

Abstract
The synthesis, isolation, and reactivity of a cationic, geometrically constrained σ3-P compound in the hexaphenyl-carbodiphosphoranyl-based pincer-type ligand (1+) are reported. 1+ reacts with electron-poor fluoroarenes via an oxidative addition-type reaction of the C–F bond to the PIII-center, yielding new fluorophosphorane-type species (PV). This reactivity of 1+ was used in the catalytic hydrodefluorination of Ar–F bonds with PhSiH3, and in a catalytic C–N bond-forming cross-coupling reactions between fluoroarenes and aminosilanes. Importantly, 1+ in these catalytic reactions closely mimics the mode of action of the transition metal-based catalysts.
Introduction
The past decade witnessed a growing interest in the chemistry of geometrically constrained main-group centers and their reactivity.1 A lot of work in this field is focused on the chemistry of geometrically constrained PIII centers due to their ability to cycle between two stable oxidation states, PIII and PV, which makes them a potent target for metallomimetic chemistry in catalysis.2 In comparison to phosphines with typical trigonal pyramidal geometry that are usually only nucleophilic, geometrically constrained phosphines have an ambiphilic, both nucleophilic and electrophilic, reactivity toward small molecules and often react by insertion of the PIII center into strong bonds via an oxidative addition-type reaction.3
In 1986, Arduengo reported the first C2v fold, phosphorus center in ONO pincer-type ligand.4 Radosevich, in 2012, reported the use of this phosphine to activate ammonia borane and used it to catalytically transfer hydrogen to azobenzene.5 Two years later, Radosevich reported a new geometrically constrained PIII-center with a Cs local symmetry (I, Figure 1),6 which showed ambiphilic reactivity in small-molecule activation.7 Kinjo reported on a diazadiphosphapentalene with a geometrically constrained phosphorus center that activated ammonia by a P-center ligand-assisted process.8 Aldridge and Goicoechea reported on a constrained PIII-center in an ONO pincer-type ligand that showed ambiphilic reactivity toward amines and alcohols.9 In 2018, we reported the synthesis of the first geometrically constrained amphiphilic phosphenium cation, which activated water, alcohols, and ammonia, while the activation of ammonia was reversible.10 Recently, we also reported on the phosphenium cation in an NNN pincer-type ligand that reacted with O–H and N–H bonds by the P-center ligand-assisted process, and with Si–H bonds by an oxidative addition-type reaction.11 In our last work, we reported on the intramolecular oxidative addition-type reactions of polar C–N bonds with a geometrically constrained PIII-center.12
Figure 1.

Metallomimetic chemistry of geometrically constrained PIII species. Previously reported stepwise, noncatalytic hydrodefluorination of fluoroarenes (top); this work, catalytic hydrodefluorination and amination of fluoroarenes (bottom).
Importantly, despite the progress made in the field of geometrically constrained PIII compounds,2−12 which led to a number of PIII/PV catalytic transformations,13 their catalytic application in a metallomimetic fashion, i.e., following “oxidative addition” (OA) → “ligand metathesis” (LM) → “reductive elimination” (RE) steps, is scarce.5,11 In fact, to the best of our knowledge there are only two recent reports in which metallomimetic cycles of this type were shown.7b,11 In 2020, a noncatalytic hydrodefluorination reaction of pentafluoropyridine and octafluorotoluene following the OA → LM → RE steps using geometrically constrained PIII triamide species (I) was reported (Figure 1).7b In 2022, we reported a metallomimetic catalytic hydrosylilation of benzaldehyde using a geometrically constrained PIII cation.11 It is important to mention here that much progress has been recently done in the metallomimetic catalysis involving bismuth-based catalysts,14 a heavier analogue of phosphorus. A noteworthy example of BiI/BiIII metallomimetic catalysis following the OA → LM → RE steps was recently reported for hydrodefluorination of fluoroarenes.15
Continuing with our efforts to synthesize new geometrically constrained PIII cations and study their chemistry in small-molecule activation, we report here the synthesis of a cationic, geometrically constrained PIII species (1+) in a hexaphenyl-carbodiphosphoranyl-based CCC pincer-type ligand. The reactivity of 1+ in activation of electron-poor fluoroarenes by oxidative addition-type reaction of C–F bonds to the PIII center and its use as a catalyst in hydrodefluorination and C–N bond-forming cross-coupling reactions is reported (Figure 1). The mechanism of these catalytic reactions was studied both experimentally and by density functional theory (DFT) computations and closely mimics the transition metal-based catalysis.16
Results and Discussion
1+ was prepared using a similar methodology recently reported by Sundermeyer, which showed that hexaphenyl-carbodiphosphorane can be doubly deprotonated by nBuLi, producing a dilithiated hexaphenyl-carbodiphosphorane ligand,17 which can be used as a dianionic tridentate CCC pincer-type ligand.17,18 Thus, hexaphenyl-carbodiphosphorane (2) was treated with 2 equiv of nBuLi followed by addition of PCl3, which resulted in precipitation of the desired [1+][Cl] (Scheme 1). The 31P NMR of [1+][Cl] in CDCl3 was measured, showing two broad singlets with chemical shifts of δ = 29.80 and 54.24 ppm with the integration ratio of 2:1, respectively. Thus, the signal at 29.80 ppm was attributed to two phosphorus centers of the carbodiphosphorane unit and the signal at 54.24 ppm to the new PIII-center. The attempts to crystallize [1+][Cl] failed; however, after Cl– exchange with the weakly coordinating [PF6]− anion, and by reaction of [1+][Cl] with [K][PF6] (Scheme 1), [1+][PF6] was crystallized from a saturated CH2Cl2/hexane (1:10) solution, and its molecular structure was determined using X-ray crystallography (Figure 2).
Scheme 1. Synthesis of [1+][Cl] and [1+][PF6].

Figure 2.
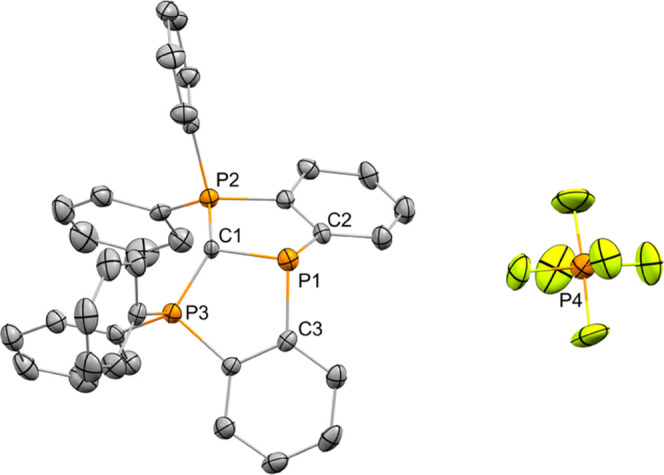
POV-ray depiction of [1+][PF6]. Thermal ellipsoids at 30% probability; hydrogen atoms were omitted for clarity.
Importantly, the 31P NMR spectrum of [1+][PF6] is nearly identical to that of [1+][Cl] with the exception of a chemical shift at δ = −144.73 ppm appearing as a septet, which corresponds to the [PF6]− anion. This means that similarly to [1+][PF6], [1+][Cl] is probably a separated ion pair in solution. Noteworthy, in 2018, Uhl reported the synthesis of a neutral geometrically constrained phosphine in CCC pincer-type ligand that exhibited unusual reactivity in coordination chemistry19 and synthesis of new heterocyclic molecules.20
The geometry around P1 in 1+ is significantly distorted from the trigonal pyramidal geometry of the analogous, not geometrically constrained carbodiphosphorane-PPh2 adduct [(Ph3P)2C–PPh2]+ that adopts a local C3v symmetry.21 The rigid, tridentate carbodiphosphorane-based ligand enforces a strained geometry around the P1 center in 1+ with a significant distortion along the P1–C1 axis, resulting in a local Cs symmetry. The two bond angles ∠C1–P1–C3 = 95.66° and ∠C1–P1–C2 = 94.67° in 1+ are essentially similar and significantly narrower than those of the previously reported bond angles in [(Ph3P)2C–PPh2]+.21 The ∠C2–P1–C3 bond angle in 1+ of 105.88° is wider than the other two angles and is in the range of previously reported species.21 The geometrical distortion of P1 in 1+ is however less pronounced than for the previously reported geometrically constrained PIII center in the CCC trianionic pincer-type ligand.19 Overall, the local geometry around the P1 center in 1+ approximates a cis-divacant pseudo trigonal bipyramid in which C2 and C3 atoms of the carbodiphosphorane unit are at the equatorial positions and the central C1 is at the axial one. The P1–C1 bond length of 1.833 Å is typical to P–C single bonds, while P2–C1 and P3–C1 bond lengths of 1.713 and 1.719 Å, respectively, are shorter than a typical P–C single bond.
To get a deeper insight into the structural features of 1+, DFT calculations at the BP86-D3/def2TZVP22 level of theory were performed. The calculated geometrical parameters of 1+ were in good agreement with the ones obtained from a single-crystal X-ray molecular structure analysis. Natural bond orbital (NBO) analysis revealed the presence of one s-type lone pair (1.86 e– occupancy) at the P1 phosphorus center residing on a sp hybrid (51.00% s and 48.96% p), and a p-type lone pair (1.65 e– occupancy) on the C1 carbon center (97.43% p). The lower electron occupancy at the C1 center is a result of the negative hyperconjugation of its p-type lone pair mostly into the parallel σ*(P(2)/P(3)–CPh) orbitals (0.12965 e– and 0.12445 e– occupancies), which also explains the short P2–C1 and P3–C1 bond lengths. The NBO charges of −1.37962, +0.89314, +1.62167, and +1.62827 on C1, P1, P2, and P3 centers, respectively, were calculated. The Wiberg bond index (WBI) values for C1–P1, C1–P2, and C1–P3 bonds of 0.9089, 1.0450, and 1.0533, respectively, indicate a single-bond nature of these three bonds. The Baders quantum theory of atoms in molecules (QTAIM) was performed to gain insight into the topology of the electron density in 1+ (Figure 3). A negative Laplacian (∇2ρ(r)) at the bond critical points (BCP) BCP1 (−0.160802), BCP2 (−0.047073), and BCP3 (−0.039161) (Figure 3) indicates the covalent nature of these bonds, which is also supported by the negative H(rc) (Figure 3). Based on these data, we suggest that the structure of 1+ is best described with covalent C1–P1, C1–P2, and C1–P3 single bonds, with +1 formal charges at the P2 and P3 centers, −1 formal charge at the C1 center, and a neutral P1 center.
Figure 3.

QTAIM analysis of 1+.
Molecular orbitals (MO) of the computed 1+ were analyzed. Thus, the highest occupied molecular orbital (HOMO) is localized mostly on P1 and C1 atoms, while the lowest unoccupied molecular orbitals (LUMO) to LUMO + 11 are sets of degenerate orbitals that are mostly localized on the phenyl rings attached to P2 and P3 atoms, which is to be expected, due to the formal positive charge at these two P-centers. The first energetically accessible empty orbital at the central P1 atom is LUMO + 12. The HOMO–LUMO + 12 energy gap in 1+ is 3.85 eV, which is 0.85 eV larger than the HOMO–LUMO gap in I (Figure 1), computed at the same level of theory (see Supporting Information for more details).
The preliminary reactivity of [1+][PF6] with small molecules was tested. While [1+][PF6] did not react with Et2NH, MeOH, and Et3SiH even at elevated temperatures, it did react with electron-poor fluoroarenes. Thus, first [1+][PF6] was mixed with excess of pentafluoropyridine (3) in 1,2-difluorobenzene (oDFB) and heated to 80 °C and the reaction progress was monitored by NMR. After 3 h, two new signals were measured by 31P NMR, one as a doublet (1J(PF) = 666 Hz) of triplets (2J(PP) = 52 Hz) at δ = −48.87 ppm, and the other as a doublet (2J(PP) = 52 Hz) at δ = 13.91 ppm (Scheme 2, inset). In 19F NMR, a complementary doublet (1J(PF) = 666 Hz) at δ = 1.92 ppm and two new singlets at δ = −89.32 and −133.71 ppm were measured (Figures S8–S11). Based on the PF and PP coupling in both 31P and 19F NMR spectra as well as the region of the chemical shifts in 31P NMR, we confidently attributed these new signals to the formal oxidative addition product of the C–F bond (at fourth position) in 3 to the PIII center in [1+][PF6], i.e., compound [4+][PF6] (Scheme 2).
Scheme 2. Reaction between [1+][PF6] and Excess of 3 Producing the Product of a Formal Oxidative Addition-type Reaction, [4+][PF6].

The 31P NMR spectrum of [4+][PF6] is shown in the inset.
Notably, while the oxidative addition-type reaction of Ar–F bonds is not known for typical σ3-P compounds, which undergo addition to fluoroarenes by SNAr without forming stable σ5-P adducts,23 a geometrically constrained I was shown to react with Ar–F bonds via an oxidative addition-type reaction, producing stable PV compounds (Figure 1).7b [4+][PF6] was then isolated (90% yields) by evaporation of solvents and further characterized by multinuclear NMR and high-resolution mass spectrometry (HRMS). Crystallization of [4+][PF6] was attempted from a variety of solvents and at different conditions; however, all of these attempts failed.24a
Interestingly, when isolated [4+][PF6] dissolved in oDFB was heated to 110 °C for 10 h, [1+-F2][PF6] and perfluoro-4,4′-bipyridine (5) were obtained (Scheme 3).24b Although the mechanism for this reaction is not entirely clear, based on the recent report on the heavier Bi analogue,15 in which a similar reaction proceeded via disproportionation for series of ligand exchange reactions followed by elimination of 5,15 we assume a similar reaction path occurs in our case.
Scheme 3. Thermally Induced Reaction of [4+][PF6] Producing [1+-F2][PF6] and 5.

[1+-F2][PF6] was isolated by crystallization from CH2Cl2/hexane (1:10). The molecular structure of [1+-F2][PF6] was determined using X-ray crystallography (Figure 4). The P1 in 1+-F2 has a slightly distorted trigonal bipyramidal geometry with bond angles ∠C1–P1–C2 = 92.50°; ∠C1–P1–C3 = 92.32°; ∠C2–P1–C3 = 173.53°; ∠F1–P1–F2 = 108.57°; ∠C1–P1–F1 = 131.62°; ∠C1–P1–F2 = 119.80°. Interestingly, the rigid carbodiphosphoranyl ligand forces the two F–, the most electronegative atoms, to occupy the equatorial positions (Figure 4), which is atypical for difluorophosphoranes in which the fluorides occupy the axial positions.21,23 It is important to note here that PV dihalides (with Cl, Br, and I) prepared previously by the reaction of the geometrically constrained PIII-center in ONO9b or CCC19 pincer-type ligand with dihalogens had either square pyramidal geometries with one halogen at basal and other at apical positions for the ONO system,9b or a heavily distorted trigonal bipyramidal geometry with halides (F, Cl, Br) at the equatorial position.19
Figure 4.
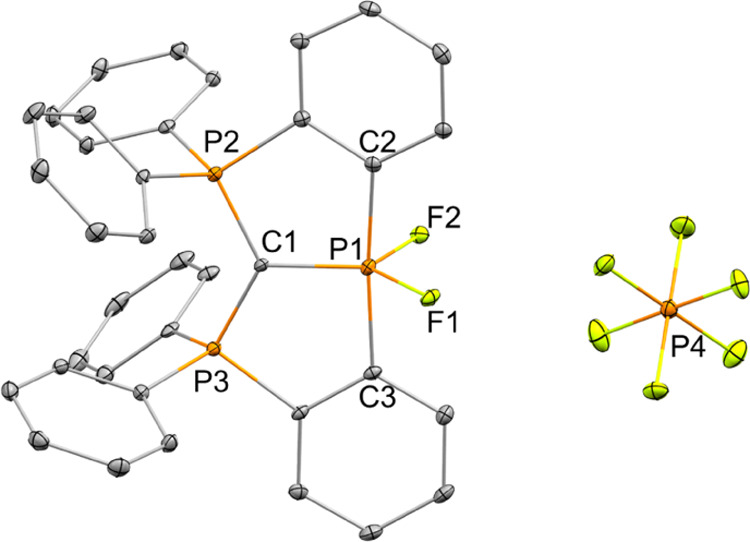
POV-ray depiction of [1+-F2][PF6]. Thermal ellipsoids at 30% probability; hydrogen atoms were omitted for clarity.
We next reacted [4+][PF6] with PhSiH3 in order to reduce P–F to P–H and obtain hydrophosphorane [6+][PF6]. This however led after 3 h at 80 °C directly to [1+][PF6], PhSiF3, and the product of hydrodefluorination, 7, while [6+][PF6] was not obtained at all.24c This we assumed was a result of a fast reductive elimination-type reaction of the C–H bond from the P-center in the intermediate [6+][PF6] (Scheme 4). It is important to note that in a previously reported similar reaction, the product of P–F to P–H exchange was stable and could be isolated in the reaction with DIBAL-H (Figure 1).7b To make sure that the inability to obtain [6+][PF6] in our case was not related to the reaction with PhSiH3, we performed the reaction of [4+][PF6] with DIBAL-H; however, the outcome was similar and [6+][PF6] was not obtained (Figure S29).
Scheme 4. Stoichiometric Reaction between [4+][PF6] and PhSiH3 Producing the Product of Hydrodefluorination 7, [1+][PF6], and PhSiF3 via Intermediate [6+][PF6].

The inability of [1+][PF6] to react with hydrosilanes and at the same time the hydrodefluorination reaction described in Scheme 4 provide an opportunity to use [1+][PF6] as the catalyst in the hydrodefluorination reaction of fluoroarenes. Thus, a reaction of 3 with PhSiH3 in the presence of a catalytic amount of [1+][PF6] (10 mol %) was done in oDFB at 80 °C, which after 3 h led to 7 in 95% yield (Table 1). The reaction with pentafluorobenzonitrile with PhSiH3 proceeded similarly in the presence of [1+][PF6] (10 mol%) and gave after 3 h at 80 °C product 8 (90%) (Table 1). The catalytic hydrodefluorination reaction of methyl pentafluorobenzoate using PhSiH3 and [1+][PF6] as the catalyst (10 mol %) led to product 9 after 16 h at 120 °C (Table 1). Remarkably, the pyridine in 7, the nitrile group in 8, and ester group in 9 all remained intact in these hydrodefluorination reactions, meaning that this method is tolerant toward these functional groups. The [1+][PF6]-catalyzed hydrodefluorination reactions of octafluorotoluene and decafluorobiphenyl with PhSiH3 leading to 10 and 11, respectively, proceeded much slower (9 and 19 h, respectively) and required a higher temperature of 120 °C (Table 1). Notably, the reaction of tris(pentafluorophenyl)phosphine with PhSiH3 in the presence of [1+][PF6] (10 mol %) led after 48 h at 120 °C to product 12, in which all of the fluorides at the para-position were substituted by hydrides (Table 1).
Table 1. Hydrodefluorination of Fluoroarenes Using PhSiH324c Catalyzed by [1+][PF6]a,b.


Isolated yields in parentheses.
The reactions of some of the substrates with PhSiH3 were attempted without the presence of [1+][PF6] and did not produce any reactivity even after prolonged heating.
Interestingly, the selectivity of [1+][PF6] in the catalytic hydrodefluorination reactions shown above is completely different from that of a previously reported dicationic PIII species, which catalyzed the hydrodefluorination of alkyl fluorides only via the Lewis acidic path,25 pointing to a different mechanism of these hydrodefluorination reactions. Although, as was previously mentioned, the oxidative addition-type reaction of the electron-poor Ar–F to PIII centers was reported (Figure 1),7b the catalytic hydrodefluorination reaction of fluoroarenes in a metallomimetic PIII/PV redox cycle has not yet been shown to the best of our knowledge. While the reason for the inability to perform catalysis with the previously mentioned I (Figure 1) was not specifically mentioned,7b it is likely that I reacts with DIBAL-H leading to the deactivation of the catalyst; this assumption is supported by the fact that I reacts with B–H bonds via the P-center/ligand-assisted path.7a As mentioned previously, however, the metallomimetic catalytic hydrodefluorination reaction of fluoroarenes using the BiI-based catalyst with a lower catalyst loading and a larger scope, milder conditions, and shorter reaction times (compared to the catalysis using [1+][PF6]) was recently achieved.15
Next, we were interested in applying the reactivity of 1+ with ArF–F bonds in the catalytic C–N bond-forming cross-coupling reactions. Thus, we first reacted 3 with Et3Si–NEt2 (13) in the presence of 10 mol% of [1+][PF6] in oDFB, which led after 2.5 h at 80 °C to the product of C/N cross-coupling (14) in 92% yield (Table 2). A similar result was obtained for the catalytic reaction of pentafluorobenzonitrile with 13, which after 2.5 h led to complete conversion to the product of C/N cross-coupling (15) (Table 2). Methyl pentafluorobenzoate reacted with 13 and the catalytic amount of [1+][PF6] (10 mol %) very slowly, leading after 15 days to a mixture of two C/N cross-coupling products at ortho and para positions (16 and 17, respectively) in 1:4 ratio (Table 2). The cross-coupling reaction between 13 and octafluorotoluene catalyzed by [1+][PF6] gave after 23 h at 120 °C 18 in 93% yield. Decafluorobiphenyl reacted extremely slowly with 13 in the presence of [1+][PF6], producing 19 (90%) after 25 days at 120 °C (Table 2). The catalytic C–N bond-forming cross-coupling reaction of the primary silylamine, Et3Si–N(H)Bn (13a), with 3 and pentafluorobenzonitrile catalyzed by [1+][PF6] (10 mol%) was performed as well, leading to 14a (87%) and 15a (95%), respectively, after 16–22 h at 80 °C (Table 2). [1+][PF6] (10 mol%)-catalyzed amination of tris(pentafluorophenyl)phosphine using 13 (1 equiv) led to product 20 after 55 h at 120 °C (Table 2). It is important to note here that the transition metal-free catalytic amination of fluoroarenes using the magnesium-based catalyst was recently reported; however, the mechanism for this amination reaction proceeded through an SNAr-type reaction.26
Table 2. [1+][PF6]-Catalyzed Amination of Fluoroarenesa,b.


Isolated yields in parentheses.
The reactions of some of the substrates with Et3SiNEt2 were attempted without the presence of [1+][PF6], and did produce products in a very low conversion ratio (below 10%) after much longer heating.
We believe that both catalytic processes, hydrodefluorination and C–N bond-forming cross-coupling, proceed via similar steps in which 1+ mimics the transition metal catalyst’s behavior. Thus, the first step is the oxidative addition-type reaction (OA) of the C–F bond to the geometrically constrained PIII center in 1+, giving a stable intermediate (INT1) (Scheme 5). The next step is the ligand metathesis (LM)-type reaction, in which the fluoride at the P-center is replaced by either a hydride or an amino group leading to the intermediate (INT2) (Scheme 5). The last step of this catalytic cycle is the reductive elimination-type reaction (RE) of C–H or C–N bonds from the P-center in INT2 producing the products of the hydrodefluorination or the C–N bond-forming cross-coupling and regenerating the catalyst (1+) (Scheme 5).
Scheme 5. Hydrodefluorination and C–N Bond-Forming Cross-Coupling Reactions Catalyzed by 1+.

To study further the mechanism of these catalytic reactions, DFT computations of the potential energy surface (PES) for the hydrodefluorination reaction of pentafluoropyridine (3) using PhSiH3 catalyzed by 1+ were performed at the BP86-D3/def2TZVP level theory.22 As a result, the first step of the reaction, which is the oxidative addition-type reaction of the C–F bond to the PIII center leading to 4+, is exergonic (ΔG = −17.9 kcal·mol–1) and strongly exothermic (ΔH = −33.2 kcal·mol–1), which proceeds through the free Gibbs energy barrier of ΔG‡= 20.1 kcal·mol–1 (TS1) (Figure 5). The next step, a ligand metathesis process, presumably producing 6+, is again exergonic and exothermic with ΔG = −6.8 and ΔH = −5.9 kcal·mol–1 and proceeds via the transition state TS2 with ΔG‡ = 33.5 kcal·mol–1 (Figure 5). Based on the calculated geometry of TS2, the ligand exchange is a concerted σ-bond metathesis process. Importantly, the ligand metathesis (4+ to 6+) is a rate-determining step, which explains the inability to observe 6+ in the reaction. The last step of the reaction, which is the reductive elimination-type reaction of the C–H bond from the PV center in 6+ leading to 7 and 1+, is strongly exergonic and exothermic (ΔG = −26.4 and ΔH = −12.6 kcal·mol–1) and proceeds via transition state TS2 with ΔG‡ = 27.9 kcal·mol–1 (Figure 5). This mechanistic picture is also supported by the variable temperature (VT) NMR experiment, in which only [1+][PF6] was measured by 31P-NMR during the catalysis, meaning that it is indeed the resting state of this catalytic cycle (Figure S28). We believe that the reaction with aminosilanes proceeds via similar mechanistic steps (Figure S61 for DFT calculation).
Figure 5.
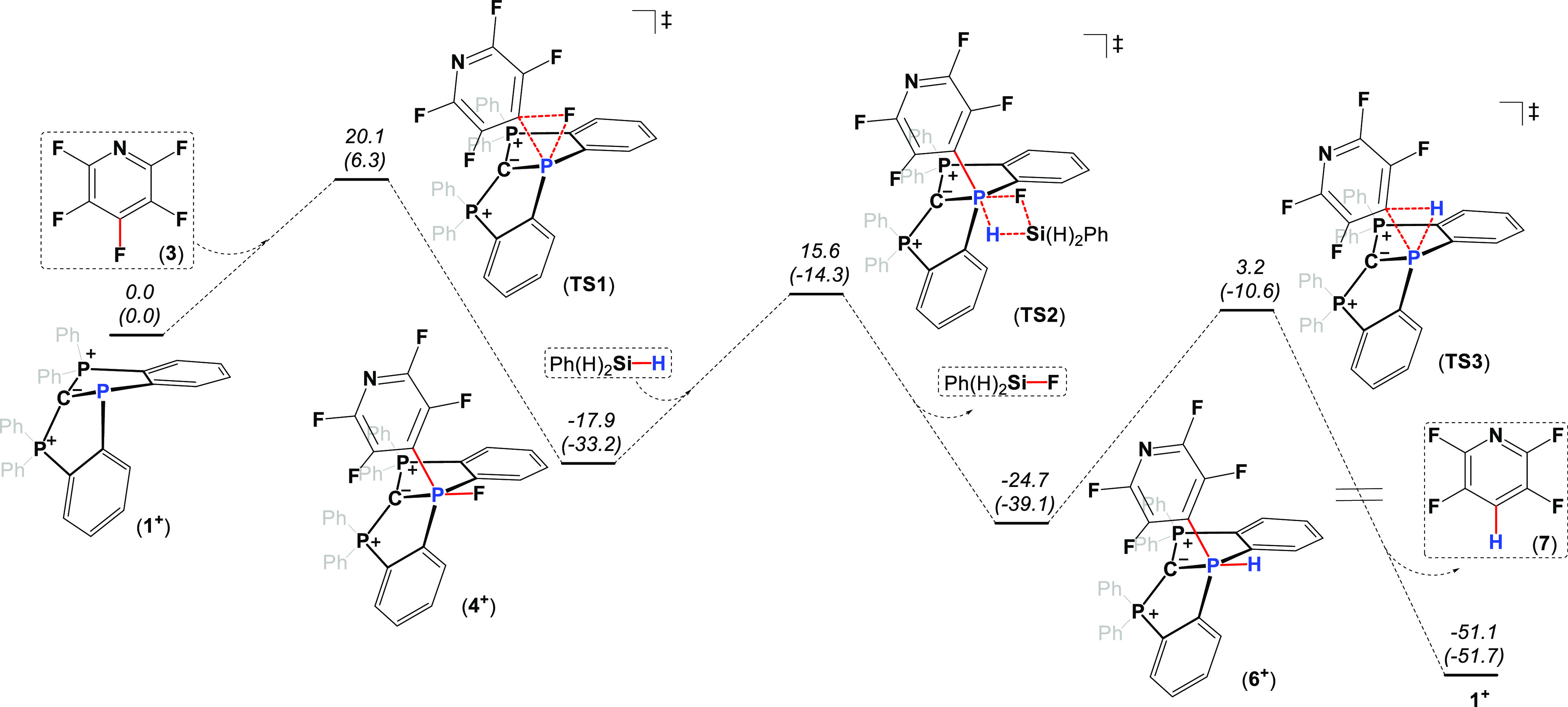
DFT-calculated (BP86-D3/def2TZVP)22 potential energy surface of the proposed mechanism of 1+-catalyzed hydrodefluorination of 3 by PhSiH3. Free Gibbs energies (enthalpies) are given relative to the starting materials.
Conclusions
To conclude, a new cationic, geometrically constrained PIII species supported by a carbodiphosphorane-based pincer-type ligand [1+][PF6] was synthesized. [1+][PF6] reacted with electron-poor fluoroarenes via oxidative addition-type reaction of the C–F bonds to the central PIII center. This reactivity of [1+][PF6] was used for catalytic hydrodefluorination and the C–N bond, forming cross-coupling reactions. The mechanism of these two catalytic processes was investigated both experimentally and computationally and proceeds in a metallomimetic fashion by following the OA → LM → RE steps. We continue to study the chemistry of the geometrically constrained P centers and their potential in catalysis.
Acknowledgments
This work was supported by the Israeli Science Foundation, Grant 237/18 and the Israel Ministry of Science Technology & Space, Grant 65692.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c13318.
NMR; experimental; and computational details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Sigmund L. M.; Maiera R.; Greb L. The inversion of tetrahedral p-block element compounds: general trends and the relation to the second-order Jahn–Teller effect. Chem. Sci. 2022, 13, 510–521. 10.1039/D1SC05395G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ruppert H.; Sigmund L. M.; Greb L. Calix[4]pyrroles as ligands: recent progress with a focus on the emerging p-block element chemistry. Chem. Commun. 2021, 57, 11751 10.1039/D1CC05120B. [DOI] [PubMed] [Google Scholar]; c Greb L.; Ebner F.; Ginzburg Y.; Sigmund L. M. Element-Ligand Cooperativity with p-Block Elements. Eur. J. Inorg. Chem. 2020, 2020, 3030–3047. 10.1002/ejic.202000449. [DOI] [Google Scholar]; d Ebner F.; Greb L. An isolable, crystalline complex of square-planar silicon(IV). Chem 2021, 7, 2151–2159. 10.1016/j.chempr.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ebner F.; Wadepohl H.; Greb L. Calix[4]pyrrole Aluminate: A Planar Tetracoordinate Aluminum(III) Anion and Its Unusual Lewis Acidity. J. Am. Chem. Soc. 2019, 141, 18009–18012. 10.1021/jacs.9b10628. [DOI] [PubMed] [Google Scholar]; f Ben Saida A.; Chardon A.; Osi A.; Tumanov N.; Wouters J.; Adjieufack A. I.; Champagne B.; Berionni G. Pushing the Lewis Acidity Boundaries of Boron Compounds With Non-Planar Triarylboranes Derived from Triptycenes. Angew. Chem., Int. Ed. 2019, 58, 16889–16893. 10.1002/anie.201910908. [DOI] [PubMed] [Google Scholar]; g Ruppert H.; Greb L. Calix[4]pyrrolato Stannate(II): A Tetraamido Tin(II) Dianion and Strong Metal-Centered σ-Donor. Angew. Chem., Int. Ed. 2022, e202116615 10.1002/anie.202116615. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Volodarsky S.; Malahov I.; Bawari D.; Diab M.; Malik N.; Tumanskii B.; Dobrovetsky R. Geometrically constrained square pyramidal phosphoranide. Chem. Sci. 2022, 13, 5957–5963. 10.1039/D2SC01060G. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kindervater M. B.; Marczenko K. M.; Werner-Zwanziger U.; Chitnis S. S. A Redox-Confused Bismuth(I/III) Triamide with a T-Shaped Planar Ground State. Angew. Chem., Int. Ed. 2019, 58, 7850–7855. 10.1002/anie.201903354. [DOI] [PubMed] [Google Scholar]; j Marczenko K. M.; Zurakowski J. A.; Kindervater M. B.; Jee S.; Hynes T.; Roberts N.; Park S.; Werner-Zwanziger U.; Lumsden M.; Langelaan D. N.; Chitnis S. S. Periodicity in Structure, Bonding, and Reactivity for p-Block Complexes of a Geometry Constraining Triamide Ligand. Chem. - Eur. J. 2019, 25, 16414–16424. 10.1002/chem.201904361. [DOI] [PubMed] [Google Scholar]; k Hynes T.; Masuda J. D.; Chitnis S. S. Mesomeric Tuning at Planar Bi centres: Unexpected Dimerization and Benzyl C-H Activation in [CN2]Bi Complexes. ChemPlusChem 2022, 87, e202200244 10.1002/cplu.202200244. [DOI] [PubMed] [Google Scholar]; l Karnbrock S. B. H.; Golz C.; Mata R. A.; Alcarazo M. Ligand-Enabled Disproportionation of 1,2-Diphenylhydrazine at a PV-Center. Angew. Chem., Int. Ed. 2022, 61, e202207450 10.1002/anie.202207450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Abbenseth J.; Goicoechea J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis. Chem. Sci. 2020, 11, 9728–9740. 10.1039/D0SC03819A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kundu S. Pincer-Type Ligand-Assisted Catalysis and Small-Molecule Activation by non-VSEPR Main-Group Compounds. Chem. Asian J. 2020, 15, 3209–3224. 10.1002/asia.202000800. [DOI] [PubMed] [Google Scholar]
- Lipshultz J. M.; Li G.; Radosevich A. T. Main Group Redox Catalysis of Organopnictogens: Vertical Periodic Trends and Emerging Opportunities in Group 15. J. Am. Chem. Soc. 2021, 143, 1699–1721. 10.1021/jacs.0c12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arduengo A. J.; Stewart C. A.; Davidson F.; Dixon D. A.; Becker J. Y.; Culley S. A.; Mizen M. B. The synthesis, structure, and chemistry of 10-Pn-3 systems: tricoordinate hypervalent pnicogen compounds. J. Am. Chem. Soc. 1987, 109, 627–647. 10.1021/ja00237a001. [DOI] [Google Scholar]
- Dunn N. L.; Ha M.; Radosevich A. T. Main Group Redox Catalysis: Reversible PIII/PV Redox Cycling at a Phosphorus Platform. J. Am. Chem. Soc. 2012, 134, 11330–11333. 10.1021/ja302963p. [DOI] [PubMed] [Google Scholar]
- McCarthy S. M.; Lin Yi.-C.; Devarajan D.; Chang J. W.; Yennawar H. P.; Rioux R. M.; Ess D. H.; Radosevich A. T. Intermolecular N–H Oxidative Addition of Ammonia, Alkylamines, and Arylamines to a Planar σ3-Phosphorus Compound via an Entropy-Controlled Electrophilic Mechanism. J. Am. Chem. Soc. 2014, 136, 4640–4650. 10.1021/ja412469e. [DOI] [PubMed] [Google Scholar]
- a Lin Y.-C.; Hatzakis E.; McCarthy S. M.; Reichl K. D.; Lai T.-Y.; Yennawar H. P.; Radosevich A. T. P–N Cooperative Borane Activation and Catalytic Hydroboration by a Distorted Phosphorous Triamide Platform. J. Am. Chem. Soc. 2017, 139, 6008–6016. 10.1021/jacs.7b02512. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lim S.; Radosevich A. T. Round-Trip Oxidative Addition, Ligand Metathesis, and Reductive Elimination in a PIII/PV Synthetic Cycle. J. Am. Chem. Soc. 2020, 142, 16188–16193. 10.1021/jacs.0c07580. [DOI] [PubMed] [Google Scholar]
- a Cui J.; Li Y.; Ganguly R.; Inthirarajah A.; Hirao H.; Kinjo R. Metal-Free σ-Bond Metathesis in Ammonia Activation by a Diazadiphosphapentalene. J. Am. Chem. Soc. 2014, 136, 16764–16767. 10.1021/ja509963m. [DOI] [PubMed] [Google Scholar]; b Cui J.; Li Y.; Ganguly R.; Kinjo R. Reactivity Studies on a Diazadiphosphapentalene. Chem. - Eur. J. 2016, 22, 9976–9985. 10.1002/chem.201600935. [DOI] [PubMed] [Google Scholar]
- a Robinson T. P.; De Rosa D. M.; Aldridge S.; Goicoechea J. M. E–H Bond Activation of Ammonia and Water by a Geometrically Constrained Phosphorus(III) Compound. Angew. Chem., Int. Ed. 2015, 54, 13758–13763. 10.1002/ange.201506998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Robinson T. P.; De Rosa D.; Aldridge S.; Goicoechea J. M. On the Redox Reactivity of a Geometrically Constrained Phosphorus(III) Compound. Chem. - Eur. J. 2017, 23, 15455–15465. 10.1002/chem.201703119. [DOI] [PubMed] [Google Scholar]
- Volodarsky S.; Dobrovetsky R. Ambiphilic geometrically constrained phosphenium cation. Chem. Commun. 2018, 54, 6931–6934. 10.1039/C8CC02423E. [DOI] [PubMed] [Google Scholar]
- Volodarsky S.; Bawari D.; Dobrovetsky R. Dual Reactivity of a Geometrically Constrained Phosphenium Cation. Angew. Chem., Int. Ed. 2022, 61, e202208401 10.1002/anie.202208401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawari D.; Volodarsky S.; Ginzburg Y.; Jaiswal K.; Joshi P.; Dobrovetsky R. Intramolecular C–N bond activation by a geometrically constrained PIII-centre. Chem. Commun. 2022, 58, 12176–12179. 10.1039/D2CC04359A. [DOI] [PubMed] [Google Scholar]
- Xie C.; Smaligo A. J.; Song X.-R.; Kwon O. Phosphorus-Based Catalysis. ACS Cent. Sci. 2021, 7, 536–558. 10.1021/acscentsci.0c01493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H. W.; Cornella J. Bismuth Redox Catalysis: An Emerging Main-Group Platform for Organic Synthesis. ACS Catal. 2022, 12, 1382–1393. 10.1021/acscatal.1c04897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y.; Leutzsch M.; Nöthling N.; Katzenburg F.; Cornella J. Catalytic Hydrodefluorination via Oxidative Addition, Ligand Metathesis, and Reductive Elimination at Bi(I)/Bi(III) Centers. J. Am. Chem. Soc. 2021, 143, 12487–12493. 10.1021/jacs.1c06735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Organometallic Chemistry of the Transition Metals; Crabtree R. H., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp 163–184. [Google Scholar]
- Buchner M. R.; Pan S.; Poggel C.; Spang N.; Müller M.; Frenking G.; Sundermeyer J. Di-ortho-beryllated Carbodiphosphorane: A Compound with a Metal–Carbon Double Bond to an Element of the s-Block. Organometallics 2020, 39, 3224–3231. 10.1021/acs.organomet.0c00434. [DOI] [Google Scholar]
- In the process of writing this manuscript a bismuth analogue of 1+ was reported:; Obi A. D.; Dickie D. A.; Tiznado W.; Frenking G.; Pan S.; Gilliard R. J. Jr. A Multidimensional Approach to Carbodiphosphorane–Bismuth Coordination Chemistry: Cationization, Redox-Flexibility, and Stabilization of a Crystalline Bismuth Hydridoborate. Inorg. Chem. 2022, 48, 19452–19462. 10.1021/acs.inorgchem.2c03337. [DOI] [PubMed] [Google Scholar]
- a Hentschel A.; Brand A.; Wegener P.; Uhl W. A Sterically Constrained Tricyclic PC3 Phosphine: Coordination Behavior and Insertion of Chalcogen Atoms into P-C Bonds. Angew. Chem., Int. Ed. 2018, 57, 832–835. 10.1002/anie.201711373. [DOI] [PubMed] [Google Scholar]; b Brand A.; Hentschel A.; Hepp A.; Uhl W. Dihalides of Sterically Constrained Tricyclic Phosphines, Lewis Acidity and Fluoride Affinity, Chloride Abstraction, and a Phosphonium Cation, Dimethylphosphorane. Eur. J. Inorg. Chem. 2020, 2020, 361–369. 10.1002/ejic.201901231. [DOI] [Google Scholar]
- Brand A.; Schulz S.; Hepp A.; Weigand J. J.; Uhl W. Sterically constrained tricyclic phosphine: redox behaviour, reductive and oxidative cleavage of P–C bonds, generation of a dilithium phosphaindole as a promising synthon in phosphine chemistry. Chem. Sci. 2021, 12, 3460–3474. 10.1039/D0SC06155G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogendra S.; Hennersdorf F.; Bauzá A.; Frontera A.; Fischer R.; Weigand J. J. Carbodiphosphorane mediated synthesis of a triflyloxyphosphonium dication and its reactivity towards nucleophiles. Chem. Commun. 2017, 53, 2954–2957. 10.1039/C7CC00982H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 34, 7406 10.1103/PhysRevB.34.7406. [DOI] [PubMed] [Google Scholar]; b Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; c Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- a Arévalo A.; Tlahuext-Aca A.; Flores-Alamo M.; García J. J. On the Catalytic Hydrodefluorination of Fluoroaromatics Using Nickel Complexes: The True Role of the Phosphine. J. Am. Chem. Soc. 2014, 136, 4634–4639. 10.1021/ja412268y. [DOI] [PubMed] [Google Scholar]; b Facundo A. A.; Arévalo A.; Fundora-Galano G.; Flores-Álamo M.; Orgaz E.; García J. J. Hydrodefluorination of Functionalized Fluoroaromatics with Triethylphosphine: A Theoretical and Experimental Study. New J. Chem. 2019, 43, 6897–6908. 10.1039/C9NJ00721K. [DOI] [Google Scholar]; c Weiss J.-V.; Schmutzler R. Formation of a Carbon–Phosphorus Bond between a C–F Compound and Phosphorus(III) Fluorides: A Fluorine Analogue of the Kinnear–Perren Reaction Furnishing 1-Adamantylfluorophosphoranes. J. Chem. Soc. Chem. Commun. 1976, 0, 643–644. 10.1039/C39760000643. [DOI] [Google Scholar]; d Burton D. J.; Shinya S.; Howells R. D. The Role of α and β Flu- orine in Product Determination of Fluoro Olefin-Tertiary Phosphine Reactions. Ylide vs. Vinylphosphorane Formation. J. Am. Chem. Soc. 1979, 101, 3689–3690. 10.1021/ja00507a060. [DOI] [Google Scholar]; e Plack V.; Goerlich J. R.; Thönnessen H.; Jones P. G.; Schmutzler R. Air-Stable Trifluorophosphoranes: Preparation, X-Ray Crystal Structure Determinations, and Reactions. Z. Anorg. Allg. Chem. 1999, 625, 1278–1286. . [DOI] [Google Scholar]; f Keßler M.; Neumann B.; Stammler H.-G.; Hoge B. Fluorotrimethyl[(Z)-Pentafluoropropen1-Yl]Phosphorane: Structure, Bonding, and Reactivity. Z. Anorg. Allg. Chem. 2020, 646, 1 10.1002/zaac.202070011. [DOI] [Google Scholar]
- a Over time [4+][PF6] stored in glass vial converted to [1=O+][PF6], which was isolated and fully characterized (see SI). Crystallization in a Teflon vial did not produce crystals of sufficient quality;; b [1=O+][PF6] was also obtained in this reaction as one of the products, however, was not isolated from this reaction. The formation of this compound is still not entirely understood, possibly reaction of one of the intermediates with the glass could lead to its formation;; c All the reactions, both stoichiometric and catalytic, were performed with 1 equiv of PhSiH3, while only PhSiF3 was obtained after reaction completion, meaning that 0.3 equiv of PhSiH3 is consumed.
- Chitnis S. S.; Krischer F.; Stephan D. W. Catalytic Hydrodefluorination of C–F Bonds by an Air-Stable PIII Lewis Acid. Chem. - Eur. J. 2018, 24, 6543–6546. 10.1002/chem.201801305. [DOI] [PubMed] [Google Scholar]
- Bole L. J.; Davin L.; Kennedy A. R.; McLellan R.; Hevia E. Magnesium-mediated arylation of amines via C–F bond activation of fluoroarenes. Chem. Commun. 2019, 55, 4339–4342. 10.1039/C9CC01670H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.