


Abstract
The vast majority of disease-associated single nucleotide polymorphisms (SNP) identified from genome-wide association studies (GWAS) are localized in non-coding regions. A significant fraction of these variants impact transcription factors binding to enhancer elements and alter gene expression. To functionally interrogate the activity of such variants we developed snpSTARRseq, a high-throughput experimental method that can interrogate the functional impact of hundreds to thousands of non-coding variants on enhancer activity. snpSTARRseq dramatically improves signal-to-noise by utilizing a novel sequencing and bioinformatic approach that increases both insert size and the number of variants tested per loci. Using this strategy, we interrogated known prostate cancer (PCa) risk-associated loci and demonstrated that 35% of them harbor SNPs that significantly altered enhancer activity. Combining these results with chromosomal looping data we could identify interacting genes and provide a mechanism of action for 20 PCa GWAS risk regions. When benchmarked to orthogonal methods, snpSTARRseq showed a strong correlation with in vivo experimental allelic-imbalance studies whereas there was no correlation with predictive in silico approaches. Overall, snpSTARRseq provides an integrated experimental and computational framework to functionally test non-coding genetic variants.
INTRODUCTION
Germline genetic variants contribute to numerous diseases from COVID-19 (1) to cancer development (2–6). Disease-associated SNPs are primarily identified from GWAS (7). While those SNPs that occur in protein-coding regions have a predictable impact on protein sequence, the vast majority of disease-associated SNPs are located in non-coding regions (8,9). There is increasing evidence that these non-coding variants affect disease initiation and progression by altering critical cis-regulatory elements (CRE) that are involved in the spatiotemporal expression of target genes (10–14). These variants commonly occur at enhancers where they can alter transcription factors (TF) binding and gene transcription (15–19). For instance, a SNP in the PCAT19 locus disrupts NKX3.1 and YY1 binding which alters enhancer activity causing dysregulation of oncogene expression and prostate cancer (PCa) progression (14,20,21). While most SNPs identified from GWAS studies are found within the non-coding region of the genome, it remains difficult to mechanistically characterize the impact of these variants (22,23).
Several in silico and experimental approaches are commonly used to characterize potential pathogenic non-coding variants. Current in silico methods apply machine learning that is trained on previously published data to predict activity without experimental input (23–26). Given their relative ease, these bioinformatic approaches are used to stratify candidates for validation. Yet, they do have several major limitations. Previous benchmarking studies demonstrated considerable variability between each computational method (27) with a very high error rate (28–30). In contrast, experimental approaches are much more robust. One promising method utilizes in vivo chromatin immunoprecipitation with sequencing (ChIPseq) to measure the impact of non-coding variants on allelic-imbalance of TF binding (31–34). Demonstrating the potential utility, recent work combined >7000 ChIPseq from 649 cell lines and identified 270 000 SNPs with altered TF binding affinity (25). While promising, this experimental approach is limited by the frequency of each variant in the tested population which requires a substantial number of clinical samples for accurate functional genomic testing. These challenges, therefore, limit allelic-imbalance studies to only a handful of specific experimental models, tissues, and disease states (35). To overcome these challenges, massively parallel reporter assay (MPRA) (36) including self-transcribing active regulatory region sequencing (STARRseq) (37) has been used to directly quantify the activity of thousands of non-coding sequences in a single experiment (38–41). Importantly, these methodologies do not require clinical samples and are amenable to functional perturbations. Several studies have adopted STARRseq to systematically screen the impact of SNPs on enhancer function (42–52). These methods can be broadly separated based on the source of the target non-coding sequence with variants. While a few obtained mutant and variant sequences from mixed DNA libraries (43,47), many used oligonucleotide synthesis to obtain enhancer sequences (42,44,45,49–53). While synthesis-based methods can generate sequences harboring targeted variants that allow fine-mapping of complex haplotypes (51,52), pooled methods incorporate longer fragments that provide additional sequence context of co-regulator binding (54) and produce more reproducible enhancer activity quantification (55). Despite the feasibility of the large-scale functional screens, their performance has been limited by library representation which causes poor signal-to-noise, false positives, or limited statistical robustness. Further, it is unclear how these plasmid-based methods compare to in silico predictions and other experimental approaches.
In this work, we developed a standardized experimental and computational STARRseq framework to identify disease-associated genetic variants that impact enhancer activity. To address the previous limitations due to short enhancer sequences, we developed asymmetrical Illumina sequencing and covered enhancer fragments up to 841 bp (mean: 543 bp). This can accurately identify critical non-coding SNPs and test hundreds to thousands of variants in a single experiment Using this approach we functionally tested 68 SNPs from known prostate cancer (PCa) risk-associated loci and demonstrated that 36 (51%) of them significantly altered enhancer activity. Combining these results with chromosomal looping we provided a mechanism of action for 20 PCa GWAS risk regions. Our methodology, snpSTARRseq, provides streamlined bioinformatic analysis and is amenable to different genomic regions, diseases, and sequencing approaches including PacBio Long Reads. Overall, snpSTARRseq functionally characterizes hits from GWAS studies and provides a mechanistic understanding of critical genetic variants.
MATERIALS AND METHODS
Detailed information can be found in Supplementary Methods.
snpSTARRseq capture library design and additional SNP expansion
We tested disease-associated 252 SNPs (Supplementary Table S1), which are located in enhancer regions that have H3K27Ac signal and chromosomal looping to a gene promoter (56) as well as 50 control regions (25 positive and 25 negative control). Positive control regions (strong enhancers) were identified from previously published whole genome STARRseq (57). Negative-control regions contained an androgen response element (ARE) motif but no AR binding or enhancer activity in published work (41). Chromosomal locations of all capture regions can be found in (Supplementary Table S2).
Generation of snpSTARRseq capture library
Pooled human genomic DNA (NA13421; consisted of 27 males and 27 females from CEPH Utah pedigrees) obtained from Coriell Institute for Medical Research (58) (Supplementary Table S3) was fragmented (500–800 bp), end-repaired and ligated with xGen stubby adaptors (IDT) containing random i7 3bp UMI. The captured regions were enriched using xGen biotinylated oligonucleotide probe pool (IDT) (59) and Dynabeads M-270 Streptavidin beads (IDT). Post-capture was PCR-amplified with STARR_in-fusion_F primer and STARR_in-fusion_R primer, and then cloned into AgeI-HF (NEB) and SalI-HF (NEB) digested hSTARR-ORI plasmid (Addgene plasmid #99296) with NEBuilder HiFi DNA Assembly Master Mix (NEB). The snpSTARRseq capture library was then transformed into MegaX DH10B T1R electrocompetent cells (Invitrogen) and the plasmid DNA was extracted using the Qiagen Plasmid Maxi Kit. The sequences of all the primers used for generating the snpSTARRseq capture library were listed (Supplementary Table S4).
Experimental method for snpSTARRseq and sequencing
The cloned snpSTARRseq library (100 ug plasmid DNA/replica) was transiently transfected into LNCaP cells (5 × 107 cells/replica; three biological replicas) using the Neon Transfection System (Invitrogen). Cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and collected 48hrs post electroporation. These cells were lysed with Precellys CKMix Tissue Homogenizing Kit (Bertin Technologies) and total RNA was extracted using RNeasy Maxi Kit (Qiagen). mRNA was isolated with Oligo (dT) 25 Dynabeads (Thermo Fisher) and the reverse transcription was done with the plasmid-specific primer. The synthesized snpSTARRseq cDNA was treated with RNaseA (Thermo Fisher) and amplified by a junction PCR (15 cycles) with the RNA_jPCR_f primer and the jPCR_r primer. The snpSTARRseq capture library was PCR-amplified with DNA-specific junction PCR primer (DNA_jPCR_f primer) and jPCR_r primer. All primer sequences were listed in Supplementary Table S4. After purification with Ampure XP beads (Beckman Coulter), both the snpSTARRseq samples were PCR-amplified with TruSeq dual indexing primers (Illumina) to generate Illumina-compatible libraries. RNA samples were sequenced with a HiSeq4000 (150 bp; paired-end (PE)) while the DNA STARRseq capture library was asymmetrically sequenced with Illumina MiSeq PE reads. In the latter sequencing, we did two rounds of PE sequencing with round 1 being forward 75 cycles/reverse 425 cycles reverse and round 2 being forward 425 cycles/reverse 75 cycles reverse.
PacBio long-read sequencing
snpSTARRseq input DNA library was digested with NotI-HF (NEB) enzyme for linearization of the plasmid DNA. After that sequence library was sequenced by the PacBio SMRT link. Raw sequencing data (subreads.bam) was processed by the ccs function of SMRT tools (version 9.03). Output CCS file was processed by our pipeline (see the section below) to validate reconstructed enhancer fragments.
Reconstruction of enhancer sequences
We developed our computational framework to reconstruct enhancer sequencing by using ‘long’ pairs of asymmetrical reads that cover the full enhancer sequence. Briefly, UMI attached reads from ‘long-short’ and ‘short-long’ asymmetrical sequencing are first clustered with Calib's cluster (60) based on their UMI and sequence context (Figure 1B-Clustering). This step gathers reads belonging to the same enhancer fragments from each independent run. This step is followed up by consensus sequence generation to correct any random error due to sequencing (Figure 1B-Consensus). Having the high-quality reads generated for each enhancer fragment, in the next step, ‘long’ reads of ‘long-short’ and ‘short-long’ were matched using 12 bp sequences from 5’ and 3’ of the enhancer fragments. These short sequences represent enhancer fragments and are used as unique barcodes (Figure 1B-Match). Consequently, matched long reads then collapsed to reconstruct enhancer fragment (Figure 1B-Collapse) using bbmerge software (61). A detailed explanation of each step of the analysis pipeline can be found in the Supplementary Methods section. The asymmetrical processing pipeline can be found at https://github.com/mortunco/snp-starrseq.
Figure 1.

Schematic representation of enhancer fragment sequence reconstruction. (A) Experimental steps of snpSTARRseq method. (B) Computational analysis of asymmetrical reads. (C) Following the clustering step, the number of reads supporting each reconstructed fragment was investigated for each asymmetric run. The top histogram demonstrates the number of single reads supported fragments that are removed before the consensus step whereas the bottom histogram represents those reads included in the further analysis. (D) DNA input library was sequenced by PacBio CCS sequencing to validate the presence of reconstructed sequences
Testing bi-allelic activity
To identify SNPs with allelic-specific enhancer activity, we conducted a Negative-Binomial Regression analysis to compare the expression of alternative allele-supporting fragments with reference allele-supporting fragments. Fragments overlapping at a SNP position were assigned as alternative or reference types based on the allele they carry. Only those SNPs with >15 unique plasmids for both alternative and reference type alleles were included for analysis. For each SNP, a negative Binomial regression was performed with the following model by using the glm.nb() function in the MASS R package (version 7.3–54) (62):
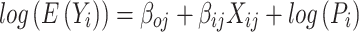 |
where 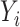 is the RNA read counts of fragment i,
is the RNA read counts of fragment i, 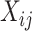 is the allele type of SNP j carried by fragment i, where
is the allele type of SNP j carried by fragment i, where 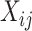 =0 when the allele on fragment i is the reference allele-supporting type and
=0 when the allele on fragment i is the reference allele-supporting type and 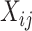 =1 when the allele on fragment i is an alternative allele-supporting type,
=1 when the allele on fragment i is an alternative allele-supporting type, 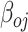 is the log expression per plasmid of the reference allele and
is the log expression per plasmid of the reference allele and 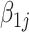 is the log fold change of expression per plasmid comparing alternative type allele versus reference type allele,
is the log fold change of expression per plasmid comparing alternative type allele versus reference type allele, 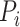 is the plasmid DNA read count of the barcode serving as an offset term.
is the plasmid DNA read count of the barcode serving as an offset term.
Empirical type-I error for NBR
As the fragment enhancer activity can be affected by not only the variants they carry but also the specific genomic region they cover, therefore, enough coverage of the SNP to be tested is required to reduce the impact of the position bias. We conducted an empirical analysis to investigate the relationship between the number of fragments and FDR. First, we selected 10 independent SNPs randomly with at least 30 fragments for each of the VAR and REF alleles, absolute alternative allele effect smaller than 0.1, and p-value larger than 0.5 to treat them as true null SNPs. For each SNP, we downsampled the fragments of each allele type to N (where N = 5, 10, 15, 20, 25, 30) and conducted the NBR to test the allelic-specific enhancer activity. We repeated the process 100 times to compute the proportion of tests with p-value < 0.05 as the empirical type-I error at a significance level of 0.05.
In silico method comparisons
We obtained five different prediction scores tables for each method (ncER (23), CADD (24), DVAR (63), LINSIGHT (64), deltaSVM (26)) and compared them with respect to snpSTARRseq absolute allelic effect abs(Log2(ALT/REF)) and adjusted P-values (FDR). Detailed information about every step can be found in Supplementary Methods and the visualization code can be found at https://github.com/mortunco/snp-starrseq. We stored corresponding snpSTARRseq allelic-effect and in silico impact scores in supplementary table (Supplementary Table S5).
Comparison with in vivo methods
We obtained pre-processed allelic imbalance datasets from three different studies such as H3K27Ac ChIPseq (n = 200) (65), AR ChIPseq (n = 131) (65) or ATACseq (n = 26) (66). We matched all datasets (H3K27Ac and AR ChIPseq, ATACseq, and snpSTARRseq) using rsID therefore, we dropped 12 Coriell DNA SNPs which did not have the corresponding rsID in dbsnp150 common VCF. We shared corresponding AF for snpSTARRseq and in vivo methods as well as significance annotation (Supplementary Table S6).
PCa risk loci analysis
We extracted 147 index SNPs from (5) manuscript's Supplementary Table S7 (European SNPs only) and Supplementary Table S8. Using rsID (rsXXX), we extracted SNP positions from dbsnp150 common VCF file (hg19). Later intersected with 252 custom capture probe locations. We accepted all interactions within a 150 kb distance (Supplementary Table S7).
SNP and gene overlap analysis
We obtained HiChIP-H3k27Ac chromosome interaction data paired-end BED file (BEDPE) from previous work (67) and used it for annotating our SNPs. In addition to this, from the same study, we also obtained COLOC annotation based TCGA (prostate) and (68) FUSION annotation based on TWAS genes. The final SNP-gene association table is deposited in (Supplementary Table S8).
RESULTS
Design considerations and enhancements
To develop a methodology that functionally characterizes the impact of genetic variants on enhancer activity we adopted STARRseq, given both the ease of use and demonstrated experimental feasibility (47–49). We designed our experimental approach with the following features: (i) to scale efficiently allowing hundreds or thousands of variants to be tested, (ii) to maintain a large insert size to ensure a high enhancer signal, (iii) to have a high experimental signal to noise ratio by increasing the number of tested plasmids, (iv) to reduce systemic false-positives associated with the earlier STARR plasmids. With this framework, we optimized several key parameters with the following improvements. To reduce false-positives we utilized the second-generation STARRseq plasmid and ensured that the experimental models had minimal IFN-gamma response, which can strongly influence STARRseq signal (69). Next, we utilized a DNA-capture to enrich the number of plasmids per variant and increase the signal to noise. Given that insert size influences enhancer signal, testing a low number of plasmids per variant is extremely error-prone as you cannot separate the impact of insert size from the impact of the genetic variant (69,70). Further, the use of DNA capture also solves the fragment size limitations that are intrinsic to oligonucleotide synthesis (150–200 bp) (71,72). Increasing the library insert size leads to an increase in relative signal strength, which reduces false positives and negatives (50,70,73). We also incorporated a unique molecular identifier (UMI) for each cloned fragment to increase statistical strength and limit amplification artifacts. Finally, we used pooled genomic material from a healthy population (Coriell DNA library; (58), n = 54) to introduce genetic diversity and increase the number of SNPs tested.
SNP selection criteria
With the modified design we chose to test 252 PCa risk-associated SNPs, which are located on 184 non-overlapping segments that include 52% (68/130) of the previously published PCa risk-associated loci (Supplementary Table S1; Supplementary Table S7) (5). These regions were selected as they are potential enhancers that have both H3K27Ac and chromosomal looping to a gene promoter (56). In addition to these risk-associated SNPs we designed the DNA capture to also target 25 strong enhancers (57) (positive control) and 25 inactive regions (negative control) as experimental controls (Figure 1.A; Supplementary Table S2; Methods). Using this capture approach, we enriched randomly fragmented DNA (400–600 bp) that was hybridized with adapters each containing flanking 3bp UMI (59). This was PCR amplified and then cloned into a second-generation STARRseq plasmid. Due to the relatively large size of the insert (median = 543 bp), it was not feasible to use conventional short-read Illumina paired-end sequencing as variants in these larger inserts potentially will not be sequenced. Therefore, to ensure that the entire insert is sequenced, we modified a 500-cycle Illumina sequencing protocol and did two rounds of asymmetric sequencing to include both a ‘long (425)-short (75)’ and ‘short (75)-long (425)’ reads (Figure 1B, top) (48). As we sequenced the same plasmid library twice, both asymmetric runs covered opposite ends of the potential enhancer fragments. We hypothesized that the full enhancer sequence could be reconstructed by matching the long sequences of the same fragment. To process this complex dataset, we developed a novel computational pipeline that clusters, collapses, and matches asymmetrical reads (Figure 1B, bottom). We first clustered the enhancer fragments using the UMI (6 bp) and sequence context of the enhancers to group the reads supporting each enhancer fragment. Next, we collapsed the short and long mates of asymmetric runs and took the consensus sequence (median 36 reads/enhancer; SEM = 0.03) (Figure 1C). By matching the unique fragment barcode from the UMI (6bp) and randomly captured insert (18 bp), we clustered each pair of asymmetric reads to obtain the full enhancer sequence. In total, we reconstructed 32 620 fragments that were located in the capture regions. These asymmetric sequencing results were confirmed with PacBio circular consensus sequencing (CCS) long reads, with 90% (29106/32620) of reconstructed fragments found with PacBio (Figure 1D). In addition to the PCa risk-associated germline variants, captured enhancer fragments also included variants from common population SNPs (dbSNP150 common VCF) (9) as well as SNPs that were specific to the Coriell DNA population (58). Overall, our library was represented by a median of 67 unique plasmids per SNP containing 41 reference (REF) alleles and 26 alternative alleles (ALT) respectively. We failed to capture 50 SNPs that were either not present in the Coriell DNA library (3/50) or were not sufficiently represented to pass variant filtration (47/50) (Supplementary Figure S1A). To limit the impact of the potential enhancer position and size bias we modelled the impact of unique plasmids thresholds on experimental data (Materials and Methods). We found that > 15 unique plasmids for both REF and ALT SNP minimizes the Type-I error to 0.05. Therefore, we focused our analysis on the 308 SNPs that have > 30 unique plasmids with REF and ALT allele supporting inserts (Supplementary Figure S1B; Table S9) and also a minimum of 1000 mRNA reads, which is 100× higher than previous work (47). These SNPs include 102 (of 252) PCa risk-associated SNPs (PCa), 194 SNPs commonly found in the 1000 Genome Project (G5), and 12 Coriell DNA library-specific SNPs (Supplementary Table S10). When we compared the variant-allele frequency (VAF) with these SNPs we observed a higher but not significant VAF of PCa risk-associated SNPs (P = 0.56) and G5 SNPs (P = 0.18) compared to Coriell SNP (Supplementary Figure S1C).
Characterization of genetic variants that impact enhancer activity
To test the impact of PCa-associated SNPs on enhancer activity, we electroporated the snpSTARRseq library into LNCaP, an androgen receptor (AR)-dependent prostate cancer cell line, and harvested at 24, 48 and 72 h (n = 3 biological replicas). With this, we quantified the insert self-transcription, a surrogate for enhancer activity, at all high-confidence REF and ALT fragments. Following normalization to the input library, we observed a clear increase in self-transcription at known enhancers (positive control) but not negative controls with a high correlation of allelic enhancer activity across each experimental time point (Figure 2A; Supplementary Figure S2A). Principal component analysis (PCA) of all samples demonstrated that 48- and 72-h samples had a closer enhancer profile compared to the 24-h samples (Supplementary Figure S2B). We next investigated how each SNP affected the activity of the enhancers. Using a differential allelic enhancer activity test based on a negative binomial regression model (NBR) we identified 78 unique SNPs across the three-time points that showed bi-allelic activity at the nominal significance level (P-value < 0.05) (60, 63 and 43 at 24, 48 and 72 h, respectively) (Figure 2B; Supplementary Figure S2C; Table S11). Of these a total of 31 (39%) nominally significant SNPs passed multiple hypothesis testing correction (FDR < 0.05) with 23 being PCa disease-associated SNPs identified from GWAS. Interestingly, the majority of SNPs that affected enhancer activity (36/78) were PCa risk-associated SNPs. Supporting our PCA analysis, we observed a higher correlation in bi-allelic activity across all significant SNPs in the 48 and 72 h samples (Pearson = 0.94) (Supplementary Figure S2D). Focusing on the 48 h samples we observed a similar frequency of activating (n = 31) and repressive (n = 32) events. Of the specific PCa-associated SNPs we observed that rs11083046 (chr18:51781019) alternative C allele had a 30% increased enhancer activity (P-value = 0.00367) compared to the reference T allele (Figure 2C). In contrast, the enhancer activity of rs13215402 (chr6:153447550) decreased by 50% (P-value = 0.00349) when the reference allele G was substituted with the alternative allele A (Figure 2C). Supporting these plasmid-based results, the SNPs with bi-allelic enhancer activity commonly affected target gene expression. Using previously published enhancer-promoter interactions from H3K27Ac-HiChIP in LNCaP cells (56), 20 of 36 ‘hit’ PCa risk-associated SNPs correlated with altered expression of the target gene (Supplementary Table S8). We found that these 20 SNPs also overlapped with PCa-specific expression quantitative trait loci (eQTL) identified by both tumor-adjacent normal samples (n = 471) and multi-tissue transcriptome-wide association study (TWAS) (n-tissue = 45, n-individual = 4448), using COLOC (74) and FUSION (75) tools, respectively (Supplementary Table S8). Interestingly, we found two significant SNPs (rs13265330; 2.2-fold, FDR = 9.25 × 10−6, rs11782388; 1.47-fold FDR = 0.07) that were located ∼10 kb downstream of NKX3-1, a gene involved in early prostate tumorigenesis (76). While not significant, we also found supporting evidence of two risk loci enhancers that loop to the PCa-associated genes CTBP2 and PCAT19. Similar to published work the two SNPs (rs11672691 and rs887391) near PCAT19 caused increased enhancer activity in all of the time points (14). At the CTBP2 loci, rs4962416 and rs12769019 caused repression and activation respectively in accordance with previous work (19). Taken together, these results demonstrate that snpSTARRseq can identify those SNPs that alter enhancer activity. When combined with chromosomal confirmation data these results can provide a mechanism of action for non-coding disease-associated SNPs.
Figure 2.
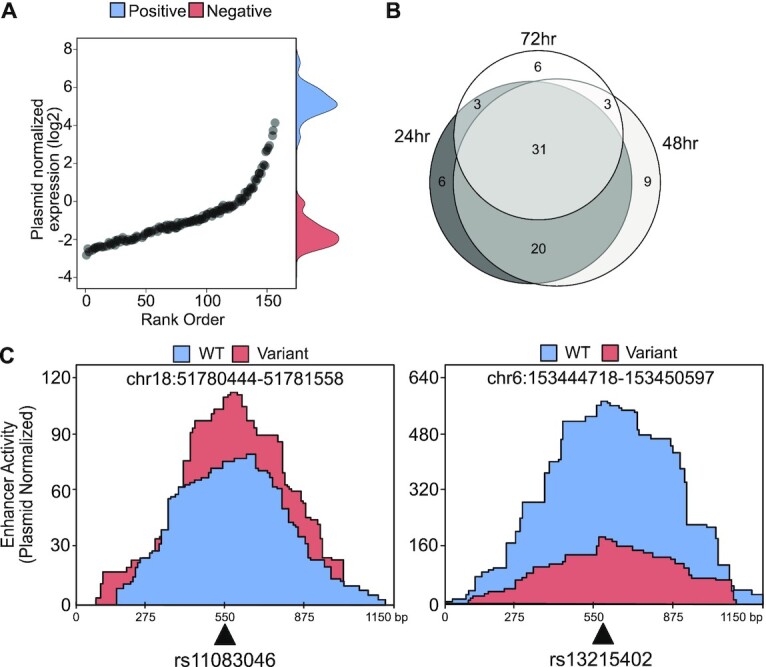
Characterisation of genetic variants that impact enhancer activity. (A) Quality control analysis of 48-hour time point demonstrates the SNP, positive control and negative control capture regions normalized count (mRNA/input DNA) distribution. Each black dot represents a single SNP capture region, whereas the density plot on the right-hand side represents the control capture regions (blue = positive control, red = negative control). (B) NBR model was used to determine SNPs with significant bi-allelic activity. As a result of the calculation, 78 unique SNPs were found among all time points. The number of overlapping SNPs from each time point is depicted by the Venn diagram. (C) Activating (rs11083046) and repressive (rs13215402) SNPs that cause bi-allelic enhancer activity were visualized.
Comparison of snpSTARRseq to in silico methodologies
In silico based pathogenicity predictions are commonly used to stratify non-coding variants for functional characterization studies (44,77,78). To benchmark the performance of these methods to snpSTARRseq, we obtained the variant impact scores at the tested PCa disease-associated SNPs from ncER (23), CADD (24), DVAR (63), LINSIGHT (64) and deltaSVM (26). When comparing these in silico predictions to our experimental snpSTARRseq results we observed no positive correlation between the deleteriousness (CADD and DVAR), impact on chromatin accessibility (deltaSVM), or essentiality score (ncER, LINSIGHT) to the experimental (snpSTARRseq) bi-allelic effect (log2(ALT/REF)) or statistical significance (FDR) (Figure 3A, Supplementary Figure S3A, Supplementary Figure S3.B). In addition, we observed low negative correlation with ncER (Pearson = –0.12; P-value = 0.023) (Figure 3A). Further, when we separated SNPs into binary groups based on in silico annotations there were no significant changes in the enhancer activity (Supplementary Figure S3C). Our findings are consistent with the current literature (28,29) highlighting the challenges of in silico methods to accurately predict how variants predict enhancer activity (Supplementary Table S5).
Figure 3.

Comparison of snpSTARRseq to orthogonal methodologies. (A) snpSTARRseq allelic-effects (ALT/REF; Method; Log2FoldChange) was compared to DeltaSVM (Essentiality Score), ncER (Essentiality Score) and CADD (PHRED score) and no significant relation found. (B) snpSTARRseq allele frequency (AF) values were compared against in-vivo AF obtained by H3k27Ac and AR ChIPseq and ATACseq. The top row contains all SNP values without any filtration whereas the bottom row has only significant SNPs found by snpSTARRSeq (nominal p-value < 0.05). Two anecdotal examples of repressive (purple) and activating (blue) SNPs were colored to which were found by all 4 methodologies. (C) SNPs with activating and repressive effects found by previous in vivo work and snpSTARRseq were demonstrated. snpSTARRseq captures the bi-allelic effect of these SNPs accurately.
snpSTARRseq correlates with clinical allelic-imbalance
We next compared our experimental snpSTARRseq results with in vivo allelic-imbalance from H3K27Ac (n = 200) and AR (n = 131) ChIPseq (65), as well as the assay for transposase-accessible chromatin using sequencing (ATACseq) (n = 26) in prostate tumors (66). This approach utilizes the endogenous heterogeneous allelic pool from clinical functional genomic studies to determine if there is an allelic preference for specific histone modifications, TFs, and chromosome accessibility. Of the 308 SNPs tested by snpSTARRseq, H3K27Ac ChIPseq had the highest coverage and captured 50% (148/308) of all SNPs while AR ChIPseq and ATACseq only captured 18% (57/308) of all SNPs tested. Based on our initial comparison without filtration, we observed low to moderate correlation between samples (Pearson; H3K27Ac = 0.17, AR = 0.44, ATACseq = 0.42) (Figure 3B, top; Supplementary Table S6). However, when non-significant SNPs were filtered out we observed a marked increase in correlation among all comparisons (Pearson; H3K27Ac = 0.31, AR = 0.75, ATACseq = 0.80) (Figure 3B, bottom; Supplementary Table S6). Based on 48 hr sample, 33% (26/78) of significant bi-allelic SNPs were supported by one, 24% (19/78) by two, and 2% (2/78) were supported by all of the allelic-imbalance in vivo methods. Those two SNPs that were captured by all methodologies (rs13215402 and rs11083046) demonstrated parallel repression (Figure 3C, left) or activation (Figure 3C, right) of enhancer activity and allelic imbalance. Overall, the in vitro snpSTARRseq results broadly correlate with in vivo allelic imbalance but not in silico predictions, highlighting the need for experimental validation of non-coding variants.
DISCUSSION
The impact of genetic variants on protein-coding amino acid sequences is generally well understood. However, the diversity of activity greatly limits large-scale testing, as each protein requires a specialized assay. Paradoxically, while non-coding variants are poorly understood, the common activity of enhancer CREs makes them extremely amenable to high-throughput screening. In a single experiment, hundreds to thousands of non-coding variants can be functionally tested. Further, when combined with chromosome conformation capture methods these massively multi-parallel assays offer a promising approach to systematically characterize the mechanism of disease-associated non-coding SNPs. However, previous adaptations were either designed to identify new variants (48,49) or were prone to false-positives (47). Therefore, we optimized snpSTARRseq to functionally test non-coding genetic variants.
In this work, we utilized a larger insert fragment (400–600 bp; ∼543 bp) to maximize TF and co-regulator interactions on CREs. This is the longest average fragment used in comparable MPRA methodologies (Supplementary Table S12). While larger fragments (>600 bp) increase signal strength, they cannot be fully sequenced with standard short-read Illumina sequencing. To address this limitation, we modified existing Illumina technology to allow sequencing of DNA fragments lengths up to 850 bp using an asymmetric approach. Next, our design utilized a second-generation STARRseq plasmid, which provides a reduced background signal as compared to earlier reporter assays (69). Most importantly, by using a capture-based enrichment of specific genomic loci, we significantly increased the number of target fragments per genetic variant. This is critical as the unique number of plasmids can strongly influence Type-I error due to both position and insert size heterogeneity (Supplementary Figure S1B). To reduce these problems, we filtered all SNPs with less than 15 REF and ALT unique plasmids from our library. As a result, SNPs tested by our method had a minimum of 30 (REF + ALT) unique plasmids supported by >1000 mRNA reads. This is significantly higher than previous work (44,45,51,53), excluding one publication (45) (Supplementary Table S12; Figure S1A). This increased plasmid coverage dramatically reduces the overall noise caused by variable insert size (47). Overall, these modifications provide a robust platform for functional testing of GWAS hits.
With this approach, we targeted the bi-allelic enhancer activities of 252 PCa risk-associated SNPs from 184 non-overlapping regions that contain both a H3K27Ac mark and a chromatin loop to a gene promoter. Due to our very conservative threshold of REF + ALT plasmids (>30), we covered 35% (102/252) of PCa risk-associated variants. This could be improved with increased plasmid numbers during library generation. With this threshold, we observed that bi-allelic SNPs were highly concordant across multiple time points with minor differences between the earlier (24 h) and later (48 and 72 h) time points. Potentially, this may be due to cells reaching equilibrium in the later time points between snpSTARRseq mRNA transcription and degradation. From the PCa risk-associated SNPs tested we observed that 35% (36/102) had significantly altered enhancer activity. Many of these significant SNPs were located in 26 previously published PCa risk-associated loci (5). Moreover, 55% (20/36) of those with altered enhancer activity were associated with previously published eQTL (Supplementary Table S8). Consistent with the literature, we found supporting evidence for previously published SNPs that alter enhancer activity which impacts the expression of NKX3-1 (rs13215045 and rs11782388) (80–82), CTBP2 (rs4962416 and rs12769019), PCAT19 (rs11672691 and rs887391) and RGS17 (rs13215045, 6p25 RGS17 intron variant) genes (79).
We also compared the performance of snpSTARRseq to multiple in silico methodologies. While several of these approaches were designed to predict protein-coding mutations, we compared these results as we observed that in silico pathogenicity scores are commonly implemented for supporting enhancer variant annotation (80), GWAS prioritization (78,81,82) and driver gene calculation (83). This is particularly concerning as our experimental results did not strongly correlate with any in silico method. These differences could be potentially attributed to methodological differences (30) or a paucity of datasets that represent our experimental conditions. Specifically, there is an overall lack of representation of prostate models in public databases. For instance, ncER was trained on 38 databases that contain 9 targeted enhancer activity screens. However, none of these enhancer quantifications were based on PCa cell lines. Regardless of the cause, the low correlation between different in silico techniques (∼1%) suggests that there is a need for improved accuracy with these approaches (84). Recent studies have utilized semi-supervised methods to improve results by calibrating calculations and generating cell-type-specific predictions (85). For instance, MPRA datasets were incorporated to optimize feature weights for maximum tissue-specific separation (85,86). Contrasting these in silico methods, we observed a significant correlation between snpSTARRseq and clinical allelic imbalance of ATACseq, H3K27Ac and AR ChIPseq from tumor tissues. Overall, this supports the necessity of experimental validation of non-coding variants.
There are limitations to this methodology. Specifically, we missed 18% (47/252) of the targeted PCa risk-associated SNPs due to the low VAF of these variants in the DNA population. This can be easily overcome by increasing the number of plasmids during the cloning of the STARRseq library or genetic diversity of individuals in the DNA library (45). Further, as we are working with a pooled population, linkage disequilibrium (LD) makes GWAS traits to be harder to be finely mapped. Those events with low LD, conserved loci, and overlapping TF binding regions are more likely to be validated (87). To identify such complex events or multi-allelic events that are not present in the population, mutant enhancer sequences were previously generated either with saturation mutagenesis (29,88) or oligonucleotide synthesis (45,89). However, these methods lack control over the position of the mutations or lower enhancer activity due to the short fragment size. Shorter synthesized oligonucleotides can be used to characterize TF binding and co-regulator proteins on the target variants (90,91). For example, the binding affinity of TFs to oligonucleotides with reference and alternative alleles (26–40 bp) have been used to infer bi-allelic TF binding using SELEX (90,91). While there is little correlation between motifs and enhancer activity, these methods could be potentially incorporated to characterize the mechanism of variants identified from snpSTARRseq (41). Lastly, we did not characterize 36 INDELS as our snpSTARRseq computational pipeline exclusively focused on SNPs due to the ambiguity in INDELs calling (92).
Functionally characterization of non-coding variants is an emerging field, and we are now just beginning to learn the strengths and limitations of the various methodologies. For instance, in this work, 55% (20/36) of our PCa risk-associated variants were identified as eQTL. Given the availability of public databases (GTEx (93), eQTLgen (94)), these orthogonal results are important to validate our snpSTARRseq findings. However, eQTL-based studies also have limitations. For instance, eQTL studies only measure steady-state transcript levels. Consequently, the literature is now reporting that eQTLs explain only 11% of the heritability for an average trait (95,96) or up to 25% when transcription is profiled in disease-relevant tissue (97). Moreover, steady-state eQTLs are depleted near genes that are likely to contribute to complex phenotypes, including transcription factors, developmental genes, and highly conserved or essential genes (98). Finally, recent work demonstrated GWAS and eQTL studies are systematically biased toward different types of variants (99). Because of these limitations, there is a need for robust experimental approaches that can complement eQTL studies. In addition, the in vitro STARRseq can measure the activity of the enhancers generated by functional perturbations that can delineate complex regulatory mechanisms which is not possible in eQTL tissue-based approaches.
Herein, we developed snpSTARRSeq to improve the sensitivity and accuracy of large-scale non-coding enhancer assays. By increasing fragment length and reducing signal to noise, this approach can precisely identify functional variants. Potentially, the insert sizes could be further increased with long-read PacBio CCS sequencing (100,101). While not the goal of this work, genetic perturbations of snpSTARRseq systems could be used to identify how specific transcription factor binding is altered by SNPs. Further while focused on germline genetic variants, this same approach can be adopted to study somatic variants. Overall, snpSTARRseq offers an integrated experimental and computational approach to test the bi-allelic activity of hundreds to thousands of genetic variants in a single experiment.
DATA AVAILABILITY
We deposited the snpSTARRSeq computational framework at GitHub (https://github.com/mortunco/snp-starrseq). All visualization parameters and scripts could be found in publication-figures.rmd file in our repository. All datasets generated during this study along with other processed files are available at SRA under accession PRJNA791664.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Baraa Orabi and Fatih Karaoğlanoğlu for their contribution on the implementation of Calib and Snakemake, and Dr Yen-yi Lin for his useful discussions during manuscript preparation.
Contributor Information
Tunc Morova, Vancouver Prostate Centre, Vancouver, BC V6H 3Z6, Canada.
Yi Ding, Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Chia-Chi F Huang, Vancouver Prostate Centre, Vancouver, BC V6H 3Z6, Canada.
Funda Sar, Vancouver Prostate Centre, Vancouver, BC V6H 3Z6, Canada.
Tommer Schwarz, Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Claudia Giambartolomei, Central RNA Lab, Istituto Italiano di Tecnologia, Genova 16163, Italy; Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Sylvan C Baca, Department of Medical Oncology, The Center for Functional Cancer Epigenetics, Dana Farber Cancer Institute, Boston, MA 02215, USA.
Dennis Grishin, Department of Medical Oncology, The Center for Functional Cancer Epigenetics, Dana Farber Cancer Institute, Boston, MA 02215, USA.
Faraz Hach, Vancouver Prostate Centre, Vancouver, BC V6H 3Z6, Canada; Department of Urologic Science, University of British Columbia, Vancouver, BC V5Z 1M9, Canada.
Alexander Gusev, Department of Medical Oncology, The Center for Functional Cancer Epigenetics, Dana Farber Cancer Institute, Boston, MA 02215, USA; Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA 02115, USA.
Matthew L Freedman, Department of Medical Oncology, The Center for Functional Cancer Epigenetics, Dana Farber Cancer Institute, Boston, MA 02215, USA; The Center for Cancer Genome Discovery, Dana Farber Cancer Institute, Boston, MA 02215, USA.
Bogdan Pasaniuc, Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Computational Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Nathan A Lack, Vancouver Prostate Centre, Vancouver, BC V6H 3Z6, Canada; Department of Urologic Science, University of British Columbia, Vancouver, BC V5Z 1M9, Canada; School of Medicine, Koç University, Istanbul 34450, Turkey; Koç University Research Centre for Translational Medicine (KUTTAM), Koç University, Rumelifeneri Yolu, Istanbul 34450, Turkey.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
TUBITAK 1001 [119Z279]; Turkish Science Academy's Young Scientist Award Program (BAGEP). Funding for open access charge: Koç University School of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1. Pairo-Castineira E., Clohisey S., Klaric L., Bretherick A.D., Rawlik K., Pasko D., Walker S., Parkinson N., Fourman M.H., Russell C.D.et al.. Genetic mechanisms of critical illness in COVID-19. Nature. 2021; 591:92–98. [DOI] [PubMed] [Google Scholar]
- 2. Freedman M.L., Monteiro A.N.A., Gayther S.A., Coetzee G.A., Risch A., Plass C., Casey G., De Biasi M., Carlson C., Duggan D.et al.. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011; 43:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schumacher F.R., Berndt S.I., Siddiq A., Jacobs K.B., Wang Z., Lindstrom S., Stevens V.L., Chen C., Mondul A.M., Travis R.C.et al.. Genome-wide association study identifies new prostate cancer susceptibility loci. Hum. Mol. Genet. 2011; 20:3867–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al Olama A.A., Kote-Jarai Z., Berndt S.I., Conti D.V., Schumacher F., Han Y., Benlloch S., Hazelett D.J., Wang Z., Saunders E.et al.. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat. Genet. 2014; 46:1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schumacher F.R., Al Olama A.A., Berndt S.I., Benlloch S., Ahmed M., Saunders E.J., Dadaev T., Leongamornlert D., Anokian E., Cieza-Borrella C.et al.. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018; 50:928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hazelett D.J., Rhie S.K., Gaddis M., Yan C., Lakeland D.L., Coetzee S.G., consortium Ellipse/GAME-ON, consortium Practical, Henderson B.E., Noushmehr H.et al.. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014; 10:e1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pomerantz M.M., Freedman M.L.. The genetics of cancer risk. Cancer J. 2011; 17:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A.. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smigielski E.M., Sirotkin K., Ward M., Sherry S.T.. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000; 28:352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qian Y., Zhang L., Cai M., Li H., Xu H., Yang H., Zhao Z., Rhie S.K., Farnham P.J., Shi J.et al.. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 2019; 5:eaaw6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cong Z., Li Q., Yang Y., Guo X., Cui L., You T.. The SNP of rs6854845 suppresses transcription via the DNA looping structure alteration of super-enhancer in colon cells. Biochem. Biophys. Res. Commun. 2019; 514:734–741. [DOI] [PubMed] [Google Scholar]
- 12. Wasserman N.F., Aneas I., Nobrega M.A.. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010; 20:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kandaswamy R., Sava G.P., Speedy H.E., Beà S., Martín-Subero J.I., Studd J.B., Migliorini G., Law P.J., Puente X.S., Martín-García D.et al.. Genetic predisposition to chronic lymphocytic leukemia is mediated by a BMF super-enhancer polymorphism. Cell Rep. 2016; 16:2061–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hua J.T., Ahmed M., Guo H., Zhang Y.. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell. 2018; 174:564–575. [DOI] [PubMed] [Google Scholar]
- 15. Panigrahi A., O’Malley B.W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 2021; 22:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morova T., McNeill D.R., Lallous N., Gönen M., Dalal K., Wilson D.M. 3rd, Gürsoy A., Keskin Ö., Lack N.A.. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 2020; 11:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou S., Hawley J.R., Soares F., Grillo G., Teng M., Tonekaboni S.A.M., Hua J.T., Kron K.J., Mazrooei P., Ahmed M.et al.. Noncoding mutations target cis-regulatory elements of the FOXA1 plexus in prostate cancer. Nat. Commun. 2020; 11:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pomerantz M.M., Ahmadiyeh N., Jia L., Herman P., Verzi M.P., Doddapaneni H., Beckwith C.A., Chan J.A., Hills A., Davis M.et al.. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009; 41:882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takayama K.-I., Suzuki T., Fujimura T., Urano T., Takahashi S., Homma Y., Inoue S.. CtBP2 modulates the androgen receptor to promote prostate cancer progression. Cancer Res. 2014; 74:6542–6553. [DOI] [PubMed] [Google Scholar]
- 20. Gao P., Xia J.-H., Sipeky C., Dong X.-M., Zhang Q., Yang Y., Zhang P., Cruz S.P., Zhang K., Zhu J.et al.. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell. 2018; 174:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spisák S., Lawrenson K., Fu Y., Csabai I., Cottman R.T., Seo J.-H., Haiman C., Han Y., Lenci R., Li Q.et al.. CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat. Med. 2015; 21:1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Y.A., Chang M.M., Skanderup A.J.. MutSpot: detection of non-coding mutation hotspots in cancer genomes. NPJ Genom Med. 2020; 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wells A., Heckerman D., Torkamani A., Yin L., Sebat J., Ren B., Telenti A., di Iulio J.. Ranking of non-coding pathogenic variants and putative essential regions of the human genome. Nat. Commun. 2019; 10:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abramov S., Boytsov A., Bykova D., Penzar D.D., Yevshin I., Kolmykov S.K., Fridman M.V., Favorov A.V., Vorontsov I.E., Baulin E.et al.. Landscape of allele-specific transcription factor binding in the human genome. Nat. Commun. 2021; 12:2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S., Beer M.A.. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015; 47:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drubay D., Gautheret D., Michiels S.. A benchmark study of scoring methods for non-coding mutations. Bioinformatics. 2018; 34:1635–1641. [DOI] [PubMed] [Google Scholar]
- 28. Liu L., Sanderford M.D., Patel R., Chandrashekar P., Gibson G., Kumar S.. Biological relevance of computationally predicted pathogenicity of noncoding variants. Nat. Commun. 2019; 10:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kircher M., Xiong C., Martin B., Schubach M., Inoue F., Bell R.J.A., Costello J.F., Shendure J., Ahituv N.. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 2019; 10:3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Z., Zhao G., Li B., Fang Z., Chen Q., Wang X., Luo T., Wang Y., Zhou Q., Li K.et al.. Performance comparison of computational methods for the prediction of the function and pathogenicity of non-coding variants. Genomics Proteomics Bioinformatics. 2022; 7:S1672-0229(22)00016-X. [DOI] [PubMed] [Google Scholar]
- 31. Kasowski M., Kyriazopoulou-Panagiotopoulou S., Grubert F., Zaugg J.B., Kundaje A., Liu Y., Boyle A.P., Zhang Q.C., Zakharia F., Spacek D.V.et al.. Extensive variation in chromatin states across humans. Science. 2013; 342:750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McVicker G., van de Geijn B., Degner J.F., Cain C.E., Banovich N.E., Raj A., Lewellen N., Myrthil M., Gilad Y., Pritchard J.K.. Identification of genetic variants that affect histone modifications in human cells. Science. 2013; 342:747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng Z., Vermeulen M., Rollins-Green M., DeVeale B., Babak T.. Cis-regulatory mutations with driver hallmarks in major cancers. Iscience. 2021; 24:102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schizophrenia Working Group of the Psychiatric Genomics Consortium Gusev A., Mancuso N., Won H., Kousi M., Finucane H.K., Reshef Y., Song L., Safi A., McCarroll S.et al.. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 2018; 50:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Project Consortium ENCODE, Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G. Jr, Kinney J.B.et al.. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012; 30:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnold C.D., Gerlach D., Stelzer C., Boryń Ł.M., Rath M., Stark A.. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013; 339:1074–1077. [DOI] [PubMed] [Google Scholar]
- 38. Zacher B., Michel M., Schwalb B., Cramer P., Tresch A., Gagneur J.. Accurate promoter and enhancer identification in 127 ENCODE and roadmap epigenomics cell types and tissues by GenoSTAN. PLoS One. 2017; 12:e0169249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang T., Zhang Z., Dong Q., Xiong J., Zhu B.. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020; 21:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inoue F., Kircher M., Martin B., Cooper G.M., Witten D.M., McManus M.T., Ahituv N., Shendure J.. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017; 27:38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang C.-C.F., Lingadahalli S., Morova T., Ozturan D., Hu E., Yu I.P.L., Linder S., Hoogstraat M., Stelloo S., Sar F.et al.. Functional mapping of androgen receptor enhancer activity. Genome Biol. 2021; 22:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patwardhan R.P., Lee C., Litvin O., Young D.L., Pe’er D., Shendure J.. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009; 27:1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vockley C.M., Guo C., Majoros W.H., Nodzenski M., Scholtens D.M., Hayes M.G., Lowe W.L. Jr, Reddy T.E. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 2015; 25:1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ulirsch J.C., Nandakumar S.K., Wang L., Giani F.C., Zhang X., Rogov P., Melnikov A., McDonel P., Do R., Mikkelsen T.S.et al.. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell. 2016; 165:1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tewhey R., Kotliar D., Park D.S., Liu B., Winnicki S., Reilly S.K., Andersen K.G., Mikkelsen T.S., Lander E.S., Schaffner S.F.et al.. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016; 165:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ernst J., Melnikov A., Zhang X., Wang L., Rogov P., Mikkelsen T.S., Kellis M.. Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat. Biotechnol. 2016; 34:1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu S., Liu Y., Zhang Q., Wu J., Liang J., Yu S., Wei G.-H., White K.P., Wang X.. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017; 18:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., He L., Goggin S.M., Saadat A., Wang L., Sinnott-Armstrong N., Claussnitzer M., Kellis M.. High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nat. Commun. 2018; 9:5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang P., Xia J.-H., Zhu J., Gao P., Tian Y.-J., Du M., Guo Y.-C., Suleman S., Zhang Q., Kohli M.et al.. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018; 9:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Klein J.C., Keith A., Rice S.J., Shepherd C., Agarwal V., Loughlin J., Shendure J.. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019; 10:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choi J., Zhang T., Vu A., Ablain J., Makowski M.M., Colli L.M., Xu M., Hennessey R.C., Yin J., Rothschild H.et al.. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 2020; 11:2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abell N.S., DeGorter M.K., Gloudemans M.J., Greenwald E., Smith K.S., He Z., Montgomery S.B.. Multiple causal variants underlie genetic associations in humans. Science. 2022; 375:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weiss C.V., Harshman L., Inoue F., Fraser H.B., Petrov D.A., Ahituv N., Gokhman D.. The cis-regulatory effects of modern human-specific variants. Elife. 2021; 10:e63713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yáñez-Cuna J.O., Kvon E.Z., Stark A.. Deciphering the transcriptional cis-regulatory code. Trends Genet. 2013; 29:11–22. [DOI] [PubMed] [Google Scholar]
- 55. Klein J.C., Agarwal V., Inoue F., Keith A., Martin B., Kircher M., Ahituv N., Shendure J.. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods. 2020; 17:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Giambartolomei C., Seo J.-H., Schwarz T., Freund M.K., Johnson R.D., Spisak S., Baca S.C., Gusev A., Mancuso N., Pasaniuc B.et al.. H3K27ac HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. J. Hum. Genet. 2021; 108:2284–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Y., Yu S., Dhiman V.K., Brunetti T., Eckart H., White K.P.. Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol. 2017; 18:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carpen J.D., Archer S.N., Skene D.J., Smits M., von Schantz M.. A single-nucleotide polymorphism in the 5’-untranslated region of the hPER2 gene is associated with diurnal preference. J. Sleep Res. 2005; 14:293–297. [DOI] [PubMed] [Google Scholar]
- 59. MacConaill L.E., Burns R.T., Nag A., Coleman H.A., Slevin M.K., Giorda K., Light M., Lai K., Jarosz M., McNeill M.S.et al.. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genomics. 2018; 19:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Orabi B., Erhan E., McConeghy B., Volik S.V., Le Bihan S., Bell R., Collins C.C., Chauve C., Hach F.. Alignment-free clustering of UMI tagged DNA molecules. Bioinformatics. 2019; 35:1829–1836. [DOI] [PubMed] [Google Scholar]
- 61. Bushnell B., Rood J., Singer E.. BBMerge – accurate paired shotgun read merging via overlap. PLoS One. 2017; 12:e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Venables W.N., Ripley B.D.. Modern applied statistics with S. 2002; NY: Springer. [Google Scholar]
- 63. Yang H., Chen R., Wang Q., Wei Q., Ji Y., Zheng G., Zhong X., Cox N.J., Li B.. De novo pattern discovery enables robust assessment of functional consequences of non-coding variants. Bioinformatics. 2019; 35:1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huang Y.-F., Gulko B., Siepel A.. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017; 49:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Baca S.C., Singler C., Zacharia S., Seo J.-H., Morova T., Hach F., Ding Y., Schwarz T., Huang C.-C.F., Anderson J.et al.. Genetic determinants of chromatin reveal prostate cancer risk mediated by context-dependent gene regulation. Nat. Genet. 2022; 54:1364–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W.et al.. The chromatin accessibility landscape of primary human cancers. Science. 2018; 362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giambartolomei C., Seo J.-H., Schwarz T., Freund M.K., Johnson R.D., Spisak S., Baca S.C., Gusev A., Mancuso N., Pasaniuc B.et al.. H3k27ac-HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. J. Hum. Genet. 2021; 108:2284–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thibodeau S.N., French A.J., McDonnell S.K., Cheville J., Middha S., Tillmans L., Riska S., Baheti S., Larson M.C., Fogarty Z.et al.. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nat. Commun. 2015; 6:8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Muerdter F., Boryń Ł.M., Woodfin A.R., Neumayr C., Rath M., Zabidi M.A., Pagani M., Haberle V., Kazmar T., Catarino R.R.et al.. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat. Methods. 2018; 15:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee D., Shi M., Moran J., Wall M., Zhang J., Liu J., Fitzgerald D., Kyono Y., Ma L., White K.P.et al.. STARRPeaker: uniform processing and accurate identification of STARR-seq active regions. Genome Biol. 2020; 21:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Song L.-F., Deng Z.-H., Gong Z.-Y., Li L.-L., Li B.-Z.. Large-scale de novo oligonucleotide synthesis for whole-genome synthesis and data storage: challenges and opportunities. Front. Bioeng. Biotechnol. 2021; 9:689797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Palluk S., Arlow D.H., de Rond T., Barthel S., Kang J.S., Bector R., Baghdassarian H.M., Truong A.N., Kim P.W., Singh A.K.et al.. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018; 36:645–650. [DOI] [PubMed] [Google Scholar]
- 73. Vockley C.M., D’Ippolito A.M., McDowell I.C., Majoros W.H., Safi A., Song L., Crawford G.E., Reddy T.E. Direct GR binding sites potentiate clusters of TF binding across the human genome. Cell. 2016; 166:1269–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Giambartolomei C., Zhenli Liu J., Zhang W., Hauberg M., Shi H., Boocock J., Pickrell J., Jaffe A.E., Consortium CommonMind, Pasaniuc B.et al.. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018; 34:2538–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W.J.H., Jansen R., de Geus E.J.C., Boomsma D.I., Wright F.A.et al.. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016; 48:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Song H., Zhang B., Watson M.A., Humphrey P.A., Lim H., Milbrandt J.. Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene. 2009; 28:3307–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chalmers Z.R., Connelly C.F., Fabrizio D., Gay L., Ali S.M., Ennis R., Schrock A., Campbell B., Shlien A., Chmielecki J.et al.. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jang Y.J., LaBella A.L., Feeney T.P., Braverman N., Tuchman M., Morizono H., Ah Mew N., Caldovic L.. Disease-causing mutations in the promoter and enhancer of the ornithine transcarbamylase gene. Hum. Mutat. 2018; 39:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Han Y., Hazelett D.J., Wiklund F., Schumacher F.R., Stram D.O., Berndt S.I., Wang Z., Rand K.A., Hoover R.N., Machiela M.J.et al.. Integration of multiethnic fine-mapping and genomic annotation to prioritize candidate functional SNPs at prostate cancer susceptibility regions. Hum. Mol. Genet. 2015; 24:5603–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Claringbould A., Zaugg J.B.. Enhancers in disease: molecular basis and emerging treatment strategies. Trends Mol. Med. 2021; 27:1060–1073. [DOI] [PubMed] [Google Scholar]
- 81. Lee J., Suh Y., Jeong H., Kim G.-H., Byeon S.H., Han J., Lim H.T.. Aberrant expression of PAX6 gene associated with classical aniridia: identification and functional characterization of novel noncoding mutations. J. Hum. Genet. 2021; 66:333–338. [DOI] [PubMed] [Google Scholar]
- 82. Watanabe K., Taskesen E., van Bochoven A., Posthuma D.. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017; 8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shuai S.PCAWG Drivers and Functional Interpretation Working Group PCAWG Consortium PCAWG Drivers and Functional Interpretation Working Group Gallinger S., Stein L.. Combined burden and functional impact tests for cancer driver discovery using DriverPower. Nat. Commun. 2020; 11:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li J., Drubay D., Michiels S., Gautheret D.. Mining the coding and non-coding genome for cancer drivers. Cancer Lett. 2015; 369:307–315. [DOI] [PubMed] [Google Scholar]
- 85. He Z., Liu L., Wang K., Ionita-Laza I.. A semi-supervised approach for predicting cell-type specific functional consequences of non-coding variation using MPRAs. Nat. Commun. 2018; 9:5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dong S., Boyle A.P.. Prioritization of regulatory variants with tissue-specific function in the non-coding regions of human genome. Nucleic Acids Res. 2022; 50:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gorlova O.Y., Xiao X., Tsavachidis S., Amos C.I., Gorlov I.P.. SNP characteristics and validation success in genome wide association studies. Hum. Genet. 2022; 141:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kvon E.Z., Zhu Y., Kelman G., Novak C.S., Plajzer-Frick I., Kato M., Garvin T.H., Pham Q., Harrington A.N., Hunter R.D.et al.. Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell. 2020; 180:1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schöne S., Bothe M., Einfeldt E., Borschiwer M., Benner P., Vingron M., Thomas-Chollier M., Meijsing S.H.. Synthetic STARR-seq reveals how DNA shape and sequence modulate transcriptional output and noise. PLoS Genet. 2018; 14:e1007793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yan J., Qiu Y., Ribeiro Dos Santos A.M., Yin Y., Li Y.E., Vinckier N., Nariai N., Benaglio P., Raman A., Li X.et al.. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021; 591:147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bray D., Hook H., Zhao R., Keenan J.L., Penvose A., Osayame Y., Mohaghegh N., Chen X., Parameswaran S., Kottyan L.C.et al.. CASCADE: high-throughput characterization of regulatory complex binding altered by non-coding variants. Cell Genom. 2022; 2:100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang N., Lysenkov V., Orte K., Kairisto V., Aakko J., Khan S., Elo L.L.. Tool evaluation for the detection of variably sized indels from next generation whole genome and targeted sequencing data. PLoS Comput. Biol. 2022; 18:e1009269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. The GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S.et al.. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021; 53:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Umans B.D., Battle A., Gilad Y.. Where are the disease-associated eQTLs?. Trends Genet. 2021; 37:109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yao D.W., O’Connor L.J., Price A.L., Gusev A. Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet. 2020; 52:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chun S., Casparino A., Patsopoulos N.A., Croteau-Chonka D.C., Raby B.A., De Jager P.L., Sunyaev S.R., Cotsapas C.. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet. 2017; 49:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang X., Goldstein D.B.. Enhancer domains predict gene pathogenicity and inform gene discovery in complex disease. Am. J. Hum. Genet. 2020; 106:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mostafavi H., Spence J.P., Naqvi S., Pritchard J.K.. Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. 2022; bioRxiv doi:08 May 2022, preprint: not peer reviewed 10.1101/2022.05.07.491045. [DOI]
- 100. Ardui S., Ameur A., Vermeesch J.R., Hestand M.S.. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018; 46:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D.et al.. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019; 37:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We deposited the snpSTARRSeq computational framework at GitHub (https://github.com/mortunco/snp-starrseq). All visualization parameters and scripts could be found in publication-figures.rmd file in our repository. All datasets generated during this study along with other processed files are available at SRA under accession PRJNA791664.