limited RPMI medium, 0 mg/l D‐glucose and 300 mg/l L‐glutamine were added to prepare the glucose‐free RPMI medium, and 2000 mg/l D‐glucose and 0 mg/l L‐glutamine were added to prepare the glutamine‐free RPMI medium. The cell death rate was estimated by the trypan blue dye exclusion method.
XF24 extracellular flux analysis
The extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) analyses were performed with an XF24 extracellular flux analyzer (Seahorse Biosciences, North Billerica, MA). A total of 4.0 × 104 cells per well were seeded in 24‐well cell culture plates (Seahorse Biosciences, North Billerica, MA) in RPMI 1640 medium supplemented with 10% FBS and incubated at 37°C overnight in 5% CO2 incubator. The next day, the growth medium was replaced with bicarbonate‐free RPMI 1640 medium, and the cells were incubated at 37°C for 1 h in a CO2‐free incubator to equilibrate the medium temperature and pH. By utilizing a Seahorse XF24 analyzer, ECAR and OCR were measured under baseline conditions and under treatment with 2‐deoxyglucose (2‐DG; 50 μM) and rotenone (1 μM). The values are presented as the mean values ± standard errors of the mean.
DNA extraction and sequencing
Genomic DNA was extracted from cell lysates using Blood & Cell Culture DNA extraction Kits (QIAGEN, Venlo, The Netherlands). After DNA extraction, each sample was sequenced by the 150 bp paired‐end sequencing method on the Illumina NovaSeq 6000 platform (Illumina, Inc.). The quality of the paired fastq files was analyzed using FastQC (v0.11.7; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; Cock et al, 2010). In brief, Sequence reads were trimmed using Trimmomatic software (v0.38; Bolger et al, 2014). The trimmed reads were aligned to the indexed reference genome using bwa‐mem (v0.7.17‐r1188; Li & Durbin, 2009). SAM files were converted into BAM files and sorted using Picard SortSam (v2.18.11, http://broadinstitute.github.io/picard/). The number of duplicates was calculated using Picard MarkDuplicates After removing the duplicates, reads were realigned around the known indels provided by the Genome Analysis Toolkit (GATK) group (Van der Auwera et al, 2013). Single nucleotide variants (SNVs) and short indels were called using GATK HaplotypeCaller (v4.0.8.1) with parameters “‐ERC GVCF” to find genetic variants between the sample DNA sequence compared with the reference sequence. We filtered out variants that were < 10 sequencing depths in all samples. Identified variants were annotated using snpEff (v4.3t) to identify the putative effects on protein translation and high impact mutations (Cingolani et al, 2012).
Genetic perturbation of IDH2 expression
Transient IDH2 transfections were performed in the three cell lines, T24, UMUC3, and J82. Stable and transient transfections were carried out by using Xfect transient Reagent (Clontech) according to the manufacturer's instructions. J82 cells were transfected with pcDNA/IDH2 constructed for overexpression and pcDNA/GFP as a control.
Cell viability assay
T24WT, T24GR, UMUC3WT, and UMUC3GR cells were seeded on 96‐well plates, allowed to attach for 24 h, and then treated with various concentrations of GEM (Nippon Kayaku, Tokyo, Japan), CDDP (Nippon Kayaku, Tokyo, Japan), and IDH2 inhibitor (AGI6780, Cayman Chemical, Ann Arbor, MI, USA). After 48 h of exposure to the drugs, WST‐8 reagents (Cayman Chemical, Ann Arbor, MI, USA) were added to each well, and the cells were incubated for 2 h. Cell viability was estimated by reading the color intensity in a plate reader at 450 and 620 nm.
RT–PCR analysis
Total RNA was isolated by using RNA easy columns (QIAGEN) following the manufacturer's instructions. Total RNA was reverse transcribed to cDNA by using a High Capacity Reverse Transcription Kit (Applied Biosystems, Tokyo, Japan). qPCR was performed with gene‐specific primers with 40 cycles at 95°C for 10 s and 60°C for 60 s in a 10 μL reaction mixture containing 1 μL of cDNA, 1 μL of each primer and 8 μL of TaqMan® Fast Universal PCR Master Mix (2×) (Applied Biosystems, Waltham, MA, USA). Beta‐actin was used as the internal control. Quantification was performed with the ΔΔC t method (Schmittgen et al, 2008). All primers used for reverse transcription and amplification were listed in Reagents and Tools table.
Western blotting
Whole‐cell extracts were obtained using RIPA buffer composed of 50 mM Tris–HCl (pH 7.5), 15 mM NaCl, 1% NP‐40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate and protease inhibitors. In the Western blotting analysis, 30 μg of total protein was loaded onto a 12.5% sodium dodecyl sulfate‐polyacrylamide gel and then transferred to a nitrocellulose membrane. The membrane was blocked at 4°C overnight in TBS containing 5% Phospho Blocker Blocking Reagent and 0.2% Tween‐20 and then incubated at 4°C overnight with the following primary antibodies (Abs): anti‐IDH2 (1:500 dilution), anti‐Hif‐1α (1:200 dilution, Santa Cruz Biotechnologies, Dallas, TX, USA), anti‐PHD2 (1:500 dilution, Invitrogen, Waltham, MA, USA), anti‐GLUT1 (1:200 dilution), anti‐TIGAR (1:1,000 dilution), anti‐anti‐TKT (1:2,500 dilution), anti‐CTPS1 (1:1,000 dilution), anti‐PKM2 (1:1,000 dilution), anti‐LDHA (1:2,000 dilution), anti‐IDH1 (1:500 dilution; Abcam, Tokyo, Japan), anti‐ME1 (1:500 dilution), and anti‐G6PD (1:100 dilution). The blots were incubated with a peroxidase‐labeled secondary Ab for 1 h. After PBS washing, the signals were detected using enhanced chemiluminescence reagents with the ECL plus Western Blotting Detection System and analyzed using the LAS 3000 system (GE Healthcare, Los Angeles, CA, USA).
Small interfering RNA (siRNA) and transfection
T24GR and UMUC3GR cells were transfected with HIF1A‐‐targeting siRNA (siHIF1A#1 and #2) and with IDH2‐targeting siRNA (siIDH2#1 and #2). A mock‐transfected control was prepared using the transfection reagent with nontargeting control (NTC) siRNA.
siRNA#1(s6539) HIF1A sense: 5′‐CCAUAUAGAGAUACUCAAA‐3′ antisense: 5′‐UUUGAGUAUCUCUAUAUGG‐3 siRNA#2(s6541) HIF1A sense: 5′‐CCUCAGUGUGGGUAUAAGA‐3′ antisense: 5′‐UCUUAUACCCACACUGAGG‐3′.
siRNA#2(s227) IDH2 sense: 5′‐CAAGAACACCAUACUGAAA‐3′ antisense: 5′‐UUUCAGUAUGGUGUUCUUG‐3′ siRNA#2(106706) IDH2 sense: 5′‐GGAGCAGUGCGUUUUACCU‐3′ antisense: 5′AGGUAAAACGCACUGCUCCtg‐3′.
siRNA non‐targeting control (AM4611) sense: 5′‐UAGCGACUAAACACAUCAA‐3′ antisense: 5′‐UUGAUGUGUUUAGUCGCUA‐3′.
Cells were transiently transfected with 5 nmol of the respective siRNAs using Dermafect (Dharmacon, Lafayette, CO, USA). After 24 h, the siRNAs were removed by replacing the culture medium with fresh RPMI 1640 containing 10% FBS, and cells were incubated for another 24 h.
In vitro NADPH and NADP assay
The intracellular NADPH content was determined by the colorimetric determination method using an NADP/NADPH determination kit according to the manufacturer's manual (Biovision, Milpitas, CA, USA). T24WT, T24GR, UMUC3WT, and UMUC3GR cells (5 × 106 each) were used for NADPH determination.
Measurement of GSH and ROS levels
For cellular GSH measurements, 1 × 104 cells in 100 μl of culture medium were plated on a 96‐well white plate and allowed to attach for 24 h. Following the addition of 100 μl of GSH‐Glo Reagent (Promega Corp, Tokyo, Japan) at room temperature for 30 min, 100 μl of the luciferin detection reagent was added at room temperature for an additional 15 min. The luminescence intensity of each well was recorded on a GloMax™ 96 Microplate Luminometer.
Intracellular ROS levels were assessed using CellROX orange reagent (C10443; Invitrogen, USA). Cells were seeded at a density of 1 × 104 cells in 100 μl of culture medium per well in a 96‐well black plates to reach 80% confluence. For cellular ROS measurements, T24 (WT, GR, GR #siNTC, GR #siIDH2#1, GR #siIDH2#2, and GR with AGI6780) and UMUC3 (WT, GR, GR #siNTC, GR #siIDH2#1, GR #siIDH2#2, and GR with AGI6780) cells were treated with control vehicle, CDDP 1 μM, and CDDP 10 μM for 3 h and stained with 5 μM CellROx Orange reagent at 37°C for 30 min. The cells were washed twice with PBS and replaced with fresh medium for living image of intracellular ROS measurement. ROS levels were quantified as a measure of fluorescence at 545/565 nm by using 2300 EnSpire Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA).
UC murine xenograft models
Four‐ to 6‐week‐old female BALB/c‐nu/nu mice with an average body weight of 20 g were obtained from Sankyo Lab Service Co. (Tokyo, Japan). UMUC3GR cells (2 × 106 cells) suspended in 100 μl of Matrigel (Becton Dickinson Labware, Bedford, MA, USA) were implanted subcutaneously into the flank of each nude mouse. When a mouse developed a palpable tumor, it was randomly assigned to one of four groups. Each of these groups of five mice was treated with DMSO, GEM alone (biweekly i.p. injection of 50 mg/kg), AGI6780 alone (daily i.p. injection of 50 mg/kg) or a combination of AGI6780 and GEM. Furthermore, each of these groups of five mice was treated with CDDP alone (biweekly i.p. injection of 15 mg/kg), AGI6780 alone (daily i.p. injection of 50 mg/kg) or a combination of AGI6780 and CDDP to confirm the sensitivity to CDDP. Tumor volumes were calculated according to the formula a2 × b × 0.52, where a and b are the smallest and largest diameters, respectively. Twenty‐six days after drug administration, the mice were sacrificed, and the subcutaneous tumors were harvested. For the IHC study, primary Abs against the following targets were utilized: IDH2 (Abcam, Cambridge, UK), Ki‐67 (Dako, Santa Clara, CA, USA), CAIX (Bioscience, San Jose, CA, USA), TIGAR (Invitrogen, Waltham, MA, USA), TKT (Sigma–Aldrich, Munich, Germany), and CTPS1 (Sigma–Aldrich H, Munich, Germany). All of the procedures involving animals and their care in this study were approved by the Animal Care Committee of Keio University School of Medicine (Approval number #6496).
Tissue samples
Two hundred and fourteen patients who underwent radical nephroureterectomy (RNU) for upper tract urothelial carcinoma (UTUC) at Keio University Hospital between 2000 and 2017 were identified (Table 1). Furthermore, among the 121 muscle‐invasive bladder cancer (MIBC) patients who underwent radical cystectomy (RC) between 2004 and 2017, we identified 32 MIBC patients who received GEM plus CDDP (GC) neoadjuvant chemotherapy (Table 2).
Table 1.
Clinicopathological characteristics of upper tract urothelial carcinoma patients who underwent radical nephroureterectomy.
| Patient characteristics | Patients who underwent RNU | Total n = 214, (%) |
|---|---|---|
| Follow‐up duration | Mean ± SD | 62.7 ± 39.2 |
| Time to death | Mean ± SD | 57.9 ± 42.3 |
| Time to metastasis | Mean ± SD | 39.1 ± 43.6 |
| Time to bladder tumor recurrence | Mean ± SD | 20.0 ± 7.87 |
| Age | < 75 | 107 (50.0) |
| ≥ 75 | 107 (50.0) | |
| Sex | Male | 158 (73.8) |
| Female | 56 (26.2) | |
| Tumor location | Pelvis | 105 (49.1) |
| Ureter | 109 (50.9) | |
| Tumor histology | Pure UC | 172 (80.4) |
| Non‐pure UC | 42 (19.6) | |
| Pathological T stage | < 3 | 98 (45.8) |
| ≥ 3 | 116 (54.2) | |
| Pathological N stage | 0 | 199 (93.0) |
| 1,2 | 15 (7.0) | |
| Lymph node dissection | No | 150 (70.1) |
| Yes | 64 (29.9) | |
| Surgical margin | Negative | 189 (88.3) |
| Positive | 25 (11.7) | |
| Tumor grade | Low | 64 (29.9) |
| High | 150 (70.1) | |
| LVI | Absent | 118 (55.1) |
| Present | 96 (44.9) | |
| Concomitant CIS | No | 170 (79.4) |
| Yes | 44 (20.6) | |
| Tumor multifocality | No | 182 (85.0) |
| Yes | 32 (15.0) | |
| Systemic adjuvant chemotherapy | No | 140 (65.4) |
| Yes | 74 (34.6) | |
| Previous history of bladder cancer | No | 149 (69.6) |
| Yes | 65 (30.4) |
CIS, carcinoma in situ; LVI, lymphovascular invasion; RNU, radical nephroureterectomy; SD, standard deviation; UC, urothelial carcinoma.
Tissue samples were obtained from consenting patients in the present study, which was approved by the Keio University Ethics Committee (Ethical Committee number: 2013‐0095). All specimens were fixed in 10% formalin and embedded in paraffin, and all slides were re‐reviewed by a genitourinary pathologists. Two board‐certified pathologists (S.M., with 34 years of experience in pathology, and K.K., with 34 years of experience in pathology) evaluated all IHC stains and calculated H‐score in blinded manner. Tumors were staged according to the American Joint Committee on Cancer‐Union Internationale Contre le Cancer tumor‐node‐metastasis (TNM) classification. Tumor grading was assessed according to the 2004 WHO/International Society of Urology Pathology consensus classification (Soukup et al, 2017). Lymphovascular invasion (LVI) was defined as the presence of tumor cells within an endothelium‐lined space without underlying muscular walls (Kikuchi et al, 2009).
Immunohistochemistry (IHC)
Sections (thickness of 4 μm) of formalin‐fixed and paraffin‐embedded material were evaluated. Sections were deparaffinized in xylene and rehydrated in graded alcohol and distilled water. After antigen retrieval with citric acid (pH = 6.0) at 120°C for 10 min, endogenous peroxidase activity was blocked with 1% hydrogen peroxide for 15 min followed by washing with distilled water. To bind nonspecific antigens, sections were incubated at room temperature for 15 min with 5% skim milk in phosphate‐buffered saline (PBS). Sections were then incubated at 4°C overnight with anti‐IDH2 mouse monoclonal Ab (1:100 dilution; Abcam, Cambridge, UK), anti‐carbonic anhydrase IX (CAIX) mouse Ab (1:200 dilution; Bioscience, San Jose, CA, USA), anti‐GLUT1 mouse Ab (1:100 dilution; Sigma‐Aldrich, Munich, Germany), anti‐TIGAR rabbit polyclonal Ab (1:250 dilution; Invitrogen, Waltham, MA, USA), anti‐anti‐TKT rabbit polyclonal Ab (1:500 dilution; Sigma‐Aldrich, Munich, Germany), anti‐CTPS1 rabbit polyclonal Ab (1:500 dilution; Sigma‐Aldrich H, Munich, Germany), anti‐PKM2 rabbit monoclonal Ab (1:800 dilution; Cell Signaling Technology, Danvers, MA, USA), anti‐LDHA rabbit polyclonal Ab (1:500 dilution; Proteintech, Rosemont, IL, USA), anti‐IDH1 rabbit polyclonal Ab (1:100 dilution; Abcam, Cambridge, UK), and anti‐G6PD mouse monoclonal Ab (1:100 dilution; Santa Cruz Biotechnologies, Dallas, TX, USA).
After washing with PBS, the tissue sections were incubated with secondary Abs (1:1,000 dilution; Dako, Tokyo, Japan) for 60 min. Color was developed with 3,30‐diaminobenzidine tetrahydrochloride in 50 mmol/L Tris–HCl (pH 7.5) containing 0.005% hydrogen peroxide. Sections were then counterstained with hematoxylin.
The stained sections were imaged at 100× under an inverted microscope. Cancer cells with positive staining in the cell cytoplasm were counted in at least five representative fields, and the mean percentage of positive cancer cells and staining intensity scored from 0 to 3 (0: no staining 1: low staining, 2: moderate staining, and 3: strong staining) were estimated. The histoscore (H‐score) was calculated by applying the following formula: mean percentage × intensity (range 0–300).
Statistical analysis
The difference between two groups in the in vitro and in vivo study was assessed using the two‐tailed Student's t‐test. For the in vitro study, each value represents the mean ± standard deviation (SD) or mean ± standard error (SE) of at least three individual experiments. For the in vivo study, each value represents the mean ± SD of at least three individual experiments. Nonparametric one‐way ANOVA (Kruskal–Wallis tests) was utilized to compare differences between animal groups. Regarding patient prognosis, cancer‐specific survival (CSS) was estimated using the Kaplan–Meier method and compared with the log‐rank test. The level of significance was set at P < 0.05. All statistical tests were performed by utilizing GraphPad Prism 8, Excel Soft‐ware, and Statistical Package of Social Sciences software, version 24.0 (SPSS, Chicago, Illinois, USA) for all experiments.
Author contributions
Keisuke Shigeta: Conceptualization; resources; data curation; software; formal analysis; funding acquisition; validation; visualization; methodology; writing—original draft. Masanori Hasegawa: Conceptualization; resources; data curation; formal analysis; supervision; writing—original draft; writing—review and editing. Takako Hishiki: Data curation; formal analysis; investigation; methodology. Yoshiko Naito: Data curation; formal analysis; investigation; methodology. Yuto Baba: Resources; validation; methodology. Shuji Mikami: Data curation; formal analysis; methodology. Kazuhiro Matsumoto: Resources; supervision. Ryuichi Mizuno: Validation. Akira Miyajima: Resources; supervision. Eiji Kikuchi: Resources; supervision. Hideyuki Saya: Supervision. Takeo Kosaka: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; writing—original draft; writing—review and editing. Mototsugu Oya: Resources; supervision; funding acquisition; writing—original draft; project administration; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
The authors thank Noriyo Hayakawa and Tomomi Matsuura for technical assistance with metabolome analysis (Department of Clinical and Translational Research center, Keio University School of Medicine). This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grant numbers 21K20811 and 22K16809 to K.S. and grant number 20H03817 and 21K19579 to T.K.), by the Keio Medical Association Grant (to K.S.), by the 2019 Japanese Foundation for Research and Promotion of Endoscopy (JFE) Grant (to K.S.), by 2020 Keio University Graduate School Doctoral Program Grant (to K.S.), and by 2020 Keio Medical Otsuka Fumon/Fusako Fellowship Grant (to K.S.).
The EMBO Journal (2023) 42: e110620
Contributor Information
Masanori Hasegawa, Email: hasem@tsc.u-tokai.ac.jp.
Takeo Kosaka, Email: takemduro@gmail.com.
Mototsugu Oya, Email: moto-oya@sc.itc.keio.ac.jp.
Data availability
All raw sequencing reads generated for this study are available for download from the NCBI SRA database under BioProject; accession number: PRJNA863932 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA863932).
References
- Afonso J, Santos LL, Longatto‐Filho A, Baltazar F (2020) Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat Rev Urol 17: 77–106 [DOI] [PubMed] [Google Scholar]
- Ai Z, Lu Y, Qiu S, Fan Z (2016) Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF‐1‐regulated cancer metabolism. Cancer Lett 373: 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabas GD, Lee JS, Shami T, Harel M, Beck L, Selitrennik M, Jerby‐Arnon L, Erez N, Ruppin E, Geiger T (2021) Serine biosynthesis is a metabolic vulnerability in IDH2‐driven breast cancer progression. Cancer Res 81: 1443–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK et al (2017) Pembrolizumab as second‐line therapy for advanced urothelial carcinoma. N Engl J Med 376: 1015–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Mak TW (2013) Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3: 730–741 [DOI] [PubMed] [Google Scholar]
- Calabro F, Lorusso V, Rosati G, Manzione L, Frassineti L, Sava T, Di Paula ED, Alonso S, Sternberg CN (2009) Gemcitabine and paclitaxel every 2 weeks in patients with previously untreated urothelial carcinoma. Cancer 115: 2652–2659 [DOI] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces‐Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA, Majeti R (2015) Isocitrate dehydrogenase 1 and 2 mutations induce BCL‐2 dependence in acute myeloid leukemia. Nat Med 21: 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, Rabinowitz JD (2019) NADPH production by the oxidative pentose‐phosphate pathway supports folate metabolism. Nat Metab 1: 404–415 [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yang J, Wei Q, Weng L, Wu F, Shi Y, Cheng X, Cai X, Hu C, Cao P (2020) Identification of a selective inhibitor of IDH2/R140Q enzyme that induces cellular differentiation in leukemia cells. Cell Commun Signal 18: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, Vousden KH (2020) Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37: 168–182.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin) 6: 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM (2010) The sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 38: 1767–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Wang K, Fan J, Liu H, Fan X, Lin Q, Chen Y, Chen H, Li Y, Liu H et al (2022) Nrf2 transcriptional upregulation of IDH2 to tune mitochondrial dynamics and rescue angiogenic function of diabetic EPCs. Redox Biol 56: 102449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis M, Bellmunt J, Mead G, Kerst JM, Leahy M, Maroto P, Gil T, Marreaud S, Daugaard G, Skoneczna I et al (2012) Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin‐based chemotherapy: EORTC study 30986. J Clin Oncol 30: 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20 [DOI] [PubMed] [Google Scholar]
- Du X, Hu H (2021) The roles of 2‐Hydroxyglutarate. Front Cell Dev Biol 9: 651317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Rabinowitz JD, Shlomi T (2013) Fatty acid labeling from glutamine in hypoxia can be explained by isotope exchange without net reductive isocitrate dehydrogenase (IDH) flux. J Biol Chem 288: 31363–31369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Jezek P (2020) 2‐Hydroxyglutarate in cancer cells. Antioxid Redox Signal 33: 903–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B et al (2016) Reductive carboxylation supports redox homeostasis during anchorage‐independent growth. Nature 532: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B, Cheng G, Hardy M, Ouari O, Bennett B, Zielonka J (2018) Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol 15: 347–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi E, Margulis V, Karakiewicz PI, Roscigno M, Mikami S, Lotan Y, Remzi M, Bolenz C, Langner C, Weizer A et al (2009) Lymphovascular invasion predicts clinical outcomes in patients with node‐negative upper tract urothelial carcinoma. J Clin Oncol 27: 612–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, He Y, Tan Z, Lu J, Li L, Song X, Shi F, Xie L, You S, Luo X et al (2018) Wild‐type IDH2 promotes the Warburg effect and tumor growth through HIF1alpha in lung cancer. Theranostics 8: 4050–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sun Y, Ge W, Zhang F, Gan L, Zhu Y, Guo T, Liu K (2022) DIA‐based proteomics identifies IDH2 as a targetable regulator of acquired drug resistance in chronic myeloid leukemia. Mol Cell Proteomics 21: 100187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Maase H, Hansen SW, Roberts JT, Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A, Lippert CM et al (2000) Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol 18: 3068–3077 [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L et al (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF (2018) Wild‐type and mutated IDH1/2 enzymes and therapy responses. Oncogene 37: 1949–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481: 385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM et al (2014) Oxidation of alpha‐ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7: 1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro C, Ortega A, Santeliz R, Garrido B, Chacin M, Galban N, Vera I, De Sanctis JB, Bermudez V (2022) Metabolic reprogramming in cancer cells: emerging molecular mechanisms and novel therapeutic approaches. Pharmaceutics 14: 1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin EB, Giaccia AJ (2016) Hypoxic control of metastasis. Science 352: 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, Zhang DD (2018) NRF2 and the hallmarks of cancer. Cancer Cell 34: 21–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roupret M, Babjuk M, Burger M, Capoun O, Cohen D, Comperat EM, Cowan NC, Dominguez‐Escrig JL, Gontero P, Hugh Mostafid A et al (2021) European Association of Urology guidelines on upper urinary tract urothelial carcinoma: 2020 update. Eur Urol 79: 62–79 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS, Chen C (2008) Real‐time PCR quantification of precursor and mature microRNA. Methods 44: 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtes MP, de Jong FC, Zuiverloon TCM, Theodorescu D (2021) Role of bladder cancer metabolic reprogramming in the effectiveness of immunotherapy. Cancers (Basel) 13: 288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2010) Defining the role of hypoxia‐inducible factor 1 in cancer biology and therapeutics. Oncogene 29: 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2013) HIF‐1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 123: 3664–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, He Y, Li J, Tang M, Li Y, Xie L, Zhao L, Hu J, Luo X, Zhou M et al (2020) Wild‐type IDH2 contributes to Epstein‐Barr virus‐dependent metabolic alterations and tumorigenesis. Mol Metab 36: 100966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta K, Hasegawa M, Kikuchi E, Yasumizu Y, Kosaka T, Mizuno R, Mikami S, Miyajima A, Kufe D, Oya M (2020) Role of the MUC1‐C oncoprotein in the acquisition of cisplatin resistance by urothelial carcinoma. Cancer Sci 111: 3639–3652 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shintani T, Iwabuchi T, Soga T, Kato Y, Yamamoto T, Takano N, Hishiki T, Ueno Y, Ikeda S, Sakuragawa T et al (2009) Cystathionine beta‐synthase as a carbon monoxide‐sensitive regulator of bile excretion. Hepatology 49: 141–150 [DOI] [PubMed] [Google Scholar]
- Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A et al (2017) MUC1 and HIF‐1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32: 71–87.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup V, Capoun O, Cohen D, Hernandez V, Babjuk M, Burger M, Comperat E, Gontero P, Lam T, MacLennan S et al (2017) Prognostic performance and reproducibility of the 1973 and 2004/2016 World Health Organization grading classification Systems in non‐muscle‐invasive Bladder Cancer: a European Association of Urology non‐muscle invasive bladder cancer guidelines panel systematic review. Eur Urol 72: 801–813 [DOI] [PubMed] [Google Scholar]
- Sternberg CN, de Mulder PH, Schornagel JH, Theodore C, Fossa SD, van Oosterom AT, Witjes F, Spina M, van Groeningen CJ, de Balincourt C et al (2001) Randomized phase III trial of high‐dose‐intensity methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) chemotherapy and recombinant human granulocyte colony‐stimulating factor versus classic MVAC in advanced urothelial tract tumors: European Organization for Research and Treatment of cancer protocol no. 30924. J Clin Oncol 19: 2638–2646 [DOI] [PubMed] [Google Scholar]
- Sun RC, Denko NC (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y et al (2013) Distinct metabolic flow enables large‐scale purification of mouse and human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell 12: 127–137 [DOI] [PubMed] [Google Scholar]
- Tohyama S, Fujita J, Hishiki T, Matsuura T, Hattori F, Ohno R, Kanazawa H, Seki T, Nakajima K, Kishino Y et al (2016) Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab 23: 663–674 [DOI] [PubMed] [Google Scholar]
- Tseng CW, Kuo WH, Chan SH, Chan HL, Chang KJ, Wang LH (2018) Transketolase regulates the metabolic switch to control breast cancer cell metastasis via the alpha‐Ketoglutarate signaling pathway. Cancer Res 78: 2799–2812 [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al‐Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O'Connor KW et al (2014) Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle‐invasive urothelial carcinoma. Cancer Discov 4: 1140–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy‐Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J et al (2013) From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.1–11.10.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuilen CS, van Gennep EJ, Einerhand SMH, Vegt E, Donswijk ML, Bruining A, van der Poel HG, Horenblas S, Hendricksen K, van Rhijn BWG et al (2021) Staging (18)F‐fluorodeoxyglucose positron emission tomography/computed tomography changes treatment recommendation in invasive bladder cancer. Eur Urol Oncol 5: 366–369 [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard‐Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C et al (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340: 622–626 [DOI] [PubMed] [Google Scholar]
- Warburg O (1956) On the origin of cancer cells. Science 123: 309–314 [DOI] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB (2011) Hypoxia promotes isocitrate dehydrogenase‐dependent carboxylation of alpha‐ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA 108: 19611–19616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjes JA, Bruins HM, Cathomas R, Comperat EM, Cowan NC, Gakis G, Hernandez V, Linares Espinos E, Lorch A, Neuzillet Y et al (2021) European Association of Urology guidelines on muscle‐invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur Urol 79: 82–104 [DOI] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT et al (2011) Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of alpha‐ketoglutarate‐dependent dioxygenases. Cancer Cell 19: 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Li Y, Lv N, Jing Y, Xu Y, Li Y, Li W, Yao Z, Chen X, Huang S et al (2017) Correlation between Isocitrate dehydrogenase gene aberrations and prognosis of patients with acute myeloid leukemia: a systematic review and meta‐analysis. Clin Cancer Res 23: 4511–4522 [DOI] [PubMed] [Google Scholar]
- Yokoi K, Fidler IJ (2004) Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res 10: 2299–2306 [DOI] [PubMed] [Google Scholar]
- Zeng P, Lu W, Tian J, Qiao S, Li J, Glorieux C, Wen S, Zhang H, Li Y, Huang P (2022) Reductive TCA cycle catalyzed by wild‐type IDH2 promotes acute myeloid leukemia and is a metabolic vulnerability for potential targeted therapy. J Hematol Oncol 15: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GF, Jensen MV, Gray SM, El K, Wang Y, Lu D, Becker TC, Campbell JE, Newgard CB (2021) Reductive TCA cycle metabolism fuels glutamine‐ and glucose‐stimulated insulin secretion. Cell Metab 33: 804–817.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu Y, Lu W, Li J, Yu S, Brown EJ, Stanger BZ, Rabinowitz JD, Yang X (2022) G6PD‐mediated increase in de novo NADP(+) biosynthesis promotes antioxidant defense and tumor metastasis. Sci Adv 8: eabo0404 [DOI] [PMC free article] [PubMed] [Google Scholar]