

Abstract
Purpose of review:
Bone marrow adipose tissue (BMAT) in the skeleton likely plays a variety of physiological and pathophysiological roles that are not yet fully understood. In elucidating the complex relationship between bone and BMAT, glucocorticoids (GCs) are positioned to play a key role, as they have been implicated in the differentiation of bone marrow mesenchymal stem cells (BMSCs) between osteogenic and adipogenic lineages. The purpose of this review is to illuminate aspects of both endogenous and exogenous GC signaling, including the influence of GC receptors, in mechanisms of bone aging including relationships to BMAT.
Recent findings:
Harmful effects of GCs on bone mass involve several cellular pathways and events that can include BMSC differentiation bias toward adipogenesis and the influence of mature BMAT on bone remodeling through crosstalk. Interestingly, BMAT involvement remains poorly explored in GC-induced osteoporosis and warrants further investigation.
Summary:
This review provides an update on the current understanding of the role of glucocorticoids in the biology of osteoblasts and bone marrow adipocytes (BMAds).
Keywords: glucocorticoid, BMAT, BMAd, dexamethasone, osteoporosis, BMSC
1. Introduction
Aging is typically characterized by the deterioration of the structural and functional characteristics of an organism over time (1). One of the hallmarks of aging is an accumulation of senescent cells, leading to a decline in regenerative capabilities and the creation of a pro-inflammatory environment (2) that favors the development of disease states (3). Osteoporosis is a metabolic bone disease associated with aging in both men and women, although women are typically more deleteriously affected due to hormonal changes that occur during menopause (4,5). Aberrant bone metabolism is a key feature in the pathophysiology of osteoporosis, in which an imbalance between bone formation and resorption activity occurs (6,7). On the cellular level, this imbalance can be caused by decreased activity or abundance of bone-forming cells (osteoblasts) and/or overactivation or increased abundance of bone resorptive cells (osteoclasts), resulting in a lower bone mineral density (BMD), destruction of bone microstructure, and subsequent bone fracture (4,5).
Bone marrow mesenchymal stem cells (BMSCs) are multipotent cells that can differentiate into various cell lineages including osteoblasts and adipocytes (8). Accordingly, BMSCs are positioned to have a profound influence on bone metabolism and play a key role in metabolic bone diseases including osteoporosis (8-10). BMSC differentiation pathways can be guided via signaling cascades like Wnt/β catenin and Notch (11). Certain transcription factors also play an important role in BMSC differentiation, such that runt-related transcription factor 2 (Runx2) and osterix (Osx) are critical for osteoblastogenesis, and peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/enhancer-binding protein alpha (C/EBPα) enhance adipogenesis (9,11,12). It has been reported that the cellular composition of bone marrow changes with age (13,14). Bone marrow adipocytes (BMAds) are sparse in the bone marrow at early stages of life, but gradually accumulate in long bones beginning around puberty (13). With older age, bone marrow adiposity increases significantly resulting in the development of a fatty marrow (13). On a molecular level, expression levels of adipogenic transcription factors such as PPARγ2 are elevated with advancing age (13). Furthermore, older osteoblasts have been found to accumulate more intracellular lipids than younger osteoblasts, which could contribute to their diminished function and, in turn, to greater marrow adiposity with age (15,16). This accumulation may in part be facilitated by epigenetic changes. For example, we showed that expression of the epigenetic enzyme Hdac3 was reduced in aged wild-type osteoblasts (16), and BMSCs from Hdac3-insufficient mice cultured in an osteogenic medium accumulated more lipid droplets compared to cells from control mice (16). At the organ level, conditional deletion of Hdac3 in Osterix-expressing cells of young mice resulted in a phenotype of low bone mass and elevated abundance of bone marrow adipose tissue (BMAT) (17), which may have been related to these cellular-level changes.
Both osteoblasts and bone marrow adipocytes (BMAds) are derived from the same pool of multipotent mesenchymal stem cell progenitors, and bone marrow space is a unique location where skeletal and adipose tissues directly interface (18), although emerging data from large-scale single-cell RNA-sequencing (scRNA-seq) studies has shown that preadipocyte-like cells represent a distinct state from osteoblasts and their precursors among BMSCs (19-22). These studies identified unique clusters of adipocyte precursor cells (preadipocyte-like cells) that express the leptin receptor (Lepr), in addition to cell clusters that might correspond to skeletal stem and progenitor cells, preosteoblasts, and other stromal cells (23). BMAT can be further characterized into two distinct subpopulations: regulated BMAT (rBMAT) located in regions such as the proximal tibia and distal femur, and constitutive BMAT (cBMAT) which can be found in locations like the distal tibia and caudal vertebra (24,25). By definition, rBMAT is more responsive to stimuli than cBMAT (25). While the functions of BMAT are still being elucidated, key studies have shown that BMAT is an important endocrine organ in that it produces physiologically relevant levels of adiponectin during caloric restriction (26), aids in regulation of glucose homeostasis (27), and serves as an important energy source for the skeleton during metabolic challenges such as injury repair, cold exposure, and caloric restriction (28). An inverse relationship between bone and BMAT abundance has been reported in a variety of disease states and natural phenomena, including aging, estrogen deficiency, anorexia, calorie restriction, diabetes, and physical disuse (24,29-34). BMAT also expands in cases of spinal cord injury, high-fat diet, and exogenous glucocorticoid treatment (12,35-37), and an increase in BMAT is negatively correlated with outcomes in multiple hematopoietic disorders (38). Historically speaking, this often-observed inverse relationship between BMAT and bone density was interpreted to indicate that BMAT is a negative regulator of bone mass (25,30,31,39), either due to preferential differentiation of BMSC into the BMAd lineage at the expense of osteoblastogenesis, or due to crosstalk between osteoblast and BMAds. For example, when osteoblasts are cocultured with adipocytes or adipocyte-conditioned media, osteoblastic differentiation is inhibited (40,41). Interestingly, however, several prior investigations have shown that BMAT is not necessarily a causal factor for osteoporosis, as elimination of BMAds did not improve bone mass (42-44), and bone loss can occur independently of BMAT expansion (45). As a result, and considered in light of the emerging roles for BMAT that have been uncovered in recent studies (26-28), a more accurate interpretation is that BMAT likely plays a variety of physiological and pathophysiological roles which are yet to be fully understood.
In elucidating the complex relationship between bone and BMAT, glucocorticoids (GCs) are positioned to play a key role, as they have been implicated in the decision of BMSCs to differentiate into osteogenic or adipogenic lineages (9). GCs are essential steroid hormones that orchestrate the stress response of the body and circadian rhythm (9). They are functionally involved in many physiological processes including growth, immune responses, and bone metabolism (9). Although great attention has been paid to the deleterious skeletal effects of exogenous GC in causing glucocorticoid-induced osteoporosis (GIO), endogenous physiological GC have been much less extensively studied (46). Notably, it has been proposed that increases in circulating endogenous glucocorticoids drive BMAT expansion during the metabolic challenge of caloric restriction (45). The purpose of this review is to illuminate aspects of both endogenous and exogenous GC signaling, including the influence of GC receptors, in bone aging and the relationship of this signaling to marrow adiposity. In particular, this review will attempt to provide an update on the current understanding of the role of glucocorticoids in the biology of osteoblasts and BMAds.
2. Exogenous glucocorticoids in osteoblasts and BMAds
Exogenous GC are chemically synthesized molecules used therapeutically to treat inflammatory diseases such as rheumatoid arthritis (47). Commonly used GC therapies include dexamethasone, prednisone, methylprednisolone, and hydrocortisone, which vary in their relative potencies and activity (Table 1) (48-51). In vitro and in vivo studies have established that high levels of pharmacological GCs exert detrimental effects on all stages of osteoblastogenesis (12,52,53). The underlying molecular mechanism involves suppression of key signaling pathways that are essential for differentiation of BMSCs to osteoblasts, such as the WNT pathway. In fact, exogenous GC can induce osteoblast apoptosis, which decreases bone density and elevates the risk of fracture in animal models and in humans (54). In addition, previous studies have shown that humans and rodents that are exposed to exogenous GCs have fewer osteoblast progenitor cells and increased BMAT (12,55). Mechanistically, GCs inhibit osteoblast progenitor cells through upregulating cell cycle inhibitors such as p21, p27, and p53, and downregulating cell cycle activators including cyclin D3, cyclin-dependent kinase (CDK) 4 and CDK6 (56-58). Moreover, it has been shown that exposure to supra-physiological levels of GCs leads to significant suppression of bone formation. Accordingly, serum markers of osteocalcin, procollagen type I N-terminal propeptide (P1NP), and bone-specific alkaline phosphatase (ALP) are reduced upon GC treatment (59). The secretion of collagen and osteocalcin is also reduced upon exposure to exogenous GCs, which indicates that bone mineralization was negatively influenced. Furthermore, GCs increase bone matrix degradation by upregulating the synthesis of metalloproteinases (59).
Table 1:
Pharmacological characteristics of exogenous GCs
| Name | Equivalent glucocorticoid dose (mg) |
Duration of action in humans (hrs) |
Potency relative to hydrocortisone (cortisol) |
|---|---|---|---|
| Cortisone | 25 | Short (8-12) | 0.8 |
| Hydrocortisone (cortisol) | 20 | Short (8-12) | 1 |
| Prednisone | 5 | Intermediate (12-36) | 4 |
| Prednisolone | 5 | Intermediate (12-36) | 4 |
| Triamcinolone | 4 | Intermediate (12-36) | 5 |
| Methylprednisolone | 4 | Intermediate (12-36) | 5 |
| Dexamethasone | 0.75 | Long (36-72) | 30 |
| Betamethasone | 0.6 | Long (36-72) | 30 |
The use of exogenous GCs also affects osteocytes, which are terminally differentiated osteoblasts fully embedded in mineralized matrix. Several in vivo and in vitro studies have demonstrated that exogenous GCs increase apoptosis and reduce the lifespan of osteocytes (60-63). However, other studies failed to correlate reduced bone mass upon GC exposure with higher osteocyte apoptosis (64). One of the possible explanations for this discrepancy is that exogenous GCs exert their effects on bone cells in a dose and time-dependent manner (54,63). Dose-dependent effects can be due to the modulation of autophagy and antioxidant gene expression which eventually leads to apoptosis (54,63,65). Exogenous GCs influence osteocytes in several ways, including up-regulating the production and secretion of sclerostin and dickkopf-related protein-1 (Dkk-1) (66). These two proteins inhibit bone formation by blocking the osteoblastic Wnt/β-catenin signaling pathway (67). Importantly, sclerostin has been shown to increase adipogenesis in a variety of experimental models, which could contribute to the mechanisms by which GC influence BMAds (68-70). GCs also compromise bone strength via effects on the osteocyte lacunocanalicular system; GCs increase the lacunar size and change the bone matrix (26). In vitro studies have found that exogenous GCs have detrimental effects on connectivity between osteocytes by disrupting gap junctions between cells (71).
It has been well established that GC treatment reduces bone mass and elevates BMAT (12). Both osteoblasts and adipocytes are derived from progenitor populations within the broader BMSC pool, and previous studies have suggested that when BMSCs commit to one lineage, they may begin to lose the ability to differentiate along another lineage (72). To understand the molecular mechanism underlying dexamethasone-induced osteoporosis and fat accumulation, Li et al. treated mice with dexamethasone to create an osteoporotic model (12). They found that upon dexamethasone treatment, BMSC differentiation shift towards adipocytes rather than osteoblasts. They also demonstrated that dexamethasone inhibits the methylation of the C/EBPα promoter (a key transcription factor for adipocyte differentiation) through the Wnt/beta-catenin pathway. As a result, C/EBPα expression is significantly higher in dexamethasone-treated mice, consistent with their increased bone marrow adiposity (12). Other studies have shown that adipocytes secrete substances that negatively influence osteoblast differentiation and function (73-75). For example, Wang et al. co-cultured adipocytes and osteoblasts obtained from a human mesenchymal stem cell line with dexamethasone, finding that six days of co-culture in the presence of dexamethasone reduces alkaline phosphatase activity, bone mineralization and expression of osteogenic markers like Runx2 and osteocalcin (74). The conditioned media of dexamethasone-treated co-cultures contains higher fatty acid levels compared to the control co-cultures (74). Further investigation revealed that fatty acids are produced by adipocytes, leading to increased reactive oxygen species levels, which eventually results in osteoblast apoptosis (74). A recent clinical study also indicated that patients with glucocorticoid-induced osteoporosis present with altered BMAT lipid profiles as compared to patients with non-GC-related osteoporosis (76).
MicroRNAs are single-stranded non-coding RNAs that regulate a plethora of biological functions (77). Many microRNAs have been associated with bone health including miR-106b (77), and miR-188 (78). Kang et al. found that the synthetic GC dexamethasone regulates microRNA-34a-5p, which is crucial to the dexamethasone-mediated inhibition of BMSC proliferation and osteogenic differentiation (79). This effect is established through targeting of CDK4, CDK6, and Cyclin D1, and activation of Notch signaling with the ultimate outcome of inhibiting osteogenic differentiation (79). Wang et al. showed that dexamethasone induces high miR-133a expression, and a miR-133a antagomir significantly enhances osteoblast differentiation and inhibits adipocyte differentiation, highlighting the role of synthetic GC’s effects on microRNAs in the process (80). Dexamethasone was also found to inhibit let-7f-5p expression, and Shen et al. showed that let-7f-5p reverses dexamethasone-induced bone loss in mice (81). MicroRNAs transported in exosomes (extracellular vesicles containing cargoes of proteins, lipids, and nucleic acids) (82), may play a role in cellular crosstalk between adipocytes and osteoblasts (83,84), and there is growing evidence showing that certain exosome cargoes can counter deleterious effects of GC in bone (52,85,86). However, it remains unknown whether GCs regulate exosome secretion and function in various bone cells and bone marrow adipocytes and if this plays a role in GC-induced osteoporosis.
3. Endogenous glucocorticoids in osteoblasts and BMAds
Endogenous GCs are steroid hormones synthesized and released by the adrenal glands in response to biological, physiological, and environmental cues. The hypothalamic-pituitary-adrenal axis regulates GC secretion (46,87). Physiological and biological triggers like changes in metabolism, cardiac output, or inflammation trigger the release of corticotropin-releasing hormone (CRH) from the hypothalamus (87). This hormone stimulates the anterior pituitary to release adrenocorticotropic hormone (ACTH), which in turn acts on the zona fasciculata of the adrenal cortex to synthesize GC from its precursor cholesterol (9,87). Cells of zona fasciculata produce and release active GCs (corticosterone in rodents and mainly cortisol in humans) within 5 min of an ACTH pulse (88,89). However, more than 90% of GCs are bound by corticosteroid-binding globulins, which renders them biologically inactive (90). When released from protein binding, free GCs can exert their effects in target tissues (90). Local activity of GCs is regulated through the enzymes 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and -HSD2. 11β-HSD2 inactivates cortisol and corticosterone through an oxidation reaction which converts them to cortisone and 11-dehydrocorticosterone (11DHC), respectively (91). This is especially important in mineralocorticoid (aldosterone) target tissues, to prevent GCs from occupying mineralocorticoid receptors (MRs) (92). As a result, 11β-HSD2 protects organs such kidney (93), placenta (94), and colon (95) from overactivation by GCs. In contrast, 11β-HSD1 is present throughout most tissues in the body (9,96); it reactivates cortisone and 11DHC through a reduction reaction to yield functional (active) cortisol and corticosterone (96,97). In fact, 11β-HSD1’s action in a given cell was proven to be predominantly GC recycling, acting as a local signaling amplifier (98,99) to render local levels of both active and inactive GCs equally potent in activating target receptors, as described below (99).
Once inside cells, active GCs can exert their function through genomic or non-genomic pathways. In the genomic pathway, GCs bind to a target receptor such as the glucocorticoid receptor (GR) (9). Subsequently, the receptor-GC complex is translocated to the nucleus, where it binds directly to a glucocorticoid response element (GRE) sequence in the DNA of target genes and acts as a transcription factor to elicit altered gene expression and protein synthesis (9). Non-genomic GC-signaling, in contrast, occurs rapidly and does not necessarily involve protein synthesis (100). Instead, once GC are bound to their target receptor, chaperone proteins are released from the complex and play a role in secondary signaling cascades utilizing the activity of various kinases like mitogen-activated protein kinases (MAPKs) and AKT (100).
Although less extensively studied than exogenous GCs, several previous articles make the case that endogenous GCs play a key role in the differentiation of BMSC-lineage cells. Sher et al. created a transgenic mouse model that overexpressed 11β-HSD2 in osteoblasts and demonstrated that GCs are needed for proper skeletal development, as local inactivation of GC led to vertebral osteopenia and decreased cortical bone area and thickness (101). Interestingly, overexpression of 11β-HSD2 in this manner displayed a sexually dimorphic effect, with female mice more adversely affected than males (101). In another report, a similar mouse model displayed delayed cranial bone formation and could not properly resorb parietal cartilage postnatally (102). These effects were attributed to reduced expression of Wnt9a and Wnt10b (102). In addition, β-catenin levels were reduced in BMSCs, chondrocytes and osteoblasts, which disrupted the activity of matrix metalloproteinase 14 and reduced cranial cartilage removal (102). Therefore, the authors concluded that endogenous GCs are important regulators of bone formation, acting in a paracrine mechanism not only to direct BMSC commitment towards the osteoblastic lineage but also to control cartilage dissolution postnatally (102). Similarly, Kalak et al. studied a Col2.3-11β-HSD2 transgenic mouse model and showed that young (3- and 7-week-old) mice with 11β-HSD2 overexpression in osteoblasts had reduced mechanical bone strength compared to wild-type mice, highlighting the important role of GCs in bone development and the maintenance of a healthy bone phenotype (103). Several other studies showed comparable findings, in which the overexpression of 11β-HSD2 reduced bone mass, trabecular number, and bone strength (104,105). Ex vivo studies demonstrated lower alkaline phosphatase staining, reduced levels of bone sialoprotein, and decreased osteocalcin mRNA expression in cultures derived from transgenic mice with 11β-HSD2 overexpression (104). Examining another aspect of GC activation, Justensen et al. showed that mice with germline (whole body) deletion of 11β-HSD1 presented with little to no BMAT compared to wild-type mice, but their bone formation rate and bone mass were comparable (96). Consistent with this role for 11β-HSD1, we showed that the loss of Hdac3 in Osterix-expressing osteoprogenitors increased expression of 11β-HSD1 and lipid storage genes like Plin1 and Fsp27/Cidec (16), as well as increasing bone marrow adiposity. These results suggest that high endogenous GC activity contributes to increased marrow adiposity (16). In contrast, Zhou et al. noted that osteogenic cultures from mice that overexpress 11β-HSD2 in osteoblasts had a higher number of adipocytes compared to cultures from wild-type mice (106). They also indicated that Wnt signaling was reduced in transgenic mice with 11β-HSD2 overexpression, explaining the shift from osteogenic to adipogenic lineage commitment (106). Taken together, these studies demonstrate that tight regulation of endogenous GCs is important in the balance of osteogenesis and adipogenesis.
Despite the evidence showing that endogenous GCs are needed for osteoblastogenesis, when GC levels increase substantially in diseases such as Cushing’s syndrome, they negatively influence bone (107). A mouse model for Cushing’s syndrome exhibits low bone mineral density, osteoblast numbers, and bone formation rates along with higher numbers of BMAds in the bone marrow (107). Similarly, Belaya et al. revealed that hypercortisolism suppresses osteoblast function and maturation through downregulating genes like BMP2 and RUNX2, while upregulating the expression of Wnt-signaling antagonists such as Dkk1 and SOST (108). Levels of certain miRNAs known to suppress osteoblastogenesis including miR-125b-5p, miR-218-5p, miR-34a-5p are also altered (108). Tauchmanova et al. designed a cross-sectional case control study to evaluate the effect of excess endogenous GCs on vertebral fractures and bone mass in human subjects (53). Their results showed that the prevalence of vertebral fractures is significantly higher in patients with GC excess as compared to healthy controls (53). Among the different etiologies studied, cortisol levels are highest in patients with ectopic ACTH secretion, which is accompanied by low lumbar bone mineral density, and high fracture frequency (53). The results also indicated that women are more adversely affected by excess cortisol secretion than men and that androgens exhibit protective effect in both genders (53). Another retrospective study on 104 patients with Cushing’s syndrome reported that the increased fracture risk is higher before diagnosis and therapy, but it is minimized after therapy (109). This suggested that bone loss is reversible upon correcting the abnormality, and that reducing GC levels by adrenal or pituitary surgery might reduce the risk of bone fractures (109). Relating more specifically to BMAT, a recent clinical study of young individuals revealed that daily adrenal GC secretion levels are inversely related to bone marrow density, i.e., directly correlated with BMAT abundance (36). Collectively, these studies described above suggest that the impact of GC on bone and BMAT depends on the concentrations, with low amounts favoring bone formation, but excessive levels adversely affecting bone and increasing BMAT.
During aging, the beneficial impact of endogenous glucocorticoids on bone may be blunted and instead contribute to bone loss in the aged skeleton (46). Dysregulated glucocorticoid signaling may contribute to age-related bone loss through effects on osteoblastic senescence and osteoprogenitor lineage selection bias (i.e., affecting the ratio of osteoblastic versus adipogenic differentiation of progenitor cells) even in young individuals (36,37,110). In aging individuals, local activation of endogenous GCs by 11β-HSD1 is substantially enhanced; this phenomenon contributes to increased cortisol levels and altered circadian cortisol variation, both of which may be associated with bone frailty (111-113). Weinstein et al. demonstrated that the production of GCs from the adrenal gland increases in aged mice, and that the effect of GCs is potentiated due to higher local expression of 11β-HSD1 (114). The resulting high level of active GCs adversely influences bone since it reduces the lifespan of osteoblasts and osteocytes, decreases bone vasculature, and impairs the transport of solutes from peripheral circulation to the lacunar-canalicular system (114). The authors concluded that GCs are causative in the observed bone phenotype because transgenic mice with overexpression of 11β-HSD2 are protected against the deleterious effects of GCs in bone and show higher bone mass with age (114). This finding contrasts with results in younger animals, in which bone density, strength and histomorphometry are not different between 11β-HSD2 and wild-type mice (54). A more recent study demonstrated that skeletal GC signaling is involved in bone’s role as an endocrine organ, as Col2.3-11β-HSD2-overexpressing mice are protected against the development of age-related obesity and insulin resistance, but intriguingly found a contrasting phenotype to that of Weinstein et al, in that 11β-HSD2-overexpressing mice have a mild phenotype of decreased trabecular bone mass in the spine (115). Thus, the role of endogenous GCs in skeletal aging, at least at the level of the active GCs ligand, remains somewhat unclear.
4. GC Receptors in osteoblasts and BMAds:
GCs can exert downstream effects via activating the glucocorticoid receptor (GR), a transcription factor encoded by the NR3C1 gene (116,117). GR is a member of the nuclear receptor (NR) superfamily of intracellular receptors belonging to subclass 3C of the steroid/thyroid hormone receptor superfamily (118). Nuclear receptors, including mineralocorticoid receptor (MR), progesterone receptor (PR), estrogen receptor (ER), and androgen receptor (AR), contain a variable N-terminal domain (NTD), a DNA binding domain (DBD), a hinge region, a conserved ligand-binding domain (LBD), and a variable C-terminal domain (119). Phylogenetic analysis by Vlachakis et al. revealed a novel evolutionary relationship between the LBD of NRs, indicating a potential functional overlap in LBD between members of the NR superfamily (120). Indeed, GCs can bind to both GR and MR as target receptors, with MR having much higher affinity for GC than GR (121). Consequently, GR tends to be bound by GCs during stress and at the circadian peak of GC secretion, while MR is occupied at basal hormone levels (121,122). Nonetheless, in MR-rich tissues, the effect of GCs is often limited through 11β-HSD2, which unidirectionally converts active GCs to an inactive form (9,96,121).
The role of GR-mediated signaling of endogenous GCs in bone health has been investigated by us and others through the use of osteoblast-targeted disruption of GR signaling in rodent models. Rapp et al. showed that global deletion of GR interferes with fracture healing by disrupting post-fracture endochondral ossification (123). Furthermore, Rauch et al. disrupted GC-GR signaling axis in the osteoblasts by utilizing a GRRunx2Cre transgenic mouse line, revealing a significant decrease of trabecular bone density in the GR knockout animals compared to their WT littermates (64). Similarly, our laboratory demonstrated that conditional deletion of GR in Osterix (Osx1) expressing cells (GRosx1Cre) leads to a significant decrease in cortical and trabecular bone mass and a significant increase in BMAT in young, adult, and aged female mice (39,124). Moreover, BMSC-derived osteoblasts from these GR-deficient animals surprisingly still demonstrate excessive lipid storage in the presence of GCs, a phenomenon that can be reversed by treating the cells with MR antagonists (39). Interestingly, Fumoto et al. showed that the deleterious effects of prednisolone are negated either by pharmacological inhibition or knocking down MR in osteocytes (125).
The GC effects that are mediated through GR can be disrupted using GR antagonists (126) (Table 2). Mifepristone, also known as RU486, was first reported in 1981 as a GR antagonist but later recognized as also antagonizing the PR (126-128). Mifepristone was approved by the US Food and Drug Administration (FDA) based on its PR antagonist effects for medical termination of pregnancy through 70 days gestation (129), and later granted approval for the treatment of Cushing’s syndrome due to its GR-targeting effects (130). Despite showing efficacy as a GR antagonist, mifepristone treatment has notable limitations; overtreatment may lead to adrenal insufficiency, increased cortisol levels, endometrial hyperplasia in females, and interference with negative feedback mechanisms of GCs (131,132). Other GR antagonists and selective GR modulators designed to have no effect on PR activity have been investigated and are in various stages of development including PT150 (formerly known as ORG-34517) (133-135). CORT125281 (136), ORIC-101 (137), CORT118335 (miricorilant) (138), CORT108297 (139), CORT125134 (relacorilant) (140), dicyclohexyl phthalate (DCHP) and FX5 (141,142). Among the selective GR modulators, CORT118335 has been found to antagonize MR as well as GR (139). Interestingly, although the deleterious effects of exogenous corticosteroids on bone have been widely reported, comparatively less is known about the skeletal effects of the above listed pharmacological GR antagonists. Mifepristone’s skeletal effects were mainly reported in vitro as counteracting/reversing the effects of exogenous GC (dexamethasone, prednisolone) (143,144), rather than describing its effects independently of GC co-treatment. Mifepristone’s lack of selectivity for the GR, and the relative lack of available FDA approved GR antagonists other than mifepristone have likely contributed to the knowledge gap surrounding the skeletal effects of GR antagonists (128,130).
Table 2:
List of pharmacological GR antagonists
| Name | Nuclear receptor specificity | Reference |
|---|---|---|
| Mifepristone (RU486) | PR antagonist | (128) |
| GR antagonist | (129) | |
| AR agonist/antagonist | (137,157) | |
| PT150 (ORG-34517) | GR antagonist | (133) |
| AR antagonist | (135) | |
| ORIC-101 | GR antagonist | (137) |
| CORT125281 | GR antagonist | (136) |
| CORT118335 (Miricorilant) | GR antagonist | (158) |
| MR antagonist | ||
| CORT108297 | GR antagonist | (139) |
| CORT125134 (Relacorilant) | GR antagonist | (140) |
| Dicyclohexyl phthalate (DCHP) | GR antagonist | (141) |
| FX5 | GR antagonist | (142) |
GR: glucocorticoid receptor, PR: progesterone receptor, MR: mineralocorticoid receptor, AR: androgen receptor
Conclusions:
Although physiological concentrations of GCs are important for healthy bone development and homeostasis, prolonged exposure to elevated GC levels (both endogenous and exogenous) can be harmful to the skeleton through promotion of altered bone remodeling activity, including reduced bone formation. It is important to remember that endogenous GC are counter-regulatory hormones, released during times of stress to help mobilize nutrients including glucose, amino acids, and fatty acids from less immediately critical body systems (e.g., peripheral fat, muscle, bone) to maintain energy homeostasis in systems essential for survival (e.g., brain, heart) (145). To this end, BMAT may represent an important fuel source for hematopoietic and immune cell populations within the bone marrow, as supported by recent publications (28,146). In addition, recent work has shown that BMAds may act as a critical fuel source for bone remodeling cells during times of stress; for instance, lipolysis by BMAds was important for the maintenance of trabecular bone mass under conditions of caloric restriction in male (but not female) mice, as well as for proper bone cell function in the face of cold stress and during energetically-expensive processes of bone repair / regeneration (28,147). These findings are consistent with the idea that fatty acids are an important fuel source for both osteoblasts (148,149) and osteoclasts (150). However, targeted deletion of BMAds provided a net benefit, rather than detriment, to the skeleton, enhancing cortical and trabecular bone formation in cBMAT-rich regions of the skeleton, and protecting against cortical bone loss induced by caloric restriction (146). These studies highlight the important role of BMAds not only as a nearby fuel source for osteoblasts, but also the potential impact of their role as a paracrine source of adipokines and cytokines in times of stress. The specific role of GCs in mediating these dynamics of crosstalk between BMAds and bone remodeling cells in the presence and absence of stress is not yet clear, and represents an exciting area for future research. Deleterious effects of GCs in the skeleton involve several cellular pathways and events and include a complex interplay of both local and systemic factors that bias BMSC differentiation toward adipogenesis (7,11,31,44,151-153), affecting cellular crosstalk between mature BMAds and osteoblasts (154). Several published studies highlight the positive influence of GCs on BMAT development; however, a clear connection between GC effects on BMAT and bone health has yet to be defined (9,12,36,39,40,155,156). Future studies focused on the effects of GCs on crosstalk between bone and BMAT, including the role of various GC-binding nuclear receptors in these mechanisms, are needed to fully elucidate these relationships and their impact on skeletal aging and frailty.
Figure 1:
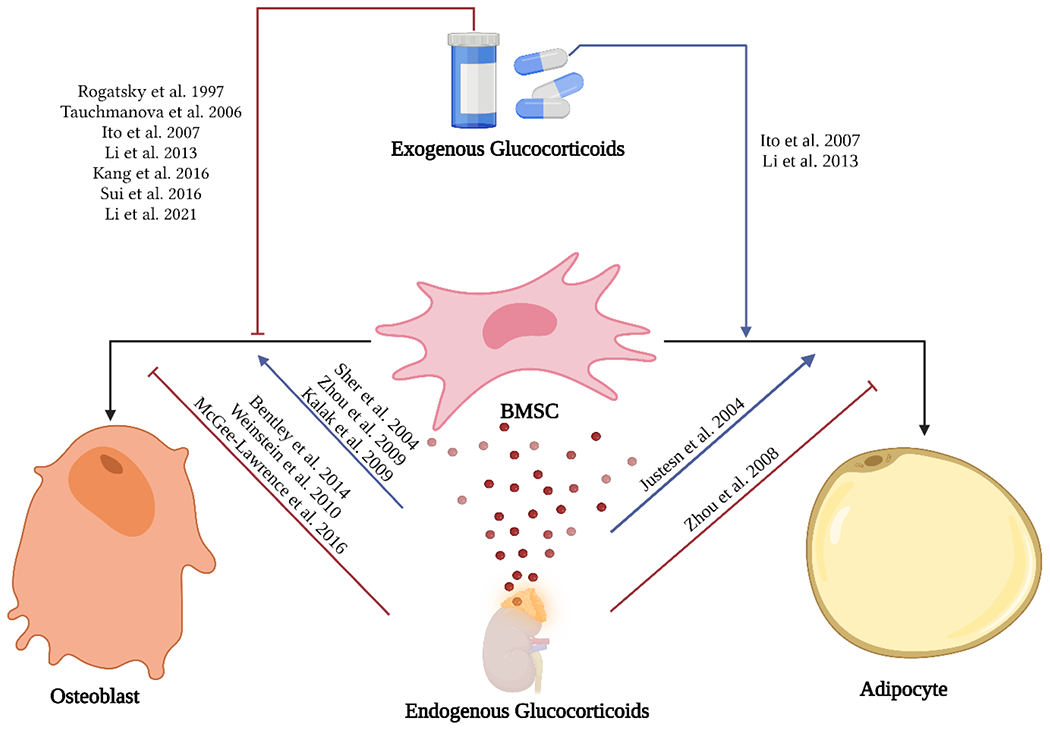
Reported effects of exogenous and endogenous GC on bone marrow stem cells differentiation into osteoblast and adipocyte lineages. Figure created with BioRender.com.
Declarations
Funding and/or conflicts of interests / competing interests: The authors are supported by funding provided by the National Institute on Aging (NIA P01 AG036675, R01 AG 067510 and T35 AG 067577). The contents of this publication do not represent the views of the Department of Veterans Affairs or the United States Government. The authors state that they do not have existing conflicts of interest.
References:
- 1.Hernandez-Segura A, Nehme J, and Demaria M (2018) Hallmarks of Cellular Senescence. Trends in Cell Biology 28, 436–453 [DOI] [PubMed] [Google Scholar]
- 2.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013) The Hallmarks of Aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lecot P, Alimirah F, Desprez PY, Campisi J, and Wiley C (2016) Context-dependent effects of cellular senescence in cancer development. Br J Cancer 114, 1180–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macías I, Alcorta-Sevillano N, Rodríguez CI, and Infante A (2020) Osteoporosis and the Potential of Cell-Based Therapeutic Strategies. Int J Mol Sci 21, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Cai W, Wang G, and Shen X (2020) Prevalence and contributing factors of osteoporosis in the elderly over 70 years old: an epidemiological study of several community health centers in Shanghai. Ann Palliat Med 9, 231–238 [DOI] [PubMed] [Google Scholar]
- 6.Raisz LG (2005) Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest 115, 3318–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell RGG, Espina B, and Hulley P (2006) Bone biology and the pathogenesis of osteoporosis. Current Opinion in Rheumatology 18 [DOI] [PubMed] [Google Scholar]
- 8.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, and Marshak DR (1999) Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 284, 143. [DOI] [PubMed] [Google Scholar]
- 9.Sharma AK, Shi X, Isales CM, and McGee-Lawrence ME (2019) Endogenous Glucocorticoid Signaling in the Regulation of Bone and Marrow Adiposity: Lessons from Metabolism and Cross Talk in Other Tissues. Current Osteoporosis Reports 17, 438–445 [DOI] [PubMed] [Google Scholar]
- 10.Paspaliaris V, and Kolios G (2019) Stem cells in Osteoporosis: From Biology to New Therapeutic Approaches. Stem Cells Int 2019, 1730978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han L, Wang B, Wang R, Gong S, Chen G, and Xu W (2019) The shift in the balance between osteoblastogenesis and adipogenesis of mesenchymal stem cells mediated by glucocorticoid receptor. Stem Cell Res Ther 10, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Zhang N, Huang X, Xu J, Fernandes JC, Dai K, and Zhang X (2013) Dexamethasone shifts bone marrow stromal cells from osteoblasts to adipocytes by C/EBPalpha promoter methylation. Cell Death Dis 4, e832–e832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lecka-Czernik B, Rosen CJ, and Kawai M (2010) Skeletal aging and the adipocyte program: New insights from an “old” molecule. Cell Cycle 9, 3648–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veldhuis-Vlug AG, and Rosen CJ (2018) Clinical implications of bone marrow adiposity. J Intern Med 283, 121–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bethel M, Chitteti BR, Srour EF, and Kacena MA (2013) The changing balance between osteoblastogenesis and adipogenesis in aging and its impact on hematopoiesis. Curr Osteoporos Rep 11, 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGee-Lawrence ME, Carpio LR, Schulze RJ, Pierce JL, McNiven MA, Farr JN, Khosla S, Oursler MJ, and Westendorf JJ (2016) Hdac3 Deficiency Increases Marrow Adiposity and Induces Lipid Storage and Glucocorticoid Metabolism in Osteochondroprogenitor Cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 31, 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, Secreto FJ, Knutson SK, Hiebert SW, and Westendorf JJ (2010) Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS one 5, e11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devlin MJ, and Rosen CJ (2015) The bone–fat interface: basic and clinical implications of marrow adiposity. The Lancet Diabetes & Endocrinology 3, 141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, Guillamot M, Gutkin MC, Zhang Y, Marier C, Diefenbach C, Kousteni S, Heguy A, Zhong H, Fooksman DR, Butler JM, Economides A, Frenette PS, Adams RH, Satija R, Tsirigos A, and Aifantis I (2019) The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, Dionne D, Papazian A, Lee D, Ashenberg O, Subramanian A, Vaishnav ED, Rozenblatt-Rosen O, Regev A, and Scadden DT (2019) A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 177, 1915–1932.e1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, Agarwal P, Bhatia R, Tenen DG, Klein AM, and Welner RS (2019) Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Reports 28, 302–311.e305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolgalev I, and Tikhonova AN (2021) Connecting the Dots: Resolving the Bone Marrow Niche Heterogeneity. Frontiers in Cell and Developmental Biology 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushita Y, Ono W, and Ono N (2022) Toward Marrow Adipocytes: Adipogenic Trajectory of the Bone Marrow Stromal Cell Lineage. Frontiers in Endocrinology 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trudel G, Payne M, Mädler B, Ramachandran N, Lecompte M, Wade C, Biolo G, Blanc S, Hughson R, Bear L, and Uhthoff HK (2009) Bone marrow fat accumulation after 60 days of bed rest persisted 1 year after activities were resumed along with hemopoietic stimulation: the Women International Space Simulation for Exploration study. Journal of Applied Physiology 107, 540–548 [DOI] [PubMed] [Google Scholar]
- 25.Shen W, Chen J, Punyanitya M, Shapses S, Heshka S, and Heymsfield SB (2007) MRI-measured bone marrow adipose tissue is inversely related to DXA-measured bone mineral in Caucasian women. Osteoporos Int 18, 641–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, Ning X, Bree AJ, Schell B, Broome DT, Soliman SS, DelProposto JL, Lumeng CN, Mitra A, Pandit SV, Gallagher KA, Miller JD, Krishnan V, Hui SK, Bredella MA, Fazeli PK, Klibanski A, Horowitz MC, Rosen CJ, and MacDougald OA (2014) Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab 20, 368–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suchacki KJ, Tavares AAS, Mattiucci D, Scheller EL, Papanastasiou G, Gray C, Sinton MC, Ramage LE, McDougald WA, Lovdel A, Sulston RJ, Thomas BJ, Nicholson BM, Drake AJ, Alcaide-Corral CJ, Said D, Poloni A, Cinti S, Macpherson GJ, Dweck MR, Andrews JPM, Williams MC, Wallace RJ, van Beek EJR, MacDougald OA, Morton NM, Stimson RH, and Cawthorn WP (2020) Bone marrow adipose tissue is a unique adipose subtype with distinct roles in glucose homeostasis. Nat Commun 11, 3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Bowers E, Zhu J, Yu H, Hardij J, Bagchi DP, Mori H, Lewis KT, Granger K, Schill RL, Romanelli SM, Abrishami S, Hankenson KD, Singer K, Rosen CJ, and MacDougald OA (2022) Lipolysis of bone marrow adipocytes is required to fuel bone and the marrow niche during energy deficits. Elife 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz AV, Sigurdsson S, Hue TF, Lang TF, Harris TB, Rosen CJ, Vittinghoff E, Siggeirsdottir K, Sigurdsson G, Oskarsdottir D, Shet K, Palermo L, Gudnason V, and Li X (2013) Vertebral Bone Marrow Fat Associated With Lower Trabecular BMD and Prevalent Vertebral Fracture in Older Adults. The Journal of Clinical Endocrinology & Metabolism 98, 2294–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandra A, Lagnado AB, Farr JN, Schleusner M, Monroe DG, Saul D, Passos JF, Khosla S, and Pignolo RJ (2022) Bone Marrow Adiposity in Models of Radiation- and Aging-Related Bone Loss Is Dependent on Cellular Senescence. J Bone Miner Res 37, 997–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Lu L, Liu Y, and Yu X (2022) Bone marrow adiposity during pathologic bone loss: molecular mechanisms underlying the cellular events. Journal of Molecular Medicine 100, 167–183 [DOI] [PubMed] [Google Scholar]
- 32.Bredella MA, Fazeli PK, Miller KK, Misra M, Torriani M, Thomas BJ, Ghomi RH, Rosen CJ, and Klibanski A (2009) Increased Bone Marrow Fat in Anorexia Nervosa. The Journal of Clinical Endocrinology & Metabolism 94, 2129–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, Baron R , Rosen CJ, and Bouxsein ML (2010) Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 25, 2078–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baum T, Yap SP, Karampinos DC, Nardo L, Kuo D, Burghardt AJ, Masharani UB, Schwartz AV, Li X, and Link TM (2012) Does vertebral bone marrow fat content correlate with abdominal adipose tissue, lumbar spine bone mineral density, and blood biomarkers in women with type 2 diabetes mellitus? Journal of Magnetic Resonance Imaging 35, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorgey AS, Poarch HJ, Adler RA, Khalil RE, and Gater DR (2013) Femoral bone marrow adiposity and cortical bone cross-sectional areas in men with motor complete spinal cord injury. PM & R : the journal of injury, function, and rehabilitation 5, 939–948 [DOI] [PubMed] [Google Scholar]
- 36.Esche J, Shi L, Hartmann MF, Schonau E, Wudy SA, and Remer T (2019) Glucocorticoids and Body Fat Inversely Associate With Bone Marrow Density of the Distal Radius in Healthy Youths. J Clin Endocrinol Metab 104, 2250–2256 [DOI] [PubMed] [Google Scholar]
- 37.Vande Berg BC, Malghem J, Lecouvet FE, Devogelaer JP, Maldague B, and Houssiau FA (1999) Fat conversion of femoral marrow in glucocorticoid-treated patients: A cross-sectional and longitudinal study with magnetic resonance imaging. Arthritis & Rheumatism 42, 1405–1411 [DOI] [PubMed] [Google Scholar]
- 38.Li Z, and MacDougald OA (2021) Preclinical models for investigating how bone marrow adipocytes influence bone and hematopoietic cellularity. Best Practice & Research Clinical Endocrinology & Metabolism 35, 101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierce JL, Sharma AK, Roberts RL, Yu K, Irsik DL, Choudhary V, Dorn JS , Bensreti H, Benson RD Jr, Kaiser H, Khayrullin A, Davis C, Wehrle CJ, Johnson MH, Bollag WB, Hamrick MW, Shi X, Isales CM, and McGee-Lawrence ME (2022) The Glucocorticoid Receptor in Osterix-Expressing Cells Regulates Bone Mass, Bone Marrow Adipose Tissue, and Systemic Metabolism in Female Mice During Aging. Journal of Bone and Mineral Research 37, 285–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romacho T, Elsen M, Röhrborn D, and Eckel J (2014) Adipose tissue and its role in organ crosstalk. Acta Physiol (Oxf) 210, 733–753 [DOI] [PubMed] [Google Scholar]
- 41.Maurin AC, Chavassieux PM, Frappart L, Delmas PD, Serre CM, and Meunier PJ (2000) Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone 26, 485–489 [DOI] [PubMed] [Google Scholar]
- 42.Iwaniec UT, and Turner RT (2013) Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone 53, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Botolin S, and McCabe LR (2006) Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. Journal of cellular physiology 209, 967–976 [DOI] [PubMed] [Google Scholar]
- 44.Almeida M, Kim HN, Han L, Zhou D, Thostenson J, Porter RM, Ambrogini E, Manolagas SC, and Jilka RL (2020) Increased marrow adipogenesis does not contribute to age-dependent appendicular bone loss in female mice. Aging Cell 19, e13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cawthorn WP, Scheller EL, Parlee SD, Pham HA, Learman BS, Redshaw CM, Sulston RJ, Burr AA, Das AK, Simon BR, Mori H, Bree AJ, Schell B, Krishnan V, and MacDougald OA (2016) Expansion of Bone Marrow Adipose Tissue During Caloric Restriction Is Associated With Increased Circulating Glucocorticoids and Not With Hypoleptinemia. Endocrinology 157, 508–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou H, Cooper MS, and Seibel MJ (2013) Endogenous Glucocorticoids and Bone. Bone Research 1, 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buttgereit F, Burmester GR, Straub RH, Seibel MJ, and Zhou H (2011) Exogenous and endogenous glucocorticoids in rheumatic diseases. Arthritis Rheum 63, 1–9 [DOI] [PubMed] [Google Scholar]
- 48.Meikle AW, and Tyler FH (1977) Potency and duration of action of glucocorticoids. Effects of hydrocortisone, prednisone and dexamethasone on human pituitary-adrenal function. Am J Med 63, 200–207 [DOI] [PubMed] [Google Scholar]
- 49.Parente L. (2017) Deflazacort: therapeutic index, relative potency and equivalent doses versus other corticosteroids. BMC Pharmacology and Toxicology 18, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caplan A, Fett N, Rosenbach M, Werth VP, and Micheletti RG (2017) Prevention and management of glucocorticoid-induced side effects: A comprehensive review: A review of glucocorticoid pharmacology and bone health. Journal of the American Academy of Dermatology 76, 1–9 [DOI] [PubMed] [Google Scholar]
- 51.Zoorob RJ, and Cender D (1998) A different look at corticosteroids. Am Fam Physician 58, 443–450 [PubMed] [Google Scholar]
- 52.Li Z, Liu C, Li S, Li T, Li Y, Wang N, Bao X, Xue P, and Liu S (2021) BMSC-Derived Exosomes Inhibit Dexamethasone-Induced Muscle Atrophy via the miR-486-5p/FoxO1 Axis. Frontiers in Endocrinology 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tauchmanova L, Pivonello R, Di Somma C, Rossi R, De Martino MC, Camera L, Klain M, Salvatore M, Lombardi G, and Colao A (2006) Bone demineralization and vertebral fractures in endogenous cortisol excess: role of disease etiology and gonadal status. J Clin Endocrinol Metab 91, 1779–1784 [DOI] [PubMed] [Google Scholar]
- 54.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, and Weinstein RS (2004) Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145, 1835–1841 [DOI] [PubMed] [Google Scholar]
- 55.Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X, Zheng C, Li M, Wu L, Zhao X, and Jin Y (2016) Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci Rep 6, 30186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogatsky I, Trowbridge JM, and Garabedian MJ (1997) Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol 17, 3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang JK, Li CJ, Liao HJ, Wang CK, Wang GJ, and Ho ML (2009) Anti-inflammatory drugs suppress proliferation and induce apoptosis through altering expressions of cell cycle regulators and pro-apoptotic factors in cultured human osteoblasts. Toxicology 258, 148–156 [DOI] [PubMed] [Google Scholar]
- 58.Li H, Qian W, Weng X, Wu Z, Li H, Zhuang Q, Feng B, and Bian Y (2012) Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS One 7, e37030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gado M, Baschant U, Hofbauer LC, and Henneicke H (2022) Bad to the Bone: The Effects of Therapeutic Glucocorticoids on Osteoblasts and Osteocytes. Front Endocrinol (Lausanne) 13, 835720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, Kinney JH, and Bonewald LF (2006) Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 21, 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinstein RS, Jilka RL, Parfitt AM, and Manolagas SC (1998) Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 102, 274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato AY, Tu X, McAndrews KA, Plotkin LI, and Bellido T (2015) Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone 73, 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, Bonewald L, Jiang JX, and Lane NE (2011) Glucocorticoid dose determines osteocyte cell fate. Faseb j 25, 3366–3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, Ostermay S, Schinke T, Spanbroek R, Zaiss MM, Angel PE, Lerner UH, David JP, Reichardt HM, Amling M, Schutz G, and Tuckermann JP (2010) Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab 11, 517–531 [DOI] [PubMed] [Google Scholar]
- 65.Zhang S, Liu Y, and Liang Q (2018) Low-dose dexamethasone affects osteoblast viability by inducing autophagy via intracellular ROS. Mol Med Rep 17, 4307–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao W, Dai W, Jiang L, Lay EY, Zhong Z, Ritchie RO, Li X, Ke H, and Lane NE (2016) Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 27, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krishnan V, Bryant HU, and Macdougald OA (2006) Regulation of bone mass by Wnt signaling. J Clin Invest 116, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, Mohanty ST, Croucher P, Kramer I, Kneissel M, Rosen CJ, and Reagan MR (2018) The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. Journal of cellular physiology 233, 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim SP, Da H, Wang L, Taketo MM, Wan M, and Riddle RC (2021) Bone-derived sclerostin and Wnt/beta-catenin signaling regulate PDGFRalpha(+) adipoprogenitor cell differentiation. FASEB J 35, e21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balani DH, Trinh S, Xu M, and Kronenberg HM (2021) Sclerostin Antibody Administration Increases the Numbers of Sox9creER+ Skeletal Precursors and Their Progeny. J Bone Miner Res 36, 757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao J, Cheng TS, Qin A, Pavlos NJ, Wang T, Song K, Wang Y, Chen L, Zhou L, Jiang Q, Takayanagi H, Yan S, and Zheng M (2016) Glucocorticoid impairs cell-cell communication by autophagy-mediated degradation of connexin 43 in osteocytes. Oncotarget 7, 26966–26978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abdallah BM, and Kassem M (2012) New factors controlling the balance between osteoblastogenesis and adipogenesis. Bone 50, 540–545 [DOI] [PubMed] [Google Scholar]
- 73.Maurin AC, Chavassieux PM, Vericel E, and Meunier PJ (2002) Role of polyunsaturated fatty acids in the inhibitory effect of human adipocytes on osteoblastic proliferation. Bone 31, 260–266 [DOI] [PubMed] [Google Scholar]
- 74.Wang D, Haile A, and Jones LC (2013) Dexamethasone-induced lipolysis increases the adverse effect of adipocytes on osteoblasts using cells derived from human mesenchymal stem cells. Bone 53, 520–530 [DOI] [PubMed] [Google Scholar]
- 75.Elbaz A, Wu X, Rivas D, Gimble JM, and Duque G (2010) Inhibition of fatty acid biosynthesis prevents adipocyte lipotoxicity on human osteoblasts in vitro. J Cell Mol Med 14, 982–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martel D, Leporq B, Saxena A, Belmont HM, Turyan G, Honig S, Regatte RR, and Chang G (2019) 3T chemical shift-encoded MRI: Detection of altered proximal femur marrow adipose tissue composition in glucocorticoid users and validation with magnetic resonance spectroscopy. J Magn Reson Imaging 50, 490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu K, Jing Y, Zhang W, Fu X, Zhao H, Zhou X, Tao Y, Yang H, Zhang Y, Zen K, Zhang C, Li D, and Shi Q (2017) Silencing miR-106b accelerates osteogenesis of mesenchymal stem cells and rescues against glucocorticoid-induced osteoporosis by targeting BMP2. Bone 97, 130–138 [DOI] [PubMed] [Google Scholar]
- 78.Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY, Xia ZY, Zhou H , Cao X, Xie H, Liao EY, and Luo XH (2015) MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest 125, 1509–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang H, Chen H, Huang P, Qi J, Qian N, Deng L, and Guo L (2016) Glucocorticoids impair bone formation of bone marrow stromal stem cells by reciprocally regulating microRNA-34a-5p. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 27, 1493–1505 [DOI] [PubMed] [Google Scholar]
- 80.Wang G, Wang F, Zhang L, Yan C, and Zhang Y (2021) miR-133a silencing rescues glucocorticoid-induced bone loss by regulating the MAPK/ERK signaling pathway. Stem Cell Res Ther 12, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen GY, Ren H, Shang Q, Zhao WH, Zhang ZD, Yu X, Huang JJ, Tang JJ, Yang ZD, Liang D, and Jiang XB (2019) Let-7f-5p regulates TGFBR1 in glucocorticoid-inhibited osteoblast differentiation and ameliorates glucocorticoid-induced bone loss. Int J Biol Sci 15, 2182–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalluri R, and LeBleu Valerie S (2020) The biology, function, and biomedical applications of exosomes. Science 367, eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu Y, Wang C, Wei M, Yang G, and Yuan L (2021) Multifaceted Roles of Adipose Tissue-Derived Exosomes in Physiological and Pathological Conditions. Front Physiol 12, 669429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quan M, and Kuang S (2020) Exosomal Secretion of Adipose Tissue during Various Physiological States. Pharm Res 37, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tao SC, Yuan T, Rui BY, Zhu ZZ, Guo SC, and Zhang CQ (2017) Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway. Theranostics 7, 733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nan K, Zhang Y, Zhang X, Li D, Zhao Y, Jing Z, Liu K, Shang D, Geng Z, and Fan L (2021) Exosomes from miRNA-378-modified adipose-derived stem cells prevent glucocorticoid-induced osteonecrosis of the femoral head by enhancing angiogenesis and osteogenesis via targeting miR-378 negatively regulated suppressor of fused (Sufu). Stem Cell Res Ther 12, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kadmiel M, and Cidlowski JA (2013) Glucocorticoid receptor signaling in health and disease. Trends in Pharmacological Sciences 34, 518–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fudulu DP, Horn G, Hazell G, Lefrantçois-Martinez AM, Martinez A, Angelini GD, Lightman SL, and Spiga F (2021) Co-culture of monocytes and zona fasciculata adrenal cells: An in vitro model to study the immune-adrenal cross-talk. Mol Cell Endocrinol 526, 111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pon LA, Hartigan JA, and Orme-Johnson NR (1986) Acute ACTH regulation of adrenal corticosteroid biosynthesis. Rapid accumulation of a phosphoprotein. J Biol Chem 261, 13309–13316 [PubMed] [Google Scholar]
- 90.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, and Stewart PM (2004) 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 25, 831–866 [DOI] [PubMed] [Google Scholar]
- 91.Almanzar G, Mayerl C, Seitz JC, Hofner K, Brunner A, Wild V, Jahn D, Geier A, Fassnacht M, and Prelog M (2016) Expression of 11beta-hydroxysteroid-dehydrogenase type 2 in human thymus. Steroids 110, 35–40 [DOI] [PubMed] [Google Scholar]
- 92.Albiston AL, Obeyesekere VR, Smith RE, and Krozowski ZS (1994) Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol 105, R11–17 [DOI] [PubMed] [Google Scholar]
- 93.Luft FC (2016) 11 beta-Hydroxysteroid Dehydrogenase-2 and Salt-Sensitive Hypertension. Circulation 133, 1335–1337 [DOI] [PubMed] [Google Scholar]
- 94.Salvante KG, Milano K, Kliman HJ, and Nepomnaschy PA (2017) Placental 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2) expression very early during human pregnancy. J Dev Orig Health Dis 8, 149–154 [DOI] [PubMed] [Google Scholar]
- 95.Whorwood CB, Ricketts ML, and Stewart PM (1994) Epithelial cell localization of type 2 11 beta-hydroxysteroid dehydrogenase in rat and human colon. Endocrinology 135, 2533–2541 [DOI] [PubMed] [Google Scholar]
- 96.Justesen J, Mosekilde L, Holmes M, Stenderup K, Gasser J. r., Mullins JJ, Seckl JR, and Kassem M (2004) Mice Deficient in 11β-Hydroxysteroid Dehydrogenase Type 1 Lack Bone Marrow Adipocytes, but Maintain Normal Bone Formation. Endocrinology 145, 1916–1925 [DOI] [PubMed] [Google Scholar]
- 97.Boucher E, Provost PR, and Tremblay Y (2014) Ontogeny of adrenal-like glucocorticoid synthesis pathway and of 20alpha-hydroxysteroid dehydrogenase in the mouse lung. BMC Res Notes 7, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chapman K, Holmes M, and Seckl J (2013) 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev 93, 1139–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS, and Chapman KE (2006) Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol 176, 7605–7611 [DOI] [PubMed] [Google Scholar]
- 100.Ramamoorthy S, and Cidlowski JA (2016) Corticosteroids: Mechanisms of Action in Health and Disease. Rheum Dis Clin North Am 42, 15–31, vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sher LB, Woitge HW, Adams DJ, Gronowicz GA, Krozowski Z, Harrison JR, and Kream BE (2004) Transgenic expression of 11 beta-hydroxysteroid dehydrogenase type 2 in osteoblasts reveals an anabolic role for endogenous glucocorticoids in bone. Endocrinology 145, 922–929 [DOI] [PubMed] [Google Scholar]
- 102.Zhou H, Mak W, Kalak R, Street J, Fong-Yee C, Zheng Y, Dunstan CR, and Seibel MJ (2009) Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development 136, 427–436 [DOI] [PubMed] [Google Scholar]
- 103.Kalak R, Zhou H, Street J, Day RE, Modzelewski JR, Spies CM, Liu PY, Li G, Dunstan CR, and Seibel MJ (2009) Endogenous glucocorticoid signalling in osteoblasts is necessary to maintain normal bone structure in mice. Bone 45, 61–67 [DOI] [PubMed] [Google Scholar]
- 104.Yang M, Trettel LB, Adams DJ, Harrison JR, Canalis E, and Kream BE (2010) Col3.6-HSD2 transgenic mice: a glucocorticoid loss-of-function model spanning early and late osteoblast differentiation. Bone 47, 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sher LB, Harrison JR, Adams DJ, and Kream BE (2006) Impaired cortical bone acquisition and osteoblast differentiation in mice with osteoblast-targeted disruption of glucocorticoid signaling. Calcif Tissue Int 79, 118–125 [DOI] [PubMed] [Google Scholar]
- 106.Zhou H, Mak W, Zheng Y, Dunstan CR, and Seibel MJ (2008) Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J Biol Chem 283, 1936–1945 [DOI] [PubMed] [Google Scholar]
- 107.Bentley L, Esapa CT, Nesbit MA, Head RA, Evans H, Lath D, Scudamore CL, Hough TA, Podrini C, Hannan FM, Fraser WD, Croucher PI, Brown MA, Brown SD, Cox RD, and Thakker RV (2014) An N-ethyl-N-nitrosourea induced corticotropin-releasing hormone promoter mutation provides a mouse model for endogenous glucocorticoid excess. Endocrinology 155, 908–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Belaya ZE, Grebennikova TA, Melnichenko GA, Nikitin AG, Solodovnikov AG, Brovkina OI, Grigoriev AU, Rozhinskaya LY, and Dedov II. (2018) Effects of endogenous hypercortisolism on bone mRNA and microRNA expression in humans. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 29, 211–221 [DOI] [PubMed] [Google Scholar]
- 109.Vestergaard P, Lindholm J, Jorgensen JO, Hagen C, Hoeck HC, Laurberg P, Rejnmark L, Brixen K, Kristensen LO, Feldt-Rasmussen U, and Mosekilde L (2002) Increased risk of osteoporotic fractures in patients with Cushing’s syndrome. Eur J Endocrinol 146, 51–56 [DOI] [PubMed] [Google Scholar]
- 110.Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, and Tuckermann JP (2016) Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol Rev 96, 409–447 [DOI] [PubMed] [Google Scholar]
- 111.Zhao ZY, Lu FH, Xie Y, Fu YR, Bogdan A, and Touitou Y (2003) Cortisol secretion in the elderly. Influence of age, sex and cardiovascular disease in a Chinese population. Steroids 68, 551–555 [DOI] [PubMed] [Google Scholar]
- 112.Reynolds RM, Dennison EM, Walker BR, Syddall HE, Wood PJ, Andrew R, Phillips DI, and Cooper C (2005) Cortisol secretion and rate of bone loss in a population-based cohort of elderly men and women. Calcified tissue international 77, 134–138 [DOI] [PubMed] [Google Scholar]
- 113.Johar H, Emeny RT, Bidlingmaier M, Reincke M, Thorand B, Peters A, Heier M, and Ladwig KH (2014) Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab 99, E464–468 [DOI] [PubMed] [Google Scholar]
- 114.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, and Manolagas SC (2010) Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging cell 9, 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henneicke H, Kim S, Swarbrick MM, Li J, Gasparini SJ, Thai J, Foong D, Cavanagh LL, Fong-Yee C, Karsten E, Lin RCY, Cooper MS, Zhou H, and Seibel MJ (2020) Skeletal glucocorticoid signalling determines leptin resistance and obesity in aging mice. Molecular metabolism 42, 101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vandewalle J, Luypaert A, De Bosscher K, and Libert C (2018) Therapeutic Mechanisms of Glucocorticoids. Trends in Endocrinology & Metabolism 29, 42–54 [DOI] [PubMed] [Google Scholar]
- 117.Timmermans S, Souffriau J, and Libert C (2019) A General Introduction to Glucocorticoid Biology. Frontiers in Immunology 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, and De Bosscher K (2013) How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol 380, 41–54 [DOI] [PubMed] [Google Scholar]
- 119.Porter BA, Ortiz MA, Bratslavsky G, and Kotula L (2019) Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers (Basel) 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mitsis T, Papageorgiou L, Efthimiadou A, Bacopoulou F, Vlachakis D, Chrousos GP, Eliopoulos E, Mitsis T, Papageorgiou L, Efthimiadou A, Bacopoulou F, Vlachakis D, Chrousos GP, and Eliopoulos E (2019) A comprehensive structural and functional analysis of the ligand binding domain of the nuclear receptor superfamily reveals highly conserved signaling motifs and two distinct canonical forms through evolution. World Acad Sci J. 1, 264–274 [Google Scholar]
- 121.Meijer OC, Buurstede JC, and Schaaf MJM (2019) Corticosteroid Receptors in the Brain: Transcriptional Mechanisms for Specificity and Context-Dependent Effects. Cellular and Molecular Neurobiology 39, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koning A, Buurstede JC, van Weert L, and Meijer OC (2019) Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. JEndocr Soc 3, 1917–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rapp AE, Hachemi Y, Kemmler J, Koenen M, Tuckermann J, and Ignatius A (2018) Induced global deletion of glucocorticoid receptor impairs fracture healing. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 32, 2235–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pierce JL, Ding KH, Xu J, Sharma AK, Yu K, Del Mazo Arbona N, Rodriguez-Santos Z, Bernard P, Bollag WB, Johnson MH, Hamrick MW, Begun DL, Shi XM, Isales CM, and McGee-Lawrence ME (2019) The glucocorticoid receptor in osteoprogenitors regulates bone mass and marrow fat. J Endocrinol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fumoto T, Ishii KA, Ito M, Berger S, Schutz G, and Ikeda K (2014) Mineralocorticoid receptor function in bone metabolism and its role in glucocorticoid-induced osteopenia. Biochemical and biophysical research communications 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 126.Castinetti F, Conte-Devolx B, and Brue T (2010) Medical Treatment of Cushing’s Syndrome: Glucocorticoid Receptor Antagonists and Mifepristone. Neuroendocrinology 92(suppl 1), 125–130 [DOI] [PubMed] [Google Scholar]
- 127.Mahajan DK, and London SN (1997) Mifepristone (RU486): a review. Fertil Steril 68, 967–976 [DOI] [PubMed] [Google Scholar]
- 128.Adashi EY, Rajan RS, O’Mahony DP, and Cohen IG (2022) The next two decades of mifepristone at FDA: History as destiny. Contraception 109, 1–7 [DOI] [PubMed] [Google Scholar]
- 129.Beaman J, Prifti C, Schwarz EB, and Sobota M (2020) Medication to Manage Abortion and Miscarriage. Journal of General Internal Medicine 35, 2398–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Molitch ME (2022) Glucocorticoid receptor blockers. Pituitary [DOI] [PubMed] [Google Scholar]
- 131.Meijer OC, Koorneef LL, and Kroon J (2018) Glucocorticoid receptor modulators. Ann Endocrinol (Paris) 79, 107–111 [DOI] [PubMed] [Google Scholar]
- 132.Spitz IM, Grunberg SM, Chabbert-Buffet N, Lindenberg T, Gelber H, and Sitruk-Ware R (2005) Management of patients receiving long-term treatment with mifepristone. Fertility and Sterility 84, 1719–1726 [DOI] [PubMed] [Google Scholar]
- 133.Morice C, Baker DG, Patel MM, Nolen TL, Nowak K, Hirsch S, Kosten TR, and Verrico CD (2021) A randomized trial of safety and pharmacodynamic interactions between a selective glucocorticoid receptor antagonist, PT150, and ethanol in healthy volunteers. Scientific Reports 11, 9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rocha SM, Fagre AC, Latham AS, Cummings JE, Aboellail TA, Reigan P, Aldaz DA, McDermott CP, Popichak KA, Kading RC, Schountz T, Theise ND, Slayden RA, and Tjalkens RB (2022) A Novel Glucocorticoid and Androgen Receptor Modulator Reduces Viral Entry and Innate Immune Inflammatory Responses in the Syrian Hamster Model of SARS-CoV-2 Infection. Front Immunol 13, 811430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tatman P, Fringuello A, Graner M, Lillehei K, Parasido E, Albanese C, Tewari AK, Chakravarty D, Nair S, and Theise N (2020) Pan-cancer analysis to identify a novel class of glucocorticoid and androgen receptor antagonists with potent anti-tumor activity. Journal of Clinical Oncology 38, e15663–e15663 [Google Scholar]
- 136.Hunt HJ, Belanoff JK, Walters I, Gourdet B, Thomas J, Barton N, Unitt J, Phillips T, Swift D, and Eaton E (2017) Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. Journal of Medicinal Chemistry 60, 3405–3421 [DOI] [PubMed] [Google Scholar]
- 137.Rew Y, Du X, Eksterowicz J, Zhou H, Jahchan N, Zhu L, Yan X, Kawai H, McGee LR, Medina JC, Huang T, Chen C, Zavorotinskaya T, Sutimantanapi D, Waszczuk J, Jackson E, Huang E, Ye Q, Fantin VR, and Sun D (2018) Discovery of a Potent and Selective Steroidal Glucocorticoid Receptor Antagonist (ORIC-101). Journal of Medicinal Chemistry 61, 7767–7784 [DOI] [PubMed] [Google Scholar]
- 138.Khom S, Rodriguez L, Gandhi P, Kirson D, Bajo M, Oleata CS, Vendruscolo LF, Mason BJ, and Roberto M (2022) Alcohol dependence and withdrawal increase sensitivity of central amygdalar GABAergic synapses to the glucocorticoid receptor antagonist mifepristone in male rats. Neurobiol Dis 164, 105610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Buurstede JC, Umeoka EHL, da Silva MS, Krugers HJ, Joëls M, and Meijer OC (2022) Application of a pharmacological transcriptome filter identifies a shortlist of mouse glucocorticoid receptor target genes associated with memory consolidation. Neuropharmacology 216, 109186. [DOI] [PubMed] [Google Scholar]
- 140.Pivonello R, Bancos I, Feelders RA, Kargi AY, Kerr JM, Gordon MB, Mariash CN, Terzolo M, Ellison N, and Moraitis AG (2021) Relacorilant, a Selective Glucocorticoid Receptor Modulator, Induces Clinical Improvements in Patients With Cushing Syndrome: Results From A Prospective, Open-Label Phase 2 Study. Front Endocrinol (Lausanne) 12, 662865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leng Y, Sun Y, Huang W, Lv C, Cui J, Li T, and Wang Y (2021) Identification of dicyclohexyl phthalate as a glucocorticoid receptor antagonist by molecular docking and multiple in vitro methods. Molecular Biology Reports 48, 3145–3154 [DOI] [PubMed] [Google Scholar]
- 142.Xu X, Chen Y, Zhu D, Zhao T, Xu R, Wang J, Hu L, and Shen X (2020) FX5 as a non-steroidal GR antagonist improved glucose homeostasis in type 2 diabetic mice via GR/HNF4α/miR-122-5p pathway. Aging (Albany NY) 13, 2436–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Feger M, Ewendt F, Strotmann J, Schäffler H, Kempe-Teufel D, Glosse P, Stangl GI, and Föller M (2021) Glucocorticoids dexamethasone and prednisolone suppress fibroblast growth factor 23 (FGF23). J Mol Med (Berl) 99, 699–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pierce JL, Sharma AK, Roberts RL, Yu K, Irsik DL, Choudhary V, Dorn JS, Bensreti H, Benson RD Jr., Kaiser H, Khayrullin A, Davis C, Wehrle CJ, Johnson MH, Bollag WB, Hamrick MW, Shi X, Isales CM, and McGee-Lawrence ME (2022) The Glucocorticoid Receptor in Osterix-Expressing Cells Regulates Bone Mass, Bone Marrow Adipose Tissue, and Systemic Metabolism in Female Mice During Aging. J Bone Miner Res 37, 285–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jacobson L, Ansari T, Potts J, and McGuinness OP (2006) Glucocorticoid-deficient corticotropin-releasing hormone knockout mice maintain glucose requirements but not autonomic responses during repeated hypoglycemia. Am J Physiol Endocrinol Metab 291, E15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li Z, Bagchi DP, Zhu J, Bowers E, Yu H, Hardij J, Mori H, Granger K, Skjaerlund JD, Mandair GS, Abrishami S, Singer K, Hankenson KD, Rosen CJ, and MacDougald OA (2022) Constitutive bone marrow adipocytes suppress local bone formation. JCI Insight [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rendina-Ruedy E, and Rosen CJ (2020) Lipids in the Bone Marrow: An Evolving Perspective. Cell metabolism 31, 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, Ryan KA, Goh BC, Scafidi S, Pickett JE, Faugere MC, Kershaw EE, Thorek DLJ, Clemens TL, Wolfgang MJ, and Riddle RC (2017) Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dirckx N, Moorer MC, Clemens TL, and Riddle RC (2019) The role of osteoblasts in energy homeostasis. Nat Rev Endocrinol 15, 651–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kushwaha P, Alekos NS, Kim SP, Li Z, Wolfgang MJ, and Riddle RC (2022) Mitochondrial fatty acid beta-oxidation is important for normal osteoclast formation in growing female mice. Front Physiol 13, 997358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Phinney DG (2012) Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem 113, 2806–2812 [DOI] [PubMed] [Google Scholar]
- 152.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, and Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 [DOI] [PubMed] [Google Scholar]
- 153.Li J, Zuo B, Zhang L, Dai L, and Zhang X (2018) Osteoblast versus Adipocyte: Bone Marrow Microenvironment-Guided Epigenetic Control. Case Reports in Orthopedic Research 1, 2–18 [Google Scholar]
- 154.Flehr A, Källgård J, Alvén J, Lagerstrand K, Papalini E, Wheeler M, Vandenput L, Kahl F, Axelsson KF, Sundh D, Mysore RS, and Lorentzon M (2022) Development of a novel method to measure bone marrow fat fraction in older women using high-resolution peripheral quantitative computed tomography. Osteoporosis International 33, 1545–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.He HP, and Gu S (2021) The PPAR-γ/SFRP5/Wnt/β-catenin signal axis regulates the dexamethasone-induced osteoporosis. Cytokine 143, 155488. [DOI] [PubMed] [Google Scholar]
- 156.Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C, and Haché RJ (2003) Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. Embo j 22, 2135–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Song LN, Coghlan M, and Gelmann EP (2004) Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol 18, 70–85 [DOI] [PubMed] [Google Scholar]
- 158.Mammi C, Marzolla V, Armani A, Feraco A, Antelmi A, Maslak E, Chlopicki S, Cinti F, Hunt H, Fabbri A, and Caprio M (2016) A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. International Journal of Obesity 40, 964–972 [DOI] [PubMed] [Google Scholar]