

Summary
Biobanks of linked clinical patient histories and biological samples are an efficient strategy to generate large cohorts for modern genetics research. Biobank recruitment varies by factors such as geographic catchment and sampling strategy, which affect biobank demographics and research utility. Here, we describe the Michigan Genomics Initiative (MGI), a single-health-system biobank currently consisting of >91,000 participants recruited primarily during surgical encounters at Michigan Medicine. The surgical enrollment results in a biobank enriched for many diseases and ideally suited for a disease genetics cohort. Compared with the much larger population-based UK Biobank, MGI has higher prevalence for nearly all diagnosis-code-based phenotypes and larger absolute case counts for many phenotypes. Genome-wide association study (GWAS) results replicate known findings, thereby validating the genetic and clinical data. Our results illustrate that opportunistic biobank sampling within single health systems provides a unique and complementary resource for exploring the genetics of complex diseases.
Graphical abstract
Highlights
-
•
The Michigan Genomics Initiative (MGI) is a biobank of Michigan Medicine patients
-
•
MGI participants are primarily recruited during surgical procedures
-
•
The surgical enrollment enriches clinical outcomes relative to population-based biobanks
-
•
Summary statistics for GWASs performed in MGI are available to interested researchers
Zawistowski et al. present the Michigan Genomics Initiative, a biobank of linked genotype and clinical data among patients at Michigan Medicine. This health-system-based biobank opportunistically recruits primarily surgical patients, yielding higher case counts than much larger population-based biobanks, enabling genetic research for a wide range of traits.
Introduction
Genome-wide association studies (GWASs) have identified thousands of genetic variants associated with a wide range of human phenotypes.1 Traditionally, GWASs have been designed with a specific phenotype or handful of related outcomes in mind. Participants are specifically recruited on the basis of that phenotype and data collection restricted to the specific outcome of interest and relevant confounding variables. This design strategy optimizes power for a single particular phenotype but has limited reuse potential for studying additional outcomes.
The recent wave of biobank repositories linking individual-level genetic data with dense clinical health history has dramatically changed the phenotyping paradigm for genetic studies.2 Biobanks allow broad phenotyping across a common set of genotyped samples, often by leveraging existing patient electronic health records (EHRs), allowing the investigation of a wide range of clinically important outcomes within the same cohort. Rather than being optimized for a single phenotype, the biobank design creates a resource for repeated use across diverse phenotypes and study questions. The rich clinical data allows fine-tuned inclusion criteria and phenotype definitions on a per-study basis using combinations of diagnoses, clinical lab results, medication usage, imaging results, and more. Thus, the same biobank cohort can yield GWASs for thousands of phenotypes, with each GWAS being highly cost and time effective since participant recruitment, consent, and genotyping are completed in advance and phenotyping is performed on existing clinical data. In addition, biobanks have spawned novel analytic methods that leverage the unique feature of having the entire phenome measured on the same set of samples. For example, the phenome-wide association study (PheWAS) tests individual genetic variants for associations across the phenome, allowing investigation of comorbid outcomes and pleiotropic genetic effects, again without the need for additional participant recruitment or data collection.3
Although biobanks share a common theme of linked clinical and biological data, they are otherwise remarkably heterogeneous. Differences in target population demographics, recruitment strategy and criteria, consent procedures, and data sharing introduce distinct benefits and limitations. Large national biobanks such as UK Biobank (UKB),4 BioBank Japan,5 and All of Us6 aim to capture a diverse set of individuals across their respective nations using broad geographical recruitment strategies. This “population-based” approach to recruitment is effective at generating very large sample sizes, with UKB notably containing >500,000 samples and All of Us aiming for >1 million samples. To achieve these massive sizes, participants are potentially recruited from across multiple health systems, and it can require substantial effort to merge the heterogeneous sources of clinical data.
An alternative biobank design is localized recruitment within a single healthcare system.7,8,9 In this article, we describe the Michigan Genomics Initiative (MGI), a single-health-system biobank recruited from adult patients receiving care at Michigan Medicine, the University of Michigan’s health system. MGI recruitment began in 2012 with the goal of creating a resource to accelerate biomedical and precision health research at the University of Michigan. Recruitment has primarily occurred through the Department of Anesthesiology during inpatient surgical procedures at Michigan Medicine. Recruiting during a surgical encounter provides a convenient opportunity to obtain patient consent, complete questionnaires, and collect a blood sample biospecimen. MGI participants consent to linkage of their blood sample, which is subsequently stored in the University of Michigan Central Biorepository, to their existing and future clinical data, including their Michigan Medicine EHR. The consent form, which covers broad research purposes and recontact potential, is intentionally brief and accompanied by an easy-to-read pamphlet describing the risks and benefits in layman’s terms and pictorial descriptions to maximize participant understanding of the project (https://precisionhealth.umich.edu/our-research/michigangenomics/). The resulting dataset is a rich resource freely available to University of Michigan researchers. Already, MGI has yielded a wide range of research contributions including novel variant discovery for clinical laboratory measurements10; PheWAS-based identification of polygenic risk score-trait associations11; pharmacogenetic analysis of chemotherapeutic toxicity12; integration of MGI participants as “external” controls into GWAS13; and pre-operative phenotypic characterization and opioid usage for surgical patients.14
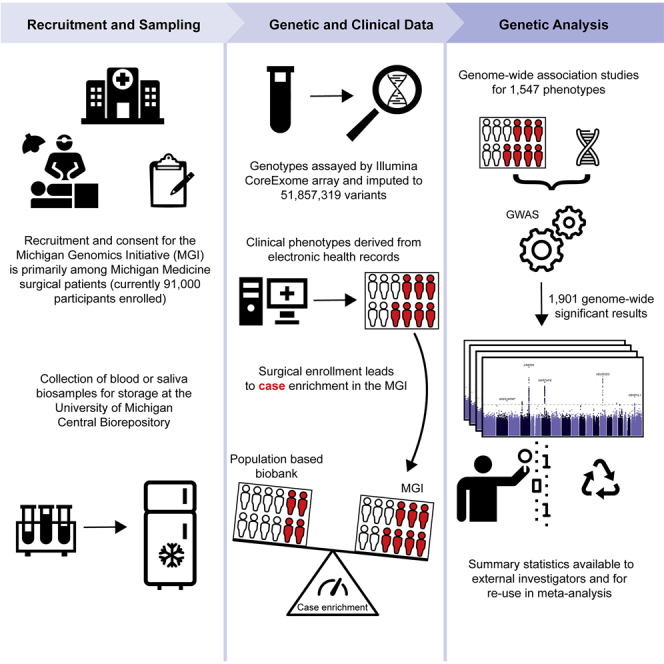
This article provides a description of the MGI cohort, details our rigorous quality control procedures, and provides proof-of-principle GWAS results for 1,547 phenotypes based on diagnosis codes (Figure 1). We investigate the impact of the opportunistic recruitment of inpatient surgical patients in MGI by comparing case counts for a broad range of clinical phenotypes with the much larger population-based UKB. We demonstrate the valuable contribution that single-health-system biobanks can provide to the broader genetic research community by sharing the complete set of GWAS results presented in this article through an interactive “PheWeb” application15 that includes Manhattan plots, regional association plots, and PheWAS analysis (https://pheweb.org/MGI/). The corresponding GWAS summary statistics are available to the research community for replication analysis, meta-analysis, and hypothesis-driven look ups. Information on requesting summary statistics is available at https://precisionhealth.umich.edu/our-research/documents-for-researchers/.
Figure 1.

Overview of the Michigan Genomics Initiative (MGI) resource and analysis
MGI currently consists of ∼91,000 participants recruited while seeking care at the Michigan Medicine health system. Recruitment is predominantly through the Department of Anesthesiology during inpatient surgical encounters. Participants agree to link a blood sample obtained during consent with their electronic health records for broad research purposes. Genotypes for ∼570,000 genetic variants are obtained from DNA extracted from the blood sample using a customized Illumina Infinium CoreExome-24 array. In this article, we describe the MGI “Freeze 3” cohort consisting of ∼57,000 samples having passed sample-level quality control filtering and imputed for >50 million variants using the TOPMed reference panel. We extracted all available International Classification of Disease (ICD) diagnosis codes from patient electronic health records and mapped to broader dichotomous phecode traits using the PheWAS software. We performed GWASs within a subset of ∼51,000 European-inferred samples from the Freeze 3 cohort using a linear mixed-effect regression model implemented in the SAIGE software. We report results and share GWAS summary statistics for 1,547 traits with ≥60 cases.
Results
As of April 30, 2022, 91,695 patients receiving care at the Michigan Medicine health system have consented to participate in the MGI. Participants are recruited on a rolling basis and genotyped in batches at the university’s Advanced Genomics Core. Enrollment has steadily increased since project initiation, beginning at approximately 730 samples per month in 2013 to just over 1,000 samples per month in 2019, prior to suspension of enrollment in 2020 due to the pandemic (Figure 2A). Notably, enrollment of individuals who self-report their race as something other than White has likewise increased from 71 individuals per month in 2013 to 292 samples per month in 2019 (note: throughout the main text, “White” has been used in place of “Caucasian,” which was the terminology used in the self-reported data). In this article, we describe the genetic and clinical data for MGI “Freeze 3” (March 23, 2020) comprised of 57,055 participants and present results from GWASs for 1,547 traits in a set of 51,583 European samples.
Figure 2.

MGI recruitment, demographics, and clinical follow-up
(A) MGI recruitment over time. The solid line is overall recruitment, and the dashed line is participants with self-reported race other than White.
(B) Age and sex distribution of MGI participants.
(C) Clinical follow-up time for MGI participants. Follow-up is the amount of time between a participant’s first and most recent diagnosis codes in the Michigan Medicine electronic health records (EHRs).
(D) Distribution of ages for MGI participants is nearly identical across follow-up times.
Demographic and clinical description of the cohort
MGI participants range in age from 18 to over 90 years (Figure 2B; Table S1). There are slightly more females (53%), with male participants being slightly older (58.4 versus 54.7 years; Figure 2B). The majority of participants self-report race as White (n = 49,605, 87%), with African American (n = 3,223, 5.6%) and Asian (n = 1,324, 2.3%) the next most common, and 805 individuals indicating Hispanic or Latino ethnicity (Table S1).
The number of International Classification of Disease (ICD) codes differ across participants, reflecting inter-individual differences in overall health and utilization of the health system. To measure the length of time each participant has interacted with the Michigan Medicine healthcare system, we compute follow-up time, defined as the difference in time between the oldest and most recent ICD diagnoses for an individual. The distribution of follow-up time is U-shaped, with the most frequent follow-up times being <1 year and ∼19 years (Figure 2C). The upper bound of approximately 20 years corresponds to the beginning of electronic capture of diagnosis codes beginning at Michigan Medicine in 2000. The distribution of participant age is almost identical across follow-up times, suggesting that follow-up time is largely independent of participant age (Figure 2D).
Phecode traits
Due to the granularity and redundancy of ICD codes, we mapped individual ICD codes to broader binary phecode traits using the PheWAS software.16 Individual phecode traits can be grouped into 17 general categories of clinically similar traits. For example, hypertension (phecode 401), myocardial infarction (411.2), and myocarditis (420.1) are each mapped to the “circulatory system” phecode group. In total, we observed case samples for 1,817 phecode traits, with 1,712 traits having at least 20 cases (Table 1). The most common traits are related to high-prevalence diseases (Figure 3A), including hypertension (phecodes 401 and 401.1); lipid disorders (272 and 272.1); obesity (278 and 278.1); esophagus/gastroesophageal reflux disease (GERD; 530, 530.1, and 530.11); and mental health disorders (mood disorders: 296; anxiety: 300, 300.1; depression: 296.2). Several pain-related traits (pain in joint: 745; abdominal pain: 785; pain: 338) also appear among the most common phecodes, likely due in part to the enrollment of surgical patients through anesthesiology. The number of phecodes per sample was strongly right skewed (median: 31, mean: 44.2, max: 435) and positively correlated with both age (Figure 3B) and follow-up time (Figure 3C).
Table 1.
Summary of phecode traits and GWAS results in European MGI participants
| Phecode category | Total phecode traits | Analyzed traits (≥60 cases) | Traits with ≥1 GWS loci (MAF >1%) | Number of GWS loci (MAF >1%) | Strongest association (MAF >1%) |
|---|---|---|---|---|---|
| Circulatory system | 171 | 160 | 108 (43) | 200 (72) | atrial fibrillation (427.21), p = 1.2e−37, chr4:110,762,205 |
| Congenital anomalies | 56 | 44 | 18 (3) | 36 (3) | genitourinary congenital anomalies (751), p = 4.0e−09, chr2:161,318,326 |
| Dermatologic | 95 | 77 | 53 (17) | 93 (22) | psoriasis vulgaris (696.41), p = 4.7e−28, chr6:31,274,954 |
| Digestive | 162 | 149 | 95 (39) | 198 (59) | other chronic non-alcoholic liver disease (571.5), p = 3.0e−54, chr22:43,928,975 |
| Endocrine/metabolic | 169 | 129 | 92 (65) | 277 (180) | type 1 diabetes (250.1), p = 4.2e−106, chr6:32,658,525 |
| Genitourinary | 173 | 157 | 101 (25) | 191 (39) | nephritis and nephropathy in diseases classified elsewhere (580.31), p = 1.4e−19, chr6:32,706,117 |
| Hematopoietic | 62 | 45 | 32 (16) | 65 (26) | primary hypercoagulable state (286.81), p = 2.8e−157, chr1:169,549,811 |
| Infectious diseases | 69 | 54 | 28 (8) | 37 (8) | aspergillosis (117.4), p = 4.3e−17, chr7:117,559,590 |
| Injuries and poisonings | 122 | 93 | 49 (5) | 79 (6) | salicylates causing adverse effects in therapeutic use (965.3), p = 2.4e−10, chr6:33,091,097 |
| Mental disorders | 76 | 63 | 39 (11) | 64 (12) | dementias (290.1), p = 2.1e−18, chr19:44,908,684 |
| Musculoskeletal | 132 | 114 | 71 (19) | 121 (20) | ankylosing spondylitis (715.2), p = 2.9e−35, 6:31,357,491 |
| Neoplasms | 141 | 129 | 76 (29) | 194 (85) | other non-epithelial cancer of skin (172.2), p = 1.8e−38, chr6:396,321 |
| Neurological | 85 | 74 | 50 (11) | 79 (14) | restless legs syndrome (327.71), p = 6.8e−29, chr2:66,523,432 |
| Pregnancy complications | 46 | 28 | 18 (7) | 23 (7) | rhesus isoimmunization in pregnancy (654.2), p = 1.4e−54, chr1:25,257,119 |
| Respiratory | 85 | 78 | 57 (22) | 96 (26) | cystic fibrosis (499), p = 9.8e−49, chr7:117,559,590 |
| sense organs | 127 | 112 | 65 (18) | 105 (25) | Fuchs’ dystrophy (364.51), p = 2.0e−31, chr18:55,543,071 |
| Symptoms | 46 | 41 | 25 (2) | 43 (2) | fever of unknown origin (783), p = 2.9e−08, chr7:37,808,912 |
| Total | 1,817 | 1,547 | 977 (340) | 1,901 (606) |
We report results for phecode traits with at least sixty cases. The strongest association column contains the phecode trait name (numerical phecode), p value, and chromosomal location for the association with smallest p value in each phecode category. A threshold of p = 5e−8 was used for genome-wide significance (GWS).
Figure 3.

MGI clinical data
(A) Most common phecode traits among MGI participants.
(B) Number of phecode case assignments per sample increases with participant age.
(C) Number of phecode case assignments per sample increases with participant follow-up time. Outlier values were excluded from boxplots to improve readability.
We compared phecode traits between MGI and the substantially larger UKB. Overall, MGI has higher prevalence for nearly all phecode traits (Figure S1). We observed 1,772 phecode traits for which either MGI or UKB had at least one case. Of these, UKB has no cases for 354 phecodes, and MGI has no cases for 22, many of which are common conditions. For example, there are no phecode-defined cases in UKB for basal cell carcinoma (172.21), insulin pump user (250.3), and hypo- (275.51) and hypercalcemia (275.6). The missing cases for these traits reflect different ICD code systems or differential use of ICD codes between the two biobanks rather than an actual lack of these traits in the cohorts.
As the power of association studies depends most strongly on the number of cases, it is more informative to compare the overall number of cases between MGI and UKB: MGI has a higher case count for 557 (41%) of the 1,358 phecodes for which both biobanks have cases (Figure 4). MGI has traits with greater case counts across all phecode categories, particularly within endocrine/metabolic and neurological categories. There are 48 phecode traits for which MGI has over 10-fold number of cases found in UKB (Table S2), including “vitamin D deficiency” (phecode: 261.4); “pain” (phecode: 338); “migraine with aura” (phecode: 340.1); “insomnia” (phecode: 327.4); and “varicella infection” (phecode: 079.1). Phecode traits for which MGI has more cases than UKB and a case count >10,000 include overweight/obesity (278, 278.1); mood disorders (296); depression (296.2); anxiety (300, 300.1); sleep apnea (327.3); allergic rhinitis (476); other symptoms of respiratory system (512); pain (338); pain in joint (745); and back pain (760).
Figure 4.

Comparison of phecode case counts between MGI and UKB by disease category
MGI has phecode traits with more cases than UKB across all disease categories.
Genetic data
Overall, genetically inferred ancestry is consistent with self-reported race and ethnicity obtained from appointment intake surveys (Figure 5A). The majority of participants that self-report as White clustered with European Human Genome Diversity Project (HGDP) populations at the top of the familiar continental principal-component analysis (PCA) plot. Nearly all self-reported African American participants in MGI cluster between the HGDP African and European reference populations, consistent with admixture between those populations. Self-reported Asian participants show two distinct clusters corresponding to Western Asian and Central/Southern Asian HGDP populations. As expected, participants that reported Hispanic/Latino ethnicity overwhelmingly appear between European and Asian continental populations.17 We identified numerous genetically inferred familial configurations among MGI participants (Figure 5B). Overall, 10,246 (18%) participants have at least one third-degree or closer relationship with another MGI participant, including 1,496 parent-offspring pairs and 838 full-sibling pairs. Various complex, multi-generational configurations are observed when considering second- and third-degree relationships (Figure S2).
Figure 5.

Summary of genetically inferred ancestry and relatedness in MGI participants
(A) Comparison of self-reported race/ethnicity and genetically inferred ancestry. MGI samples are projected in the principal component (PC) reference space created by worldwide samples from the Human Genome Diversity Project (HGDP). Each panel shows all MGI participants, with participants colored by the indicated self-reported race or ethnicity.
(B) Unique genetically inferred familial configurations containing parent-offspring and full-sibling relationships among MGI participants. The numbers are the observed count for each configuration.
(C) Comparison of TopMed and HRC imputation accuracy by inferred ancestry groups. TopMed provides more accurate imputation in all populations with notable gains among non-European participants.
We compared the number and quality of imputed genotypes in MGI participants between the TOPMed and HRC reference panels. Imputation using TOPMed produces 51,857,319 variants post quality control (QC) filtering compared with 32,477,751 using the HRC reference panel, with the largest gain in imputable variants at the lower end of the allele frequency spectrum (Figure S3); TOPMed imputation results in 45,399,294 variants with minor allele frequencies (MAFs) between 0.01% and 5% and imputation Rsq >0.3 compared with 26,769,074 of such variants based on HRC. Moreover, TOPMed-imputed variants are more accurate across the frequency spectrum, particularly for variants with MAFs <5% (Figure S4). Comparing the reference panels across samples from different ancestries reveals that the increased diversity in TOPMed reference haplotypes leads to increased imputation accuracy in all non-European samples (Figure 5C). The majority African ancestry samples showed the largest improvement in imputation accuracy, even for common variants, reflecting the large proportion of African American individuals in TOPMed compared with in HRC. We observe a more modest increase in accuracy among Asian MGI samples, likely because TOPMed contains comparatively fewer Asian haplotypes.
GWAS results
We initially conducted GWASs of the 1,712 phecode traits with at least 20 cases in the set of 51,583 MGI samples with genetically inferred European ancestry across 51,857,319 SNPs with MAFs >0.01% and imputation score Rsq >0.3. Evaluation of genomic control values indicated that traits with less than 60 cases were highly susceptible to inflation (Figure S5). Thus, we present results for the 1,547 traits with ≥60 cases (Table 1). We identified 1,901 distinct genome-wide significant loci across 977 phecode traits, including at least one genome-wide significant association for each of the 17 phecode categories. Many of the associations occur at low-frequency SNPs, which have higher false-positive rates at the standard 5e−8 threshold for genome-wide significance.18 Among SNPs with MAFs >1%, we observe 606 associations in 340 traits.
To assess the quality of our genetic data and phecode traits, we compared our 30 most significant associations at MAF >1% variants with previously identified associations reported in the GWAS Catalog (Table 2). Among this list, there are 15 unique SNPs because several were associated with multiple related phecode traits, reflecting the hierarchical nature of ICD coding. For 10 of the SNPs, we observed an association with a related trait in the GWAS Catalog at the exact chromosomal location. Four SNPs had a relevant association within a 50 kb window. The one association for which we did not observe a close phenotypically relevant association within the GWAS Catalog was for the insertion or deletion (indel) rs113993960 (chr7:117,559,590:ATCT:A) and cystic fibrosis (phecode 499). The indel, however, is a low-frequency, pathogenic in-frame shift within CFTR.19
Table 2.
Top thirty strongest associations among MAF >1% SNPs in MGI Freeze 3 GWASs
| rsid; chromosome position | Alleles | Allele 2 frequency | Trait description (phecode) | Cases/controls | Log odds ratio | p value | Relevant GWAS catalog citation |
|---|---|---|---|---|---|---|---|
| rs6025; chr1:169,549,811 | C/T | 0.0282 | primary hypercoagulable state (286.81) | 727/43,826 | 6.41 | 2.81e−157 | venous thromboembolism20 |
| – | – | – | hypercoagulable state (286.8) | 755/43,826 | 6.13 | 1.19e−153 | – |
| – | – | – | coagulation defects (286) | 2,693/43,826 | 2.03 | 1.80e−83 | – |
| – | – | – | other and unspecified coagulation defects (286.7) | 1,942/43,826 | 1.86 | 6.73e−50 | – |
| – | – | – | congenital deficiency of other clotting factors, including factor VII (286.12) | 94/43,826 | 11.12 | 5.24e−39 | – |
| – | – | – | other venous embolism and thrombosis (452) | 4,201/36,930 | 0.98 | 1.82e−36 | – |
| – | – | – | deep vein thrombosis (452.2) | 3,162/36,930 | 1.10 | 3.01e−34 | thrombosis21 |
| rs72660908; chr1:25,257,119 | C/G | 0.3856 | rhesus isoimmunization in pregnancy (654.2) | 145/26,348 | 2.25 | 1.40e−54 | blood protein levels22 |
| rs4148325; chr2:233,764,663 | C/T | 0.3272 | disorders of bilirubin excretion (277.4) | 321/48,830 | 1.84 | 6.00e−82 | bilirubin levels23 |
| rs143260331; chr4:110,762,205 | T/C | 0.1226 | atrial fibrillation (427.21) | 4,825/31,060 | 0.49 | 1.17e−37 | atrial fibrillation24,a |
| – | – | – | atrial fibrillation and flutter (427.2) | 4,978/31,060 | 0.48 | 2.42e−37 | atrial fibrillation/atrial flutter25,a |
| rs1800562; chr6:26,092,913 | G/A | 0.0602 | disorders of iron metabolism (275.1) | 201/47,321 | 4.33 | 1.07e−51 | hemoglobin26 |
| rs185937162; chr6:31,357,491 | T/G | 0.0428 | ankylosing spondylitis (715.2) | 190/35,793 | 4.34 | 2.92e−35 | ankylosing spondylitis27,a |
| rs2040410; chr6:32,634,921 | C/T | 0.1260 | celiac disease (557.1) | 407/37,236 | 1.63 | 5.91e−39 | celiac disease28,a |
| rs9273364; chr6:32,658,525 | T/G | 0.2769 | type 1 diabetes (250.1) | 2,266/36,631 | 0.80 | 4.23e−106 | medication use: drugs used in diabetes29 |
| – | – | – | type 2 diabetes with ophthalmic manifestations (250.23) | 1,522/36,631 | 0.54 | 1.32e−34 | – |
| rs9273368; chr6:32,658,698 | G/A | 0.2713 | type 1 diabetes with ophthalmic manifestations (250.13) | 760/36,631 | 1.41 | 2.91e−101 | latent autoimmune diabetes versus type 1 diabetes30 |
| – | – | – | type 1 diabetes with renal manifestations (250.12) | 509/36,631 | 1.55 | 4.02e−80 | – |
| – | – | – | type 1 diabetes with neurological manifestations (250.14) | 559/36,631 | 1.43 | 6.99e−76 | – |
| – | – | – | type 1 diabetes with ketoacidosis (250.11) | 205/36,631 | 1.75 | 1.23e−40 | – |
| rs1794269; chr6:32,706,117 | C/T | 0.3760 | diabetic retinopathy (250.7) | 1,544/43,849 | 0.60 | 4.53e−52 | type 2 diabetes31,a |
| – | – | – | insulin pump user (250.3) | 3,155/36,631 | 0.37 | 1.04e−39 | – |
| rs12203592; chr6:396,321 | C/T | 0.1616 | Other non-epithelial cancer of skin (172.2) | 6,627/41,896 | 0.36 | 1.83e−38 | basal cell carcinoma32 |
| – | – | – | skin cancer (172) | 8,228/41,896 | 0.32 | 1.65E-36 | – |
| – | – | – | basal cell carcinoma (172.21) | 3,509/41,896 | 0.47 | 2.36e−36 | – |
| rs113993960; chr7:117,559,590 | ATCT/A | 0.0146 | cystic fibrosis (499) | 97/51,358 | 18.90 | 9.80e−49 | lung function: FEV1/FVC33,b |
| rs28929474; chr14:94,378,610 | C/T | 0.0179 | alpha-1-antitrypsin deficiency (270.34) | 60/48,887 | 21.05 | 1.71e−52 | serum albumin level34 |
| rs1421085; chr16:53,767,042 | T/C | 0.4156 | morbid obesity (278.11) | 7,255/32,074 | 0.25 | 1.65e−36 | body mass index33 |
| rs3747207; chr22:43,928,975 | G/A | 0.2296 | other chronic non-alcoholic liver disease (571.5) | 2,973/41,006 | 0.52 | 2.95e−54 | alanine transaminase levels in high alcohol intake35 |
| – | – | – | chronic liver disease and cirrhosis (571) | 3,150/41,006 | 0.50 | 7.98e−53 | – |
The GWASs were conducted on 1,712 phecode traits with at least 20 cases in the set of 51,583 MGI samples with genetically inferred European ancestry across 51.8 million SNPs with MAFs >0.01% and imputation score Rsq >0.3. The relevant GWAS catalog citation column provides a phenotype and citation identified in the GWAS Catalog for a related trait at the indicated SNP in MGI.
GWAS Catalog association is within 50 kb of the indicated SNP.
GWAS Catalog association is within 1 Mb of the indicated SNP.
Our strongest association occurred between rs6025 (chr1:169,549,811, also known as the Factor V Leiden mutation p.Arg506Gln) and primary hypercoagulable state (phecode: 286.81). This SNP is among our top associations for multiple phecode traits related to coagulation (286.8: hypercoagulable state; 286: coagulation defects; 286.7: other and unspecified coagulation defects; 286.12: congenital deficiency of other clotting factors [including factor VII]). Associations between rs6025 and venous thromboembolism20 and thrombosis have previously been reported.21 rs143260331 was associated with two nested atrial fibrillation phecode traits (427.2 and 427.21) and was nearby previous associations for atrial fibrillation and flutter.
We also observed several strong associations between SNPs in the HLA locus and phecodes related to type 1 diabetes. These associations have been reported for related traits in the GWAS Catalog. For example, we observed an association between chr6:32,658,525, near HLA-DQB1, with the phecode 250.1: type 1 diabetes (4.23e−106), which has been previously reported for diabetes medication use.29 Broadly, our results replicate known signals, indicating that phenotyping and genotyping in MGI enable well-calibrated GWASs.
Discussion
This article describes the recruitment, data collection, and quality metrics for the MGI biobank and contrasts it with UKB, a large population-based biobank. It validates the design of a biobank based on localized recruitment within a tertiary healthcare center, primarily during pre-surgical inpatient encounters. The emphasis on surgical patients introduces a selection bias, which distorts population measures such as disease prevalence but provides distinct advantages for a genetic research resource. Specifically, MGI is enriched for nearly all disease outcomes compared with the general health system population as well as larger population-sampled biobanks. Although some of the observed case count differences between MGI and UKB are likely the result of differing diagnostic coding criteria, they nevertheless reflect the ability to identify cases within the respective biobanks. This case enrichment mirrors non-random sampling techniques routinely used in GWASs, for example case-control and extreme phenotype selection, specifically designed to increase statistical power. The result is that MGI provides powerful GWAS testing despite being substantially smaller than biobanks with national recruitment. This is confirmed by our GWAS analysis of ICD-derived phecode traits, which yielded 1,901 genome-wide significant associations, the strongest of which replicate known genotype-phenotype associations. Not surprisingly, the localized recruitment also led to the enrollment of many related participants in MGI, including various complex, multi-generational configurations. Depending on the analysis, related samples can either be informative or introduce statistical challenges to a genetic study. The degree of relatedness among MGI participants highlights the importance of using methods that properly account for sample relatedness when performing GWAS on biobank data.36
Single-health-system biobanks provide numerous benefits to genetic research, most importantly at the local institution but also to the broader community. At the local institution, biobanks democratize genetic research by providing open access to a state-of-the-art resource containing individual-level genotypes and rich clinical data. Investigators at the University of Michigan (UM) are required to obtain Institutional Review Board approval for proposed projects, but the MGI data are otherwise free to use. Moreover, UM investigators are supported with a free-of-charge HIPAA-secure computing environment to store and analyze data and genetic analysis support. This equitable access to a large-scale, multi-use cohort with centralized QC has the potential to dramatically accelerate research efforts. It is particularly empowering to junior researchers who may lack the funding to recruit their own cohort and collect genetic and phenotypic data. Moreover, the resource encourages investigators with limited genetics experience to engage in genetic research without the daunting tasks of collecting and performing QC on data with which they are unfamiliar.
The benefits of single-health-system biobanks extend to researchers outside of the local institution despite the fact that access to individual-level data is usually restricted to investigators at the institution. For example, we have provided access to all GWAS summary statistics reported in this article through an interactive PheWeb website. Downloadable MGI summary statistics are available to external investigators through a data usage agreement (see resource availability in the STAR Methods). Most importantly, the data generated for single-health-system biobanks benefit the broader community through incorporation in meta-analyses and consortium. Notably, nearly a quarter of the >2 million participants in the Global Biobank Meta-Analysis are from health-system-based biobanks.37
As biobanks continue to increase in number, they will remain major contributors to the large-scale GWAS meta-analyses that drive genetic discovery. As such, it is important to understand the distinct features of individual biobanks. Here, we have shown that a biobank recruited within a single health system can strategically recruit sufficiently large sample sizes for powerful genetic analysis and provides a valuable multi-use institutional resource that is complementary to large national biobank projects. With a sample size expected to top 100,000 participants during 2023, we anticipate that MGI will play an important role in future research both at the UM and the broader genetics community.
Limitations of the study
Our analysis revealed some limitations to MGI and similarly designed single-health-system biobanks. These biobanks have the potential for gaps in the health history of participants resulting from events that occur outside the respective health system. The bimodal distribution of participant follow-up time suggests that MGI is a mixture of long-time users of the health system with lengthy follow-up times and newer patients to Michigan Medicine with follow-up times of less than one year. Participants with short follow-up times are likely individuals who receive primary care from a different health system and are utilizing Michigan Medicine for the first and potentially only time during the surgical procedure in which they enrolled in MGI. We found that patients with longer follow-up times had higher numbers of phecode case assignments despite the fact that patient age was relatively consistent across follow-up times. It is possible that participants with longer follow-up times, despite being of similar age, simply have more health problems. A more plausible explanation is that participants with shorter follow-up times are out-of-system enrollees receiving temporary, specialized care at UM and therefore missing aspects of their medical history in the Michigan Medicine EHRs. For these participants, we are likely misclassifying them as controls for disease outcomes with missing diagnoses in the Michigan Medicine EHRs.
An additional limitation of the single-health-system design is that the demographics of the biobank naturally reflect the patient population served by the health system. In the case of MGI, the cohort largely comes from the surrounding Ann Arbor community and thus overrepresents individuals of European ancestry relative to both the population of Michigan and the US. Moreover, the MGI cohort itself is less diverse in terms of age, sex, race, ethnicity, and socioeconomic status than the overall clinical population at Michigan Medicine.38 Underrepresentation of minority individuals in particular can result in non-generalizable results and exacerbate existing health inequities.39,40 There is a clear need to improve enrollment of underrepresented populations beyond what is obtained in the current recruitment strategy. To meet this need, MGI is initiating recruitment efforts that leverage epidemiological studies in minority populations and targeted recruitment using the Michigan Health Care patient portal. Given that major differences exist in recruitment strategies between single-health-system biobanks, a careful analysis is required to evaluate the unique limitations and blind spots of a biobank.
The limitations of single-health-system biobanks underscore the continued importance of large, national biobanks with population-based recruitment in medical and public health research. In addition to the large sample sizes afforded by these biobanks, which is critical in collection of cases for very rare diseases, population-based recruitment of broader geographic and demographic segments of the populations increase biobank diversity. Further, gaps in health records from individual health systems can potentially be addressed by combining health histories from multiple sources.
Finally, phenotype development from EHRs requires making sense of dense, imperfect data. The wealth of available clinical data means there is no definitive definition for any phenotype of interest. In fact, one of the main strengths of EHR-based phenotyping is the ability to fine-tune case definitions. In this article, we used the PheWAS software, which provides a convenient approach to map granular ICD codes to phecode traits. The advantage of this technique is the rapid and automated generation of the phenome across all individuals in a biobank. Given the ubiquity of ICD codes, the PheWAS software provides a realistic strategy for consistent and harmonized large-scale phenotyping across biobanks. Thus, the phenotype definitions in this article are well defined and, importantly, replicable in other biobanks. That our strongest association results replicate known signals indicate that phecodes are an effective tool for broad phenotyping at the phenome scale. The phecode mappings, however, are not sufficiently precise to correctly identify cases or controls with perfect sensitivity. The phecode system also neglects clinical data sources like laboratory results, physician notes, and medication history that can be informative for elucidating true disease status. To maximize power and obtain unbiased effect size estimates for specific traits, it may be advantageous to carefully extract all relevant information from the EHR data and apply more complex validated electronic phenotype algorithms, for example, as described by the Phenotype KnowledgeBase (https://phekb.org).
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information should be directed to and fulfilled by the lead contact, Matthew Zawistowski (mattz@umich.edu).
Materials availability
This study did not generate new unique reagents or material.
Experimental model and subject details
Human subjects
Participants in the Michigan Genomics Initiative (MGI) consent to allow research on both their biospecimens and EHR data, as well as linking their EHR data to national data sources such as medical and pharmaceutical claims data. As of April 30th 2022, 91,695 participants have enrolled in the study. Participants are primarily recruited through the MGI - Anesthesiology Collection Effort (n = 72,461) while awaiting a diagnostic or interventional procedure either at a pre-operative appointment or on the day of their operative procedure at the University of Michigan Health System (Michigan Medicine). Additional participants are recruited through the Michigan Predictive Activity and Clinical Trajectories (MiPACT) Study (n = 7,616), the Michigan Genomics Initiative-Metabolism, Endocrinology, and Diabetes (MGI-MEND) Study (n = 4,153), the Mental Health BioBank (MHB2; n = 2,361), The Michigan and You – Partnering to Advance Research Together (MY PART) Study (n = 2,037), Providing Mental Health Precision Treatment (PROMPT; n = 1619) and the Biobank to Illuminate the Genomic Basis of Pediatric Disease (BIGBiRD; n = 226) among others.
We collect various self-reported demographic data provided by participants as part of routine appointment questionnaires for the health system. Participant age is computed based on self-reported date of birth and defined as age as of April 2020 or age at death if the participant is deceased. Self-reported race is based on a multiple-choice question with available options: Caucasian, African American, Asian, American Indian or Alaska Native, Native Hawaiian or Other Pacific Islander, Other/Unknown. Likewise, self-reported ethnicity is based on a multiple-choice question with available options: Hispanic or Latino, Not Hispanic or Latino, Unknown). Data were collected according to the Declaration of Helsinki principles.51 MGI study participants’ consent forms and protocols were reviewed and approved by the University of Michigan Medical School Institutional Review Board (IRB IDs HUM00071298, HUM00148297, HUM00099197, HUM00097962, and HUM00106315). Opt-in written informed consent was obtained. Additional details about MGI can be found online (https://precisionhealth.umich.edu/our-research/michigangenomics/).
Method details
Genetic data
Samples were genotyped by the University of Michigan Advanced Genomics Core on one of two customized versions of the Illumina Infinium CoreExome-24 bead array platform. These array versions have nearly identical 570K marker backbones synthesized in two batches. The array design contains customized probes incorporated to detect candidate variants from GWAS for multiple diseases and traits (∼2,700), nonsense and missense variants (∼49,000), ancestry informative markers (∼3,300), and Neanderthal variants (∼5,300).52
Genetic quality control procedures
We perform sample-level quality control (QC) on a rolling basis as batches of samples are genotyped. We estimate pairwise relatedness using KING (v2.1.3),45 and cross-sample contamination using VICES.46 Using PLINK (v1.9), we determine sample level call-rates.44 We exclude individual samples for any of the following: (1) the participant withdraws from the study, (2) genotype-inferred sex does not match the self-reported gender or self-reported gender was missing, (3) sample has an atypical sex chromosomal aberration, (4) kinship coefficient >0.45 with another participant with a different study ID, (5) sample-level call-rate <99%, (6) sample is a technical duplicate or twin of another sample with a higher call-rate either within the same array or across arrays, (7) estimated contamination level exceeds 2.5%, (8) missingness on any chromosome exceeds 5%, or (9) sample is processed in a DNA extraction batch that is flagged for severe technical problems.
We estimate the genetic ancestry of participants passing QC using principal component analysis (PCA) and admixture analysis using SNP data for 938 unrelated samples of known worldwide ancestry from the Human Genome Diversity Panel (HGDP) as ancestry reference samples.41,53 We define continental labels for the individual populations based on mappings available from the Center for the Study of Human Polymorphism’s website (https://cephb.fr/en/hgdp_panel.php). We first calculate a reference space of worldwide principal components (PCs) for the HGDP samples using PLINK. We then project MGI samples into this space and broadly infer the genetic ancestry of MGI samples based on their proximity to the known HGDP continental labels. We define MGI participants to be of European ancestry if their first two PCs are contained within a circle defined by a radius ⅛ the distance between the centroid formed by European HGDP samples and the centroid formed between European, East Asian, and African HGDP samples in the PC1 vs. PC2 space.11 We estimate the fraction of each MGI participant’s genome that originates from European, African, East Asian, Central/South Asian, West Asian, Native American, or Oceaninan ancestral HGDP continental populations using ADMIXTURE (v1.3.0)48 (Figure S6). We merge genotypes of MGI participants and HGDP reference samples prior to running ADMIXTURE in supervised mode using the total number of HGDP continental population labels (K = 7) as a template. We define the ADMIXTURE-based majority global ancestry for each MGI participant as the largest Q value (ancestry fraction) reported by ADMIXTURE.
We merge samples across genotyping batches and apply SNP-level QC procedures. We exclude SNPs with poor intensity separation based on metrics from the GenomeStudio Genotyping Module (GenTrain score <0.15 or Cluster Separation score <0.3) and drop SNPs with overall call-rate <99% or Hardy Weinberg p < 10−4 within each array. To identify potential batch effects between arrays, we test for differences in allele frequency between array versions using the Fisher Exact Test and exclude variants with p value <10−3, then merge the genotype data from the two arrays.
We phase the genotypes of the full set of merged samples using EAGLE (v2.4.1)54 without the use of a reference panel (“within-cohort” phasing). We then impute samples with both the Haplotype Reference Consortium (HRC) reference panel (64,940 predominantly European haplotypes containing 40,457,219 genetic variants)42 and the Trans-Omics for Precision Medicine (TOPMed) reference panel (194,512 ancestrally diverse haplotypes containing 308,107,085 genetic variants).43 We measure imputation quality using the estimate of imputation accuracy (Rsq) and the squared correlation between imputed and true genotypes (EmpRsq) metrics produced by the imputation software Minimac4 (v1.0.0).55
Clinical phenotype data
We extract all available ICD 9 and 10 diagnosis codes for MGI participants from the Michigan Medicine EHR. These codes are mapped to binary phecode phenotypes based on ICD inclusion and exclusion criteria using the PheWAS R package v0.99.5.-5.16 We use the default PheWAS package requirements for case and control definitions: cases require two instances of an inclusion ICD code and controls have neither inclusion nor exclusion ICD codes. We also account for sex-specific phenotypes using the restrictPhecodesByGender() function and the genotype-inferred sex.
Genetic analysis
We perform GWAS in MGI samples of genetically inferred European ancestry on a total of 1,712 phecode traits with case count ≥20. The GWAS cohort contains 51,583 MGI participants, including 49,689 with inferred European ancestry by the HGDP projection PCA and an additional 1,894 participants with inferred majority European ancestry by ADMIXTURE, but not identified as East Asian or African by the projection PCA. GWAS are run on the TOPMed-imputed genetic dataset using a mixed model implemented in SAIGE v0.43.3 to account for relatedness and case-control imbalance.36 For each phecode trait, we analyze variants with minor allele frequency (MAF) > 0.01% and adjusted for age, inferred sex, genotyping array, and the first ten genetic PCs. We compute the genomic control inflation factor for the GWAS of each phecode trait to assess stratification and test inflation.56
We identify quasi-independent genome-wide significant loci for each GWAS in the following manner: For each trait, we extract all SNPs with GWAS p value < 5e-8 and create 1Mb intervals centered around each SNPs. Overlapping intervals are combined and we report the SNP with the lowest p value from each of the resulting intervals as the genome-wide significant peak SNP.
We compared the 30 associations with smallest p value for variants with MAF>1% with associations reported in the GWAS Catalog (flat file downloaded August 16, 2021).1 We considered only associations in the GWAS Catalog that had a minimum reported p value < 5e-10 to decrease potential false positives within the Catalog. We defined an exact regional match as Catalog associations reported at the same chromosomal position location as the peak SNP. If an exact positional match was found, we manually scanned the list of Catalog associations for the same or a clinically similar phenotype to the corresponding phecode trait that produced the genome-wide significant association in MGI. If multiple related traits were reported in the Catalog for that SNP, we reported the trait with lowest p value except in one case where the top association appeared to be a sub-analysis that was more specific than our definition (e.g. for rs4148325 associated with "Disorders of bilirubin excretion," we reported "Bilirubin levels" as the GWAS Catalog match which had p = 5e-62 in the Catalog, even though the Catalog also listed this SNP for "Bilirubin levels in extreme obesity" at p = 5e-93). If an exact positional match was not found, we expanded our search to a 50kb window surrounding the peak SNP and followed the same protocol. In only one case was an association not found within a 50kb window and we expanded to a 1Mb region for this association.
Phecodes in UK biobank
We computed phecodes for a cohort of 408,595 individuals of White British ancestry with high-quality genetic data in the UK Biobank (UKB). We used ICD codes and genotyped derived data from open-access UK Biobank data. UK Biobank received ethical approval from the NHS National Research Ethics Service North West (11/NW/0382). The present analyses were conducted under UK Biobank data application number 24460.
We excluded samples which were flagged by the UK Biobank quality control documentation (Resource 531) as (1) “het.missing.outliers”, (2) “putative.sex.chromosome.aneuploidy”, (3) “excess.relatives”, (4) “excluded.from.kinship.inference”, (5) the reported gender (“Submitted.Gender”) did not match the inferred sex (“Inferred.Gender”), (6) withdrew from the UKB study and (7) were not included in the phased and imputed genotype data of chromosomes 1–22, and X (“in.Phasing.Input.chr1_22 and in.Phasing.Input.chrX”). Furthermore, we reduced the data to samples of White British ancestry (see UK Biobank Resource 531, “in.white.British.ancestry.subset”). We used the PheWAS R package to aggregate the ICD9 and ICD10 codes into phecode traits, requiring one inclusion code for case definitions.
Acknowledgments
The authors acknowledge the MGI participants, Precision Health at the UM, the UM Medical School Central Biorepository, and the UM Advanced Genomics Core for providing data and specimen storage, management, processing, and distribution services and the Center for Statistical Genetics in the Department of Biostatistics at the School of Public Health for genotype data curation, imputation, and management in support of the research reported in this publication. The MGI is funded through the Precision Health Initiative at the UM. S.Z. was funded through R01 HG011031. We thank the Global Biobank Meta-Analysis Initiative internal reviewers Ruth Johnson and Ben Neale as well as the anonymous reviewers for valuable feedback that improved and clarified the manuscript.
Author contributions
Conceptualization, C.M.B., S.K., and G.R.A.; data curation, M.Z., L.G.F., A.P., B.V., E.M.S., and C.J.W.; formal analysis, M.Z., L.G.F., A.P., B.V., and E.M.S.; investigation, M.Z., L.G.F., A.P., B.V., S.P., and E.M.S.; project administration, C.M.B., S.K., X.Z., M.B., G.R.A., and S.Z.; software, P.V. and S.P.; supervision, M.Z., L.G.F., X.Z., M.B., G.R.A., and S.Z.; visualization, M.Z., A.P., and B.V.; writing – original draft, all authors; writing – review & editing, M.Z., B.V., C.J.W., M.B., and S.Z.
Declaration of interests
G.R.A. and A.P. work for Regeneron Pharmaceuticals. C.J.W. took a position at Regeneron Pharmaceuticals after the initial submission of this manuscript.
Published: January 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100257.
Contributor Information
Matthew Zawistowski, Email: mattz@umich.edu.
Sebastian Zöllner, Email: szoellne@umich.edu.
Supplemental information
Data and code availability
The individual level genetic and clinical MGI data reported in this study cannot be deposited in a public repository because of patient confidentiality. Summary statistics of Genome-Wide Association Studies reported in this study can be viewed through an interactive pheweb website (https://pheweb.org/MGI/) that requires registration with a gmail e-mail address and acknowledgment that users will not (i) attempt to scrape genetic data from the pheweb website, (ii) attempt to identify or contact individuals upon whom these analyses are based, (iii) use the summary statistics contained on the website for commercial use. Summary statistics can be requested for download by emailing a completed Data Use Agreement form available at https://precisionhealth.umich.edu/our-research/documents-for-researchers/ to phdatahelp@umich.edu.
This paper does not report original code. DOIs for pre-existing code and external data sources used in this paper are listed in the key resources table.
References
- 1.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beesley L.J., Salvatore M., Fritsche L.G., Pandit A., Rao A., Brummett C., Willer C.J., Lisabeth L.D., Mukherjee B. The emerging landscape of health research based on biobanks linked to electronic health records: existing resources, statistical challenges, and potential opportunities. Stat. Med. 2020;39:773–800. doi: 10.1002/sim.8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denny J.C., Ritchie M.D., Basford M.A., Pulley J.M., Bastarache L., Brown-Gentry K., Wang D., Masys D.R., Roden D.M., Crawford D.C. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. Genome-wide genetic data on ∼500,000 UK Biobank participants. bioRxiv. 2017 doi: 10.1101/166298. Preprint at. [DOI] [Google Scholar]
- 5.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. Overview of the BioBank Japan project: study design and profile. J. Epidemiol. 2017;27:S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.All of Us Research Program Investigators. Denny J.C., Rutter J.L., Goldstein D.B., Philippakis A., Smoller J.W., Jenkins G., Dishman E. The “All of Us” research program. N. Engl. J. Med. 2019;381:668–676. doi: 10.1056/NEJMsr1809937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roden D.M., Pulley J.M., Basford M.A., Bernard G.R., Clayton E.W., Balser J.R., Masys D.R. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008;84:362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey D.J., Fetterolf S.N., Davis F.D., Faucett W.A., Kirchner H.L., Mirshahi U., Murray M.F., Smelser D.T., Gerhard G.S., Ledbetter D.H. The geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet. Med. 2016;18:906–913. doi: 10.1038/gim.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson R., Ding Y., Venkateswaran V., Bhattacharya A., Chiu A., Schwarz T., Freund M., Zhan L., Burch K.S., Caggiano C., et al. Leveraging genomic diversity for discovery in an EHR-linked biobank: the UCLA ATLAS Community Health Initiative. medRxiv. 2021 doi: 10.1101/2021.09.22.21263987. Preprint at. [DOI] [Google Scholar]
- 10.Goldstein J.A., Weinstock J.S., Bastarache L.A., Larach D.B., Fritsche L.G., Schmidt E.M., Brummett C.M., Kheterpal S., Abecasis G.R., Denny J.C., Zawistowski M. LabWAS: novel findings and study design recommendations from a meta-analysis of clinical labs in two independent biobanks. PLoS Genet. 2020;16:e1009077. doi: 10.1371/journal.pgen.1009077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritsche L.G., Gruber S.B., Wu Z., Schmidt E.M., Zawistowski M., Moser S.E., Blanc V.M., Brummett C.M., Kheterpal S., Abecasis G.R., Mukherjee B. Association of polygenic risk scores for multiple cancers in a phenome-wide study: results from the Michigan genomics initiative. Am. J. Hum. Genet. 2018;102:1048–1061. doi: 10.1016/j.ajhg.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shakeel F., Fang F., Kwon J.W., Koo K., Pasternak A.L., Henry N.L., Sahai V., Kidwell K.M., Hertz D.L. Patients carrying DPYD variant alleles have increased risk of severe toxicity and related treatment modifications during fluoropyrimidine chemotherapy. Pharmacogenomics. 2021;22:145–155. doi: 10.2217/pgs-2020-0154. [DOI] [PubMed] [Google Scholar]
- 13.Li Y., Lee S. Novel score test to increase power in association test by integrating external controls. Genet. Epidemiol. 2021;45:293–304. doi: 10.1002/gepi.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilliard P.E., Waljee J., Moser S., Metz L., Mathis M., Goesling J., Cron D., Clauw D.J., Englesbe M., Abecasis G., Brummett C.M. Prevalence of preoperative opioid use and characteristics associated with opioid use among patients presenting for surgery. JAMA Surg. 2018;153:929–937. doi: 10.1001/jamasurg.2018.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagliano Taliun S.A., VandeHaar P., Boughton A.P., Welch R.P., Taliun D., Schmidt E.M., Zhou W., Nielsen J.B., Willer C.J., Lee S., et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat. Genet. 2020;52:550–552. doi: 10.1038/s41588-020-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll R.J., Bastarache L., Denny J.C. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–2376. doi: 10.1093/bioinformatics/btu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryc K., Velez C., Karafet T., Moreno-Estrada A., Reynolds A., Auton A., Hammer M., Bustamante C.D., Ostrer H. Colloquium paper: genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 2):8954–8961. doi: 10.1073/pnas.0914618107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Annis A., Pandit A., LeFaive J., Taliun S.G., Fritsche L., VandeHaar P., Boehnke M., Zawistowski M., Abecasis G., Zöllner S. 2021. False Discovery Rates for Genome-wide Association Tests in Biobanks with Thousands of Phenotypes. [DOI] [Google Scholar]
- 19.VCV000007105.43 - ClinVar - NCBI (2021). https://www.ncbi.nlm.nih.gov/clinvar/variation/7105/.
- 20.Klarin D., Busenkell E., Judy R., Lynch J., Levin M., Haessler J., Aragam K., Chaffin M., Haas M., Lindström S., et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat. Genet. 2019;51:1574–1579. doi: 10.1038/s41588-019-0519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinds D.A., Buil A., Ziemek D., Martinez-Perez A., Malik R., Folkersen L., Germain M., Mälarstig A., Brown A., Soria J.M., et al. Genome-wide association analysis of self-reported events in 6135 individuals and 252 827 controls identifies 8 loci associated with thrombosis. Hum. Mol. Genet. 2016;25:1867–1874. doi: 10.1093/hmg/ddw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun B.B., Maranville J.C., Peters J.E., Stacey D., Staley J.R., Blackshaw J., Burgess S., Jiang T., Paige E., Surendran P., et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bielinski S.J., Chai H.S., Pathak J., Talwalkar J.A., Limburg P.J., Gullerud R.E., Sicotte H., Klee E.W., Ross J.L., Kocher J.-P.A., et al. Mayo Genome Consortia: a genotype-phenotype resource for genome-wide association studies with an application to the analysis of circulating bilirubin levels. Mayo Clin. Proc. 2011;86:606–614. doi: 10.4065/mcp.2011.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roselli C., Chaffin M.D., Weng L.-C., Aeschbacher S., Ahlberg G., Albert C.M., Almgren P., Alonso A., Anderson C.D., Aragam K.G., et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gudbjartsson D.F., Arnar D.O., Helgadottir A., Gretarsdottir S., Holm H., Sigurdsson A., Jonasdottir A., Baker A., Thorleifsson G., Kristjansson K., et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 26.Vuckovic D., Bao E.L., Akbari P., Lareau C.A., Mousas A., Jiang T., Chen M.-H., Raffield L.M., Tardaguila M., Huffman J.E., et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–1231.e11. doi: 10.1016/j.cell.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Australo-Anglo-American Spondyloarthritis Consortium TASC. Reveille J.D., Sims A.-M., Danoy P., Evans D.M., Leo P., Pointon J.J., Jin R., Zhou X., Bradbury L.A., et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 2010;42:123–127. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubois P.C.A., Trynka G., Franke L., Hunt K.A., Romanos J., Curtotti A., Zhernakova A., Heap G.A.R., Adány R., Aromaa A., et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y., Byrne E.M., Zheng Z., Kemper K.E., Yengo L., Mallett A.J., Yang J., Visscher P.M., Wray N.R. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 2019;10:1891. doi: 10.1038/s41467-019-09572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cousminer D.L., Ahlqvist E., Mishra R., Andersen M.K., Chesi A., Hawa M.I., Davis A., Hodge K.M., Bradfield J.P., Zhou K., et al. First genome-wide association study of latent autoimmune diabetes in adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care. 2018;41:2396–2403. doi: 10.2337/dc18-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vujkovic M., Keaton J.M., Lynch J.A., Miller D.R., Zhou J., Tcheandjieu C., Huffman J.E., Assimes T.L., Lorenz K., Zhu X., et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020;52:680–691. doi: 10.1038/s41588-020-0637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liyanage U.E., Law M.H., Han X., An J., Ong J.-S., Gharahkhani P., Gordon S., Neale R.E., Olsen C.M., et al. 23andMe Research Team Combined analysis of keratinocyte cancers identifies novel genome-wide loci. Hum. Mol. Genet. 2019;28:3148–3160. doi: 10.1093/hmg/ddz121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kichaev G., Bhatia G., Loh P.-R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinnott-Armstrong N., Tanigawa Y., Amar D., Mars N., Benner C., Aguirre M., Venkataraman G.R., Wainberg M., Ollila H.M., Kiiskinen T., et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Innes H., Buch S., Hutchinson S., Guha I.N., Morling J.R., Barnes E., Irving W., Forrest E., Pedergnana V., Goldberg D., et al. Genome-wide association study for alcohol-related cirrhosis identifies risk loci in MARC1 and HNRNPUL1. Gastroenterology. 2020;159:1276–1289.e7. doi: 10.1053/j.gastro.2020.06.014. [DOI] [PubMed] [Google Scholar]
- 36.Zhou W., Zhao Z., Nielsen J.B., Fritsche L.G., LeFaive J., Gagliano Taliun S.A., Bi W., Gabrielsen M.E., Daly M.J., Neale B.M., et al. Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nat. Genet. 2020;52:634–639. doi: 10.1038/s41588-020-0621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W., Kanai M., Wu K.-H.H., Rasheed H., Tsuo K., Hirbo J.B., Wang Y., Bhattacharya A., Zhao H., Namba S., et al. Global biobank meta-analysis initiative: powering genetic discovery across human disease. Cell Genomics. 2022;2:100192. doi: 10.1016/j.xgen.2022.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spector-Bagdady K., Tang S., Jabbour S., Price W.N., Bracic A., Creary M.S., Kheterpal S., Brummett C.M., Wiens J. Respecting autonomy and enabling diversity: the effect of eligibility and enrollment on research data demographics. Health Aff. 2021;40:1892–1899. doi: 10.1377/hlthaff.2021.01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landry L.G., Ali N., Williams D.R., Rehm H.L., Bonham V.L. Lack of diversity in genomic databases is a barrier to translating precision medicine research into practice. Health Aff. 2018;37:780–785. doi: 10.1377/hlthaff.2017.1595. [DOI] [PubMed] [Google Scholar]
- 40.Sirugo G., Williams S.M., Tishkoff S.A. The missing diversity in human genetic studies. Cell. 2019;177:26–31. doi: 10.1016/j.cell.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K., et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zajac G.J.M., Fritsche L.G., Weinstock J.S., Dagenais S.L., Lyons R.H., Brummett C.M., Abecasis G.R. Estimation of DNA contamination and its sources in genotyped samples. Genet. Epidemiol. 2019;43:980–995. doi: 10.1002/gepi.22257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldstein J.I., Crenshaw A., Carey J., Grant G.B., Maguire J., Fromer M., O’Dushlaine C., Moran J.L., Chambert K., Stevens C., et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28:2543–2545. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abraham G., Qiu Y., Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017;33:2776–2778. doi: 10.1093/bioinformatics/btx299. [DOI] [PubMed] [Google Scholar]
- 51.World Medical Association World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 52.Surakka I., Fritsche L.G., Zhou W., Backman J., Kosmicki J.A., Lu H., Brumpton B., Nielsen J.B., Gabrielsen M.E., Skogholt A.H., et al. MEPE loss-of-function variant associates with decreased bone mineral density and increased fracture risk. Nat. Commun. 2020;11:4093. doi: 10.1038/s41467-020-17315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang C., Zhan X., Bragg-Gresham J., Kang H.M., Stambolian D., Chew E.Y., Branham K.E., Heckenlively J., FUSION Study. Fulton R., et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet. 2014;46:409–415. doi: 10.1038/ng.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loh P.-R., Palamara P.F., Price A.L. Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet. 2016;48:811–816. doi: 10.1038/ng.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The individual level genetic and clinical MGI data reported in this study cannot be deposited in a public repository because of patient confidentiality. Summary statistics of Genome-Wide Association Studies reported in this study can be viewed through an interactive pheweb website (https://pheweb.org/MGI/) that requires registration with a gmail e-mail address and acknowledgment that users will not (i) attempt to scrape genetic data from the pheweb website, (ii) attempt to identify or contact individuals upon whom these analyses are based, (iii) use the summary statistics contained on the website for commercial use. Summary statistics can be requested for download by emailing a completed Data Use Agreement form available at https://precisionhealth.umich.edu/our-research/documents-for-researchers/ to phdatahelp@umich.edu.
This paper does not report original code. DOIs for pre-existing code and external data sources used in this paper are listed in the key resources table.