


Abstract
The DNA mismatch repair protein MutSα recognizes wrongly incorporated DNA bases and initiates their correction during DNA replication. Dysfunctions in mismatch repair lead to a predisposition to cancer. Here, we study the homozygous mutation V63E in MSH2 that was found in the germline of a patient with suspected constitutional mismatch repair deficiency syndrome who developed colorectal cancer before the age of 30. Characterization of the mutant in mouse models, as well as slippage and repair assays, shows a mildly pathogenic phenotype. Using cryogenic electron microscopy and surface plasmon resonance, we explored the mechanistic effect of this mutation on MutSα function. We discovered that V63E disrupts a previously unappreciated interface between the mismatch binding domains (MBDs) of MSH2 and MSH6 and leads to reduced DNA binding. Our research identifies this interface as a ‘safety lock’ that ensures high-affinity DNA binding to increase replication fidelity. Our mechanistic model explains the hypomorphic phenotype of the V63E patient mutation and other variants in the MBD interface.
INTRODUCTION
Error-free DNA replication is essential to maintain genome integrity in dividing cells. To ensure replication quality, the DNA mismatch repair (MMR) proteins scan the DNA for wrongly inserted bases or short insertion/deletion loops after replication and initiate their correction (1,2). Heterozygous germline mutations in the human MMR genes lead to a predisposition to colorectal and endometrial cancer known as Lynch syndrome (LS) (3). Homozygous germline mutations occur more rarely and lead to constitutional mismatch repair deficiency syndrome (CMMRD) (4). Moreover, in various tumor types, sporadic loss of MMR protein expression has been demonstrated (5,6). In most cases, MMR deficiency can be identified by the instability of microsatellite sequences in the genome.
In hereditary cancer syndromes, such as LS and CMMRD, good understanding of the consequences of particular germline mutations is critical. Variants of uncertain significance are therefore studied in vitro and in mouse models to understand the effect of mutations on MMR function and to be able to diagnose LS/CMMRD (7–9). Since MMR-deficient tumors exhibit increased sensitivity to immunotherapy (10,11) but reduced response to certain types of chemotherapy (12–15), this information can be important for treatment decisions. Conversely, mutations found in patients can provide valuable insights into the MMR mechanism.
The general process of MMR is conserved throughout all kingdoms of life, and involves three main steps: mismatch recognition, strand removal and resynthesis. In the latter two steps, an array of proteins is involved and its composition and specific activity differ between organisms. The first step, mismatch recognition, however, is highly conserved and carried out by the ATPase protein MutS (1,2) in bacteria, or its heterodimeric MutS homologs (MSHs) in eukaryotes (16). Simple mismatches and short loops of unpaired bases are recognized by MutSα, a heterodimer of the proteins MSH2 and MSH6, while longer unpaired loops are recognized by MutSβ (MSH2/MSH3) (17). In canonical MMR, the MutS homolog thereafter recruits MutLα, a heterodimer of MLH1 and PMS2. Together with the processivity clamp PCNA, MutLα discriminates between the old and the new strand, where it can nick the newly synthesized strand (18,19). The erroneous strand is then displaced in an exonuclease 1-dependent or -independent way (20). The gap is subsequently filled by DNA polymerase δ or ϵ (21) and sealed by DNA ligase (1).
From extensive biophysical and structural studies on bacterial MutS, its mismatch recognition and repair signal propagation are well understood. MutS acts as a homodimer that recognizes the mismatch by stacking a phenylalanine parallel to the mispairing bases leading to a 60° kink of the DNA (22,23). The complex is stable until ATP is bound. ATP engagement leads to a large conformational change of MutS into the so-called sliding clamp state, where the two monomers of the protein together provide an interface for loading MutL onto DNA, which allows activation of downstream repair factors (24). In the eukaryotic heterodimers, each monomer has a defined function. Mismatch recognition is facilitated through the mismatch binding domain of MSH6 (MBD6), while MSH2 is important for the dimer stability and the recruitment of downstream factors such as MutLα. The mismatch binding domain of MSH2 (MBD2, domain I) is currently not thought to influence DNA binding.
Here, we studied a homozygous mutation in MSH2 leading to substitution of valine at position 63 for glutamate (V63E), which was found in a suspected CMMRD patient who developed colorectal cancer (CRC) in early adulthood. In vitro assays and mouse models of the V63E variant established a mildly pathogenic phenotype. The mutation is located in MBD2 and disrupts the interaction with MBD6. The inability to form a stable interface between the MBDs leads to a loss of high-affinity mismatch binding. Using cryogenic electron microscopy (cryoEM), we could see that the MBD2 needs to move into place for MutSα to form a stable high-affinity complex on DNA, suggesting that the interface between the MBDs is relevant for effective MMR in humans.
MATERIALS AND METHODS
Mice and treatments
Mice were housed in individually ventilated cages (IVC; Innocage®, Innovive) at room temperature with relative humidity of 55%. Mice were fed RM3 (HDE) PL MIN (Special Diet Services, England) pellet nutrition and water (Aquavive®) ad libitum. Msh2V63E mice were created by CRISPR/Cas9 injection into zygotes along with a 130 bp repair template containing the GTG>GAA substitution. Msh2−/− and WT mice were obtained from intercrossing in-house bred Msh2+/− mice (25). Lgr5-CreERT2;Msh2flox/− (Msh2-Lynch) and Lgr5-CreERT2;Msh2flox/V63E (V63E-Lynch) were induced with tamoxifen and treated for 5 or 10 days with 100 mg/kg temozolomide (TMZ) as previously established (26). Experimental mice were identified by genotyping using allele-specific primers on DNA extracted from toe biopsies. Primer sequences are listed in Supplementary Table S2. Both males and females were used in experiments. All mice were on the FVB background. Mice were sacrificed using CO2. Experiments were performed in accordance with Dutch and European guidelines and were approved by the local Animal Ethical Committee at the Netherlands Cancer Institute and the National Commission for Animal Experimentation (Centrale Commissie Dierproeven) of The Netherlands.
Histopathological analysis
Mice were sacrificed using CO2 and tissues were fixed in 4% formaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin. Immunohistochemical staining for MSH2 was performed as previously described (26).
DNA isolation and qPCR
Proximal jejunum was isolated, snap frozen and stored at −80°C for later processing. DNA was isolated using the Isolate II Genomic DNA Kit (Bioline). DNA concentration was measured on the NanoDrop ND-100 spectrophotometer and samples were diluted to 25 ng/µl. qPCR was performed using LightCycler® 480 SYBR Green I Master Mix (Roche) with allele-specific primers for the inactivated Msh2 allele. A schematic overview of the PCR strategy can be found in Supplementary Figure S1B. Genomic primers targeting part of Msh2 exon 3 were used to normalize for input DNA. Primer sequences are listed in Supplementary Table S2.
Organoid culture
Small intestinal crypts were isolated and cultured into organoids as described with 10% R-spondin conditioned medium and 10% Noggin conditioned medium (27). For the 6-thioguanine (6-TG) cytotoxicity assay, organoids were passaged by mechanical dissociation, plated and exposed to 500 nM 6-TG in the culture medium for 3 days. Organoids were imaged on an Axio Observer Z1 (Zeiss) using the tile scan option to visualize the complete well. Z-stacks ranging the entire depth of the gel were made in order to capture all organoids. For image analysis, Z stacks were merged using the Stack Focuser plugin in Fiji. Organoid area was measured by delineating organoid edges manually. The organoid area for each genotype was calculated relative to mock-treated controls.
In vitro DNA repair
The functional consequences of the MSH2 missense substitutions V63E, V3D and del2–7 were assessed using the cell-free in vitro mismatch repair activity (CIMRA) assay, using previously described methods (9), with a change of nuclear extract (28). All variants were tested at least in quadruplicates.
Expression constructs and mutagenesis
Synthetic genes of MSH2 and MSH6, codon optimized for Escherichia coli and GC content optimized, were purchased from General Biosystems Inc. (USA) and subcloned into the pFastBac Dual vector (Thermo Fisher Scientific), to allow expression of MutSα from one single vector. MSH2 was cloned under the P10 promotor using Xho1 and Kpn1 restriction sites; MSH6 was inserted under the polyhedrin promotor using BamH1 and EcoR1 sites. A dual-affinity tag of 10His-TwinStrep with an HRV 3C protease site was fused at the N-terminus of MSH6.
MSH2 and MSH6 mutations were introduced using site-directed mutagenesis. In short, PCR reactions were run for 10 cycles with samples for forward and reverse primers separated, then combined and run for another 15 cycles. PCR samples were treated with Dpn1 (NEB) to digest template plasmid DNA. All genes were fully sequenced.
Protein expression
WT MutSα and all mutants were expressed in Sf9 cells using identical conditions. Production of recombinant bacmid and baculoviruses was carried out based on the manual of Invitrogen’s Bac-to-Bac system using EMBacY bacmids (Geneva Biotech). Sf9 cells were cultured in Insect-Xpress media (Lonza) supplemented with penicillin and streptomycin. MutSα bacmid DNA was used to transfect Sf9 cells in a six-well plate. After 4 days, the supernatant (P0) was transferred into a 25 ml suspension culture with 1.0 Mio cells/ml. After 3 days, the P1 virus was harvested and used for large-scale protein expression. P1 virus showed reduced expression levels if stored for >2 weeks. Per batch of MutSα, eight 500 ml suspension cultures at densities of 1.2–1.5 Mio cells/ml were transfected with 150 μl P1 virus each and grown in a shaker at 28°C for 70 h. Cells were harvested by centrifugation at 1200 × g for 10 min. The resulting pellet was carefully resuspended in 100 ml of 25 mM HEPES, pH 7.5, and 150 mM KCl, flash frozen in liquid nitrogen and stored at −20°C.
Protein purification
All steps were performed at 4°C. Frozen cell pellet suspension from 4 l cell culture was thawed in a water bath and supplemented with two protease inhibitor tablets (Thermo Fisher Scientific), 5 mM PMSF and 1 mM TCEP. Cells were lysed by pulse sonication at 30 Hz for 2.5 min. Glycerol and KCl were slowly added to final concentrations of 20% and 500 mM, respectively, and the suspension was stirred for 10 min. Debris was sedimented by centrifugation at 53 340 × g for 30 min. The supernatant was incubated with 3 ml Ni-chelated Sepharose beads (Cytiva) equilibrated with Nickel Wash Buffer (25 mM HEPES, pH 7.5, 150 mM KCl, 5 mM MgCl2, 5 mM imidazole, 10% glycerol, 1 mM TCEP) at 4°C for 30 min. Beads were sedimented by centrifugation at 200 × g for 2 min, washed repeatedly with Nickel Wash Buffer, transferred into a gravity flow column and eluted with 25 mM HEPES, pH 7.5, 300 mM KCl, 5 mM MgCl2, 300 mM imidazole, 15% glycerol, 1 mM TCEP and 1 mM ADP. The eluate was immediately loaded on a self-packed Strep-Tactin XT (IBA) column (GE Healthcare) equilibrated with Strep Wash Buffer (25 mM HEPES, pH 7.5, 150 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT). The beads were thoroughly washed with Strep Wash Buffer and the same buffer containing 1 M KCl. Protein was eluted with 25 mM HEPES, pH 7.5, 100 mM KCl, 5 mM MgCl2, 20% glycerol, 1 mM DTT, 1 mM ADP and 50 mM biotin. The eluting protein was immediately loaded onto a 5 ml Heparin column equilibrated with 25 mM HEPES, pH 7.5, 120 mM KCl, 5 mM MgCl2, 20% glycerol and 1 mM TCEP, washed with the same buffer and then eluted in a step with 400 mM KCl. Peak fractions were pooled and supplemented with 1 mM ADP. A typical yield from 4 l culture for MutSα WT was 6 mg.
For surface plasmon resonance (SPR) experiments, we cleaved the His tag before concentration by incubation overnight with His-tagged HRV 3C protease. Remaining uncleaved protein, the cleaved tag and protease were removed using Talon beads (Takara Bio) and Strep-Tactin XT beads both equilibrated with 25 mM HEPES, pH 7.5, 200 mM KCl, 5 mM MgCl2, 20% glycerol and 1 mM TCEP.
Protein was concentrated in a Centriprep 10 kDa cutoff concentrator (Merck) at 1850 × g to reduce the volume to 500 μl, followed by an AmiconUltra 0.5 ml centrifugal filter (Merck) to concentrate the protein to ∼40 μM. Samples were aliquoted and flash frozen in liquid nitrogen.
Prior to experiments, protein aliquots were subjected to gel filtration to remove glycerol and ADP (buffer: 25 mM HEPES, pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM DTT) using a Superose 6 Increase 3.2/300 (GE Healthcare).
DNA oligonucleotides
For nano differential scanning fluorimetry (nanoDSF) and cryoEM, 32 or 50 bp oligonucleotides with a GT mismatch in the middle (32GT16 and 50GT25, respectively; GGATCATCGAGGATCGAGCTCGGTGCAATTCA or CTTAGCTTAGGATCATCGAGGATCGAGCTCGGTGCAATTCAGCGGTACCC) were ordered from IDT. For SPR experiments, double-stranded oligonucleotides of 21 bp with a GT mismatch at position 10 containing a biotin-conjugated (dT)20 linker at the 5′ end and digoxigenin modification at the 3′ end (Btn-TTTTTTTTTTTTTTTTTTTTAGTCGCCAGGGACCAGTGTCA-Dig and TGACACTGGTCTCTGGCGACT) were ordered from Eurogentec.
In both cases, forward and reverse strands were combined, heated to 95°C for 10 min and cooled down slowly to allow annealing. DNA was subsequently purified using size exclusion chromatography (Superdex 200 10/300, GE Healthcare) or anion exchange chromatography (MiniQ) for nanoDSF and cryoEM or SPR, respectively.
Nano differential scanning fluorimetry
For protein stability measurements, MutSα peak fractions from size exclusion chromatography were pooled and diluted to 0.3 mg/ml. If appropriate, samples were supplemented with 1 mM ADP and/or 2-fold excess of 32GT16 DNA oligo directly before the measurement. Samples were measured in duplicates using a Prometheus NT.48 (Nano Temper) heating from 20 to 90°C with 1°C/min. Readout is the shift in tryptophan fluorescence. The instrument measures fluorescence at 330 and 350 nm and the change in the ratio of 350/330 nm is used to detect protein unfolding where the maximum of its derivative is defined as the melting temperature Tm(unfolding).
Surface plasmon resonance
SPR binding experiments were performed on a Biacore T200 instrument (Cytiva) at 25°C. Biotinylated DNA oligos were immobilized on a streptavidin-coated SA chip (Cytiva) keeping immobilization levels between 5 and 20 RUs. Running Buffer was 25 mM HEPES, pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.05% Tween and 1 mg/ml BSA. Samples were kept at 15°C until injection.
For kinetic measurements, E. coli MutS D835R [0.5–256 nM (dimer concentration)] or MutSα (1–256 or 1–128 nM) was injected for 60 s and dissociation was monitored for 120 s at a flow rate of 50 μl/min. The chip surface was regenerated with a solution of 0.5% SDS at the end of each injection cycle. If appropriate, Running Buffer was supplemented with 1 mM ADP or ATP.
For the control experiments, injection time and flow rate were varied according to the experimental description. For the ATP release and MutLα recruitment experiments, dual injection mode was used with two consecutive injections of 120 s. For the ATP release assay, injection of E. coli MutS D835R (200 nM) or MutSα (50 nM) is followed by an ATP injection of 0–204.8 μM in 4- or 2-fold dilutions, respectively. In the MutLα recruitment experiment, MutSα WT or MSH2 V63E was injected at 100 nM concentration alone first and then together with MutLα in concentration range of 0–320 nM in 2-fold dilution. The latter experiments were qualitative and were not used for MutLα stoichiometry determination. In the presence of ATP, multiple MutSα molecules were loaded onto the 100 bp oligo, which did not leave sufficient space to fully accommodate MutLα on each of these.
Kinetic analysis
All the data from the SPR measurement were analyzed using Biacore T200 Evaluation software v3.2. Subsequently, datasets for kinetic analysis were exported to CLAMP XP Biosensor Data Analysis v3.50 (29), where the data for MutSα WT, MSH2 V63E and MSH2 V3D were fitted simultaneously with a two-state reaction model. During the fitting procedure, the dissociation rate constants (k−1) were kept identical for all three datasets. Then, the k−1 value obtained from this fit was used to fit the rest of the MutSα mutants using a two-state reaction model. The E. coli MutS kinetic data were fitted using a 1:1 binding model.
During the experiments, we checked that calculated maximum binding was in line with immobilized oligonucleotide. This indicated that active fractions were comparable between wild-type (WT) and mutant MutSα. Moreover, the difference in the binding between WT and mutant came primarily from the difference in dissociation rates, which is concentration independent, and therefore also independent from potential differences in protein active fractions between WT and mutant.
The half-life values on DNA for MutSα WT and MSH2 V63E experiments in the presence of ADP were calculated by fitting the dissociation phase of the binding using the one-phase exponential decay model in GraphPad Prism v9 (GraphPad Prism Software, LLC): 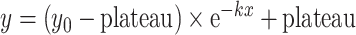 .
.
The final graphs were created using GraphPad Prism v9.
CryoEM sample preparation
MutSα peak fractions from size exclusion chromatography were diluted to 2 μM and supplemented with a 2-fold excess of 50GT25 DNA oligonucleotide. For the double ADP-bound MutSα dataset, the sample was additionally supplied with 1 mM ADP. Cu 300 Holey Carbon R1.2/1.3 grids (Quantifoil) were freshly glow discharged using a PELCO easiGlow (Ted Pella) for the ADP samples or a GloQube Plus (Quorum) for the ADP-free samples. All samples were vitrified using a Vitrobot Mark IV (Thermo Fisher Scientific) at 4°C and 100% humidity. Three microliter samples were applied, blotted with 595 blotting paper (Ted Pella) (blot force 10, 2 or 3 s) and flash frozen into liquid ethane.
CryoEM data collection
Data for MutSα WT and V63E without ADP were collected on a 300 kV Titan Krios instrument (Thermo Fisher Scientific) with a post-column energy filter (slit width 20 eV) and a K3 direct electron detector (Gatan). EPU-2.10.0 was used for automated data collection in super-resolution mode using AFIS centering. Movies with 50 frames were acquired at a pixel size of 0.418 Å/pixel with a total dose of 50 e−/Å2 (20.035 e−/pixel/s). Defocus values range between 0.9 and 2.4 μm. Data quality was monitored in real time using Warp (30).
The MutSα dataset with ADP was collected on a 200 kV Talos Arctica microscope (Thermo Fisher Scientific) with a post-column energy filter (slit width 20 eV) and a K2 summit direct electron detector (Gatan). EPU (Thermo Fisher Scientific) was used for automated data collection in counted mode. Movies with 26 frames were acquired at a pixel size of 1.041 Å/pixel with a total dose of 60 e/A2 (8.09 e−/pixel/s). Defocus values range between 1.6 and 3.2 μm.
CryoEM processing MutSα WT (no ADP) (Supplementary Figure S5A)
Movie stacks were imported into Relion 4.0 (31), motion corrected with 5 × 5 patches and binned (2×). Data were CTF estimated using exhaustive search in CTFFind 4.0 (32). Images with an estimated resolution >6 Å and an astigmatism >700 Å were discarded. Images with pronounced ice rings were manually removed. Particle picking was performed with SPHIRE-crYOLO (33) on JANNI-denoised micrographs using a model trained on a subset of micrographs (picked 5.4 Mio particles). Particles were imported into Relion, extracted 4× binned (original box size 256 pixels) and subjected to four rounds of 2D classification with an angular sampling of 5° in round 1 and 1° thereafter. The cleaned particles were used to generate an initial model in cryoSPARC using ab initio reconstruction (34,35). The resulting map was imported back into Relion and used for consensus 3D refinement. Particles were re-extracted 2× binned (original box size 360 pixels) with updated coordinates. One round of 3D classification removed particles with less pronounced MSH2 clamp density (16% of particles). Afterward, particles were re-extracted unbinned and subjected to three rounds of CTF refinement and Bayesian polishing interspersed with 3D refinements. One round of 2D classification removed remaining junk particles. The final consensus map was generated by two more rounds of 3D classification (first one with mask, second one alignment free) with a final stack of 289 289 particles. A final round of refinement resulted in a map with 2.8 Å resolution. Map sharpening for better interpretation was done with LocSpiral (36). The final pixel spacing was calibrated using the X-ray structure (PDB ID: 2o8b) (see below). This sharpened map and the half maps from the last refinement were deposited to the Electron Microscopy Data Bank (EMDB) (EMDB ID: EMD-15417).
CryoEM processing MutSα V63E (Supplementary Figure S5B)
Motion correction, CTF estimation, particle picking and initial particle extraction for the V63E set were performed analogously to WT (initial stack size 4.5 Mio particles). Because many broken particles were present in the dataset, eight rounds of 2D classification were necessary. Since there was still substantial remaining heterogeneity that was not removed efficiently in 2D, an attempt to generate an initial model did not give a reasonable map. Therefore, the initial alignment of particles in 3D was performed using the WT initial model as reference map. 3D classification attempts with (two rounds) and without alignment (three rounds) resulted in different sets of good particles (only 30% overlap). The resulting particle stacks were recombined and duplicates removed. At this stage, an initial model could be generated in Relion 4.0 to get a bias-free reference map, which was used for the remainder of the processing. Particles were re-extracted unbinned and subjected to three rounds of CTF refinement and Bayesian polishing interspersed with 3D refinements. One round of 2D classification removed some more junk particles. Following one more 3D refinement to align all particles in 3D, cryoDRGN (37) was used to reveal remaining conformational heterogeneity. This analysis showed a subset with an open and a closed clamp. In order to deconvolute these two subsets, a last round of 3D classification was performed in Relion using three input models: two cryoDRGN maps that are representative of each main state plus one junk model. This classification sorted good particles into subsets with an open and a closed clamp, respectively. CryoDRGN was used for both subsets individually to assess further conformational rearrangements. Both subsets showed significant conformational variability of MBD2 (Supplementary Figure S4G and H). The maps of the complete refined particle stack and the subsets of the open and closed clamps were deposited to the EMDB (EMDB ID: EMD-15518).
CryoEM processing MutSα WT with ADP (Supplementary Figure S5C)
This dataset was processed with Relion 3.0. Movie stacks were imported into Relion, motion corrected with 5 × 5 patches and binned (2×) using MotionCor2 (38). Data were CTF estimated using CTFFind (32). Images with pronounced ice rings were manually removed. Particle picking was done using Relion autopick. Therefore, a 3D template was generated from manually picked particles using a subset of 20 micrographs. 1.7 Mio particles were extracted 4× binned (original box size 256 pixels) and subjected to five rounds of 2D classification with an angular sampling of 5° in round 1 and 1° thereafter. Particles were then re-extracted unbinned (box size of 256 pixels was kept throughout processing) with updated coordinates and an initial model was calculated in cryoSPARC using ab initio reconstruction (34). One round of 3D classification removed particles with missing density for MSH2. Subsequently, one round of alignment-free 3D classification using a mask revealed two major classes, with differently kinked DNA but indistinguishable protein parts. Particles from both classes were pooled and subjected to three rounds of CTF refinement and Bayesian polishing interspersed with 3D refinements. A final 2D classification removed remaining junk particles. The final resolution was 4.1 Å. This final stack of 111 096 particles was used to visualize heterogeneity by cryoDRGN and cryoSPARC 3D variability analysis. The sharpened map and the half maps from the last refinement were deposited to the EMDB (EMDB ID: EMD-15519).
CryoEM model building and refinement
The atomic model of crystal structure 2o8b was used as a starting model and rigid body fitted into the LocSpiral sharpened map using UCSF ChimeraX (39). Coot (40) was then used to adjust the model by jiggle fit, morph fit and manual curation. We improved the existing model by also considering the AlphaFold structure predictions (41) of MSH2 and MSH6 and the crystal structure of MutSβ (PDB ID: 3thx), revealing register shifts in both proteins: (i) MSH6 residues 936–961 of the clamp domain were shifted by two residues, resulting in a better density with hydrophobic residues buried in the coiled coil, and (ii) MSH2 residues 231–243 were shifted by one residue in a connector domain loop. Previously unseen density at the C-termini allowed us to extend the MutSα model. Predicted models by ColabFold (42) of residues MSH2 856–934 and MSH6 1335–1360 were docked into the map and manually adjusted (Supplementary Figure S4A). The C-termini fold into alpha helices similar to E. coli MutS and MutSβ. We see no density for most of the long N-terminus of MSH6, confirming its flexible nature. However, we observed some extra helical density on top of the MSH2 lever domains. Although the density was too weak for inclusion in the final model, ColabFold predicts MSH6 residues 332–343 to form a helix and predicts binding at that position where the extra density localized (Supplementary Figure S4B). This indicated that part of the MSH6 N-terminus wrapped around MSH2 before becoming fully unstructured. We could build 25 out of 50 bp of the DNA in Coot, applying extra B-DNA and planar restraints. Several rounds of phenix.real_space_refine (43) and Coot adjustments using the MolProbity validation were used to improve the model. A pixel size calibration resulted in a reduced clash score. This calibration was done in Chimera by calculating the cross-correlation between a map calculated from the X-ray structure of MutSα (PDB ID: 2o8b) using molmap and the experimental cryoEM map at different pixel spacings. The final calibrated pixel spacing was 0.839 Å. Finally, the model was submitted to the PDB-REDO server (44), which further improved model geometry and clash score. A final manual sanity check was done in Coot before deposition to the PDB [PDB ID: 8AG6]. Model statistics (Table 2) were calculated with phenix.molprobity with the exception of the base pair geometry (rmsZbpG), which was extracted from PDB-REDO. Figures were made using UCSF ChimeraX.
Table 2.
Data collection and refinement statistics
| MutSα WT | MutSα V63E | MutSα WT ADP | |
|---|---|---|---|
| Data collection and processing | |||
| Microscope | Titan Krios | Titan Krios | Talos Arctica |
| Camera | K3 | K3 | K2 summit |
| Magnification | 105 000 | 105 000 | 130 000 |
| Voltage (kV) | 300 | 300 | 200 |
| Total dose (e−/Å2) | 50 | 50 | 60 |
| Defocus range (μm) | 0.9–2.4 | 0.9–2.4 | 1.6–3.2 |
| Number of fractions | 50 | 50 | 26 |
| Movies | 4821 | 4456 | 2608 |
| Pixel spacing (Å) | 0.836 | 0.836 | 1.041 |
| Symmetry imposed | C1 | C1 | C1 |
| Number of Mio particles, initial/final | 5.4/387 692 | 4.4/28 948 | 1.7/111 096 |
| Map resolution (Å) | 2.8 | 3.8 | 4.1 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map sharpening B factor (Å2) | Local (LocSpiral) | None | None |
| Model validation | |||
| Map pixel size (Å) | 0.839 | ||
| Real-space correlation (mask) | 0.85 | ||
| MolProbity score | 1.58 | ||
| Clash score | 5.11 | ||
| Ramachandran plot (%) | |||
| Favored | 96.51 | ||
| Allowed | 3.44 | ||
| Outliers | 0.05 | ||
| Ramachandran plot Z-score | −0.61 | ||
| Rotamer outliers (%) | 1.15 | ||
| Base pair geometry (rmsZbpG) | 0.93 | ||
| Mean model B-factors | |||
| Protein | 71.88 | ||
| DNA | 109.32 | ||
| ADP | 59.78 | ||
RESULTS
MSH2 V63E is a mildly deleterious variant
Homozygous germline mutations in MMR genes give rise to CMMRD. Patients suffering from CMMRD caused by fully inactivating MMR mutations typically develop cancer during early childhood or even prenatally (45).
The MSH2 mutation V63E (c.T188A; p.V63E) was homozygously identified in the germline of a young adult CRC patient who was below 30 years of age, suggesting that the homozygous presence of the MSH2 V63E mutation may cause CMMRD. Two sisters (no genomic material available) also developed CRC before the age of 30 (Supplementary Figure S1A). In these patients, no other tumor types or phenotypes typical for CMMRD were seen. The relatively older age of tumor onset in the proband and the absence of overt LS in the maternal and paternal families suggested that the V63E variant is only weakly pathogenic and may have residual MMR activity.
We previously identified the MSH2 V63E variant as defective in a cellular functional assay termed oligonucleotide-directed mutation screening. This technique utilizes single-stranded DNA oligonucleotides to create site-specific point mutations in cells that are hemizygous for one of the MMR genes; the appearance of MNNG-resistant colonies indicates that the mutation abrogates MMR (8). Indeed, mouse embryonic stem cells (mESCs) containing the MSH2 V63E variant showed increased resistance to methylating agents, albeit not as high as fully MSH2-deficient mESCs. The rate of microsatellite instability (MSI) was 100-fold higher than in WT cells, but 10-fold lower than in fully MSH2-deficent cells (8).
To study MMR capacity of the V63E variant in a more physiological setting, we utilized CRISPR/Cas9 to introduce the GTG>GAA codon change into mouse zygotes. Successfully edited offspring was interbred to obtain Msh2V63E/V63E mice. Immunohistochemical staining for MSH2 did not reveal noticeable differences in protein expression and nuclear localization in intestinal tissues derived from WT and Msh2V63E/V63E mice (Figure 1A). We next compared the toxicity of 6-TG in intestinal organoids derived from Msh2V63E/V63E, Msh2V63E/+, WT and Msh2−/− mice as MMR-deficient cells lose sensitivity to methylating agents (25). As expected, WT organoids were killed by 6-TG exposure, while Msh2−/− organoids remained unaffected (Figure 1B and C). Consistent with mESCs, Msh2V63E/V63E organoids showed intermediate resistance to 6-TG, while Msh2V63E/+ organoids behaved like WT, indicating that the V63E variant is not acting in a dominant negative fashion (Figure 1B and C).
Figure 1.

The MSH2 V63E variant shows a weak pathogenic phenotype in vivo. (A) Immunohistochemical staining for MSH2 on small intestines from WT or Msh2V63E/V63E mice (n = 3). (B) Representative images of WT, Msh2−/−, Msh2V63E/V63E and Msh2V63E/+ organoids treated with control or 500 nM 6-TG. (C) Quantification of panel (B). Bars represent the average and standard deviation (SD) of four independent experiments. Asterisks indicate a significance level of P < 0.01 and P < 0.0001, respectively (one-way ANOVA with Tukey’s correction for multiple comparisons). (D) Experimental setup. Loss of WT MSH2 activity in Msh2-Lynch mice and V63E-Lynch mice was induced with tamoxifen. Then, mice were treated with control solution (n = 3) or TMZ (n = 12) for 10 days. Mice were sacrificed 2 weeks after the treatment and a qPCR for the Msh2floxOFF allele was performed on small intestinal DNA to measure intestinal crypt expansion. (E) Relative crypt expansion after TMZ treatment in Msh2-Lynch and V63E-Lynch mice. Bars represent average and SD. Asterisks indicate a significance level of P < 0.05 (one-way ANOVA with Tukey’s correction for multiple comparisons). (F) Tumor-free survival of WT, Msh2−/− and Msh2V63E/V63E mice. (G) Tumor-free survival of WT, Msh2−/− and Msh2V63E/V63E mice after 5-day TMZ treatment.
Finally, we studied the response of V63E-expressing intestinal crypts to a methylating agent in vivo. During the lifetime of an LS patient carrying a heterozygous deleterious MMR gene mutation, loss of the remaining functional allele in the intestinal epithelium gives rise to fully MMR-deficient intestinal crypts, which may eventually progress into tumors (46). In order to study the fate of these precursor lesions, we previously generated a tamoxifen-inducible mouse model Lgr5-CreERT2;Msh2−/flox (further mentioned as Msh2-Lynch) that allows for the generation of a minor number of MSH2-deficient crypts among a pool of MSH2 heterozygous crypts in the intestinal epithelium, thereby closely mimicking the clinical scenario. Using this model, we have previously shown that these MMR-deficient crypts expanded in numbers upon exposure to the methylating agent TMZ (26). To evaluate whether similar responses occur in small intestinal crypts that are solely dependent on the MSH2 V63E variant, we crossed the V63E allele into the Msh2-Lynch background to obtain Lgr5-CreERT2;Msh2V63E/flox (further mentioned as V63E-Lynch) mice. Cre-mediated recombination of the Msh2flox allele generates a small number of Msh2V63E/− crypts that solely express the MSH2 V63E protein, surrounded by Msh2V63E/flox crypts that still express WT MSH2. The fraction of Msh2V63E/− crypts was monitored in control conditions and upon TMZ treatment using a qPCR strategy on intestinal DNA detecting the Msh2− allele (Figure 1D and Supplementary Figure S2B). Interestingly, the TMZ-induced expansion of the recombined allele was similar between Msh2-Lynch and V63E-Lynch mice. Thus, upon treatment with a methylating agent, the MSH2 V63E variant conferred a similar proliferative advantage to intestinal crypts as full MSH2 deficiency (Figure 1E).
MSH2 V63E only slightly accelerates tumorigenesis
To study the effect of the V63E variant on tumorigenesis, we followed cohorts of Msh2V63E/V63E mice along with WT and Msh2−/− controls. Msh2V63E/V63E mice were only slightly more tumor-prone than WT mice, and showed drastically prolonged tumor-free survival as compared to Msh2−/− mice, which all developed lymphomas within 4 months of age (Figure 1F and Supplementary Table S1) (25). Unfortunately, we could not study intestinal tumorigenesis in Msh2-Lynch and V63E-Lynch mice because the microbiota in our animal facility was unfavorable for intestinal tumor development (47). Instead, we investigated whether Msh2V63E/V63E mice were susceptible to TMZ-induced lymphomagenesis. In line with our in vitro findings, Msh2V63E/V63E mice displayed intermediate tumor-free survival after treatment with TMZ as compared to Msh2−/− and WT mice (Figure 1G).
Together, our in vitro and in vivo characterization of the MSH2 variant p.V63E demonstrated that the variant has residual MMR capacity and leads to a mild cancer predisposition in mice. This is consistent with the relatively late time of onset of cancer in the suspected CMMRD patient carrying the biallelic V63E mutation. To understand this hypomorphic phenotype, we decided to investigate the mechanism of reduced MMR activity by the V63E variant.
MutSα MSH2 V63E shows reduced DNA binding
Previously, we reported that mESCs expressing the V63E variant show reduced MSH2 protein levels (8). It could be that the V63E variant causes aberrant peptide folding and thereby triggers proteasomal degradation. Treatment with proteasomal inhibitor MG-132 did not dramatically enhance MSH2 levels in Msh2V63E/Δ mESCs (Supplementary Figure S2A). We therefore wondered whether reduced stability of the MSH2/MSH6 complex could be the cause of decreased protein levels and less functional MMR.
To test this hypothesis, we recombinantly expressed and purified both MutSα WT and MutSα MSH2 V63E (further mentioned as MSH2 V63E) proteins and assessed their thermostability in vitro using nanoDSF, which monitors protein unfolding in a temperature gradient. In the absence of interaction partners such as DNA or ATP/ADP, MutSα WT and MSH2 V63E proteins displayed a similar melting temperature of 42.5 and 42.0°C, respectively (Figure 2A). Although the difference of 0.5°C is above the machine noise, this alone is unlikely to explain the reduced MMR activity. However, in the presence of a DNA oligomer containing a GT mismatch, MutSα WT showed an increased stability by 4.9°C. In contrast, MSH2 V63E was stabilized by only 2.8°C (Figure 2B). This DNA-dependent difference in stability suggests impaired DNA binding of MSH2 V63E.
Figure 2.

MutSα DNA binding involves a conformational change (A, B) Thermostability of MutSα WT versus V63E in nanoDSF assay: (A) without additives and (B) in the presence of GT mismatched DNA. (C) Comparison of DNA binding and release for MutSα WT, MSH2 V63E and E. coli MutS WT visualized from normalized SPR profiles at 128 nM protein concentration [full titration in panels (E) and (F), and Supplementary Figure S2G]. (D–F) Curve profiles of protein titrations ranging from 1 to 256 nM in 2-fold dilutions (from light to dark color). Binding and dissociation were fitted using CLAMP (29) (black curves) and resulting values shown in scheme below. (D) Escherichia coli MutS D835R curves fitted with 1:1 model. MutSα curves required fitting with a two-state reaction model (see Supplementary Figure S2K–M for controls of the model): (E) MutSα WT and (F) MSH2 V63E.
The presence of ADP stabilized both WT and V63E variant to the same extent (Supplementary Figure S2B) as nucleotides improve dimerization at the ATPase sites (48). This stabilization effect prevails in the presence of DNA, as in the presence of ADP and mismatched DNA both proteins are similarly stabilized (Supplementary Figure S2C).
MutSα DNA binding involves a conformational change
To compare the DNA binding kinetics of WT and mutant MutSα, we used SPR with a DNA oligomer containing a single GT mismatch. Indeed, the MSH2 V63E protein released the mismatched DNA substantially faster than the WT protein (Figure 2C). Similar results were obtained for a DNA oligomer with a single unpaired base (Supplementary Figure S2D), for fully matched homoduplex DNA (Supplementary Figure S2E) and for single-stranded DNA (Supplementary Figure S2F). Interestingly, E. coli MutS released the DNA much faster than the human homolog (Figure 2C and Supplementary Figure S2G). However, because of its unique feature to tetramerize at increasing concentrations, binding kinetics of E. coli MutS WT is difficult to analyze (49). We therefore used E. coli MutS D835R to prevent tetramer formation (49,50) for kinetic analysis and compared it to human MutSα (hMutSα). Surprisingly, we found that they could not be fitted with the same model. Mismatch binding by E. coli MutS D835R could be fitted with a simple 1:1 binding model, resulting in a KD of 31 nM (Figure 2D). In contrast, a 1:1 fit for the human protein did not adequately describe the curves (Supplementary Figure S2H). Upon ATP titration, hMutSα and E. coli MutS D835R formed sliding clamps and rapidly released from mismatched DNA (Supplementary Figure S2I), demonstrating that the comparably slow release of hMutSα from DNA was not caused by unspecific binding due to poor protein quality. We could also fully exclude effects of mass transfer limitation, since (i) the binding profile of the curves did not change upon injection of MutSα at different flow rates (Supplementary Figure S2J) and (ii) we used very low immobilizing levels. Furthermore, the 1:1 binding model takes the possibility of mass transfer into account, but nevertheless the fits for this model were poor. Next, we tested whether heterogeneity in the DNA or double interaction sites on MutSα could explain the data, but neither of these could explain the observed SPR curves (Supplementary Figure S2K).
The only model that gave excellent fits to all recorded human MutSα SPR curves was a two-state reaction model, where initial DNA interaction is followed by a conformational change of MutSα (Supplementary Figure S2L). We confirmed this interpretation by altering MutSα injection periods at different MutSα concentrations and observed that all traces could be fitted simultaneously using this model (Supplementary Figure S2M). We thus conclude that MutSα mismatch binding involves a conformational change that is not observed in E. coli MutS.
According to this new model, we can discriminate between two different DNA-bound states of MutSα. The kinetic parameters indicate that the second DNA-bound state is more stable, mainly because its back-reaction (k−2 in Table 1) is slow. Due to this stability, the affinity of MutSα to the mismatched DNA is high (1 nM) (Figure 2E). Therefore, human MutSα binds substantially tighter to mismatches than E. coli MutS.
Table 1.
Kinetic analysis of DNA mismatch binding by MutS(α) mutants
| k 1 (μM−1 s−1) | k −1 (ms−1) | k 2 (ms−1) | k −2 (ms−1) | K D (nM) | |
|---|---|---|---|---|---|
| MutS D835R | 1.13 ± 0.005 | 35 ± 0.083 | N/A | N/A | 30.85 ± 0.08 |
| MutSα WT | 3.01 ± 0.020 | 110 ± 0.053# | 50.2 ± 0.551 | 1.53 ± 0.026 | 1.08 ± 0.02 |
| MSH2 V63E | 1.34 ± 0.005 | 110 ± 0.053# | 31.3 ± 0.147 | 10.13 ± 0.027 | 19.99 ± 0.16 |
| MSH2 V3D | 3.09 ± 0.011 | 110 ± 0.053# | 35.9 ± 0.167 | 7.53 ± 0.020 | 6.17 ± 0.05 |
| MSH2 dMBD2 | 1.27 ± 0.025 | 110 ± N/A* | 27.6 ± 1.03 | 6.44 ± 0.196 | 16.37 ± 0.86 |
| MSH6 S472Y | 1.76 ± 0.007 | 110 ± N/A* | 46.8 ± 0.377 | 3.12 ± 0.024 | 3.90 ± 0.05 |
| MSH2 V3D V63E | 2.44 ± 0.007 | 110 ± N/A* | 35.5 ± 0.212 | 17.56 ± 0.054 | 14.88 ± 0.13 |
| MSH2 del2–7 | 4.47 ± 0.067 | 110 ± N/A* | 70.8 ± 2.026 | 10.92 ± 0.163 | 3.28 ± 0.12 |
E. coli MutS fitted with 1:1 binding model and human MutSα fitted with a two-state binding model (Figure 2E). SE, standard error of the model; #, shared values; *, fixed value in fitting (see the ‘Materials and Methods’ section).
Our SPR assay revealed that the dissociation of the MSH2 V63E mutant from a GT mismatch was substantially faster than that for WT MutSα (Figure 2E and F). Nevertheless, fitting of the MSH2 V63E curves still required a two-state model. The mutant’s GT mismatch affinity was substantially lower with a KD of 20 nM. Kinetic analysis revealed that this effect was due to a 10-fold faster back-reaction in the conformational change component (k−1 in Table 1). Hence, the MSH2 V63E mutation seemed to reduce the time of MutSα in the second high-affinity state, consequently decreasing DNA binding.
Since the addition of ADP canceled the difference in thermostability between MutSα WT and MSH2 V63E (Supplementary Figure S2B and C), we tested how ADP affects the kinetics of DNA binding. SPR in the presence of ADP resulted in similar curve profiles for WT and mutant protein with fast dissociations (Supplementary Figure S2N). The fitting of the dissociation phase with a one-phase exponential decay model revealed a slightly longer lifetime of MutSα WT on mismatched DNA (11 s) compared to MSH2 V63E (8 s) (Supplementary Figure S2O). Thus, the MSH2 V63E mutant was still less stable on DNA in the presence of ADP.
In addition, we checked whether MSH2 V63E can recruit MutLα. Both MutSα WT and MSH2 V63E were able to recruit MutLα in the presence of ATP; hence, the mutation did not affect the interaction with MutLα (Supplementary Figure S2P). We therefore focused on the effect of decreased DNA binding.
Interface between the MBDs is responsible for high-affinity DNA binding
Structurally, valine 63 is located in the MBD of MSH2 (domain I) (Figure 3A); however, this residue is not directly involved in DNA binding. Structural analysis revealed that it is buried in the hydrophobic core of the MBD, close to an interface with the MBD of MSH6, first observed in the MutSα crystal structure (51). Mutation of the small hydrophobic valine to a charged glutamate could disturb the hydrophobic core of the domain and affect the interaction between the MBDs. Intriguingly, this interface between MBDs is not present in E. coli MutS (Figure 3A). We wondered whether formation of the interface contributes to the conformational change that we observed in SPR.
Figure 3.

Interface between the MBDs is responsible for high affinity DNA binding (A) In MutSα, the MBD of MSH2 (dark blue) and MBD of MSH6 (light blue) form an interface. V63 is located in the MBD of MSH2 and highlighted in magenta. It is not involved in DNA binding but part of a hydrophobic network that forms the interface with MBD6. DNA is colored in gold. Escherichia coli MutS, with monomers in light and dark green, has no interface between the MBDs. (B) MBD interface residues and selected mutations. Residues involved in the interface according to PISA are shown as sticks. V3 (red) on MSH2 side points toward MSH6. S472 (yellow) is in the middle of the MSH6 interface helix. MSH2 residues 2–7 are highlighted in cyan. (C, D) SPR mismatch DNA binding profiles of protein titrations ranging from 1 to 256 nM (C) or from 1 to 128 nM (D) in 2-fold dilutions (from light to dark color). Binding and dissociation periods were fitted using CLAMP and values given in scheme below. Curves were fitted with a two-state reaction model (black curves): (C) MutSα MSH2 V3D and (D) MutSα MSH6 S472Y. (E) Comparison of SPR normalized dissociation curves of all tested mutants at 128 nM protein concentration.
To test this hypothesis, we removed the complete MBD2 and tested for DNA binding. MutSα ΔMBD2 protein was stably expressed and bound mismatched DNA. Although a two-state fit was still necessary to explain the binding profile, its affinity was 16 nM and thus similar to E. coli MutS and MSH2 V63E (Supplementary Figure S3A).
Next, we perturbed the interface between the MBDs by introducing other mutations at the MBD interface. We identified MSH2 valine 3 as a suitable target as it makes hydrophobic contacts with MBD6, and mutated it to a negatively charged aspartic acid (V3D) (Figure 3B). SPR analysis showed faster dissociation from mismatched DNA compared to MutSα WT, similar to MSH2 V63E (Figures 2E and 3C). Consequently, the overall affinity for the GT mismatch was reduced to a KD of 6 nM, again due to a faster back-reaction in the conformational change component (Figure 3C). In addition, a double mutant of MSH2 V3D and V63E exhibited an even faster off-rate (Supplementary Figure S3B). Although the curves could still not be fitted by a one-state reaction, this behavior was reminiscent of the E. coli MutS curve profile rather than that of MutSα WT. Reduced DNA binding was also seen for another mutant, where MSH2 residues 2–7 were deleted (Supplementary Figure S3C).
The MSH6 side of the interface is primarily formed by main chain atoms, and hence is harder to disrupt by mutations. We mutated serine 472 to a tyrosine in an attempt to perturb the interface by increasing the steric bulk while keeping the polarity intact (Figure 3B). This rather mild mutation displayed a subtle decrease in DNA affinity with the same trend as the MSH2 mutants (Figure 3D). Thus, disturbing the MBD interface on the MSH6 site also created a protein with reduced DNA binding ability. We conclude that disturbing either side of the interface between the two MBDs affected the conformational change after initial DNA binding, leading to a lower affinity for DNA (Figure 3E and Table 1).
Based on this analysis, we hypothesize that MBD2 moves toward MBD6 after initial mismatch binding, generating an interface between the MBDs and locking MutSα in a high-affinity state on the mismatch.
We then assessed the functional consequences of interface mutants by a biochemical assay: the complete in vitro mismatch repair activity (CIMRA) assay (9). In the CIMRA assay, the V63E variant was mildly impaired compared to WT protein (Supplementary Figure S3D), whereas MSH2 V3D and del2–7 proteins showed almost WT levels of repair. Apparently, the sensitivity or conditions of this in vitro repair assay are not sufficient to reveal the mild effects of these mutants.
CryoEM structures reveal mobility of MBD2 in V63E
We took advantage of recent developments in cryoEM single particle analysis to study the effect of the V63E mutation and compare it to MutSα WT. We recorded cryoEM datasets of full-length MutSα WT and MSH2 V63E on a 50 bp DNA oligomer with a GT mismatch and were able to obtain reconstructions at 2.8 and 3.8 Å, respectively.
The MutSα WT cryoEM dataset can be explained by a single major conformation with only minor variations, and we were able to build an atomic model into the resulting map (Figure 4A and B). The core of our MutSα WT model is similar to the crystal structure (PDB ID: 2o8b) (51), with MSH2 and MSH6 forming a clamp around the DNA and MSH6 F432 stacking on the mismatch resulting in kinked DNA.
Figure 4.

CryoEM structures reveal mobility of MBD2 in V63E. CryoEM density maps and model for MutSα WT and MSH2 V63E. (A) CryoEM map of MutSα WT colored as in Figure 3A. (B) Atomic model of MutSα, colored as in Figure 3A. (C) Map (left) and model (right) of MBD interface with MBD2 in dark purple and MBD6 in light purple for better visibility. (D) The subclasses of the MSH2 V63E dataset show flexibility of the MBD2 domain. Left panel: closed MBD2 reminiscent of MutSα WT; middle panel: disordered MBD2; right panel: disordered MBD2 and dragged-out connector domain.
The MutSα cryoEM map allowed us to resolve surface-exposed loops, several residues at the MSH6 N-termini and both C-terminal domains (residues MSH2 856–934 and MSH6 1335–1360) (Supplementary Figure S4A), which are folded analogously to the C-terminal domains of MutSβ (PDB ID: 3thx) (52). We could not detect density for most of the long N-terminus of MSH6 (except for a short helix; see the ‘Materials and Methods’ section and Supplementary Figure S4B) confirming that it is mostly unstructured without binding partner. In accordance with the X-ray structure (PDB ID: 2o8e) (51), ADP was bound to MSH2 even though it was not added to the sample, while the MSH6 nucleotide binding site did not have density for a nucleotide (Supplementary Figure S4C). Due to longer DNA, we observe an additional contact of the MBD2 with the DNA (Supplementary Figure S4D). However, we did not build that loop because it is poorly resolved indicating that it is not ordered well.
The interface between the MBDs in our cryoEM map was similar to the crystal structure. It involves primarily residues 1–6, 59 and 83 of MBD2 and residues 446–448, 468, 469, 471, 472, 475 and 476 of MBD6 (PISA calculations; Figures 3B and 4C). The conservation of this interface in the cryoEM density shows that it was not a crystallographic artifact.
The single conformation adopted by MutSα WT is likely the stable high-affinity state 2 as this is the long-lasting state in our SPR experiments. Focused classification and principal component analysis [3D variability analysis in cryoSPARC (35)] revealed only some minor ‘breathing’ motion of MutSα WT that coincided with varying degrees of DNA kinking (Supplementary Movie S1). The interaction between MBD2 and MBD6 remained unaffected in all sampled conformations (Figure 4C).
Analysis of the MSH2 V63E cryoEM dataset was complicated, as many of the particles were partially unfolded. Although the dataset was of the same initial size as the WT set, 90% of the particles showed a partial loss of MSH2, most likely due to partial protein denaturation at the air–water interface (Supplementary Figure S4E). This was already visible in the 2D classes (Supplementary Figure S4F).
We carefully classified particles that showed density for both clamp domains and selected these for detailed analysis. In contrast to MutSα WT, a range of conformational states was visible, with especially a substantial conformational heterogeneity for MSH2. For example, the clamp domain of MSH2 opened up in a subset of particles (Supplementary Figure S4G). Analysis of 3D movement in the final closed subset of particles (by cryoDRGN and cryoSPARC 3D variability analysis) showed both the MBD2 and connector domain were moving, showing a flexible turned-out state (Supplementary Figure S4H and Supplementary Movie S2), compared to the MutSα WT state where the MBD2 was bound to DNA. Further 3D classification identified three distinct intermediate subpopulations: WT-like closed MBDs, invisible and therefore flexible MBD2 and one where the connector domain was misplaced as well, suggesting that it was dragged out by the movement of MBD2 (Figure 4D). The conformational change of the MBD2 and the connector domain was also visible in the subset with the open MSH2 clamp (Supplementary Figure S4G) indicating that the MBD2/connector domain and the clamp domain movements are independent.
In summary, the cryoEM analysis revealed major differences between MutSα WT and MSH2 V63E. While MutSα WT adopted a single primary DNA-bound conformation with only subtle movements in the MBD2, MSH2 V63E displayed strong conformational flexibility on DNA. The major movements were seen in the clamp and MBD2 domain of MSH2 V63E, which presumably led to the destruction of the protein at the air–water interface. The intact particles showed a highly flexible MBD2 and connector domain. The particle distribution suggested a movement of the MBD2 swinging between an MBD6-bound and an outward turned state at the extremes (Supplementary Movie S2).
In addition to the MutSα WT set, we also collected a dataset of MutSα WT with additional ADP (1 mM). The available crystal structure of DNA-bound human MutSα with two ADPs bound (PDB ID: 2o8b) showed the same conformation as with just one ADP bound (PDB ID: 2o8e), but recent data on DNA-free E. coli MutS suggested that the protein became more open and flexible when ADP was bound (53). We solved the DNA-bound cryoEM structure of MutSα WT in the presence of ADP, revealing a closed clamp but a more flexible conformational landscape (Supplementary Figure S4I). Especially, the MBD2 showed a high degree of flexibility. It seems that the addition of ADP produced a similar effect to the V63E mutation, mobilizing MutSα on DNA.
DISCUSSION
In this work, characterization of a weakly pathogenic MMR mutant, MSH2 V63E, led to the discovery of a new molecular principle in DNA mismatch binding of MutSα. We found that MutSα undergoes an additional conformational change upon mismatch binding. As a consequence of the movement, an additional interface between the MBDs of MSH2 and MSH6 is formed, which results in high-affinity DNA binding. Distortion of the interface by mutations leads to reduced DNA binding, decreased repair efficiency in vitro and increased tumor development in mice. Our experiments provide a mechanism for the pathogenicity of the V63E mutation.
Based on our findings, we propose an updated model of MutSα DNA mismatch binding: MutSα initially binds a mismatch with low affinity by the MBD of MSH6, kinking the DNA by insertion of phenylalanine 432 (PDB ID: 2o8b) (51). This step is followed by the closing of the MBD2 to generate the additional MBD interaction and results in the high-affinity DNA binding by MutSα. Thus, the MBD2 acts as a ‘safety lock’ that needs to pivot into position (in a motion involving the MSH2 connector domain) in order to generate the additional interface and guarantee a stable interaction on the mismatch (Figure 5). Without the ‘safety lock’, MutSα can still recognize mismatches and recruit MutLα, but has lower affinity for DNA (and faster off-rate), which eventually causes reduced MMR capacity and predisposes to cancer development in the long term.
Figure 5.

Model of MutSα DNA mismatch binding involving a conformational change (A) MutSα scans DNA and (B) initial mismatch binding through MBD6. The insertion of phenylalanine generates the low-affinity DNA-bound state. (C) In a second step, a conformational change occurs. In this step, closing of the MBD2 forms the interface with MBD6 stabilizing the high-affinity DNA-bound state. In all states, MSH2 has ADP bound, while MSH6 is nucleotide-free.
The genetic variant MSH2 V63E was homozygously present in the germline of a patient who suffered from CRC around 30 years of age. Biallelic MSH2 mutations generally result in embryonic lethality or cancer in early childhood, which suggested that the V63E variant may have residual MMR activity (45). This moderate cancer phenotype was reflected in our in vivo tumor studies showing that Msh2V63E/V63E mice were only slightly more prone to spontaneous or TMZ-induced tumorigenesis than WT animals.
An interesting question is whether the MSH2 V63E variant would also predispose heterozygous carriers to tumor development and thus give rise to LS. Given the significantly delayed tumorigenesis in Msh2V63E/V63E as compared to Msh2−/− mice, it seems likely that in heterozygous V63E mutation carriers, tumors would take more time to develop upon loss of the WT MMR allele than in fully pathogenic MMR mutation carriers. Therefore, the age of cancer onset may overlap with that of sporadic CRC. However, we did find that exposure of mice to the methylating agent TMZ caused expansion of intestinal crypts that only have the V63E variant. Therefore, caution is also warranted when using methylating chemotherapeutics on patients with suspected hypomorphic mutations in MMR proteins. Additionally, (low levels of) methylating agents in the form of dietary components, bacterial metabolites or cigarette smoke may enhance cancer risk in patients with a single germline MSH2 V63E mutation (54,55). Unfortunately, we were not able to study this possibility as the microbiota composition in our current mouse facility did not support intestinal tumorigenesis (47).
Studying the biophysics of human MSH2 V63E mutation helped to reveal the importance of the interface between the MSH2 and MSH6 MBDs. In yeast, MBD deletion experiments showed that MBD2 is dispensable for MMR by MutSα and shows almost full repair (56). The relatively high MMR activity of the MBD2 interface mutant proteins in the CIMRA assay and the intermediate resistance of V63E to methylating agents and reduced MSI in MSH2 V63E-expressing cells indicate that also in higher organisms the ‘safety lock’ by MBD2 is not absolutely essential for mismatch correction. The CIMRA assay may also not accurately mimic relevant concentrations and therefore it may not have sufficient sensitivity to reveal the mild differences that would be expected for the MBD interface mutants. It is also possible that somewhat retarded MMR remained unnoticed in the CIMRA assay. The late tumor onset in Msh2V63E/V63E mice suggests that the residual MMR capacity is sufficient to largely prevent tumorigenesis in an organism with a relatively short life span. However, in case of humans, subtle effects in DNA affinity are augmented over the life span of the patient and a 20-fold reduced DNA binding eventually takes its toll and accelerates cancer development.
The interface between MBDs provides additional affinity for mismatched DNA, which stabilizes the complex. The V63E mutation leads to a destabilization of this interface, as supported by mutagenesis of other interface residues. In practice, the cryoEM analysis indicated substantial movement of the MBD2, to the point that the entire MSH2 became unstable under the cryoEM freezing conditions. For those particles that did contain complete MutSα, we observe that the MBD2 can drag the adjacent connector domain outward. Interestingly, the connector domain also moved during the mismatch release step upon ATP binding, though it is unclear whether these two movements are structurally related or not (24,57).
In the cryoDRGN subclasses of the cryoEM data, we see slightly different degrees of DNA kinking for both WT and V63E mutant, with kinking angle being less pronounced for V63E. However, the DNA kinking is induced even when the MBD2 is flexible indicating that MBD2 is not significant for DNA bending. The interface with the DNA contributed by MBD2 is small and transient compared to the MBD6 and the clamp domains. Contacts with the DNA by MBD2 through K7 or K110 presumably help stabilizing the interaction. The V63E mutation might prevent these interactions through destabilization of the domain leading to faster dissociations from the mismatch. Therefore, the proportion of binding events that lead to successful repair may be limited in the interface mutants.
Since ADP levels in the cell are generally low, we hypothesize that the double ADP-bound state is primarily generated toward the end of the MutSα conformational cycle through ATP hydrolysis, initiating DNA release after MutLα is recruited. Our SPR data indeed show a faster off-rate upon addition of ADP. Interestingly, the effect of the V63E mutation was much stronger in the absence of external nucleotides, when MutSα is in a state where only MSH2 has ADP bound. In line with this, our cryoEM data showed a release of the MSH2 safety lock even in the WT protein upon ADP addition, which explains the reduced effect of the V63E mutation. ADP remained bound to MSH2, but not to MSH6 even though it was not added to the sample (Supplementary Figure S4B). This is a major difference with E. coli MutS, where it is the mismatch-bound MutS monomer that binds ADP (22).
We show here that the formation of the MBD2–MBD6 interface acts as a safety lock for DNA engagement in human MutSα, which is not seen in E. coli MutS. We postulate the formation of this interface is the main contributor to the stability of the second high-affinity binding state seen in our SPR experiments. Nevertheless, complete removal of MBD2 did not remove the second state completely. It therefore seems that there is some residual contribution to this state outside the MBD2. Interestingly, Taq MutS also shows two-state DNA binding kinetics (58) (like human MutSα) and large movements in the MBDs upon DNA engagement (59) but (like E. coli MutS) lacks an extended MBD interface, reinforcing the notion that additional conformational changes may contribute to the two-state equilibrium outside the MBD interface. The higher affinity of human MutSα, however, is completely explained by the ‘safety lock’ and removing it destabilizes the high-affinity state.
While we cannot provide a definite explanation for the affinity difference between the species, there could be several reasons why the ‘safety lock’ is not present in E. coli. In general, single-cell organisms and bacteria have less stringent requirements for replication fidelity as the demise of individual cells does not harm the population as much as it might in a tissue environment. The longer the life span and the bigger the size of an organism, the more important error-free duplication of the genome becomes. In addition, eukaryotic MutSα is thought to be traveling with the replication fork by interaction with PCNA (1,60,61). This might require a more thorough stop during scanning. Moreover, eukaryotic MMR happens in the context of chromatin, causing proteins to deal with nucleosomes on DNA.
Taken together, we here identified the molecular mechanism underlying the pathogenicity of MSH2 V63E. Patient data helped us to explain a difference in DNA binding between human MutSα and bacterial MutS. A combination of mutagenesis, binding studies and cryoEM analyses provided mechanistic insights. It seems likely that other mildly pathogenic MMR mutations will be equally informative about molecular mechanisms of MMR. Conversely, future biochemical and structural analyses may contribute to understanding of the underlying defect in genetic variants of uncertain significance.
DATA AVAILABILITY
Atomic coordinates and cryoEM maps for the reported cryoEM data have been deposited with the EMDB under accession numbers EMD-15417, EMD-15518, EMD-15519 and PDB ID 8AG6. Other data are available upon request from the corresponding authors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Sixma, Perrakis and te Riele labs for valuable discussions, Andrea Muracchelli for help in the cryoEM processing pipeline, Rafael Fernandez Leiro for advice on cryoEM data processing, Robbie Joosten for support in PDB-REDO refinement, the staff at NeCEN for help with data collection, and staff of the animal facility and animal pathology of the Netherlands Cancer Institute for outstanding technical support.
Author contributions: W.P., H.t.R., S.R.B., A.F. and T.K.S. conceived the project. W.P., E.R., D.K., L.D., M.D., E.D.-G. and C.d.K. performed cell-based and mouse experiments and analyzed the data. H.J.D. and A.A. provided patient data. E.R. and S.v.H.-S. performed CIMRA assay, supervised by N.d.W. H.H. and N.d.W. provided critical scientific input and reagents. S.R.B. established protein purification protocol. S.R.B. and S.S. expressed and purified all proteins. S.R.B. performed stability assays. S.R.B. and A.F. performed SPR experiments and analysis. S.R.B. solved the cryoEM structure, including sample preparation, data processing, and refinement. A.M.L. and F.F. supervised initial cryoEM analysis and gave advice during the entire cryoEM workflow. T.K.S. and H.t.R. supervised the study. S.R.B, W.P., T.K.S, and H.t.R. wrote the manuscript and generated the figures with help from all coauthors.
Contributor Information
Susanne R Bruekner, Division of Biochemistry, Netherlands Cancer Institute and Oncode Institute, 1066 CX Amsterdam, The Netherlands.
Wietske Pieters, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Alexander Fish, Division of Biochemistry, Netherlands Cancer Institute and Oncode Institute, 1066 CX Amsterdam, The Netherlands.
A Manuel Liaci, Structural Biochemistry, Bijvoet Centre for Biomolecular Research, Utrecht University, 3584CH Utrecht, The Netherlands.
Serge Scheffers, Division of Biochemistry, Netherlands Cancer Institute and Oncode Institute, 1066 CX Amsterdam, The Netherlands.
Emily Rayner, Department of Human Genetics, Leiden University Medical Center, PO Box 9600 2300RC Leiden, The Netherlands.
Daphne Kaldenbach, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Lisa Drost, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Marleen Dekker, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Sandrine van Hees-Stuivenberg, Department of Human Genetics, Leiden University Medical Center, PO Box 9600 2300RC Leiden, The Netherlands.
Elly Delzenne-Goette, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Charlotte de Konink, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Hellen Houlleberghs, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Hendrikus Jan Dubbink, Department of Pathology, Erasmus Medical Center, PO Box 2040 3000CA Rotterdam, The Netherlands.
Abeer AlSaegh, Sultan Qaboos Comprehensive Cancer Care and Research Center, PO Box 787, 117 Muscat, Oman.
Niels de Wind, Department of Human Genetics, Leiden University Medical Center, PO Box 9600 2300RC Leiden, The Netherlands.
Friedrich Förster, Structural Biochemistry, Bijvoet Centre for Biomolecular Research, Utrecht University, 3584CH Utrecht, The Netherlands.
Hein te Riele, Division of Tumor Biology and Immunology, Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands.
Titia K Sixma, Division of Biochemistry, Netherlands Cancer Institute and Oncode Institute, 1066 CX Amsterdam, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Dutch Research Council [NWO-TOP 714.016.002 to T.K.S]; European Commission [H2020-MSCA-ITN-2016 722433 DNAREPAIRMAN to T.K.S]; Dutch Cancer Society [KWF 2014-7176 to H.t.R., KWF 2016-10645 to H.t.R. and N.d.W.]; European Commision [ERC Consolidator grant 724425 to F.F]. Funding for open access charge: Netherlands Cancer Institute.
Conflict of interest statement. The authors declare no competing interests.
REFERENCES
- 1. Kunkel T.A., Erie D.A.. DNA mismatch repair. Annu. Rev. Biochem. 2005; 74:681–710. [DOI] [PubMed] [Google Scholar]
- 2. Jiricny J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch H.T., Snyder C.L., Shaw T.G., Heinen C.D., Hitchins M.P.. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer. 2015; 15:181–194. [DOI] [PubMed] [Google Scholar]
- 4. Abedalthagafi M. Constitutional mismatch repair-deficiency: current problems and emerging therapeutic strategies. Oncotarget. 2018; 9:35458–35469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nik-Zainal S., Morganella S.. Mutational signatures in breast cancer: the problem at the DNA level. Clin. Cancer Res. 2017; 23:2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poynter J.N., Siegmund K.D., Weisenberger D.J., Long T.I., Thibodeau S.N., Lindor N., Young J., Jenkins M.A., Hopper J.L., Baron J.A.et al.. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol. Biomarkers Prev. 2008; 17:3208–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouvet D., Bodo S., Munier A., Guillerm E., Bertrand R., Colas C., Duval A., Coulet F., Muleris M.. Methylation tolerance-based functional assay to assess variants of unknown significance in the MLH1 and MSH2 genes and identify patients with Lynch syndrome. Gastroenterology. 2019; 157:421–431. [DOI] [PubMed] [Google Scholar]
- 8. Houlleberghs H., Dekker M., Lantermans H., Kleinendorst R., Dubbink H.J., Hofstra R.M.W., Verhoef S., te Riele H.. Oligonucleotide-directed mutagenesis screen to identify pathogenic Lynch syndrome-associated MSH2 DNA mismatch repair gene variants. Proc. Natl Acad. Sci. U.S.A. 2016; 113:4128–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drost M., Tiersma Y., Thompson B.A., Frederiksen J.H., Keijzers G., Glubb D., Kathe S., Osinga J., Westers H., Pappas L.et al.. A functional assay-based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet. Med. 2019; 21:1486–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dudley J.C., Lin M.-T., Le D.T., Eshleman J.R.. Microsatellite instability as a biomarker for PD-1 blockade. Clin. Cancer Res. 2016; 22:813–820. [DOI] [PubMed] [Google Scholar]
- 11. Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D.et al.. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl. J. Med. 2015; 372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swann P.F., Waters T.R., Moulton D.C., Xu Y.-Z., Zheng Q., Edwards M., Mace R.. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996; 273:1109–1111. [DOI] [PubMed] [Google Scholar]
- 13. Nevala-Plagemann C., Hidalgo M., Garrido-Laguna I.. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020; 17:108–123. [DOI] [PubMed] [Google Scholar]
- 14. Pani E., Stojic L., El-Shemerly M., Jiricny J., Ferrari S.. Mismatch repair status and the response of human cells to cisplatin. Cell Cycle. 2007; 6:1796–1802. [DOI] [PubMed] [Google Scholar]
- 15. Topping R.P., Wilkinson J.C., Scarpinato K.D.. Mismatch repair protein deficiency compromises cisplatin-induced apoptotic signaling. J. Biol. Chem. 2009; 284:14029–14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Acharya S., Wilson T., Gradia S., Kane M.F., Guerrette S., Marsischky G.T., Kolodner R., Fishel R.. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl Acad. Sci. U.S.A. 1996; 93:13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schofield M.J., Hsieh P.. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 2003; 57:579–608. [DOI] [PubMed] [Google Scholar]
- 18. Kadyrov F.A., Dzantiev L., Constantin N., Modrich P.. Endonucleolytic function of MutLα in human mismatch repair. Cell. 2006; 126:297–308. [DOI] [PubMed] [Google Scholar]
- 19. Kadyrov F.A., Holmes S.F., Arana M.E., Lukianova O.A., O’Donnell M., Kunkel T.A., Modrich P. Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. J. Biol. Chem. 2007; 282:37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goellner E.M., Putnam C.D., Kolodner R.D.. Exonuclease 1-dependent and independent mismatch repair. DNA Repair. 2015; 32:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kunkel T.A., Erie D.A.. Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 2015; 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lamers M.H., Perrakis A., Enzlin J.H., Winterwerp H.H.K., de Wind N., Sixma T.K.. The crystal structure of DNA mismatch repair protein MutS binding to a G·T mismatch. Nature. 2000; 407:711–717. [DOI] [PubMed] [Google Scholar]
- 23. Obmolova G., Ban C., Hsieh P, Yang W.. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000; 407:703–710. [DOI] [PubMed] [Google Scholar]
- 24. Groothuizen F.S., Winkler I., Cristóvão M., Fish A., Winterwerp H.H.K., Reumer A., Marx A.D., Hermans N., Nicholls R.A., Murshudov G.N.et al.. MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA. eLife. 2015; 4:e06744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Wind N., Dekker M., Berns A., Radman M., te Riele H.. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995; 82:321–330. [DOI] [PubMed] [Google Scholar]
- 26. Wojciechowicz K., Cantelli E., Gerwen B.V., Plug M., Wal A.V.D., Delzenne-Goette E., Song J.-Y., De Vries S., Dekker M., te Riele H.. Temozolomide increases the number of mismatch repair-deficient intestinal crypts and accelerates tumorigenesis in a mouse model of Lynch syndrome. Gastroenterology. 2014; 147:1064–1072. [DOI] [PubMed] [Google Scholar]
- 27. Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E., van Es J.H., Abo A., Kujala P., Peters P.J.et al.. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009; 459:262–265. [DOI] [PubMed] [Google Scholar]
- 28. Drost M., Tiersma Y., Glubb D., Kathe S., van Hees S., Calléja F., Zonneveld J.B.M., Boucher K.M., Ramlal R.P.E., Thompson B.A.et al.. Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome. Genet. Med. 2020; 22:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Myszka D.G., Morton T.A.. CLAMP: a biosensor kinetic data analysis program. Trends Biochem. Sci. 1998; 23:149–150. [DOI] [PubMed] [Google Scholar]
- 30. Tegunov D., Cramer P.. Real-time cryo-EM data pre-processing with Warp. Nat. Methods. 2019; 16:1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimanius D., Dong L., Sharov G., Nakane T., Scheres S.H.W.. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 2021; 478:4169–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rohou A., Grigorieff N.. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015; 192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner T., Merino F., Stabrin M., Moriya T., Antoni C., Apelbaum A., Hagel P., Sitsel O., Raisch T., Prumbaum D.et al.. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2019; 2:218–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A.. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017; 14:290–296. [DOI] [PubMed] [Google Scholar]
- 35. Punjani A., Fleet D.J.. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 2021; 213:107702. [DOI] [PubMed] [Google Scholar]
- 36. Kaur S., Gomez-Blanco J., Khalifa A.A.Z., Adinarayanan S., Sanchez-Garcia R., Wrapp D., McLellan J.S., Bui K.H., Vargas J.. Local computational methods to improve the interpretability and analysis of cryo-EM maps. Nat. Commun. 2021; 12:1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong E.D., Bepler T., Berger B., Davis J.H.. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods. 2021; 18:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng S.Q., Palovcak E., Armache J.-P., Verba K.A., Cheng Y., Agard D.A.. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 41. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M.. ColabFold: making protein folding accessible to all. Nat. Methods. 2022; 19:679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joosten R.P., Long F., Murshudov G.N., Perrakis A.. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014; 1:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Felton K., Gilchrist D., Andrew S.. Constitutive deficiency in DNA mismatch repair. Clin. Genet. 2007; 71:483–498. [DOI] [PubMed] [Google Scholar]
- 46. Kloor M., Huth C., Voigt A.Y., Benner A., Schirmacher P., von Knebel Doeberitz M., Bläker H.. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol. 2012; 13:598–606. [DOI] [PubMed] [Google Scholar]
- 47. Pieters W., Hugenholtz F., Kos K., Cammeraat M., Moliej T.C., Kaldenbach D., Klarenbeek S., Davids M., Drost L., de Konink C.et al.. Pro-mutagenic effects of the gut microbiota in a Lynch syndrome mouse model. Gut Microbes. 2022; 14:2035660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lamers M.H., Georgijevic D., Lebbink J.H., Winterwerp H.H.K., Agianian B., de Wind N., Sixma T.K. ATP increases the affinity between MutS ATPase domains. Implications for ATP hydrolysis and conformational changes. J. Biol. Chem. 2004; 279:43879–43885. [DOI] [PubMed] [Google Scholar]
- 49. Groothuizen F.S., Fish A., Petoukhov M.V., Reumer A., Manelyte L., Winterwerp H.H.K., Marinus M.G., Lebbink J.H.G., Svergun D.I., Friedhoff P.et al.. Using stable MutS dimers and tetramers to quantitatively analyze DNA mismatch recognition and sliding clamp formation. Nucleic Acids Res. 2013; 41:8166–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Manelyte L., Urbanke C., Giron-Monzon L., Friedhoff P.. Structural and functional analysis of the MutS C-terminal tetramerization domain. Nucleic Acids Res. 2006; 34:5270–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Warren J.J., Pohlhaus T.J., Changela A., Iyer R.R., Modrich P.L., Beese L.S.. Structure of the human MutSα DNA lesion recognition complex. Mol. Cell. 2007; 26:579–592. [DOI] [PubMed] [Google Scholar]
- 52. Gupta S., Gellert M., Yang W.. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2012; 19:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Borsellini A., Kunetsky V., Friedhoff P., Lamers M.H.. Cryogenic electron microscopy structures reveal how ATP and DNA binding in MutS coordinates sequential steps of DNA mismatch repair. Nat. Struct. Mol. Biol. 2022; 29:59–66. [DOI] [PubMed] [Google Scholar]
- 54. Groenen P.J., Busink E.. Alkylating activity in food products—especially sauerkraut and sour fermented dairy products—after incubation with nitrite under quasi-gastric conditions. Food Chem. Toxicol. 1988; 26:215–225. [DOI] [PubMed] [Google Scholar]
- 55. Winkels R.M., Botma A., Duijnhoven F.J.B.V., Nagengast F.M., Kleibeuker J.H., Vasen H.F.A., Kampman E.. Smoking increases the risk for colorectal adenomas in patients with Lynch syndrome. Gastroenterology. 2012; 142:241–247. [DOI] [PubMed] [Google Scholar]
- 56. Lee S.D., Surtees J.A., Alani E.. Saccharomyces cerevisiae MSH2–MSH3 and MSH2–MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J. Mol. Biol. 2007; 366:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fernandez-Leiro R., Bhairosing-Kok D., Kunetsky V., Laffeber C., Winterwerp H.H., Groothuizen F., Fish A., Lebbink J.H.G., Friedhoff P., Sixma T.K.et al.. The selection process of licensing a DNA mismatch for repair. Nat. Struct. Mol. Biol. 2021; 28:373–381. [DOI] [PubMed] [Google Scholar]
- 58. Sharma A., Doucette C., Biro F.N., Hingorani M.M.. Slow conformational changes in MutS and DNA direct ordered transitions between mismatch search, recognition and signaling of DNA repair. J. Mol. Biol. 2013; 425:4192–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qiu R., DeRocco V.C., Harris C., Sharma A., Hingorani M.M., Erie D.A., Weninger K.R.. Large conformational changes in MutS during DNA scanning, mismatch recognition and repair signalling. EMBO J. 2012; 31:2528–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Umar A., Buermeyer A.B., Simon J.A., Thomas D.C., Clark A.B., Liskay R.M., Kunkel T.A.. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996; 87:65–73. [DOI] [PubMed] [Google Scholar]
- 61. Kleczkowska H.E., Marra G., Lettieri T., Jiricny J.. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001; 15:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and cryoEM maps for the reported cryoEM data have been deposited with the EMDB under accession numbers EMD-15417, EMD-15518, EMD-15519 and PDB ID 8AG6. Other data are available upon request from the corresponding authors.