

Abstract
We report here the reprogramming of non-heme iron enzymes to catalyze an abiological C(sp3)—H azidation reaction via iron-catalyzed radical relay. This biocatalytic transformation uses amidyl radicals as hydrogen atom abstractors and Fe(III)—N3 intermediates as radical trapping agents. A high-throughput screening platform based on click chemistry was established for rapid optimization of the catalytic performance of enzymes identified. The final optimized variants function in whole Escherichia coli cells and deliver a range of azidation products with up to 10600 total turnovers and 93% enantiomeric excess. Given the high prevalence of radical relay reactions in organic synthesis and the large diversity of non-heme iron enzymes, we envision that this discovery will stimulate future development of metalloenzyme catalysts for synthetically useful transformations unexplored by natural evolution.
One Sentence Summary,
Non-heme iron enzymes engineered by directed evolution enable abiological radical relay chemistry for enantioselective C—N3 bond formation via remote C(sp3)—H functionalization.
Introducing abiological chemical transformations to natural proteins represents a powerful approach to advance enzymatic catalysis to reaction territories unexplored by natural evolution (1). This strategy enables enzyme reprogramming to achieve challenging synthetic reactions regio- and enantioselectively, while maintaining genetic tunability (2, 3). Representative examples include taming heme and non-heme metalloenzymes to mediate carbene- and nitrene-transfer reactions (4), reprogramming flavoenzymes for photoredox catalysis (5, 6), and reconfiguring carbonic anhydrase to perform metal-hydride chemistry (7). Despite this progress, the vast majority of reactions in organic synthesis have no biological counterparts, as the mechanisms that empower these transformations were not developed by nature during the course of natural evolution (1). To unleash the full potential of enzymes for modern chemical synthesis, it is pivotal to introduce fundamental reaction modes in synthetic chemistry to the catalytic repertoire of biology.
We consider a mechanism-driven approach to expand the scope of biocatalysis. We envisioned that natural metalloenzymes that share mechanistic elements with synthetic metal-catalyzed reactions would exhibit promiscuous activity towards these reactions, from which new catalytic functions can be evolved. Guided by this design principle, we envisaged that non-heme iron enzymes could be reprogrammed to perform radical-relay C—H functionalization—an important class of unnatural reactions widely used in organic synthesis (8–11). The defining feature of a metal-catalyzed radical relay is the use of a reactive radical (X•) to activate a C(sp3)—H bond via hydrogen atom transfer (HAT) and the interception of the resulting carbon-centered radical by a redox-active metal complex (Fig. 1A). This process mechanistically resembles the C(sp3)—H halogenation reactions catalyzed by non-heme iron halogenases, in which an iron(IV)-oxo complex is used to activate substrates via HAT and an iron(III)-azide/halide intermediate intercepts substrate radicals to form carbon—halogen/azide bonds (Fig. 1B) (12–16). Enlightened by this mechanistic similarity, we proposed that a non-heme iron enzyme could mediate a radical relay process via an initial substrate activation at Fe(II) center to generate a reactive amidyl radical for HAT and subsequent transfer of a Fe(III)-bound ligand to the carbon-centered radical (Fig. 1C). This new biocatalytic reaction proceeds through a redox neutral pathway involving an FeII/FeIII redox couple, which offers a complementary approach to the native FeII/IV catalytic cycle of non-heme iron enzymes for C—H functionalization.
Figure 1.
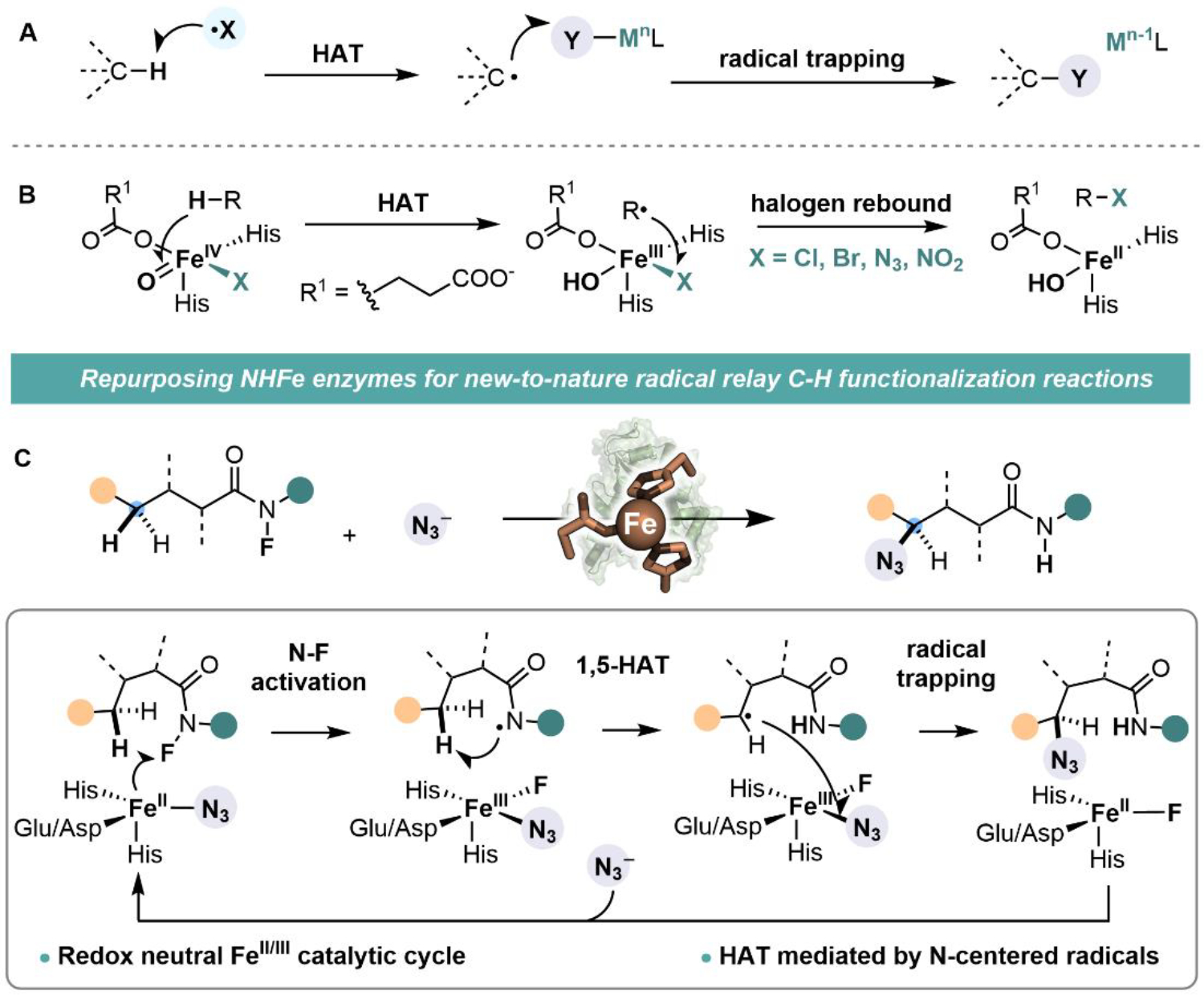
(A) Radical relay C—H functionalization involves an initial hydrogen atom transfer (HAT) mediated by a het2roatom-centered radical (X•) followed by the trapping of the carbon-centered radical with a redox-active metal complex. (B) Mechanism employed by natural non-heme iron enzymes for C(sp3)—H halogenation/azidation. (C) Integration of radical relay chemistry into non-heme iron enzymes enables unnatural C—H functionalization reactions.
As a proof-of-concept, we employed this enzymatic radical-relay strategy to develop new biocatalysts to perform a non-natural C(sp3)—H azidation reaction. Current synthetic approaches for this reaction are limited in turnovers, not enantioselective, and require acidic azide source (Scheme S4) (17). We presumed that these challenges can be met by leveraging the genetic tunability and high catalytic efficiency of non-heme iron enzymes. We began our investigation by testing a panel of nine functionally diverse non-heme iron enzymes with an N-fluoroamide substrate 1NF under whole-cell conditions. A (4-hydroxyphenyl)pyruvate dioxygenase from Streptomyces avermitilis (Sav HppD) provided the desired azidation product with a total turnovers (TTN) of 250, an enantiomeric ratio (e.r.) of 63:37, and a chemoselectivity of 9:1 for azidation over fluorination product (Entry 1, Table S1). Only trace amount of azidation product was obtained in a reaction lacking Sav HppD (Table S1 and S2). Moreover, mutating the two iron-coordinating histidines to alanines abolished the enzyme activity while retaining the fold of wt Sav HppD (Entry 11, Table S1 and Fig. S1), supporting the proposal that reaction occurs at the 2-His-1-carboxylate iron center. The unazidated amide product was also detected in trace amount, but was likely formed via an unidentified non-enzymatic process, as the double alanine mutant afforded this product in a yield comparable to that of the wild-type enzyme (Table S1 and S2).
We set out to improve the performance of Sav HppD via directed evolution. We performed computational modeling on the wt enzyme with both azide and 1NF substrate bound and chose fifteen residues for optimization (Fig. S10, S11, S13, and S14). These residues mainly reside in three regions: C—terminal α-helix, β barrel of the C-terminal domain, and loops surrounding the active site (Fig. 2A). We have also established a high-throughput screening (HTS) platform based on copper-catalyzed azide-alkyne cycloaddition (CuAAC) (18) and achieved reliable quantification of enzymatic azidation products with a coefficient of variation of 9% and a detection limit of 4 μM (Scheme S1 and Fig. S2). With this HTS platform, we evaluated more than 5,000 clones generated through error-prone PCR or site-saturation mutagenesis (Fig. 2B and 2C). A sextuple mutant Sav HppD V189A F216A P243A N245Q Q255A L367I (denoted as Sav HppD Az1) furnished the product with 1340 TTN and 87:13 e.r.. In this evolution campaign, we could not identify an enzyme variant with an e.r. higher than 87:13. This result indicates that mutations that were beneficial for improving activity might not necessarily lead to an increase in enantioselectivity, which might be due to the differences in substrate positioning and geometric requirement for the rate-determining N—F activation step and the enantio-determining azide rebound step as revealed by molecular dynamics simulation (Fig. S18). In this regard, we reevaluated some of the libraries with chiral HPLC and performed additional rounds of evolution aided by computational modelling (details see Fig. S13 and S14). We found a septuple mutant Sav HppD V189A N191A S230L P243G N245F Q255P L367I (denoted as Sav HppD Az2) which showed an enantioselectivity of 96:4 e.r. and 490 TTN. Kinetic analyses with purified enzymes showed that Az1 variant exhibited a 4.1-fold increase in kcat and a 1.7-fold increase in KM over the wild-type enzyme (29.4 min−1 (Az1) vs 7.20 min−1 (wt) for kcat and 790 μM (Az1) vs 470 μM (wt) for KM), whereas the more enantioselective Az2 variant displayed a 9-fold decrease in kcat (3.39 min−1) and a 6.6-fold decrease in KM (120 μM). Overall, both variants showed around 2-fold improvement in catalytic efficiency (kcat/KM) compared to that of the wild-type enzyme (Fig. S3).
Figure 2.
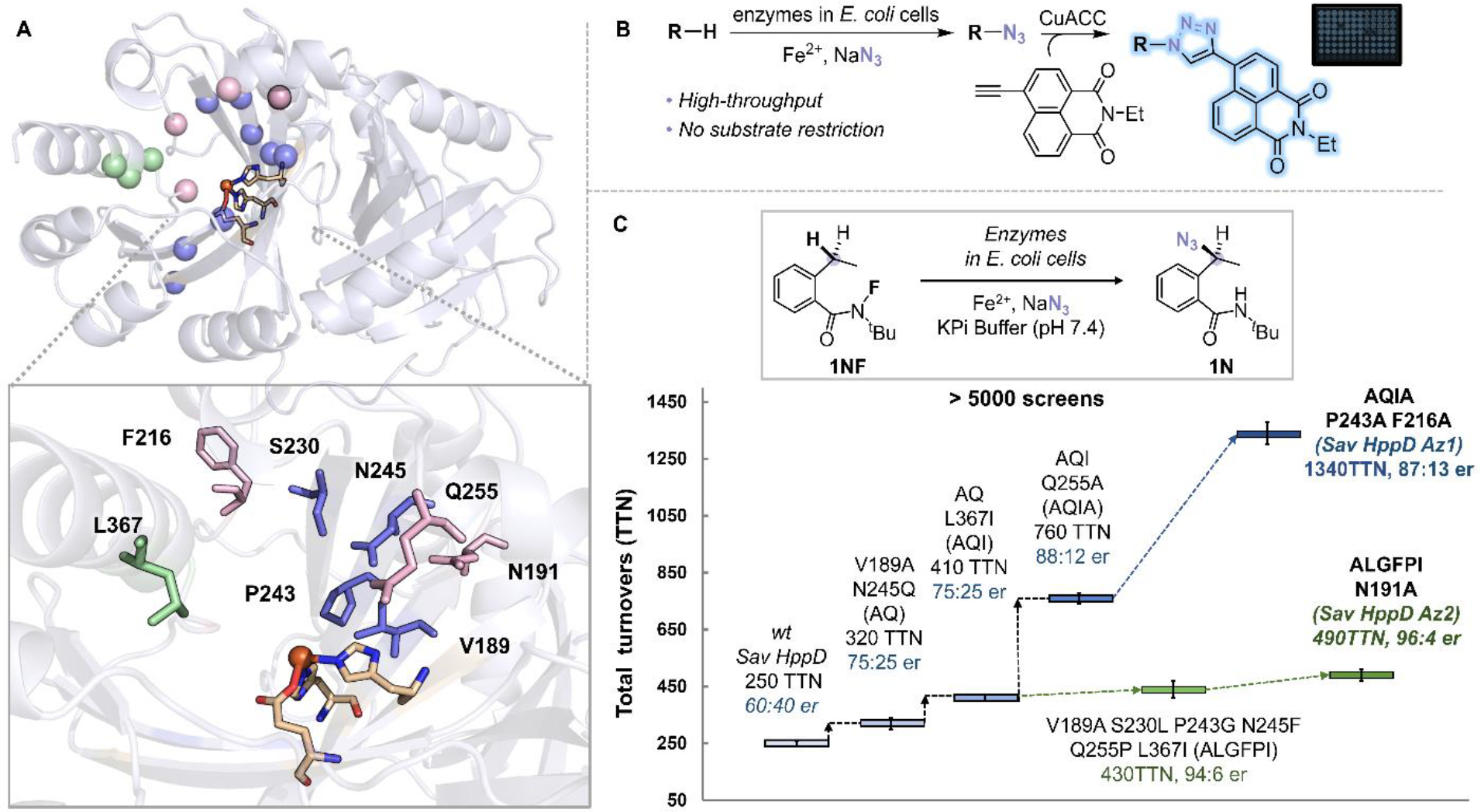
(A) Protein residues selected for mutagenesis (pink: loop residues surounded the active site (N191, F216, Q255, F359), green: residues on the C—terminal α-helix (K361, L367, N363), blue: residues on the β barrel of the C-terminal domain (V189, S230, P243, N245, Q269, Q334, F336, R353) (PDB: 1T47). (B) A high-throughput screening platform for detection of enzymatic azidation products. (C) Representative variants identified during the directed evolution of Sav HppD. Experiments were performed at analytical scale using suspensions of E. coli expressing Sav HppD variants (OD600 = 10), 10 mM substrate 1NF, 25 mM NaN3, 2.5 mM Fe2+ in KPi buffer (pH 7.4) at room temperature under anaerobic conditions for 24 hours (Table S2).
With these two final variants in hand, we optimized reaction conditions (Table S3) and assayed a range of N-fluoroamide substrates to explore the scope and limitation of this reaction (Fig. 3A). Sav HppD Az1 generally exhibited higher activity but lower enantioselectivity than Sav HppD Az2. The enzymatic reaction tolerates a range of aromatic substitution patterns with total turnovers up to 10060 and enantiomeric ratio up to 96.5:3.5 (product 5N). Substrates with an extended alkyl chain at the benzylic position were well tolerated, providing products in moderate-to-good TTNs and enantioselectivity (products 8N-10N). The amide nitrogen substituent also impacts enzyme performance, as evidenced by a decrease in activity when a larger N-tert-amyl group is substituted for the N-tert-butyl group (1N and 6N, 15N and 17N). We also tried to extend the scope of N-radical precursors and replace azide with other halide or pseudohalide anions. (Scheme S2 and Table S4). However, these efforts have not been successful with starting materials being recovered in most cases. As suggested by Mössbauer studies, the inability of our method to incorporate other anionic ligands might be due to a much weaker binding of these anions to the Fe(II) center of the enzymes. In a larger scale reaction, Sav HppD Az1 furnished 1N in 65% isolated yield at 120 mg scale with undiminished enantioselectivity (Fig. 3B). We also obtained single crystals of 1N and assign its absolute configuration as S by X-ray crystallography. We also produced primary organic azide 11N at preparative scale and subsequently converted it into an estrone derivative 18 via a CuAAC reaction (Fig. 3C). This chemoenzymatic two-step synthesis yielded the triazole product 19 in 55% isolated yield, demonstrating the potential of our platform to produce highly functionalized molecules when used in tandem with biocompatible reactions.
Figure 3.
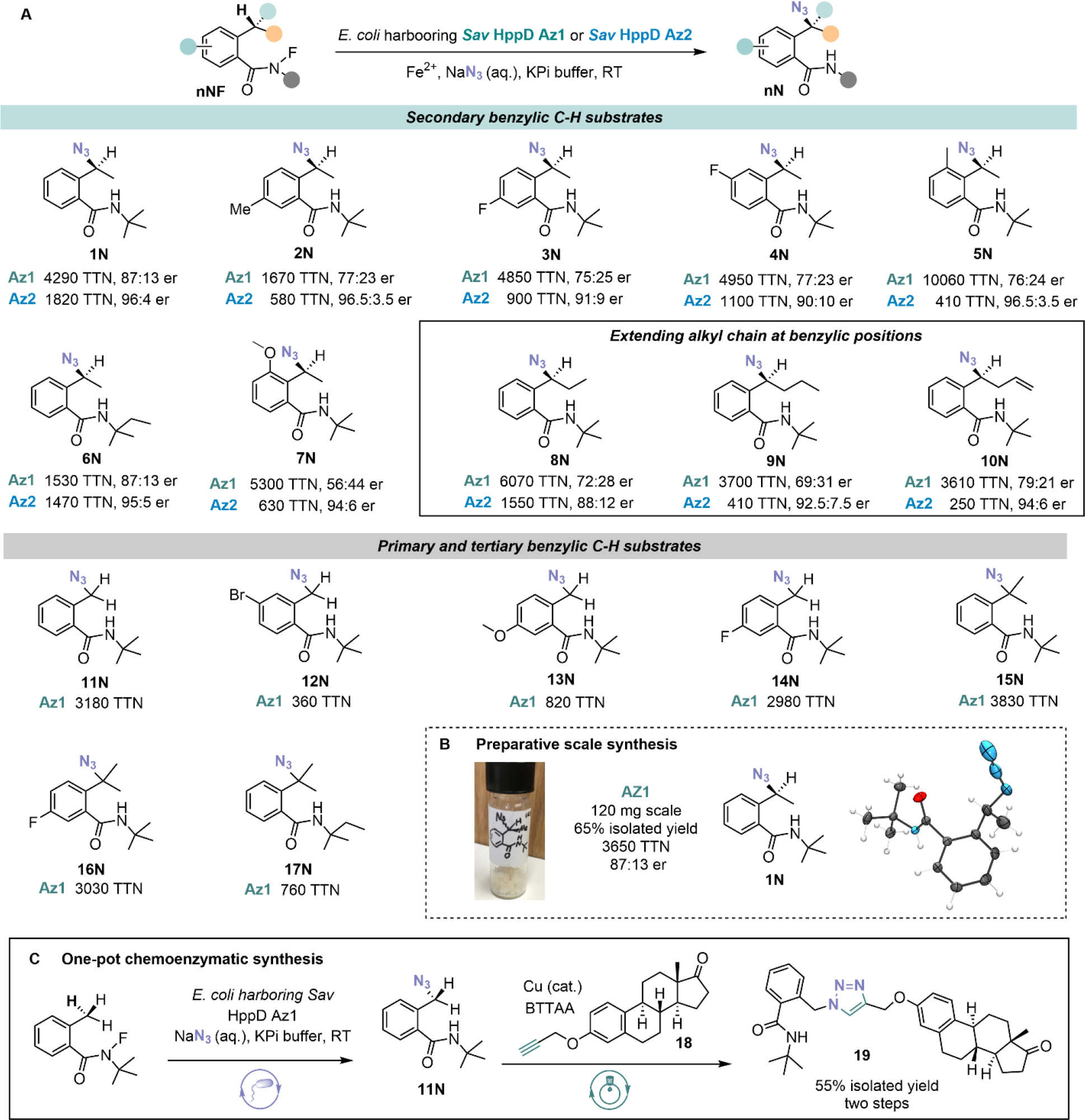
(A) Substrate scope of Sav HppD Az1 and Sav HppD Az2. Experiments were performed at analytical scale using suspensions of E. coli expressing Sav HppD variants in KPi buffer (pH 7.4) at room temperature under anaerobic conditions for 24 hours (detailed conditions see Table S3). The absolute configuration of enzymatically synthesized azidation product 1 was determined to be S via X-ray crystallography. The absolute configurations of all other azidation products were inferred by analogy. (B) Preparative scale synthesis and absolute configuration determination. (C) One-pot chemoenzymatic synthesis by in situ derivatization of enzymatic azidation products via CuAAC. Detailed conditions see section IX of the SI.
We conducted a few mechanistic studies to investigate the mechanism proposed in Fig. 1C. Addition of N3− to Sav HppD Az1•Fe(II) complex induced the formation of two quadrupole doublets in Mössbauer spectrum with isomer shifts (δ) of 1.20 and 1.17 mm/s and quadrupole splittings (ΔEQ) of 2.29 and 2.97 mm/s, respectively. The observation of two quadrupole doublets may reflect different azide binding configurations to the Fe(II) center (Fig. 4A, S5, S8 and S10, see section X of the SI for more discussion). We then carried out electron paramagnetic resonance (EPR) measurements on nitric oxide (NO)-bound Sav HppD Az1•Fe(II) complex whose prominent g ~ 4 EPR resonance can be used to monitor the interactions between the substrate and the non-heme iron center (19). Adding azide to Sav HppD Az1•Fe(II)•NO complex increased the rhombicity (E/D) of the g ~ 4 signal from 0.014 to ~0.017, the further addition of 1NF continued increasing the signal rhombicity (E/D = 0.023). These observations suggest that both N3 and 1NF interact with the Fe(II) center of Sav HppD Az1 (Fig. 4A, Fig. S6, and Table S5). To demonstrate an Fe(III)-N3 species is involved in the reaction, we incubated Sav HppD Az1•Fe(II)•N3 with an N-fluoroamide 18NF that lacked the reactive benzylic C—H bonds. A slow accumulation of a red species was observed with an optical absorption centered at 505 nm (Fig. 4B), which likely originated from the Fe(III)-N3 ligand-to-metal charge transfer band (20–22). The EPR signal of this red species was located at g ~ 4.3, further confirming its oxidation state was high spin (S = 5/2) Fe(III) (see section X of the SI). In this study, we also observed the formation of a minor stable organic radical centered at g = 2 (Scheme S6). Although further studies are needed to characterize this radical species, we speculated it to be a secondary radical formed via the quench of the initial amidyl radical, as this g = 2 signal was not observed when incubating Sav HppD Az1•Fe(II)•N3 with the model N-fluoraoamide 1NF (Scheme S6).
Figure 4.
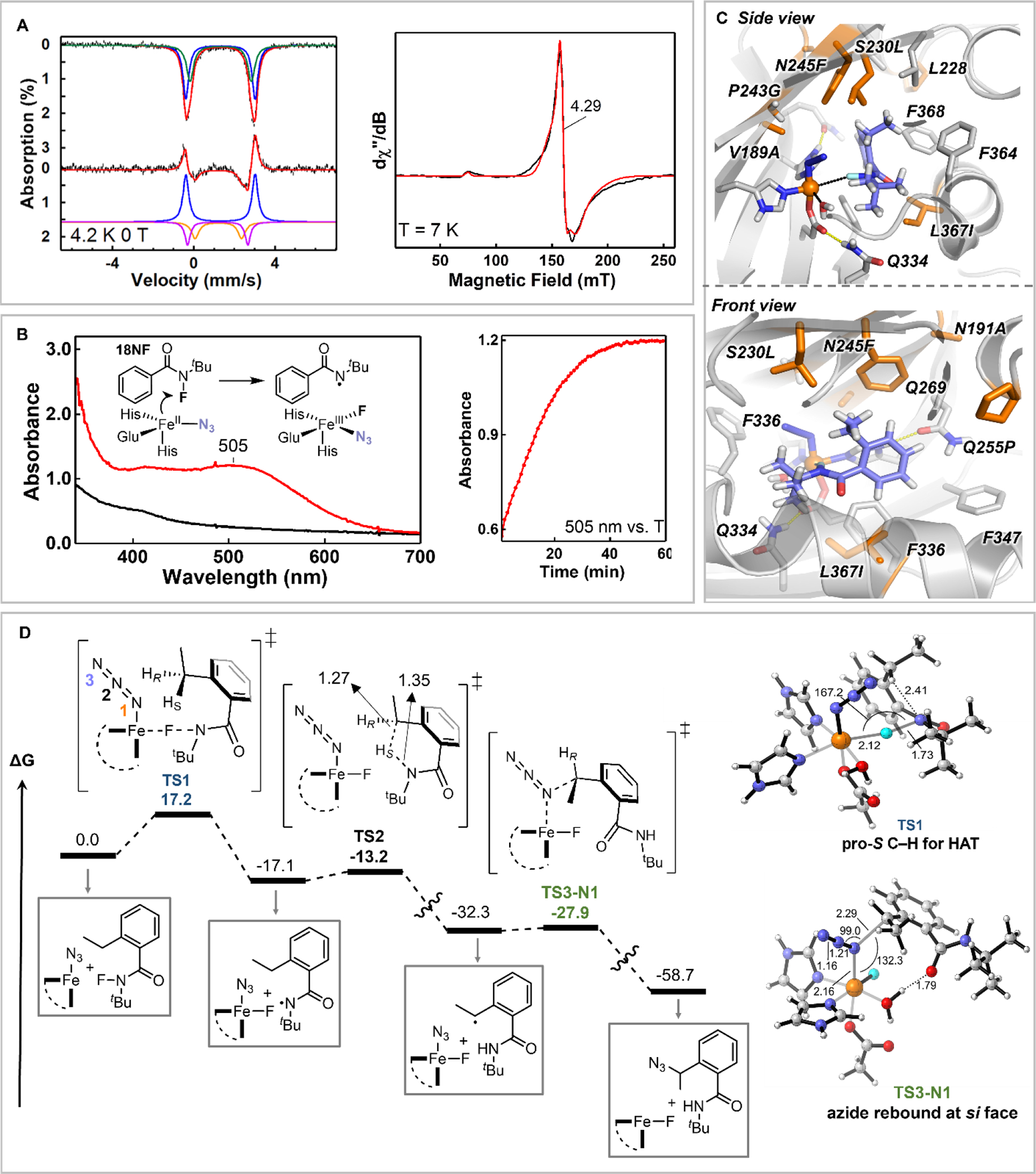
(A) Left: Mössbauer spectrum of Sav HppD Az1•Fe(II) complex (top, black) and the spectroscopic changes upon azide addition (bottom, black). The upward and the downward absorption peaks represent the disappeared and the appeared spectral components after the addition of azide. The colored solid lines represent spectral simulations (see SI for detailed discussion); Right: EPR spectrum of Sav HppD Az1•Fe(II)•N3 complex after incubation with 18NF for 60 min (black) and the spectral simulation (red). (B) Left: Optical absorption spectra of Sav HppD Az1•Fe(II)•N3 complex with 18NF (black) and after incubation with 18NF for 60 min (red). The inset shows the reaction scheme; Right: The time dependent change the 505 nm feature. (C) Active site arrangement of Az2 variant with 1NF substrate bound in a near-attack conformation for N—F activation characterized from MD simulations (see SI for details, Fig. S13). (D) Reaction mechanism obtained from DFT calculations employing a truncated active-site model build from MD simulations (see Fig. S18 for details) (energies in kcal/mol, distances in Å, and angles in deg.).
We also performed computational modelling to understand the molecular basis of this reaction. Focusing on enantioselective variant Sav HppD Az2, MD simulations showed that V189A and P243G generated more space to accommodate iron-bound azide in the active site (Fig. 4C, Figs. S8–S12). In wt Sav HppD, N191, N245 and S230 participated in a hydrogen bonding network with Q269 for native substrate positioning (23). Introducing the mutations N191A, S230L, and P243G disrupted this network. These mutations together with N245F and L367I created a hydrophobic environment to accommodate N-fluoroamide substrates for N—F activation and position the ethyl group of the substrate closer to the iron-bound azide in a restricted and preorganized conformation for the subsequent reaction steps (Fig. 4C, Fig. S8). Model DFT calculations (Fig. 4D) indicated that the initial N—F activation step (ΔG‡ = 17.2 kcal·mol−1) was rate limiting and was followed by a fast 1,5-HAT (ΔG‡ = 4.0 kcal·mol−1) to the N-centered radical. This mechanistic scheme was similar to that reported in an iron-catalyzed fluoroamide-directed fluorination (24) and was consistent with the absence of a kinetic isotope effect (KIE) via measuring independent initial rates for reactions with 1NF and 1NF-d2 (Fig. S4). The major conformation of substrate 1NF in Az2 preorganized the pro-S benzylic C—H bond for HAT, with the benzylic carbon projected in proximity to the iron-bound azide (Figs. 4C and Fig. S8). Therefore, after 1,5-HAT, the resulting C-radical was well positioned for azide recombination in a stereo-retentive manner with low energy barriers (ΔG‡ = 4.4 kcal·mol−1). Intriguingly, although fluorine transfer had an intrinsically low activation barrier (ΔG‡ = 5.0 kcal·mol−1, Fig. S18), steric constraints imposed within the active site likely prevented the substrate repositioning to enable fluorine recombination. Such control of radical rebound via substrate positioning has also been observed in native reactions catalyzed by non-heme iron halogenases, in which the proximity of substrate radical towards the iron-bound halide facilitated the C—Cl bond formation over C—OH bond formation (25, 26).
The biocatalytic system reported here performs C(sp3)—H functionalization reactions via a metal-catalyzed radical relay mechanism, expanding the scope of non-heme iron enzyme catalysis. We envision that the merger of various radical generation processes in synthetic chemistry and the capability of metalloenzymes for radical trapping will provide a powerful and general strategy to advance the frontier of radical biocatalysis.
Supplementary Material
Acknowledgments
We thank Prof. Marc Greenberg for helpful discussions and comments on the manuscript. We thank Dr. Maxime A. Siegler and JHU X-ray Crystallography Facility for analytical support. We also thank Dr. Katie Tripp and JHU Center for Molecular Biophysics (CMB) for assistance in performing CD experiments.
Funding:
Financial support was provided by the Johns Hopkins University and National Institute for General Medical Sciences R00GM129419 (to X. H.). Y. G. thanks the financial support from National Science Foundation, CHE1654060. This work was also supported by the Generalitat de Catalunya AGAUR Beatriu de Pinós H2020 MSCA-Cofund 2018-BP-00204 project (to M.G.B.), the Spanish MICINN (Ministerio de Ciencia e Innovación) PID2019–111300GA-I00 project (to M.G.B) and the Ramón y Cajal program via the RYC 2020–028628-I fellowship (to M.G.B), and the Spanish MIU (Ministerio de Universidades) predoctoral fellowship FPU18/02380 (to J.S.).
Footnotes
Competing Interests: A provisional patent application covering enantioselective biocatalytic C—N3 bond formation has been filed through the Johns Hopkins University with J.R., Q.Z., X.H., A.J.H., Z.C. as inventors. Authors J.S., J.C.P., V.R., Y.Y., Y.G., M.G.B. declare no competing financial interests.
Data and materials availability:
All data needed to evaluate the conclusions in this study are present in the main paper or the supplementary materials. The crystal structure of 1N is available from the Cambridge Crystallographic Data Centre under reference numbers CCDC 2163783.
References and notes
- 1.Arnold FH, Angew. Chem. Int. Ed. 57, 4143–4148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornscheuer UT, Philos. Trans. A Math. Phys. Eng. Sci. 376, 20170063 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Chen K, Arnold FH, Nat. Catal. 3, 203–213 (2020). [Google Scholar]
- 4.Brandenberg OF, Fasan R, Arnold FH, Curr. Opin. Biotechnol. 47, 102–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biegasiewicz KF, Cooper SJ, Gao X, Oblinsky DG, Kim JH, Garfinkle SE, Joyce LA, Sandoval BA, Scholes GD, Hyster TK, Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X, Wang B, Wang Y, Jiang G, Feng J, Zhao H, Nature 584, 69–74 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Ji P, Park J, Gu Y, Clark DS, Hartwig JF, Nat. Chem. 13, 312–318 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stateman LM, Nakafuku KM, Nagib DA, Synthesis 50, 1569–1586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang C, Li Z-L, Gu Q-S, Liu X-Y, Nat. Commun. 12, 475 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Chen P, Liu G, Acc. Chem. Res. 51, 2036–2046 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Groendyke BJ, AbuSalim DI, Cook SP, J. Am. Chem. Soc. 138, 12771–12774 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Crowe C, Molyneux S, Sharma SV, Zhang Y, Gkotsi DS, Connaris H, Goss RJM, Chem. Soc. Rev. 50, 9443–9481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthews ML, Chang WC, Layne AP, Miles LA, Krebs C, Bollinger JM, Nat. Chem. Biol. 10, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neugebauer ME, Sumida KH, Pelton JG, McMurry JL, Marchand JA, Chang MCY, Nat. Chem. Biol. 15, 1009–1016 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Kim CY, Mitchell AJ, Glinkerman CM, Li F-S, Pluskal T, Weng J-K, Nat. Commun. 11, 1867 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Groves JT, J. Biol. Inorg. 22, 185–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sivaguru P, Ning Y, Bi X, Chem. Rev. 121, 4253–4307 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Meldal M, Tornøe CW, Chem. Rev. 108, 2952–3015 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Orville AM, Chen VJ, Kriauciunas A, Harpel MR, Fox BG, Munck E, Lipscomb JD, Biochemistry 31, 4602–4612 (1992). [DOI] [PubMed] [Google Scholar]
- 20.Bull C, Fee JA, J. Am. Chem. Soc. 107, 3295–3304 (1985). [Google Scholar]
- 21.Meyer K, Bill E, Mienert B, Weyhermüller T, Wieghardt K, J. Am. Chem. Soc. 121, 4859–4876 (1999). [Google Scholar]
- 22.Grapperhaus CA, Mienert B, Bill E, Weyhermüller T, Wieghardt K, Inorg. Chem. 39, 5306–5317 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Brownlee JM, Johnson-Winters K, Harrison DHT, Moran GR, Biochemistry 43, 6370–6377 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Pinter EN, Bingham JE, AbuSalim DI, Cook SP, Chemical Science 11, 1102–1106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM, Proc. Natl. Acad. Sci. U. S. A. 106, 17723–17728 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blasiak LC, Drennan CL, Acc. Chem. Res. 42, 147–155 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in this study are present in the main paper or the supplementary materials. The crystal structure of 1N is available from the Cambridge Crystallographic Data Centre under reference numbers CCDC 2163783.