

Abstract
Cytokines have pivotal roles in immunity, making them attractive as therapeutics for a variety of immune-related disorders. However, the widespread clinical use of cytokines has been limited by their short blood half-lives and severe side effects caused by low specificity and unfavourable biodistribution. Innovations in bioengineering have aided in advancing our knowledge of cytokine biology and yielded new technologies for cytokine engineering. In this Review, we discuss how the development of bioanalytical methods, such as sequencing and high-resolution imaging combined with genetic techniques, have facilitated a better understanding of cytokine biology. We then present an overview of therapeutics arising from cytokine re-engineering, targeting and delivery, mRNA therapeutics and cell therapy. We also highlight the application of these strategies to adjust the immunological imbalance in different immune-mediated disorders, including cancer, infection and autoimmune diseases. Finally, we look ahead to the hurdles that must be overcome before cytokine therapeutics can live up to their full potential.
Subject terms: Cytokines, Cell delivery, Protein design, Nanoparticles, Nucleic-acid therapeutics
Cytokines are key regulators of the immune system and can be recombinantly designed as therapeutics for immune-related disorders. However, the severe toxicity of recombinant cytokines limits their clinical translation. In this Review, the authors highlight bioengineering approaches for the design of clinically applicable and safe cytokine-based therapeutics.
Key points
Cytokines are crucial regulators of the immune system and have important roles in health and disease.
Recombinant cytokine therapy is hampered by the short blood half-life and severe side effects of cytokines.
Increased knowledge of cytokine biology converges with new bioengineering approaches to develop engineered cytokine therapeutics.
Cytokine therapeutics can be applied to modulate dysregulated immune responses in disorders such as cancer or autoimmune diseases.
Introduction
Finely tuned cooperation between diverse cell types is essential for the immune system to effectively exert its various functions. The immune system protects the host against pathogens and supports tissue repair, while safeguarding homeostasis in different tissues1. Communication during immune responses occurs through either direct cell-to-cell interaction or the release of biomolecules, the most important of which are small proteins known as cytokines2–4. Immune cells are the main source of cytokines, secreted in response to infections, inflammation or endogenous stimuli5.
Several major cytokine classes can be distinguished according to their functions. Pro-inflammatory cytokines activate antimicrobial and immunostimulatory programmes and include tumour necrosis factor (TNF), IL-1β, IL-6 (ref. 6), IL-17 and IL-22 (ref. 7). Anti-inflammatory cytokines, such as IL-1 receptor antagonist (IL-1RA) and transforming growth factor-β (TGFβ), resolve inflammation and promote wound healing8,9. Chemokines, such as IL-8 and CC-chemokine ligand 2 (CCL2), comprise another class of cytokines that direct immune cell migration10. Interferons are vital to antiviral immunity11, and colony-stimulating factors, such as granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF), regulate homeostasis and immune cell progenitor proliferation12. Although immune cells produce most cytokines, non-immune cells and tissues can also secrete cytokines5. For example, endothelial and epithelial cells in the skin and the lungs express IL-33 and IL-25, which are involved in allergic inflammation13,14. Adipose tissue can release cytokines known as adipokines, which, among many capacities, regulate the body’s metabolic state. Although leptin and resistin are adipokines, they also exert proinflammatory functions15, illustrating the complex and diverse functions of cytokines.
Cytokines induce intracellular signalling after binding to their receptor on a target cell16. Cytokine receptors are as diverse as their ligands and are often multimers consisting of multiple transmembrane domains17. Upon binding, intracellular signalling prompts specific biological processes that, depending on the cytokine and cell type, can range from activating enzymes that regulate epigenetic modifications18, cytokine synthesis19 and augmented metabolism15 to triggering cellular proliferation20,21 or apoptosis22. A defining characteristic of cytokines is their ability to induce different phenotypic traits23. This phenomenon is known as pleiotropy, where a specific cytokine receptor can be present on various cell types, resulting in a variety of biological consequences24 (Fig. 1a). Additionally, some cytokines can bind to multiple receptors, which in turn can produce diverse downstream effects, such as inducing cell differentiation or inactivating effector functions25. Different cytokines can also function as co-activators, generating different effects depending on how they synergize26,27. Moreover, cytokines show partial functional redundancy, where more than one cytokine can have the same biological function. For example, both IL-2 and IL-15 induce T cell proliferation28, whereas IL-1α and TNF induce endothelial cell activation29. On the one hand, this ensures the immune system’s robustness; on the other hand, it complicates both therapeutic immunoregulation and the design of cytokine-based immunotherapies.
Fig. 1. Cytokine biology and mechanisms.

a, Cytokines are key regulators of the immune system and can be classified according to their function, for example, as pro-inflammatory or anti-inflammatory. Upon binding to the (multimeric) cytokine receptor on a target cell, cytokines can activate enzymes that regulate epigenetic modifications, cytokine synthesis, augmented metabolism, cellular proliferation and apoptosis. Cytokine pleiotropy refers to the ability to induce different phenotypic traits, resulting in a variety of biological consequences. The ability of cytokines to act on the same receptor indicates their redundancy. b, Insights into cytokine biology owing to advances in bioengineering, including greater knowledge about cytokine sequence and structure, receptor binding mechanisms, signalling pathways and function. Data are taken from refs. 47,56,58,63. ITC, isothermal titration calorimetry; JAK, Janus kinase; MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; NMR, nuclear magnetic resonance; SPR, surface plasmon resonance.
Because cytokines have critical roles in many immune-mediated diseases, they have been investigated as therapeutic targets30. Inhibiting cytokine function by either monoclonal antibodies or receptor blockers is particularly successful in conditions characterized by exacerbated cytokine production, such as autoimmune and inflammatory diseases. Blocking TNF is an effective treatment strategy for rheumatoid arthritis and Crohn’s disease31. Psoriasis can be treated by suppressing IL-17 or IL-23 (refs. 4,31). IL-6 and IL-1 antagonists can treat COVID-19 (ref. 33). Cytokines can also be therapeutically administered to direct immune responses. For example, recombinant protein technology has generated some approved cytokine drugs, such as colony-stimulating factors for haematological disorders34 or type I interferons for viral diseases such as hepatitis35. However, developing cytokine-based therapeutics remains challenging. Cytokines’ short blood half-lives, pleiotropism and unfavourable tissue distribution all contribute to their narrow therapeutic range36. In the late 1980s, a clinical trial with IFNα resulted in severe side effects such as WHO grade III flu-like symptoms in patients treated for hairy-cell leukaemia37. An IL-12 phase II clinical trial for treatment of renal cell carcinoma resulted in two deaths in 1995 (ref. 38), and IL-2 clinical trials for metastatic melanoma lead to six deaths39. Here, we first discuss how advances in bioanalytical technologies can be deployed to deepen our understanding of cytokine biology. Then, we describe innovations in protein engineering, nanomedicine, RNA technology and cellular engineering in relation to cytokine therapeutics. We show that bioengineers can gain and harness knowledge to develop safe, effective cytokine-based therapies for a variety of immune-mediated diseases.
Understanding cytokine biology
Advances in bioengineering have greatly increased our understanding of cytokine biology over the past 40 years. Our knowledge has expanded on various levels, ranging from cytokine structure and receptor binding to cytokine pleiotropy and redundancies in signalling pathways, all of which ultimately help us to comprehend different cytokine functions. Early elucidation of protein identity was achieved after the development of polymerase chain reaction (PCR)40 and Sanger sequencing41. Innovations in analytical modalities, including matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for macromolecules, such as proteins, in the 1980s42,43, helped us to study cytokine protein structure, including post-translational modifications.
Observing the 3D structure of cytokines is important to discern their function. Innovations in protein nuclear magnetic resonance (NMR)44, X-ray crystallography45 and cryogenic electron microscopy (cryo-EM)46 have greatly advanced the cytokine research field. For example, structural analyses of cytokine–receptor complexes allowed us to understand the interleukin IL-2’s interactions with its receptor (IL-2R) in detail. High-affinity IL-2R is composed of three subunits: IL-2Rα, IL-2Rβ and γc. IL-2Rβ and γc are sufficient for effective signal transduction through the formation of intermediate-affinity IL-2R, and IL-2Rα acts as an important affinity modulator that increases the sensitivity of cells to circulating IL-2 (refs. 47,48). Supplementary to structural knowledge, techniques such as surface plasmon resonance (SPR)49 and isothermal calorimetry (ITC)50 can reveal binding kinetics and thermodynamics. Combined with insights obtained from structural biology, these bioanalytical techniques have enabled rational modifications of cytokines to regulate receptor binding and thus function. Comprehending complex cytokine signalling routes can help us to develop ‘smart’ cytokine therapeutics to overcome pleiotropy and thus off-target effects51.
Ultimately, the binding of a cytokine to its receptors results in activation of the receiving cell and triggering of an intracellular signalling cascade. Various methods have been devised to study cytokines’ functional effects, such as genetic techniques, imaging and single-cell technologies.
Advances in genetic techniques such as gene ablation allowed the generation of knockout mice52. Knockout and knock-in technologies have been crucial in understanding how cytokines and their receptors participate in host defence. For example, studies using Il6-knockout mice have identified IL-6’s key role in resisting infections53. Cytokine knockout mice facilitated research into redundancies in the IL-1β pathway and helped to define distinct cytokine pathways for different fever-inducing stimuli54. Since the early 2010s, organ-specific conditional knockout of cytokine genes is used to investigate cytokines’ role in physiological processes, such as reproduction and T cell differentiation in the thymus55,56.
Innovative imaging techniques also contribute to our understanding of cytokines in pathological processes. For example, intravital microscopy is used to investigate cytokines’ roles in infections57. Nuclear imaging modalities, such as positron emission tomography with computed tomography (PET–CT), allow longitudinal tracking of labelled proteins in vivo, which can be used to study immune cells’ spatial distribution58.
Developments in single-cell technologies have considerably increased knowledge of cytokine functions in immune cells. One of the most important tools for exploring immunity is flow cytometry, which can be used to study immune cell activation and differentiation. Spectral flow cytometry allows the effects that cytokines exert on up to 40 parameters of a single cell to be probed59. Engineers have further expanded this with a new approach that combines flow cytometry with mass spectrometry: mass cytometry (cytometry by time of flight (CyTOF))60. In this modality, cell surface markers are labelled with lanthanide metals, which implies that theoretically 120 cellular markers can be detected simultaneously, although in practice the panel size is typically limited to roughly 60 parameters61. Single-cell RNA sequencing and transcriptomic analyses also contribute to understanding cytokine signalling and how cytokines affect immune cells. For example, proteomics combined with RNA sequencing datasets are applied to study naive and memory T cell responses to cytokines, revealing the heterogeneity of T cell reactivity in mounting a rapid cytokine response62. Transcriptomic profile datasets can also be used to model the interplay between cytokines. The complexity of cytokine biology means that computational modelling of cytokine signalling pathways should help to uncover new therapeutic targets, such as IL-8 in the case of severe COVID-19 (ref. 63).
The bioanalytical innovations discussed above illustrate the sizeable leaps researchers have made in understanding cytokine biology (Fig. 1b). However, very recent discoveries such as trained immunity64 and the existence of orphan cytokine receptors65 indicate that we still do not completely comprehend the immune system’s complexity. New analytical technologies will continue to deepen our insight into cytokine biology, which in turn will empower the development of cytokine therapeutics.
Re-engineering cytokine therapeutics
Our increased understanding of cytokine biology warrants revisiting their therapeutic exploitation. Together with advances in analytical techniques, innovative protein engineering tools have been developed to modify cytokine function, signalling and distribution. The introduction of PCR facilitated small protein modifications through point mutations40,66. Since then, the protein engineering field has progressed tremendously. In this section, we discuss how cytokine-based therapeutics (with an emphasis on IL-2 as an example) are designed using different protein engineering modalities.
Directed evolution
Two strategies can be employed for developing protein mutants; directed evolution and rational protein design. In directed evolution, mutations are introduced into a gene of interest and subsequently a variety of selection methods, such as yeast and phage display, are used to obtain a protein with the desired function. In contrast to rational design, directed evolution does not rely on detailed structural knowledge and protein 3D-structure prediction because it is based on functional selection of mutants. Thus, directed evolution is a popular and successful strategy for engineering proteins67. The method can be applied to create an IL-2 mutein with improved affinity for the IL-2Rβγ complex51. This dimeric form of the IL-2 receptor is mainly present on CD4+ and CD8+ memory T cells and natural killer (NK) cells, owing to low levels of IL-2Rα expression68. The toxicity of high-dose IL-2, which is a US Food and Drug Administration (FDA)-approved cancer therapy, is frequently associated with the activation of the high-affinity trimeric IL-2Rαβγ complex68. Moreover, the IL-2Rαβγ complex is abundantly expressed on regulatory T (Treg) cells68, and stimulating these immunosuppressive cells is counterproductive for anticancer approaches. In designing the mutein, error-prone PCR is used to create a mutagenic library of IL-2 variants that are screened for IL-2Rβ affinity. This eventually yields an IL-2 ‘superkine’ with 200-fold higher affinity for IL-2Rβ. The superkine erases the affinity discrepancy between the dimeric IL-2Rβγ and the trimeric IL-2Rαβγ complex and results in greater antitumour responses with reduced toxicity in an in vivo mouse model. To improve its pharmacokinetic properties, this IL-2 superkine was fused to albumin (MDNA11) and is now undergoing a phase I/II clinical trial (NCT05086692) in patients with advanced solid tumours (Fig. 2).
Fig. 2. Re-engineering cytokines.
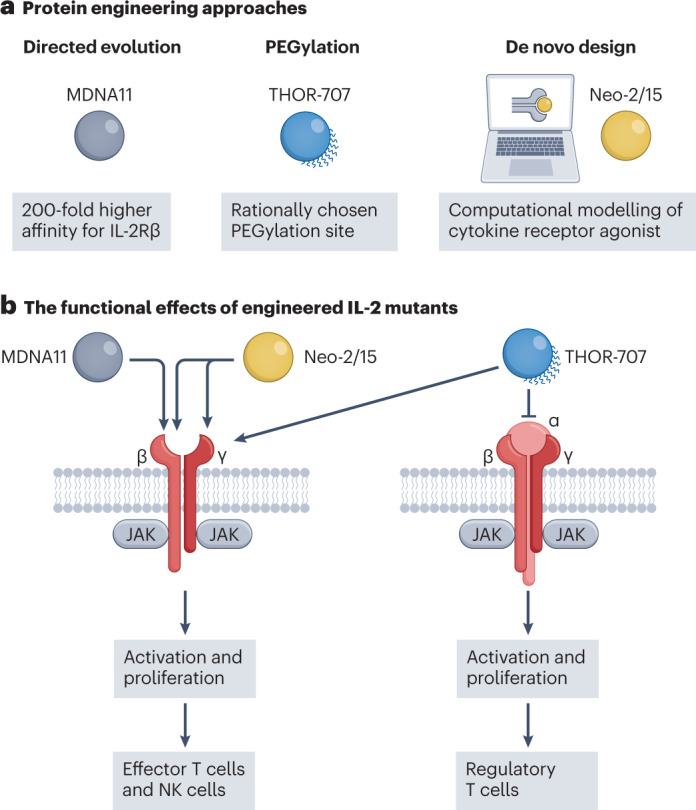
a, Bioengineering methods for improving the function and pharmacokinetic properties of IL-2 include directed evolution, rational PEGylation and de novo design signalling. Taking advantage of these engineering tools, three different cytokine-based drugs (MDNA11, THOR-707 and Neo-2/15) have been developed to enhance IL-2Rβγ-mediated signalling. MDNA11 is a variant of IL-2, developed using directed evolution, with 200-fold higher binding affinity for IL-2 receptor subunit-β (IL-2Rβ) compared to IL-2. To develop THOR-707, rational PEGylation is employed to block IL-2’s affinity for IL-2Rα to eliminate IL-2’s innate bias towards cells that express IL-2Rα. Neo-2/15 is produced by computationally modelling peptide motifs in IL-2Rβγ without interacting with IL-2Rα, resulting in a de novo protein that selectively mediates IL-2Rβγ signalling. b, High specificity of cytokine-based drugs increases effector T cell and natural killer (NK) cell activation and proliferation, but prevents signalling through the trimeric form (IL-2Rαβγ), thereby minimizing undesired activation of regulatory T cells. JAK, Janus kinase; PEG, polyethylene glycol.
Directed evolution has been applied to modify a variety of cytokines beyond IL-2. An IL-4 superkine has been developed to decouple its pleiotropic effects69. After binding to IL-4Rα, the IL-4/IL4Rα complex can engage either γc or IL-13Rα1 to form a type I or type II complex70, respectively. One of the engineered IL-4 superkines (termed Super-4) displayed reduced pleiotropy owing to its 3,700-fold higher affinity for γc, effectively steering activity towards the type I receptor rather than towards the type II receptor. To improve IL-10’s toxicity profile, an affinity-matured cytokine variant with an improved bioactivity profile at low dosages was developed71. In addition to improving affinity for receptors, directed evolution approaches can also improve specificity for receptors. IL-18 has potential as an anticancer drug, but its interactions with the inhibitory decoy receptor IL-18 binding protein (IL-18BP), which is frequently upregulated in tumours, has limited its clinical utility as an antitumour agent. Thus, directed evolution, using positive IL-18Rα and negative IL-18BP screening, can be applied to engineer an IL-18 mutein that evades its decoy receptor but still mediates inflammation72.
Rational design
Instead of using directed evolution, methods have been developed to rationally redesign cytokine properties. These techniques rely heavily on detailed knowledge of cytokine structure and advanced protein modelling73. For example, IL-22’s crystal structure bound to its receptor complex IL-22Rα/IL-10Rβ made it possible to reduce affinity for IL-10Rβ by mutating four key amino acid residues74. This quadruple IL-22 mutant has higher receptor and cell specificity, thereby diminishing undesirable pro-inflammatory properties and preserving its regenerative functions in vivo. Seeking to reduce pleiotropy, similar approaches yield variants of therapeutically potent cytokines, such as IL-10 (ref. 75), IFNγ76, IL-12 (ref. 77) and IL-4 (ref. 69). Interestingly, IL-2’s pleiotropy can also be exploited to rebalance immunity towards tolerance. Concurrent with IL-2 re-engineering efforts for cancer immunotherapy, engineers have developed IL-2 muteins that selectively activate Treg cells for treating autoimmune diseases78–84.
PEGylation
In addition to their binding properties, the circulation half-life of cytokines determines the in vivo effects of cytokine-based therapeutics. Functionalizing proteins with polyethylene glycol (PEG) is a popular and effective way to prolong their persistence in the circulation85. PEGylation increases a protein’s molecular weight and creates a hydrophilic corona that expands the hydrodynamic diameter, thus reducing renal clearance, and decreases interactions with plasma constituents, thereby diminishing immunogenicity86. PEGylation has successfully been applied to colony-stimulating factors and interferons (Table 1). However, although these changes facilitate alternate dosage regimens through a modified pharmacokinetic profile, they fail to reduce IL-2 toxicity87,88. PEGylation can also be applied for other purposes. IL-10 is known for its anti-inflammatory properties and is being clinically investigated as a therapy for inflammatory diseases (Table 1). Interestingly, at higher concentrations, IL-10 activates cytotoxic T cells, making it potentially useful for cancer immunotherapy89. Re-engineering was required to achieve elevated local IL-10 concentrations without causing severe side effects. This prompted the development of an IL-10 PEG-conjugate (pegilodecakin)90, with an improved pharmacokinetic profile, which is now undergoing clinical trials for treating various cancers (Table 2). Although PEGylation has historically been used to improve the blood half-life of proteins, the new PEGylated cytokines are designed to additionally alter receptor-binding91–93. In an attempt to take advantage of IL-2’s dual role in immunity, Nektar Therapeutics developed both Treg cell-selective (NKTR-358)93 and NK cell- and effector T cell-selective (BEMPEG/NKTR-214)92 IL-2 PEG-conjugates. However, all clinical trials of BEMPEG have been terminated owing to lack of clinical benefit94. Besides PEGylated IL-2 therapeutics, Nektar Therapeutics also engineered an IL-15-PEG variant (NKTR-255) with natural receptor binding and improved pharmacokinetic properties, aiming to circumvent the toxicity caused by the frequent dosing required in conventional IL-15 cancer therapy95 (Table 2).
Table 1.
Approved and failed cytokine therapies
| Cytokine | Name | Route of administration | Disease | Company | Outcome |
|---|---|---|---|---|---|
| Approved | |||||
| IL-1RA | Kineret/anakinra | s.c. | Range of autoimmune disorders | Swedish Orphan Biovitrum | Approved by the FDA in 2001 and the EMA in 2002 |
| IL-2 | Proleukin/aldesleukin | i.v. (high dose) or s.c. (low dose) | Metastatic melanoma and renal cell carcinoma | Clinigen Group | Approved by the FDA in 1992 |
| Ontak/denileukin diftitox/IL2-diftitox | i.v. | T cell lymphoma | Eisai | Approved by the FDA in 2008c | |
| IL-11 | Neumega/oprelvekin | s.c. | Thrombocytopaenia following myelosuppressive chemotherapy | Wyeth (now Pfizer) | Approved by the FDA in 1997d |
| TNF | Beromun/tasonermin | Isolated-limb perfusion | Limb soft-tissue carcinoma | Belpharma | Approved by the EMA in 1999 |
| G-CSF | Neupogen/filgrastim | s.c. | Neutropenia | Amgen | Approved by the FDA in 1991 |
| Neulasta/pegfilgrastim | Approved by the FDA and the EMA in 2002 | ||||
| Lonquex/lipegfilgrastima | Teva Pharmaceuticals | Approved by the EMA in 2013 | |||
| GM-CSF | Leukine/sargramostim | i.v. or s.c. (after myelosuppressive radiotherapy) | Post radiotherapy and chemotherapy, and bone marrow and peripheral blood cell transplantation | Partner Therapeutics | Approved by the FDA in 1991 |
| IFNα2a | Roferon A | s.c. | Chronic hepatitis C, hairy cell leukaemia and chronic myelogenous leukaemia | F. Hoffmann-La Roche | Approved by the FDA in 1995d |
| Pegasysa | Chronic hepatitis B and C | Approved by the FDA in 2002 | |||
| IFNα2b | Intron A | i.v., i.m. or s.c. | Chronic hepatitis B and C, hairy cell leukemia, malignant melanoma, follicular lymphoma, Kaposi’s sarcoma and genital warts | Schering-Plough (now Merck)b | Approved by the FDA in 1995d |
| PegIntron/Sylatrona | s.c. | Chronic hepatitis C, melanoma | Approved by the FDA in 2001 and the EMA in 2000e | ||
| Besremia | Polycythaemia vera | PharmaEssentia | Approved by the FDA in 2021 | ||
| IFN mix | Multiferon | s.c. | Malignant melanoma | Swedish Orphan Biovitrum | Approved in several European countries since 2005 |
| IFNβ1a | Avonex/Rebif/Cinnovex/Soluferon | i.m. or s.c. | Multiple sclerosis | Biogen, Merckb and CinnaGen | Approved by the FDA in 1996/2002 |
| Plegridya | Biogen | Approved by the FDA in 2014 | |||
| IFNβ1b | Betaseron/Extavia | s.c. | Multiple sclerosis | Bayer and Novartis | Approved by the FDA in 1993 |
| IFNγ1b | Actimmune | s.c. | Chronic granulomatous disease | Horizon Pharma | Approved by the FDA in 1990 |
| Epoetin alfa | Epogen/Procrit | i.v. or s.c. | Anaemia | Amgen and Johnson & Johnson | Approved by the FDA in 1989 |
| Aranesp/darbepoetin alfa | Amgen | Approved by the FDA in 2001 | |||
| Mirceraa | F. Hoffmann-La Roche | Approved by the FDA and the EMA in 2007 | |||
| Omontys/peginesatidea | Affymax and Takeda Pharmaceutical | Approved by the FDA in 2012f | |||
| Epoetin beta | NeoRecormon | i.v. or s.c. | Anaemia | F. Hoffmann-La Roche | Approved by the EMA in 2001 |
| Epoetin theta | Biopoin | s.c. | Anaemia | Teva Pharmaceuticals | Approved by the EMA in 2009 |
| Failed | |||||
| IL-4 | NA | s.c. | Psoriasis | Schering-Plough (now Merck)b | Limited clinical benefit207 |
| AIDS-related Kaposi’s sarcoma | NIAID | Limited clinical benefit208 | |||
| IL-7 | NA | s.c. | Metastatic melanoma, locally advanced or metastatic kidney cancer, refractory solid tumours | Revimmune and National Cancer Institute | Limited clinical benefit, formation of neutralizing antibodies209,210 |
| IL-10 | Tenovil | s.c. | Crohn’s disease | Schering-Plough (now Merck)b | No clinical benefit211,212 |
| NA | i.v. | Rheumatoid arthritis | Tufts University School of Medicine | No clinical benefit and complications upon multiple dosing213 | |
| IL-12 | NA | i.v. | Renal cell carcinoma | Wyeth (now Pfizer) | Severe toxicity issues: 2 deaths upon high dosing38 |
| IL-15 | NA | i.v. | Metastatic malignant melanoma or metastatic renal cell cancer | National Cancer Institute | Toxicity issues214 |
| IL-21 | NA | i.v. | Recurrent or metastatic melanoma | ZymoGenetics (now Bristol Myers Squibb) | No clinical benefit215 |
| TRAIL | Dulanermin/AMG 951 | i.v. | Colorectal cancer, non-Hodgkin’s lymphoma, non-small-cell lung cancer | Amgen and Genentech (F. Hoffmann-La Roche) | No clinical benefit and adverse events216–218 |
EMA, European Medicines Agency; FDA, US Food and Drug Administration; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFN, interferon; IL-1RA, IL-1 receptor antagonist; i.m., intramuscular; i.v., intravenous; NA, not applicable; NIAID, National Institute of Allergy and Infectious Diseases; PEG, polyethylene glycol; s.c., subcutaneous; TNF, tumour-necrosis factor; TRAIL, tumour-necrosis factor-related apoptosis-inducing ligand. aThese are PEGylated cytokines. bMerck Sharp and Dohme outside the USA and Canada. cDiscontinued in 2014 following manufacturing issues. dDiscontinued. eInduces severe side-effects (for example, suicidal depression). fDiscontinued in 2013 owing to severe adverse effects.
Table 2.
Engineered cytokine therapeutics in clinical trials
| Approach | Cytokine | Aim | Administration route | Disease | Company | Clinical trials |
|---|---|---|---|---|---|---|
| Re-engineered cytokines | ||||||
| Amino acids | BAY 50-4798/ N88R mutein | Non-β-IL-2 | s.c. | HIV | Bayer | NCT00059462 |
| PT101/MK-6194 | Ulcerative colitits | Pandion Therapeutics (acquired by Merck)a | NCT04924114 | |||
| Glycosylation | CYT107 | Prevent IL-7 antibody neutralization | i.m. | COVID-19, refractory nontuberculous mycobacterial lung disease, locally advanced, inoperable or metastatic urothelial carcinoma | Revimmune | NCT03513952, NCT04154826, NCT04379076 |
| PEGylation | THOR-707 | Non-α-IL-2 | i.v. | Advanced or metastatic solid tumours | Synthorx (acquired by Sanofi) | NCT04009681 |
| NKTR-358 | Non-β-IL-2 | Systemic lupus erythematosus | Nektar Therapeutics | NCT03556007 | ||
| NKTR-255 | Improve pharmacokinetics IL-15 | i.v. | Colorectal cancer and head and neck squamous cell carcinoma | NCT04616196 | ||
| Pegilodecakin | Improve IL-10 | s.c. | Advanced solid tumours | Eli Lilly and Company | NCT02009449 | |
| Peginterferon lambda-1a | Improve pharmacokinetics IFNλ1a | Chronic hepatitis D, COVID-19 | Eiger BioPharmaceuticals | NCT05070364, NCT04967430 | ||
| Directed evolution | MDNA11/IL-2 ‘superkine’–albumin | non-ɑ-IL-2 | i.v. | Advanced solid tumours | Medicenna Therapeutics | NCT05086692 |
| MDNA55/circular permuted IL4–PE | Tumour (IL-4R) targeting of PE | Recurrent/progressive glioblastoma | Medicenna Therapeutics | NCT02858895 | ||
| De novo | NL-201 | Non-ɑ-IL-2/IL-15 | i.v. | Relapsed or refractory cancer | Neoleukin Therapeutics | NCT04659629 |
| STK-012 | λ/β-selective PEGylated IL-2 mutein | s.c. | Advanced solid tumours | Synthekine | NCT05098132 | |
| Targeting and delivery | ||||||
| Immunocytokines | CEA-IL2v/RG7813 | Tumour (CEA) targeting + non-ɑ-IL-2 fusion | i.v. | Advanced or metastatic solid tumours | F. Hoffmann-La Roche | NCT02350673 |
| Simlukafusp alfa/RG7461/FAP–IL-2v | Tumour (FAP) targeting + non-ɑ-IL-2 fusion | NCT03386721 | ||||
| ANV419/IL-2A–IL-2 | Non-α-IL-2 | Advanced solid tumours and multiple myeloma | Anaveon | NCT04855929 | ||
| F16-IL2 | Tumour (TNC A1) targeting | Acute myeloid leukemia | Philogen | NCT02957032 | ||
| Darleukin/L19–IL-2 | Tumour (EDB fibronectin) targeting | Metastatic non-small-cell lung cancer | Maastricht University | NCT03705403 | ||
| DI-Leu16-IL2 | Tumour (CD20) targeting | B cell non-Hodgkin lymphoma | Alopexx | NCT02151903 | ||
| RG6279/RO7284755/PD1–IL-2v | Tumour (PD1) targeting | Advanced or metastatic solid tumours | F. Hoffmann-La Roche | NCT04303858 | ||
| hu14.18-IL2 | Tumour (GD2) targeting | i.t. | Advanced melanoma | Apeiron Biologics | NCT03958383 | |
| Efavaleukin alfa/AMG 592/Fc–IL-2v | Non-β-IL-2 | s.c. | Autoimmune diseases | Amgen | NCT04987333 | |
| XmAb564/Fc-IL-2 | Xencor | NCT04857866 | ||||
| Efineptakin alfa/GX-I7/NT-I7/IL-7–hyFc/hyleukin-7 | Improve pharmacokinetics of IL-7 | i.p. | COVID-19, Kaposi sarcoma, squamous cell carcinoma | Genexine and NeoImmuneTech | NCT04730427, NCT04893018, NCT04588038 | |
| Dekavil/F8–IL-10 | Inflamed tissue (EDA fibronectin) targeting | s.c. | Rheumatoid arthritis | Philogen | NCT02270632 | |
| Dodekin/IL-12–L19L19 | Tumour (EDB fibronectin) targeting | i.v. | Advanced solid tumours | Philogen | NCT04471987 | |
| NHS–IL12 | Tumour (NHS76) targeting | Solid tumours | Merck and Coa | NCT01417546 | ||
| Fibromun/L19TNF | Tumour (EDB fibronectin) targeting | i.v. | Metastatic soft tissue sarcoma, grade III/IV glioma, glioblastoma | Philogen | NCT03420014, NCT03779230, NCT04443010 | |
| TAK-573/CD38–IFNα2b | Tumour (CD38) targeting | Refractory multiple myeloma | Takeda Pharmaceutical | NCT03215030 | ||
| Fusion protein: IL-2–E7–HLA | CUE-101 | Activation of tumour-specific (HPV16) T cells | i.v. | Recurrent/metastatic head and neck quamous cell carcinoma | Cue Biopharma | NCT03978689 |
| Fusion protein: cpIL-2–IL2Rα | ALKS 4230/nemvaleukin alfa | Non-ɑ-IL-2 | i.v. | Advanced or metastatic solid tumours | Alkermes | NCT04592653 |
| Fusion protein: IL-15–IL-15Rα (sushi) heterodimer | NIZ958/IL-15–sIL-15Rα | IL-15 superagonist | s.c. | Metastatic cancers | Novartis | NCT02452268 |
| XmAb24306/RO7310729 | IL-15 superagonist–Fc complex | i.v. | Advanced or metastatic solid tumours | Genentech (F. Hoffmann-La Roche) | NCT04250155 | |
| Anktiva/N-803/ALT-803 | Advanced solid tumours | Altor BioScience | NCT01946789 | |||
| Prodrug | TransCon IL-2β/γ | Long-acting non-ɑ-IL-2 | i.v. | Advanced or metastatic solid tumours | Ascendis Pharma | NCT05081609 |
| XTX202 | TME-selective prodrug | Advanced solid tumours | Xilio Therapeutics | NCT05052268 | ||
| Nucleic acid therapeutics | ||||||
| Plasmid | NNC0361-0041/TOPPLE T1D | Antigen-specific tolerance through IL-2, preproinsulin, TGFβ1 and IL-10 expression | s.c. | Type 1 diabetes | Novo Nordisk | NCT04279613 |
| pIL-12 electroporation | IL-12 expression in tumour | i.t. | Metastatic melanoma | H. Lee Moffitt Cancer Center and Research Institute | NCT00323206 | |
| mRNA | mRNA-6231 | IL-2–human albumin fusion protein expression | s.c. | Autoimmune diseases | ModernaTX | NCT04916431 |
| BNT-151 | Optimized IL-2 expression | i.v. | Advanced or metastatic solid tumours | BioNTech | NCT04455620 | |
| BNT-153 | IL-2 expression | NCT04710043 | ||||
| BNT-152 | IL-7 expression | NCT04710043 | ||||
| MEDI1191 | IL-12 expression in tumour | i.t. | ModernaTX and Medimmune (now AstraZeneca) | NCT03946800 | ||
| SAR441000/BNT-131 | IL-12, IL-15Rα (sushi), IFNα and GM-CSF expression in tumour | Sanofi and BioNTech | NCT03871348 | |||
| mRNA-2752 | IL-23 and IL-36γ expression in tumour | ModernaTX | NCT03739931 | |||
| Cell therapy | ||||||
| TCR T cell | LMBP2-specific IL-12 expressing TCR T cell | Improve TCR therapy with IL-12 | i.v. | EBV-positive metastatic/refractory nasopharyngeal carcinoma | TCRCure Biopharma | NCT04509726 |
| CAR NK cell | IL-15 expressing CAR NK cells | Enhance in vivo expansion of CAR NK cells with IL-15 | i.v. | CD19-positive lymphoid tumours | M.D. Anderson Cancer Center | NCT03056339 |
CAR, chimeric antigen receptor; CEA, carcinoembryonic antigen; EBV, Epstein–Barr virus; EDA, extra-domain A; EDB, extra-domain B; FAP, fibroblast activation protein; HPV16, human papillomavirus 16; IFN, interferon; i.m., intramuscular; i.p., intraperitoneal; i.t., intratumoral; i.v., intravenous; NK cell, natural killer cell; PD1, programmed cell death 1; PE, Pseudomonas exotoxin; PEG, polyethylene glycol; s.c., subcutaneous; TCR, T cell receptor; TGFβ1, transforming growth factor β1; TME, tumour microenvironment; TNC, tenascin C; TNF, tumour necrosis factor. aMerck Sharp and Dohme outside the USA and Canada.
Expanding the genetic code with unnatural amino acids allows a unique reactive group to be introduced at a specific site within the protein, granting full control over the PEG chains’ position and stoichiometry96. Using this approach, Synthorx has designed a new PEG-IL-2 variant (THOR-707)91, which is currently in a phase I/II clinical trial (NCT04009681) as a potential therapy against advanced or metastatic solid tumours. The PEG moieties of THOR-707 specifically block the binding site with the IL-2Rα subunit, but maintain interaction with the other receptor subunits (Fig. 2). As a result, THOR-707 improves effector T cell and NK cell expansion and activation, and displays improved pharmacokinetic and safety profiles in cancer models compared to wild-type IL-2. A possible limitation of PEGylated cytokines is that the patient may develop anti-PEG antibodies that could inactivate the cytokine or initiate an inflammatory immune response97. This phenomenon has not been observed in studies with PEGylated cytokines, but the widespread application of mRNA COVID-19 vaccines shows that PEG can cause anaphylaxis in rare cases98.
Computational methods
With the aid of computational and data-driven methods and recent advances in hardware and algorithmic power, proteins can be designed de novo99–101. Computational tools promise to help to overcome key limitations of cytokine drugs, including pleiotropy, redundancy, poor pharmacokinetics and toxicity102. There are multiple approaches to computer-assisted protein design, for example, relying on predicted changes in free energy103, multiple sequence alignments103,104, backbone redesign105,106 or deep learning107,108. Moreover, computational tools initially designed for protein structure prediction, such as Rosetta109 or AlphaFold110, can provide valuable insights into protein design. Such computer-assisted tools could be used to customize cytokine agonists that are not necessarily related in amino acid sequence or topology to their natural equivalents. Therefore, the same function can be achieved with a completely different sequence. De novo protein design can be employed to create synthetic cytokines (synthekines) that engage unnatural receptor domain pairs, for example, IL-2Rβ/IL-10Rβ111, IL-2Rβ/IL-4Rα or IL-4Rα/IFNαR2, and therefore activate distinct signalling pathways112. De novo design methods also provide a strategy to effectively decouple cytokine pleiotropy and target specific pathways, as illustrated by phase I clinical trials with Synthekine’s STK-012 (ref. 113) and Neoleukin Therapeutics’s NL-201 (ref. 106) (Table 2). NL-201 has been developed by de novo computational design, using the crystal structure of IL-2 and IL-15 bound to the IL-2Rβγ receptor subunits as a blueprint. Computational design algorithms have been subsequently employed to engineer a new protein that mimics natural cytokine interactions with IL-2Rβγ without binding to IL-2Rα. Further stability optimizations generate the final variant Neo-2/15, which prolongs native IL-2’s inactivation half-time (Fig. 2 and Box 1). Although these computational-based cytokine redesigns target the cytokine–receptor interface, new affinity-matured cytokines can also be engineered solely through computational-driven protein stabilization, as showcased in a proof-of-concept study with a new IL-2Rβ-selective variant114. This computational protein stabilization approach requires less experimental intervention compared to the receptor-interface-focused designs, such as STK-012 and NL-201, potentially decreasing the costs and time involved in computational cytokine engineering. De novo-designed proteins, such as NL-201, are inherently foreign and thus may present immunogenicity issues. Although not observed in preclinical studies of Neo-2/15 (ref. 106), this remains an important consideration for the clinical translation of de novo cytokines.
Box 1 Translational considerations.
Protein engineering methods in academic laboratories differ vastly from those used in scaled-up good manufacturing practice (GMP) production. In research laboratories, protein therapeutics, including cytokine-based drugs, are often recombinantly expressed with purification tags in bacterial hosts, such as Escherichia coli. Although this approach allows swift development and downstream purification, it makes downstream processing challenging219. Bacterial endotoxins can be detrimental, particularly for drugs that regulate immune responses220. Therefore, for translational purposes, mammalian expression workflows are preferred. In addition to host organism considerations, production scale must be considered, in part because preclinical (mouse) models are much smaller than clinical (human) models. Moreover, stability studies, bioburden characterization and analytical methods often require large quantities of therapeutics. Substantial amounts of protein must also be generated to optimize the upstream manufacturing processes and the downstream processing221 required to purify the protein and minimize potentially harmful contaminants, such as endotoxins.
Upon establishing a robust, GMP-ready manufacturing process, investigational new drug (IND)-enabling studies must include dose range finding and toxicity experiments. The choice of species here is crucial. Unlike for small-molecule drugs, differences in cytokine biology and cross-species reactivity must be carefully considered when developing cytokine-based therapeutics. If the cytokine is not cross-reactive between human and murine systems, a mouse variant of the human cytokine must be developed and then evaluated in vitro and in vivo. Using the mouse surrogate protein, the pharmacological and immunological properties of the molecule can be studied in animal models of the disease. Optimizing binding affinity and specificity is most efficiently done using in vitro assays. Although in vitro models may provide some information on potential dosing and toxicity, full-scale toxicity data must be obtained in animal models, preferably in non-human primates. Drug biodistribution and in vivo bioavailability can only be studied in animal models.
The shelf-life, storage conditions and worldwide distribution of drugs are typically not considered in the early development stages; however, these considerations are crucial as development progresses. For IND-enabling dose range finding and toxicity studies, the formulation and storage conditions must resemble those used for subsequent phase I and II studies in humans. For example, for protein-based drugs with limited stability that require storage at low temperatures, cryoprotectants must be added to the formulation. Such considerations are best established early in the development trajectory.
Targeting and delivery
Routing cytokines with fusion proteins
Genetically fusing a cytokine to other proteins can help to reshape the cytokine’s biodistribution profile. This approach can be applied to target cytokines to a specific site or cell type. Cytokine fusion constructs help to promote tumour localization and overcome poor pharmacokinetic properties and unfavourable biodistribution profiles of cytokine-based drugs.
Immunocytokines
The idea of using antibodies to improve cytokine cancer therapies was introduced at the end of the twentieth century115,116. Over the past 15 years, multiple antibody–cytokine fusion constructs, known as immunocytokines, with IL-2 have been engineered and are undergoing clinical trials for treating various types of cancer (Table 2). In addition to improving currently approved cytokine therapies such as IL-2, designing immunocytokines could revitalize failed cytokine therapy approaches.
Although IL-12’s potency as an antitumour agent was recognized three decades ago, early clinical trials failed and resulted in two treatment-related deaths38 (Table 1). However, genetically fusing IL-12 to tumour-targeting antibodies can increase this cytokine’s therapeutic window and limit toxicity, as demonstrated by fusing IL-12 to an L19 antibody117. The L19 antibody is specific to fibronectin’s extra-domain B (EDB), a marker overexpressed on angiogenic blood vessels, which are abundant in many cancers118. In another study, two IL-12 heterodimers were fused to a histone-binding antibody (NHS76)119 to form the NHS–IL-12 immunocytokine that targets extracellular DNA in regions of tumour necrosis and apoptosis (Fig. 3a). The genetic fusion increases IL-12’s blood half-life, improves its therapeutic window, improves tumour localization and augments antitumour activity in murine Lewis lung carcinoma, MC38 colon carcinoma and B16 melanoma models. Both of these IL-12-based immunocytokines are currently undergoing clinical trials (NCT01417546 and NCT04471987) as treatments against solid tumours. Although most immunocytokines aim to deliver a pro-inflammatory cytokine to the tumour, the platform’s versatility can also direct anti-inflammatory cytokines to inflamed tissues. This is illustrated by the IL-22 immunocytokine (ABBV-022; out-licensed by Abbvie) which targets the gut mucosa for treatment of ulcerative colitis120.
Fig. 3. Cytokine targeting and delivery.

a, Cytokine fusion proteins and their roles in redirecting cytokines to a specific site or cell type. Immunocytokines are cytokines fused to antibodies that target cytokines to distinct locations. This strategy can, for example, be used with the pro-inflammatory cytokine IL-12 to actively target exposed DNA found in necrotic areas within the tumour microenvironment (TME). Tumour localization can be achieved by directing cytokines to the tumour’s extracellular components, such as collagen. Fusing a cytokine to collagen-binding proteins can anchor it in collagen and increase its retention in the tumour. Cytokine prodrugs can be engineered by fusion protein technology. These prodrugs usually exploit tumour-associated proteases that remove the inactivation domain from the cytokines through cleavage to release tumour-specific active cytokines. b, Routing cytokines with nanomedicine. Cytokine receptors are expressed on the cell surface and, thus, nanomedicine strategies must avoid uptake by immune cells. Nanoparticles can be designed with surface-displayed cytokines to target T cells139. In another approach, polymeric nanoparticles target collagen in the extracellular matrix, aiming for local sustained release of cytokines. COL-IV, type IV collagen.
Despite these improvements in cytokine localization, immunocytokine interactions with off-target cells can still cause side effects. For example, IL-2 immunocytokines mainly associate with IL-2R-expressing innate immune cells instead of accumulating in the tumour microenvironment (TME)121. To improve receptor and cellular specificity, new engineered therapeutics combine the immunocytokine approach with re-engineered cytokine mutants (Table 2). IL-2 can be fused to the anti-IL-2 antibody JES6-1, which blocks IL-2 interactions with the IL-2Rβ and γc domains, but displays a unique allosteric exchange mechanism between the antibody binding site and IL-2Rα, selectively activating IL-2Rα-expressing Treg cells122. Orionis Biosciences is developing a special class of immunocytokines, called Activity-on-Target cytokines (AcTakines), which combine a targeting antibody and an engineered cytokine mutant with reduced receptor affinity. The reduced affinity of AcTakines hampers cytokine activity until the fusion protein accumulates near the cytokine receptor on a target cell, lowering off-target effects123,124. Despite these advances, immunocytokines contain foreign epitopes that can induce immunogenicity, as reported in a phase I clinical trial (NCT03958383) of an IL-2 immunocytokine for treatment against melanoma and neuroblastoma125. Nevertheless, numerous immunocytokines are progressing to phase II clinical trials, demonstrating the promise of this therapeutic approach (Table 2).
Tumour localization
Rather than targeting protein antigens on cells, alternative fusion protein strategies can be adopted to overcome cytokine therapeutics’ limitations. Collagen is the major component of the TME and is therefore an interesting target for cytokine tumour localization126. Two collagen-binding protein fusion constructs have been engineered with either IL-2 or IL-12, anchoring these cytokines to collagen in the TME upon intratumoral administration127 (Fig. 3a). This approach increases cytokine retention in the tumour, consequently reducing systemic toxicity and boosting cellular antitumour immunity. In a similar strategy, IL-12 fused to a phosphorylated aluminium hydroxide (alum, the FDA-approved vaccine adjuvant) binding peptide tag shows weeks-long retention in the tumour in mouse models128. An alternative tumour retention strategy, that is, fusing IL-12 to the A3 collagen-binding domain of von Willebrand factor, can increase the cytokine’s tumour accumulation and decrease systemic toxicity upon intravenous administration129. The same strategy can be applied to IL-2, highlighting this approach’s adaptability for improving cytokine tumour accumulation130.
Prodrug strategies
One approach to overcoming off-target effects is to design prodrug constructs, whereby a peptide or protein linked to the cytokine renders it inactive. The inactivating unit can be cleaved, for example, by proteases overexpressed at the disease site, to reactivate the cytokine131. This strategy is particularly useful in the context of cancer treatment, for which several tumour-specific proteases have been identified132. Cytokine prodrug designs are often combined with re-engineered cytokines. For example, ProIL2 is an inactive form of the IL-2 superkine and is activated through cleavage by tumour-associated proteases133. Another non-IL-2Rα prodrug with a similar mechanism134 (Table 2 and Fig. 3a) is currently being tested in a clinical trial (NCT05052268) treating advanced solid tumours. Arrow Immune135 and Werewolf Therapeutics136,137 are employing this tumour protease approach to develop prodrugs of various cytokines, including IL-2, IL-12 and IFNα2b. Ascendis Pharma uses a different prodrug strategy with TransCon IL-2β/γ involving transient conjugation138. In addition to elevating IL-2Rβγ bias, this sustained-release prodrug is designed to prevent IL-2 spike concentrations and prolong blood half-life and has advanced to a phase I/II clinical trial (Table 2).
Routing cytokines with nanomedicine
Nanomedicine approaches have the potential to modulate the bioavailability and biodistribution profile of cytokines by routing them to specific organs or (immune) cells. However, as most cytokines act through interactions with cell surface receptors, nanomedicine strategies should generally avoid uptake by immune cells to facilitate interactions at the cell surface (Fig. 3b). To accomplish this, two approaches have been pursued: cytokines can be presented on the nanoparticle surface to enable direct interactions with their respective receptors, or nanomedicines can be directed to non-cellular targets, such as collagen in the extracellular matrix, where cytokines are released for local interaction with their receptors. The first approach was demonstrated by surface-functionalization of PEGylated liposomes with IL-2 and anti-CD137 to route IL-2 to effector T cells139 (Fig. 3b). Upon intravenous injection, the liposomes preferentially accumulate in the tumour, expand CD8+ T cells and induce an antitumour response in mice. A non-cellular targeting approach involves a type-IV-collagen-binding polymeric nanoparticle delivering anti-inflammatory IL-10 to atherosclerotic plaques140 (Fig. 3b). Upon collagen binding of these particles, polymer degradation slowly releases IL-10, allowing its subsequent interaction with inflammatory cells. A therapeutic regimen consisting of four injections over a four-week period prevented plaque formation in a murine model of atherosclerosis140.
mRNA-based cytokine therapeutics
The COVID-19 pandemic has propelled the development, clinical approval and widespread deployment of mRNA-based vaccines and RNA nanotherapeutics in general141. As a result, there is potential for therapeutic strategies based on regulating cellular cytokine expression following intracellular mRNA delivery by nanoparticles. However, systemically administering mRNA leads to its rapid degradation by endonucleases and recognition by Toll-like receptors, such as TLR7 and TLR8, resulting in innate immune activation and the concurrent secretion of pro-inflammatory cytokines, such as TNF and IL-6 (ref. 142). To avoid TLR7 activation, and the resulting induction of cellular apoptosis, mRNA chemical modifications, such as N1-methylpseudouridine, enable efficient protein translation with reduced innate immune activation143–146. Additionally, mRNA must be stably delivered and transported across the cellular membrane into the cytosol. Efforts to accomplish this led to lipid nanoparticle (LNP) technology for mRNA delivery147 (Fig. 4a). In brief, LNP–mRNA systems are composed of carrier phospholipids, PEGylated lipids, cholesterol and ionizable cationic lipids to complex negatively charged mRNA. Clinical data from the COVID-19 mRNA vaccines indicate consistent innate immune activation, which serves as a de facto adjuvant strategy148,149. Although advantageous in vaccine technology, innate immune activation can hamper the application of mRNA for conditions in which exacerbated immune responses need to be tempered, such as autoimmune diseases. These challenges are particularly relevant to cytokine therapy and innovations are necessary to overcome them.
Fig. 4. mRNA encoding cytokines and cell therapeutics.

a, mRNA encoding cytokines is packaged in lipid nanoparticles and taken up by cells. The mRNA is then released into the cytosol and translated into cytokines. b, Local cytokine delivery to tumours using lipid nanoparticles encapsulating mRNA-encoding cytokines that locally induce inflammation against the tumour. Systemic mRNA therapy strategies include: harnessing the liver as a production factory to generate re-engineered cytokines, such as an IL-2–albumin fusion protein; treating liver cancer with mRNA encoding pro-inflammatory cytokines; and passively targeting mRNA encoding pro-inflammatory cytokines to the tumour to reshape the tumour microenvironment (TME). Chimeric antigen receptor (CAR) T cells can include a gene to express cytokines, thereby enhancing immune activation. This technique can also be used to engineer other immune cells, for example, genetically engineered myeloid cells, dendritic cells and natural killer cells. These engineered immune cells can modulate the immunosuppressive tumour environment, after systemic adminstration.
Current mRNA nanotherapeutic strategies involve local and systemic administration routes (Fig. 4b). In this section, we describe how applying these two strategies to cytokine therapeutics can surmount their current limitations.
Local mRNA administration to tumours
To improve IL-12’s tumour retention, an LNP–mRNA has been designed that encodes IL-12 fused to the collagen-binding protein lumican150. An ionizable lipid in the LNP formulation provokes an immune response and improves the fusion protein’s translation. Upon intratumoral injection, the expressed IL-12 fusion protein mediates antitumour effects and systemic immune memory activation, leading to distal tumour regression in a mouse melanoma model. A similar mRNA therapy encoding IL-12, IL-27 and GM-CSF suppresses tumour growth without substantial toxicity in the same melanoma model151. Moreover, intratumorally injectable mRNA therapies encoding a cytokine cocktail can reshape the TME152,153. LNP–mRNA encoding IL-23, IL-36γ and OX40L can boost T cell responses against cancer cells152 (Table 2). This expressed cytokine mixture improves the response rate compared to treatment with mRNA coding for any of these cytokines separately. Furthermore, owing to the sustained release of cytokines encoded by mRNA, this mixture has higher antitumour efficacy than a cocktail of recombinant cytokines in a murine colon carcinoma model.
Despite their local administration, nanoparticles can still leak from the tumour into the vasculature, reach the liver and cause off-target effects154. Alternatively, a microRNA-122 (miR-122) binding site can be introduced into the mRNA sequence. Upon miR-122 binding, the complementary mRNA strand is cleaved, and protein expression is halted. This strategy exploits the abundance of miR-122 in the liver, preventing mRNA translation in this organ154. A different cytokine mRNA mixture encoding GM-CSF, IFNγ, IL-15 and IL-12 without LNP encapsulation153 (Table 2) shows higher IFNγ response and increases CD4+ and CD8+ T cell proliferation compared to separate mRNA-encoded cytokines in immune cells isolated from cancer patients153. Several companies are currently conducting phase I clinical trials to explore the safety of these new mRNA-encoding cytokine-based immuno-oncology treatments (Table 2). Although local delivery strategies are clearly promising, this approach faces challenges for treating deep-seated tumours, which necessitate systemic mRNA therapy strategies155.
Systemic LNP–mRNA therapy
Using systemically administered mRNA therapeutics to modulate the immune landscape can help to avoid the toxicities of recombinant cytokine therapies. One strategy for systemic mRNA therapy exploits high LNP accumulation in the liver to express cytokines and turn the liver into a cytokine production ‘factory’. This approach improves cytokines’ blood half-life and decreases their toxicity with lower and less frequent dosing156. In another example, the translation of a therapeutic mRNA encoding an IL-2–human albumin fusion protein in the liver systemically stimulates T cells to eradicate tumours157. This drug is currently undergoing a phase I/IIa clinical trial (NCT04455620) for treating solid tumours (Table 2). In a similar way, LNP accumulation in the liver could be harnessed to deliver mRNA encoding pro-inflammatory cytokines to treat hepatocellular cancer. LNP–mRNA encoding IL-12 can activate and recruit immune cells in the liver158, reducing tumour growth in mice without inducing systemic toxicity.
Targeting other sites beyond the liver with mRNA is complex but holds potential for a broader range of therapeutic applications. In this context, a passive tumour-targeting approach, harnessing the enhanced permeability and retention effect, could be applied to express cytokines that can reshape the TME. For example, an LNP–mRNA encoding IL-15 can treat pulmonary metastasis tumours159. This mRNA therapy inhibits tumour growth without systemic toxicity following intravenous injection in a colorectal cancer model159. Despite promising preclinical results, mRNA therapeutics’ clinical translation still faces challenges, such as immunogenicity triggered by cationic ionizable lipids148,149,160. Although these immunogenic effects complicate the use of mRNA in autoimmune disorders, LNP–mRNAs are being generated for treating inflammatory bowel disease. Adopting an active targeting approach, Il10 mRNA combined with a Ly6C-antibody-anchored LNP can specifically target inflammatory monocytes161. The therapy induces IL-10 expression in monocytes and effectively tunes the inflammatory state in an inflammatory bowel disease mouse model.
The biodistribution profile of the LNP–mRNA remains similar when the mRNA sequence is altered, which facilitates switching between encoded therapeutic proteins162. However, changing the LNP composition alters the biodistribution profiles and can help to skew accumulation to specific organs163. This has potential in either promoting or dampening immune activation by targeting mRNA-encoded cytokines to haematopoietic organs such as the spleen and bone marrow164.
Cell therapeutics
The translation of ex vivo engineered cells as therapeutics is progressing, as illustrated by the approval of chimeric antigen receptor (CAR) T cell therapy165. CAR T cells are genetically engineered to recognize tumour antigens to specifically eliminate cancer cells. Using autologous cells reduces the chance of immune-mediated host rejection but adds complexity to the manufacturing process and substantially increases costs166. Despite CAR T cell therapy’s clinical success in haematological malignancies, it is only effective in a fraction of patients167. Combining cellular engineering and cytokine therapeutics will help circumvent the current difficulties facing both cell and cytokine therapies (Fig. 4b).
Improving immune cell therapy
One issue hampering CAR T cell therapy’s effectiveness is the inability of engineered T cells to overcome the immunosuppressive TME168. Trials to reverse this suppressive state have been initiated by engineering IL-12-expressing CAR T cells, which effectively deplete tumour-associated macrophages169,170. The therapeutic efficacy of this approach has been demonstrated in a clinical trial but caused dose-limiting toxicities171. Additionally, CAR T cell therapy is often restricted by low cell expansion and survival172 as well as poor CAR T cell accumulation in the TME168. IL-2 potently stimulates T cell proliferation and improves persistence of adoptively transferred T cells173, whereas IL-7 and CCL19 are critical for maintaining a T cell zone in lymphoid organs174. However, directly administering these cytokines in combination with CAR T cells can induce cytokine-mediated toxicity175. To avoid the need to administer cytokines, CAR T cells can be engineered to express IL-2 (ref. 176) and IL-7 with CCL19 (ref. 174).
Cytokine re-engineering can also be integrated into cellular engineering. For example, an orthogonal IL-2 variant can be created to activate only the co-developed IL-2Rβ mutant by a lock-and-key approach177. The IL-2 variant can selectively target T cells expressing orthogonal hIL-2Rβ, without interfering with native T cells and natural IL-2 signalling. The orthogonal IL-2–IL-2Rβ pair improves the efficacy of adoptive T cell therapy in a mouse melanoma model, with negligible toxicity and off-target effects. The same strategy has been tested for a more challenging mouse leukaemia model, achieving anticancer effects178. The orthogonal IL-2–IL-2Rβ pair can also be translated to adoptive Treg therapy179. Treg therapy improves bone marrow acceptance in animal allo-engraftment models180, but these positive results are dampened by limited cell expansion. Therefore, an orthogonal IL-2–IL-2Rβ pair was introduced into Treg cells to enable selective signalling in vivo. Using orthogonal IL-2–IL-2Rβ to selectively expand adoptive Treg cells improves heart allograft acceptance in a murine haematopoietic mixed chimerism model.
Protein engineering can be employed to overcome challenges arising before engineered T cells are administered. For example, a new IL-2 variant can stimulate ex vivo T cell expansion and maintain them in a more stem-cell-like state181. This effectively prevents T cells from terminally differentiating before in vivo transfer, thereby improving therapeutic efficiency.
Engineered cells for cytokine delivery
Cellular engineering can also be employed for cytokine delivery. For example, GM-CSF-expressing T cells can target cytokines to central nervous system malignancies182, possibly providing new opportunities for cytokine delivery across the blood–brain barrier. As another example, synthetic Notch receptors can be introduced into engineered T cells183. The synNotch receptors recognize specific cancer-related antigens, and the T cells display customizable protein expression. This approach can be exploited for the local selective expression of therapeutic cytokines, such as IL-2 and TNF-related apoptosis-inducing ligand (TRAIL), in the tumour.
Importantly, cellular engineering is not limited to T cells; for example, IL-12-expressing dendritic cells can be designed to activate T cells for tumour elimination184,185. Dendritic cells with combined IL-12 and IL-18 expression more effectively induce T cell-mediated antitumour immunity than dendritic cells expressing either IL-12 or IL-18 alone186. In addition to expressing IL-12, dendritic cells can be engineered to additionally produce and present tumour-associated antigens to activate T cells187. A similar strategy has been applied to a cancer vaccine by including GM-CSF expression to improve the vaccine’s efficiency in inducing cytotoxic antitumour activity188. Dendritic cells can also be engineered to activate NK cells instead of antitumour T cells through expression of IL-15. Cancer cells lose major histocompatibility complex (MHC) class I molecules to avoid being killed by CD8+ T cells as an immune-evading mechanism. As part of immune surveillance and second defence line, NK cells recognize and kill cells that downregulate MHC class I189. In vitro experiments show that NK cells derived from healthy donors and acute myeloid leukaemia patients acquire cytotoxic activity against tumour cells after co-culture with IL-15-transpresenting dendritic cells190.
NK cells themselves can also be genetically modified in a more direct approach; for example, anti-CD19 CAR NK cells as treatment for CD19-positive cancers191. These CAR NK cells are modified to additionally express IL-15 to improve their expansion and persistence. A majority of patients in a phase I and II clinical trial (NCT03056339) of IL-15 CAR NK cells therapy showed response to the treatment, with seven out of eleven patients showing complete tumour remission, illustrating that CAR therapy can be developed with immune cell types beyond T cells. Similarly, CAR-macrophages can be engineered to express IFNγ along with the CAR construct, mediating a shift towards an M1 phenotype in the CAR-macrophages, which inhibits tumour growth192,193.
Myeloid cells drive the inhibitory environment in pre-metastatic niches, allowing metastases to develop. IL-12-expressing genetically engineered myeloid cells can modulate the immunosuppressive TME and prevent metastasis194. IL-12-expressing genetically engineered myeloid cells improve survival in multiple metastatic tumour models by inducing antigen presentation and T cell activation. Alternatively, a hydrogel encapsulating IL-2-producing retinal pigmented epithelial cells can be delivered in alginate-based capsules195, which can be implanted in the intraperitoneal cavity. Upon IL-2 release, the local concentration of IL-2 reaches therapeutically relevant levels, whereas systemic concentrations remain low enough to avoid IL-2-mediated toxicity. This approach results in decreased tumour volume and improved survival in ovarian and colorectal mouse models. Pharmacokinetic studies after intraperitoneal administration in non-human primates showed limited toxicity and an increase in CD8+ T cells195. Engineered mesenchymal stem cells are suitable for cytokine delivery in cancer immunotherapy because of their inherent tropism and preferential homing to tumours and low immunogenicity196,197.
Outlook
Immunotherapy has become a mainstay of treatment for numerous immune-mediated diseases, ranging from autoimmune and inflammatory disorders to cancer. New immune-based approaches such as monoclonal antibodies198, checkpoint inhibitor drugs199 and CAR T cells165 have improved and extended the lives of millions of patients. Cytokines are one of the most intensively studied immunology domains, and engineered cytokine-based therapies represent a new evolution of immunotherapeutics.
By the end of the twentieth century, cytokine-based drugs were among the earliest immunotherapies being developed for advanced treatment of cancer. Unfortunately, poor pharmacokinetic properties and systemic side effects have limited the applications of cytokine drugs, with a few notable exceptions, such as interferons for chronic hepatitis200 or multiple sclerosis201, rIL-2 in renal carcinoma202 and colony growth factors in neutropaenia203. However, the bioengineering advances described in this Review provide a developmental framework for major progress in the number and efficacy of cytokine-based therapies in the coming years. Advances in cytokine therapies are expected to emerge from two directions; first, technological innovations to re-engineer cytokines and deliver them to the site of pathological progress; and second, improved trial designs, sophisticated patient stratification and precision-medicine-based approaches. Both directions are needed to take advantage of new cytokine-based therapies in various diseases. As these research directions progress, scientists must bear in mind that protein engineering that deviates from endogenous molecules runs the risk of immunogenicity. For example, immunogenicity has been reported for an IL-2 immunocytokine125, and modifications to proteins, such as PEGylation, can cause similar problems204.
More approaches are expected to emerge to improve the activity, lifetime, pharmacokinetics and delivery of cytokines for therapeutic purposes. Current efforts focus mainly on developing cytokine receptor agonists; however, bioengineering can also improve therapies seeking to silence cytokine expression or dampen cytokine-receptor activation. Anakinra (recombinant IL-1RA) is a cytokine receptor antagonist that inhibits inflammation in a variety of diseases, such as rheumatoid arthritis, and hyperinflammation in severe infections, such as COVID-19 (ref. 33). This success paves the way for similar approaches targeting cytokine receptor pathways. Protein engineering technologies can be applied to design cytokine antagonists, such as an IL-2 mutein binding IL-2Rβ with improved affinity to prevent receptor dimerization and thereby effectively impair IL-2 and IL-15 activation205. This approach of preventing receptor dimerization can be used as a general strategy for antagonizing cytokine activity. In addition, although monoclonal antibodies, such as anti-TNFα31, anti-IL-6 (ref. 33) and anti-IL-17 (ref. 32), are established therapies for blocking cytokines, they often have poor pharmacokinetic profiles206. Technologies enabling the control of cytokine expression on a genetic level will probably arise as more effective cytokine therapy variants. Furthermore, emerging technologies, including single-cell analytical technologies or artificial intelligence (AI)-guided design, should continue to improve cytokine-based therapies (Box 1). These developments will lead to more rapid and more effective designs for cytokine-receptor agonists. Finally, increasing knowledge of nanoparticle structure–function relationships will lead to developments in nanomedicine-based drugs with improved biodistribution profiles and targeting capabilities.
More clinical trials on cytokine therapeutics are expected to be conducted, based on the recent successful experimental studies on cytokine-based technologies. The success of new therapies is expected to come not only from improved formulations, but also from innovative approaches to performing clinical trials. Furthermore, cytokine-based treatments will probably be combined with established therapies, such as checkpoint inhibitors or CAR T cells, or following surgery or radiation therapy. Going forward, patient stratification and precision medicine will represent one of the most important developments in modern treatment, and cytokine-based therapies are well suited to such approaches. Moreover, the technological challenges of clinical translation of cytokine-based therapies, such as cross-species reactivity, and regulatory processes, such as GMP manufacturing, should be considered (Box 1). Accompanying diagnostics to identify the patient subgroups most likely to respond to a certain type of cytokine therapy should be a goal of all commercial entities aiming at developing cytokine-based treatments. Additionally, precision medicine will probably be integrated into future technological developments, for example, for the design of personalized mRNA therapeutics. Together, bioengineering developments, innovative approaches to clinical testing and precision medicine will help to realize the potential of cytokine-based therapies for treating various diseases.
Author contributions
J.D., T.A., A.M.H., M.G.N. and W.J.M.M. wrote the manuscript. All authors researched data for the article, provided substantial contributions to discussion of content and reviewed and edited the manuscript before submission.
Peer review
Peer review information
Nature Reviews Bioengineering thanks Yizhou Dong, Frances Balkwill, Beatrice Malacrida and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
W.J.M.M., L.A.B.J. and M.G.N. are scientific co-founders of and have equity in Trained Therapeutix Discovery. W.J.M.M. and M.G.N. have consulting agreements with Trained Therapeutix Discovery. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jeroen Deckers, Tom Anbergen, Ayla M. Hokke.
Change history
3/27/2023
A Correction to this paper has been published: 10.1038/s44222-023-00057-1
Contributor Information
Mihai G. Netea, Email: mihai.netea@radboudumc.nl
Willem J. M. Mulder, Email: w.j.m.mulder@tue.nl
References
- 1.Chaplin DD. Overview of the immune response. J. Allergy Clin. Immunol. 2010;125:S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altan-Bonnet G, Mukherjee R. Cytokine-mediated communication: a quantitative appraisal of immune complexity. Nat. Rev. Immunol. 2019;19:205–217. doi: 10.1038/s41577-019-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley AC, Lacy P. Pathways for cytokine secretion. Physiology. 2010;25:218–229. doi: 10.1152/physiol.00017.2010. [DOI] [PubMed] [Google Scholar]
- 4.Leonardi CL, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–1674. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 5.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol. 2010;31:354–361. doi: 10.1016/j.it.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117:1162–1172. doi: 10.1378/chest.117.4.1162. [DOI] [PubMed] [Google Scholar]
- 9.Gharee-Kermani M, Pham S. Role of cytokines and cytokine therapy in wound healing and fibrotic diseases. Curr. Pharm. Des. 2001;7:1083–1103. doi: 10.2174/1381612013397573. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285:2944–2971. doi: 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 13.Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J. Clin. Invest. 2019;129:1441–1451. doi: 10.1172/JCI124606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith DE. IL-33: a tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin. Exp. Allergy. 2010;40:200–208. doi: 10.1111/j.1365-2222.2009.03384.x. [DOI] [PubMed] [Google Scholar]
- 15.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihle JN. Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 17.O’Shea, J. J., Gadina, M. & Siegel, R. M. in Clinical Immunology 5th edn Ch. 9 (eds Rich, R. R. et al.) 127–155.e1 (2019).
- 18.Arts RJW, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018;23:89–100.e5. doi: 10.1016/j.chom.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Guo L, Junttila IS, Paul WE. Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends Immunol. 2012;33:598. doi: 10.1016/j.it.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rousset F, Garcia E, Banchereau J. Cytokine-induced proliferation and immunoglobulin production of human B lymphocytes triggered through their CD40 antigen. J. Exp. Med. 1991;173:705–710. doi: 10.1084/jem.173.3.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 22.Ross ME, Caligiuri MA. Cytokine-induced apoptosis of human natural killer cells identifies a novel mechanism to regulate the innate immune response. Blood. 1997;89:910–918. doi: 10.1182/blood.V89.3.910. [DOI] [PubMed] [Google Scholar]
- 23.Lin JX, et al. The role of shared receptor motifs and common stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 24.Berraondo P, et al. Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120:6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozaki K, Leonard WJ. Cytokine and cytokine receptor pleiotropy and redundancy. J. Biol. Chem. 2002;277:29355–29358. doi: 10.1074/jbc.R200003200. [DOI] [PubMed] [Google Scholar]
- 26.Ruddy MJ, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimoto T, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-γ production. J. Immunol. 1998;161:3400–3407. doi: 10.4049/jimmunol.161.7.3400. [DOI] [PubMed] [Google Scholar]
- 28.Waldmann TA. The shared and contrasting roles of interleukin-2 (IL-2) and IL-15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol. Res. 2015;3:219. doi: 10.1158/2326-6066.CIR-15-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Royall JA, et al. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 1989;257:L399–L410. doi: 10.1152/ajplung.1989.257.6.L399. [DOI] [PubMed] [Google Scholar]
- 30.Hafler DA. Cytokines and interventional immunology. Nat. Rev. Immunol. 2007;7:423–423. doi: 10.1038/nri2101. [DOI] [Google Scholar]
- 31.Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF therapy: past, present and future. Int. Immunol. 2015;27:55–62. doi: 10.1093/intimm/dxu102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ly K, et al. Anti IL-17 in psoriasis. Expert Rev. Clin. Immunol. 2019;15:1185–1194. doi: 10.1080/1744666X.2020.1679625. [DOI] [PubMed] [Google Scholar]
- 33.van de Veerdonk FL, et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022;28:39–50. doi: 10.1038/s41591-021-01643-9. [DOI] [PubMed] [Google Scholar]
- 34.Herman, A. C., Boone, T. C. & Lu, H. S. in Formulation, Characterization, and Stability of Protein Drugs (eds Pearlman, R. & Wang, Y. J.) 303–328 (Springer, 2002).
- 35.Rasenack J, et al. Peginterferon alpha-2a (40kD) [Pegasys] improves HR-QOL outcomes compared with unmodified interferon alpha-2a [Roferon-A] Pharmacoeconomics. 2003;21:341–349. doi: 10.2165/00019053-200321050-00005. [DOI] [PubMed] [Google Scholar]
- 36.Baldo BA. Side effects of cytokines approved for therapy. Drug Safety. 2014;37:921–943. doi: 10.1007/s40264-014-0226-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golomb HM, et al. Alpha-2 interferon therapy of hairy-cell leukemia: a multicenter study of 64 patients. J. Clin. Oncol. 1986;4:900–905. doi: 10.1200/JCO.1986.4.6.900. [DOI] [PubMed] [Google Scholar]
- 38.Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908. doi: 10.1126/science.270.5238.908.a. [DOI] [PubMed] [Google Scholar]
- 39.Atkins MB, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 40.Saiki RK, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 41.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka K, et al. Protein and polymer analyses up to mlz 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass. Spectrom. 1988;2:151–153. doi: 10.1002/rcm.1290020802. [DOI] [Google Scholar]
- 44.Williamson MP, Havel TF, Wüthrich K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J. Mol. Biol. 1985;182:295–315. doi: 10.1016/0022-2836(85)90347-X. [DOI] [PubMed] [Google Scholar]
- 45.Woolfson MM. The development of structural x-ray crystallography. Phys. Scr. 2018;93:032501. doi: 10.1088/1402-4896/aa9c30. [DOI] [Google Scholar]
- 46.Matadeen R, Hon WC, Heath JK, Jones EY, Fuller S. The dynamics of signal triggering in a gp130-receptor complex. Structure. 2007;15:441–448. doi: 10.1016/j.str.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science. 2005;310:1159–1163. doi: 10.1126/science.1117893. [DOI] [PubMed] [Google Scholar]
- 48.Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14:63–77. doi: 10.1006/cyto.2001.0862. [DOI] [PubMed] [Google Scholar]
- 49.Johnson K, Granzow R, Creasey A, Ciardelli T. Ligand binding analyses and functional activity of interleukin-2 receptor ectodomains. Methods. 1994;6:199–205. doi: 10.1006/meth.1994.1022. [DOI] [Google Scholar]
- 50.Truneh A, et al. Temperature-sensitive differential affinity of TRAIL for its receptors: DR5 is the highest affinity receptor. J. Biol. Chem. 2000;275:23319–23325. doi: 10.1074/jbc.M910438199. [DOI] [PubMed] [Google Scholar]
- 51.Levin AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484:529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lumsden A, Wilkinson D. The promise of gene ablation. Nature. 1990;347:335–336. doi: 10.1038/347335a0. [DOI] [PubMed] [Google Scholar]
- 53.Kopf M, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 54.Kluger MJ, Kozak W, Leon LR, Conn CA. The use of knockout mice to understand the role of cytokines in fever. Clin. Exp. Pharmacol. Physiol. 1998;25:141–144. doi: 10.1111/j.1440-1681.1998.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 55.Ingman WV, Jones RL. Cytokine knockouts in reproduction: the use of gene ablation to dissect roles of cytokines in reproductive biology. Hum. Reprod. Update. 2008;14:179–192. doi: 10.1093/humupd/dmm042. [DOI] [PubMed] [Google Scholar]
- 56.McCaughtry TM, et al. Conditional deletion of cytokine receptor chains reveals that IL-7 and IL-15 specify CD8 cytotoxic lineage fate in the thymus. J. Exp. Med. 2012;209:2263–2276. doi: 10.1084/jem.20121505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Archambault LS, Trzilova D, Gonia S, Gale C, Wheeler RT. Intravital imaging reveals divergent cytokine and cellular immune responses to Candida albicans and Candida parapsilosis. mBio. 2019;10:e00266-19. doi: 10.1128/mBio.00266-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van de Donk PP, et al. Interleukin-2 PET imaging in patients with metastatic melanoma before and during immune checkpoint inhibitor therapy. Eur. J. Nucl. Med. Mol. Imaging. 2021;48:4369–4376. doi: 10.1007/s00259-021-05407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park LM, Lannigan J, Jaimes MC. OMIP‐069: forty‐color full spectrum flow cytometry panel for deep immunophenotyping of major cell subsets in human peripheral blood. Cytometry. 2020;97:1044. doi: 10.1002/cyto.a.24213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang T, Warden AR, Li Y, Ding X. Progress and applications of mass cytometry in sketching immune landscapes. Clin. Transl Med. 2020;10:e206. doi: 10.1002/ctm2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iyer A, Hamers AAJ, Pillai AB. CyTOF® for the masses. Front. Immunol. 2022;13:815828. doi: 10.3389/fimmu.2022.815828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cano-Gamez E, et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4+ T cells to cytokines. Nat. Commun. 2020;11:1801. doi: 10.1038/s41467-020-15543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang P, et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat. Methods. 2021;18:1181–1191. doi: 10.1038/s41592-021-01274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Netea MG, et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hahn N, et al. The orphan cytokine receptor CRLF3 emerged with the origin of the nervous system and is a neuroprotective erythropoietin receptor in locusts. Front. Mol. Neurosci. 2019;12:251. doi: 10.3389/fnmol.2019.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carter P. Site-directed mutagenesis. Biochemistry. 1986;237:1–7. doi: 10.1042/bj2370001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015;16:379–394. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 68.Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl Acad. Sci. USA. 2010;107:11906–11911. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Junttila IS, et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol. 2012;8:990–998. doi: 10.1038/nchembio.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mueller TD, Zhang J-L, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim. Biophys. Acta Mol. Cell Res. 2002;1592:237–250. doi: 10.1016/S0167-4889(02)00318-X. [DOI] [PubMed] [Google Scholar]
- 71.Gorby C, et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci. Signal. 2020;13:eabc0653. doi: 10.1126/scisignal.abc0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou T, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583:609–614. doi: 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine–receptor interactions from cytokine engineering. Annu. Rev. Immunol. 2015;33:139–167. doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saxton RA, et al. The tissue protective functions of interleukin-22 can be decoupled from pro-inflammatory actions through structure-based design. Immunity. 2021;54:660–672.e9. doi: 10.1016/j.immuni.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saxton RA, et al. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science. 2021;371:eabc8433. doi: 10.1126/science.abc8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mendoza JL, et al. Structure of the IFNγ receptor complex guides design of biased agonists. Nature. 2019;567:56–60. doi: 10.1038/s41586-019-0988-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glassman CR, et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell. 2021;184:983–999.e24. doi: 10.1016/j.cell.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carmenate T, et al. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J. Immunol. 2013;190:6230–6238. doi: 10.4049/jimmunol.1201895. [DOI] [PubMed] [Google Scholar]
- 79.Chen X, et al. A novel human IL-2 mutein with minimal systemic toxicity exerts greater antitumor efficacy than wild-type IL-2. Cell Death Dis. 2018;9:989. doi: 10.1038/s41419-018-1047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shanafelt AB, et al. A T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat. Biotechnol. 2000;18:1197–1202. doi: 10.1038/81199. [DOI] [PubMed] [Google Scholar]
- 81.Peterson LB, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J. Autoimmun. 2018;95:1–14. doi: 10.1016/j.jaut.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghelani A, et al. Defining the threshold IL-2 signal required for induction of selective Treg cell responses using engineered IL-2 muteins. Front. Immunol. 2020;11:1106. doi: 10.3389/fimmu.2020.01106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Visweswaraiah J, et al. Generation of PT101, a highly selective IL-2 mutein for treatment of autoimmune diseases. Ann. Rheum. Dis. 2021;80:13–13. doi: 10.1136/annrheumdis-2021-eular.2097. [DOI] [Google Scholar]
- 84.Khoryati L, et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 2020;5:eaba5264. doi: 10.1126/sciimmunol.aba5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Milton Harris J, Chess RB. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 86.Molineux G. Pegylation: engineering improved biopharmaceuticals for oncology. Pharmacotherapy. 2003;23:3S–8S. doi: 10.1592/phco.23.9.3S.32886. [DOI] [PubMed] [Google Scholar]
- 87.Meyers FJ, Paradise C, Scudder SA, Goodman G, Konrad M. A phase I study including pharmacokinetics of polyethylene glycol conjugated interleukin-2. Clin. Pharmacol. Ther. 1991;49:307–313. doi: 10.1038/clpt.1991.33. [DOI] [PubMed] [Google Scholar]
- 88.Mattijssen V, et al. Intratumoral PEG-interleukin-2 therapy in patients with locoregionally recurrent head and neck squamous-cell carcinoma. Ann. Oncol. 1994;5:957–960. doi: 10.1093/oxfordjournals.annonc.a058739. [DOI] [PubMed] [Google Scholar]
- 89.Emmerich J, et al. IL-10 directly activates and expands tumor-resident CD8+ T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72:3570–3581. doi: 10.1158/0008-5472.CAN-12-0721. [DOI] [PubMed] [Google Scholar]
- 90.Naing A, et al. PEGylated IL-10 (pegilodecakin) induces systemic immune activation, CD8+ T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. 2018;34:775–791.e3. doi: 10.1016/j.ccell.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ptacin JL, et al. An engineered IL-2 reprogrammed for anti-tumor therapy using a semi-synthetic organism. Nat. Commun. 2021;12:4785. doi: 10.1038/s41467-021-24987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Charych DH, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res. 2016;22:680–690. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 93.Dixit N, et al. NKTR-358: a novel regulatory T-cell stimulator that selectively stimulates expansion and suppressive function of regulatory T cells for the treatment of autoimmune and inflammatory diseases. J. Transl Autoimmun. 2021;4:100103. doi: 10.1016/j.jtauto.2021.100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bristol Myers Squibb. Nektar and Bristol Myers Squibb announce update on clinical development program for bempegaldesleukin (BEMPEG) in pdivo (nivolumab). BMShttps://news.bms.com/news/details/2022/Nektar-and-Bristol-Myers-Squibb-Announce-Update-on-Clinical-Development-Program-for-Bempegaldesleukin-BEMPEG-in-Combination-with-Opdivo-nivolumab/default.aspx (2022).
- 95.Miyazaki T, et al. NKTR-255, a novel polymer-conjugated rhIL-15 with potent antitumor efficacy. J. Immunother. Cancer. 2021;9:e002024. doi: 10.1136/jitc-2020-002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 97.Yang Q, Lai SK. Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdisc. Rev. Nanomed. Nanobiotechnol. 2015;7:655–677. doi: 10.1002/wnan.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sellaturay P, Nasser S, Islam S, Gurugama P, Ewan PW. Polyethylene glycol (PEG) is a cause of anaphylaxis to the Pfizer/BioNTech mRNA COVID-19 vaccine. Clin. Exp. Allergy. 2021;51:861–863. doi: 10.1111/cea.13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anishchenko I, et al. De novo protein design by deep network hallucination. Nature. 2021;600:547–552. doi: 10.1038/s41586-021-04184-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM. Low-N protein engineering with data-efficient deep learning. Nat. Methods. 2021;18:389–396. doi: 10.1038/s41592-021-01100-y. [DOI] [PubMed] [Google Scholar]
- 101.Pan X, Kortemme T. Recent advances in de novo protein design: principles, methods, and applications. J. Biol. Chem. 2021;296:100558. doi: 10.1016/j.jbc.2021.100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang P-S, Boyken SE, Baker D. The coming of age of de novo protein design. Nature. 2016;537:320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 103.Goldenzweig A, et al. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell. 2016;63:337–346. doi: 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Khersonsky O, et al. Automated design of efficient and functionally diverse enzyme repertoires. Mol. Cell. 2018;72:178–186.e5. doi: 10.1016/j.molcel.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Correia BE, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507:201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Silva DA, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565:186–191. doi: 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J, Cao H, Zhang JZH, Qi Y. Computational protein design with deep learning neural networks. Sci. Rep. 2018;8:6349. doi: 10.1038/s41598-018-24760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin E, Lin CH, Lane HY. De novo peptide and protein design using generative adversarial networks: an update. J. Chem. Inf. Model. 2022;62:761–774. doi: 10.1021/acs.jcim.1c01361. [DOI] [PubMed] [Google Scholar]
- 109.Leman JK, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods. 2020;17:665–680. doi: 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yen M, et al. Facile discovery of surrogate cytokine agonists. Cell. 2022;185:1414–1430.e19. doi: 10.1016/j.cell.2022.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moraga I, et al. Synthekines are surrogate cytokine and growth factor agonists that compel signaling through non-natural receptor dimers. eLife. 2017;6:22882. doi: 10.7554/eLife.22882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Emmerich J, et al. Abstract 1744: STK-012, an α/β selective IL-2 mutein for the activation of the antigen-activated T cells in solid tumor. Cancer Res. 2021;81:1744–1744. doi: 10.1158/1538-7445.AM2021-1744. [DOI] [Google Scholar]
- 114.Ren J, et al. Interleukin-2 superkines by computational design. Proc. Natl Acad. Sci. USA. 2022;119:e2117401119. doi: 10.1073/pnas.2117401119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harvill ET, Morrison SL. An IgG3–IL2 fusion protein activates complement, binds FcγRI, generates LAK activity and shows enhanced binding to the high affinity IL-2R. Immunotechnology. 1995;1:95–105. doi: 10.1016/1380-2933(95)00009-7. [DOI] [PubMed] [Google Scholar]
- 116.Peng LS, Penichet ML, Morrison SL. A single-chain IL-12 IgG3 antibody fusion protein retains antibody specificity and IL-12 bioactivity and demonstrates antitumor activity. J. Immunol. 1999;163:250–258. doi: 10.4049/jimmunol.163.1.250. [DOI] [PubMed] [Google Scholar]
- 117.Halin C, et al. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat. Biotechnol. 2002;20:264–269. doi: 10.1038/nbt0302-264. [DOI] [PubMed] [Google Scholar]
- 118.Castellani P, et al. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. Int. J. Cancer. 1994;59:612–618. doi: 10.1002/ijc.2910590507. [DOI] [PubMed] [Google Scholar]
- 119.Fallon J, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5:1869–1884. doi: 10.18632/oncotarget.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang L, et al. Imaging the alternatively spliced D domain of tenascin C in preclinical models of inflammatory bowel disease. Mol. Imaging Biol. 2022 doi: 10.1007/s11307-022-01758-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc. Natl Acad. Sci. USA. 2015;112:3320–3325. doi: 10.1073/pnas.1416159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spangler JB, et al. Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J. Immunol. 2018;201:2094–2106. doi: 10.4049/jimmunol.1800578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huyghe L, et al. Safe eradication of large established tumors using neovasculature-targeted tumor necrosis factor-based therapies. EMBO Mol. Med. 2020;12:e11223. doi: 10.15252/emmm.201911223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cauwels A, Tavernier J. Tolerizing strategies for the treatment of autoimmune diseases: from ex vivo to in vivo strategies. Front. Immunol. 2020;11:674. doi: 10.3389/fimmu.2020.00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hank JA, et al. Immunogenicity of the Hu14.18-IL2 immunocytokine molecule in adults with melanoma and children with neuroblastoma. Clin. Cancer Res. 2009;15:5923–5930. doi: 10.1158/1078-0432.CCR-08-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xu S, et al. The role of collagen in cancer: from bench to bedside. J. Transl Med. 2019;17:309. doi: 10.1186/s12967-019-2058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Momin N, et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl Med. 2019;11:2614. doi: 10.1126/scitranslmed.aaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Agarwal Y, et al. Intratumourally injected alum-tethered cytokines elicit potent and safer local and systemic anticancer immunity. Nat. Biomed. Eng. 2022;6:129–143. doi: 10.1038/s41551-021-00831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mansurov A, et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng. 2020;4:531–543. doi: 10.1038/s41551-020-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ishihara J, et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl Med. 2019;11:3259. doi: 10.1126/scitranslmed.aau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018;17:559–587. doi: 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- 132.López-Otín C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 133.Hsu EJ, et al. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat. Commun. 2021;12:2768. doi: 10.1038/s41467-021-22980-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.O’Neil J, et al. Tumor-selective activity of XTX202, a protein-engineered IL-2, in mice without peripheral toxicities in nonhuman primates. J. Clin. Oncol. 2021;39:2563–2563. doi: 10.1200/JCO.2021.39.15_suppl.2563. [DOI] [Google Scholar]
- 135.Mansurov A, et al. Masking the immunotoxicity of interleukin-12 by fusing it with a domain of its receptor via a tumour-protease-cleavable linker. Nat. Biomed. Eng. 2022;6:819–829. doi: 10.1038/s41551-022-00888-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nirschl C, et al. WTX-124 is a novel IL-2 pro-drug that is conditionally activated in tumors and drives antitumor immunity in murine syngeneic cancer models. J. Immunother. Cancer. 2021;9:A747–A747. doi: 10.1136/jitc-2021-SITC2021.718. [DOI] [Google Scholar]
- 137.Steiner P, et al. Conditionally activated IL-12 or IFNα indukineTM molecules inhibit syngeneic lymphoma tumor growth in mice, induce anti-tumor immune responses and are tolerated in non-human primates. Blood. 2021;138:2258–2258. doi: 10.1182/blood-2021-152758. [DOI] [Google Scholar]
- 138.Rosen DB, et al. TransConTM IL-2 β/γ: a novel long-acting prodrug of receptor-biased IL-2 designed for improved pharmocokinetics and optimal activation of T cells for the treatment of cancer [abstract 4507] Immunology. 2020;80:4507–4507. [Google Scholar]
- 139.Zhang Y, Li N, Suh H, Irvine DJ. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat. Commun. 2018;9:6. doi: 10.1038/s41467-017-02251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kamaly N, et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano. 2016;10:5280–5292. doi: 10.1021/acsnano.6b01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barbier AJ, Jiang AY, Zhang P, Wooster R, Anderson DG. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022;40:840–854. doi: 10.1038/s41587-022-01294-2. [DOI] [PubMed] [Google Scholar]
- 142.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front. Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nance KD, Meier JL. Modifications in an emergency: the role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent. Sci. 2021;7:748–756. doi: 10.1021/acscentsci.1c00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Andries O, et al. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release. 2015;217:337–344. doi: 10.1016/j.jconrel.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 145.Karikó K, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 147.Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021;6:1078–1094. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Alameh M-G, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–2892.e7. doi: 10.1016/j.immuni.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li C, et al. Mechanisms of innate and adaptive immunity to the Pfizer–BioNTech BNT162b2 vaccine. Nat. Immunol. 2022;23:543–555. doi: 10.1038/s41590-022-01163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li Y, et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer. 2020;1:882–893. doi: 10.1038/s43018-020-0095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu JQ, et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. J. Control. Release. 2022;345:306–313. doi: 10.1016/j.jconrel.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hewitt SL, et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36g, and OX40L mRNAs. Sci. Transl Med. 2019;11:9143. doi: 10.1126/scitranslmed.aat9143. [DOI] [PubMed] [Google Scholar]
- 153.Hotz C, et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci. Transl Med. 2021;13:eabc7804. doi: 10.1126/scitranslmed.abc7804. [DOI] [PubMed] [Google Scholar]
- 154.Jain R, et al. MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid. Ther. 2018;28:285–296. doi: 10.1089/nat.2018.0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Melero I, Castanon E, Alvarez M, Champiat S, Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 2021;18:558–576. doi: 10.1038/s41571-021-00507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Beck JD, et al. mRNA therapeutics in cancer immunotherapy. Mol. Cancer. 2021;20:69. doi: 10.1186/s12943-021-01348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT04455620 (2023).
- 158.Lai I, et al. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer. 2018;6:125. doi: 10.1186/s40425-018-0431-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lei S, et al. Efficient colorectal cancer gene therapy with IL-15 mRNA nanoformulation. Mol. Pharm. 2020;17:3378–3391. doi: 10.1021/acs.molpharmaceut.0c00451. [DOI] [PubMed] [Google Scholar]
- 160.Tahtinen S, et al. IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat. Immunol. 2022;23:532–542. doi: 10.1038/s41590-022-01160-y. [DOI] [PubMed] [Google Scholar]
- 161.Veiga N, et al. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018;9:4493. doi: 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kulkarni JA, et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021;16:630–643. doi: 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- 163.Cheng Q, et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020;15:313–320. doi: 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.van Leent MMT, et al. Regulating trained immunity with nanomedicine. Nat. Rev. Mater. 2022;7:465–481. doi: 10.1038/s41578-021-00413-w. [DOI] [Google Scholar]
- 165.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 166.Seimetz D, Heller K, Richter J. Approval of first CAR-Ts: have we solved all hurdles for ATMPs? Cell Med. 2019;11:215517901882278. doi: 10.1177/2155179018822781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Morotti M, et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer. 2021;124:1759–1776. doi: 10.1038/s41416-021-01353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gajewski TF, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 169.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017;7:10541. doi: 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang L, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl Acad. Sci. USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Adachi K, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 175.Andersen R, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin. Cancer Res. 2016;22:3734–3745. doi: 10.1158/1078-0432.CCR-15-1879. [DOI] [PubMed] [Google Scholar]
- 176.Lee JM, et al. Direct and indirect antitumor effects by human peripheral blood lymphocytes expressing both chimeric immune receptor and interleukin-2 in ovarian cancer xenograft model. Cancer Gene Ther. 2010;17:742–750. doi: 10.1038/cgt.2010.30. [DOI] [PubMed] [Google Scholar]
- 177.Sockolosky JT, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359:1037–1042. doi: 10.1126/science.aar3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang Q, et al. A human orthogonal IL-2 and IL-2Rβ system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl Med. 2021;13:6986. doi: 10.1126/scitranslmed.abg6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hirai T, et al. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J. Clin. Invest. 2021;131:e139991. doi: 10.1172/JCI139991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Taylor PA, et al. L-selectinhi but not the L-selectinlo CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. 2004;104:3804–3812. doi: 10.1182/blood-2004-05-1850. [DOI] [PubMed] [Google Scholar]
- 181.Mo F, et al. An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature. 2021;597:544–548. doi: 10.1038/s41586-021-03861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pohl-Guimarães F, et al. RNA-modified T cells mediate effective delivery of immunomodulatory cytokines to brain tumors. Mol. Ther. 2019;27:837–849. doi: 10.1016/j.ymthe.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Roybal KT, et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell. 2016;167:419–432.e16. doi: 10.1016/j.cell.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yao W, et al. Intratumoral injection of dendritic cells overexpressing interleukin-12 inhibits melanoma growth. Oncol. Rep. 2019;42:370–376. doi: 10.3892/or.2019.7165. [DOI] [PubMed] [Google Scholar]
- 185.Minkis K, et al. Type 2 bias of T cells expanded from the blood of melanoma patients switched to type 1 by IL-12p70 mRNA-transfected dendritic cells. Cancer Res. 2008;68:9441–9450. doi: 10.1158/0008-5472.CAN-08-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tatsumi T, et al. Intratumoral delivery of dendritic cells engineered to secrete both interleukin (IL)-12 and IL-18 effectively treats local and distant disease in association with broadly reactive Tc1-type immunity 1. Cancer Res. 2003;63:6378–6386. [PubMed] [Google Scholar]
- 187.Bontkes HJ, et al. Dendritic cells transfected with interleukin-12 and tumor-associated antigen messenger RNA induce high avidity cytotoxic T cells. Gene Ther. 2007;14:366–375. doi: 10.1038/sj.gt.3302874. [DOI] [PubMed] [Google Scholar]
- 188.Naka T, et al. Tumor vaccine therapy against recrudescent tumor using dendritic cells simultaneously transfected with tumor RNA and granulocyte macrophage colony-stimulating factor RNA. Cancer Sci. 2008;99:407–413. doi: 10.1111/j.1349-7006.2007.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ljunggren HG, Kärre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-S. [DOI] [PubMed] [Google Scholar]
- 190.van den Bergh J, et al. Transpresentation of interleukin-15 by IL-15/IL-15Rα mRNA-engineered human dendritic cells boosts antitumoral natural killer cell activity. Oncotarget. 2015;6:44123–44133. doi: 10.18632/oncotarget.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu E, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kang M, et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv. Mater. 2021;33:2103258. doi: 10.1002/adma.202103258. [DOI] [PubMed] [Google Scholar]
- 193.Wang S, et al. CAR-macrophage: an extensive immune enhancer to fight cancer. eBioMedicine. 2022;76:103873. doi: 10.1016/j.ebiom.2022.103873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kaczanowska S, et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell. 2021;184:2033–2052.e21. doi: 10.1016/j.cell.2021.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nash AM, et al. Clinically translatable cytokine delivery platform for eradication of intraperitoneal tumors. Sci. Adv. 2022;8:1032. doi: 10.1126/sciadv.abm1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mosallaei M, et al. Genetically engineered mesenchymal stem cells: targeted delivery of immunomodulatory agents for tumor eradication. Cancer Gene Ther. 2020;27:854–868. doi: 10.1038/s41417-020-0179-6. [DOI] [PubMed] [Google Scholar]
- 197.Razeghian E, et al. Mesenchymal stem/stromal cells as a vehicle for cytokine delivery: an emerging approach for tumor immunotherapy. Front. Med. 2021;8:1405. doi: 10.3389/fmed.2021.721174. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 198.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 199.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 200.Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 201.Panitch HS. Interferons in multiple sclerosis. Drugs. 1992;44:946–962. doi: 10.2165/00003495-199244060-00004. [DOI] [PubMed] [Google Scholar]
- 202.Fyfe G, et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995;13:688–696. doi: 10.1200/JCO.1995.13.3.688. [DOI] [PubMed] [Google Scholar]
- 203.Bennett CL, Djulbegovic B, Norris LB, Armitage JO. Colony-stimulating factors for febrile neutropenia during cancer therapy. N. Engl. J. Med. 2013;368:1131–1139. doi: 10.1056/NEJMct1210890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhang P, Sun F, Liu S, Jiang S. Anti-PEG antibodies in the clinic: current issues and beyond PEGylation. J. Control. Release. 2016;244:184–193. doi: 10.1016/j.jconrel.2016.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mitra S, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity. 2015;42:826–838. doi: 10.1016/j.immuni.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24:23–39. doi: 10.2165/11530560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 207.Ghoreschi K, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat. Med. 2002;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 208.Tulpule A, et al. Interleukin-4 in the treatment of AIDS-related Kaposi’s sarcoma. Ann. Oncol. 1997;8:79–83. doi: 10.1023/A:1008205424763. [DOI] [PubMed] [Google Scholar]
- 209.Sportès C, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 2008;205:1701–1714. doi: 10.1084/jem.20071681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rosenberg SA, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 2006;29:313–319. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Buruiana FE, Solà I, Alonso-Coello P. Recombinant human interleukin 10 for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2010;2012:CD005109. doi: 10.1002/14651858.CD005109.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Colombel JF, et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut. 2001;49:42–46. doi: 10.1136/gut.49.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chernoff AE, et al. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J. Immunol. 1995;154:5492–5499. doi: 10.4049/jimmunol.154.10.5492. [DOI] [PubMed] [Google Scholar]
- 214.Conlon KC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015;33:74–82. doi: 10.1200/JCO.2014.57.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Petrella TM, et al. Final efficacy results of NCIC CTG IND.202: a randomized phase II study of recombinant interleukin-21 (rIL21) in patients with recurrent or metastatic melanoma (MM) J. Clin. Oncol. 2013;31:9032–9032. doi: 10.1200/jco.2013.31.15_suppl.9032. [DOI] [Google Scholar]
- 216.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00873756 (2016).
- 217.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00508625 (2016).
- 218.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00400764 (2011).
- 219.Pina AS, Lowe CR, Roque ACA. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol. Adv. 2014;32:366–381. doi: 10.1016/j.biotechadv.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Malyala P, Singh M. Endotoxin limits in formulations for preclinical research. J. Pharm. Sci. 2008;97:2041–2044. doi: 10.1002/jps.21152. [DOI] [PubMed] [Google Scholar]
- 221.Freitag, R. in Animal Cell Biotechnology Vol. 1104 (ed. Pörtner, R.) 419–458 (Humana, 2014).