

Abstract
Aberrant expression of protein kinase C (PKC) isozymes is a hallmark of cancer. The different members of the PKC family control cellular events associated with cancer development and progression. Whereas the classical/conventional PKCα isozyme has been linked to tumor suppression in most cancer types, here we demonstrate that this kinase is required for the mitogenic activity of aggressive human prostate cancer cells displaying aberrantly high PKCα expression. IHC analysis showed abnormal upregulation of PKCα in human primary prostate tumors. Interestingly, silencing PKCα expression from aggressive prostate cancer cells impairs cell-cycle progression, proliferation, and invasion, as well as their tumorigenic activity in a mouse xenograft model. Mechanistic analysis revealed that PKCα exerts a profound control of gene expression, particularly over genes and transcriptional networks associated with cell-cycle progression and E2F transcription factors. PKCα RNAi depletion from PC3 prostate cancer cells led to a reduction in the expression of proinflammatory cytokine and epithelial-to-mesenchymal transition (EMT) genes, as well as a prominent downregulation of the immune checkpoint ligand PD-L1. This PKCα-dependent gene expression profile was corroborated in silico using human prostate cancer databases. Our studies established PKCα as a multifunctional kinase that plays pleiotropic roles in prostate cancer, particularly by controlling genetic networks associated with tumor growth and progression. The identification of PKCα as a protumorigenic kinase in human prostate cancer provides strong rationale for the development of therapeutic approaches toward targeting PKCα or its effectors.
Significance:
PKCα was found to be aberrantly expressed in human prostate cancer. Silencing the expression of this kinase from aggressive prostate cancer cell lines reduces their proliferative, tumorigenic, and invasive properties. In addition, our findings implicate PKCα as a major node for transcriptional regulation of tumorigenic, inflammatory, and EMT networks in prostate cancer, highlighting its potential relevance as a therapeutic target.
Introduction
Prostate cancer is the second leading cause of cancer-related deaths among men in the United States, with approximately 268,500 new cases and approximately 34,600 projected deaths in the United States for 2022 (1). Although patients with prostate cancer generally respond favorably to available therapies, the disease eventually progresses to metastatic castration-resistant prostate cancer (mCRPC), which is hard to eradicate and is ultimately lethal (2, 3). The initiation and progression of prostate cancer is driven by genetic and epigenetic changes leading to dysregulation of oncogenic signal transduction pathways mostly linked to the acquisition of proliferative and invasive traits. Hallmarks of prostate cancer include the functional inactivation and/or deletion of tumor suppressors (e.g., PTEN, NKX3.1), genomic rearrangements (e.g., TMPRSS2-ERG), altered growth factor signaling (e.g., IGF-1R, FGFR, ErbB receptors), and aberrant signaling (e.g., PI3K/Akt, Src; refs. 4–6).
The protein kinase C (PKC family) has been recognized as a major player in the progression of multiple cancers, including prostate cancer. Members of the three PKC classes (“conventional/classical” cPKCs α, β, and γ; “novel” nPKCs δ, ε, η, and θ; and “atypical” aPKCs ζ and ι) have been widely implicated in fundamental cancer-driving events, such as cell proliferation, survival, motility, and invasion. cPKCs and nPKCs, the PKCs activated by the lipid second messenger diacylglycerol (DAG) and phorbol esters, have a remarkably high functional complexity. Indeed, these kinases could wield either tumor-promoting or tumor-suppressive activities depending on the cell type (7–9). While PKC isozymes are rarely mutated in cancer, upregulation of prooncogenic PKCs and downregulation of tumor-suppressing PKCs are common events in epithelial tumors. This distorted pattern may result in rerouting DAG signals through abnormally overexpressed PKCs, ultimately boosting mitogenic and survival responses through oncogenic signaling pathways such as ERK, NFκB, PI3K, and STAT3 (10). Aberrant PKC isozyme expression has been also linked to epithelial-to-mesenchymal transition (EMT), a dynamic process by which cancer cells acquire invasive properties (11–14). In prostate cancer, PKCε expression is causally associated with disease initiation and progression, as previously established using transgenic mouse models. Conversely, overexpression of other DAG-regulated PKCs in the mouse prostate, namely PKCα and PKCδ, failed to confer a tumorigenic phenotype (15–17). Consistent with these in vivo studies, PKCα and PKCδ mediate proapoptotic responses in androgen-dependent prostate cancer cellular models, such as LNCaP cells (18–20). Indeed, there is a vast literature describing PKCα as a tumor-suppressive kinase in several cancers, such as lung, colon, endometrial, and skin cancer (21–26). Intriguingly, PKCα has also been reported to be tumor-promoting kinase, for example in triple-negative breast cancer (26–28). This dichotomy, together with the limited information available in models of prostate cancer, prompted us to investigate the role of DAG-regulated PKCα in this malignancy in further detail.
Here, we show that PKCα is upregulated in primary prostate cancer, and its elevated levels in aggressive prostate cancer cell lines causally associate with proliferative, tumorigenic, and invasive behaviors. We found PKCα to be at the core of gene transcriptional networks associated with the progression of prostate cancer, playing a stringent control over mitogenic, invasive, inflammatory, and tumor evasion gene expression signatures.
Materials and Methods
Cell Lines and RNAi Interference
Authenticated human prostate cancer cells were obtained from ATCC and cultured in RPMI medium supplemented with 10% FBS, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin). Cells are tested for Mycoplasma at least twice a year, and normally used at low passage (generally <10 passages).
For the generation of stably depleted PKCα cell lines, we used a short hairpin RNA (shRNA) lentiviral approach. Cells were infected with MISSION shRNA lentiviruses (Sigma-Aldrich) designed for the human PRKCA gene (TRCN0000196730, TRCN0000233512), and followed by puromycin selection, as described previously (29).
For transient silencing of PKCα, we used three different siRNA duplexes (#1 and #2 as in ref. 12, #3 from Dharmacon, catalog no. J-003523-18-0005). As non-target control (NTC), we used D-001810-02-05 ON-TARGETplus nontargeting siRNA. siRNA duplexes were transfected using Lipofectamine RNAi Max (Invitrogen), as described previously (30, 31).
Western Blots
Western blots were done essentially as described previously (31). Briefly, cells were harvested in lysis buffer containing 50 mmol/L Tris-HCl, pH 6.8, 10% glycerol, and 2% β-mercaptoethanol. Cell lysates were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore Corporation). After blocking with 5% milk or 5% BSA in TBS/0.1% Tween for 1 hour, membranes were incubated overnight with the following primary antibodies: anti-PKCα anti-PKCδ, anti-PKCε (Cell Signaling Technology, catalog nos. 2056, #2058, and #2083, respectively), anti-phospho-Rb (Cell Signaling Technology, catalog no. 2181), anti-caspase-3 (Cell Signaling Technology, catalog no. 9662), anti-caspase-9 (Cell Signaling Technology, catalog no. 9508), anti-PARP1 (Cell Signaling Technology, catalog no. 9532), anti-vimentin (Cell Signaling Technology, catalog no. 5741), anti-Zeb1 (Cell Signaling Technology, catalog no. 3396), anti-AXL (Cell Signaling Technology, catalog no. 8661), anti-E-cadherin (R&D, catalog no. AF748), anti-PD-L1 (Cell Signaling Technology, catalog no. 13684), anti-vinculin (Sigma-Aldrich, catalog no. V9131), or β-actin (Sigma-Aldrich, catalog no. A5441). Membranes were then incubated for 1 hour with either anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase (Bio-Rad Laboratories). Bands were visualized and subjected to densitometric analysis using an Odyssey Fc system (LI-COR Biotechnology).
Cell Proliferation and Viability Assay
Cell number was determined using a Bio-Rad TC20 automated cell counter. Cell viability was assessed using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay in 96-well plates. Absorbance at 570 nm was determined in a plate-reader spectrophotometer (EMax Plus Microplate Reader, Molecular Devices Inc.).
Invasion Assay
A Boyden chamber assay was used, as described previously (17, 29). Briefly, cells were trypsinized, suspended in 0.1% BSA/RPMI, and seeded (2.5 × 104 cells/well) in the upper compartment of a Boyden chamber (NeuroProbe). A 12-μm-pore Matrigel-coated polycarbonate membrane was used to separate the upper and lower compartments. In the lower chamber, RPMI medium containing 10% FBS was used. After an incubation period of 16 hours at 37°C, membranes were recovered and cells on the upper side of the membrane (nonmigrating) were wiped off the surface. Migrating cells on the lower side of the membrane were fixed and stained with the Hema 3 Staining kit (Thermo Fisher Scientific). Migrating cells in each well were counted in five random fields by contrast microscopy using an Eclipse E200 Nikon microscopy (4X magnification) and the ImageJ/Fiji software.
Tumorigenesis in Nude Mice
Male athymic nude mice (Foxn1nu/Foxn1nu) were purchased from Harlan Laboratories. Animals were maintained in a temperature-controlled room located at the University of Pennsylvania School of Veterinary Medicine (Philadelphia, PA) and fed ad libitum. All animal studies were carried out in strict accordance with the University of Pennsylvania Institutional Animal Care and Use Committee guidelines.
PC3 cells (1 × 105) were injected subcutaneously into the flanks of 6-week-old male athymic nude mice (Foxn1nu/Foxn1nu). Tumor formation was monitored for 37 days. Tumor volume was determined with caliper measurements and calculated using the formula L1 × L2 × H × 0.5238, where L1 is the long diameter, L2 is the short diameter, and H is the height of the tumor.
RNA Isolation and Real-time Quantitative PCR
Total RNA was extracted from subconfluent plates using the RNeasy kit as directed by the manufacturer (Qiagen), as described previously (31). Briefly, 1 μg of RNA per sample was reverse transcribed using the TaqMan reverse transcription reagent kit and random hexamers as primers (Applied Biosystems). Primers for individual genes were purchased from Applied Biosystems. PCR amplifications were performed using an ABI PRISM 7300 Detection System in a total volume of 20 μL containing Taqman Universal PCR Master Mix (Applied Biosystems), commercial target primers (300 nmol/L), the fluorescent probe (200 nmol/L), and 1 μL of cDNA. PCR product formation was continuously monitored using the Sequence Detection System software version 1.7 (Applied Biosystems). The FAM signal was normalized to endogenous UBC (housekeeping gene).
RNA Sequencing Data Analysis
RNA was isolated from triplicate subconfluent plates using the RNeasy kit. RNA concentration and integrity were measured on an Agilent 2100 Bioanalyzer (Agilent Technologies). Only RNA samples with RNA integrity values (RIN) over 8.0 were considered for subsequent analysis. One of the triplicate samples in the PKCα2 siRNA group behaved as an outlier based on unsupervised analysis of the transcriptomic data, and therefore this group remained as duplicate samples. RNA samples were processed for directional RNA sequencing (RNA-seq) library construction and sequencing at the Next-Generation Sequencing (NGS) Core facility of the Perelman School of Medicine, University of Pennsylvania (Philadelphia, PA). We performed 100 nt singled-end sequencing using an Illumina HiSeq4000 platform and obtained approximately 20 million reads per sample. The short-sequenced reads were mapped to the human reference genome (hg19) with the splice junction aligner Rsubread R/Bioconductor package (v2.6.3). We employed featureCounts function to calculate the gene expression abundance using the aligned BAM files. To identify differentially expressed genes (log2 fold change > ±1, FDR < 0.001) between the three different siRNA duplexes (α1, α2, or α3) with the NTC siRNA and parental cells, we employed the edgeR R/Bioconductor package based on the normalized log2 based count per million values.
For functional enrichment analyses (FEA), we used ClueGo Cytoscape's plug-in (http://www.cytoscape.org/) and the InnateDB resource (http://www.innatedb.com/) based on the list of dysregulated transcripts. Gene set enrichment analysis (GSEA) of transcription factor binding sites (TFBS) was performed with the R/Bioconductor package clusterProfiler based on the TFBS signature obtained from MSigDB (c3.tft.v7.4.entrez.gmt). RNA-seq expression profiles of 497 primary prostate adenocarcinomas from The Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) project were downloaded from the UCSD-Xena resource (https://xena.ucsc.edu/). Prostate carcinomas were divided into low (n = 75) or high (n = 134) PRKCA expression levels according to the StepMiner one-step algorithm for further analysis of PKCα-modulated genes. Data integration and visualization of differentially expressed transcripts were done with R/Bioconductor and the MultiExperiment Viewer software (MeV v4.9).
Correlation Analyses
To explore associations between PRKCA mRNA levels and EMT phenotype of primary prostate carcinomas, EMTome resource (http://www.emtome.org/) was employed to extract three relevant EMT gene expression signatures identified in invasive prostate carcinomas (32–34). PKCα mRNA expression levels, the EMT gene expression signatures, and their derived EMT scores from TCGA-PRAD dataset were directly retrieved and visualized using the UCSC Xena resource (https://xenabrowser.net/). PKCα mRNA levels and EMT scores for each of the EMT signatures across all primary invasive prostate carcinomas were used for correlation analysis with R software. EMT scores were computed as an average weighted sum of the gene expression levels that constitute each EMT signature.
In a separate association analysis between PKCα expression and androgen receptor (AR), we carried out a meta-analysis of correlation coefficients obtained from eight independent prostate cancer datasets using the DerSimonian-Laird (DSL) random-effect method with correlation coefficients as effect sizes. Briefly, PRKCA and AR mRNA profiles from primary prostate carcinomas were retrieved from Gene Expression Omnibus (GSE41967 and GSE70768), CancerTool (GSE3325, GSE3933, GSE21032, GSE35988, and MSKCC) and UCSC Xena (GDC-TCGA-PRAD) resources. Pearson correlation coefficients were independently computed from each study in R and subsequently integrated into a meta-analysis using metacor R package.
Cytokine Measurements
PC3 cells were transfected with siRNA duplexes for PKCα or NTC. After 48 hours, medium was replaced, and conditioned medium (CM) was collected after 12 hours (IL8/CXCL8) or 24 hours (GRO). Cytokines in the CM were determined using the Quantikine ELISA kit for human IL8 (R&D Systems) and the human GRO ELISA kit (Invitrogen), following the protocols provided by the manufacturers.
Tissue Microarray and IHC
For each case of prostatic adenocarcinoma, one slide most representative of the overall Gleason score and stage was identified. The corresponding paraffin-embedded tissue block was used to construct a tissue microarray (TMA) using 1 mm coring needle (Beecher Tissue Arrayer MTA-1). All 88 cases were represented at least in triplicate resulting in production of four TMA blocks that contained a total of 284 cores. Non-neoplastic liver, kidney, spleen, and tonsil of patients operated for diseases other than cancer was also included in the same blocks as controls. For staining, we used an anti-PKCα antibody (Abcam, ab32376, clone Y124, a validated antibody for IHC in human specimens) at a 1:1,000 dilution. Staining was performed on a Leica Bond-IIITM instrument using the Bond Polymer Refine Detection System (Leica Microsystems DS9800). Heat-induced epitope retrieval was performed for 20 minutes with ER2 solution (Leica Microsystems AR9640). All the experiments were performed at room temperature. For calculation of the H score, the intensity of staining was graded on a scale of 0 to 3, with 0 being no staining and 3 being strongest staining. The percentage of tumor cells staining was calculated over multiple high-power fields in the region over the entire tumor. The staining was normalized to compare different tumors by multiplying the intensity of staining with the percentage of tumor cells staining.
Statistical Analysis
For most experiments, statistical analysis (Student t test, ANOVA) was performed using GraphPad Prism software built-in analysis tools. A P value < 0.05 was considered statistically significant.
Data Availability Statement
Raw data for this study were generated at the NGS Core facility of the Perelman School of Medicine, University of Pennsylvania (Philadelphia, PA). Derived data supporting the findings of this study are available in Supplementary Table S1. Other data generated in this study are available within the article and its Supplementary Data.
Results
Upregulation of PKCα in Human Prostate Cancer
The aberrant PKC signaling observed in multiple cancer types foresees important roles for this pathway in disease progression (7, 8), as we previously established for PKCε in prostate cancer (15–17). PKCα, a DAG-responsive PKC with dual effects either as a tumor promoter or suppressor (21–28), has been poorly studied in prostate cancer. Examination of PKCα expression in established prostate cancer cell models showed a prominent upregulation in PC3, PC3-ML, and DU145 prostate cancer cell lines compared with less aggressive LNCaP cells and its sublines (C4, C4-2) or 22RV1 cells (Fig. 1A).
FIGURE 1.

PKCα upregulation in aggressive prostate cancer cells. A, Left, PKCα expression in prostate cancer cells, as determined by Western blot analysis. Right, Densitometric analysis. Results are expressed as mean ± SEM (n = 4). B, Representative IHC staining of PKCα in prostate cancer specimens. Left, Prostate cancer with low PKCα expression. Right, Prostate cancer with high PKCα expression. C, Enhanced view of PKCα staining in a prostate cancer specimen. D,H score is plotted for normal and cancer areas stainings with an anti-PKCα antibody. The mean and P value are indicated.
Next, we took advantage of a TMA comprising prostate cancer specimens from 88 patients and evaluated PKCα staining by IHC using an anti-PKCα antibody previously validated in PKCα wild-type versus knockout mice (25), followed by a quantitative analysis reported as H score. We observed heterogenous PKCα staining across different specimens, with cases displaying very high staining and others with weak or no staining. Representative examples of TMA micrographs are shown in Fig. 1B. A detailed analysis revealed low or no intensity staining in nontumor areas, whereas PKCα staining was primarily detected in those areas defined as adenocarcinoma. An enhanced view is shown in Fig. 1C. Quantitative analysis of PKCα immunostaining in tumor areas relative to normal areas is depicted in Fig. 1D.
PKCα Mediates Human Prostate Cancer Cell Growth
To begin elucidating the functional consequences of PKCα upregulation in aggressive cellular models of prostate cancers, we first analyzed the effect of PKCα RNAi silencing on proliferation. Three different siRNA duplexes were used to knockdown PKCα in PC3 and DU145 cell lines. These duplexes caused >80% PKCα depletion in all cases without affecting the expression of PKCδ and PKCε, the other DAG-responsive PKCs expressed in these cells (Fig. 2A). PKCα depletion lasted for at least >120 hours posttransfection (Supplementary Fig. S1). Silencing PKCα conferred slower growth rate properties to both PC3 (Fig. 2B, top) and DU145 cells (Fig. 2B, bottom) when compared with parental cells or cells subjected to NTC RNAi. Cell-cycle distribution analysis using FACS revealed an accumulation of PKCα-depleted PC3 and DU145 cells in G0–G1, with a subsequent reduction in S-phase (Fig. 2C). 5-Ethynyl-2′-deoxyuridine (EdU) incorporation assays showed a significant decrease in the fraction of cells undergoing DNA synthesis in PKCα-depleted PC3 or DU145 cells relative to control cells (Fig. 2D). Consistent with the inhibition in G1 to S transition, a prominent Rb dephosphorylation was observed in PKCα knockdown cells (Supplementary Fig. S2A). We were unable to detect any significant changes in the expression of apoptotic markers, namely cleaved PARP1, cleaved caspase-3, and cleaved caspase-9, in PC3 or DU145 cells subjected to PKCα RNAi depletion (Fig. 2E). There was no significant sub-G0–G1 population in the FACS analysis in PKCα-depleted cells, consistent with the absence of apoptotic cells (Supplementary Fig. S2B). In addition, there was no additive growth inhibitory effect between PKCα RNAi and treatment with either the dual Cdk4/6 inhibitor palbociclib/PD 0332991 (Supplementary Fig. S3A) or the Cdk4 inhibitor NSC 625987 (Supplementary Fig. S3B). Altogether, these results strongly support the requirement of PKCα for mitogenesis in aggressive prostate cancer cells.
FIGURE 2.

Silencing PKCα from aggressive prostate cancer cells induces cell growth arrest. PC3 or DU145 cells were transfected with three different PKCα (α1, α2, or α3) or NTC siRNA duplexes. Twenty-four hours later, cells were serum starved for 24 hours and experiments carried out at the indicated times. P, parental. A, Expression of PKC isozymes was determined by Western blot analysis at day 3 after transfection. Representative Western blots are shown. B, Effect of PKCα RNAi depletion on cell number. Left, Representative micrographs 1–5 days after transfection of siRNA duplexes. Right, Cell number was expressed as mean ± SD (n = 3). An additional experiment gave similar results. C, Percentage of cells in S-phase 48 hours after transfection with siRNA duplexes, as determined by flow cytometry in EdU-labeled cells. Results are expressed as mean ± SE (n = 3). D, Cell-cycle distribution was determined by flow cytometry 48 hours after transfection with siRNA duplexes. Left, Representative flow cytometry charts. Right, Cell-cycle distribution in a representative experiment. E, Expression of apoptosis markers (cleaved PARP1, cleaved caspase 3, and cleaved caspase 9). A representative experiment is shown. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 versus NTC.
Next, to examine whether PKCα is involved in in vivo prostate cancer tumor growth, we generated stably depleted PKCα PC3 cell lines using shRNA lentiviruses. Like the siRNA-mediated PKCα transient approach, stable knockdown was highly specific for PKCα without significant changes in the expression of other DAG-regulated PKCs (Fig. 3A). Upon subcutaneous inoculation of parental PC3 cells into nude mice, a tumor growth response was readily observed. Notably, PKCα-depleted PC3 cells displayed a significant reduction in tumor growth compared with parental PC3 cells or cells subjected to NTC RNAi, as determined by measurements of tumor volume (Fig. 3B and C) and weight (Fig. 3D). PKCα-depleted tumors exhibit reduced Ki67 staining, a marker for mitotic index, as well as diminished phospho-Erk staining compared to tumors from parental or NTC PC3 cells. We detected TUNEL-positive areas in PKCα-depleted tumors (Fig. 3E), suggesting that PKCα may confer to some degree an anti-apoptotic response in an in vivo setting.
FIGURE 3.

Silencing PKCα reduces PC3 cell tumorigenic activity in nude mice. PC3 cells were subjected to stable PKCα depletion using two different shRNA lentiviruses, followed by puromycin selection. Cells were inoculated subcutaneously into nude mice, and tumor formation was followed for the time indicated in the figure. A, Expression of PKC isozymes in the inoculated cell lines, as determined by Western blot analysis. B, Pictures of tumors isolated from all mouse cohorts. C, Time-course analysis of tumor formation Results are expressed as mean ± SD (n = 8). D, Tumor weight was determined after sacrificing mice at day 37 postinoculation with the different PC3 cell lines. Results are expressed as mean ± SD (n = 8). E, Representative IHC images for Ki67, phospho-ERK, and TUNEL. NTC, non-target control shRNA lentivirus. ****, P < 0.0001 versus NTC.
PKCα Expression Correlates with EMT Markers and is Required for Prostate Cancer Cell Invasion
Recent studies linked PKC isozymes to EMT in a variety of tumor types (11–14, 35). We used the Cancer Cell Line Encyclopedia (CCLE) to investigate whether there is an association between the expression of PKCα and markers of the mesenchymal phenotype in prostate cancer cell lines. This analysis revealed a striking positive correlation between the expression of PKCα (PRKCA gene) and mesenchymal markers vimentin, ZEB1, ZEB2, and AXL in prostate cancer cell lines (Fig. 4A). A similar positive trend between PKCα and EMT markers was detected in human prostate cancer specimens upon inquire of TCGA-PRAD dataset (Fig. 4B). To further explore the association between PRKCA and EMT phenotype, EMTome resource (http://www.emtome.org/) was employed to extract three relevant EMT gene expression signatures identified in prostate carcinomas (32–34). A significant positive correlation between PRKCA and prostate cancer EMT signatures was observed in all cases (Fig. 4C). Proof of principle for the association between PKCα expression and EMT was established by Western blot analysis using LNCaP cell lines and 22RV1 as an “epithelial” model and PC3/PC3-ML/DU145 cells as “mesenchymal” models. PC3, PC3-ML, and DU145 cell lines, which aberrantly express PKCα, displayed high expression of mesenchymal markers vimentin and AXL as well as E-cadherin downregulation, whereas the opposite was true for LNCaP/22RV1 cell lines (Fig. 4D).
FIGURE 4.

PKCα association with prostate cancer cell line EMT and invasiveness. A, Correlations between PKCα (PRKCA) and EMT markers from the CCLE database. ●, DU145; ▲, PC3; ■, NCI H660; ●, VCaP; ▲, 22RV1; ■, LNCaP. B, Correlations between PKCα (PRKCA) and EMT markers from TCGA-PRAD human dataset (498 patients). D, Representative Western blot analysis for PKCα and EMT markers in prostate cancer cell lines. E, Cell invasion was determined in PC3 and DU145 cells using a Boyden chamber with Matrigel, 48 hours after transfection with siRNA duplexes for PKCα (α1, α2) or NTC. Experiments were done in the absence or presence of 5% FBS in the lower chamber. Left, Representative invasion experiment. Right, Quantification of Boyden chamber migration using 5% FBS. Results are normalized to invasion in parental cells (no siRNA, dotted lines), and expressed as mean ± SEM of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 versus NTC.
Because the mesenchymal phenotype of cancer cells is linked to highly invasive traits, we next investigated the effect of knocking down PKCα on prostate cancer cell invasion. Assessment of PC3 and DU145 invasion through Matrigel using a Boyden chamber assay showed a major reduction in transwell migration in PKCα knockdown PC3 and DU145 cells relative to their corresponding parental cells or cells transfected with NTC siRNA (Fig. 4E). Similar results were observed using stably PKCα depleted PC3 and DU145 prostate cancer cells (Supplementary Fig. S4).
Next, we explored a potential relationship between PKCα and AR expression by analyzing eight prostate cancer datasets (see Materials and Methods for details). Although a general pattern could not be defined, a trend for negative correlation was observed in some cases, reaching statistically significance in four datasets. No negative correlation between PRKCA and AR expression could be found in TCGA-PRAD. Nonetheless, a meta-analysis of the eight datasets showed a statistically significant negative correlation between PRKCA and AR (Supplementary Fig. S5).
Characterization of the PKCα Transcriptome in Prostate Cancer Cells
Previous studies from our laboratory established key roles for DAG-regulated PKCs in the control of gene expression (17, 31, 36, 37). To begin elucidating the molecular changes associated with PKCα proliferative and invasive activities in prostate cancer, we carried out a whole transcriptome analysis in PC3 cells. We used five cohorts (parental, NTC siRNA, PKCα siRNA #1, #2, and #3). RNA was extracted 48 hours after transfection of the corresponding siRNA duplexes and subjected to RNA-seq. A comparison of transcriptome profiles was conducted to identify differentially regulated genes using the edgeR test (FDR < 0.001). A 2-fold change relative to parental cells was used as a cutoff. Cluster dendrogram revealed significant overlapping between parental and NTC cells, as well as between the three different PKCα knockdown cell lines (Fig. 5A). Using the described stringent cutoffs, we identified 848 upregulated genes and 602 downregulated genes overlapped by all three PKCα RNAi sequences relative to NTC (P < 0.05; Fig. 5B and C). A complete list of PKCα-regulated genes is shown in Supplementary Table S1. As expected, the PKCα gene (PRKCA) was a top downregulated gene (>99% depletion), whereas expression of other PKC genes remained unchanged. The AR gene was not among the PKCα-regulated genes (see Supplementary Table S1). Validation of the RNA-seq analysis was done by Q-PCR using specific probes for the three top downregulated genes (ESM1, BCL2A1, and PRKCA) and the three top upregulated genes (DMBT1, GLYATL2, and ABCB11; Fig. 5D).
FIGURE 5.

PKCα regulates gene expression in prostate cancer cells. A, Dendrogram for deregulated genes in PC3 cells subjected to PKCα silencing using three different siRNA duplexes (α1, α2, or α3), and comparison with NTC siRNA and parental cells. B, Number of genes regulated by PKCα, as determined by RNA-seq, which represents the overlapped signature with α1, α2, and α3 siRNA duplexes. C, Heatmap for PKCα-regulated genes. D, Q-PCR validation the top three upregulated and downregulated genes.
By means of pathway enrichment analysis using REACTOME and Kyoto Encyclopedia of Genes and Genomes (KEGG), we were able to identify prominent enrichment of pathways associated with cell-cycle progression, DNA replication, and regulation of the extracellular matrix (ECM; Fig. 6A). Gene Ontology (GO) analysis provided similar enrichment in gene sets associated with mitogenesis and ECM function, as well as processes related to cell migration and immune responses (Fig. 6B). Complete lists of pathway enrichment analysis and GO enrichment analysis regulated by PKCα can be found in Supplementary Table S2. Further analysis using GSEA for quantitative determination of activated versus suppressed functional enrichments, clearly revealed a major suppression of pathways associated with cell proliferation in PKCα-silenced cells, as depicted in Fig. 6C and D. Top suppressed pathways include “chromosome segregation,” “mitotic cell-cycle process,” “DNA replication,” “mitotic cell-cycle phase transition,” and “DNA replication.”
FIGURE 6.

FEAs for PKCα regulates gene expression in prostate cancer cells. A, FEA of pathways identified as affected by PKCα silencing in PC3 cells. B, FEA of GO biological processes affected by PKCα silencing in PC3 cells. C, GSEA of the activated and suppressed bioprocesses in PKCα silencing in PC3 cells. D, GSEA of the activated and suppressed KEGG pathways in PKCα silencing in PC3 cells. E, GSEA of the activated and suppressed TFBS upon PKCα silencing in PC3 cells based on the TFBS Molecular Signatures Database (MsigDB).
An analysis of TFBS enrichment in PKCα-regulated genes was performed using the R/Bioconductor package clusterProfiler and the TFBS signatures available at the Molecular Signatures Database. Results depicted in Fig. 6E showed a striking enrichment for E2F TFBSs in the promoters of genes which are downregulated when PKCα is silenced. We also noticed an enrichment for Oct-1 TFBSs in the promoters of genes upregulated upon PKCα silencing. A complete list of transcriptional binding sites regulated by PKCα is presented in Supplementary Table S3. Notably, this E2F signature is suppressed in PKCα knockdown cells, a finding that fits with the expected G1 arrest and enhanced E2F activity resulting from Rb dephosphorylation upon PKCα silencing. These results undoubtedly place PKCα as a major hub for the control of transcriptional pathways associated with prostate cancer cell proliferation.
Differential Expression of EMT Genes in Human Prostate Tumors Based on PKCα Expression
To further ascertain the biological relevance of the PKCα gene signature identified by RNA-seq in PC3 cells, we next analyzed TCGA-PRAD data collection. Briefly, a group of 498 patients with primary prostate adenocarcinomas were classified into “Low PKCα” or “High PKCα” mRNA expression levels according to the StepMiner one-step algorithm, establishing 134 tumors and 75 tumors in each category, respectively (Fig. 7A). Differentially expressed genes were scrutinized with the MultiExperiment Viewer software (MeV v4.9), revealing 116 genes in the “High PKCα” group which are downregulated in PKCα silenced PC3 cells, and 196 genes in the “Low PKCα” group which are upregulated in PKCα-depleted PC3 cells (Fig. 7B). Hence, a significant overlap exists between PKCα-regulated genes in the RNA-seq analysis and a human prostate cancer database.
FIGURE 7.
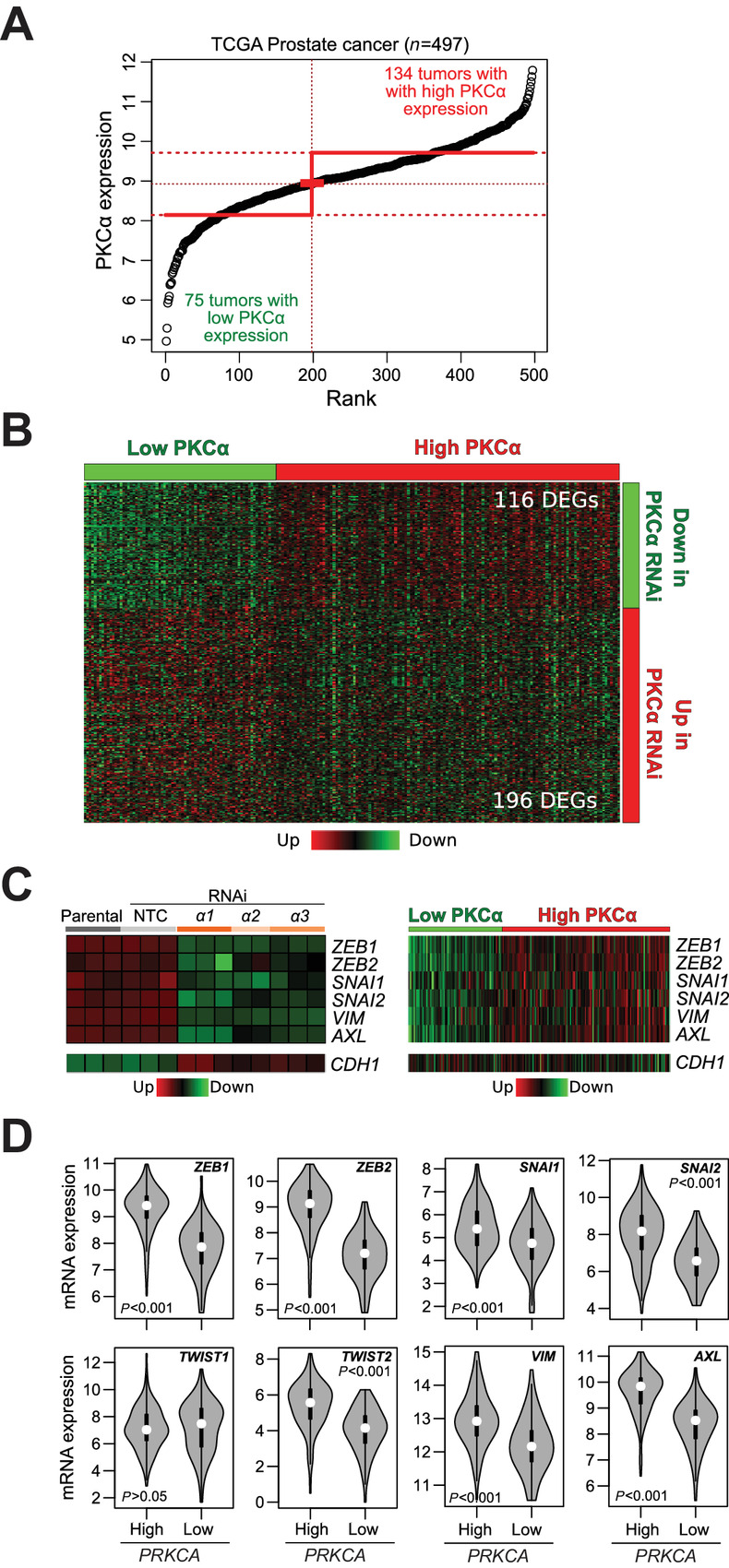
PKCα controls the expression of EMT markers in human prostate cancer. A, Classification of primary prostate adenocarcinomas (TCGA-PRAD, n = 498) into “High PKCa” or “Low PKCa” expression levels according to the StepMiner one-step algorithm. B, Heatmap showing the differential gene expression from RNA-seq analysis in comparison with the same genes in “High PKCa” or “Low PKCa” human prostate tumors. C, Heatmaps for the expression of EMT-TFs and mesenchymal markers. Left, Expression upon PKCα RNAi silencing in PC3 cells, as determined by RNA-seq. Right, Expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD. The epithelial marker E-cadherin (CDH1) is also shown. D, Violin plots indicating P values for the comparison of EMT-TF and mesenchymal marker expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD.
Next, we carried out an analysis for selected genes associated with epithelial and mesenchymal phenotypes. Interestingly, RNA-seq analysis showed significant downregulation of ZEB1, ZEB2, SNAI1, and SNAI2, as also determined in the correlation analysis presented in Fig. 4B. Therefore, PKCα upregulation is associated with the expression of these EMT transcription factors. No changes were observed in TWIST1, and TWIST2 was not detected in the RNA-seq profiling. The RNA-seq in PC3 cells also revealed prominent downregulation of AXL and vimentin (VIM) upon PKCα silencing (Fig. 7C, left). These results display a remarkable overlap with expression data from TCGA-PRAD, which shows a major upregulation of these EMT markers in “High PKCα” human prostate adenocarcinomas relative to the “Low-PKCα” patient group (Fig. 7C, right). A significant upregulation of CDH1 was detected upon PKCα silencing in the RNA-seq profile; however, this was not observed in the human TCGA-PRAD database. Violin plots depicted in Fig. 7D indicate the expression of mesenchymal markers in “High PKCα” versus “Low-PKCα” groups with the corresponding P values.
PKCα Controls the Expression of Proinflammatory Cytokines and PD-L1 in Human Prostate Cancer
A focused analysis of cytokine expression from the RNA-seq data revealed prominent downregulation of proinflammatory cytokines in PKCα-depleted PC3 cells, including IL1β, IL6, IL8 (CXCL8), CXCL1 (GRO1), CXCL3 (GRO3), and CXCL6 (Fig. 8A, left). This is remarkable because most of these cytokines have important tumor-promoting roles in prostate cancer and have been associated with high-grade disease (38–40). Interestingly, “High PKCα” human prostate tumors displayed elevated expression of these cytokines relative to “Low PKCα” tumors, as depicted in heatmaps (Fig. 8A, right) and violin plots (Fig. 8B). Proof of principle for the PKCα-dependent production of cytokines (IL8 and GRO) was determined by ELISA in conditioned medium (Supplementary Fig. S6).
FIGURE 8.

Association of PKCα with proinflammatory and/or tumorigenic cytokines and PD-L1 expression in human prostate cancer. A, Heatmaps for the cytokines and/or chemokines regulated by PKCα. Left, Expression upon PKCα RNAi silencing in PC3 cells, as determined by RNA-seq. Right, Expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD. NTC, non-target control. B, Violin plots indicating P values for the comparison of cytokine expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD. C, Representative Western blot analysis for PD-L1 expression in prostate cells. D, Inhibition of PD-L1 expression in PC3 cells by PKCα RNAi silencing, as determined by RNA-seq. E, Heatmaps for PD-L1. Top, Expression upon PKCα RNAi silencing in PC3 cells, as determined by RNA-seq. Bottom, Expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD. F, Violin plots indicating P value for PD-L1 expression in “High PKCa” and “Low PKCa” human prostate tumors from TCGA-PRAD. G, PD-L1 protein expression was determined by Western blot analysis in PC3 cells, 48 hours after transfection with siRNA duplexes for PKCα (α1, α2, α3) or NTC.
Another important finding from the RNA-seq analysis was the identification of the immune checkpoint PD-L1/CD274 as a PKCα-regulated gene. In agreement with other studies (41, 42), we observed PD-L1 upregulation in PC3 and DU145 cell lines compared to LNCaP cell lines (Fig. 8C). Notably, our RNA-seq analysis showed downregulated PD-L1 mRNA levels in PKCα-silenced PC3 cells, ranging between 60% and 80% (Fig. 8D and E, top). Moreover, analysis in TCGA-PRAD revealed significantly elevated PD-L1 levels in “High PKCα” versus “Low PKCα” human prostate adenocarcinomas (Fig. 8E, bottom; violin plot in Fig. 8F). We verified the causal association between PD-L1 and PKCα expression at the protein level, with a clear PD-L1 downregulation in PC3 cells upon PKCα RNAi-mediated silencing, as determined by Western blot analysis (Fig. 8G). In conclusion, high PKCα levels associate with upregulation of proinflammatory and/or tumorigenic cytokines as well as with immune checkpoint PD-L1 in human prostate cancer.
Discussion
Our study established PKCα as a protumorigenic kinase in human prostate cancer. PKCα is prominently upregulated in aggressive cellular models of prostate cancer, and is required for their growth in culture and as xenografts in mice. Using a human prostate cancer TMA, we found PKCα to be highly expressed in a significant fraction of tumors. We hypothesize that abnormally expressed PKCα reroutes DAG inputs by boosting oncogenic PKCα effector pathways, favoring prostate cancer cells to become addicted to this pro-oncogenic axis. The tumorigenic role of PKCα in prostate cancer emphasizes its remarkable functional complexity and epitomizes an example of a DAG/phorbol ester-regulated kinase having dual effects. Indeed, PKCα has been shown to be downregulated and to act as a tumor suppressor kinase in many cancer types. The dichotomous roles of PKCα likely reflect unique links with effector pathways and transcriptional programs depending on expression levels and context, as thoroughly reviewed recently (26), possibly exposing distinctive functional PKCα interactions with oncogenic and tumor-suppressing signals.
Our findings, together with preceding studies, underscore precise roles for PKC isozymes in different stages of prostate cancer progression. We previously generated prostate-specific transgenic mouse models for PKC isozyme overexpression under the control of the probasin (PB) promoter. PB-PKCε mice develop a dysplastic phenotype characteristic of prostatic intraepithelial neoplasia (PIN; ref. 15), underlining a role for PKCε in tumor initiation, as we also postulated in lung cancer models (37). PIN lesions in PB-PKCε mice exhibit early signs of deregulated oncogenic signaling, including hyperactivation of Akt, ERK, STAT3, and NFκB (15–17). However, PB-PKCα mice did not develop any obvious phenotype, indicating its lack of involvement in tumor initiation (15). On the other hand, the PKCα requirement for proliferation strongly supports its involvement in the expanding phase of tumor growth. In this regard, our results show that silencing PKCα expression from PC3 or DU145 cells leads to the accumulation of cells in G0–G1 and a discernible Rb hypophosphorylation. While the mechanistic aspects of this growth delay remain to be determined, reduced growth upon PKCα inhibition has been linked to p21cip1 induction in other cancer types (26, 43–45). The weakened phospho-ERK signal in PKCα-deficient PC3 xenografts fits with the established role of the MEK/ERK cascade in the control of transcriptional events leading to DNA synthesis and mitogenesis (46). Moreover, the enrichment in E2F transcription binding sites in PKCα-regulated genes is a clear indication of the impact that this kinase has on transcriptional events associated with proliferative programs. On the other hand, in scenarios of G1 arrest driven by PKCα (i.e., tumor suppression), this kinase drives a transcriptional program of cell-cycle exit, as extensively described by Black and co-workers in models of intestinal epithelial cells (26, 47). In that model, PKCα suppresses the expression of inhibitor of DNA binding (Id) transcription factors through an ERK-dependent mechanism (23, 26). Because Id transcriptions factors have been associated to mitogenesis and oncogenicity, and they are heavily regulated by phosphorylation (48), it would be interesting to determine whether aberrant PKCα signaling contributes to Id activation and/or upregulation, which has been reported in hormone refractory prostate cancer (49, 50). Of note, our RNA-seq analysis reveals a major downregulation of Id1 upon PKCα silencing in PC3 cells (see Supplementary Table S1). Disentangling this enthralling dichotomy represents a significant challenge in PKCα signaling.
Intriguingly, PKCα expression levels do not unequivocally associate with AR responsiveness status. While cell lines displaying aberrant PKCα expression (PC3, PC3-ML, DU145) are AR negative, LNCaP variants that lose androgen responsiveness display low PKCα levels. Notably, PKCα-depleted PC3 cells did not reveal changes in AR levels, an indication that PKCα per se does not regulate AR expression. Moreover, analysis of PKCα and AR expression from multiple databases did not show an obvious association, despite a trend for negative correlation in some datasets and a statistically significant negative correlation between PRKCA and AR in our meta-analysis. It would be interesting to determine the molecular basis behind the PKCα upregulation in AR negative cells, which presumably involves reprogrammed transcriptional mechanisms that occur in mCRPC.
Our bioinformatics analysis using the CCLE database and the TCGA-PRAD tumor database revealed positive correlations between PKCα and the expression of EMT markers. Notably, “High PKCα” tumors express high levels of mesenchymal phenotype markers, including EMT transcription factors. A causal association seems to be in place based on the noted downregulation of these markers upon PKCα silencing, as shown in our RNA-seq analysis. It remains to be determined whether PKCα is causally related to the transition from the epithelial to mesenchymal state in prostate cancer. Our previous studies in lung cancer cells revealed that silencing the expression of individual PKC isozymes, including PKCα, did not prevent the acquisition of the mesenchymal phenotype in response to TGFβ (51). Still, a partial reversal of the mesenchymal phenotype upon PKCα inhibition is seen in breast and lung cancer models (12, 52). A likely possibility is that PKCα upregulation is secondary to the acquisition of a mesenchymal state rather than contributing to the EMT transformation process. Elevated PKCα signaling may aid in the maintenance of the mesenchymal phenotype by upregulating EMT transcription factors (14, 35) and upholding invasive capacity, as revealed in our Boyden chamber analysis using PKCα-silenced cells. Along this line, our previous study in lung cancer models established PKCα as a determining factor for the production of metalloproteases required for ECM degradation (31). PKCα may also control fundamental cellular processes associated with cell migration, a conclusion supported by the enrichment in PKCα-regulated genes associated with cell motility and ECM function. PKCα may regulate basic mechanisms driving cell motility, such as the coordinated processes involved in actin cytoskeleton assembly and disassembly. In fact, PKC signaling regulates Rho GTPases involved in the formation of promotility actin-rich peripheral protrusions (e.g., lamellipodia, ruffles) and stress fibers (29, 30, 49, 53–55). Dissecting PKCα downstream effectors is key to untangle the molecular basis of these regulatory processes.
Finally and unexpectedly, our study identified PKCα as a cancer cell intrinsic factor responsible for PD-L1 upregulation in aggressive prostate cancer cells. It is well known that oncogenes and loss of tumor suppressor genes induce PD-L1 expression in cancer cells, often involving PKC effector pathways such as MEK/ERK (56). Our finding may have significant impact on tumor immune evasion, because elevated PKCα levels in prostate cancer cells may potentially contribute to the abrogation of T-cell antitumor responses. Our RNA-seq analysis also established PKCα as a key signaling node for proinflammatory cytokine expression. Many of the identified cytokines, including IL6, IL8/CXCL8, and IL1β, are known to stimulate cell-autonomous protumorigenic mechanisms and shape the local microenvironment to support growth, survival, and invasion of primary tumors (38–40). An additional inference relates to the ability of PD-L1 to transduce intrinsic signals independent of T-cell PD-1 ligation. Indeed, PD-L1 contributes to the protumoral activities of cancer cells by increasing proliferation and suppressing apoptotic responses. In addition, overexpressed PD-L1 promotes EMT, and augments migratory and invasive properties of cancer cells (57), Thus, our observation raises the question whether a PKCα/PD-L1 link contributes to proliferative and invasive activities of prostate cancer cells in addition to promoting an inflammatory and/or immunosuppressive landscape.
In summary, the identification of PKCα as a “multifunctional” kinase underlines its relevance in the control of myriad of events associated with prostate cancer. Considering the reported antitumorigenic activity of PKC inhibitors, including those targeting cPKCs (10), PKCα may represent an attractive therapeutic target for aggressive prostate cancer. Overall, the extraordinary functional complexity of PKCα signaling merits a dedicated mechanistic analysis in a cancer type–specific manner, determining their eligibility for a rationale pharmacological approach targeting this kinase or its effectors.
Supplementary Material
Time-dependent expression of PKCalpha in prostate cancer cells after RNAi silencing with three different siRNA duplexes.
Cell cycle analysis upon PKCalpha RNAi silencing. Representative Rb phosphorylation as well as cell cycle distribution analysis by FACS in PKCalpha silenced PC3 cells are shown.
Effect of treatment with Cdk4/6 and Cdk4 inhibitors in combination with PKCα RNAi silencing on prostate cancer cell viability.
Effect of PKCalpha depletion using shRNA lentiviruses on PC3 cell invasion, as determined with a Boyden chamber.
Relationship between PRKCA and AR expression in prostate cancer. Correlation graphs for individual datasets as well as a meta-analysis are shown.
Reduced production of cytokines in PC3 cells subjected to PKCalpha RNAi depletion. IL-8 and GRO were measured in conditioned medium of PC3 cells by ELISA.
Differentially expressed genes in PKCalpha silenced PC3 cells.
Pathway and GO enrichment analysis.
Gene Set Enrichment analysis (GSEA) of transcription factor binding sites (TFBS).
Acknowledgments
This work was supported by grants R01-CA189765 and R01-CA196232 from the NIH (M.G. Kazanietz), and PICT-2018-01403 from the National Agency of Scientific and Technological Promotion (M.C. Abba).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
No disclosures were reported.
Authors’ Contributions
M. Cooke: Conceptualization, data curation, formal analysis, supervision, validation, investigation, visualization, methodology, writing-original draft, project administration, writing-review and editing. X. Zhang: Conceptualization, formal analysis, investigation, methodology. S. Zhang: Formal analysis, investigation, methodology. E. Eruslanov: Investigation, methodology. P. Lal: Conceptualization, resources, formal analysis, supervision, investigation, visualization, methodology, writing-original draft. R.E. Daniel: Formal analysis, validation, investigation, visualization, methodology. M.D. Feldman: Resources, project administration. M.C. Abba: Conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, writing-original draft. M.G. Kazanietz: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, visualization, methodology, writing-original draft, project administration, writing-review and editing.
References
- 1. Siegel R, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakazawa M, Paller C, Kyprianou N. Mechanisms of therapeutic resistance in prostate cancer. Curr Oncol Rep 2017;19:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev 2018;32:1105–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers 2021;7:9. [DOI] [PubMed] [Google Scholar]
- 6. Joshi G, Singh PK, Negi A, Rana A, Singh S, Kumar R. Growth factors mediated cell signalling in prostate cancer progression: implications in discovery of anti-prostate cancer agents. Chem Biol Interact 2015;240:120–33. [DOI] [PubMed] [Google Scholar]
- 7. Cooke M, Kazanietz MG. Overarching roles of diacylglycerol signaling in cancer development and antitumor immunity. Sci Signal 2022;15:eabo0264. [DOI] [PubMed] [Google Scholar]
- 8. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene 2014;33:5225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newton AC, Brognard J. Reversing the paradigm: protein kinase C as a tumor suppressor. Trends Pharmacol Sci 2017;38:438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cooke M, Magimaidas A, Casado-Medrano V, Kazanietz MG. Protein kinase C in cancer: the top five unanswered questions. Mol Carcinog 2017;56:1531–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain K, Basu A. The multifunctional protein kinase C-ε in cancer development and progression. Cancers 2014;6:860–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abera MB, Kazanietz MG. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol Pharmacol 2015;87:832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kyuno D, Kojima T, Yamaguchi H, Ito T, Kimura Y, Imamura M, et al. Protein kinase Cα inhibitor protects against downregulation of claudin-1 during epithelial-mesenchymal transition of pancreatic cancer. Carcinogenesis 2013;34:1232–43. [DOI] [PubMed] [Google Scholar]
- 14. Llorens MC, Rossi FA, García IA, Cooke M, Abba MC, Lopez-Haber C, et al. PKCα modulates epithelial-to-mesenchymal transition and invasiveness of breast cancer cells through ZEB1. Front Oncol 2019;9:1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benavides F, Blando J, Perez CJ, Garg R, Conti CJ, DiGiovanni J, et al. Transgenic overexpression of PKCε in the mouse prostate induces preneoplastic lesions. Cell Cycle 2011;10:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garg R, Blando JM, Perez CJ, Lal FMD, Smyth EM, et al. COX-2 mediates pro-tumorigenic effects of PKCε in prostate cancer. Oncogene 2018;37:4735–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garg R, Blando JM, Perez CJ, Abba MC, Benavides F, Kazanietz MG. Protein kinase C epsilon cooperates with PTEN loss for prostate tumorigenesis through the CXCL13-CXCR5 pathway. Cell Rep 2017;19:375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujii T, García-Bermejo ML, Bernabó JL, Caamaño J, Ohba M, Kuroki T, et al. Involvement of protein kinase C delta (PKCdelta) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells. Lack of proteolytic cleavage of PKCdelta. J Biol Chem 2000;275:7574–82. [DOI] [PubMed] [Google Scholar]
- 19. Sumitomo M, Ohba M, Asakuma J, Asano T, Kuroki T, Asano T, et al. Protein kinase C delta amplifies ceramide formation via mitochondrial signaling in prostate cancer cells. J Clin Invest 2002;109:827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia-Bermejo ML, Leskow FC, Fujii T, Wang Q, Blumberg PM, Ohba M, et al. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J Biol Chem 2002;277:645–55. [DOI] [PubMed] [Google Scholar]
- 21. Hara T, Saito Y, Hirai T, Nakamura K, Nakao K, Katsuki M, et al. Deficiency of protein kinase C alpha in mice results in impairment of epidermal hyperplasia and enhancement of tumor formation in two-stage skin carcinogenesis. Cancer Res 2005;65:7356–62. [DOI] [PubMed] [Google Scholar]
- 22. Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res 2006;66:6955–63. [DOI] [PubMed] [Google Scholar]
- 23. Hao F, Pysz MA, Curry KJ, Haas KN, Seedhouse SJ, Black AR, et al. Protein kinase Cα signaling regulates inhibitor of DNA binding 1 in the intestinal epithelium. J Biol Chem 2011;286:18104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hill KS, Erdogan E, Khoor A, Walsh MP, Leitges M, Murray NR, et al. Protein kinase Cα suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFβ signaling axis. Oncogene 2014;33:2134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsu AH, Lum MA, Shim KS, Frederick PJ, Morrison CD, Chen B, et al. Crosstalk between PKCα and PI3K/AKT signaling is tumor suppressive in the endometrium. Cell Rep 2018;24:655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Black AR, Black JD. The complexities of PKCα signaling in cancer. Adv Biol Regul 2021;80:100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013;24:347–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Humphries B, Wang Z, Oom AL, Fisher T, Tan D, Cui Y, et al. MicroRNA-200b targets protein kinase Cα and suppresses triple-negative breast cancer metastasis. Carcinogenesis 2014;35:2254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caino MC, Lopez-Haber C, Kissil JL, Kazanietz MG. Non-small cell lung carcinoma cell motility, rac activation and metastatic dissemination are mediated by protein kinase C epsilon. PLoS One 2012;7:e31714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cooke M, Zhou X, Casado-Medrano V, Lopez-Haber B, MJ GR, et al. Characterization of AJH-836, a diacylglycerol-lactone with selectivity for novel PKC isozymes. J Biol Chem 2018;293:8330–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cooke M, Casado-Medrano V, Ann J, Lee J, Blumberg PM, Abba MC, et al. Differential regulation of gene expression in lung cancer cells by diacyglycerol-lactones and a phorbol ester via selective activation of protein kinase C isozymes. Sci Rep 2019;9:6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klarmann GJ, Hurt EM, Mathews LA, Zhang X, Duhagon MA, Mistree T, et al. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis 2009;26:433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stylianou N, Lehman ML, Wang C, Fard AT, Rockstroh A, Fazli L, et al. A molecular portrait of epithelial-mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene 2019;38:913–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Becker-Santos DD, Guo Y, Ghaffari M, Vickers ED, Lehman M, Altamirano-Dimas M, et al. Integrin-linked kinase as a target for ERG-mediated invasive properties in prostate cancer models. Carcinogenesis 2012;33:2558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tedja R, Roberts CM, Alvero AB, Cardenas C, Yang-Hartwich Y, Spadinger S, et al. Protein kinase Cα-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J Biol Chem 2019;294:5082–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caino MC, von Burstin VA, Lopez-Haber C, Kazanietz MG. Differential regulation of gene expression by protein kinase C isozymes as determined by genome-wide expression analysis. J Biol Chem 2011;286:11254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garg R, Cooke M, Benavides F, Abba MC, Cicchini M, Feldser DM, et al. PKCε is required for KRAS-driven lung tumorigenesis. Cancer Res 2020;80:5166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waugh DJ, Wilson C, Seaton A, Maxwell PJ. Multi-faceted roles for CXC-chemokines in prostate cancer progression. Front Biosci 2008;13:4595–604. [DOI] [PubMed] [Google Scholar]
- 39. Liu Q, Russell MR, Shahriari K, Jernigan DL, Lioni MI, Garcia FU, et al. Interleukin-1β promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res 2013;73:3297–305. [DOI] [PubMed] [Google Scholar]
- 40. Culig Z, Puhr M. Interleukin-6 and prostate cancer: current developments and unsolved questions. Mol Cell Endocrin 2018;462:25–30. [DOI] [PubMed] [Google Scholar]
- 41. Bernstein MB, Garnett CT, Zhang H, Velcich A, Wattenberg MM, Gameiro SR, et al. Radiation-induced modulation of costimulatory and coinhibitory T-cell signaling molecules on human prostate carcinoma cells promotes productive antitumor immune interactions. Cancer Biother Radiopharm 2014;29:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee JH, Lee DY, Lee HJ, Im E, Sim DY, Park JE, et al. Inhibition of STAT3/PD-L1 and activation of miR193a-5p are critically involved in apoptotic effect of compound K in prostate cancer cells. Cells 2021;10:2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deeds L, Teodorescu S, Chu M, Yu Q, Chen CY. A p53-independent G1 cell cycle checkpoint induced by the suppression of protein kinase C alpha and theta isoforms. J Biol Chem 2003;278:39782–93. [DOI] [PubMed] [Google Scholar]
- 44. Wu TT, Hsieh YH, Hsieh YS, Liu JY. Reduction of PKC alpha decreases cell proliferation, migration, and invasion of human malignant hepatocellular carcinoma. J Cell Biochem 2008;103:9–20. [DOI] [PubMed] [Google Scholar]
- 45. Haughian JM, Reno EM, Thorne AM, Bradford AP. Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int J Cancer 2009;125:2556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta 2007;1773:1285–98. [DOI] [PubMed] [Google Scholar]
- 47. Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol 2000;151:763–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roschger C, Cabrele C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun Signal 2017;15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Casado-Medrano V, Barrio-Real L, Wang A, Cooke M, Lopez-Haber C, Kazanietz MG. Distinctive requirement of PKCε in the control of Rho GTPases in epithelial and mesenchymally transformed lung cancer cells. Oncogene 2019;38:5396–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ling MT, Wang X, Lee DT, Tam PC, Tsao SW, Wong YC. Id-1 expression induces androgen-independent prostate cancer cell growth through activation of epidermal growth factor receptor (EGF-R). Carcinogenesis 2004;25:517–25. [DOI] [PubMed] [Google Scholar]
- 51. Sharma P, Patel D, Chaudhary J. Id1 and Id3 expression is associated with increasing grade of prostate cancer: id3 preferentially regulates CDKN1B. Cancer Med 2012;1:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yue CH, Liu LC, Kao ES, Lin H, Hsu LS, Hsu CW, et al. Protein kinase C α is involved in the regulation of AXL receptor tyrosine kinase expression in triple-negative breast cancer cells. Mol Med Rep 2016;14:1636–42. [DOI] [PubMed] [Google Scholar]
- 53. Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 2001;276:22614–20. [DOI] [PubMed] [Google Scholar]
- 54. Byers HR, Boissel SJ, Tu C, Park HY. RNAi-mediated knockdown of protein kinase C-alpha inhibits cell migration in MM-RU human metastatic melanoma cell line. Melanoma Res 2010;20:171–8. [DOI] [PubMed] [Google Scholar]
- 55. Elhalem E, Bellomo A, Cooke M, Scravaglieri A, Pearce LV, Peach ML, et al. Design, synthesis, and characterization of novel sn-1 heterocyclic DAG-lactones as PKC activators. J Med Chem 2021;64:11418–431. [DOI] [PubMed] [Google Scholar]
- 56. Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother 2018;67:1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yadollahi P, Jeon YK, Ng WL, Choi I. Current understanding of cancer-intrinsic PD-L1: regulation of expression and its protumoral activity. BMB Rep 2021;54:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-dependent expression of PKCalpha in prostate cancer cells after RNAi silencing with three different siRNA duplexes.
Cell cycle analysis upon PKCalpha RNAi silencing. Representative Rb phosphorylation as well as cell cycle distribution analysis by FACS in PKCalpha silenced PC3 cells are shown.
Effect of treatment with Cdk4/6 and Cdk4 inhibitors in combination with PKCα RNAi silencing on prostate cancer cell viability.
Effect of PKCalpha depletion using shRNA lentiviruses on PC3 cell invasion, as determined with a Boyden chamber.
Relationship between PRKCA and AR expression in prostate cancer. Correlation graphs for individual datasets as well as a meta-analysis are shown.
Reduced production of cytokines in PC3 cells subjected to PKCalpha RNAi depletion. IL-8 and GRO were measured in conditioned medium of PC3 cells by ELISA.
Differentially expressed genes in PKCalpha silenced PC3 cells.
Pathway and GO enrichment analysis.
Gene Set Enrichment analysis (GSEA) of transcription factor binding sites (TFBS).
Data Availability Statement
Raw data for this study were generated at the NGS Core facility of the Perelman School of Medicine, University of Pennsylvania (Philadelphia, PA). Derived data supporting the findings of this study are available in Supplementary Table S1. Other data generated in this study are available within the article and its Supplementary Data.