

Summary
Despite modest clinical improvement with anti-vascular endothelial growth factor antibody (AVA) therapy in ovarian cancer, adaptive resistance is ubiquitous and additional options are limited. A dependence on glutamine metabolism, via the enzyme glutaminase (GLS), is a known mechanism of adaptive resistance and we aimed to investigate the utility of a GLS inhibitor (GLSi). Our in vitro findings demonstrated increased glutamine abundance and a significant cytotoxic effect in AVA-resistant tumors when GLSi was administered in combination with bevacizumab. In vivo, GLSi led to a reduction in tumor growth as monotherapy and when combined with AVA. Furthermore, GLSi initiated after the emergence of resistance to AVA therapy resulted in a decreased metabolic conversion of pyruvate to lactate as assessed by hyperpolarized magnetic resonance spectroscopy and demonstrated robust antitumor effects with a survival advantage. Given the increasing population of patients receiving AVA therapy, these findings justify further development of GLSi in AVA resistance.
Subject areas: Oncology, Cellular physiology, Cancer
Graphical abstract
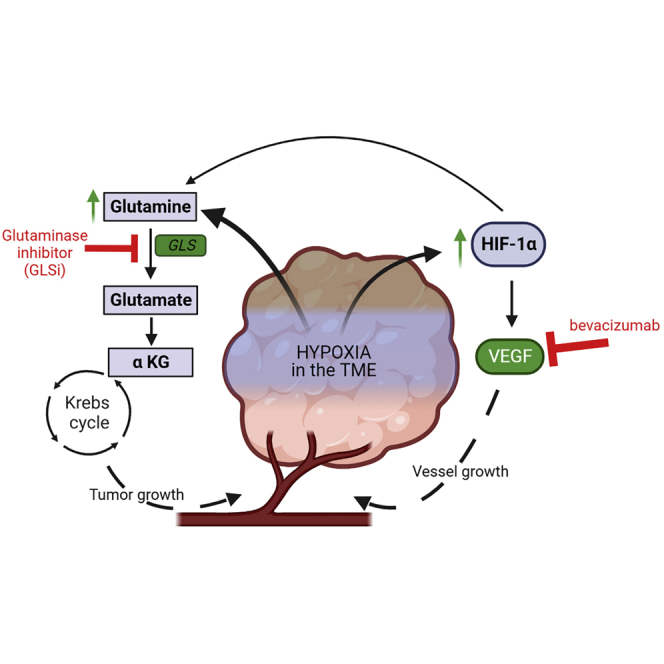
Highlights
-
•
Resistance to anti-vascular endothelial growth factor antibody (AVA) is common
-
•
We examine a small molecule inhibitor of glutaminase (GLSi) to combat resistance
-
•
GLSi enhanced the efficacy of AVA in vitro and in vivo, even in AVA-resistant tumors
-
•
This work supports further development of GLSi in AVA-resistant ovarian cancer
Oncology; Cellular physiology; Cancer.
Introduction
Ovarian cancer presents at late stages and is the most lethal gynecologic malignancy with more than 14,000 deaths per year.1 The tumor microenvironment (TME) of ovarian cancer, like those of other solid tumors, is characterized by hypoxia and increased levels of vascular endothelial growth factor (VEGF) which promotes angiogenesis, tumor growth, and progression.2,3 While the mainstay of treatment of ovarian cancer consists of tumor-reductive surgery and chemotherapy,4 advances in therapy have been aimed at targeting angiogenesis.5 Treatment with bevacizumab, a humanized monoclonal antibody against VEGF, has resulted in modest improvements in progression-free survival6,7,8; and it represents the first effective biologically targeted therapy for ovarian cancer. It is now approved by the U.S. Food and Drug Administration for use in both the up-front and recurrent settings.9 Practically speaking, this means that nearly all patients with ovarian cancer will be treated with anti-VEGF antibody (AVA) therapy at some point in their care. Unfortunately, the effects of this treatment are transient; virtually all tumors adapt a resistance to it and patients eventually succumb to their disease.10 Thus, alternative treatment strategies for AVA-resistant ovarian tumors represent a growing unmet clinical need.
Among the possible opportunities for overcoming adaptive resistance to AVA drugs, the dynamic changes in metabolism that are due, in part, to the hypoxic stress imposed by AVA therapy are considered here. In concert with elevated VEGF expression, tumor cells exhibit not only the Warburg effect with high rates of glucose consumption but also a dependence on glutamine metabolism, particularly with long-term exposure to bevacizumab.11 Glutamine is an essential amino acid that plays a critical role in anaplerosis, cancer initiation, and progression. Metabolic utilization of glutamine requires conversion to glutamate via the enzyme glutaminase (GLS). Researchers have exploited glutamine dependence in clinical trials of the treatment of solid tumors with GLS inhibitors (GLSi) (clinicaltrials.gov).12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32 However, the role of GLSi in overcoming adaptive resistance to AVA therapy is not known. We therefore investigated the impact of inhibiting GLS in the setting of glutamine dependence experienced by tumors under hypoxic stress imposed by AVA therapy. We hypothesize that hypoxia-mediated glutamine dependence in AVA-resistant ovarian cancer will make it susceptible to GLS inhibition.
Results
Treatment with, and adaptive resistance to, AVA therapy induces alterations in glutamine metabolism
Due to the abnormal vasculature inherent in tumors during malignant growth, the physiologic oxygen tension of solid tumors is hypoxic (1%–2%) compared to the surrounding parenchyma.33 Additional hypoxic stress to the tumor is imposed by treatment with AVA therapy (i.e. bevacizumab). After prolonged AVA exposure, tumor oxygen tension levels are estimated to be lower than 1%33 and we therefore set out to understand the impact of hypoxia extremes on glutamine metabolism. To do so, we first evaluated in vitro GLS expression in the ovarian cancer cell line OVCAR8 and immortalized endothelial cell line RF24 at 1% oxygen (aka hypoxic) conditions. We did this in concert with measuring the hypoxia-inducible factor-1 alpha (HIF-1α) expression level. We identified that GLS and HIF-1α expression in both cell lines was higher under hypoxic rather than under normoxic conditions (Figures 1A and 1B).
Figure 1.

Metabolic alterations in hypoxia and resulting from AVA therapy
(A and B) RF24 and OVCAR8 cells were cultured in normoxia (N) or hypoxia (H) for 12 and 24 h. GLS and HIF-1α protein (A) and GLS mRNA (B) expression levels in both cell lines were measured. Error bars indicate SD, ∗∗∗p < 0.001.
(C) Images of nude mice inoculated with 1 × 106 SKOV3ip1 cells and given bevacizumab (6.25 mg/kg twice weekly) until resistance emerged.
(D) IHC staining of frozen mouse ovarian tumors for the hypoxia marker CA9 in control vs. bevacizumab-treated (AVA-treated) tumors.
(E) Quantification of CA9 staining in control vs. AVA-treated tumors per high power field, error bars indicate SD ∗∗p < 0.01.
(F) IHC staining of frozen mouse ovarian tumors for endothelial cell marker CD31 in control vs. AVA-sensitive or AVA-resistant tumors.
(G) Quantification of vessel densities in control, AVA-sensitive, and AVA-resistant tumor samples. Error bars indicate SD. ∗∗∗p < 0.001 compared with the control group (Student’s t test). AVA, anti-VEGF antibody (bevacizumab); AVA-sens, AVA-sensitive; AVA-resis, AVA-resistant.
To evaluate the downstream metabolic effects of altered GLS expression, we performed an in vivo investigation using an orthotopic SKOV3ip1 model of ovarian cancer with adaptive resistance to AVA therapy with bevacizumab. In this experiment, mice received either a vehicle control or AVA therapy and all AVA-treated mice underwent weekly bioluminescent imaging in order to stratify them based on sensitivity or resistance to AVA therapy as outlined in the Materials and Methods (Figure 1C). We harvested tumors after they became resistant to treatment and assessed markers of hypoxic stress using immunohistochemical (IHC) staining. Compared with control tumors, all AVA-treated tumors had higher levels of the hypoxia marker CA9 (p < 0.01, Figures 1D and 1E) and the AVA-sensitive tumors demonstrated lower levels of the endothelial cell marker CD31 compared with those that were resistant to AVA treatment (p < 0.001, Figures 1F and 1G).
After confirming that AVA therapy results in hypoxic stress, we set out to define the specific alterations in tumor metabolism that occur in response to this stress. We first performed a quantitative metabolic analysis of the AVA-treated tumor tissues using liquid chromatography-mass spectrometry (LC-MS). These analyses revealed greater alterations in the purine synthesis pathways in AVA-resistant samples than in AVA-sensitive and vehicle-treated control samples (Figure 2A). Specifically, we found a greater abundance of metabolites such as urate, xanthine, xanthosine, and 3-ureidopropionate in the AVA-resistant group than in the control group (Figure 2B, p < 0.05 and Figure S3). We then performed desorption electrospray ionization mass spectrometry (DESI-MS) imaging34,35 of the same tumor tissues to further investigate treatment-specific metabolic changes within the heterogeneous TME. Spatially resolved molecular information was selectively extracted from the DESI-MS data obtained from tumor regions within each sample, excluding adjacent stroma or necrotic tissue. Significant alterations in the relative abundance of a variety of molecular species were identified when comparing control to AVA-treated tumors, including multiple key intermediates in amino acid metabolism, glycolysis, and purine synthesis (Figure 2C, FDR < 0.05). Higher relative abundances of xanthine and hypoxanthine were detected in AVA-resistant tumor tissue regions relative to control, in agreement with LC-MS experiments Figure 2B and 2C; p < 0.01 and p < 0.03, respectively). Additionally, significant alterations in glutaminolysis metabolites were identified with increased relative abundances of glutamate relative to glutamine in AVA-resistant compared to AVA-sensitive tissues, but not when compared to control (normalized ion abundance: 7.4 vs. 5.3 and 9.1, respectively; Figures 2D and S1, p < 0.05). Similar trends were observed for fumarate and glutathione, both of which are downstream metabolites of glutamate. Overall, our comprehensive mass spectral analyses of AVA-treated tumors indicate a significant role of glutamine metabolism in these tumors.
Figure 2.

Adaptive resistance of ovarian cancer to AVA therapy induces alterations in glutamine metabolism
(A) Plot of metabolic pathway impact analysis results after quantitative metabolic analysis by LC-MS of ovarian tumor from an orthotopic mouse model with SKOV3ip1 cells comparing control vs. AVA-resistant groups showing that the purine and pyrimidine metabolism differed markedly (FDR < 0.05). Data were generated using a MetaboAnalyst plot made with Prism software.
(B) Simplified diagram of the purine and pyrimidine metabolic pathways showing the key metabolites involved in purine and pyrimidine nucleotide metabolism. The inserted scatterplots show significantly higher levels of downstream metabolites indicated in the dashed arrow lines (urate, xanthine, xanthosine, and 3-ureidopropionate) in the resistant group (R) than in the control group (C), p < 0.05. UMP, uridine monophosphate; OMP, orotidine 5′-monophosphate; CMP, cytidine monophosphate; PRPP, phosphoribosyl pyrophosphate; GMP, guanosine monophosphate; XMP, xanthosine monophosphate; IMP, inosine monophosphate; AMP, adenosine monophosphate. Key intermediates in amino acid metabolism, glycolysis, and purine synthesis are indicated in bold.
(C) Intensity heatmap for metabolites selected by SAM as significantly altered among control, AVA-resistant, and AVA-sensitive tissues (FDR <5%) identified by DESI-MS imaging. Features were clustered using a Euclidean-distance formula according to the average signal intensity of the corresponding m/z value measured from tumor-specific regions. The color scale reflects Z score standard deviations from the mean relative abundance measured for each ion. For fatty acid (FA) species, X:Y indicates the number of carbons and double bonds, respectively.
(D) DESI-MS images showing an increase in glutaminolysis in AVA-treated tumors compared to control tumors, with an increased relative abundances of glutamate relative to glutamine in AVA-resistant compared to AVA-sensitive tissues and a decreased abundance of glutamate and glutamine in all AVA-treated tumors.
Hypoxia induces GLS expression in a hypoxia-inducible factor-1α-dependent manner
We next set out to further investigate the mechanisms by which GLS expression is altered after exposure to AVA therapy. In many cancers, GLS expression is correlated with HIF-1α expression.33,36,37 To determine whether this is a direct correlation, we analyzed OVCAR8 ovarian cancer cells and RF24 endothelial cells that were transiently transfected with small interfering RNA (siRNA) targeting HIF-1α or HIF-2α (Figure 3A). We observed that increased GLS expression under hypoxic conditions (1% O2) was abrogated by HIF-1α siRNA but not by HIF-2α or non-targeting siRNA (p < 0.001). These data indicate that enhanced upregulation of GLS in hypoxic conditions occurs in a HIF-1α-dependent manner.
Figure 3.

Effect of HIF-1α on GLS expression and baseline GLS expression levels in ovarian cancer and endothelial cells
(A) RF24 and OVCAR8 cells were transfected with HIF-1α or HIF-2α siRNA following exposure to 1% O2 for 24 h. Non-targeting siRNA (NS) was used for control. GLS mRNA relative expression level is shown with error bars indicating SD, ∗∗∗p < 0.001.
(B) Western blot of GLS expression level in multiple untreated ovarian cancer cell lines.
(C) Western blot of GLS expression in a series of endothelial cell lines cultured in both normoxic (N, 20% O2) or hypoxic (H, 1% O2) conditions; cell lines tested included a bevacizumab-resistant endothelial cell line (RF24-bev) and parental RF24 cells along with murine ovarian endothelial cells (MOEC) and human microvascular endothelial cells (HMEC).
GLS is ubiquitously expressed in both ovarian cancer and endothelial cell lines
Before performing additional in vitro experiments to assess the effects of GLS inhibition on ovarian cancer and endothelial cell lines, we first confirmed GLS expression in the ovarian cancer cell lines A2780, HeyA8, SKOV3, OVCAR3, OVCAR4, OVCAR5, OVCAR8, OVCA432, and IGROV (Figure 3B). We cultured the cells under normoxic conditions and measured GLS protein and mRNA expression in them (Figure 3B). GLS expression levels were highest in the SKOV3 cells; therefore, we chose this cell line for further in vivo work. We subsequently confirmed that GLS expression was present in a series of endothelial cell lines to include murine ovarian endothelial cells (MOEC), human microvascular endothelial cells (HMEC), and RF24 endothelial cells as well as RF24 cells with acquired resistance to bevacizumab (RF24-bev) (Figure 3C).
Altered glutamine metabolism under hypoxic conditions enhances sensitivity to GLS inhibition for both ovarian cancer and endothelial cells in vitro
Given the observed increase in glutaminolysis in a hypoxic environment, we next targeted this potential vulnerability with an internally synthesized GLSi known as IACS-012031,38,39 subsequently referred to as GLSi throughout the manuscript. We first tested the viability of OVCAR5, OVCAR8, and SKOV3 cells after culturing with escalating doses of this GLSi in vitro. Under normoxic conditions, the viability of all three cell lines was inhibited in a time-dependent manner (Figure 4A). Culture of the parental RF24 endothelial cell line with AVA, GLSi, or both demonstrated a reduction in cell viability when treated with GLSi as monotherapy and when it was combined with AVA under normoxic conditions (Figure 4B). We also observed dose-dependent inhibition of viability in all endothelial lines when cultured with GLSi, including MOEC, HMEC, and both the RF24 parental (RF24) and bevacizumab-resistant (RF24-bev) endothelial cells (Figure 4C). The HMEC and RF24-bev cells exhibited a greater sensitivity to GLSi-based therapy than either the MOEC or the RF24 parental cells (Figure 4C) when cultured under hypoxic conditions with the same dose of GLSi.
Figure 4.

Hypoxia enhances sensitivity to GLS inhibition in both cancer and endothelial cells in vitro
(A) The viability of three ovarian cancer cell lines (SKOV3, OVCAR5, and OVCAR8) upon culture with GLSi alone for 24–48 h in normoxic conditions (p < 0.05).
(B) The viability of the RF24 endothelial cell line after culturing with a positive control (VEGF), bevacizumab (AVA), GLSi monotherapy, or a combination of both therapies for 24 h in normoxic conditions (∗p < 0.05, ∗∗∗p < 0.001).
(C) The viability of four endothelial cell lines (MOEC, HMEC, parental RF24, and RF24-bev) after culturing in increasing concentrations of GLSi in either normoxic (20% O2) or hypoxic (1% O2) conditions.
(D) Bright-field microscopic images at 50X showing the vessel loop formation and the effect of bevacizumab (AVA) and GLSi monotherapy or in combination (AVA + GLSi) on the angiogenic capability of RF24-par cells in culture in normoxia (top row) versus hypoxia (bottom row).
(E) Average branch counts of the vessel loops described in the tube formation assay in (D) detected at 6 h as measured in triplicate experiments; ∗∗p < 0.01, ∗∗∗p < 0.001. VEGF, vascular endothelial growth factor; AVA, anti-VEGF antibody; GLSi, glutaminase inhibitor.
We also observed a significant reduction in the angiogenic capability (tube formation) of RF24 parental endothelial cells after treatment with GLSi (Figures 4D and 4E, p < 0.01). Under normoxia, the combination of GLSi and AVA resulted in significantly less tube formation than did monotherapy with either drug (branch count 9.2 vs. 21.0 and 13.2, respectively; p < 0.01) or no treatment (control 27.4; p < 0.001). This effect of reduced tube formation ability was even more pronounced in hypoxia than in normoxia, again demonstrating that GLSi in combination with bevacizumab had a significantly greater reduction in tube formation than control therapy or monotherapy with either GLSi or bevacizumab (branch count 2.6 vs. 16.8, 14.9, and 8.2, respectively; p ≤ 0.001). Ultimately, we observed increased sensitivity of both ovarian cancer and endothelial cell lines to GLSi therapy under hypoxic conditions, an effect that was even more pronounced when combined with AVA, and particularly in the endothelial cells that were resistant to AVA.
GLSi has robust antitumor effects in combination with AVA in vivo
To further assess the in vitro findings of increased GLSi sensitivity in hypoxia for both ovarian cancer and endothelial cells, we performed an in vivo experiment with an orthotopic SKOV3ip1 ovarian cancer mouse model. In this experiment, we randomized 60 nude mice to receive treatment with a vehicle control, GLSi, AVA, or a combination of AVA and GLSi given at the same doses as in monotherapy (Figure 5A). After seven weeks of treatment, the group of mice treated with the combination of AVA and GLSi exhibited a robust antitumor effect, with a lower mean tumor weight (0.05 g vs. 0.62 g and 0.64 g; p < 0.01) and tumor nodule number (3.3 vs. 12.8 and 13.9; p < 0.05) than those in the control and GLSi monotherapy groups, respectively (Figure 5B). The mean body weights were not significantly different between the combination therapy, GLSi monotherapy, and control groups (23.6 g, 22.0 g, and 22.1 g, respectively; p = 0.16, Figure S4). The mean tumor vessel density, as assessed by IHC staining for CD31, an endothelial cell marker, in the combination therapy group was lower than those in the control, GLSi monotherapy, and AVA monotherapy groups (11 vs. 30, 15, and 15, respectively; p < 0.001, Figure 5C). The tumors in the combination group also exhibited less cell proliferation than those in the GLSi and bevacizumab monotherapy treatment groups or control groups, respectively, as assessed using Ki67 staining (mean cell count/hpf: 108 vs. 151, 200, and 292; p < 0.01, Figure 5D).
Figure 5.

Robust antitumor effect of GLSi combined with AVA in an SKOV3ip1 mouse model as assessed using gross necropsy and immunohistochemistry
(A) In vivo schema of experimental protocol.
(B) Nodule numbers and tumor weights of SKOV3ip1 mouse model after either no treatment (Control), bevacizumab (AVA), IACS-012031 (GLSi), or a combination of bevacizumab and GLSi (AVA + GLSi). Error bars, SD. ∗p < 0.05; ∗∗p < 0.01 (compared with the control group using the Student’s t test).
(C) Blood vessel densities in ovarian tumor tissues harvested from mice in each treatment group as assessed by endothelial cell marker CD31. ∗p < 0.05; ∗∗∗p < 0.001.
(D) Cell proliferation assay according to immunohistochemistry staining of mouse ovarian cancer tissues stained with anti-Ki67. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We also subjected a subset of tissues obtained from this experiment (n = 5 per treatment group) to DESI-MS imaging analysis to investigate treatment-specific alterations. The relative abundances of 74 m/z features were significantly altered among treatment groups, corresponding to a broad range of small metabolites and lipid species (Figure S2, FDR < 5%). In particular, an overall reduction in the relative abundances of glutamate, malate, and hexose was identified in the combination therapy group compared to the control or monotherapy groups with either GLSi or AVA, respectively (normalized ion abundance 9.2, 13.1, 11.2 vs. 16.3, respectively; p < 0.05, Figures 6A and 6B). The relative abundances of glutamine and citrate were significantly increased in the GLSi monotherapy and combination groups compared to the control and AVA monotherapy groups (normalized ion abundance 13.4 and 9.0 vs. 1.9 and 3.6, respectively p < 0.0001, Figure 6B). When evaluating the conversion of pyruvate to lactate in the treatment groups, we observed a reduced relative abundance of lactate relative to pyruvate in all treatment groups compared to control, but most notably in the GLSi monotherapy and combination therapy groups, suggesting that the most substantial impact arising from GLSi therapy is on lactate metabolism (70.9, 72.6, 85.7 vs. 101.4, p < 0.05, Figures 6C and 6D).
Figure 6.

GLSi therapy has robust antitumor effects in combination with bevacizumab (AVA)
(A) Representative DESI-MS ion images of pyruvate, lactate, glutamine, and glutamate in mouse tumors after treatment with a control vehicle, AVA, GLSi, or a combination of the two. H&E, hematoxylin and eosin.
(B) Median normalized intensity of the relative abundance of glutamate metabolism and TCA cycle intermediates in control, AVA monotherapy, GLSi monotherapy, and combination therapy groups (Combo) mice in the SKOV3ip1 orthotopic ovarian cancer model. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(C) Median normalized intensity of the lactate-to-pyruvate ratio in the control and treatment groups. ∗p < 0.05; ns, not significant.
(D) Median normalized intensity ratio of glutamate to glutamine in the control, AVA monotherapy, GLSi monotherapy, and combination therapy treatment groups. Statistical significance was determined by Tukey’s HSD test, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Detection of GLSi therapy response using hyperpolarized magnetic resonance spectroscopy
To more accurately assess the therapeutic efficacy of GLSi therapy, we performed hyperpolarized magnetic resonance spectroscopy (HP-MRS) to quantify metabolic changes in lactate metabolism associated with this therapy in real time. We did so using a total of 10 mice from the aforementioned SKOV3ip1 model of adaptive resistance of ovarian cancer to AVA therapy, with five mice receiving a vehicle control and five receiving GLSi. HP-MRS is a novel technique that enhances the sensitivity of traditional magnetic resonance imaging up to 10,000-fold and facilitates a direct, noninvasive analysis of the flux of pyruvate metabolism with spatial resolution of tumors in situ.40 Baseline MRI images were obtained with mice under anesthesia to confirm tumor location in both groups. After tail vein injection of hyperpolarized [1-13C] pyruvate as outlined in the Materials and Methods, we quantitatively calculated each tumor’s normalized lactate over pyruvate ratio, defined as the 13C resonance signal of lactate divided by the 13C resonance signal of pyruvate. Our analysis demonstrated that pyruvate-to-lactate conversion in AVA-resistant tumors was significantly reduced in vivo by GLSi therapy (0.337 vs. 0.178; p ≤ 0.001, Figures 7A and 7B). These findings are concordant with the changes in relative abundance measured for pyruvate and lactate using DESI-MS imaging discussed above (Figure 6C).
Figure 7.

Detection of GLSi therapy response by HP-MRS and evaluating changes in AVA sensitivity when GLSi is given after emergence of AVA resistance
(A) Representative T2-weighted MRI (coronal slice) and real-time in vivo13C magnetic resonance spectroscopy images of AVA-resistant ovarian tumors in mice after intravenous hyperpolarized pyruvate injection for two treatment groups: vehicle control (left) and GLSi (right) with spectra collected from the MRI slabs on the ovarian tumors over 2 s.
(B) The normalized lactate/pyruvate ratios for vehicle- and GLSi-treated mice. ∗∗∗p < 0.001.
(C) Schema of the orthotopic SKOV3ip1 mouse model in which AVA resistance was established prior to the initiation of GLSi therapy. AVA, anti-VEGF antibody, (bevacizumab).
(D) Body weights, tumor weights, and nodule numbers at the time of necropsy for mice treated with AVA combined with a vehicle vs. those given AVA combined with GLSi. Error bars, SD. ∗p < 0.05; ∗∗∗p < 0.001 (compared with the control group using the Student’s t test). ns, not significant.
(E) Kaplan-Meier survival curve showing survival advantage with GLSi therapy after AVA resistance is established (p = 0.048, HR 2.07; 95% CI 1.03–4.15). AVA, anti-VEGF antibody, (bevacizumab); GLSi, glutaminase inhibitor.
GLS inhibition at the emergence of AVA resistance restores sensitivity in an adaptive resistance model of ovarian cancer
After observing the robust antitumor effects of GLSi combined with AVA therapy when given in combination up front, we set out to determine whether GLSi can restore sensitivity of ovarian cancer to AVA therapy if initiated after AVA resistance is established. For this experiment, we again used the SKOV3ip1 orthotopic mouse model but administered AVA monotherapy with bevacizumab after tumor establishment and continued it until resistance was confirmed by bioluminescent imaging (Figure 7C). Afterward, we maintained all AVA-resistant mice on AVA therapy and subsequently randomized them to receive either a vehicle control or GLSi (both at 200 μL via oral gavage twice a day) until they became moribund. GLSi therapy after establishment of adaptive resistance to AVA therapy resulted in a partial abrogation of tumor growth as evidenced by a 57% reduction in mean tumor weight (0.43 g vs. 1.01 g; p = 0.04) and a 68% lower mean tumor nodule number in the GLSi group than in the control group (5 vs. 15; p = 0.0003, Figure 7D). The administration of GLSi therapy after the emergence of AVA resistance not only provided an antitumor response but it also resulted in a significant survival benefit (mean survival 69 vs. 63 days; p = 0.048). Continuing AVA therapy in the resistant setting with a vehicle control was associated with a 2-fold increased risk of death compared to treatment with AVA plus GLSi (HR 2.07; 95% CI 1.03–4.15; Figure 7E).
Discussion
Overall, the key finding of our investigation is that increased glutamine metabolism is a metabolic adaptation that occurs in an orthotopic ovarian cancer model of adaptive resistance to AVA therapy. We exploited this metabolic vulnerability of glutamine dependence by combining AVA therapy (bevacizumab) with GLSi which produced robust antitumor efficacy in both in vitro and in vivo models of ovarian cancer.
Reliance on glutamine metabolism for growth and development is an established hallmark of cancer.3 However, the fate of glutamine and its metabolism is complex.41 Previously, we reported the adaptive nature of the TME under nutrient stress, and in particular, we elucidated the role of reactive stromal cells under glutamine deprivation and the ability of malignant tumors to harness carbon and nitrogen from alternative sources to maintain glutamine stores.42 Use of these glutamine stores is heterogeneous, as they can be consumed during protein synthesis, serve as building blocks for the antioxidant glutathione, or be converted into α-ketoglutarate for tricarboxylic acid cycle anaplerosis via GLS.41,43 The role of GLS in cancer growth and progression varies among human cancers and is dependent on the cancer phenotype, with strong evidence of an oncogenic role for GLS in colon, liver, and ovarian cancers but not in other cancers, such as non-small-cell lung cancer.41 Thus, GLS expression and the prognostic implications of its expression vary by cancer type, with higher GLS expression associated with worse prognosis for ovarian cancer.36
Since the U.S. Food and Drug Administration approved the AVA, bevacizumab, for use in patients with ovarian cancer in 2014, modest improvements in the progression-free survival of this cancer have occurred.6,7,8 Subsequently, bevacizumab was approved for patients with relapsed ovarian cancer.9 These broad categories of approval translate into the reality that nearly every patient receiving treatment for ovarian cancer will be exposed to AVA with bevacizumab. Unfortunately, due in part to the metabolic adaptations discussed above, the benefits of bevacizumab are modest and adaptive resistance develops nearly universally within months of initiation. Therefore, additional agents are needed to target the population with adaptive resistance of ovarian cancer to AVA therapy.
In the present study, we identified ubiquitous expression of GLS in ovarian cancer cell lines and confirmed that treatment with bevacizumab is associated with an upregulation of GLS as a metabolic adaptation by both cancer cells and endothelial cells. This upregulation occurs in an HIF-1α-dependent fashion in both ovarian cancer cells and endothelial cells where bevacizumab functions. We targeted this hypoxia-mediated increase in GLS expression using GLSi therapy. In vitro, we observed significant vulnerability of both cancer cells and endothelial cells to GLSi treatment, with increased sensitivity to GLSi treatment in the bevacizumab-resistant RF24 cells (RF24-bev). We subsequently noted a profound antitumor effect of GLSi in the SKOV3ip1 ovarian cancer orthotopic mouse model of adaptive resistance to bevacizumab suggesting that this combination of therapy may be of clinical benefit. Interestingly, we demonstrated antitumor efficacy of GLSi when given together with bevacizumab as up-front therapy and also demonstrated a survival benefit when GLSi was added to bevacizumab after the emergence of resistance. This suggests that GLSi therapy is of value when given in combination or sequentially with bevacizumab. Therefore, GLSi therapy may prove to be beneficial for patients whose disease relapses after exposure to bevacizumab or is AVA resistant. Our in vivo studies suggest that this combination therapy is well tolerated. Further investigations evaluating biologic markers that could predict response of ovarian cancer to GLSi therapy as well as evaluating the combination of GLSi with additional chemotherapy drugs are warranted.
Limitations of the study
Limitations of this study include the utilization of a single ovarian cancer cell line for the in vivo investigations. However, the authors selected the SKOV3ip1 ovarian cancer cell line due to the expression profile of GLS making it the most suitable candidate for study. Additionally, while the study provided a well-rounded assessment of metabolic adaptations of ovarian cancer cell lines and tumors exposed to hypoxic environments and GLSi treatment, definitive evaluation of the mechanisms of resistance and therapy response(s) were limited and merit further investigation.
Conclusion
GLSi therapy, when combined with AVA therapy, has robust antitumor effects in preclinical ovarian cancer models both in vitro and in vivo. This combined therapy warrants further exploration given the potential to expand the utility and efficacy of bevacizumab in treatment of ovarian cancer, particularly in the AVA-resistant setting.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse CD31 monoclonal antibody | BD Pharmingen | Cat. #553370; RRID: AB394816 |
| Rabbit anti-mouse CA9 monoclonal antibody | Novus Biologics | Cat. # NB100-417SS; RRID: AB_788423 |
| Goat anti-rat horseradish peroxidase-linked secondary antibody | Jackson ImmunoResearch | Cat. # 112-035-167; RRID: AB_2338139 |
| Rabbit anti-mouse Ki67 monoclonal antibody | NeoMarkers | Cat. # RB-9043-P1; RRID: AB_149873 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | Invitrogen | Cat # 11668030 |
| Hank’s Balanced Salt Solution | Cellgro | Cat. # 21-021-CV |
| Luciferin | GoldBio | Cat. # LUCK-1G |
| 2-hydroxypropyl-beta-cyclodextrin | Sigma-Aldrich | Cat. # H107-100G |
| DAB | Invitrogen | Cat. # 750118 |
| 13C pyruvic acid | ISOTEC; Sigma-Aldrich | Cat. # 677175 |
| Glutaminase inhibitor (GLSi) | IACS | IACS-012031 |
| Critical commercial assays | ||
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat. # 23225 |
| Western Lightning PLUS ECL Kit | PerkinElmer | Cat. # NEL103E001EA |
| RNeasy Mini Kits | Qiagen | Cat. # 74004 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems, Inc. | Cat. # 4368814 |
| Experimental models: Cell lines | ||
| Human: RF24 | Courtesy of Dr. Lee Ellis | CVCL_AX74 |
| Human: SKOV3ip1 | ATCC | CVCL_0C84 |
| Human: OVCAR8 | MDACC cell line bank | CVCL_1629 |
| Human: OVCAR5 | ATCC | CVCL_1628 |
| Human: HeyA8 | MDACC cell line bank | CVCL_8878 |
| Human: A2780 | MDACC cell line bank | CVCL_0134 |
| Human: OVCAR3 | MDACC cell line bank | CVCL_0465 |
| Human: OVCAR 4 | MDACC cell line bank | CVCL_1627 |
| Human: IGROV | MDACC cell line bank | CVCL_8723 |
| Human: HMEC | ScienCell Researh Laboratories | Cat. # 1000 |
| Murine: MOEC | Courtesy of Dr. Robert Langley44 | Courtesy of Dr. Robert Langley44 |
| Experimental models: Organisms/strains | ||
| Mouse: athymic nude female (NCr-nude) | Taconic Biosciences | Model # NCRNU-F |
| Oligonucleotides | ||
| Human-specific GLS siRNA (GAGGAAAAGGUUGCAUGAUUAdtdT) | Proteintech | Cat. # 12855-1-AP |
| Control-non-targeting siRNA (UAAUCUGCAACCUUUCCUCTdT) | Sigma-Aldrich (St. Louis, MO) | Cat. # SIC001 |
| MISSION siRNA Universal Negative Control #1 | Sigma-Aldrich | Cat. # SIC0012NMOL |
| Software and algorithms | ||
| GraphPad Prism v 9.3.0 | GraphPad Software, Inc., San Diego, CA | https://www.graphpad.com/ |
| ImageJ | NIH Image | https://imagej.nih.gov/ij/inex.html |
| MATLAB | The MathWorks | https://www.mathworks.com/ |
| Xenogen IVIS imaging system | PerkinElmer | https://www.perkinelmer.com/product/ivis-lumina-s5-imaging-system-cls148588 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anil. K. Sood, M.D. (asood@mdanderson.org).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer agreement.
Experimental model and subject details
Cell lines and culture conditions
The human ovarian cancer cell lines SKOV3ipluc (RRID: CVCL_0C84), OVCAR5 (RRID: CVCL_1628), and OVCAR8 (RRID: CVCL_1629) were obtained from the ATCC and The University of Texas MD Anderson Cancer Center Cytogenetics and Cell Authentication Core. The immortalized human vascular endothelial cell line RF24 (RRID: CVCL_AX74) was obtained from Dr. Lee Ellis. The murine ovarian endothelial cells (MOEC) were obtained from Dr. Robert Langley and have been previously characterized and described.44 Human microvascular endothelial cells (HMEC) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained according to the manufacturer’s instructions with endothelial cell medium (ScienCell) containing 10% fetal bovine serum. For in vitro studies, HMECs were treated and applied to MTT and Western blot analysis between 5 and 8 passages. SKOV3ipluc and OVCAR8 cells were cultured in RPMI 1640 medium (HyClone) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% gentamycin. OVCAR5 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% gentamycin. RF24 cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum, sodium pyruvate, non-essential amino acids, and 1% gentamycin. MOEC cells were cultured in phenol red free DMEM supplemented with 10% fetal bovine serum and 0.1% gentamicin sulfate. All cells were cultured at 37°C with 5% CO2 with the exception of MOEC which were cultured at 33°C. All cells were cultured at ambient atmospheric O2 with the exception of hypoxia experiments in which case they were cultured 1% O2, as indicated. All cell lines were authenticated by the Cytogenetics and Cell Authentication Core using short tandem repeat fingerprinting and were tested for mycoplasma contamination using polymerase chain reaction. Cells were used within 20 passages after thawing for in vitro experiments and 10 passages after thawing for in vivo experiments. In vitro studies were done with 70-80% confluent cultures.
In vivo models of ovarian cancer
All animal protocols were approved by the MD Anderson Institutional Animal Care and Use Committee. All animal experiments were performed with 6- to 8-week-old female athymic nude mice (NCr-nude) obtained from Taconic Biosciences. The mice were housed five per cage under pathogen-free conditions at a constant temperature and humidity. All mice were fed a regular diet and water ad libitum according to the guidelines of the American Association for Laboratory Animal Science and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. Investigators sacrificed the mice via carbon dioxide euthanasia followed by cervical dislocation once the mice were moribund.
To establish xenograft models for all mouse experiments, luciferase-labeled SKOV3ip1 ovarian cancer cells were cultured to 70–90% confluence and then trypsinized, washed twice with phosphate-buffered saline, and resuspended in ice-cold Hank’s Balanced Salt Solution (cat. #21-021-CV; Cellgro). The mice were then inoculated with 1 × 106 SKOV3ip1 cells via intraperitoneal injection to the right side of the abdomen. Tumor establishment was subsequently confirmed after injection of 200 μL of 14.3 mg/mL luciferin (cat. #LUCK-1G; GoldBio) using a Xenogen IVIS in vivo imaging system. Following confirmation of disease burden (mice without tumor uptake were removed from the experiment), all mice in the therapeutic experiment were randomly assigned to the following treatment groups: vehicle control, bevacizumab (anti-human VEGF antibody, AVA, at 6.25 mg/kg, intraperitoneally, twice a week), GLSi (IACS-012031, 200 mg/kg given orally twice daily, five days a week), or a combination of the two at these doses (n = 15 for all groups). In the AVA-resistance model, twice-weekly treatment with AVA (bevacizumab) was initiated upon confirmation of tumor uptake. Tumor burden was subsequently assessed weekly via IVIS imaging (Xenogen), and the mice were placed in two groups: therapy-sensitive and -resistant. Sensitivity to AVA therapy was defined as a decrease or plateau in the relative intensity of bioluminescent signaling over three weeks of treatment. The therapy-sensitive mice were sacrificed approximately one week later to confirm that they truly had sensitive phenotypes. Resistance to AVA therapy was defined as an initial decrease then steady increase in the relative intensity of bioluminescent signaling. The resistant group was then separated into two groups: control (AVA plus vehicle control given via oral gavage twice a day, n = 20) and treatment (AVA plus GLSi given via oral gavage twice a day, n = 25). The treatment in both groups continued until each mouse became moribund and then were sacrificed. The mice were monitored daily for adverse effects of treatment, and their body weights were measured weekly. Survival time was calculated for each mouse as the number of days from the date of inoculation of SKOV3ip1 cells to the date of euthanasia.
Mouse tumor weights, nodule numbers, distribution of metastasis, and presence of ascites were recorded at the time of gross necropsy. All tumor tissues were dissected, and samples were snap-frozen for later protein or RNA analysis (e.g., DESI-MS, LC-MS), fixed in formalin for paraffin embedding, or snap-frozen in optimal cutting temperature compound (Mercedes Scientific) for frozen slide preparation.
Bevacizumab was obtained from MD Anderson. The glutaminase inhibitor, IACS-012031, was obtained from the MD Anderson Institute for Applied Cancer Science and reconstituted in 25% aqueous 2-hydroxypropyl-β-cyclodextrin (cat. #H107-100G; Sigma-Aldrich) in phosphate-buffered saline at a dose of 25 mg/mL. This was administered via oral gavage as above. Once mice in any group became moribund, all the mice were euthanized.
Method details
Cell viability assay
To evaluate the cytotoxic effects of GLSi and bevacizumab monotherapy and in combination on cell viability, ovarian cancer cells (SKOV3ip1, OVCAR5, and OVCAR8) were seeded in a 96-well plate at a density of 3,000 cells per well in a 100-μL total volume in quadruplicates. Cells were incubated for 24 hours, and after demonstration of adequate attachment, the culture medium was removed and replaced with a medium containing serial dilutions of GLSi and bevacizumab. The cells were then incubated for 24, 48, or 72 hours depending on the experimental oxygen conditions. Following incubation, the cells were treated with a 0.05% MTT solution for 2–4 hours. The supernatant was then gently removed, and the MTT formazan was dissolved in 100 μL of dimethyl sulfoxide. The absorbance was subsequently read at 570 nm using a BioTek uQuant microplate spectrophotometer. All experiments were performed in triplicate. Dose-response curves were plotted using Prism software (version 8.0.0; GraphPad Software).
Western blotting
Extraction of total protein cell lysates was performed using modified RIPA buffer with protease and phosphatase inhibitors. BCA Protein Assay Reagent (Thermo Fisher Scientific) was then used to measure protein concentrations. Protein expression for each lysate was subsequently detected via Western blotting of a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis separation gel using a primary antibody against GLS (cat. #12855-1-AP; Proteintech). The antibody was incubated overnight at 4°C and then incubated with corresponding horseradish peroxidase-linked whole secondary antibodies. A chemiluminescence assay using a Western Lightning PLUS ECL Kit (PerkinElmer) was performed to expose the membranes and protein bands were quantified using densitometry with ImageJ software (National Institutes of Health). β-actin was used as a sample loading control for all reads.
Quantitative RT-PCR (qRT-PCR)
Relative levels of mRNA expression of GLS were detected using the qRT-PCR method as described before.11 Briefly, each qRT-PCR was carried out with 1 μg of RNA isolated from cells using RNeasy Mini Kits (Qiagen, CA) and reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems Inc.) according to the manufacturer’s protocol. The cDNA synthesized was then used as a template in the qRT-PCR using the specific GLS TaqMan Gene expression probe (Hs0104020_m1, ThermoFisher). Quantitative RT-PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems, Inc.). The 18s rRNA was used as endogenous control, and relative mRNA expression was calculated using 2−ΔΔCTmethod.45
GLS gene silencing by small interfering RNA
Small interfering RNA (siRNA) targeted to GLS was purchased from Sigma-Aldrich. In vitro transient transfection was performed as described previously.46 Briefly, the cells were transfected with a GLS1-specific or scrambled (control) siRNA using lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). siRNA with a nonspecific function that shared no sequence homology with any known mammalian mRNA in a BLAST search was used to control the target siRNA (Mission siRNA Universal Negative Control#1, Sigma). At selected time intervals, cells were harvested to measure mRNA levels of GLS1 using qRT-PCR.
IHC staining
All tumor samples subjected to IHC staining were collected from the in vivo experiment. Formalin-fixed, paraffin-embedded tumor sections were used for staining for anti-Ki67 (1:100, cat #RB-9043-PI; NeoMarkers). Paraffin slides were prepared via deparaffinization and antigen retrieval, whereas frozen slides were prepared via cold acetone fixation. This was followed by endogenous peroxide blocking with 3% hydrogen peroxide and a protein block with 4% fish gelatin. All samples were incubated with a primary antibody diluted in 4% fish gelatin overnight and then incubated with either a peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibody for one hour at room temperature.
Frozen slides were used for staining for CD31 (1:800, cat. #53370; BD Pharmingen) and CA9 (1:100, cat# NB100-417SS; Novus Biologicals) using goat anti-rat and goat anti-rabbit horseradish peroxidase-linked secondary antibodies, respectively (1:250, cat. #112-035-167; Jackson ImmunoResearch). Frozen slides were prepared by first fixing slides in cold acetone for 10 minutes and then washing them in phosphate-buffered saline and blocking with endogenous peroxidase and 3% H2O2 in methanol. All samples were incubated with a primary antibody diluted in protein block (5% normal horse serum and 1% normal goat serum in phosphate-buffered saline) overnight at 4°C. The slides were then incubated with a secondary antibody for one hour and then incubated with DAB (Invitrogen, Cat. #750118). Microscopic assessment of DAB staining was performed for all slides to monitor appropriate staining density. The slides were counterstained with hematoxylin (Gill 3). Quantification of IHC staining was performed by randomly selecting five fields at 200x magnification per slide and manually counting stained nuclei in each field. Mean cell counts and SDs were calculated, and treatment groups were compared using the Student t-test.
Polar metabolite extraction from tissue
The protocol was modified from Zhou et al.47 To minimize metabolite degradation tumor tissue samples were kept on dry ice throughout the LC-MS experiment. Briefly, 30–66 mg of each tissue sample was quickly weighed in a homogenizer tube previously filled with beads. A prechilled methanol/water (1:1, v/v) solution was added to the tube based on the weight of the measured tissue (5 mL per 1 g of tissue, or 125 μL per 25 mg of tissue). The samples were then homogenized using a Precellys homogenizer (Bertin Instruments) at 4°C and 6,500 rpm using two cycles of 25 seconds with 30-second intervals. Afterward, 100 μL of the tissue homogenate was transferred to a fresh tube, and 500 μL of methanol/water (1:1, v/v) solution and 500 μL of chloroform were added to the samples and they were vortexed for two minutes at 4°C and centrifuged at 16,000 g for 10 minutes at 4°C. A total of 500 μL of polar metabolite extract was then dried in a vacuum evaporator. The dried metabolites were reconstituted in 200 μL of methanol/water (1:1, v/v) solution, sonicated for 10 minutes, and filtered through an Agilent 0.2-μm Econofilter. The filtrate was then transferred into LC vials for analysis.
LC-MS protocol and data analysis
A 10-μL tumor sample prepared in the manner outlined above was injected for analysis into an Agilent 6520 Q-TOF LC/MS machine with an ACQUITY UPLC BEH C18 Column (130 Å, 1.7 μm, 2.1 mm × 150 mm) coupled with VanGuard Pre-Columns and an XBridge BEH Amide XP Column (130 Å, 2.5 μm, 4.6 mm × 150 mm) coupled with an XBridge BEH Amide XP VanGuard Cartridge (130 Å, 2.5 μm, 2.1 mm × 5 mm). The column compartment was set at 40°C, and the analysis was performed in both positive and negative modes. One hundred microliters of sample filtrated was transferred to LC vials, and 10 μL of each filtered sample was pooled to form the quality control samples. For analysis of the C18 column, mobile phase A consisted of water with 0.1% formic acid, whereas mobile phase B contained acetonitrile in 0.1% formic acid. The gradient method using the C18 column was as follows: 0 minutes: 1% B; 1 minute: 1% B; 8 minutes: 99% B; 13 minutes: 85% B; 13.1 minutes: 1% B; 16 minutes: 1% B. For analysis with the HILIC column, the mobile phase A was 10 mM ammonium formate in water with 0.1% formic acid, whereas mobile phase B contained 10 mM ammonium formate in acetonitrile with 0.1% formic acid. The gradient method using the HILIC column was as follows: 0 minutes: 99% B; 11.8 minutes: 20% B; 12.5 minutes: 99% B; 14.7 minutes: 99% B. The metabolite peaks were extracted for the analysis using Agilent MassHunter Profinder software based on our in-house library of metabolites. Any metabolites whose relative SD was greater than 30% in the QC measurements were excluded from further analysis. The metabolite peak areas in the spectrum were normalized according to the weight of the tissue. Other downstream analyses were performed using MetaboAnalyst software version 5.0 and data were then presented with the aid of GraphPad Prism version 8.0.0.
DESI-MS imaging and SAM analysis
Tumor tissues were flash-frozen and stored at −80°C prior to analysis. Frozen tissues were sectioned at 12 μm thickness, thaw-mounted onto glass slides, and immediately analyzed using a Q Exactive Focus or Q Exactive HF Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to a 2D OmniSpray stage (Prosolia Inc.) and a laboratory-built DESI sprayer. DESI-MS imaging was performed in the negative ion mode at a spatial resolution of 200 μm using a mass resolving power of 70,000 (m/z 200) and an instrument method optimized for enhanced detection of small metabolite species from m/z 80–500. A histologically compatible solvent system comprised of methanol:acetone 4:1 (v/v) was used as the DESI spray solvent, at a flow rate of 5.0 μL/min. DESI-MS ion images were assembled and visualized using Firefly (Prosolia, Inc.) and BioMap (Novartis) software. Ions of interest were tentatively identified using high mass accuracy measurements and tandem mass spectrometry experiments.
After DESI-MS imaging, the analyzed tissue sections were stained with hematoxylin and eosin and regions of tumor, stroma, and necrosis were annotated by D.G. within each sample. Mass spectra were extracted from pixels corresponding to tumor regions within the DESI-MS dataset using MSiReader software. The resulting ion intensity matrix was processed by binning each m/z value to the nearest thousandth and removing peaks that were present in less than 20% of the extracted pixels. The extracted DESI-MS data was analyzed using significance analysis of microarrays (SAM), a modified significance test to identify statistically significant alterations in relative abundance for specific mass-to-charge (m/z) values.48 Multiclass SAM was performed in R using the “samr” package to identify features with significantly altered abundance among treatment groups. A false discovery rate (FDR) of 5% was applied to identify m/z values with significant differences in relative abundance among treatment groups. Ion intensities for selected metabolites of interest were plotted in GraphPad Prism (version 9.3.0) and subjected to one-way ANOVA with Tukey’s post-hoc test for pairwise comparisons.
Hyperpolarized 13C-pyruvate sample preparation
A solution of 20 μL of 13C pyruvic acid (ISOTEC; Sigma-Aldrich), 15 mM OX63 trityl radical (GE Healthcare), and 1.5 mM gadolinium chelate (ProHance) was polarized at 3.35 T and 1.4 K using dynamic nuclear polarization (HyperSense; Oxford Instruments) for one hour. A frozen 13C-pyruvate sample was rapidly dissolved in 4 mL of superheated alkaline buffer containing 100 mg/L EDTA, 40 mM NaOH, 40 mM TRIS buffer, and 30 mM NaCl. The final concentration of pyruvate injected into the mouse tail vein was 80 mM, with a physiologic pH of about 7.4. Next, 200 μL of hyperpolarized pyruvate was injected into mice using a tail vein catheter for 8–10 seconds.
Animal handling during magnetic resonance imaging experiments
For magnetic resonance imaging (MRI) experiments, a 7T Bruker BioSpin MRI scanner (horizontal bore) was used. Mice were anesthetized using isoflurane in an anesthesia chamber with scavenging. The animals were fixed in a holder specially designed for mice and placed in the MRI coil equipped with a nose cone for inhaled anesthesia. A respiration monitoring pad and body temperature-maintaining heated pad were used during the MRI experiments.
T2-weighted proton MRI
Conventional anatomic magnetic resonance images of mice were acquired using multislice T2-weighted rapid acquisition with a relaxation enhancement sequence. Images with different views, including axial, coronal, and sagittal views, were acquired to identify tumors and regions of interest in the flanks of the mice. The imaging parameters for the T2-weighted scans were an echo time of 15 ms, repetition time of 2.5 seconds, 4-cm field of view, 256 μm × 256 μm in-plane resolution, 10 slices with thicknesses of 1 mm, and four image averages.
In vivo13C magnetic resonance spectroscopy
A series of slab-selective 13C spectra with a slab thickness of 8 mm was acquired right after injection of mice with hyperpolarized pyruvate using an SP Flash sequence. The subcutaneous tumor locations and similar sizes of the tumors in the experimental mice used in this study increased the precision of slab placement through most of the ovarian tumors and limited the contribution of nonmalignant signals consistently throughout the experiments. A total of 90 transients were acquired using a delay time between each transient of two seconds (total time, three minutes). For each transient, a 15° flip angle excitation Gaussian pulse and 2,048 data points were used. A small 8 M 13C urea phantom injected with gadolinium-DPTA was used in each mouse experiment for chemical shift referencing. Experimental data were processed on the MATLAB programming language (The MathWorks, Inc.) and TopSpin (Bruker BioSpin) platforms. Phase correction and 10- to 15-Hz line broadening were introduced to each individual spectrum. The areas under the spectral peaks within the frequency range for pyruvate and lactate were integrated over the entire array. The lactate-to-pyruvate metabolic flux ratios (lactate/pyruvate) were estimated by calculating the individual integration of lactate and pyruvate spectral signals between treatment groups and comparing the change in signal over time.
Quantification and statistical analysis
Differences between groups were evaluated using the Student t-test or Mann-Whitney U test according to data distribution and variance homogeneity. One-way differences between two groups were evaluated using the Student t-test or Mann-Whitney U test according to data distribution and variance homogeneity, whereas one-way analysis of variance was used for multiple group comparison. For survival experiments, Kaplan-Meier analysis and a log-rank test were performed to explain differences in survival. All statistical analyses were conducted using Prism software (version 8.0.0). The p values were two-tailed and values less than 0.05 were considered significant. All results were presented as means (± SEM or SD).
Acknowledgments
The research reported here is funded by the following: T32 training grant CA101642 (D.G.); MD Anderson Cancer Center Support Grant CA016672; CPRIT grant PR 180381; National Institutes of Health/National Cancer Institute grants award numbers P30 CA016672, R01 CA227622 (D.N. and A.K.S.), CA177909 (A.K.S.), R35 CA209904 (A.K.S.), MD Anderson Ovarian Cancer Moon Shot (A.K.S.), SPORE in Ovarian Cancer CA 217685 (A.K.S.), the American Cancer Society, the Dunwoody Fund, the Gordon Fund, and the Ovarian Cancer Research Alliance FP00006137 (E.S.). Shared resources utilized for the project include the small animal imaging facility (SAIF) and research histology core laboratory (RHCL). J.K. and M.S. provided the GLS inhibitor along with insights into formulation for its use in the in vivo experiment and contributed thoughtful discussion of the study design as well as manuscript review. Editorial support was provided by Don Norwood of MD Anderson Editing Services, Research Medical Library.
Author contributions
D.G. and A.K.S. were responsible for conceptualization, data curation, formal analysis, investigation, visualization, methodology, and manuscript writing, review, and editing. M.S.K., M.T., K.I.F., E.B., Y.W., E.S., and L.S.M. helped in establishing and maintaining the mouse colony and performed necropsy and tissue analysis. P.B., P.D., and C.V.K. conceptualized, performed, and analyzed the hyperpolarized magnetic resonance spectroscopy experiments and reviewed the manuscript. D.N., O.A., J.H.J., A.A., A.J., P.K., M.N., and F.W. performed the LC-MS experiments and L.S.E., M.S., and S.B. performed the DESI-MS experiments and performed all analyses of the results and preparation of the images in the figures related to these data. S.L., T.A.Y., and S.N.W. provided guidance on experimental design and review of the manuscript. All authors read and approved the final manuscript.
Declaration of interests
A.K.S. reports consulting for KIYATEC, Merck & Co., GSK, Onxeo, ImmunoGen, Iylon, and AstraZeneca; being a stockholder in Bio-Path Holdings; and receiving research funding from MTrap. T.A.Y. reports AstraZeneca, Almac, Aduro, Artios, Athena, Atrin, Axiom, Bayer, Bristol Myers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, F-Star, GLG, Guidepoint, Ignyta, I-Mab, ImmuneSensor, Jansen, Merck, Pfizer, Repare, Roche, Schrodinger, Seattle Genetics, Varian, Zai Labs, and ZielBio. T.A.Y. is also the Medical Director of the Institute for Applied Cancer Science where the glutaminase inhibitor IACS-012031 was developed. S.N.W. reports AstraZeneca, Bayer, Clovis Oncology, Cotinga Pharmaceuticals, GSK, Mereo, Novartis, OncXerna, Roche/Genentech, Zentalis; and is a paid consultant for Agenus, AstraZeneca, Clovis Oncology, Eisai, EQRX, GSK, ImmunoGen, Lilly, Merck, Mereo, Novartis, Pfizer, Roche/Genentech, Vincerx, and Zentalis. L.S.E. is an inventor in patents owned by Purdue Research Foundation related to desorption electrospray ionization mass spectrometry imaging technology and receives royalties from its commercialization. L.S.E. has received research funding from Merck and Thermo Fisher Scientific.
Published: January 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106020.
Supplemental information
Data and code availability
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
References
- 1.Torre L.A., Trabert B., DeSantis C.E., Miller K.D., Samimi G., Runowicz C.D., Gaudet M.M., Jemal A., Siegel R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKeown S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014;87:20130676. doi: 10.1259/bjr.20130676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Seltzer V., Batson J.L., Drukker B.H., Gillespie B.W., Gossfeld L.M., Grigsby P.W., Harvey H.A., Hendricks C.B., Hummel S., Makuch R.W., et al. Ovarian-cancer - screening, treatment, and follow-up. J. Am. Med. Assoc. 1995;273:491–497. doi: 10.1001/jama.1995.03520300065039. [DOI] [Google Scholar]
- 5.Jain R.K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pujade-Lauraine E., Hilpert F., Weber B., Reuss A., Poveda A., Kristensen G., Sorio R., Vergote I., Witteveen P., Bamias A., et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J. Clin. Oncol. 2014;32:1302–1308. doi: 10.1200/JCO.2013.51.4489. [DOI] [PubMed] [Google Scholar]
- 7.Aghajanian C., Blank S.V., Goff B.A., Judson P.L., Teneriello M.G., Husain A., Sovak M.A., Yi J., Nycum L.R. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012;30:2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perren T.J., Swart A.M., Pfisterer J., Ledermann J.A., Pujade-Lauraine E., Kristensen G., Carey M.S., Beale P., Cervantes A., Kurzeder C., et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011;365:2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 9.F.D.A. U.S. Food & Drug Administration; 2018. Avastin (Bevacizumab) Information in Postmarket Drug Saftey Information for Patients and Providers.www.fda.gov [Google Scholar]
- 10.Sennino B., McDonald D.M. Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer. 2012;12:699–709. doi: 10.1038/nrc3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L., Moss T., Mangala L.S., Marini J., Zhao H., Wahlig S., Armaiz-Pena G., Jiang D., Achreja A., Win J., et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014;10:728. doi: 10.1002/msb.20134892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glutaminase inhibitor CB-839 and azacitidine in treating patients with advanced myelodysplastic syndrome. 2021. https://ClinicalTrials.gov/show/NCT03047993
- 13.Testing whether cancers with specific mutations respond better to glutaminase inhibitor, telaglenastat hydrochloride, anti-cancer treatment, BeGIN study. 2021. https://ClinicalTrials.gov/show/NCT03872427
- 14.Study of the glutaminase inhibitor CB-839 in leukemia. 2017. https://ClinicalTrials.gov/show/NCT02071927
- 15.Study of the glutaminase inhibitor CB-839 in hematological tumors. 2018. https://ClinicalTrials.gov/show/NCT02071888
- 16.Study of the glutaminase inhibitor CB-839 in solid tumors. 2020. https://ClinicalTrials.gov/show/NCT02071862
- 17.KEAPSAKE: a study of telaglenastat (CB-839) with standard-of-care chemoimmunotherapy in 1L KEAP1/NRF2-mutated, nonsquamous NSCLC. 2021. https://ClinicalTrials.gov/show/NCT04265534
- 18.CB-839 HCl in combination with carfilzomib and dexamethasone in treating patients with recurrent or refractory multiple myeloma. 2021. https://ClinicalTrials.gov/show/NCT03798678
- 19.Study of CB-839 (telaglenastat) in combination with talazoparib in patients with solid tumors. 2021. https://ClinicalTrials.gov/show/NCT03875313
- 20.A study of telaglenastat (CB-839) in combination with palbociclib in patients with solid tumors. 2021. https://ClinicalTrials.gov/show/NCT03965845
- 21.Study CB-839 in combination with nivolumab in patients with melanoma, clear cell renal cell carcinoma (ccRCC) and non-small cell lung cancer (NSCLC) 2021. https://ClinicalTrials.gov/show/NCT02771626
- 22.A comparative, pharmacokinetic study of CB-839 capsule and tablet formulations in healthy adults. 2017. https://ClinicalTrials.gov/show/NCT02944435
- 23.Novel PET/CT imaging biomarkers of CB-839 in combination with panitumumab and irinotecan in patients with metastatic and refractory RAS wildtype colorectal cancer. 2021. https://ClinicalTrials.gov/show/NCT03263429
- 24.CB-839 with radiation therapy and temozolomide in treating patients with IDH-mutated diffuse astrocytoma or anaplastic astrocytoma. 2022. https://ClinicalTrials.gov/show/NCT03528642
- 25.Study of CB-839 in combination w/paclitaxel in patients of African ancestry and non-African ancestry with advanced triple negative breast cancer (TNBC) 2021. https://ClinicalTrials.gov/show/NCT03057600
- 26.Telaglenastat + talazoparib in prostate cancer. 2021. https://ClinicalTrials.gov/show/NCT04824937
- 27.Testing of the anti cancer drugs CB-839 HCl (telaglenastat) and MLN0128 (sapanisertib) in advanced stage non-small cell lung cancer. 2021. https://ClinicalTrials.gov/show/NCT04250545
- 28.IACS-6274 with or without pembrolizumab for the treatment of advanced solid tumors. 2021. https://ClinicalTrials.gov/show/NCT05039801
- 29.Telaglenastat hydrochloride and osimertinib in treating patients with EGFR-mutated stage IV non-small cell lung cancer. 2021. https://ClinicalTrials.gov/show/NCT03831932
- 30.CANTATA: CB-839 with cabozantinib vs. cabozantinib with placebo in patients with metastatic renal cell carcinoma. 2021. https://ClinicalTrials.gov/show/NCT03428217
- 31.CB-839 in combination with niraparib in platinum resistant BRCA -wild-type ovarian cancer patients. 2021. https://ClinicalTrials.gov/show/NCT03944902
- 32.Study to evaluate the ECG effects of telaglenastat in healthy adult subjects. 2021. https://ClinicalTrials.gov/show/NCT04607512
- 33.Koh M.Y., Powis G. Passing the baton: the HIF switch. Trends Biochem. Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiseman J.M., Ifa D.R., Song Q., Cooks R.G. Tissue imaging at atmospheric pressure using desorption electrospray ionization (DESI) mass spectrometry. Angew. Chem. Int. Ed. Engl. 2006;118:7346–7350. doi: 10.1002/ange.200602449. [DOI] [PubMed] [Google Scholar]
- 35.Eberlin L.S., Ferreira C.R., Dill A.L., Ifa D.R., Cheng L., Cooks R.G. Nondestructive, histologically compatible tissue imaging by desorption electrospray ionization mass spectrometry. Chembiochem. 2011;12:2129–2132. doi: 10.1002/cbic.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang L., Mou J., Shao B., Wei Y., Liang H., Takano N., Semenza G.L., Xie G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019;10:40. doi: 10.1038/s41419-018-1291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buffa F.M., Harris A.L., West C.M., Miller C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer. 2010;102:428–435. doi: 10.1038/sj.bjc.6605450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu C., Chen L., Jin S., Li H. Glutaminase inhibitors: a patent review. Expert Opin. Ther. Pat. 2018;28:823–835. doi: 10.1080/13543776.2018.1530759. [DOI] [PubMed] [Google Scholar]
- 39.Gross M.I., Demo S.D., Dennison J.B., Chen L., Chernov-Rogan T., Goyal B., Janes J.R., Laidig G.J., Lewis E.R., Li J., et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014;13:890–901. doi: 10.1158/1535-7163.mct-13-0870. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z.J., Ohliger M.A., Larson P.E.Z., Gordon J.W., Bok R.A., Slater J., Villanueva-Meyer J.E., Hess C.P., Kurhanewicz J., Vigneron D.B. Hyperpolarized C-13 MRI: state of the art and future directions. Radiology. 2019;291:273–284. doi: 10.1148/radiol.2019182391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cluntun A.A., Lukey M.J., Cerione R.A., Locasale J.W. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3:169–180. doi: 10.1016/j.trecan.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L., Achreja A., Yeung T.L., Mangala L.S., Jiang D., Han C., Baddour J., Marini J.C., Ni J., Nakahara R., et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 2016;24:685–700. doi: 10.1016/j.cmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wise D.R., Thompson C.B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langley R.R., Fan D., Tsan R.Z., Rebhun R., He J., Kim S.J., Fidler I.J. Activation of the platelet-derived growth factor-receptor enhances survival of murine bone endothelial cells. Cancer Res. 2004;64:3727–3730. doi: 10.1158/0008-5472.CAN-03-3863. [DOI] [PubMed] [Google Scholar]
- 45.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Landen C.N., Chavez-Reyes A., Bucana C., Schmandt R., Deavers M.T., Lopez-Berestein G., Sood A.K. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Q., Zhu C., Shen Z., Zhang T., Li M., Zhu J., Qin J., Xie Y., Zhang W., Chen R., et al. Incidence and potential predictors of thromboembolic events in epithelial ovarian carcinoma patients during perioperative period. Eur. J. Surg. Oncol. 2020;46:855–861. doi: 10.1016/j.ejso.2020.01.026. [DOI] [PubMed] [Google Scholar]
- 48.Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.