

Summary
Despite the knowledge that protein translation and various metabolic reactions that create and sustain cellular life occur in the cytoplasm, the structural organization within the cytoplasm remains unclear. Recent models indicate that cytoplasm contains viscous fluid and elastic solid phases. We separated these viscous fluid and solid elastic compartments, which we call the cytosol and cytomatrix, respectively. The distinctive composition of the cytomatrix included structural proteins, ribosomes, and metabolome enzymes. High-throughput analysis revealed unique biosynthetic pathways within the cytomatrix. Enrichment of biosynthetic pathways in the cytomatrix indicated the presence of immobilized biocatalysis. Enzymatic immobilization and segregation can surmount spatial impediments, and the local pathway segregation may form cytoplasmic organelles. Protein translation was reprogrammed within the cytomatrix under the restriction of protein synthesis by drug treatment. The cytosol and cytomatrix are an elaborately interconnected network that promotes operational flexibility in healthy cells and the survival of malignant cells.
Subject areas: Molecular biology, Cell biology, Proteomics, Transcriptomics
Graphical abstract
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.isci.2023.105965.
Highlights
-
•
The cytoplasm is a two-phase system consisting of viscous fluid and elastic solid
-
•
The elastic solid can be separated, from the viscous fluid, as the cytomatrix
-
•
The cytomatrix immobilizes catalytic complexes for efficient biocatalysis
-
•
Immobilization and segregation of catalytic complexes and proteome can form organelles
Molecular biology; Cell biology; Proteomics; Transcriptomics
Introduction
The central problem of cell biology is the organization of biochemical reactions and organelles in the cytoplasm and how organelles need to be assembled in the cytoplasmic space after cell division or depending on cell conditions. Since the 19th century, scientists have made substantial efforts to understand the physical and chemical properties of the cytoplasm, and debated the nature of the cytoplasm.1 Still, the structural organization of the cytoplasm remains a contentious topic. For the past hundred years since its inception, the concept that cell cytoplasm is a gel-like substance has been broadly acknowledged.2 However, the mechanisms of chemical reaction in the cell are routinely explained based on the idea of free diffusion, as if the cells were an aqueous solution.3 The cytoplasm is a dynamic, crowded, and heterogeneous environment, but this concept gives little help in understanding the cytoplasmic structure and organization. The cytoplasm is involved in protein synthesis, energy generation, nutrient processing, transport, and metabolism, which also contributes to nuclear processes in response to external stimuli or stressors. Capable of managing a myriad of chemical reactions, the structure of the cytoplasm is complex, with enzymes and metabolic pathways organized into clusters,4 with little space available for soluble species to roam.5
Current theories suggest that the cytomatrix is an elastic solid within which intermediate filaments anchor organelles—interdependent structures that communicate through contact sites6—against fluctuating cytosolic forces.7 Further, the structure of a large portion of the intracellular water is dictated by interactions with the cell ultrastructure, driving changes in cytoplasmic states from a viscous fluid to an elastic solid.8
The cytomatrix (CMX) is thought to be a proteinaceous matrix that forms a network with the cytoskeleton to integrate metabolic pathways.1 The CMX is traversed by channels and delimited by microtubules throughout which it associates with organelles, ER ribosomes, and metabolic protein complexes.9 Structural proteins are also an integral part of the cytoplasmic matrix, wherein the nucleus and cytoplasm are connected through the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex.10,11,12 The integrity of protein structure and macromolecular complexes within the CMX are supported by the ionic microenvironment that maintains a thermodynamically favored stable equilibrium.13 The potassium ion is the principal intracellular electrolyte necessary for the function of all living cells and the most abundant intracellular cation1 involved in protein synthesis. Potassium ions are involved in the stabilization of main functional ligands such as mRNA and transfer RNAs, as well as ribosomal RNAs and ribosomal proteins.14
Mounting evidence suggests that among the solid macromolecular matrix exists a viscous liquid, consisting of both water-soluble proteins and biopolymers along with lipid membrane-associated and lipid-wrapped protein cargo. Thus, from a physicochemical standpoint, the cytoplasm could be considered a two-phase system consisting of a viscous fluid and an elastic solid. To date, however, biophysical analysis and microscopic techniques have not distinguished the viscous fluid and elastic solid elements. Moreover, the cytoplasm and cytomatrix are often defined as equal terms. Hence, the question of how to isolate the viscous fluid phase (i.e., cytosol) from the elastic solids component (CMX) remains.
The only way to reveal the elastic solid matrix (i.e., CMX) is to chemically remove the viscous fluid (i.e., cytosol). Removing the viscous fluid may not compromise the integrity of the CMX and connections with the nucleus through the LINC complex under certain conditions. Organelles differ in their molecular structure, particularly in the composition of proteins and other biopolymers that dictate their specific physicochemical characteristics. Subcellular fractionation methods are based on the ability of cell organelles to withstand specific salt and detergent concentrations. It has been shown that the inner nuclear membrane (INM) is resistant to high salt and detergent concentrations, and that the nuclear pore complex (NPC) does not require an intact outer nuclear membrane.15 However, it took the work of several generations of cell biologists to determine that treating cells with the nonionic detergents such as Triton X-100 does not cause spilling of nuclear contents due to the discovery that the INM and NPC proteins are associated with the nuclear lamina.16 To this end, it was shown that an approximately 15-nm-thick layer lamina with attached NPCs encircle the entire nucleus, but does not extend into its interior.17 Prior studies have also shown that the Golgi matrix is a detergent- and salt-resistant complex that is easily isolated from purified rat liver Golgi.18
As first reported in 1888, the effects of inorganic salts on proteins and macromolecules in aqueous solutions generally follow a Hofmeister series.19,20 Specifically, the effects of cations are important for protein folding, protein−protein interactions, and protein aggregation. Strongly hydrated cations such as lithium (Li+) are the most effective for salting proteins into solution, whereas weakly hydrated cations lead to salting-out behavior. When the interaction of different cations with the model compound, butyramide, was studied, it was shown to have statistically significant binding amid oxygen for Li+ but weak binding for Ca++ and Mg++ and exclusion for Na+ and K+.21
Since it has been established that Li+ interacts with peptide groups,22 lithium salts have been widely investigated as a member of the Hofmeister series. In model polymers such as oligoglycines, which are insoluble in water, lithium salts increase the solubility of polyglycine, a property that likely contributes to the denaturing ability of lithium salts.23 For alkali cations, the bond dissociation energies for Li-ions are the highest.24 As well, direct protein-ion interactions are responsible for the Hofmeister effects of ions on protein stability. Further, at low ion concentrations (<100-200 mM), Li-ions specifically interact with biopolymers.25 Similarly, it has been shown that Li cations exhibit a strong salting-in effect through improved solvation of the amide carbonyl group.26 The charge-dense Li ions dissolve hydrophobic molecules. Li cations, however, can also interact with a variety of functional groups, suggesting that the apparent Li-induced lowering of hydrophobicity may be a result of specific interactions between the functional groups and Li ions.
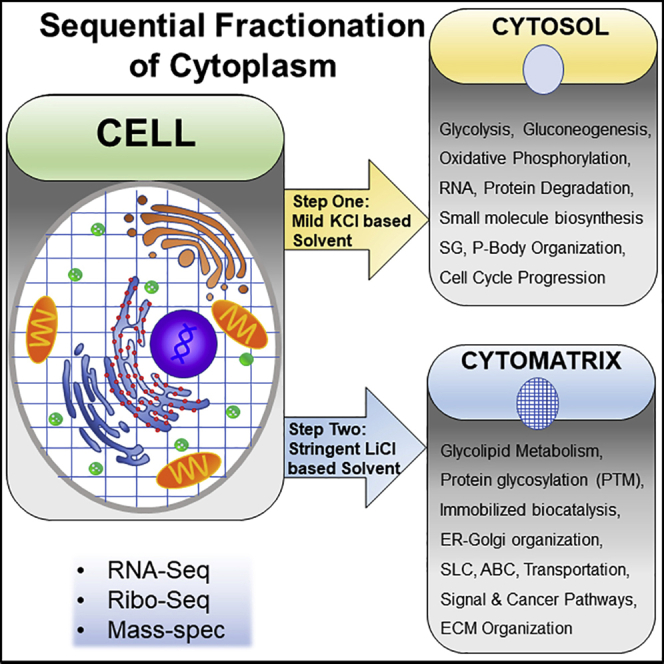
Recently, we used LiCl salt to separate a nuclear-associated fraction from the cytosolic and nuclear fractions in an embryonic fibroblast and two cancer cell lines, an approach that distinguishes our method from traditional cell fractionation methods.27 Our current work enabled us to define the nuclear-associated cytoplasmic fraction without the cytosol as the cytomatrix. In our extraction protocol, we used Hofmeister’s series of ion properties and developed an equation to calculate ionic concentrations for separating the CMX and the cytosol. The cytosol was dissolved using a detergent-containing isotonic buffer that does not cause osmotic stress due to the same osmotic pressure. The CMX was then separated from the nuclear fraction with the LiCl salt-containing stringent isotonic buffer, and the extract was solubilized with the DOX/Triton X-100. Hence, we distinguish the cytomatrix as a detergent and salt-resistant composite, the elastic solid phase of cytoplasm. In contrast, we define the cytosol as a detergent-sensitive, dynamic liquid phase of the cytoplasm that can be isolated using nonionic detergents under the physiological osmotic conditions.
Here, we determined compartmentalization of transcriptome, proteome, metabolome, and protein synthesis within the CMX and found that CMX-associated ribosomes are distinct in mRNA transcript assortment. Enrichment of biosynthetic pathways indicated the presence of immobilized biocatalysis within the cytomatrix. Efficient biocatalysis achieved by segregating and immobilizing the metabolome enzymes can overcome spatial impediments to biochemical reactions, which implies the CMX involvement in cytoplasmic organelle formation and assembly. Accordingly, the dynamics of the cytoplasmic organelles may depend on the organization of the CMX. High-throughput analysis and conventional methods revealed different responses from the cytosol and CMX during times of drug-induced protein deficiency. The reprogrammed protein translation within the cytomatrix shows operational flexibility of the cytoplasmic phases that promote cell survival under extreme conditions.
Results
The cytoplasm is a two-phase system
Our objective was to isolate the CMX and compare this elastic solid phase of the cytoplasm with the cytosolic viscous fluid, or liquid, phase of the cytoplasm. To this end, our two-step strategy for isolating the CMX (Figure S1A) included the initial separation of the dynamic, viscous fluid fraction of the cytoplasm using a nonionic detergent. To avoid osmotic stress and keep the CMX intact, we used a physiological concentration of potassium ions and a minimal solvent volume. The first dissolved cellular material was the cytoplasmic solution, otherwise known as the cytosol. In the second step, we used a stringent buffered solvent, containing lithium ions, to separate the CMX from the nucleus. Thus, using two buffers we separated the cytosolic, CMX, and nuclear fractions. Two different nonionic detergents (NP-40 and Triton X-100) were used to solubilize the cytoplasmic material. To prevent rupturing the basic structure of organelles, the cytosol and CMX were extracted under physiologically osmotic conditions. During the first step of the separation technique, the mild buffer extracted only the cytosol while the CMX was pelleted with the nucleus as a non-dissolved solid phase (Figure S1A). The CMX was subsequently isolated from the nucleus using a second, stringent solvent.
We found that the viscous fluid dynamic phase of the cytoplasm was salt and detergent sensitive and could be extracted using the nonionic detergent NP-40 or its analog. To prevent damage to the elastic solid cytomatrix, we used potassium salt, which replicates the natural ionic environment of the cell. Contrary to the cytosol, the CMX was salt and detergent resistant but sensitive to lithium salt. Thus, we used LiCl to dissociate the CMX from the nucleus and to break up the macromolecular complexes and Triton X-100/DOX for solubilization. Due to the strong salting-in or dissolving effect of Li cations, our method required a precise calculation of the lithium salt concentration to ensure that dissociating the CMX did not rupture the nucleus. The KCl concentration was also calculated to avoid the osmotic stress to keep the CMX intact during the cyto-solution (cytosol) extraction. A saline solution containing 0.9% sodium chloride (154 mM NaCl) was used as the standard to calculate the LiCl and KCl salt concentrations in our solvents.
In aqueous solutions, salts are dissociated to ions and ions are surrounded by a hydration or solvation shell. The relative ionic radius28 is a solvation shell of ions in solution that surround proteins or protein complexes in the cell. Because the ionic ratio of a protein solution depends on the relative ionic radius, we developed an equation to calculate the concentration of ions to replace sodium ions in the standard isotonic solution based on the relative ionic radius (Figure S1B). We found that solutions containing 120 mM KCl and 200 mM LiCl do not cause osmotic pressure. Further, our calculated concentration was similar to the concentration values obtained experimentally for erythrocyte hemolysis,29 in which the isotonic molarity of the LiCl solution was 0.189 M. Experimentally, it was confirmed that 0.2 M LiCl salt does not rupture the nucleus, but if this concentration were to be exceeded, the nuclear contents would spill out.
The solubilization of lipid bilayers using detergents is a slow process at 4°C,30 and can involve several steps. The sequence of events that must occur include: 1) relatively rapid penetration of detergent monomers into the outer lipid monolayer; 2) transmembrane equilibration of detergent monomers between the two monolayers; 3) saturation of the lipid bilayer by detergents and consequent permeabilization of the membrane; and 4) transition of the entire lipid bilayer to thread-like mixed micelles.31,32 Mixing the collected cells via rotation with Buffer A for 30 min provided time for the micelles, which envelop lipids and proteins immersed in lipid bilayers, to form (Figure S1C). Therefore, water-soluble extracts of the cytosolic cellular contents, and lipids of the ER, plasma membrane (PM), and outer nuclear membrane (ONM), would be contained within the suspended micelles and dispersed in a liquid colloid. Hence, the formation and stabilization of the micelles prevent post-lysis reassortment.33
To validate the fractionation method, we compared our protein extraction procedure with a widely used cell lysis buffer from Cell Signaling Technology (cat# 9803). The percent of proteins in each fraction was determined by dividing the concentration of proteins found in each fraction by the total protein concentration. Approximately 70% of the total colorectal cancer cell (HCT-15) proteins were detected in the cytosol. Further, the remaining proteins were roughly equally distributed between the nuclear and CMX fractions (Figure S1D). Our results were consistent with previous findings in four other cancer and normal cell lines.27 Notably, the concentration of proteins in the cytosol fraction was approximately 10% greater using the conventional cell lysis buffer containing 150 mM NaCl, indicating that CMX protein recovery was divided between the cytosolic and the nuclear fractions (Figure S1E).
To distinguish the cytomatrix from the cytosol we studied marker proteins of the corresponding fractions. Western blot analysis of proteins in the cytosolic, CMX, and nuclear fractions from HCT-15 colon cancer cells shows the differential distribution of well-known cellular markers (Figure 1A). The nuclear envelope proteins SUN2 and emerin were found in the CMX and nuclear fractions. With a lower molecular weight, emerin was spotted only in the CMX. Conversely, the nucleolar protein B23 was found predominantly in the nuclear fraction. The nuclear pore complex protein Nup 98 was detected exclusively in the CMX, suggesting a potential new marker for this compartment. The NPC attached to the INM17 cannot be extracted to the cytosol by the mild isotonic first buffer, even if the ONM dissolves.
Figure 1.

Illustration of cytomatrix isolation
The cytosol and cytomatrix were sequentially extracted from HCT-15 cells. The cytosol was extracted with nonionic detergent containing mild solvent, whereas the cytomatrix was separated from the nuclear fraction using a buffered, stringent solvent.
(A) Immunoblots of marker proteins in fractions.
(B) Immunofluorescence image of keratin localization in cytosol-removed cells.
(C) Immunofluorescence images of SUN2 (red) and Nesprin-1 (green) localization in HeLa cells.
(D) Nesprin-1 is predominantly localized around the nucleus in SUN2 knockdown cells.
(E-J) TEM micrographs of HCT-15 cells. Ultrastructure of the nuclear periphery of (E and F) an intact cell and (G-I) a cytosol-removed cell. (J) cytosol and cytomatrix removed core nuclei.
(K) Protein expression in the cytosol versus cytomatrix of three cell lines. The cytosol and cytomatrix were run side by side, but bands were cut based on cell line to show differences in protein expression in corresponding compartments. Western blots were run in the same gel. The three cell lines show consistent results for predominantly cytosolic proteins (upper panel), equally distributed proteins (middle panel), and predominantly cytomatrix proteins (bottom panel). Cyt – cytosol, CMX – cytomatrix, M− mitochondria, INM – inner nuclear membrane, ONM – outer nuclear membranes, PM – plasma membrane carcass, PNS – perinuclear space.
The GM130 protein was previously identified in the Golgi matrix fraction as an inter-cisternal structural component.34 Because it is known that the Golgi matrix is detergent and salt resistant,18 we did not expect GM13035 to be detected in the cytosol fraction. To this end, we found that GM130 was the most abundant protein detected within the CMX fraction, indicating that the Golgi matrix could be an integral part of the CMX. Cellular markers also were visualized by immunofluorescence. After cytosol extraction, keratin filaments and the LINC complex proteins (i.e., SUN2 and Nesprin-1) were localized to the nuclear surface (Figures 1B-1D). The nucleus is well protected by the LINC complex proteins and keratin, which allowed us to separate the core nucleus from the CMX. A previous study demonstrated a keratin cage formation in cultured monolayers of living cells by time-lapse imaging36 that supports our finding of the keratin encapsulation of the nucleus.
The nuclear membrane is resistant to high salts and detergents due to nuclear encapsulation with filamentous proteins.15 Finally, in addition to the LINC complex, approximately 200 proteins protect the INM,37 which prevented a rupture of the nucleus and allowed us to separate the CMX from the nucleus with the second Li+ containing stringent buffer.
Transmission electron microscopy (TEM) was used to visualize the outcome of fractionation at the ultrastructural level. The resulting micrographs revealed that intact HCT-15 cells had a well-defined perinuclear space, nuclear envelope, Golgi, mitochondria, and ribosomes (Figures 1E and 1F). After removing the cytosol, the nucleus had a higher electron density, likely due to cytoplasmic lipid deprivation that contrasted the nucleus on the cytoplasmic background (Figures 1G-1I). The INM, the carcass of the PM and ribosomes were clearly visible. The outer nuclear membrane was dissolved. Notably, the buffer used to extract the CMX from the nucleus caused chromatin condensation (Figure 1J). Our results indicate that the CMX buffer partially dissolved the LINC complex bridge,38 separating the CMX from the nuclear matrix. Thus, the CMX appeared to stretch from the INM to the lipid-depleted carcass of the PM.
The isolation of the CMX was validated in two additional digestive system cell lines, AGS and HepG2 cells (Figure 1K). Calnexin (CNX) and calreticulin (CalR) are chaperons that control proper protein folding in the ER in cooperation with the BiP, ERp57, PDI, and Ero-1-Lα proteins.39,40 Except for CNX (Figure 1K, middle panel), which is equally distributed between the cytosol and CMX, all other members of the protein folding process were detected predominantly in the cytosol (Figures 1A and 1K).
CalR is a soluble protein that is retained in the ER lumen, whereas CNX is localized to the cytoplasm via a single-pass transmembrane domain that couples it to the outer face of the ER membrane.39 However, CNX can translocate to the lumen during the protein folding process, as evidenced by an equal distribution of CNX within the cytosol and CMX. The ER is composed of distinct structures that include tubules, matrices, and sheets.41 Further, the ER membrane proteins tubulin, reticulon, p180, and kinectin, as well as the DP1, REEP, and Climp63 proteins and the ER luminal protein calumenin-1 maintain ER sheet morphology and shape.42,43,44 Besides well-visualized lipid membranes, the ER is an organized proteinaceous net, the hidden morphology of which can be revealed by the cryo-ET.45 We did not expect our mild isotonic buffer to rupture the proteinaceous carcass of the ER, but rather to extract the transporting luminal proteins and the lipid membrane. To this end, we detected ER luminal proteins (Figure 1K) and the dynamic lipid layers in the cytosol following extraction, as evidenced by TEM, the depletion of lipids after cytosol extraction in the CMX (Figures 1G and 1H).
Factors associated with RNA degradation, including LC3B, DCP1B, DDX6, Exportin 5, and IRE1α were also detected primarily in the cytosol,46,47 whereas proteins related to endosome biogenesis, such as EEA1, Rab5, RCAS, and AIF,48,49 were equally distributed between the cytosol and CMX. Of the additional proteins tested, the calcium-insensitive isoform of PKC delta50 and the mRNA binding proteins FUBP1/2, hnRNPK, and PTBP1/251,52,53 were enriched in the CMX.
Assessment of protein synthesis in the cytosol and the cytomatrix
To address differential mRNA translation in the cytosol and CMX, we needed a method that statistically detects ribosome biogenesis and protein biosynthesis. For ribosome biogenesis, ribosomal proteins were an obvious choice; moreover, ribosomal protein L7a (rpL7a) was a large subunit protein. The rpL7a belongs to the “druggable genome” protein family, the genome-encoding proteins that small molecules can regulate. Because the mRNA translation is controlled by 80S formation and 60S availability54 and the nucleolar hypertrophy is an indicator of ribosome biogenesis,55 the rpL7a was an appropriate choice. In addition, rpL7a is a highly conserved ribosome protein localized on the surface of the 60S56,57 and contains two distinct RNA binding domains58 and an FKBP25 binding domain,59 suggesting that rpL7a may play an essential role in 60S subunit biogenesis. Notably, the localization of rpL7a on the surface of the 60S ribosomal subunit enables the use of rpL7a as a readout for the ribosomal large subunit biogenesis and protein expression.
We preliminarily tested the effects of several compounds that suppress diverse signaling pathways: pp242, Torin 1 (mTOR inhibitors), PF-4708671 (RSK), Bio (GSK-3), Rotenone (mitochondrial electron transport), MG132 (26S proteasome), and Geldanamycin (Hsp90) on the ribosomal protein translation (Figures S2A-S2J). We expressed GFP-tagged rpL7a in HCT-15 cells and monitored the resulting fluorescence in living cells. Although the compounds inhibit different signaling pathways, all compounds upregulated rpL7a translation. Additionally, rpL7a displayed the perinuclear localization and accumulation in the nucleus. rpL7a contains a domain II (residues 52-100) that directs nuclear and nucleolar localization.60 The increase of rpL7a biosynthesis in the cytoplasm, in response to chemical agents, has likely partially delayed the nuclear entry of an excessive number of proteins at the nuclear envelope, presenting itself as a bright rim around the nucleus (Figures S2C-S2J). Consequently, the intensification of the 60S biogenesis occurring in the nucleus, was revealed as nucleolar hypertrophy, due to an excessive accumulation of rpL7a. High-throughput tests confirmed the number of nucleoli and average nucleoli size was significantly increased in treated cells (Figures S2K-S2M). Since all compounds elicited similar results, we selected the mTOR kinase inhibitor (mTOR-KI), pp242,61 for further analysis.
mTOR-KI inhibits mTOR-dependent protein translation and directly affects autophagy and PI3K-Akt signaling.62,63 Treating cell lines with pp242 shifted the localization of several proteins toward the CMX and induced posttranslational modifications (Figure 2A, right panel). Determining whether proteins are translated in the CMX or translocated is central to characterizing CMX function. To test whether motor proteins facilitate cytosol/CMX protein translocation, HCT-15 cells were treated with dynein and kinesin inhibitors, and proteins in the cytosol and CMX were compared (Figures S3A and S3B). No significant protein translocation from the cytosol to the CMX or vice versa following treatment with Dynarrestin and K858 were detected. Proteins involved in the mRNA and protein recycling pathways, including LAMP1,64 DDX6/RCK,65 DCP1B,66 IRE1α,47 and BiP,67 were exclusively localized within the cytosol (Figure 2B).
Figure 2.

Analysis of protein synthesis in the cytosol and cytomatrix
(A) Protein distribution in the cytosol and cytomatrix of three cell lines under the normal growth conditions (left panel) and 500 nM of pp242 treatment for 24 h (right panel).
(B) The effect of increasing concentration of motor protein inhibitors on protein distribution in the cytomatrix and cytosol of HCT-15 cells. Inhibitors of the dynein and kinesin proteins showed no effect on protein distribution. To minimize mixing during loading, the equal amounts of samples were run separately. Side by side running gel data for the same samples are presented in Figures S3A and S3B.
(C and D) Polysome profile of the cytomatrix and cytosol of HCT-15 cells.
(E) The incorporation of radiolabeled L-[3H]-phenylalanine into proteins of the cytosol, and the CMX in untreated and pp242-treated HCT-15 cells. L-[3H]-phenylalanine was added, and cells were incubated for 1 h. ∗p-value of one-way ANOVA, p < 0.0001. Data are given as means ± SE, n = 5.
(F) eIF4E3 is localized in the cytomatrix of HCT-15 cells.
(G) The pp242 increased eIF4E3 protein expression in the CMX of HCT-15 cells.
(H) eIF4E-BP1 phosphorylation in the cytosol and cytomatrix with and without pp242 treatment of HCT-15 cells.
(I) Hypothetical diagram of inverse relationship of the eIF4E1 and eIF4E3. eIF4E – eukaryotic initiation factor 4E. BP1 – eIF4E binding protein 1.
mTOR inhibitors remove ribosomes from polysomes and prevent polysome aggregation.68 We used polysome profiling to compare differences of the cytosol and CMX under pp242 treated conditions and normal growth conditions in HCT-15 cell lines. After inhibitor treatment, the polysome profiles in the cytosol and CMX were reversed (Figures 2C and 2D). In the CMX, polysomes and large ribosomal subunit levels increased in response to inhibitor treatment. In the cytosol, the 60S and polysome levels decreased and monosome (80S) levels increased in response to pp242 treatment. The ratio of the small subunit 18S rRNA in the cytosol and CMX, determined by RT-PCR, was 1.8 to 1 (Figure S3C). To address the 40S subunit dynamics at the normal growth condition and pp242 treatment, the mRNA of small ribosomal protein rpS6 level was monitored using single-molecule fluorescence in situ hybridization (Figure S3D). Even after pp242 treatment for 2 h, the cells had higher levels of rpS6 mRNA signal. A steady state abundance of rpL7a and the rpS6 mRNA expression evidenced the rise of ribosome biogenesis under the pp242 treatment. However, these results do not clarify the translated spectrum of the proteins.
To further address observed protein synthesis dynamics, we quantified protein synthesis using a radiolabeled amino acid to detect newly synthesized proteins. Incorporating L-[3H] phenylalanine into proteins revealed that the mTOR inhibitor pp242 decreased de novo protein synthesis in the cytosol by 20%, which aligns with prior reports of global protein translation suppression.61,69 However, the incorporation of labeled amino acids increased in CMX proteins by 50% (Figure 2E). The latter result indicated that de novo protein synthesis increased in the CMX when global protein translation was suppressed in the cytosol, the latter of which also was detected by polysome profiling (Figures 2C and 2D). Thus, using six different methods—western blotting, polysome profiling, GFP-tagged rpL7a immunofluorescent technique, high-throughput nucleolar hypertrophy test, in situ hybridization of rpS6 mRNA, and the incorporation of radiolabeled amino acids during protein synthesis—we showed that protein biosynthesis could differ in the CMX and the cytosol following pp242 treatment. These data may indicate an unknown cell survival mechanism based on the translational capabilities found in the CMX.
Next, we sought to determine whether the increased CMX protein translation following the pp242 treatment could be explained by an mTOR-independent pathway. The switch from eIF4E1-dependent to eIF4E3-dependent protein translation has been demonstrated in a few cell lines.70 After validating the specificity of an anti-eIF4E3 antibody (Figure 2F), we treated HCT-15 cells with increasing concentrations of pp242 to determine the effects of mTOR-KI on eIF4E3 expression. A dose-dependent increase in eIF4E3 protein levels in the CMX was observed (Figure 2G). Inhibiting 4E-BP1 phosphorylation using the same compound in the cytosol and CMX appears to be in the inverse relationship with the eIF4E3 level in the CMX (Figure 2H). Results indicate the possibility of activating other protein synthesis regulatory pathways in CMX (Figure 2I).
Mass-spectrometry-based proteome profiling of the cytomatrix
Large-scale protein identification with mass spectrometry-based proteomics is an appropriate way of categorizing the proteome content of the CMX. Proteins that could form the structural foundation of the CMX, the cytoskeleton-associated proteins, actins, keratins, tubulins, and other cellular microfibrils were identified (Table 1). The Golgi, ER, nuclear pore complex proteins, and nuclear and plasma membrane proteins also were enriched in CMX. Thus, mass spectrometry data confirmed immunoblotting results and supported the notion that the CMX is enriched with the Golgi and ER matrices proteome. Enzymes and substrates of protein glycosylation, prenylation, and protein-lipid modification are the types of posttranslational modification (PTM) that were identified within CMX, confirming the presence of the ER-Golgi matrix constituency. These also happen to be the most abundant types of PTM of proteins that can promote the immobilization and segregation of protein and enzyme complexes.71 Extracellular matrix proteins, desmosomes, cell junction proteins, integrins, and receptors also were enriched. Interestingly, along with the normal cell signaling pathway proteins, the CMX contained a broad spectrum of proto-oncoproteins, which suggests the essential role of CMX in cancer development. A full spectrum of the CMX’s functional proteome is represented as a pie chart (Figure S4A). The CMX proteome was grouped based on the biological function of proteins (Table S1).
Table 1.
Selected mass spectrometry based cytomatrix proteomics
| Structural proteins | Quantity | Enzymes and posttranslational protein modification | Quantity | Receptors, proto-oncogenes | Quantity | |
|---|---|---|---|---|---|---|
| 1 | Cytoskeleton-associated | 7 | Glycosyltransferase | 4 | Ras family | 15 |
| 2 | Golgi proteins | 88 | Oligosaccharyltransferase | 6 | Src family | 9 |
| 3 | ER/microtubule proteins, Receptors, Ca2+ homeostasis | 48 | Mannosyl-glycoprotein beta-1,2-N-acetylglucos-aminyl transferase | 7 | Tumor, metastasis associated proteins | 48 |
| 4 | Nuclear membrane | 22 | Mannosyltransferase | 15 | BRCA associated | 7 |
| 5 | Nucleoporins | 24 | Mannosyl-oligosaccharide glucosidase | 1 | Retinoblastoma | 9 |
| 6 | PM, membrane proteins | 166 | Mannosidase | 6 | Melanoma | 8 |
| 7 | AHNAK/desmosome | 3 | Acetylgalactosaminyl transferase | 7 | Abl Tyr-kinase | 5 |
| 8 | Transmembrane proteins | 61 | Dolichyl-saccharide-transferase | 6 | MYC associated | 7 |
| 9 | Junction proteins | 11 | O-linked GlcNAc transferase | 4 | MYB binding protein | 2 |
| 10 | Matrix proteins | 9 | Procollagen- 2-oxoGl-dioxygenase | 3 | Cbl proto-oncogene | 4 |
| 11 | Cell adhesion | 12 | Collagen b-(1-O) galactosyltransferase | 2 | Jun proto-oncogene | 2 |
| 12 | Actin, actin-binding, actin-like or related | 98 | Polysaccharide biosynthesis domain | 1 | DEK proto-oncogene | 1 |
| 13 | Tropomyosin/myosin | 29 | Phosphatidylinositol transfer protein | 5 | Growth arrest, arrestins | 6 |
| 14 | Keratins, type | 25 | Phosphatidylinositol glycan synthesis | 8 | Set proto-oncogene | 2 |
| 15 | Tubulins | 9 | α-2-HS-glycoprotein | 11 | Erb-b2 receptors | 6 |
| 16 | Profilins/cofilins | 4 | UDP-glucose glucosyl transferase | 1 | Growth factors, receptors | 29 |
| 17 | Catenin/cadherin | 5 | O-methyltransferase | 2 | G protein binding | 30 |
| 18 | Pleckstrin homology | 16 | Serine palmitoyl-transferase, long chain | 1 | Inositol-PPP-receptor | 3 |
| 19 | Stathmins | 2 | Palmitoyl-protein thioesterase | 5 | Cell division cycle, cyclins | 49 |
| 20 | Lectins | 7 | Membrane proteins, palmitoylated | 3 | Apoptosis signaling | 21 |
| 21 | Annexins | 10 | N- myristoyltransferase | 1 | p53 tumor suppressor | 4 |
| 22 | Spindle proteins | 13 | Farnesyl-PP-synthase | 1 | TNF receptors | 8 |
| 23 | Integrins | 13 | Farnesyl-PP farnesyltransferase | 3 | Calcium signaling | 19 |
| 24 | Adducin | 2 | Protein prenyltransferase | 1 | Hypoxia inducible family | 3 |
| 25 | Vinculin | 1 | Isoprenylcysteine methyltransferase | 1 | cAMP responsive element | 2 |
Cytosolic enzyme spectrum is comprised of protein and RNA degradation pathways, lipid, nucleotide, and amino acid metabolism, and glycolysis, which can be categorized as the nutrient and energy processing cycle of the cell (Figure S4B).
A peptide area-based quantification (iBAQ) showed a relative protein level of the CMX and the cytosol proteome (Table S2). Differences in the corresponding proteomes were calculated as fold change in the CMX compared with the cytosol and vice versa, the results of which indicated an inverse relationship between the cytosolic and CMX proteome. Hierarchical clustering of the proteins detected with the respective proteomes revealed differences between the cytosol and CMX proteomes, as well as between the control and TOR-KI treated cells (Figures 3A and 3B). KEGG and Reactome pathway analyses likewise revealed the effect of mTOR-KI on the proteomes (Figures 3C and 3D).
Figure 3.

Mass spectrometry proteome analysis of the cytomatrix and cytosol of HCT-15 cells
(A) Heatmap of protein expression from cytosol and cytomatrix of the control and pp242 treated HCT-15 cells.
(B) Heatmap of 25 selected proteins in each differential analysis.
(C and D) KEGG and Reactome pathway analyses of CMX proteome of the untreated and pp242 treated HCT-15 cells for 24 h. NES – normalized enrichment score.
Differences of the cytosol and cytomatrix and correspondence of RNA-seq and Ribo-seq
RNA-seq and Ribo-seq are powerful tools that identify differentially expressed genes (DEGs). DEGs permit over-representation (or enrichment) analysis (ORA), a statistical method that determines whether genes from pre-defined sets are present more than expected. ORA of the pre-defined gene sets Hallmark (Table S3), KEGG (Table S4), Reactome (Table S5), and GOBP (Table S6) permitted analysis of specific pathway distribution within the cytosol or CMX.
To distinguish pathways of the cytosol and CMX, we prepared ribosomal footprint and mRNA libraries for sequencing from untreated and pp242-treated HCT-15 cells. Using ribosome profiling (Ribo-Seq), which is based on the deep sequencing of ribosome-protected footprints of translated mRNA,72 the translation differences were compared between the cytosol and the CMX. RNA-seq and Ribo-seq datasets were used to establish a transcriptome and translatome map of the cytosol and CMX, respectively. The expression of approximately 13,000 genes was analyzed (Table S7).
Using ORA, the pathway distribution was analyzed within the cytosol and cytomatrix, and the correspondence of pathways between the RNA-seq and Ribo-seq, at normal and drug-treated conditions. The Ribo-seq and RNA-seq data obtained by CMX over cytosol (CMX) revealed that transcriptome and translatome profiles differ in the CMX (Table S8). Data acquired of pp242 treated cells also showed transcriptome and translatome differences for the CMX (Table S9) and Cytosol (Table S10). The CMX translatome pathway differences are also present when comparing cells under pp242 treated and normal growth conditions (Table S11). Results indicate the flexibility of transcription and translation, which in turn may reflect the state of cells.
Additionally, the GOBP pathway differences were analyzed to determine corresponding pathways of transcription and translation between compartments (Tables S12–S14). The highest p-values for pathways identified by Ribo-seq for CMX corresponded to the lowest or undetected p-values of the cytosol (Table S12). The opposite result was obtained by comparing cytosolic pathways with the CMX (Table S13). The parallel Ribo-seq and RNA-seq analysis of the p values of the CMX and cytosolic pathways revealed that the lowest correlation occurs with the RNA-seq dataset (Table S14). The data also showed the overlapped pathways between the cytosol and the CMX. Overall results indicated that Ribo-seq analysis could be more reliable in showing real translation than RNA-seq, which prompted us to use ribosome footprints for the pathway analysis.
Heatmaps for RNA-seq and Ribo-seq datasets, where the red color represents enriched by upregulated and the blue color is downregulated genes set, showed the distribution of transcripts in the cytosolic and CMX fractions that differed according to their respective Z score (Figures 4A and 4B). These hierarchical heatmaps also revealed differences between the pp242-treated and untreated cells for both transcriptome and translatome.
Figure 4.

Analysis of transcription and translation in the cytomatrix and cytosol of HCT-15 cells
(A) Heatmap of transcripts from RNA-seq.
(B) Heatmap of footprints from Ribo-seq.
(C) Venn diagrams of the overlaps and unique upregulated and downregulated transcript sets obtained from bulk RNA-seq and Ribosome footprint.
(D) Reactome set visualization of the enriched upregulated and downregulated pathways.
(E and F) KEGG analysis of enriched pathways identified through Ribo-seq.
Venn diagrams represent the ratio of differentially transcribed (RNA-seq), translated (Ribo-seq), and overlapped transcript sets (Figure 4C). A low overlap rate was observed in the cytosol and CMX when the dataset was acquired for pp242 treated cells. The overlapped transcripts were higher in the CMX (CMX over Cyt) under normal growth conditions. These differences may reflect translational alteration between the cytosol and CMX compartments in normal and drug-treated cell states. The dynamics of the ribosome footprint and RNA-seq analyses indicated that some of the transcribed mRNAs (15%-16%) might not have been translated, which supports the notion that Ribo-seq analysis is more reliable in the identification of the enriched (ORA) pathways.
ORA of the select pathways of the Reactome and Hallmark collections are shown in Figures 4D and S5. RNA-seq, common, and Ribo-seq pathway dynamics were compared across the three groups of experimental conditions (I-III), which revealed the distribution of upregulated (red) and downregulated (blue) pathways. Experimental condition I shows the dynamics of cytosolic pathway distribution (Figures 4D-I and S5-I), which differed from the CMX dynamics (condition II). Experimental condition III, which identified CMX pathways under normal growth conditions, revealed upregulated and downregulated pathways that were specific to the CMX. The results revealed differences in upregulated and downregulated pathways, as well as transcription and translation, between the CMX and cytosol. Thus, ribosome footprint analysis exposed the highest upregulated pathway enrichment for the CMX (condition III, Ribo-seq).
Using ribosome footprint analysis, we assessed only the upregulated pathways to reveal translation differences between the cytosol and CMX (Figures 4E, 4F, S6A, and S6B). The CMX KEGG pathway analysis showed that some pathways enriched at the normal growth conditions differ from the CMX pathways of cells treated with the mTOR inhibitor (Figure 4E). Comparing the cytosolic and CMX pathways shows that pp242 treatment prompted pathway overlaps between the cytosol and CMX (Figure 4F). Differences were observed in the spectrum of pathways between the cytosol and CMX as well as pp242 treated and the normal growth conditions. Thus, the spectrum of pathways between the cytosol and CMX is altered depending on cell conditions.
Using the upregulated Reactome pathways, we analyzed differences between the cytosol and CMX (Figures S6A and S6B). The cytosolic pathways included translation, cell cycle, and respiratory pathways, but the overall PTM pathways overlapped (Figure S6A). The CMX pathways involved the ER-Golgi function that include transport, CNX/CalR cycle, lipid and steroid metabolism, and N-linked glycosylation, but the vesicle and membrane transport pathways overlapped (Figure S6B). The compartmental differences exposed functional dynamics, which supports previous selective western blotting data (Figures 1K and 2A). Transcribed and translated mRNAs, which have been used for RNA-seq and Ribo-seq analyses, could be associated with the CMX through mRNA binding complexes such as FUBP1/2, hnRNPK, and PTBP1/2 and other RNA binding proteins (Figure 1K and Table S1).
Implications of the cytosolic and cytomatrix pathway differences
Using ribosome footprints, we compared the enriched GOBP pathways in the CMX and cytosol (Figures 5A-5D). The enrichment of RNA catabolism and the ubiquitin-proteasome pathways identified by the Ribo-seq analysis in the cytosol (Figures 5A and 5B) was supported by the mRNA and protein degradation markers detected by the western blot analysis within this compartment (Figures 1K, 2A, and 2B). Cytosolic enrichment of the upregulated pathways also included a small molecule, amide, organic acid, organophosphate, and nucleotide metabolism that can be categorized as a form of cellular nutrition. Specific transport, pathways involved in protein localization to organelles, translation, and organization of large protein complexes were enriched in the cytosol as well. The DNA repair, cell cycle transition, and apoptosis pathways were also enriched in the cytosol (Figures 5A and 5B), which corresponds with the KEGG and Reactome pathway Ribo-seq data analysis (Figures 4F and S6A).
Figure 5.

Upregulated pathways of the cytosol and cytomatrix of HCT-15 cells identified through Ribo-seq
(A and B) Categorized cytosolic GOBP pathways.
(C and D) Categorized cytomatrix GOBP pathways.
Interestingly, the enrichment of the upregulated pathways within the cytomatrix differed from the cytosol (Figures 5C and 5D). Distribution of the carbohydrate, phospholipid, glycerolipid, and glycoprotein biosynthetic pathways within the CMX indicated immobilized biocatalysis. In living cells, bio-macromolecules are exposed to a densely crowded environment where a myriad of metabolic reactions that could exclude each other coincide. The immobilization of biosynthetic pathways provides the separation of chemical reactions and efficient biocatalysis. The upregulated pathways also included lipid, glycerolipid, carbohydrate, and glycoprotein metabolism. Consequently, the upregulated pathway enrichment also involved glycosylation and O-linked glycosylation, localization of proteins to membrane, and transmembrane transport (Figure 5C). Golgi organization, endomembrane system organization, actin filament-based processes, and cell junction organization pathways were also enriched in the CMX (Figure 5D). Response to hormones and Growth Factors, Receptor signaling pathways, intracellular and extracellular cell-to-cell signaling pathways were enriched as well. Apparently, these chains of the enriched pathways within the CMX form a logical system responsible for the cell phenotype. Accordingly, cell differentiation, cell projection organization and cell morphogenesis pathways are enriched in the cytomatrix (Figure 5D).
Although upregulated pathways in the CMX and cytosol appeared to be distinct, they nonetheless complement each other, which is essential for proper cell function. The examples of complementing pathways are the transporting systems and protein localization of the cytosol and CMX (Figures 5B and 5C).
The highest number of enriched pathways in the CMX were structural, metabolic, and signaling (Figure S7). Pathways related to protein glycosylation, transport, and translocation were also abundant in the CMX. The CMX pathways derived from the GOBP ribosome footprint are presented in Table S15. The upregulated and downregulated pathways that were enriched in the CMX and the cytosol are shown in Table S16. We observed that the relationship between the CMX and cytosol was inverse—downregulated pathways enriched in the CMX corresponded to upregulated pathways enriched in the cytosol.
A good correspondence was found between the ribosome footprints (Figure 5C) and protein expression, referring to the PTM, carbohydrate, and lipid metabolism pathways for example (Table 1). Ribo-seq pathway analysis, mass spectrometry proteome profiling, and western blotting experiments confirmed that the ER and Golgi matrices are integral parts of the CMX. Along with the cytoskeleton fibers, the ER and Golgi matrices are detergent and salt-resistant and form an elastic solid phase in contrast to mobile cytosol, which is detergent sensitive. Thus, the cytomatrix proteins and enzymes catalyze glycosylation, prenylation, or lipid transfer to proteins while remaining within the cytomatrix and forming protein cargo or secretory complexes. Cells would not be efficient if these enzymes were secreted with their cargo complexes to the extracellular space. Therefore, immobilized CMX enzymes are involved in lipid, carbohydrate, and glycolipid synthesis. These immobilized enzymes could also lead to glycosylation or lipidation posttranslational modifications, followed by the endomembrane system (ER) and Golgi organizations as well as transmembrane transport and ECM organization (Figure 5D). Thus, the cytomatrix integrates ER-Golgi-specific pathways with the cell surface receptors and signaling pathways.
Discussion
In the mid-1970s, microscopy, immunocytochemistry, and cell fractionation methods revealed the cytoskeleton-associated ribosomes; however, the biological implication of this finding remained unclear due to the heterogeneous nature of the cytoskeletal fraction.73 Nevertheless, the considerable physiological significance of the mRNAs/polysomes association with the cytoskeleton has been stated. Still, it was also noted that the full implications of the molecular assembly must be awaited until detailed knowledge of the types of synthesized proteins is revealed. Keith R. Porter proposed the concept of a cytoplasmic structure termed the “microtrabecular lattice” (MTL), which was described as a three-dimensional meshwork.74,75,76 Since being proposed, however, the use of histological techniques has raised questions as to whether the MTL is a valid construct or artifact.77,78 The central principle underlying MTL was not just whether structure and order exist within the cytoplasmic matrix, but also whether the existing structure and order extend beyond the well-defined cytoskeletal fibers.79 Undoubtedly, the nano-compartments observed by HVEM74 could be the reason for the mRNA/polysome presence and distinct protein synthesis capacity of the CMX. Nano-compartmentalization concentrates the required components in a confined space, allowing an efficient formation of the end products.
Until now, there have been no significant attempts to chemically isolate the cytomatrix, which was predicted almost five decades ago.76 We used a two-step process to isolate the cytomatrix. First, the mild solvent separated the liquid phase (cytosol), and then the stringent solvent was applied to isolate the solid phase (cytomatrix) from the nucleus. The cytosol is a viscous fluid phase, consisting of water-soluble molecules and lipid-associated cargo, that is easily extracted with a nonionic detergent from the solid cytomatrix. Since the elastic solid phase is Li+ sensitive, a stringent buffer containing LiCl salt isolated the CMX. In choosing the name for the new elastic solid phase, we followed Dr. Porter’s CMX terminology.74
Coordination of the cytosol and CMX functions appear to play an essential role in fine-tuning cellular processes in healthy cells. High-throughput analysis and conventional methods revealed different responses from the cytosol and CMX during times of drug-induced protein deficiency. Of note, inhibiting mTOR evidently stimulated protein translation within CMX, suggesting the tenacity for protein synthesis. The inhibition of mTORC1 is required to initiate the autophagy process and proteolysis.80 The proteolysis that occurs in the cytosol, by providing amino acids, may stimulate protein synthesis in the CMX. The reprogrammed protein translation within the cytomatrix is likely a cell survival mechanism. Our data indicate that the CMX may potentially be essential in numerous metabolic reactions including energy generation, cell cycle, DNA replication and repair, and RNA processing depending on cell status and conditions. These functions could be a backup mechanism of the CMX in extreme conditions. Moreover, the compartmentalization of metabolic processes within the cytosol and CMX could support cancer cell survival and drug resistance as well as other pathological events. We propose that under normal growth conditions, the CMX is responsible for the cellular phenotype, whereas routine functions that support cell maintenance, such as nutrient processing and sustaining housekeeping reactions, occur in the cytosol.
To this end, the CMX appeared to be critical for performing specific tasks such as PTM of proteins, particularly glycosylation, which is the most abundant and diverse protein modification and is considered a special function of the ER and Golgi. The oligo/polysaccharide moiety could anchor (glue) the post-translationally modified proteins and protein complexes to form a matrix similar to the cell surface proteins embedded in a matrix of glycans.81 This concept of immobilized biocatalysis in the CMX is explained in Figure360 Video. We suggest that the cytomatrix filaments and motor proteins, such as actin, myosin, tubulin, and others, engages the cytosolic motion (a ripple) in reaction to extracellular receptor-ligand binding (Table 1, Figure 5D). Recent data have shown that the cytoplasmic Rho GTPases and actomyosin dynamics with the extracellular matrix can orchestrate the signaling output.82 The viscosity of the cytosol requires fluctuating cytoplasmic motion7 to transduce a cascade mechanism of immobilized biocatalysis and nuclear processes. Thus, the enzyme-substrate binding and immobilized biocatalysis could be regulated by cytosolic movement and dynamics of the specific receptor-ligand action.
The Golgi matrix is a detergent and salt-resistant complex.18 Our mild KCl-containing buffer would not dissociate Golgi, which has membrane clusters that are formed mainly by thick anastomosing tubular structures.83 The ER-to-Golgi interwoven tubular network is a stabilizing factor84 for the endoplasmic matrix organization. The ER proteinaceous carcass revealed by the cryo-ET45 also appeared to be salt and detergent-resistant. Thus, Golgi and ER networks are integral parts of the cytoplasmic matrix along with cytoskeleton fibers. The Golgi and ER matrices are detergent and salt-resistant. However, the water and lipid-soluble mobile secretory materials, or cargo, of the Golgi and ER extracted to the cytosol are detergent-sensitive. The distribution of Golgi and ER proteins in the CMX fraction and the role of detected proteins in glycosylation and transportation suggests a unique function of the CMX in the biosynthetic-secretory pathway, that affirms the ER-Golgi complex as a segment of the elastic solid phase. In addition, the role of ER-Golgi as a segment of CMX in transport and cell-surface events (cell adhesion, ECM formation) implies a connection of the intracellular matrix with the extracellular matrix. Thus, a matrix system segregating biocatalysis performs efficient reactions and signaling.
The coinciding metabolic reactions that could exclude each other may interrupt biochemical reactions in a confined space. However, the immobilization of biosynthetic pathways, via the CMX, provides separated chemical reactions and efficient biocatalysis, that overcomes spatial impediments. Efficient biocatalysis, achieved by segregating and immobilizing the metabolome enzymes through the lipid and carbohydrate PTM, may form corresponding organelles that perform specific reactions and processes within the cytoplasm. Thus, the presence or absence of organelles in tissue-specific cells and reorganization of organelles after cell division depends on biosynthetic pathway immobilization. The dynamics of the cytoplasmic organelles, apart from DNA containing mitochondria, may depend on the organization of the CMX. Consequently, the cytomatrix organizes Golgi-ER pathways into a singular integrated system along with the receptors and signaling pathways. As such, we define the CMX as the flexible solid state of the cytoplasm, and a separate but interdependent entity from the viscous fluid (liquid) state of the cytosol. Immobilized biocatalysis integrates the intracellular and extracellular matrices and receptors with the nuclear processes, thus removing the spatial hindrances for biochemical processes, which elucidates cellular mechanics and cytoplasm organization.
Limitations of the study
The high-throughput experiments (Ribo-seq, RNA-seq, and proteomics) were only performed in HCT-15 cells. Comparative studies such as between normal and cancer cells are needed to expand the results obtained using this new approach.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pan-Keratin | Cell Signaling Technology | Cat# 4545; RRID:AB_490860 |
| Emerin | Cell Signaling Technology | Cat# 5430; RRID:AB_10691714 |
| ERp57 | Cell Signaling Technology | Cat# 2881; RRID:AB_2160840 |
| B23-NPM | Cell Signaling Technology | Cat# 3542; RRID:AB_2155178 |
| Nup98 | Cell Signaling Technology | Cat# 2598; RRID:AB_2267700 |
| β-actin | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| eEF2k | Cell Signaling Technology | Cat# 3691; RRID:AB_2097313 |
| eIF4E | Cell Signaling Technology | Cat# 9742; RRID:AB_823488 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| eIF4B | Cell Signaling Technology | Cat# 3591; RRID:AB_2097522 |
| phospho-T37 4E-BP1 | Cell Signaling Technology | Cat# 2855; RRID:AB_560835 |
| phospho-T70 4E-BP1 | Cell Signaling Technology | Cat# 9455; RRID:AB_330949 |
| non-phospho-T46 4E-BP1 | Cell Signaling Technology | Cat# 4923; RRID:AB_659944 |
| phospho-S65 4E-BP1 | Cell Signaling Technology | Cat# 9451; RRID:AB_330947 |
| mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 #4517; RRID:AB_1904056 |
| p70S6K1 | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| Ero-1Lα | Cell Signaling Technology | Cat# 3264; RRID:AB_823684 |
| PDI | Cell Signaling Technology | Cat# 3501; RRID:AB_2156433 |
| DCP1B | Cell Signaling Technology | Cat# 13233; RRID:AB_2798157 |
| DDX6 | Cell Signaling Technology | Cat# 9407; RRID:AB_10556959 |
| Tubulin | Cell Signaling Technology | Cat# 2128; RRID:AB_823664 |
| CalR | Cell Signaling Technology | Cat# 2891; RRID:AB_2275208 |
| PKCζ | Cell Signaling Technology | Cat# 9368; RRID:AB_10693777 |
| PKC δ | Cell Signaling Technology | Cat# 2058; RRID:AB_10694655 |
| BiP | Cell Signaling Technology | Cat# 3177; RRID:AB_2119845 |
| LC3B | Cell Signaling Technology | Cat# 3868; RRID:AB_2137707 |
| EEA1 | Cell Signaling Technology | Cat# 3288; RRID:AB_2096811 |
| AIF | Cell Signaling Technology | Cat# 5318; RRID:AB_10634755 |
| Rab5 | Cell Signaling Technology | Cat# 3547; RRID:AB_2300649 |
| RCAS1 | Cell Signaling Technology | Cat# 12290; RRID:AB_2736985 |
| Calnexin (CNX) | Cell Signaling Technology | Cat# 2679; RRID:AB_2228381 |
| PTBP1/2 | Cell Signaling Technology | Cat# 8776 The product is discontinued |
| hnRNPK | Cell Signaling Technology | Cat# 9081; RRID:AB_11178946 |
| FUBP/ KHSRP (E2E2U) | Cell Signaling Technology | Cat# 13398; RRID:AB_2798208 |
| IRE1α | Cell Signaling Technology | Cat# 3294; RRID:AB_823545 |
| rpS6 | Cell Signaling Technology | Cat# 2217; RRID:AB_331355 |
| rpL7a | Cell Signaling Technology | Cat# 2415; RRID:AB_2182059 |
| SUN2 | Abcam | Cat# ab124916; RRID:AB_10972497 |
| Nesprin 1/Syne-1 | Abcam | Cat# ab5250; RRID:AB_2200724 |
| Exportin 5 | Abcam | Cat# ab57491; RRID:AB_941499 |
| SUN2 | Sigma-Aldrich | Cat# MABT880 |
| GRP78/HSPA5/BiP | Novus Biol | Cat# 06274; RRID:AB_1555284 |
| eIF4E3 | Proteintech | Cat# 17282-1-AP; RRID:AB_2262162 |
| Bacterial and virus strains | ||
| ShRNA_eIF4E3 lentiviral particles | Santa Cruz Biotechnology | Cat# sc-78455-V |
| Mammalian vector pCMV6-AC-GFP (Human) | Origene | Cat# PS100010 |
| Chemicals, peptides, and recombinant proteins | ||
| pp242 | Tocris Bioscience | Cat# 4257/5 |
| Kinesin inhibitor K858 | Tocris Bioscience | Cat# 4960/1 |
| Dynarrestin | Tocris Bioscience | Cat# 6526/5 |
| Torin 1 | Tocris Bioscience | Cat# 4247/10 |
| PF4708671 | Tocris Bioscience | Cat# 4032/5 |
| Bio | Tocris Bioscience | Cat# 3194/10 |
| Rotenone | Tocris Bioscience | Cat# 3616/50 |
| MG132 | Tocris Bioscience | Cat# 1748/5 |
| Geldanamycin | Tocris Bioscience | Cat# 1368/1 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201S |
| T4 PK Reaction Buffer | New England Biolabs | Cat# B0201S |
| DMEM | Thermo Fisher Scientific | Cat# A4192101 |
| RPMI 1640 | Thermo Fisher Scientific | Cat# 61870143 |
| FBS | Thermo Fisher Scientific | Cat# A4766 |
| Penicillin/Streptomycin | Sigma | Cat# P4333 |
| HEPES solution | Sigma | Cat# H0887 |
| NaCl | Sigma | Cat# S5150 |
| Glycerol | VWR | Cat# 24388-295 |
| NP-40/IPEGAL | EMD Millipore | Cat# 492016 |
| Triton X-100 | Sigma | Cat# T8787 |
| Tris-HCl pH 7.5 1M | Thermo Fisher Scientific | Cat# 15567027 |
| RNAsin RNase inhibitor | Promega | Cat# N251B |
| Sodium deoxycholate | Ameresco | Cat# 0613 |
| KCl | Thermo Fisher Scientific | Cat# AM9640G |
| MgCl2 | Thermo Fisher Scientific | Cat# AM9530G |
| LiCl | Invitrogen | Cat# L7026 |
| DTT | OmniPur | Cat# 3860 |
| Cycloheximide | Ameresco | Cat# 94271-5G |
| RNase-free water | GE Healthcare | Cat# 0226 |
| siRNA buffer | Dharmacon™ | Cat# B-002000-UB-100 |
| Protease Inhibitors | Roche | Cat# 11393100 |
| qScript cDNA Super-Mix | QuantaBio | Cat# 10142-784 |
| Formaldehyde | Sigma | Cat# F9037 |
| MNase | Sigma-Aldrich | Cat# N3755 |
| Guanidine thiocyanate | Sigma | Cat# 50983 |
| Trizol | Ambion | Cat# 10296-010 |
| GlycoBlue | ThermoFisher Scientific | Cat# AM9515 |
| Polynucleotide kinase | BioLabs | Cat# M0201S |
| PBS | Gibco | Cat# 14200-075 |
| Tween 20 | Sigma-Aldrich | Cat# H06J02 |
| Puromycin | GIBCO | Cat# J67236.8EQ |
| BSA | Sigma-Aldrich | Cat# A9647 |
| Other | ||
| Compound | Target | Concentration |
| pp242 | mTOR | 500 nM |
| K858 | Kinesin | 1–10 µM |
| Dynarrestin | Dynein | 5–25 µM |
| Torin 1 | mTOR | 250 nM |
| PF4708671 | RSK | 5 µM |
| Bio | GSK-3 | 1 µM |
| Rotenone | Mitochondrial electron transport | 1 µM |
| MG132 | 26S proteasome | 1 µM |
| Geldanamycin | Hsp90 | 1 µM |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# B-002000-UB-100 |
| Stellaris RNA FISH | Biosearch technologies | Cat# VSMF-20680-5 |
| QIAseq miRNA Library Prep Kit | QIAGEN | cat #331502 |
| MTT Cell Growth Assay Kit | Sigma-Aldrich | Cat# CT02 |
| Deposited data | ||
| mRNA and Ribo-seq data | NCBI GEO | GSE199535 |
| The mass spectrometry proteomics data | ProteomeXchange Consortium | PRIDE: PXD031043 |
| Experimental models: Cell lines | ||
| Colorectal carcinoma HCT-15 | ATCC | CCL-225 |
| Cervical adenocarcinoma HeLa | ATCC | CCL-2 |
| Gastric adenocarcinoma AGS | ATCC | CRL-1739 |
| Hepatocellular carcinoma Hep G2 | ATCC | HB-8065 |
| Experimental models: Organisms/strains | ||
| HCT-15, GFP- rpL7a expression | Origene | Cat# PS100010 |
| HeLa, siRNA gene silencing, SUN2 | Dharmacon™ | Cat# 25777 |
| HCT-15, Transduction of shRNA eIF4E3 lentiviral particles | Santa Cruz Biotechnology | Cat# sc-78455-V |
| Oligonucleotides | ||
| Human SUN2 siRNA | Dharmacon™ | Cat# 25777 |
| ShRNA_eIF4E3 lentiviral particles | Santa Cruz Biotechnology | Cat# sc-78455-V |
| Software and algorithms | ||
| Proteome Discoverer 2.1 software | Thermo Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Mascot 2.4 | Matrix Science | https://www.matrixscience.com/ |
| gpGrouper algorithm | Saltzman et al. (2018, MCP)85 | PMID: 30093420 |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al. (2005, PNAS)86 | https://www.gsea-msigdb.org/gsea/index.jsp |
| FastQC | Andrews et al. (2010)87 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic (v0.38) | Bolger et al., 201488 | Bioinformatics 30, 2114-2120 |
| Bowtie aligner (v1.3.0) | Langmead et al., 200989 | Genome Biology 10, R25. |
| The STAR aligner (v2.7.6a) | Dobin et al., 201390 | Bioinformatics 29, 15-21. |
| R package edgeR | Robinson et al., 201091 | Bioinformatics 26, 139-140 |
| RUV normalization | Risso et al., 201492 | Nature biotechnology 32, 896-902 |
| MSigDB | Liberzon et al., 201193 | Bioinformatics 27, 1739-1740 |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| Image Studio Lite | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| GraphPad Prism 9.4.1 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator CC 2022 | Adobe | https://www.adobe.com/products/illustrator.html |
| NCBI GEO RNA-seq data | GEO accession GSE199535 | https://www.ncbi.nlm.nih.gov/gds |
| ProteomeXchange Proteomic data | Dataset Identifier PXD 031043 |
http://proteomecentral.proteomexchange.org/cgi/GetDataset |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tattym Shaiken (tattyms@bcm.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines and cell culture
Colorectal carcinoma HCT-15 (CCL-225, RPM-1640), cervical adenocarcinoma HeLa (CCL-2, DMEM), gastric adenocarcinoma AGS (CRL-1739, DMEM), and hepatocellular carcinoma HepG2 (HB-8065, DMEM) cells were grown in the incubator under humidified conditions at 37°C under 5% CO2 in Dulbecco’s Modified Eagle Medium or RPMI-1640 supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin and streptomycin. The cells were treated with the indicated concentration of the compounds for 24 h or as indicated in the experiment. Cells were grown to 90-95% confluency.
Generation of fluorescently labeled cell line
HCT-15 cells were grown on 35 mm glass bottom dishes and transfected with the GFP-L7a vector pCMV6-AC-GFP at a concentration of 1 μg/dish. At 4 h post-transfection, HCT-15 cells were treated with one of the following compounds for 24 h: 500 nM of pp242, 1 μM Rotenone, 250 nM Torin 1, 1 μM Bio, 1 μM Geldanamycin, 5 μM PF4708671, and 1 μM MG132. To detect the subcellular localization of GFP-tagged proteins, confocal microscopy (Zeiss LSM 780) images of live cells were taken after 24 h of exposure to the tested compound.
siRNA gene silencing
We designed ON-TARGET plus non-targeting pool siRNA (control) and ON-TARGET plus Human SUN2 siRNA (25,777) in a SMARTpool format. HeLa cells grown at a density of 1 × 105 cells in four chamber slides were transfected with siRNAs using DharmaFECT transfection reagent according to the manufacturer’s recommended method. The final concentration of siRNA was 25 nM. After transfection, cells were incubated at 37°C for 72 h before further analysis. The efficiency of the siRNAs was determined by immunofluorescence.
Transduction of shRNA eIF4E3 lentiviral particles
We validated the specificity of an anti-eIF4E3 antibody by shRNA knock-down. Sh_eIF4E3 lentiviral particles (Santa Cruz Biotechnology Inc., Dallas TX) were used to knock-down the expression of eIF4E3 in HCT-15 cells according to manufacturer’s instruction. On day 2, HCT-15 cells were transduced with 15 μL of sh_eIF4E3 lentiviral particles. We selected stable clones expressing eIF4E3 shRNA by adding 10 μg/mL Puromycin. On day 5, the transduction efficiency was estimated by detecting cells generating green fluorescence protein under fluorescence microscope.
Method details
Buffer ionic composition calculation
Standard approaches to separating the aqueous cytoplasm94 have not been sufficient for isolating the intracellular CMX. The sequential fractionation of the cytoplasm to obtain cytosol and membrane-bound proteins95 has likewise proven insufficient for CMX separation. A hypotonic solution is often used to isolate the nucleus. The PM of mammalian cells easily ruptures with a low salt hypotonic solution.96 The osmotic shock caused by a change in the solute concentration (hypotonic or hypertonic) can rupture PM and organelles. Rupturing the PM and other organelles allows the cytoplasmic contents of the cell to leak out while leaving the nucleus intact.97 In hypertonic conditions up to 0.6M NaCl, solubilization of nucleosomes was achieved.98 High salt disrupts charged-based protein-DNA, and protein-protein interactions and chromatin-associated proteins become more soluble.99 To isolate the cytosol and CMX at physiological osmotic conditions, we developed an equation to calculate the concentration of ions needed to replace sodium ions in a buffer based on the relative ionic radii for the Hofmeister series of monovalent cations (Figure S1B). Physiological or isotonic saline is a solution of 154 mM (0.90% w/v) NaCl, 308 mOsm/L. The relative radii used for the Hofmeister series monovalent cations were as follows: Li = 90 pm., Na = 116 pm., and K = 152 pm.28 The relative ionic radius is a solvation shell of ions in solution that surround proteins or protein complexes in the cell. The ionic ratio of protein solutions depends on the relative radius of ions. The concentration of replacing cations was calculated using the equation Cr = 18/Ri, where Cr is the replacing concentration and Ri is the ionic radius. Because the dominant positive cation within the cell is potassium,100 we used potassium chloride to mimic the natural ionic environment of the cell. Using this equation, we found that 120 mM KCl and 200 mM LiCl corresponded to 154 mM NaCl. These concentrations of ions were used sequentially in the isolation of the cytosol and the CMX. We used a minimal solvent volume for cell lysis to induce less damage to the CMX and to maintain the ionic balance.
Cell fractionation
We treated HCT-15 cells with 500 nM of pp242 every 6 h for 24 h unless otherwise stated. Control cells were grown under normal growth conditions. Experiments were performed in duplicate using 10 cm dishes. At least three 10 cm dishes were used for the CMX isolation experiment.
-
1.
The growth media from control untreated HCT-15 cells at an exponential growth phase and pp242-treated cells at cell growth arrest were aspirated and 10 cm dishes with cells were placed on ice in tilted position. All the experiments were performed on ice or in cold room that slows down metabolic processes (Figure S1C).
-
2.
Cells were washed with ice-cold PBS twice allowing the PBS to flow down from tilted dishes and be aspirated.
-
3.
Cells were lysed using cell scrapers in an appropriate volume Buffer A (0.1 mL Buffer A per 10 cm dish or 0.3 mL Buffer A for 30 cm cell culture dish) and placed into Eppendorf tubes. At this volume of the Buffer A, the lysate volume would equal 0.2 mL (10 cm dish) and 0.6 mL (30 cm dish), respectively. Thus, the cell lysate would be diluted only 2-fold.
-
4.
Buffer A composition is 40 mM HEPES pH 7.4, 120 mM KCl, 0.5% Glycerol, and 0.5% NP-40 with protease inhibitors. The 120 mM KCl mimics the intracellular ionic composition that prevents the disintegration of the cytomatrix. The nonionic detergent NP-40 solubilizes the viscous fluid content of the cytoplasm, extracting water soluble biopolymers, neutral lipids, and phospholipids of the PM, ER, mitochondria, Golgi, and other lipid containing droplets, and cargo molecules from the cytomatrix.
-
5.
Scraped cell lysates rotated for 30 min at 4°C. Mixing the collected cells lysates by rotation with Buffer A for 30 min provides time for micelles to form, which is a slow process at cold conditions. The micelles envelope lipids and proteins immersed in lipid bilayers.
-
6.
Lysates were centrifuged at 500 × g for 5 min at 4°C, and the supernatants were collected as the cytosol. The cytosol represents colloid suspension of the micelles. Low speed centrifugation prevents breaking of the micelles and reabsorption to the pelleted CMX.
-
7.
The pellets were gently washed with two volumes of washing Buffer, which is 40 mM HEPES pH 7.4, 120 mM KCl, 0.5% Glycerol. The pellet was gently resuspended tapping the tube. The wash can be discarded or added to the cytosol fraction. Washing the pellet without detergent enables Buffer A to be replaced with the next buffer, Buffer B.
-
8.
The pellet was resuspended by gently pipetting it seven times in Buffer B. Buffer B has a composition of 10 mM Tris-HCl pH 7.5, 1.5 mM KCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, 2.5 mM MgCl2, 0.2 M LiCl, and protease inhibitors. The 0.2M LiCl was added to the Buffer B just before the experiment. Replacing potassium ions with lithium condenses the nuclear content, breaks LINC complex and dissolves the cytomatrix that allows for separation of the core nuclear content from the elastic solid elements of the cytoplasm.
-
9.
The resuspension of the pellet by cold rotation continued for 45 min to complete solubilization.
-
10.
The cytomatrix was isolated by centrifuging at 2000 × g for 5 min. The supernatants were collected as the CMX.
-
11.
The pellets were washed with two volumes of Buffer B and combined to CMX or discarded.
-
12.
The nuclear pellet was dissolved in 8M urea.
-
13.
Cytosol, CMX, and nuclear fractions were clarified at 10,000 × g for 10 min.
-
14.
The protein concentration was determined by Lowry. For RNA extraction and polysome profiling, all buffers were prepared in RNase-free water with the same buffer compositions and the addition of 500 U/ml RNasin. All procedures were performed at 4°C.
Polysome profiling
For the polysome profiling, 2 mM DTT and 100 μg/mL Cycloheximide (CHX) was added to the PBS. All buffers were prepared in RNase-free water. The CHX (100 μg/mL) was added to 75% confluent HCT-15 cells (control cells and cells treated with pp242) for 30 min before cell lysis. The cytosol and CMX were obtained as described. Approximately 2 mg of proteins were overlaid onto 10–50% sucrose gradients and centrifuged at 100,000 × g for 3 h. The polysome profile was monitored at 254 nm (Brandel, BR-188-176; Gradient Fractionation System).
Incorporation of labeled amino acids into proteins
HCT-15 cells were treated with 500 nM of pp242 for 24 h. Incorporation of radiolabeled amino acids into proteins was performed as described previously.101 Briefly, pp242 treated and non-treated HCT-15 cells were incubated with L-[3H] phenylalanine (5 μCi/mL) for 1 h. After incubation, the medium was removed and the cells were washed twice with ice-cold PBS. The cells were fractionated, and the protein concentration was determined by the Lowry method. Approximately 100 μg of protein from each fraction were blotted onto Whatman 3 MM paper filters and allowed to dry. The dried filters were immerged in 50 mL of 5% (w/v) TCA and boiled for 2 min. The procedure was repeated once. The filters were rinsed briefly in 100% ethanol and dried. Finally, the radioactive signal (in cpm) was counted on a Beckman LS 6500 LSC.
RNA -seq
For Next-Generation Sequencing (RNA-seq), HCT-15 cells were treated with 500 nM pp242 for 6 h, with an additional treatment at 3 h. Control cells were grown under normal growth conditions. Total RNA was isolated from the cytosol and CMX using a RNeasy Mini Kit (QIAGEN, Germantown, MD) according to the manufacturer’s instructions. Total RNA samples were normalized to 200 ng each and libraries were generated using the NEB Ultra II Directional RNA Library Prep kit according to the kit specifications. All samples were pooled equimolarly and sequenced on an Illumina NovaSeq 6000 flow cell with paired-end reads (PE150). An average of 51 million read pairs per sample were sequenced.
Ribo-seq
The cytosol and CMX from control and treated cells were obtained as previously described and digested with micrococcal nuclease (MNase, Sigma-Aldrich, St Louis, MO) at 37°C for 30 min to obtain ribosome footprints. The reaction was stopped by the addition of 1.5 volumes of 4 M guanidine thiocyanate. Footprints were extracted by adding TRIzol and isolated by precipitating with GlycoBlue (ThermoFisher Scientific, Houston, TX) overnight.102 The footprints were treated with polynucleotide kinase (ENK) at 37°C for 30 min to reverse the phosphate position. We separated 17–37 nt long footprints from the gel under UV light. We froze gel slices at −80°C and then crushed them to extract RNA.
Library Preparation
We used 10 ng of size-selected ribosome-protected RNA (17–37 nt) as the starting material for the QIAseq miRNA Library Prep Kit (cat #331502). RNA fragments were ligated to adapters at the 3′ and 5′ ends, reverse transcribed, and amplified. The resulting libraries were size selected for 185–191 bp fragments. The libraries were quantitated by qPCR using the Applied Biosystems ViiA7 qPCR instrument and a KAPA Library Quant Kit (p/n KK4824). All samples were pooled equimolarly and sequenced on a NextSeq 500 High Output v2.5 flowcell (Illumina p/n 20024906) using the Illumina NextSeq 500 sequencing instrument with a single-read configuration (75 bp). An average of 42 million reads per sample was sequenced. FastQ file generation was executed using Illumina’s cloud-based informatics platform, BaseSpace Sequencing Hub.
Bulk RNA-seq and Ribo-seq analysis
Two biological replicates of ribosomal footprint and mRNA libraries were generated and subjected to deep sequencing to obtain the Ribo-seq and RNA-seq datasets. The sequence files generated by the RNA-seq and Ribo-seq experiments were trimmed using Trimmomatic (v0.38),88 and sequence quality was assessed using FastQC (v0.11.8).87 Next, we used the bowtie aligner (v1.3.0) to remove rRNA sequences from the Ribo-seq experiment.89 The rRNA sequences used in the rRNA depletion step were downloaded from Ensembl BioMart. The STAR aligner (v2.7.6a) was then used to align and quantify gene expression for both the Ribo-seq and the RNA-seq sequence,90 using the human genome build GRCh38 p13 v35. Differential gene expression of protein coding genes was evaluated using the R package edgeR,91 with upper quartile and RUV (remove unwanted variation) normalization.92 Significance was achieved for a fold change exceeding 1.5× and an FDR-adjusted p value < 0.05.
Over-representation analysis (ORA)
ORA was performed to detect enriched gene sets corresponding to pathways and biological processes based on differential gene expression. Using the Hallmark, KEGG, Reactome, and Gene Ontology Biological Process compendia (v7.3) and the Molecular Signature Database methodology (MSigDB),93 we assessed enrichment with a hypergeometric test. Significance was achieved at an FDR-adjusted p value <0.05.
LC-MS/MS analysis
Two biological replicates were used for LC-MS/MS analysis. The cytosol and CMX fractions were concentrated and digested on an S-Trap column (Protifi, NY) per the manufacturer’s protocol. Offline high pH STAGE peptide fractionation (15 fractions combined to 5 peptide pools) for 50 μg peptides was carried out as previously described (PMID: 30093420). LC-MS/MS analysis was carried out using a nano-LC 1200 system (Thermo Fisher Scientific, San Jose, CA) coupled to Orbitrap Lumos ETD mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Peptide (1 μg) was loaded onto a 2 cm × 100 μm internal diameter (ID) pre-column switched in-line with an in-house 20 cm × 75 μm ID column (Reprosil-Pur Basic C18, 1.9 μm, Dr.Maisch GmbH, Germany) equilibrated in 0.1% formic acid/water. The column temperature was maintained at 60°C. The peptides were eluted using a 110 min gradient of 2–30% acetonitrile/0.1% formic acid at a flow rate of 200 nL/min. The mass spectrometer was operated in the data-dependent acquisition mode using the top30 method. MS1 was acquired in the Orbitrap (120,000 resolution, 350–1400 m/z) followed by MS2 in the IonTrap (HCD 32%, AGC 2E4, 30 ms ion injection) with a 15 s dynamic exclusion time.
The RAW files were processed using the Proteome Discoverer 2.1 software (Thermo Scientific) using Mascot 2.4 (Matrix Science) with percolator against human NCBI refseq database updated on 2020_0324. The precursor ion tolerance and product ion tolerance were set to 20ppm and 0.5Da, respectively. The enzyme was set to Trypsin/P with maximum missed cleavage of 2 the dynamic modifications of oxidation, carbamidomethyl on cysteine, and protein N-terminal acetylation were allowed. The peptides identified from the Mascot result file were validated with 5% false discovery rate (FDR). The gene product inference and iBAQ-based quantification was carried out using the gpGrouper algorithm.85 The median normalized iBAQ values were used for data analysis. The differentially expressed proteins were calculated using the moderated t-test to calculate p values and log2 fold changes in the R package limma. The FDR corrected p value was calculated using the Benjamini-Hochberg procedure. Gene Set Enrichment Analysis (GSEA) (PMID: 16199517) was performed using the canonical pathway gene sets derived from the KEGG and Reactome pathway databases.86
Immunoblotting
Total protein in the cell fractions was measured, equalized by concentration, and boiled at 95°C for 5 min in sample buffer. The samples were then resolved using a 4–15% Mini-PROTEAN TGX Precast Gel (Bio-Rad) at 100V for 100 min. The resolved proteins were transferred onto Immobilon PVDF membranes (Thermo Fisher Scientific) at 200 mA for 60 min at 4°C. The membranes were blocked in Odyssey Blocking Buffer (Li-Cor) and probed with the respective primary antibodies at 1:1,000 dilution overnight at 4°C. The membranes were washed three times for 5 min with Tris-buffered saline with 0.05% Tween 20 (Sigma-Aldrich) and incubated with the secondary (Santa Cruz Biotech) Horseradish Peroxidase (HRP)-antibodies (1:20,000) for 1 h at room temperature on the rocker. The blots were detected by LiCor Odyssey scanner.
Immunolabeling
Cells were immunolabeled and visualized by deconvolution (GE Healthcare) and confocal (Nikon) microscopy. Per chamber, 20,000 cells were seeded and cultured for 24 h. Cultured cells were fixed with 4% PFA, permeabilized with 0.2% (v/v) Triton X-100 in PBS for 10 min at RT, and blocked with horse or goat serum for whole cell imaging. Cell fractions (including nuclei with or without the cytosol) were deposited onto microscope slides using a cytospin at 4°C, fixed with 4% PFA, and blocked with horse serum. The slides were processed using a standard immunostaining protocol. The cells and cell fractions were incubated with the appropriate primary antibodies overnight at 4°C, washed in PBS, and incubated with Alexa 488 and/or Alexa 594-conjugated secondary antibodies for 1 h at 4°C.
Fluorescent in situ hybridization (FISH)
To detect ribosomal S6 (RPS6) mRNA, we performed single-molecule FISH using Stellaris RNA FISH probes conjugated to CAL Fluor Red 610 targeting Human RPS6 (VSMF-20680-5), Biosearch technologies, Petaluma, CA). Control and pp242 treated HCT-15 cells on cover glasses were fixed in PBS containing 3.7% (v/v) formaldehyde and permeabilized with 70% (v/v) ethanol for 1 h at 4°C. The cells were then washed with 10% formamide buffer (Stellaris Wash Buffer A) before hybridizing with the probe set in Stellaris Hybridization Buffer (SMF-HB1-10). The hybridizations were performed overnight in a moist chamber at 37°C. The following day, the cells on cover glasses were transferred to Stellaris Wash buffer A (SMF-WA1-60) and incubated for 30 min. For nuclear staining, we incubated cells with DAPI (5 ng/ml) for 30 min in the dark in Wash buffer A, washed with Stellaris Wash buffer B (SMF-WB1-20) for 5 min and mounted. Images were obtained using a deconvolution microscope (GE Healthcare, Olympus IX71).
Transmission electron microscopy (TEM)
HCT-15 cells were fractionated as described above. Cellular and subcellular fractions were fixed in 3% glutaraldehyde (PBS, pH 7.3) for two days, washed with sodium phosphate buffer (pH 7.3), post-fixed with 1% osmium tetroxide for 1 h, and dehydrated through a series of graded alcohol baths. Standard TEM preparation techniques were used to obtain sample blocks that were cut in ultra-thin sections (80 nm) for mounting onto 100 mesh copper grids. The grids were stained with 2% alcoholic uranyl acetate and Reynold’s lead citrate. The grids were examined with a Zeiss CEM 902 transmission electron microscope, and images were captured using an AMT602 digital camera.
High throughput nucleolar hypertrophy assessment
To analyze nucleolar morphology of HCT-15 cells, we performed high speed imaging with optimum focusing capabilities. We seeded 20,000 cells in a 384 well-plate (Greiner) and the next day, we treated cells with 500 nM of pp242, 1 μM of MG132, and 1 μM of Rotenone for 24 h. Cells were fixed with 4% PFA, permeabilized by 1% Triton X-100 for 1 h, blocked with goat serum, incubated with anti-L7a antibody overnight, and detected using Alexa 488-conjugated secondary antibody. Nuclei were stained with DAPI. At least 200 cell nuclei were analyzed with four repeats. Cells were imaged on an IC200 high-throughput image cytometer (VALA Sciences) using a plan APO 40× objective. A single plane was acquired for the DAPI channel, and a z stack series (10 μm, 1 μm increment) was acquired for the GFP channel. Image analysis was performed using a custom-made algorithm developed in MATLAB. We analyzed two fields-of-view (FOV) per well (average of 46 cells per FOV). For each FOV, DAPI-labelled nuclei were segmented using a watershed transform strategy. For nucleolus segmentation, the best focused GFP-labelled nucleoli plane was selected, and objects were identified by first performing morphological top-hat filtering, followed by computing the regional maxima of the H-maxima transform. The mean number of nucleoli per nucleus, average size of nucleoli, and mean fluorescence intensity of nucleoli were then calculated.
RT-PCR
For the RT-PCR, total RNA was isolated from cytosol and CMX of HCT-15 cells using RNeasy Mini Kit (QIAGEN). qScript cDNA Super-Mix (QuantaBio) was used for cDNA synthesis. Beta-actin and 18S rRNA primers (Invitrogen) were used for PCR reaction.
Quantification and statistical analysis
Data are presented as the mean ± SD. One-way ANOVA was used to detect significant differences between the means of more than two independent groups using Prism 6 (GraphPad). We independently performed t-tests to confirm statistically significant differences between two groups. A p value of <0.05 was considered as significant.
Acknowledgments
We are grateful to Drs. David Y. Graham and Nicholas T. Ingolia for thoughtful suggestions. We thank Drs. Lutfi Abu-Elheiga, Joel R. Neilson, Arindam Chaudhury, Tegy Vadakkan, Gargy Goshal, Atreyi Dasgupta, Jason T. Yustein, Fen Wang, and Ralph Nichols and Bioscience Writers team for technical assistance. We are grateful to Marzhan Urazbayeva, Marina Bissekenova, Bakhytzhan Tastulekov, DR and GP Laws Fund for their support. This project was supported in part by the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from the NIH S10 grant (1S10OD023469). Imaging for this project was in part supported by the Optical Imaging and Vital Microscopy (OIVM) core of BCM and Integrated Microscopy Core at Baylor College of Medicine and the Center for Advanced Microscopy and Image Informatics (CAMII) with funding from NIH (DK56338, CA125123, ES030285), and CPRIT (RP150578, RP170719). BCM Mass Spectrometry Proteomics Core is supported by the Dan L. Duncan Comprehensive Cancer Center NIH Award (P30 CA125123), CPRIT Core Facility Award (RP210227).
Author contributions
Conceptualization and methodology, T.E.S; formal analysis, S.L.G., M.J.R., and C.C.; investigation, T.E.S., M.S., AW, A-H.R., E.R., M.J.R., F.S., J.D., A.J.; D.K., and A.M., visualization, T.E.S., M.S., S.L.G., F.S., J.D., and A-H. R.; writing – original draft, T.E.S.; reviewing & editing, A.P.P., A.R.O., A-H. R., M.S., F.S., and T.E.S.; supervision, T.E.S.; funding acquisition, T.E.S.
Declaration of interests
The authors declare no competing interests.
Patent No: US 11,428,693 B2; Data of Patent: Aug. 30, 2022
“Method of high throughput screening of chemical compounds suppressing nucleolar hypertrophy”
Applicant: Baylor College of Medicine, Houston, TX (US)
Inventor: Tattym E. Shaiken, Houston, TX (US)
Patent materials were incorporated, in part, in the Result section “Assessment of protein synthesis in the cytosol and the cytomatrix”
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.105965.
Supplemental information
Data and code availability
This paper does not report original code. Data reported in this paper will be shared by the lead contact upon request.
The RNA sequencing data was uploaded to NCBI GEO and is publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and is publicly available as of the date of publication. The dataset identifier is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Shepherd V.A. The cytomatrix as a cooperative system of macromolecular and water networks. Curr. Top. Dev. Biol. 2006;75:171–223. doi: 10.1016/S0070-2153(06)75006-2. [DOI] [PubMed] [Google Scholar]
- 2.Taylor C.V. The contractile vacuole in Euplotes: an example of the sol-gel reversibility of cytoplasm. J. Exp. Zool. 1923;37:259–289. doi: 10.1002/jez.1400370302. [DOI] [Google Scholar]
- 3.Pollack G.H. Is the cell a gel--and why does it matter? Jpn. J. Physiol. 2001;51:649–660. doi: 10.2170/jjphysiol.51.649. [DOI] [PubMed] [Google Scholar]
- 4.Srere P.A. Macromolecular interactions: tracing the roots. Trends Biochem. Sci. 2000;25:150–153. doi: 10.1016/s0968-0004(00)01550-4. [DOI] [PubMed] [Google Scholar]
- 5.Gierasch L.M., Gershenson A. Post-reductionist protein science, or putting Humpty Dumpty back together again. Nat. Chem. Biol. 2009;5:774–777. doi: 10.1038/nchembio.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H., Carvalho P., Voeltz G.K. Here, there, and everywhere: the importance of ER membrane contact sites. Science. 2018;361:eaan5835. doi: 10.1126/science.aan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo M., Ehrlicher A.J., Jensen M.H., Renz M., Moore J.R., Goldman R.D., Lippincott-Schwartz J., Mackintosh F.C., Weitz D.A. Probing the stochastic, motor-driven properties of the cytoplasm using force spectrum microscopy. Cell. 2014;158:822–832. doi: 10.1016/j.cell.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clegg J.S. Intracellular water and the cytomatrix: some methods of study and current views. J. Cell Biol. 1984;99:167s–171s. doi: 10.1083/jcb.99.1.167s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provance D.W., Jr., McDowall A., Marko M., Luby-Phelps K. Cytoarchitecture of size-excluding compartments in living cells. J. Cell Sci. 1993;106:565–577. doi: 10.1242/jcs.106.2.565. [DOI] [PubMed] [Google Scholar]
- 10.Starr D.A., Fridolfsson H.N. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 2010;26:421–444. doi: 10.1146/annurev-cellbio-100109-104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ketema M., Wilhelmsen K., Kuikman I., Janssen H., Hodzic D., Sonnenberg A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 2007;120:3384–3394. doi: 10.1242/jcs.014191. [DOI] [PubMed] [Google Scholar]
- 12.Lu W., Schneider M., Neumann S., Jaeger V.M., Taranum S., Munck M., Cartwright S., Richardson C., Carthew J., Noh K., et al. Nesprin interchain associations control nuclear size. Cell. Mol. Life Sci. 2012;69:3493–3509. doi: 10.1007/s00018-012-1034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farías-Rico J.A., Ruud Selin F., Myronidi I., Frühauf M., von Heijne G. Effects of protein size, thermodynamic stability, and net charge on cotranslational folding on the ribosome. Proc. Natl. Acad. Sci. USA. 2018;115:E9280–E9287. doi: 10.1073/pnas.1812756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozov A., Khusainov I., El Omari K., Duman R., Mykhaylyk V., Yusupov M., Westhof E., Wagner A., Yusupova G. Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat. Commun. 2019;10:2519. doi: 10.1038/s41467-019-10409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blobel G., Potter V.R. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 16.Blobel G. Three-dimensional organization of chromatids by nuclear envelope-associated structures. Cold Spring Harb. Symp. Quant. Biol. 2010;75:545–554. doi: 10.1101/sqb.2010.75.004. [DOI] [PubMed] [Google Scholar]
- 17.Dwyer N., Blobel G. A modified procedure for the isolation of a pore complex-lamina fraction from rat liver nuclei. J. Cell Biol. 1976;70:581–591. doi: 10.1083/jcb.70.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slusarewicz P., Nilsson T., Hui N., Watson R., Warren G. Isolation of a matrix that binds medial Golgi enzymes. J. Cell Biol. 1994;124:405–413. doi: 10.1083/jcb.124.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldwin R.L. How Hofmeister ion interactions affect protein stability. Biophys. J. 1996;71:2056–2063. doi: 10.1016/S0006-3495(96)79404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okur H.I., Hladílková J., Rembert K.B., Cho Y., Heyda J., Dzubiella J., Cremer P.S., Jungwirth P. Beyond the hofmeister series: ion-specific effects on proteins and their biological functions. J. Phys. Chem. B. 2017;121:1997–2014. doi: 10.1021/acs.jpcb.6b10797. [DOI] [PubMed] [Google Scholar]
- 21.Okur H.I., Kherb J., Cremer P.S. Cations bind only weakly to amides in aqueous solutions. J. Am. Chem. Soc. 2013;135:5062–5067. doi: 10.1021/ja3119256. [DOI] [PubMed] [Google Scholar]
- 22.Bello J., Haas D., Bello H.R. Interactions of protein-denaturing salts with model amides. Biochemistry. 1966;5:2539–2548. doi: 10.1021/bi00872a008. [DOI] [PubMed] [Google Scholar]
- 23.Bykov S., Asher S. Raman studies of solution polyglycine conformations. J. Phys. Chem. B. 2010;114:6636–6641. doi: 10.1021/jp100082n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers M.T., Armentrout P.B. A thermodynamic “vocabulary” for metal ion interactions in biological systems. Acc. Chem. Res. 2004;37:989–998. doi: 10.1021/ar0302843. [DOI] [PubMed] [Google Scholar]
- 25.Sedlák E., Stagg L., Wittung-Stafshede P. Effect of Hofmeister ions on protein thermal stability: roles of ion hydration and peptide groups? Arch. Biochem. Biophys. 2008;479:69–73. doi: 10.1016/j.abb.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Gao Y.Q. Simple theory for salt effects on the solubility of amide. J. Phys. Chem. B. 2012;116:9934–9943. doi: 10.1021/jp305532h. [DOI] [PubMed] [Google Scholar]
- 27.Shaiken T.E., Opekun A.R. Dissecting the cell to nucleus, perinucleus and cytosol. Sci. Rep. 2014;4:4923. doi: 10.1038/srep04923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shannon R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A. 1976;32:751–767. [Google Scholar]
- 29.Ponder E. The tonicity-volume relations for systems containing human red cells and the chlorides of monovalent cations. J. Gen. Physiol. 1949;32:391–398. doi: 10.1085/jgp.32.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnitzer E., Kozlov M.M., Lichtenberg D. The effect of cholesterol on the solubilization of phosphatidylcholine bilayers by the non-ionic surfactant Triton X-100. Chem. Phys. Lipids. 2005;135:69–82. doi: 10.1016/j.chemphyslip.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Lichtenberg D., Ahyayauch H., Goñi F.M. The mechanism of detergent solubilization of lipid bilayers. Biophys. J. 2013;105:289–299. doi: 10.1016/j.bpj.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lichtenberg D., Ahyayauch H., Alonso A., Goñi F.M. Detergent solubilization of lipid bilayers: a balance of driving forces. Trends Biochem. Sci. 2013;38:85–93. doi: 10.1016/j.tibs.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Herbert F.C., Abeyrathna S.S., Abeyrathna N.S., Wijesundara Y.H., Brohlin O.R., Carraro F., Amenitsch H., Falcaro P., Luzuriaga M.A., Durand-Silva A., et al. Stabilization of supramolecular membrane protein-lipid bilayer assemblies through immobilization in a crystalline exoskeleton. Nat. Commun. 2021;12:2202. doi: 10.1038/s41467-021-22285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J. Pharmacol. Sci. 2010;112:255–264. doi: 10.1254/jphs.09r03cr. [DOI] [PubMed] [Google Scholar]
- 35.Rebane A.A., Ziltener P., LaMonica L.C., Bauer A.H., Zheng H., López-Montero I., Pincet F., Rothman J.E., Ernst A.M. Liquid-liquid phase separation of the Golgi matrix protein GM130. FEBS Lett. 2020;594:1132–1144. doi: 10.1002/1873-3468.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Windoffer R., Beil M., Magin T.M., Leube R.E. Cytoskeleton in motion: the dynamics of keratin intermediate filaments in epithelia. J. Cell Biol. 2011;194:669–678. doi: 10.1083/jcb.201008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berk J.M., Tifft K.E., Wilson K.L. The nuclear envelope LEM-domain protein emerin. Nucleus. 2013;4:298–314. doi: 10.4161/nucl.25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ostlund C., Folker E.S., Choi J.C., Gomes E.R., Gundersen G.G., Worman H.J. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J. Cell Sci. 2009;122:4099–4108. doi: 10.1242/jcs.057075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamriben L., Graham J.B., Adams B.M., Hebert D.N. N-Glycan-based ER molecular Chaperone and protein quality control system: the Calnexin binding cycle. Traffic. 2016;17:308–326. doi: 10.1111/tra.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L., Niu Y., Zhu L., Fang J., Wang X., Wang L., Wang C.C. Different interaction modes for protein-disulfide isomerase (PDI) as an efficient regulator and a specific substrate of endoplasmic reticulum oxidoreductin-1alpha (Ero1alpha) J. Biol. Chem. 2014;289:31188–31199. doi: 10.1074/jbc.M114.602961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng P., Obara C.J., Szczesna E., Nixon-Abell J., Mahalingan K.K., Roll-Mecak A., Lippincott-Schwartz J., Blackstone C. ER proteins decipher the tubulin code to regulate organelle distribution. Nature. 2022;601:132–138. doi: 10.1038/s41586-021-04204-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M.M., Rapoport T.A., Prinz W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–1250. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- 43.Shibata Y., Shemesh T., Prinz W.A., Palazzo A.F., Kozlov M.M., Rapoport T.A. Mechanisms determining the morphology of the peripheral ER. Cell. 2010;143:774–788. doi: 10.1016/j.cell.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen B., Zheng P., Qian N., Chen Q., Zhou X., Hu J., Chen J., Teng J. Calumenin-1 interacts with Climp63 to cooperatively determine the luminal width and distribution of endoplasmic reticulum sheets. iScience. 2019;22:70–80. doi: 10.1016/j.isci.2019.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turk M., Baumeister W. The promise and the challenges of cryo-electron tomography. FEBS Lett. 2020;594:3243–3261. doi: 10.1002/1873-3468.13948. [DOI] [PubMed] [Google Scholar]
- 46.Hwang H.J., Kim Y.K. The role of LC3B in autophagy as an RNA-binding protein. Autophagy. 2022:1–3. doi: 10.1080/15548627.2022.2111083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 48.Joza N., Pospisilik J.A., Hangen E., Hanada T., Modjtahedi N., Penninger J.M., Kroemer G. AIF: not just an apoptosis-inducing factor. Ann. N. Y. Acad. Sci. 2009;1171:2–11. doi: 10.1111/j.1749-6632.2009.04681.x. [DOI] [PubMed] [Google Scholar]
- 49.Engelsberg A., Hermosilla R., Karsten U., Schülein R., Dörken B., Rehm A. The Golgi protein RCAS1 controls cell surface expression of tumor-associated O-linked glycan antigens. J. Biol. Chem. 2003;278:22998–23007. doi: 10.1074/jbc.M301361200. [DOI] [PubMed] [Google Scholar]
- 50.Mellor H., Parker P.J. The extended protein kinase C superfamily. Biochem. J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moumen A., Masterson P., O'Connor M.J., Jackson S.P. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 52.Keppetipola N., Sharma S., Li Q., Black D.L. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit. Rev. Biochem. Mol. Biol. 2012;47:360–378. doi: 10.3109/10409238.2012.691456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J., Chen Q.M. Far upstream element binding protein 1: a commander of transcription, translation and beyond. Oncogene. 2013;32:2907–2916. doi: 10.1038/onc.2012.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brina D., Grosso S., Miluzio A., Biffo S. Translational control by 80S formation and 60S availability: the central role of eIF6, a rate limiting factor in cell cycle progression and tumorigenesis. Cell Cycle. 2011;10:3441–3446. doi: 10.4161/cc.10.20.17796. [DOI] [PubMed] [Google Scholar]
- 55.Montanaro L., Treré D., Derenzini M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008;173:301–310. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Falco S., Russo G., Angiolillo A., Pietropaolo C. Human L7a ribosomal protein: sequence, structural organization, and expression of a functional gene. Gene. 1993;126:227–235. doi: 10.1016/0378-1119(93)90371-9. [DOI] [PubMed] [Google Scholar]
- 57.Marion M.J., Marion C. Localization of ribosomal proteins on the surface of mammalian 60S ribosomal subunits by means of immobilized enzymes. Correlation with chemical cross-linking data. Biochem. Biophys. Res. Commun. 1987;149:1077–1083. doi: 10.1016/0006-291x(87)90518-3. [DOI] [PubMed] [Google Scholar]
- 58.Russo G., Cuccurese M., Monti G., Russo A., Amoresano A., Pucci P., Pietropaolo C. Ribosomal protein L7a binds RNA through two distinct RNA-binding domains. Biochem. J. 2005;385:289–299. doi: 10.1042/BJ20040371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Q., Wu G., Yang L., Lu Y.P., Liu X.X., Han F., Deng Y.P., Fu X.C., Liu Q.B., Lu Y.M. Elucidation of the FKBP25-60S ribosomal protein L7a stress response signaling during ischemic injury. Cell. Physiol. Biochem. 2018;47:2018–2030. doi: 10.1159/000491470. [DOI] [PubMed] [Google Scholar]
- 60.Russo G., Ricciardelli G., Pietropaolo C. Different domains cooperate to target the human ribosomal L7a protein to the nucleus and to the nucleoli. J. Biol. Chem. 1997;272:5229–5235. doi: 10.1074/jbc.272.8.5229. [DOI] [PubMed] [Google Scholar]
- 61.Thoreen C.C., Kang S.A., Chang J.W., Liu Q., Zhang J., Gao Y., Reichling L.J., Sim T., Sabatini D.M., Gray N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu G.Y., Sabatini D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abu-Remaileh M., Wyant G.A., Kim C., Laqtom N.N., Abbasi M., Chan S.H., Freinkman E., Sabatini D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eskelinen E.L., Schmidt C.K., Neu S., Willenborg M., Fuertes G., Salvador N., Tanaka Y., Lüllmann-Rauch R., Hartmann D., Heeren J., et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol. Biol. Cell. 2004;15:3132–3145. doi: 10.1091/mbc.e04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andrei M.A., Ingelfinger D., Heintzmann R., Achsel T., Rivera-Pomar R., Lührmann R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA. 2005;11:717–727. doi: 10.1261/rna.2340405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cougot N., Babajko S., Séraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haas I.G., Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306:387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- 68.Kubica N., Bolster D.R., Farrell P.A., Kimball S.R., Jefferson L.S. Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin-dependent manner. J. Biol. Chem. 2005;280:7570–7580. doi: 10.1074/jbc.M413732200. [DOI] [PubMed] [Google Scholar]
- 69.Huo Y., Iadevaia V., Yao Z., Kelly I., Cosulich S., Guichard S., Foster L.J., Proud C.G. Stable isotope-labelling analysis of the impact of inhibition of the mammalian target of rapamycin on protein synthesis. Biochem. J. 2012;444:141–151. doi: 10.1042/BJ20112107. [DOI] [PubMed] [Google Scholar]
- 70.Landon A.L., Muniandy P.A., Shetty A.C., Lehrmann E., Volpon L., Houng S., Zhang Y., Dai B., Peroutka R., Mazan-Mamczarz K., et al. MNKs act as a regulatory switch for eIF4E1 and eIF4E3 driven mRNA translation in DLBCL. Nat. Commun. 2014;5:5413. doi: 10.1038/ncomms6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonzom C., Hüttner S., Mirgorodskaya E., Chong S.L., Uthoff S., Steinbüchel A., Verhaert R.M.D., Olsson L. Glycosylation influences activity, stability and immobilization of the feruloyl esterase 1a from Myceliophthora thermophila. Amb. Express. 2019;9:126. doi: 10.1186/s13568-019-0852-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ingolia N.T., Hussmann J.A., Weissman J.S. Ribosome profiling: global views of translation. Cold Spring Harb. Perspect. Biol. 2019;11:a032698. doi: 10.1101/cshperspect.a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hesketh J.E., Pryme I.F. Interaction between mRNA, ribosomes and the cytoskeleton. Biochem. J. 1991;277:1–10. doi: 10.1042/bj2770001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porter K.R. The cytomatrix: a short history of its study. J. Cell Biol. 1984;99:3s–12s. doi: 10.1083/jcb.99.1.3s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gershon N.D., Porter K.R., Trus B.L. The cytoplasmic matrix: its volume and surface area and the diffusion of molecules through it. Proc. Natl. Acad. Sci. USA. 1985;82:5030–5034. doi: 10.1073/pnas.82.15.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolosewick J.J., Porter K.R. Stereo high-voltage electron microscopy of whole cells of the human diploid line, WI-38. Am. J. Anat. 1976;147:303–323. doi: 10.1002/aja.1001470305. [DOI] [PubMed] [Google Scholar]
- 77.Heuser J. Whatever happened to the 'microtrabecular concept'? Biol. Cell. 2002;94:561–596. doi: 10.1016/s0248-4900(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 78.Ris H. The cytoplasmic filament system in critical point-dried whole mounts and plastic-embedded sections. J. Cell Biol. 1985;100:1474–1487. doi: 10.1083/jcb.100.5.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schliwa M. The evolving complexity of cytoplasmic structure. Nat. Rev. Mol. Cell Biol. 2002;3:291–296. doi: 10.1038/nrm781. [DOI] [PubMed] [Google Scholar]
- 80.Dossou A.S., Basu A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers. 2019;11:1422. doi: 10.3390/cancers11101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith B.A.H., Bertozzi C.R. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat. Rev. Drug Discov. 2021;20:217–243. doi: 10.1038/s41573-020-00093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arnold T.R., Stephenson R.E., Miller A.L. Rho GTPases and actomyosin: partners in regulating epithelial cell-cell junction structure and function. Exp. Cell Res. 2017;358:20–30. doi: 10.1016/j.yexcr.2017.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trucco A., Polishchuk R.S., Martella O., Di Pentima A., Fusella A., Di Giandomenico D., San Pietro E., Beznoussenko G.V., Polishchuk E.V., Baldassarre M., et al. Secretory traffic triggers the formation of tubular continuities across Golgi sub-compartments. Nat. Cell Biol. 2004;6:1071–1081. doi: 10.1038/ncb1180. [DOI] [PubMed] [Google Scholar]
- 84.Weigel A.V., Chang C.L., Shtengel G., Xu C.S., Hoffman D.P., Freeman M., Iyer N., Aaron J., Khuon S., Bogovic J., et al. ER-to-Golgi protein delivery through an interwoven, tubular network extending from ER. Cell. 2021;184:2412–2429.e16. doi: 10.1016/j.cell.2021.03.035. [DOI] [PubMed] [Google Scholar]
- 85.Saltzman A.B., Leng M., Bhatt B., Singh P., Chan D.W., Dobrolecki L., Chandrasekaran H., Choi J.M., Jain A., Jung S.Y., et al. gpGrouper: a peptide grouping algorithm for gene-centric inference and quantitation of bottom-up proteomics data. Mol. Cell. Proteomics. 2018;17:2270–2283. doi: 10.1074/mcp.TIR118.000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andrews S., Gilley J., Coleman M.P. Difference Tracker: ImageJ plugins for fully automated analysis of multiple axonal transport parameters. J. Neurosci. Methods. 2010;193:281–287. doi: 10.1016/j.jneumeth.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 88.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Geelen M.J.H. The use of digitonin-permeabilized mammalian cells for measuring enzyme activities in the course of studies on lipid metabolism. Anal. Biochem. 2005;347:1–9. doi: 10.1016/j.ab.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 95.Baghirova S., Hughes B.G., Hendzel M.J., Schulz R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX. 2015;2:440–445. doi: 10.1016/j.mex.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nabbi A., Riabowol K. Isolation of nuclei. Cold Spring Harb. Protoc. 2015;2015:731–734. doi: 10.1101/pdb.top074583. [DOI] [PubMed] [Google Scholar]
- 97.Abmayr S.M., Yao T., Parmely T., Workman J.L. Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr. Protoc. Mol. Biol. 2006;Chapter 12 doi: 10.1002/0471142727.mb1201s75. Unit 12 11. [DOI] [PubMed] [Google Scholar]
- 98.Sanders M.M. Fractionation of nucleosomes by salt elution from micrococcal nuclease-digested nuclei. J. Cell Biol. 1978;79:97–109. doi: 10.1083/jcb.79.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Teves S.S., Henikoff S. Salt fractionation of nucleosomes for genome-wide profiling. Methods Mol. Biol. 2012;833:421–432. doi: 10.1007/978-1-61779-477-3_25. [DOI] [PubMed] [Google Scholar]
- 100.Cameron I.L., Lanctot A.C., Fullerton G.D. Maintenance of low sodium and high potassium levels in cells and in tendon/collagen. Cell Biol. Int. 2012;36:503–509. doi: 10.1042/CBI20110439. [DOI] [PubMed] [Google Scholar]
- 101.Iadevaia V., Wang X., Yao Z., Foster L.J., Proud C.G. Evaluation of mTOR-regulated mRNA translation. Methods Mol. Biol. 2012;821:171–185. doi: 10.1007/978-1-61779-430-8_10. [DOI] [PubMed] [Google Scholar]
- 102.Reid D.W., Shenolikar S., Nicchitta C.V. Simple and inexpensive ribosome profiling analysis of mRNA translation. Methods. 2015;91:69–74. doi: 10.1016/j.ymeth.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original code. Data reported in this paper will be shared by the lead contact upon request.
The RNA sequencing data was uploaded to NCBI GEO and is publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and is publicly available as of the date of publication. The dataset identifier is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.