

Abstract
Glucagon-like peptide-1 receptor (GLP1R) agonists and dipeptidyl peptidase 4 inhibitors are widely prescribed diabetes drugs due to their ability to stimulate insulin secretion from remaining β cells and to reduce caloric intake. Unfortunately, they fail to increase human β cell proliferation. Small-molecule inhibitors of dual-specificity tyrosine-regulated kinase 1A (DYRK1A) are able to induce adult human β cell proliferation, but rates are modest (~2%), and their specificity to β cells is limited. Here, we provide evidence that combining any member of the GLP1R agonist class with any member of the DYRK1A inhibitor class induces a synergistic increase in human β cell replication (5 to 6%) accompanied by an actual increase in numbers of human β cells. GLP1R agonist–DYRK1A inhibitor synergy required combined inhibition of DYRK1A and an increase in cAMP and did not lead to β cell dedifferentiation. These beneficial effects on proliferation were seen in both normal human β cells and β cells derived from individuals with type 2 diabetes. The ability of the GLP1R agonist–DYRK1A inhibitor combination to enhance human β cell proliferation, human insulin secretion, and blood glucose control extended in vivo to studies of human islets transplanted into euglycemic and streptozotocin-diabetic immunodeficient mice. No adverse events were observed in the mouse studies during a 1-week period. Because of the relative β cell specificity of GLP1R agonists, the combination provides an improved, although not complete, degree of human β cell specificity.
INTRODUCTION
Diabetes affects 422 million people globally, is increasing in prevalence (1), and results entirely or in part from inadequate numbers of functional, insulin-producing pancreatic β cells (2, 3): β Cell mass is reduced by 40 to 60% in type 2 diabetes (T2D) and by 70 to 97% in type 1 diabetes (T1D). These observations underlie current attempts at β cell replacement and regeneration, including pancreas transplantation, islet transplantation, stem cell differentiation replacement strategies, and transdifferentiation or redifferentiation of non–β pancreatic islet cells into β cells. However, with the exception of pancreas transplants, none are clinically available, and none, including pancreas transplant, are scalable to the hundreds of millions of people with T1D and T2D.
Drugs that act directly or indirectly to activate the glucagon-like peptide-1 receptor (GLP1R) are among the most widely prescribed diabetes drugs globally, reaching millions of people with T2D. The GLP1R agonist family includes glucagon-like peptide-1 (GLP-1) (7–36) itself, the more stable reptilian homolog exendin-4, and modified exenatide analogs such as liraglutide, lixisenatide, and semaglutide (4, 5). These GLP1R agonists and additional agents that prevent degradation of endogenous GLP-1 by the enzyme dipeptidyl peptidase 4 (DPP4) [exemplified by sitagliptin, vildagliptin, saxagliptin, and others (4, 6)] induce proliferation in rodent β cells but fail to activate β cell replication in adult human islets (7, 8). Instead, their beneficial clinical effects on diabetes are derived from their “incretin” effect that induces β cells to accentuate insulin secretion when blood glucose is elevated (4) and from their satiety-inducing effects. Although GLP1R agonists fail to induce human β cell proliferation, GLP1R has a restricted tissue distribution in humans and is particularly highly expressed in β cells (9–11), thus providing a currently unique, if imperfect, degree of β cell specificity.
There are currently no commercially available drugs able to induce human β cells to replicate. However, a number of experimental human β cell regeneration molecules are under study, including osteoprotegerin and denosumab, parathyroid hormone–related protein, serpin B1, the peptide TLQP21, and γ-aminobutyric acid (9, 12–15). In general, β cell proliferation, or more accurately Ki67 or 5-bromo-2′-deoxyuridine (BrdU) β cell “labeling indices,” induced by these molecules is low, under 0.5 to 1%. Recently, the dual-specificity tyrosine phosphorylation–regulated kinase 1A (DYRK1A) inhibitor class of small-molecule drugs, which includes harmine (16), INDY (16), leucettine (17), GNF4877 (18), 5-iodotubericidin (5-IT) (19), CC-401 (20), and others (21, 22), has been shown to induce human β cell labeling indices in the 1 to 3% range. DYRK1A inhibitors induce proliferation in human β cells, at least in part, via their ability to induce nuclear translocation of nuclear factors of activated T cells (NFaTs), transcription factors that then transactivate cell cycle–activating genes and repress cell cycle inhibitor genes (16, 18–22). Most recently, the combination of a transforming growth factor–β (TGFβ) superfamily (TGFβSF) inhibitor with a DYRK1A inhibitor has been shown to further increase human β cell labeling indices into the 5 to 8% range (23). Although a substantial improvement over the DYRK1A inhibitors, this approach also suffers from a lack of β cell specificity as both DYRK1A and TGFβSF receptors and signaling molecules are ubiquitous. Thus, there is a pressing need to identify drugs that provide both accelerated human β cell proliferation and improved β cell specificity.
Recognizing this need and aware that GLP1R agonists have superior β cell specificity to existing compounds (4, 10, 11) and are safe and already in widespread use, we explored the efficacy of combining GLP1R agonists with DYRK1A inhibitors. We found that this combination is synergistic, providing the best features of both classes of compounds: enhanced human β cell proliferation combined with improved human β cell specificity.
RESULTS
Harmine and GLP-1 synergistically increase human β cell proliferation
Our initial goal was to determine whether a combination of a DYRK1A inhibitor with a GLP1R agonist might synergistically induce human β cells to replicate. We selected GLP-1 as a potential partner for a DYRK1A inhibitor due to its superior human β cell targeting specificity compared to DYRK1A inhibitors. We treated adult human cadaveric islets with vehicle, harmine, GLP-1(7–36)amide (referred to hereafter as “GLP-1”), or a combination thereof and assessed Ki67 immunolabeling in insulin+ cells dispersed from human islets (Fig. 1, A and B, and fig. S1A). As previously reported (4, 7, 8), GLP-1 had negligible effects on human β cell proliferation at a dose of 5 nM. As expected (16, 18, 19), 10 μM harmine induced proliferation in ~2% of human β cells. However, the combination of harmine together with GLP-1 induced a higher rate of proliferation in each of 20 human islet donors tested, averaging 5%. Figure 1A also illustrates the widely recognized variability in proliferation encountered among pancreatic islets from different organ donors (24). We combined 10 μM harmine with a range of doses of GLP-1 and observed a dose-related, progressive increase in proliferation (Fig. 1C and fig. S1C). Because of the variability of human islet responses and the limited numbers of human islets available, not every dose of GLP-1 could be tested in every human islet preparation. Accordingly, Ki67 labeling results in Fig. 1C were normalized to the proliferation response from the single harmine dose (Fig. 1D and fig. S1D). Displayed in this manner, the harmine–GLP-1 combination showed a clear and progressive increase in proliferation of human β cells as the GLP-1 dose increased. Subsequent figures also display data normalized by the harmine dose. Collectively, these experiments indicate that doses of harmine and GLP-1 that have no effect on their own synergize in a dose-related manner to increase adult human β cell proliferation.
Fig. 1. Combination of DYRK1A inhibitors with GLP1R agonists yields synergistic increases in human β cell proliferation.
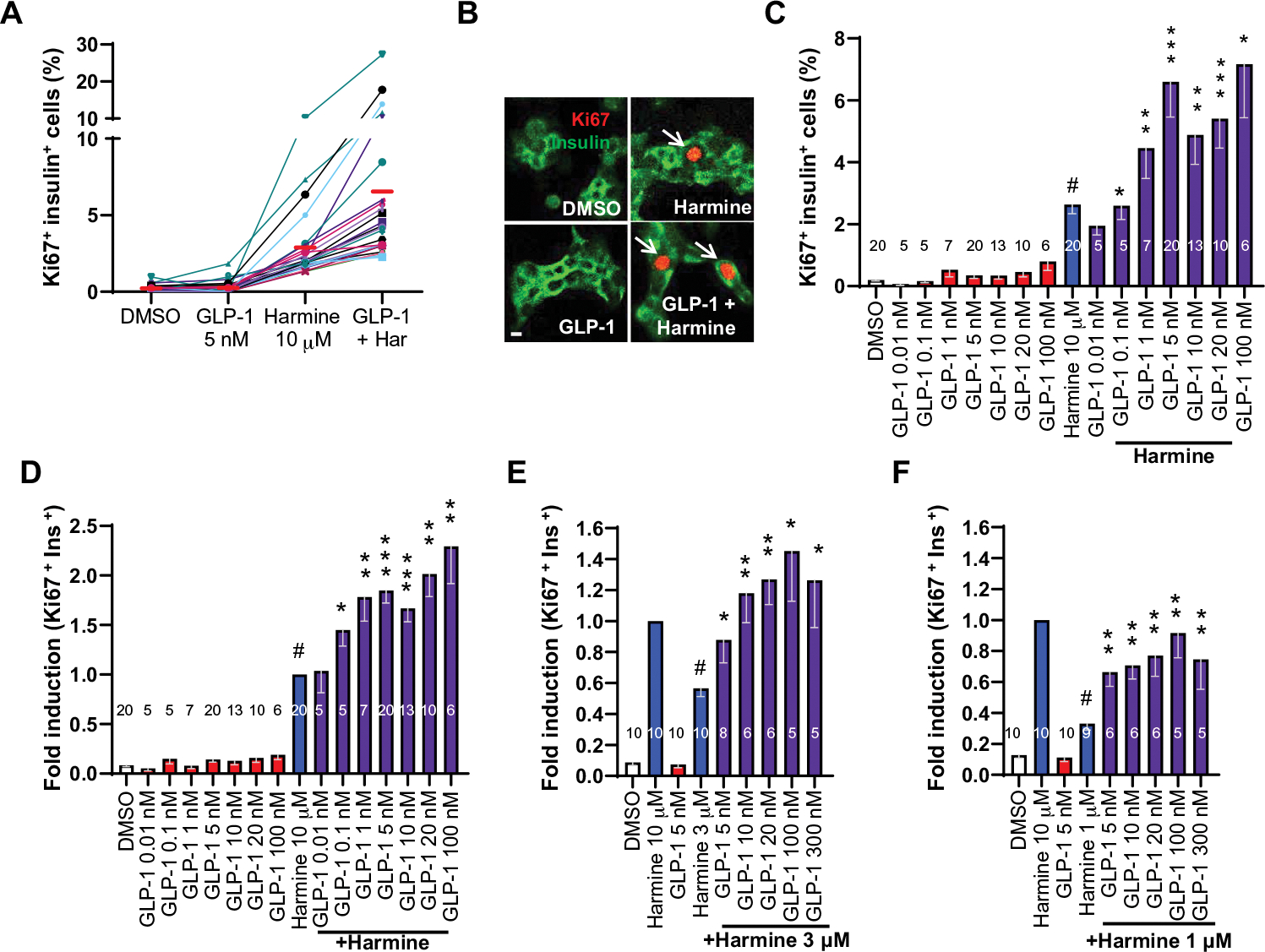
(A) Dispersed human islet cells from pancreas donors (n = 20) were treated for 96 hours with the drugs and doses indicated and immunolabeled for Ki67 and insulin. DMSO (dimethyl sulfoxide), control vehicle at 0.1%. Each colored line represents an islet preparation from one donor. Red solid bars indicate the means for each treatment condition in the 20 donors. Har, harmine. (B) An example of insulin-Ki67 coimmunolabeling on dispersed human islets under the treatment conditions indicated. Arrows illustrate Ki67+ β cells. Scale bar, 10 μm. (C) Bar graph of the 20 single-dose human islet experiments shown in (A), together with a broad range of additional doses of GLP-1 and the maximally effective mitogenic dose of harmine (10 μM). The large error bars reflect the intrinsic variability among human islet preparations shown in (A). (D) The same data (C) shown as fold difference with respect to induction by 10 μM harmine, expressed as “1.0” to adjust for the intrinsic variability in each human islet preparation. The 1.0 value for harmine reflects an actual Ki67 labeling value of 2.5%. (E and F) Comparison of 3 and 1 μM harmine to 10 μM harmine alone or in combination with a range of GLP-1 doses. Bars indicate means ± SEM. Numbers within bars indicate the number of human islet preparations studied at each dose. A minimum of 3000 β cells was counted for each bar. One-way ANOVA, #P < 0.05 versus control; *P < 0.05, **P < 0.01, and ***P < 0.001 versus harmine alone.
The maximally effective harmine dose for human β cell proliferation is 10 μM (16). The peak Ki67 labeling index was ~0.2% from GLP-1 alone (Fig. 1, C and D) and ~2% from 10 μM harmine (Fig. 1, C and D). In contrast, the labeling index from the harmine–GLP-1 combination was 5 to 6% (Fig. 1, C and D), substantially higher than observed with harmine alone, thereby fulfilling pharmacologic criteria for synergy. To explore this synergy further, we repeated these studies using lower doses of harmine, 3 and 1 μM, both individually and in combination with a range of GLP-1 doses (Fig. 1, E and F, and fig. S1, E and F). In each case, synergy was apparent, particularly at the 1 μM harmine dose. This dose had marginal effects on its own but, when combined with even trivial doses of GLP-1, induced β cell proliferation (2.7%) comparable to the maximally effective harmine dose, 10 μM (16). This suggests that doses of harmine that have no systemic effects on their own may induce β cell proliferation in individuals receiving GLP1R agonists.
Proliferative synergy is a class effect for DYRK1A inhibitors and GLP1R agonists
The synergistic ability of harmine to drive β cell proliferation in combination with GLP-1 was apparent for every DYRK1A inhibitor tested (harmine, INDY, leucettine, 5-IT, and GNF4877) (Fig. 2, A and B, and fig. S2, A and B). Thus, proliferative synergy with GLP-1 is likely a “class effect” for DYRK1A inhibitors in general. Conversely, synergy was also apparent for harmine in combination with all five GLP1R agonists tested, including several approved for clinical use: exendin-4, liraglutide, lixisenatide, and semaglutide (Fig. 2C and fig. S2C). This result indicates that mitogenic synergy with DYRK1A inhibitors is also a class effect for GLP1R agonists in general.
Fig. 2. The DYRK1A–GLP-1 increase in human β cell numbers is a class effect for both DYRK1A inhibitors and GLP1R agonists.
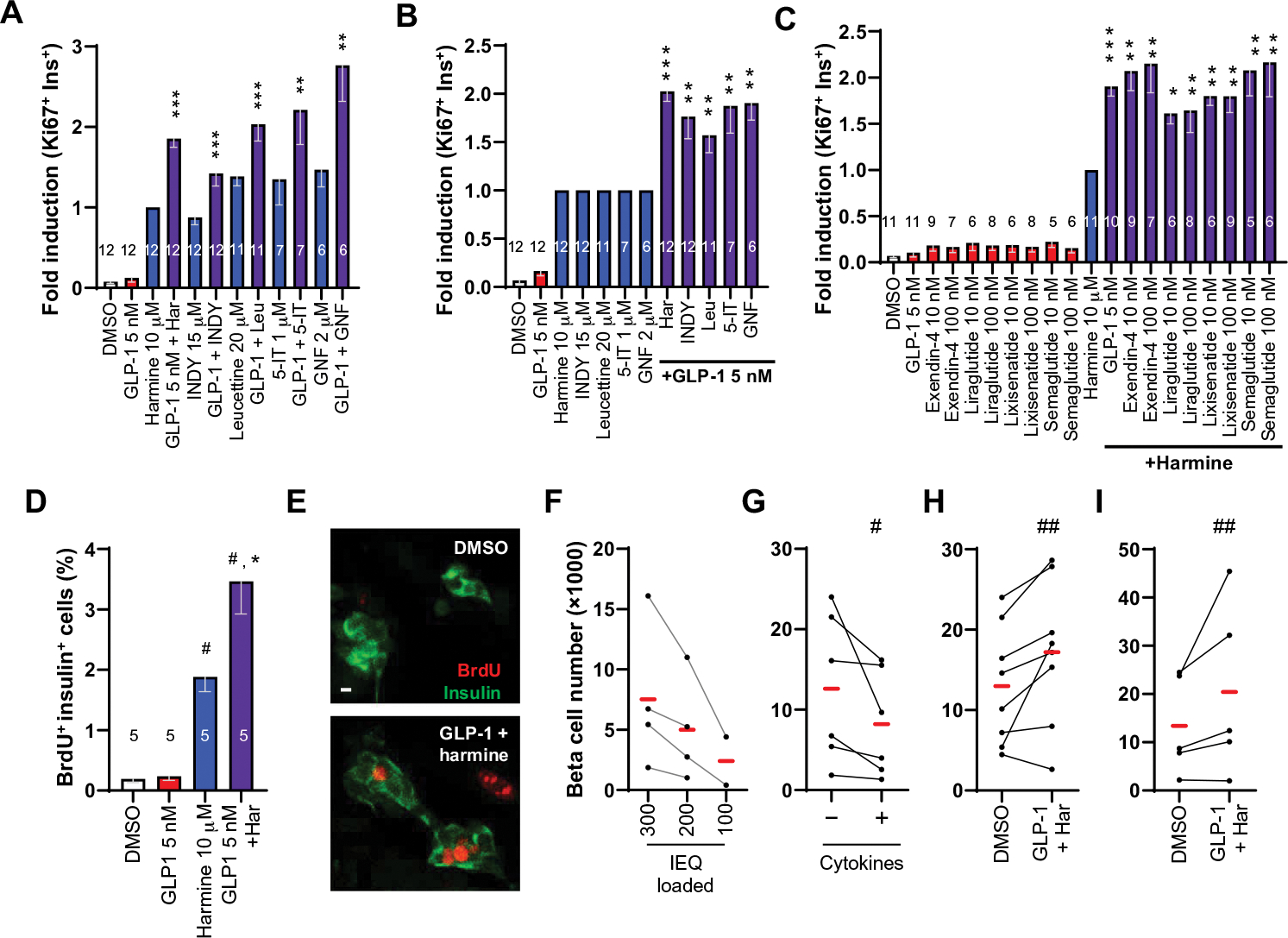
(A) The effects on human β cell on Ki67 labeling of harmine, INDY, leucettine-41 (leucettine or Leu), 5-iodotubericidin (5-IT), and GNF4877 (GNF) administered alone (blue bars) or in combination with 5 nM GLP-1 (purple bars). Results are normalized to 10 μM harmine. Ki67 percentage values were as follows: harmine, 2.1%; INDY, 1.7%; leucettine, 2.8%; 5-IT, 2.6%; GNF4877, 2.9%. (B) The same data in (A), but with each of the DYRK1A inhibitors normalized to 1.0. (C) Ki67 labeling in β cells after treatment of human islets with 10 μM harmine and a broad range of commercially available GLP-1 analogs. (D) Induction of BrdU immunolabeling in human β cells treated with harmine and GLP-1. (E) An example of BrdU incorporation in D. Scale bar, 10 μm. (F and G) Negative controls for quantification of human β cell numbers by flow cytometry in progressively lower numbers of human islets from four different donors and in response to cytokines (1000 IU/ml each, TNFα and IL-1β) in six pairs of different human islets. (H) Increases in human β cell numbers in islets from seven of eight human donors in response to 4 days of ex vivo treatment with harmine (10 μM)– GLP-1 (5 nM) or vehicle (0.1% DMSO). (I) Increases in GFP-labeled hESC-derived β-like cells in response to 7 days of combined harmine–GLP-1 treatment. Bars indicate means ± SEM. Numbers within the bars show the number of human islet preparations studied at each dose. A minimum of 3000 β cells was counted for each bar. One-way ANOVA, *P < 0.05, **P < 0.01, and ***P < 0.001 versus harmine alone; #P < 0.001 and ##P < 0.01 versus control. IEQ, islet equivalent.
Combined harmine and GLP-1 treatment increases β cell numbers
To independently confirm our results using Ki67 as a proliferation marker, we repeated the studies using a different proliferation marker, BrdU, which also revealed a synergistic increase in the labeling index (Fig. 2, D and E, and fig. S2D). Although widely used as markers for proliferation, Ki67 and BrdU also can mark DNA repair and DNA damage and, therefore, do not unequivocally prove that new cells were generated. As a more rigorous test, we developed a human β cell quantitative assay (23) wherein human islets are dispersed, β cells are labeled using an insulin promoter–driven ZsGreen fluorescent protein–expressing adenovirus (Ad.RIP-ZsGreen) (25), and ZsGreen-labeled β cells are quantified using fluorescence-activated cell sorting (FACS) with an internal microbead recovery standard to correct for β cell loading and recovery. To generate a standard curve, we labeled β cells in 300, 200, and 100 islet equivalents (IEQs) from four donors with Ad.RIP1-ZsGreen (Fig. 2F). We observed that recovered β cell numbers declined as expected in each of the four human samples. As a negative control, we treated human islets with cytokines, tumor necrosis factor–α (TNFα), and interleukin-1β (IL-1β), at doses known to induce β cell death (26). Again, we observed the expected decline in human β cell numbers in all six islet preparations (Fig. 2G). To determine whether the increase in Ki67 and BrdU labeling translated into authentic increases in human β cell numbers, we treated dispersed Ad.RIP-ZsGreen–transduced human islets with harmine–GLP-1 and observed that actual numbers of human β cells increased by an average of 40% in seven of eight donors tested over the 4 days of the experiment (Fig. 2H). Concerned that the Ad.RIP-ZsGreen transduction might affect β cell survival, proliferation, or cell numbers, we turned to human embryonic stem cells (hESCs) containing a green fluorescent protein (GFP) cassette knocked into one allele of the insulin locus (27) and differentiated these into β-like cells (28). As with the adult β cells, we observed an average 50% increase in the numbers of juvenile hESC-derived β cells in each of four experiments over the 7 days of treatment (Fig. 2I).
To explore potential β cell death in response to harmine–GLP-1 treatment of dispersed human islets, we used a terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay with insulin coimmunolabeling (Fig. 3, A and B, and fig. S3A). A positive control cytokine cocktail of TNFα and IL-1β induced a significant (P < 0.001) increase in β cell TUNEL labeling, as expected (26). In contrast, the harmine–GLP-1 combination did not increase β cell TUNEL labeling. We also found no evidence of a DNA damage in response to harmine–GLP-1 using γH2AX immunocytochemistry (Fig. 3, C and D, and fig. S3C).
Fig. 3. The effects of harmine–GLP-1 on proliferation in non–β cells, β cell death, and DNA damage.
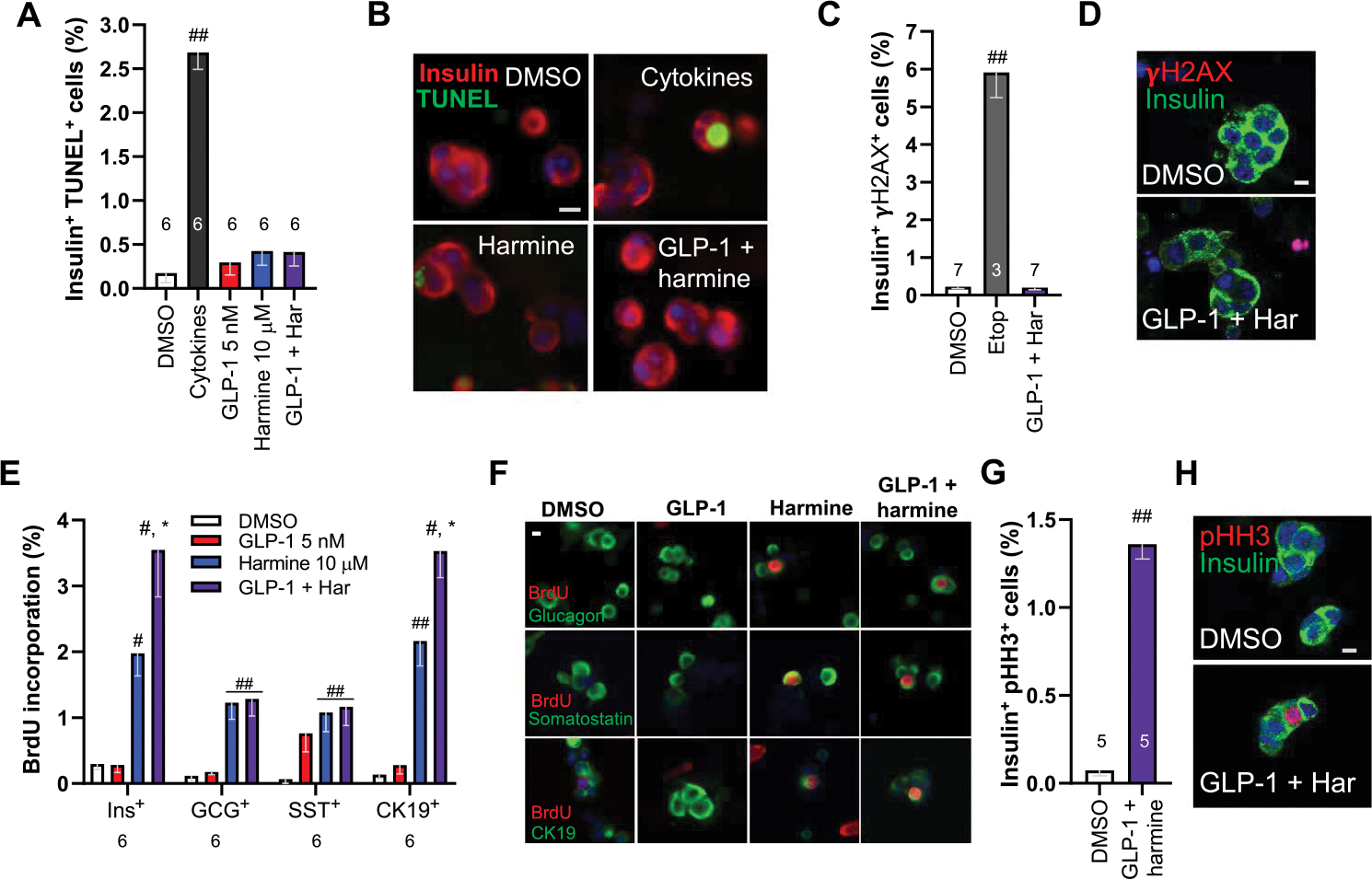
(A) The effects of harmine–GLP-1 on cell death as assessed by TUNEL assay. The cytokine cocktail in the second bar is a positive control containing TNFα and IL-1β. The numbers of islet donors are indicated within the bars. Examples of TUNEL responses under the conditions shown in (B). A minimum of 3000 β cells were counted for each bar. (C) DNA damage as assessed by γH2AX immunocytochemistry in response to harmine (10 μM) and GLP-1 (5 nM) or positive control etoposide (Etop) (20 μM). A minimum of 3000 β cells were counted for each bar. (D) An example of γH2AX immunocytochemistry. (E) Proliferation as assessed using BrdU labeling in β (INS+ ), α (GCG+ ), δ (SST+ ), and ductal (CK19+ ) cells in response to the treatments shown in the inset. Numbers of cells counted for each bar ranged from 2820 to 4060 for β cells, 2935 to 3575 for α cells, 1050 to 1220 for δ cells, and 2040 to 2315 for CK19+ cells. (F) Examples of BrdU immunolabeling in human islet cell subtypes in response to the agents shown. (G) Immunolabeling of dispersed human β cells for pHH3 and insulin. (H) An example of pHH3-insulin colabeling in dispersed human β cells. All bars indicate means ± SEM. Numbers above or below the bars indicate the sample size of human islet donors. #P < 0.01 and ##P < 0.001 versus control by two-tailed paired t test. *P < 0.01 versus harmine alone by paired t test. Scale bars, 10 μm (B, D, F, and H)
To explore the degree of β cell specificity of the proliferative response, we examined BrdU immunolabeling in β cells and other islet cell types (Fig. 3, E and F, and fig. S3E). GLP-1 alone had no effect on β, α, or ductal (CK19+) cells and a small but nonsignificant effect on δ cells. Harmine induced proliferation in all four islet cell types studied, as previously reported (16). Harmine–GLP-1 further increased proliferation in β and CK19+ cells, the two islet cell types that contain the GLP1R. To determine whether the harmine–GLP-1 combination induced β cell proliferation, we colabeled β cells for insulin and phospho-histone H3 (pHH3) and observed increased rates of β cell pHH3 immunolabeling (Fig. 3, G and H, and fig. S3G). The activation pHH3 in addition to Ki67 and BrdU indicates that all phases of the cell cycle were activated.
Harmine–GLP-1 synergy requires inhibition of DYRK1A
We silenced DYRK1A in dispersed human islets and treated them with GLP-1 or appropriate controls (Fig. 4A and fig. S4, A and E). In human islets treated with a control Ad.shRNA, adenovirus expressing a scrambled short hairpin–mediated RNA (shRNA), the combination of harmine and GLP-1 induced an increase in β cell proliferation in excess of that observed with harmine or GLP-1 alone, as expected. This could be fully mimicked by replacing harmine with an Ad.shDYRK1A adenovirus that silences DYRK1A. Conversely, Ad.DYRK1A overexpression markedly attenuated the proliferation induced by harmine or the harmine–GLP-1 combination (Fig. 4, B to D, and fig. S4, B, F, and G), whereas a control adenovirus expressing Cre recombinase had no effect on proliferation of GLP-1, harmine, or their combination. Collectively, these studies indicate that synergistic effects of GLP-1 with pharmacologic DYRK1A inhibition can be fully mimicked by genetic silencing of DYRK1A in the presence of GLP-1 and, conversely, can be blocked by genetic addition of excess DYRK1A. Thus, the harmine component of the proliferative synergy is mediated by, and requires, DYRK1A inhibition.
Fig. 4. Evaluation of the role of DYRK1A and cAMP signaling in harmine–GLP1R agonist synergy.
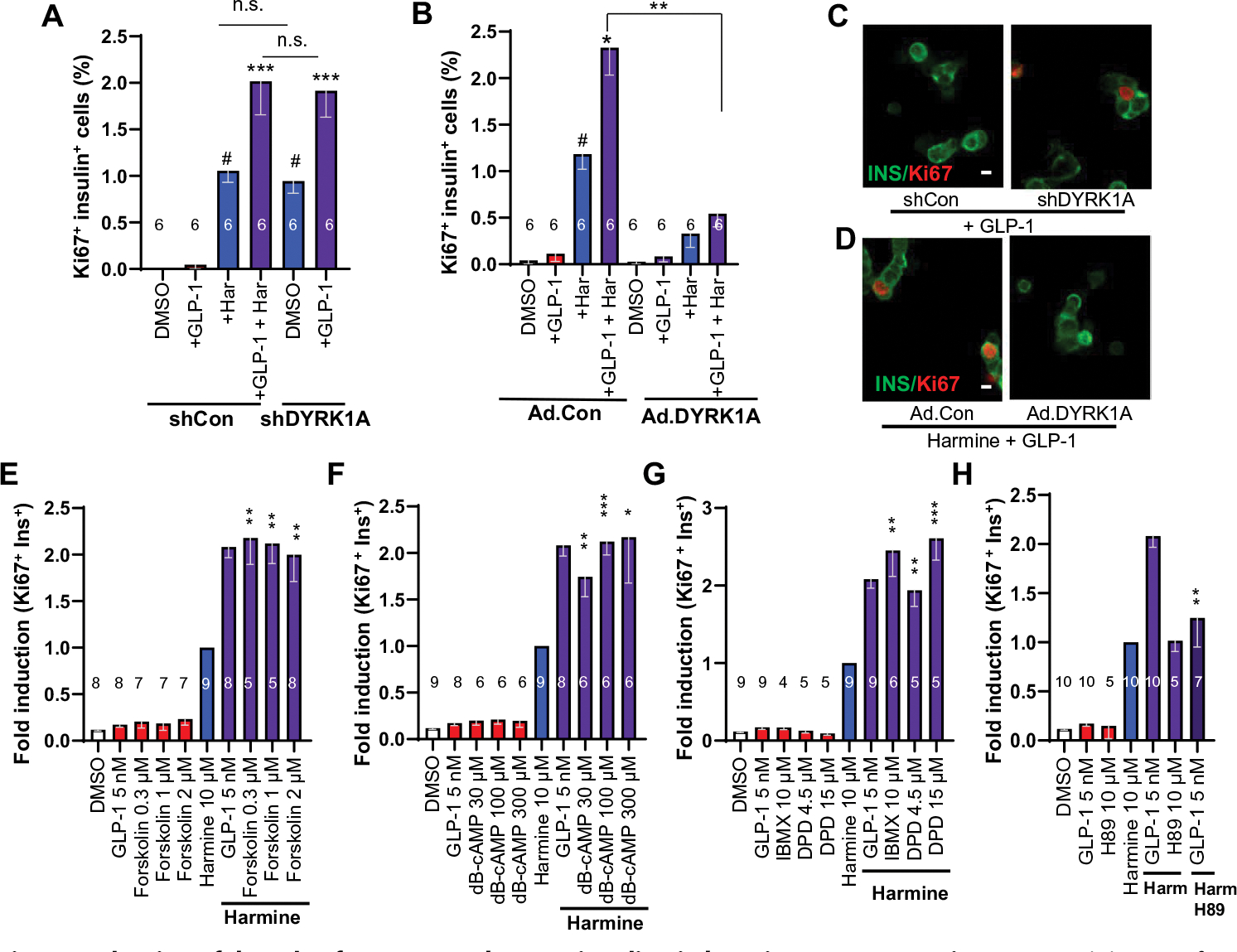
(A) Quantification of Ki67-insulin coimmunolabeling in human islets in response to adenoviral silencing of DYRK1A or harmine (10 μM) and GLP-1 (5 nM). Silencing DYRK1A can replace the effects of harmine in the presence of GLP-1. (B) Quantification of the effects of Ad.DYRK1A overexpression or a control adenovirus (Ad.Con, overexpressing Cre recombinase) in human islets treated with GLP-1 and harmine at various doses. (C) An example of Ki67 immunolabeling in an insulin-positive cell in response to Ad.shRNA silencing of DYRK1A (left), but not with a control, scrambled shRNA adenovirus (left). Scale bar, 10 μm. (D) An example of Ki67-insulin colabeling in β cells of human islets treated with harmine and GLP-1 at the doses in (A) (left) and the absence of proliferation in the presence of Ad.DYRK1A (right). (E to H) Effects on Ki67-insulin colabeling by control vehicle (DMSO), GLP-1, and the following compounds alone or in the presence of harmine: (E) forskolin, (F) dibutyryl-cAMP (dB-cAMP), (G) phosphodiesterase inhibitors isobutylmethylxanthine (IBMX) or dipyridamole (DPD). (H) Effect of PKA inhibitor H89 on Ki67-insulin colabeling on its own and combined with harmine (Harm) and GLP-1. All bars indicate means ± SEM. Numbers of individual human islet donors studied are shown within or above bars. Paired two-tailed t test, #P < 0.01 versus control; *P < 0.05, **P < 0.01, and ***P < 0.001 versus harmine alone. n.s., not significant (P > 0.05).
Harmine–GLP-1 synergy requires adenosine 3′,5′-monophosphate signaling
Because GLP-1 can evoke adenosine 3′,5′-monophosphate (cAMP) signaling in β cells (fig. S5), we asked whether replacing GLP-1 with other measures that increase cAMP in human β cells might mimic the synergy observed with harmine–GLP-1. Thus, we tested forskolin, dibutyryl-cAMP, and the phosphodiesterase inhibitors isobutylmethylxanthine (IBMX) and dipyridamole (DPD) as GLP-1 surrogates and observed that each was able to fully replace GLP-1 (Fig. 4, E to H, and fig. S6, A to D). The synergistic mitogenic effects of harmine–GLP-1 were blocked by the addition of the protein kinase A (PKA) inhibitor H89 (Fig. 4H and fig. S6D). Conversely, GLP-1 could be replaced with the PKA-specific agonist 6-bnz-cAMP (fig. S1E). cAMP effects can also be mediated by the guanyl nucleotide exchange proteins EPAC1 and EPAC2, and we found that the EPAC-specific agonist 8-CPT-cAMP could mimic the effects of GLP-1 (fig. S1F). A search of our purified human β cell RNA sequencing database (25) revealed that EPAC1 was almost undetectable in human β cells, whereas EPAC2 was readily measurable (fig. S1G). Last, the EPAC inhibitor ESI-05 also inhibited the synergistic stimulation of human β cell proliferation (fig. S1H). These studies reveal that harmine–GLP-1 synergy is, in part, mediated by cAMP and has effects downstream upon both PKA and EPAC2 pathways (fig. S5).
Signaling downstream of cAMP
Reasoning that the synergistic effect downstream of cAMP and PKA is likely mediated by members of the cAMP response element–binding protein (CREB), CREB-binding protein (CREBBP), and EP300 families, we compared the expression of this family in FACS-purified human β cells (Fig. 5A and fig. S7A) (25). This revealed that the most highly expressed members were ATF1, ATF2, ATF4, CREB3, CREBBP, and EP300. We treated human islets with harmine–GLP-1 and assessed whether mRNA expression of any of these CREB family members or the common cAMP-target CREB1 was altered by combination treatment (Fig. 5B and fig. S7B). No significant changes were observed; however, these observations at the RNA level would not capture changes in CREB family phosphorylation or interactions. To query potential cAMP/PKA signaling via association of CREB family members, we asked whether naphthol-AS-E phosphate (29) (referred to as “naphthol” hereafter), which disrupts these interactions (29), attenuated harmine–GLP-1–induced human β cell proliferation (fig. S7C). Although naphthol had no effects on basal or harmine-induced proliferation, it caused a significant, dose-related decline in the proliferation induced by harmine–GLP-1 (Fig. 5C and fig. S7D), suggesting that the GLP-1 arm of the synergy is mediated by CREB-CREBBP family members.
Fig. 5. Mechanisms downstream of the GLP1R.
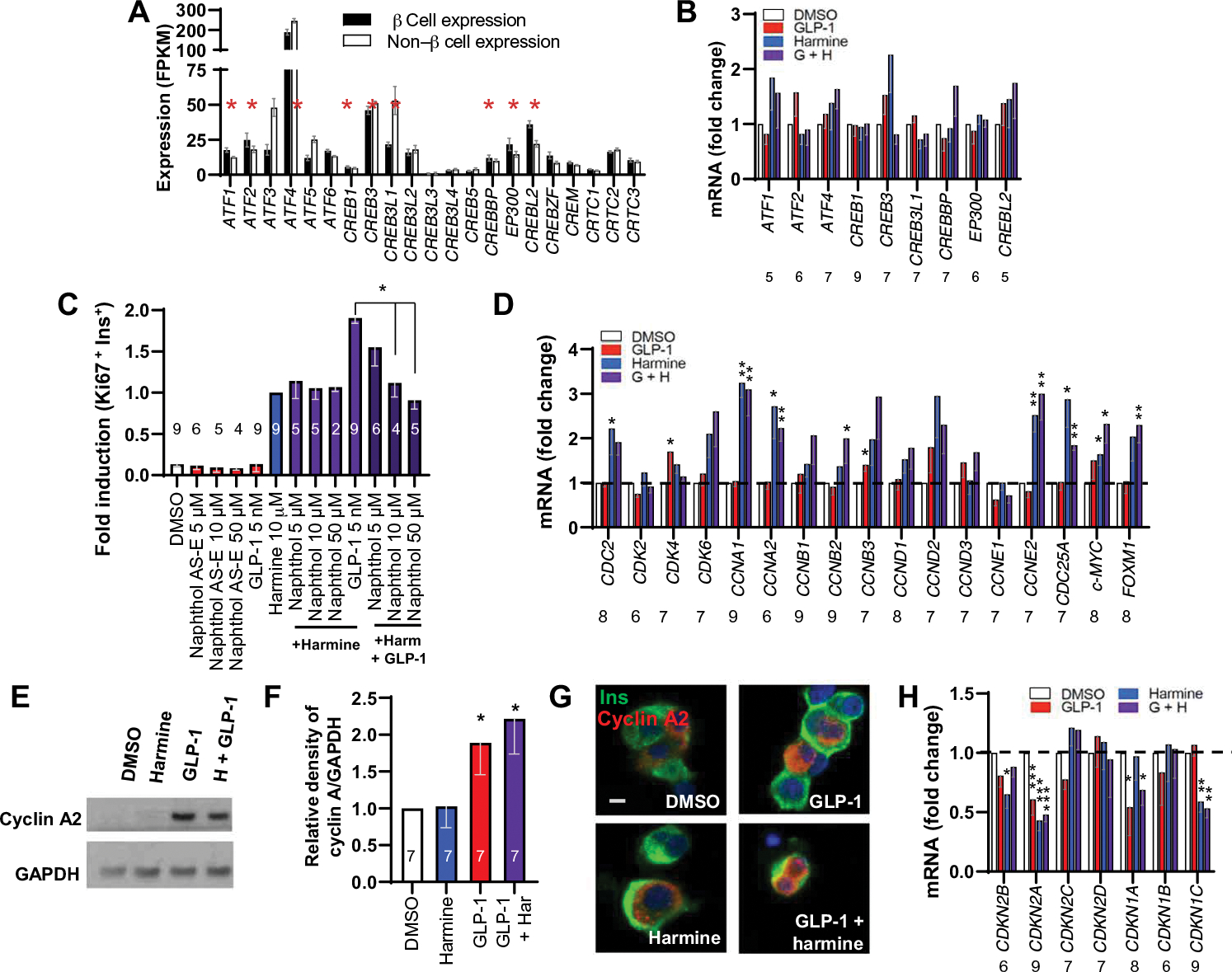
(A) Expression of ATF-CREB-CREM-CREBBP-EP300 family members in β cells and non–β islet cells FACS-sorted from human islets. Red asterisks indicate family members with FPKM values in excess of 15 in β cells, defining them as the most abundant. (B) qPCR of the ATF-CREB-CREM-CREBBP-EP300 family members in (A) marked by an asterisk, plus CREB1, in whole human islets in response to vehicle, GLP-1, harmine, or their combination. ATF, activating transcription factor; CREM, cAMP responsive element modulator; FPKM, fragments per kilobase of transcript per million mapped reads. (C) The effects of naphthol on basal and harmine–GLP1–stimulated human β cell proliferation. (D) Effects on expression of cell cycle activators 72 hours after exposure under the four conditions shown detected by qPCR. (E) An immunoblot of whole human islets for cyclin A protein in response to harmine, GLP-1, and harmine–GLP-1. Representative of experiments from seven islet preparations. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (F) Densitometric quantitation of seven experiments. (G) Immunocytochemistry of cyclin A protein in human β cells treated with harmine–GLP-1. Scale bar, 10 μm. (H) qPCR of the effects of vehicle (DMSO), GLP-1, harmine, or the combination on cell cycle inhibitors on the same islet samples. Bars indicate means ± SEM. Numbers within or above bars indicate the sample size of human islet donors. *P < 0.05 versus control by one-way ANOVA. Paired two-tailed t-test, *P < 0.05, **P < 0.01, and ***P < 0.001 for harmine versus control.
Cell cycle events
The combined mitogenic effects of harmine and GLP-1 might reflect synergistic changes in cell cycle molecule expression. We therefore explored gene expression by quantitative polymerase chain reaction (qPCR) of cyclins and cyclin-dependent kinases in whole human islets treated with control vehicle, GLP-1, harmine, or harmine–GLP-1 (Fig. 5D and fig. S7E). GLP-1 had little effect on cyclins or other cell cycle activators such as CDC25A, MYC, or FOXM1. Harmine increased CDK1, CCNAs (encoding the two cyclin A genes), CCNE2 (encoding cyclin E2), CDC25A, and FOXM1, as described previously (16, 23). There were no increases in cell cycle activators over the effect of harmine alone by the combination of harmine and GLP-1. In the absence of differences in cyclins and cyclin-dependent kinases at the mRNA level, we explored cyclin A expression at the protein level (we selected cyclin A because it displayed the highest increase in Fig. 5D). Despite the lack of difference in steady-state cyclin A mRNA expression in response to harmine alone or the harmine–GLP-1 combination, cyclin A protein abundance was higher in GLP-1–treated and harmine–GLP-1–treated β cells than controls (Fig. 5, E and F, and fig. S7G), and cyclin A nuclear abundance was also more evident (Fig. 5G).
We also examined the gene expression of cell cycle inhibitors [cyclin-dependent kinase inhibitors (CDKIs)] in response to harmine, GLP-1, or their combination (Fig. 5H and fig. S7I). GLP-1 reduced CDKN2A (encoding p16INK4), CDKN2C (encoding p18INK4), and CDKN1A (encoding p21CIP), whereas harmine induced the expected reduction in CDKN2B (encoding p15INK4), CDKN2A, and CDKN1C (encoding p57KIP), as described previously (16, 23). Although the combination of harmine and GLP-1 produced no further reductions in any single cell cycle inhibitor beyond those observed with harmine or GLP-1 alone, CDKN1A expression, which was not reduced by harmine (16), was reduced by both GLP-1 and the harmine–GLP-1 combination.
Effects of combined harmine–GLP-1 on β cell differentiation and function in normal and T2D β cells
We were concerned that pharmacologic induction of human β cell proliferation might lead to dedifferentiation. However, the harmine–GLP-1 combination did not lead to reduced expression of β cell differentiation markers; rather, it increased expression of PDX1, NKX6.1, MAFA, MAFB, GLUT2, GLP1R, and PCSK1, with the greatest increase seen with GLUT2, as assessed by qPCR of whole human islets, compatible with an increase in β cell differentiation (Fig. 6A and fig. S8A). This was accompanied by an increase in pancreatic and duodenal homeobox 1 (PDX1); musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA); and NK homeo box, family 6, member (NKX6.1) protein in individual β cells (Fig. 6B). Glucose transporter-2 (GLUT2) was also up-regulated at the protein level and appeared to traffic from cytoplasmic vesicles to the cell membrane in harmine–GLP-1–treated human islets coimmunolabeled for insulin and GLUT2 (Fig. 6C and fig. S9). Retention or maintenance of differentiation in β cells was also supported by normal glucose-stimulated insulin secretion (Fig. 6, D and E; and figs. S8, D and E and S10, A and B) and normal intracellular calcium responses to glucose and to the GLP1R agonist exendin-4 (Fig. 6, F to I), with basal intracellular calcium concentrations only modestly elevated (Fig. 6I). Thus, harmine–GLP-1 does not appear to induce β cell dedifferentiation, may even maintain or enhance differentiation, and likely does not require increases in intracellular calcium.
Fig. 6. Harmine–GLP-1 treatment maintains or enhances human β cell differentiation.
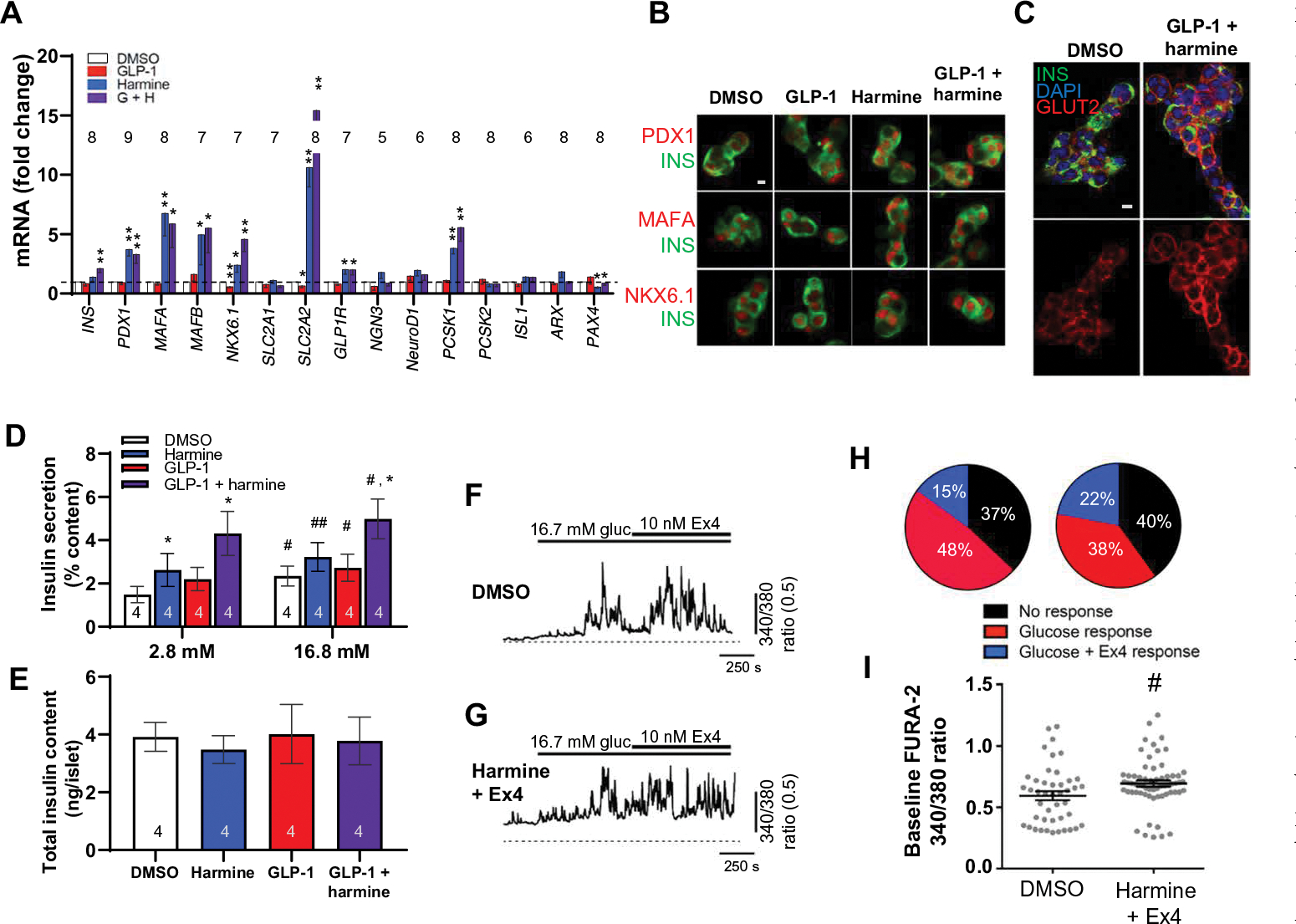
(A) The effects of control vehicle (DMSO, 0.1%), 5 nM GLP-1, 10 μM harmine, or combination harmine–GLP-1 on markers of β cell differentiation as assessed using qPCR on intact islets. *P < 0.05 and **P < 0.01 versus control by paired two-tailed t test. (B) Confocal imaging examples of immunolabeling of β cells for PDX1, MAFA, and NKX6.1 from (A). Representative of experiments in four human islet preparations. (C) Confocal imaging of GLUT2 and insulin immunocytochemistry and 4′,6-diamidino-2-phenylindole (DAPI) nuclear labeling under basal conditions and in response to harmine–GLP-1 in dispersed human islets. Top panels are merged images, and bottom images are expanded and show GLUT2 only. A larger image is shown in fig. S9. (D) Insulin secretion from human islets from four donors in response to low (2.8 mM) and high (16.8 mM) glucose after 72-hour treatment with vehicle, GLP-1 (5 nM), harmine (10 μM), or the combination. Data are shown as percent of residual insulin content. The insulin concentration (means ± SEM) in the 2.8 mM glucose control (DMSO) wells was 19.9 ± 9.1 pmol per islet, and that at 16.7 mM glucose was 33.3 ± 12.6 pmol per islet. (E) Residual insulin content after glucose-stimulated insulin secretion in the islets shown in (D). (F and G) The effect of glucose (gluc) followed by exendin-4 (Ex4) on intracellular calcium as assessed using FURA-2 AM in individual dispersed human β cells pretreated for 48 to 72 hours with 10 μM harmine plus 10 nM exendin-4 (n = 148 cells, 3 donors) or DMSO alone (n = 82 cells, 3 donors). (H) Proportions of single cells either unresponsive to stimuli (black) or responsive to glucose alone (red), or responsive to glucose and exendin-4 together (blue). (I) FURA-2 AM ratio, indicative of basal calcium; harmine plus exendin-4 (n = 63 cells, three donors) had a small but statistically significant (P < 0.05) effect compared with DMSO alone (n = 44 cells, three donors). Bars indicate means ± SEM. Numbers within or above bars indicate the sample size of human islet donors. (D to I) #P < 0.05 and ##P < 0.003 versus control by one-way ANOVA. *P < 0.05 versus harmine alone by one-way ANOVA. Scale bars, 10 μm (B and C).
Because β cell dedifferentiation has been observed in T1D and T2D (30–32), we explored the effects of combined harmine–GLP-1 on differentiation and proliferation in β cells in islets derived from human donors with T2D. As was observed in normal islets, harmine alone and in combination with GLP-1 increased the expression of PDX1, MAFB, NKX6.1, GLUT2, GLP1R, and PCSK1 (Fig. 7A and fig. S11A). MAFA did not display the considerable increase in the mRNA expression it had in normal islets. However, MAFA, NKX6.1, and PDX1 all increased in the protein level in T2D β cells as observed by immunohistochemistry (Fig. 7B). The insulin secretory response to glucose was not only normal but also augmented by harmine and harmine–GLP-1 (Fig. 7, C and D, and fig. S10, C and D). Harmine activated β cell proliferation in T2D β cells, and harmine–GLP-1 provided the same synergistic increase in Ki67 immunolabeling in T2D β cells as observed in normal β cells (Fig. 7, E and F, and fig. S11E).
Fig. 7. Effects of harmine–GLP-1 on β cells from individuals with T2D.
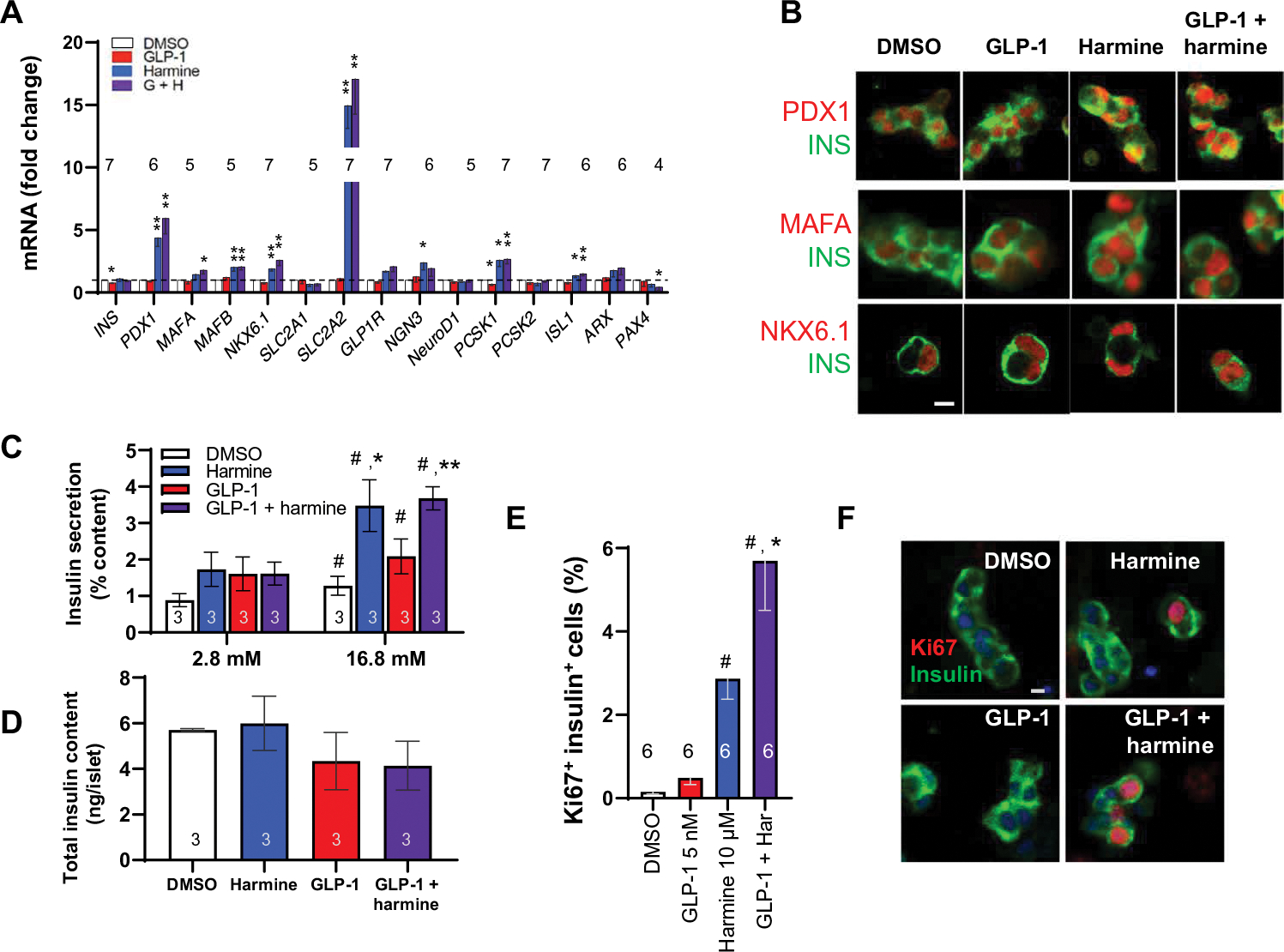
(A) The effects of harmine with or without GLP-1 on β cell differentiation markers. *P < 0.05 and **P < 0.01 versus control by paired two-tailed t test. (B) Confocal imaging examples of PDX1, MAFA, and NKX6.1 immunolabeling of dispersed human T2D islets after the treatments shown. (C) Insulin secretion in response to low (2.8 mM) and high (16.8 mM) glucose in three different T2D islet preparations pretreated with vehicle, GLP-1, harmine, or their combination for 72 hours. (D) Residual insulin content in the islets from (C). The average insulin concentration in the 2.8 mM glucose control (DMSO) wells was 18.1 ± 3.2 pmol per islet, and that at 16.7 mM glucose was 32.2 ± 4.6 pmol per islet. (E) Human T2D β cell proliferation in response to vehicle, GLP-1, harmine, or the combination. (F) Examples of insulin and Ki67 immunolabeling in β cells derived from donors with T2D. Values are means ± SEM. Numbers within or above bars indicate the sample size of human islet donors. (C to E) #P < 0.05 versus control; *P < 0.05 and **P < 0.01 versus harmine alone, both by one-way ANOVA. Scale bars, 10 μm (B and F).
Enhanced β cell proliferation and glycemic control in vivo
To determine whether the beneficial effects of harmine–GLP-1 translated from in vitro findings to enhanced proliferation and function in vivo, we used the immunoincompetent nonobese diabetic–severe combined immunodeficient–gamma (NSG) mouse model to examine the effects of the harmine–exendin-4 combination in vivo. We transplanted a standard marginal mass of 500 human IEQs or a positive control dose of 1500 IEQs into the renal capsule of NSG mice that had been rendered β cell deficient and diabetic with a single high dose of streptozotocin (STZ). One 500 IEQ group was treated with daily doses of intraperitoneal vehicle (saline) for 14 days; one 500 IEQ group received daily intraperitoneal exenatide at a dose of 0.5 μg/kg; one 500 IEQ group received daily intraperitoneal harmine at a dose of 1.0 mg/kg; and one group received daily intraperitoneal injections of exenatide (0.5 μg/kg) and harmine (1.0 mg/kg). The harmine dose selected (1.0 mg/kg) was 10-fold lower than the dose that we reported previously (10 mg/kg) (16) because the higher dose (10 mg/kg) normalizes blood glucose, which would obscure any beneficial effects of the combination on diabetes. Animals treated with saline, harmine, or exenatide remained severely diabetic during the 2 weeks of the study, whereas positive control animals transplanted with 1500 IEQs displayed normal glucose concentrations (Fig. 8A). Animals that received 500 IEQs and the exenatide-harmine combination displayed near-normal glucose values (Fig. 8A), comparable as assessed by area under the curve to those receiving 1500 IEQs (Fig. 8B and fig. S12B). Severe diabetes returned in the exenatide-harmine group and the 1500 IEQ group after unilateral nephrectomy (UNX) at day 14, making clear that the beneficial harmine-exenatide effects on glycemia were a result of their effects on the transplanted human islets and not on the endogenous mouse islets. The exenatide-harmine group also displayed an increase in circulating human insulin as compared to the saline, harmine, and exenatide groups, although not as high as in the 1500 IEQ group (Fig. 8C and fig. S12C). Further, intraperitoneal glucose tolerance testing at day 12 showed better performance in the harmine-exenatide group than the saline, exenatide, or harmine groups and was comparable to the 1500 IEQ group (Fig. 8, D and E, and fig. S12E). Collectively, these findings demonstrate that the harmine–GLP1R agonist classes are more effective in enhancing human islet function in vivo when used in combination than either agent alone.
Fig. 8. Effects of the harmine–exendin-4 combination on human β cells in vivo in immunodeficient mice.
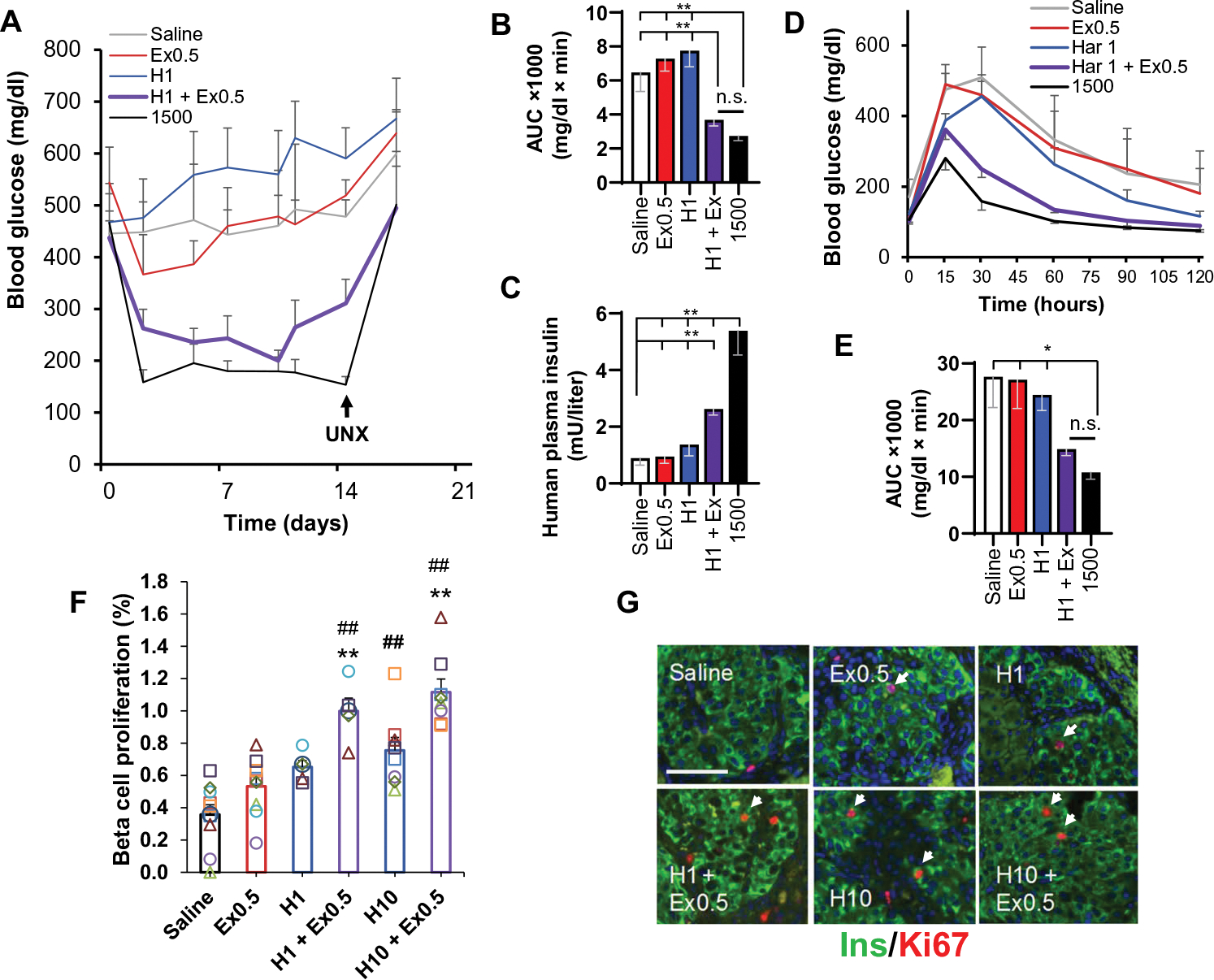
(A) Glucose control in response to harmine and GLP-1 in the STZ-diabetic NSG mouse marginal islet mass model. Five groups of n = 7 mice each received human islets from the same seven human islet donors: four groups received 500 IEQs, and the fifth group received 1500 IEQs as a positive control without drug treatment. The 500 IEQ groups were treated daily for 2 weeks with intraperitoneal saline, harmine (H; 1 mg/kg), exendin-4 (Ex; 0.5 μg/kg), or the combination. The 1500 IEQ group received saline. Unilateral nephrectomy (UNX) was performed at day 12. (B) Area under the curve (AUC) for the five groups in (A). (C) Circulating insulin measured using a human insulin-specific assay. (D) Intraperitoneal glucose-tolerance tests on day 12. (E) Area under the curve for (D). (F) Human β cell proliferation assessed by Ki67-insulin coimmunolabeling in human islet renal capsular grafts from euglycemic NSG mice. All animals received 500 IEQs and were treated once daily for 7 days with intraperitoneal saline, exendin-4 (0.5 μg/kg), harmine (1 mg/kg), both exendin-4 (0.5 μg/kg) and harmine (1 mg/kg), harmine (10 mg/kg), or both exendin-4 (0.5 μg/kg) and harmine (10 mg/kg). Each symbol represents a single human islet donor in a single mouse. (G) Examples of insulin and Ki67 immunohistochemistry in islet grafts. Arrowheads show examples of cells considered positive for both insulin and Ki67. Scale bar, 100 μm. Values are means ± SEM. (B to E) *P < 0.05 and **P < 0.01 versus without or harmine-exendin treatment as indicated, by one-way ANOVA. (F) ##P < 0.01 versus control and **P < 0.001 versus harmine alone, both by one-way ANOVA.
Last, we explored the ability of the harmine-exenatide combination to drive human β cell proliferation in vivo. Here, we selected a nondiabetic, euglycemic model to avoid the confounding effects of hyperglycemia. We transplanted human islets (500 IEQs) into the renal capsule of NSG mice and treated the mice for 1 week with daily intraperitoneal doses of vehicle (saline), exenatide (0.5 μg/kg), harmine (1 or 10 mg/kg), or combinations of either harmine (1 mg/kg) and exenatide (0.5 μg/kg) or harmine (10 mg/kg) and exenatide (0.5 μg/kg). Random blood glucose obtained on day 7 of treatment remained normal in these animals (fig. S13). As reported previously (16), the harmine dose (10 mg/kg) induced a Ki67 immunolabeling index of ~0.8% as compared to the basal value of ~0.4%. However, the 10-fold lower dose of harmine also induced labeling in human β cells in the range of 0.6% (Fig. 8, F and G). Both the low and high doses of harmine synergized with exenatide to induce further increases in proliferation, achieving 1.1% in the exenatide and harmine group (10 mg/kg). These findings support two important conclusions: that induction of human β cell proliferation assayed using in vivo settings is lower than in vitro settings, mirroring prior studies (16, 23), and that the harmine-exenatide combination affords higher rates of human β cell proliferation than maximally effective doses of either agent alone.
Safety considerations
Some reports have suggested that GLP1R agonists in clinical use may be associated with an increase in frequency of pancreatitis or pancreatic ductal hyperplasia or atypia (33–35). Accordingly, histology of multiple organs (mouse liver, kidney, pancreas, intestine, heart, and spleen) harvested from the saline, harmine (10 mg/kg), exenatide (0.5 μg/kg), or combination treatment groups in the euglycemia study was examined in a double-blinded fashion by a veterinary pathologist. Ki67 labeling of pancreatic ductal cells was also examined in a double-blinded fashion. No evidence of pancreatic ductular hyperplasia, atypia, pancreatitis, or proliferation or other abnormalities in the pancreas, liver, intestine, heart, spleen, or kidney were observed over the 1 week of this study (fig. S14).
DISCUSSION
Collectively, we report several notable observations of translational relevance. By adding an orally active, small-molecule DYRK1A inhibitor (such as harmine, INDY, leucettine, 5-IT, GNF4877, or others) to any one of a large group of currently widely used diabetes drugs that directly (GLP-1 analogs) or indirectly (DPP4 inhibitors) activate the GLP1R, one is able to convert the mitogenically inactive GLP1R agonists into potent β cell proliferative agents in vitro and in vivo. The resulting proliferation rates exceeded those of DYRK1A inhibitors alone and may be in a range that could allow for restoration of normal β cell mass in people with T2D and T1D. The increase in proliferation was synergistic in a pharmacological sense and extended to doses of harmine and GLP-1 that had no proliferative effect on their own. These findings suggest that any DYRK1A inhibitor administered with any GLP1R agonist currently in widespread use in people with T2D—and by extension with any DPP4 inhibitor drug that augments circulating GLP-1—would be able to generate substantial rates of human β cell proliferation. We note that harmine alone and in combination with a GLP1R agonist was able to induce both proliferation and differentiation in β cells derived from people with T2D, a disease associated with both inadequate numbers and dedifferentiation of β cells.
We found that harmine–GLP-1 synergy required both inhibition of the harmine target, DYRK1A, together with activation of cAMP signaling, which, in turn, appeared to activate PKA and EPAC2 signaling. Although we focused on GLP-1 analogs as cAMP agonists, our observations suggest that any molecule that activates cAMP signaling in a β cell can partner with a DYRK1A inhibitor to drive human β cell proliferation. The specific targets of PKA and EPAC downstream of cAMP remain to be fully defined but likely include CREB-CREBBP family members. Unfortunately, the lack of large numbers of healthy purified human β cells precluded further detailed exploration of the phosphorylation of each potential phosphorylation site or coimmunoprecipitation of each potential CREB family member pair in response to harmine, GLP-1, or their combination. However, our findings are consistent with the notion that cAMP- and PKA-induced interactions among CREB family members likely underlie the synergistic effects of GLP-1 on β cell proliferation.
It seems clear that harmine, acting via DYRK1A inhibition and nuclear translocation of NFaT transcription factors, activates expression of cell cycle regulatory genes such as cyclins, CDKs, FOXM1, and CDC25A and represses expression of cell cycle inhibitor (CDKI) genes such as CDKN2A/B and CDKN1C. GLP-1, presumeably acting via cAMP and PKA-EPAC2 pathways, appeared to reduce expression of a particularly abundant β cell CDKI, CDKN1A (encoding p21CIP) and selectively increased cyclin A abundance, thereby contributing to the harmine–GLP-1 mitogenic synergy. Of particular interest, although the harmine–GLP-1 combination had no differential effect on cyclin A mRNA expression, it appeared to increase the abundance of cyclin A protein, through mechanisms that remain unexplored. We speculate that the decline in p21CIP derived from GLP-1, in the setting of already reduced p15INK4 and p57KIP derived from harmine, together with the increase in cyclin A nuclear abundance, may permit further enhancement of β cell proliferation by the combination as compared to harmine or GLP-1 alone. It is also possible that additional changes in cyclin and CDK phosphorylation, abundance, and interactions may occur at the protein level; lack of an abundant supply of purified healthy human β cells, as noted earlier, makes these possibilities difficult to assess. Further dissection of the CREB-CBP and cyclin-cdk-CDKI pathways and their connection to cyclins, CDKs, and CDKI pathways in human β cells is difficult because of a lack of ideal experimental systems. Specifically, human islets are complex multicellular miniorgans, only a minority of which are β cells, and only a minority of these are proliferating. Thus, although mechanistic studies would ideally be performed in cadaveric human β cells, accruing sufficient numbers of healthy, purified human β cells with which to perform more detailed signaling studies (such as immunoblots of phosphoproteins or coimmunoprecipitation studies) for the multitude of CREB family members in response to multiple drug combinations makes this approach untenable. Rodent β cells and cell lines are not ideal systems with which to address these issues, for unlike quiescent adult human β cells, rodent β cells and cell lines display rapid and robust proliferation in response to GLP-1.
The beneficial effects of the harmine–GLP1R agonist combination translated to the in vivo setting in human islets transplanted into immunodeficient NSG mice. In particular, control STZ-diabetic NSG mice transplanted with a marginal mass of 500 human IEQs and treated with vehicle, harmine, or exenatide showed no improvement in glycemic control or human insulin secretion. In contrast, in otherwise identical mice treated with the harmine-exenatide combination, blood glucose rapidly approached the near-normal concentrations observed in mice receiving a full therapeutic dose of islets, 1500 IEQs. Moreover, glucose tolerance testing was normal, and human insulin secretion rose.
Human β cell proliferation also increased in transplanted human islets in response to the harmine-exenatide combination, and this was both synergistic and dose related. Rates of human β cell proliferation in vivo in NSG mice were lower than those in vitro but three times higher than in control β cells. Lower human β cell proliferation rates in vivo have been observed repeatedly (16, 23) and likely reflect the production of proliferation-inhibiting molecules in vivo, limited access of drugs to recently transplanted, poorly vascularized islets, and suboptimal dosing and pharmacokinetics of the drug administration paradigm selected.
We did not observe β cell dedifferentiation as a result of enhanced β cell proliferation in normal or T2D human islets. Rather, we observed retention or improvement of glucose-stimulated insulin secretion in vitro and in vivo, together with enhanced expression of canonical β cell transcription factors and identity markers. The specific mechanisms underlying these changes are poorly understood and require further study. For example, understanding whether the increase in membrane-associated GLUT2 results in increased glucose uptake or increased glucose metabolism with consequent increases in insulin secretion will require further study. It is important to emphasize that these events were observed in experiments lasting 4 to 14 days; defining whether and for how long these beneficial effects will persist will require long-term in vitro and in vivo studies.
One advantage of GLP-1 family molecules in β cell therapeutic paradigms is their relative β cell specificity (4, 10, 11). “Relative” is the key operative word. GLP1Rs are present in β cells, pancreatic ductal cells, central nervous system (CNS) nuclei, the cardiac muscle, gastric smooth muscle, and T cell subsets. This is an improvement over the ubiquitous expression of the harmine target DYRK1A or the TGFβSF members yet still may be associated with undesired risk. There is concern that GLP1R agonists have been associated with increased risk for pancreatitis, pancreatic ductal hyperplasia, and pancreatic carcinoma (33). These risks remain imperfectly understood, but meta-analyses do not support such risk (34, 35), and GLP1R agonists remain one of the most widely used classes of diabetes drugs. Our in vitro proliferation studies suggest that adding harmine to a GLP1R agonist may be a concern regarding enhanced ductal cell proliferation. On the other hand, preliminary in vivo preclinical toxicology studies do not show evidence of ductal cell proliferation or ductular atypia. We emphasize, however, that these in vivo toxicity studies lasted only 1 week. Again, longer-term and more extensive preclinical studies are required to resolve these important safety issues.
This study has a number of limitations. First, there is a need to protect newly generated β cells in people with T1D from ongoing immune attack. This challenge remains unmet at present but is not a barrier for the larger group of people with T2D who also need β cell expansion and enhanced β cell differentiation and who currently use GLP1R agonist drugs.
A second challenge is the need to improve β cell specificity by developing tools to direct regenerative drugs and imaging agents specifically and exclusively to the human β cell. At present, there is no perfect targeting molecule for delivering any therapeutic agent exclusively to the human β cell. Here, the synergistic efficacy of a low dose of harmine (which has no effects on its own on β cell proliferation) with any dose of a GLP1R agonist raises the possibility that the β cell specificity of the GLP1R may be leveraged for human β cell proliferation using low-dose harmine in people with T2D already taking GLP1R agonists.
A third challenge is the risk of oncogenic transformation in β cells or cells outside the islet (33–35). Of course, this is a concern for all forms of regenerative/replacement therapies, including pancreas and islet transplant (with a well-documented risk of lymphoma and other cancers) (36, 37), stem cell–derived therapies (which carry the risk of teratoma) (38), gene editing therapies (with the imperfect specificity of CRISPR-Cas9 targeting, off-target deletions, and the requirement for viral vector delivery) (39), and transdifferentiation of α or other islet cells (with broad, nonspecific, and potentially oncogenic effects of chromatin-modifying drugs) (40). Again, long-term preclinical studies will be needed to evaluate these possibilities for all forms of β cell regenerative therapies, including the DYRK1A inhibitor–GLP1R agonist combination.
A fourth challenge will be identifying doses of harmine or harmine-like compounds that are tolerable in humans. The effects of harmine have not been directly studied in humans. It is possible that harmine may have psychoactive effects because it is a component of ayahuasca, a psychoactive tea or infusion derived from the banasteriopsis vine (41, 42). On the other hand, ayahuasca is rich in another known human psychoactive compound, 5,5′-dimethyltryptamine, making it difficult to know whether and at what doses harmine may exert CNS effects in humans. In this context, we note that doses of harmine that have little or no effect on human β cell proliferation in vitro or in vivo were converted into effective human β cell proliferative agents when combined with GLP-1 or exenatide. Collectively, these observations suggest a need to study the pharmacokinetics and toxicity of harmine in preclinical models and humans.
A fifth challenge will be to determine whether and how long the beneficial mitogenic and differentiation effects persist over time in vivo. This is a critical question that will require 12- to 24-month dosing studies in NSG mice transplanted with human islets.
Last, it is of interest to consider cost and scalability. Transplant, stem cell, and gene editing/gene therapy are expensive and resource intense and therefore unlikely to be translated to tens or hundreds of millions of people with diabetes. The current use of GLP1R agonists in millions of people with diabetes suggests the possibility that a DYRK1A inhibitor–GLP-1–based strategy, if safe and specific, could be scalable and has broad implications for diabetes globally.
MATERIALS AND METHODS
Study design
This study evaluated the ability of GLP-1 analogs to enhance DYRK1A inhibitor–induced human β cell proliferation with superior specificity for human β cells. Because induction of β cell proliferation in rodents may not predict events in human β cells, we exclusively studied human pancreatic islets from 111 cadaveric donors, both ex vivo and transplanted into euglycemic and STZ-diabetic immunodeficient mice. Human islet sample sizes between 4 and 20 were selected for each experiment, as indicated in the figures along with numbers of replicates. We used larger sample sizes for critically important observations. Some shipments of human islets were received in poor condition due to islet isolation or shipping conditions and therefore could not be used. All data from viable human islet preparations were included. All outliers were included and are visualized in scatterplot versions of the figures in the Supplementary Materials. Human β cell proliferation assays, qPCR, mouse histopathology, and other assays were performed in a double-blinded fashion. Investigators who assessed, measured, or quantified the results were blinded to the intervention. More methods details are available in the Supplementary Materials.
Human cadaveric islets
Human islets from 111 normal donors were obtained through the National Institutes of Health (NIH)–supported Integrated Islet Distribution Program (IIDP) (https://iidp.coh.org), the Alberta Diabetes Institute Islet Core at the University of Alberta, the University of Chicago, and the Clinical Islet Laboratory, University of Alberta Hospital. Islets were used in the order in which they were received. Islets were harvested from pancreata from deceased organ donors without any identifying information and with informed consent and Institutional Review Board approval at the islet-isolating centers. Complete donor demographic information is provided in data file S1. Briefly, donors ranged in age from 15 to 76 years (means ± SEM, 45.8 ± 12.8); 33 were female and 71 were male. Mean body mass index (BMI) was 30.6 ± 5.6 (range, 17.3 to 44.4), and cold ischemia time was 592 min (range, 270 to 1007 min). Purity ranged from 55 to 98% (mean, 85.5 ± 7.9). We also obtained islets from 11 donors with T2D. Donor ages ranged from 26 to 71 years (mean, 48.2 ± 13.6; n = 3 females and n = 8 males). Mean BMI was 31.7 ± 4.4 (range, 27.3 to 38.1), and cold ischemia time was 582 min (range, 155 to 1200 min). Purity ranged from 55 to 85%. Hemoglobin A1c ranged from 6.6 to 14.1 (mean, 7.9). Three individuals were known to be on diabetes medications (metformin, n = 3; DPP4 inhibitor, n = 1). Upon arrival, islets were cultured in islet culture medium (RPMI 1640 medium containing 10% fetal bovine serum, 5.5 mM glucose, and 1% penicillin-streptomycin) at 37°C and 5% CO2 overnight.
Islet dispersion
Islets were dispersed into single cells with Accutase as previously described (16, 43, 44). Whole islets were pelleted by centrifugation at 200g and washed twice in phosphate-buffered saline. Islets were then incubated in Accutase for 15 to 17 min at 37°C and completely dispersed to single cells by pipette trituration. Single islet cells were pelleted by centrifugation at 700g and resuspended in islet culture medium.
Compound treatments
Dispersed islets were plated on poly-d-lysine–laminin–coated glass chamber slides or coverslips. Cells derived from 20 to 30 IEQ dispersed cells were seeded per well and allowed to recover for 24 hours before compound treatment. Cells were treated for 96 hours before Ki67 staining, BrdU staining, or TUNEL labeling. A cytokine cocktail containing TNFα (1000 U/ml) and IL-1β (500 U/ml) was used for TUNEL labeling.
Animal studies
Human islet transplant into 2- to 3-month-old male euglycemic or diabetic NSG mice was performed as described in detail previously (16, 23). All procedures were performed with prior approval of, and in compliance with, the Icahn School of Medicine Institutional Animal Care and Use Committee.
Diabetes was induced by a single intraperitoneal dose of STZ (200 mg/kg body weight). Glucose was measured by tail nick and glucometer measurements, and animals were confirmed to be diabetic (glucose, >400 mg/dl) for 2 to 3 days before drug treatment. In all experiments, with one exception, 500 human IEQs (1 IEQ defined as 125-μm diameter per islet) were transplanted via flank incision under the capsule of the left kidney. The one exception was the positive control group in the diabetes model, which received 1500 IEQs. Exenatide, harmine, or the combination were intraperitoneally administered daily in the doses and for the durations described in the figures. Intraperitoneal glucose tolerance tests were performed on day 12, as previously described (16, 23), after intraperitoneal administration of glucose (2 g/kg body weight). On day 14, a UNX was performed to ensure that the hyperglycemia reoccurred and that the benefit resulted from the human islet graft and not regeneration of the murine islets. Blood was obtained on day 14 before UNX, and human insulin was measured by enzyme-linked immunosorbent assay (Mercodia).
For the euglycemic model, 500 IEQs were used for all islet transplants. Animals were allowed to recover for 1 week after transplant and then received vehicle (saline), harmine (1.0 or 10 mg/kg), exenatide (0.5 μg/kg), or a harmine-exenatide combination. At the conclusion of the experiments in the euglycemic model, the kidney (including the islet graft), liver, heart, pancreas, intestine, and spleen were harvested, fixed in neutral buffered formalin, embedded in paraffin, and examined for insulin-Ki67 or CK19-Ki67 coimmunohistochemistry or submitted to for toxicology.
Toxicology studies
The kidney, liver, heart, spleen, intestine, and pancreas sections from the animals in the euglycemic study were submitted in a double-blinded fashion to the Veterinary Pathology Division at the Icahn School of Medicine such that neither the submitter (A.F.S.) nor the pathologist (V. Gillespie) was aware of the treatment group. Two slides from each organ from each treatment group were assessed as shown in fig. S14.
Statistical analysis
All experiments were repeated multiple times in multiple human islet preparations as indicated in the figure legends. Results achieved normality by Shapiro-Wilk log-normality test and were accepted as significant at P < 0.05 as determined using two-tailed Student’s t test or one-way analysis of variance (ANOVA). A minimum of 1000 β cells from a minimum of different five different donors was counted for each graph shown.
Supplementary Material
Materials and Methods
Fig. S1. Combination of DYRK1A inhibitors with GLP1R agonists yields synergistic increases in human β cell proliferation.
Fig. S2. The DYRK1A–GLP-1 increase human β cell numbers is a class effect for both DYRK1A inhibitors and GLP1R agonists.
Fig. S3. The effects of the harmine–GLP-1 combination on proliferation in non–β cells, β cell death, and DNA damage.
Fig. S4. Evaluation of the role of DYRK1A and cAMP signaling in harmine–GLP1R agonist synergy.
Fig. S5. GLP1R signaling diagram.
Fig. S6. Additional evaluation of the role cAMP pathway signaling in harmine–GLP1R agonist synergy.
Fig. S7. Mechanisms downstream of the GLP1R.
Fig. S8. Harmine–GLP-1 treatment maintains or enhances human β cell differentiation.
Fig. S9. A magnified confocal view of GLUT2 trafficking.
Fig. S10. Glucose-stimulated insulin secretion from normal and T2D islets.
Fig. S11. Effects of harmine–GLP-1 on β cells from individuals with T2D.
Fig. S12. Effects of the harmine–exendin-4 combination on human β cells in immunodeficient mice.
Fig. S13. Blood glucose values in euglycemic mice treated with saline, exendin-4, harmine, or their combination.
Fig. S14. Effects of the harmine–exendin-4 combination on normal organ histopathology and ductal proliferation in NSG mice.
Data file S1. Human islet donor demographic data.
Data file S2. Primary and secondary antisera.
Data file S3. qPCR primers.
Acknowledgments:
We thank the Bonnie and Joel Bergstein family, the Lonnie and Thomas Schwartz family, the NIH/NIDDK-supported IIDP, the Human Islet Research Network (HIRN), the Human Islet and Adenoviral Core (HIAC) of the Einstein-Mount Sinai Diabetes Research Center (DRC), the Juvenile Diabetes Research Foundation (JDRF), and the Graduate School T-32 Training grant, Integrated Training in Pharmacological Sciences. We thank V. Gillespie DVM for veterinary pathology analysis. We thank J. Lyon at the Alberta Diabetes Institute Islet Core for work on human islet isolations. We also thank the Human Organ Procurement and Exchange Program (Edmonton), the Trillium Gift of Life Network (Toronto), and other organ procurement agencies for efforts in obtaining human pancreases for research. We also thank the Mount Sinai Flow Cytometry Core for access and support. Last, we thank D. Rodriguez for technical assistance.
Funding:
This work was supported by NIH grants T32 GM062754 (to C.A.), DK105015 (to A.F.S.), DK105015-01A1S1 (to C.A.), P-30 DK 020541 (to A.F.S. and A.G.-O.), and UC4 DK104211 (to A.F.S.), by JDRF grants 2-SRA-2015-62 (to A.F.S.) and 2-SRA-2017 514-S-B (to A.F.S.), and by the Icahn School of Medicine at Mount Sinai.
Footnotes
Competing interests: The Icahn School of Medicine at Mount Sinai has filed a patent application on this work (WO 2019/136320; Method of increasing proliferation of pancreatic β cells, treatment method, and composition). B.T. is a consultant for SUN Pharma Ltd.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data related to this study can be found in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.World Health Organization, Global Reports on Diabetes; www.who.int/diabetes/global-report/en/.
- 2.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC, β-Cell deficit and increased β-cell apoptosis in humans with diabetes. Diabetes 52, 102–110 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, Atkinson MA, Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes 65, 719–731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drucker DJ, Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Guo X-H, The value of short- and long-acting glucagon-like peptide agonists in the management of type 2 diabetes mellitus: Experience with exenatide. Curr. Med. Res. Opin. 32, 67–76 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Deacon CF, Holst JJ, Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: Comparison, efficacy and safety. Expert. Opin. Pharmacother. 14, 2047–2058 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Parnaud G, Bosco D, Berney T, Pattou F, Kerr-Conte J, Donath MY, Bruun C, Mandrup-Poulsen T, Billestrup N, Halban PA, Proliferation of sorted human and rat β-cells. Diabetologia 51, 91–100 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, Phillips N, Levy SE, Greiner DL, Shultz LD, Bottino R, Kim SK, Powers AC, Age-dependent human β-cell proliferation induced by glucagon-like peptide-1 and calcineurin signaling. J. Clin. Invest. 127, 3835–3844 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephens SB, Schisler JC, Hohmeier HE, An J, Sun AY, Pitt GS, Newgard CB, A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet β-cell survival and function. Cell Metab. 16, 33–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, Hvelplund A, Bardram L, Calatayud D, Knudsen LB, GLP1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 155, 1280–1290 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ, An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacol. Ther. 139, 359–391 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocaña A, Penninger JM, Vasavada RC, Osteoprotegerin and denosumab stimulate human β-cell proliferation through inhibition of the receptor activator of NF-kB ligand pathway. Cell Metab. 22, 77–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kondegowda NG, Joshi-Gokhale S, Harb G, Williams K, Zhang XY, Takane KK, Zhang P, Scott DK, Stewart AF, Garcia-Ocaña A, Vasavada RC, Parathyroid hormone-related protein enhances human β-cell proliferation and function with simultaneous induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 59, 3131–3138 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, Boucher J, Martinez R, Gritsenko MA, De Jesus DF, Kahraman S, Bhatt S, Smith RD, Beer HD, Jungtrakoon P, Gong Y, Goldfine AB, Liew CW, Doria A, Andersson O, Qian WJ, Remold-O’Donnell E, Kulkarni RN, Serpin B1 promotes pancreatic β-cell proliferation. Cell Metab. 23, 194–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purwana I, Zheng J, Li X, Deurloo M, Son DO, Zhang Z, Liang C, Shen E, Tadkase A, Feng Z-P, Li Y, Hasilo C, Paraskevas S, Bortell R, Greiner DL, Atkinson M, Prud’homme GJ, Wang Q, GABA promotes human β-cell proliferation and modulates glucose homeostasis. Diabetes 63, 4197–4205 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Wang P, Alvarez-Perez J-C, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocaña A, Stewart AF, A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic β-cell replication. Nat. Med. 21, 383–388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahtouh T, Elkins JM, Filippakopoulos P, Soundararajan M, Burgy G, Durieu E, Cochet C, Schmid RS, Lo DC, Delhommel F, Oberholzer AE, Pearl LH, Carreaux F, Bazureau J-P, Knapp S, Meijer L, Selectivity, co-crystal structures and neuroprotective properties of leucettines, a famly of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 55, 9312–9330 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang Y-Q, Kamireddy A, Swafford A, Powers AF, Walker J, Lamb J, Bursalaya B, Donato MD, Harb G, Qiu M, Filippi CM, Deaton L, Turk CN, Suarez-Pinzon WL, Liu Y, Hao X, Mo T, Yan S, Li J, Herman AE, Hering BJ, Wu T, Seidel HM, Namara PM, Glynne R, Laffitte B, Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 6, 8372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dančík V, Burns SM, Gilbert TJ, Olson DE, Clemons PA, Kulkarni RN, Wagner BK, Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes 65, 1660–1671 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdolazimi Y, Zhao Z, Lee S, Xu H, Allegretti P, Horton TM, Yeh B, Moeller HP, Nichols RJ, Cutcheon DM, Shalizi A, Smith M, Armstrong NA, Annes JP, CC-401 promotes β-cell replication via pleiotropic consequences of DYRK1A/B inhibition. Endocrinology 159, 3143–3157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar K, Wang P, Sanchez R, Swartz EA, Stewart AF, De Vita RJ, Development of kinase-selective, harmine-based DYRK1A inhibitors that induce human pancreatic β-cell proliferation. J. Med. Chem. 61, 7687–7699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar K, Ung PM-U, Wang P, Wang H, Li H, Andrews MK, Stewart AF, Schlessinger A, De Vita RJ, Novel selective thiadiazine DYRK1A inhibitor lead scaffold with human pancreatic β-cell proliferation activity. Eur. J. Med. Chem. 157, 1005–1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, Wilson J, González BJ, Bender A, Takane KK, Ye L, Harb G, Pagliuca F, Homann D, Egli D, Argmann C, Scott DK, Garcia-Ocaña A, Stewart AF, Combined inhibition of DYRK1A, SMAD and trithorax pathways synergizes to induce robust replication in adult human β cells. Cell Metab. 29, 638–652.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart NJ, Powers AC, Use of human islets to understand islet biology and diabetes: Progress, challenges and suggestions. Diabetologia 62, 212–222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Bender A, Wang P, Karakose E, Inabnet WB, Libutti SK, Arnold A, Lambertini L, Stang M, Chen H, Kasai Y, Mahajan M, Kinoshita Y, Fernandez-Ranvier G, Becker TC, Takane KK, Walker LA, Saul S, Chen R, Scott DK, Ferrer J, Antipin Y, Donovan M, Uzilov AV, Reva B, Schadt EE, Losic B, Argmann C, Stewart AF, Insights into human β-cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat. Commun. 8, 767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takane KK, Kleinberger JW, Salim FG, Fiaschi-Taesch NM, Stewart AF, Regulated and reversible induction of adult human β-cell replication. Diabetes 61, 418–424 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Micallef SJ, Li X, Schiesser JV, Hirst CE, Yu QC, Lim SM, Nostro MC, Elliott DA, Sarangi F, Harrison LC, Keller G, Elefanty AG, Stanley EG, (GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia 55, 694–706 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sui L, Danzl N, Campbell SR, Viola R, Williams D, Xing Y, Wang Y, Phillips N, Poffenberger G, Johannesson B, Oberholzer J, Powers AC, Leibel RL, Chen X, Sykes M, Egli D, β-Cell replacement in mice using human type 1 diabetes nuclear transfer embryonic stem cells. Diabetes 67, 26–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JW, Park HS, Park S-A, Ryu S-H, Meng W, Jürgensmeier JM, Kurie JM, Hong WK, Boyer JL, Herbst RS, Koo JS, A novel small molecule inhibitor targeting CREB-CBP complex possesses anticancer effects along with cell cycle regulation, autophagy suppression and endocpasmic reticulum stress. PLOS ONE 10, e0122628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC, β-Cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab. 25, 727–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talchai C, Xuan S, Lin HV, Sussel L, Accili D, Pancreatic β-Cell dedifferentiation as a mechanism of diabetic β-cell failure. Cell 150, 1223–1234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, Accili D, Evidence of β-cell de-differentation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 1044–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC, Marked expansion of exocrine and endocrine pancreas with incretin therapy in human with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 62, 2595–2604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monami M, Nreu B, Scatena A, Cresci B, Andreozzi F, Sesti G, Mannucci E, Safety issues with glucagon-like peptide 1 receptor agonists (pancreatitis, pancreatic cancer, cholelithiasis): Data from randomized controlled trials. Diabetes Obes. Metab. 19, 1233–1241 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Liu Y, Tian Q, Yang J, Lu R, Zhan S, Haukka J, Hong T, Incretin-based therapies and risk of pancreatic cancer in patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 20, 910–920 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Major A, Kamdar M, Management of non-diffuse large B-cell lymphoma post transplant. Curr. Treat. Options Oncol. 19, 33 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Martinez OM, de Gruijl FR, Molecular and immunological mechanisms of cancer pathogenesis in solid organ transplant. Am. J. Transplant. 8, 2205–2211 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Peterson SE, Garitaonandia I, Loring JF, The tumorigenic potential of pluripotent stem cells: What can we do to minimize it? Bioessays 38, S86–S95 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Karimian A, Azizian K, Parsian H, Rafieian S, Shafiei-Irannejad V, Kheyrollah M, Yousefi M, Majidinia M, Yousefi B, CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell. Physiol. 234, 12267–12277 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, Streeter PR, Naji A, Grompe M, Kaestner KH, Epigenomic plasticity enables human pancreatic alpha to β-cell reprogramming. J. Clin. Invest. 123, 1275–1284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brierley DI, Davidson C, Developments in harmine pharmacology—Implications for ayahuasca use and drug dependence treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 39, 263–272 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Callaway JC, Raymon LP, Hearn WL, McKenna DJ, Grob CS, Brito GS, Mash DC, Quantitation of N N -dimetyltryptamine and harmala alkaloids in human plasma after oral dosing with Ayahuasca. J. Med. Toxicol. 20, 492–497 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF, Induction of β-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenoviral delivery of cyclin-dependent kinase-4 and cyclin D1. Diabetes 53, 149–159 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, Cherok E, Takane KK, Scott DK, Stewart AF, Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 59, 1926–1936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Combination of DYRK1A inhibitors with GLP1R agonists yields synergistic increases in human β cell proliferation.
Fig. S2. The DYRK1A–GLP-1 increase human β cell numbers is a class effect for both DYRK1A inhibitors and GLP1R agonists.
Fig. S3. The effects of the harmine–GLP-1 combination on proliferation in non–β cells, β cell death, and DNA damage.
Fig. S4. Evaluation of the role of DYRK1A and cAMP signaling in harmine–GLP1R agonist synergy.
Fig. S5. GLP1R signaling diagram.
Fig. S6. Additional evaluation of the role cAMP pathway signaling in harmine–GLP1R agonist synergy.
Fig. S7. Mechanisms downstream of the GLP1R.
Fig. S8. Harmine–GLP-1 treatment maintains or enhances human β cell differentiation.
Fig. S9. A magnified confocal view of GLUT2 trafficking.
Fig. S10. Glucose-stimulated insulin secretion from normal and T2D islets.
Fig. S11. Effects of harmine–GLP-1 on β cells from individuals with T2D.
Fig. S12. Effects of the harmine–exendin-4 combination on human β cells in immunodeficient mice.
Fig. S13. Blood glucose values in euglycemic mice treated with saline, exendin-4, harmine, or their combination.
Fig. S14. Effects of the harmine–exendin-4 combination on normal organ histopathology and ductal proliferation in NSG mice.
Data file S1. Human islet donor demographic data.
Data file S2. Primary and secondary antisera.
Data file S3. qPCR primers.
Data Availability Statement
All data related to this study can be found in the paper or the Supplementary Materials.