

Abstract
Fibrosis has been shown to develop in individuals with underlying health conditions, especially chronic inflammatory diseases. Fibrosis is often diagnosed in various organs, including the liver, lungs, kidneys, heart, and skin, and has been described as excessive accumulation of extracellular matrix that can affect specific organs in the body or systemically throughout the body. Fibrosis as a chronic condition can result in organ failure and result in death of the individual. Understanding and identification of specific biomarkers associated with fibrosis has emerging potential in the development of diagnosis and targeting treatment modalities. Therefore, in this review, we will discuss multiple signaling pathways such as TGF-β, collagen, angiotensin, and cadherin and outline the chemical nature of the different signaling pathways involved in fibrogenesis as well as the mechanisms. Although it has been well established that TGF-β is the main catalyst initiating and driving multiple pathways for fibrosis, targeting TGF-β can be challenging as this molecule regulates essential functions throughout the body that help to keep the body in homeostasis. We also discuss collagen, angiotensin, and cadherins and their role in fibrosis. We comprehensively discuss the various delivery systems used to target collagen, angiotensin, and cadherins to manage fibrosis. Nevertheless, understanding the steps by which this molecule drives fibrosis development can aid in the development of specific targets of its cascading mechanism. Throughout the review, we will demonstrate the mechanism of fibrosis targeting to improve targeting delivery and therapy.
1. Introduction: fibrosis disease and cell-specific targeting
Fibrosis is characterized by excessive deposition of extracellular matrix components such as collagen in tissues or organs. It is an autoimmune and rheumatology-related condition that can develop in the heart, kidneys, liver, skin, and lungs, and1 is a process that develops in response to the body’s tissue repair activity. Homeostatic conditions in the body regulate the normal tissue repair process that is self-regulated. However, when the tissue repair process deviates from the regulatory mechanisms caused by pathological conditions, fibroblasts undergo a phenotypical change into myofibroblasts, which leads to uncontrolled accumulation of fibrotic tissue causing abnormal organ function and failure.2 There are multiple types of fibrotic diseases that can be organ-specific or systemic each having different pathologies. However, they all share common molecular reactions that result in the formation of fibrotic tissue because of the uncontrolled accumulation of ECM proteins and macromolecules.2 Fig. 1 presents the differences between normal organs and organs with fibrosis by depicting the accumulation of ECM proteins in the tissues of the fibrosed organs.3
Fig. 1.
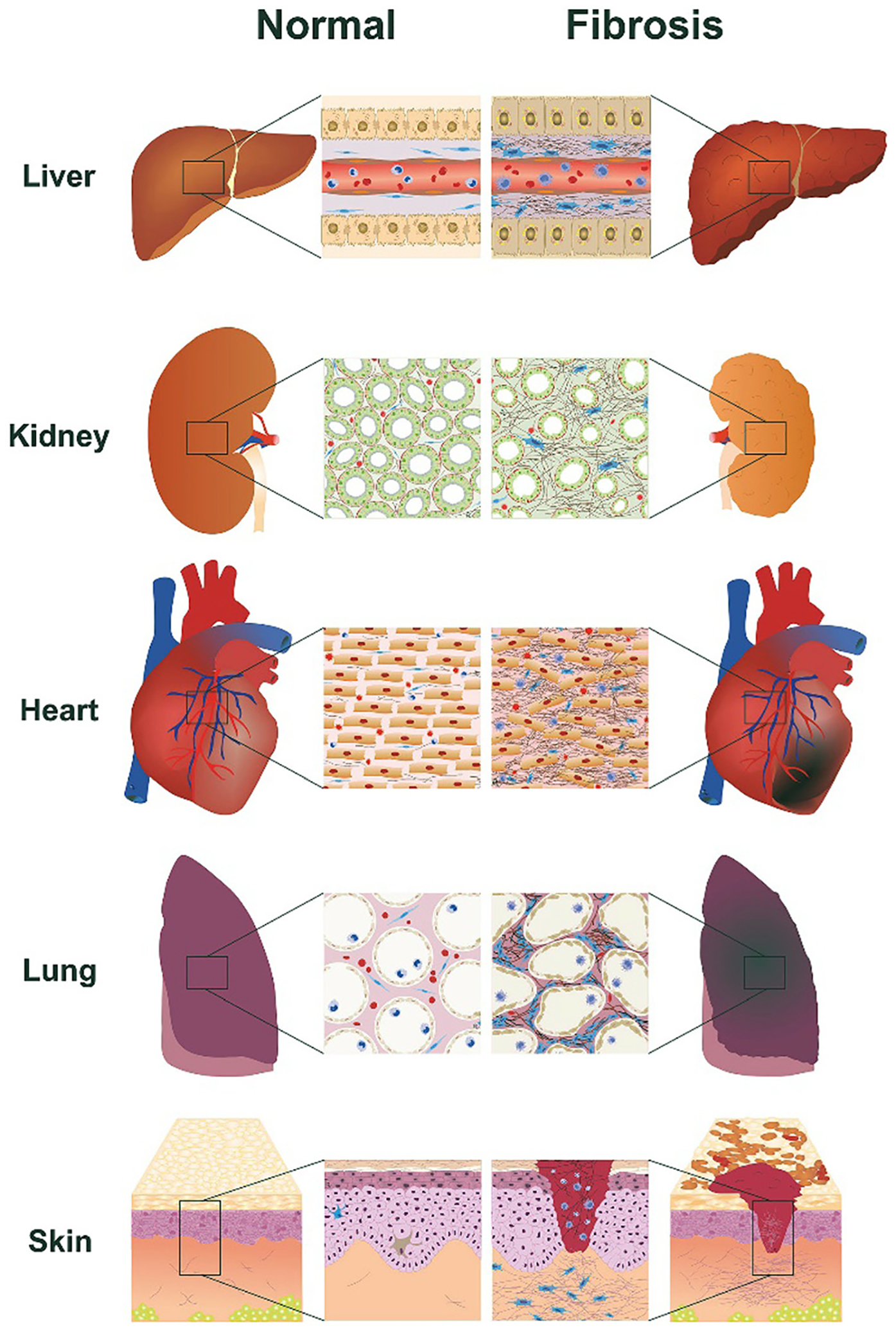
The differences between the liver, kidneys, lungs, heart and skin under normal conditions and the same organs with fibrosis. The fibrotic organs depicting the accumulation of ECM proteins in the tissues are shown in the magnified images. Reproduced with permission from ref. 3 Copyright 2018 Elsevier.
It has been widely accepted that myofibroblasts are the causative agent for the development of fibrosis. The fibroblast-myofibroblast transition (FMT) is a regulatory process where fibroblasts differentiate into myofibroblasts. This transition can be started by various stimuli, but ultimately the process will end in either myofibroblast apoptosis or fibrosis due to deviation from homeostatic conditions.4 This transition that falls out of cellular regulations leads to overexpression of ECM deposits resulting in α-SMA (alpha-smooth muscle actin) expression, leading to overproduction of fibrillar collagens driven by myofibroblasts.5
Myofibroblasts are contractile cells characterized by bundles of actin filaments that contain α-SMA which gives the myofibroblast its contractibility.6 Increased contractility facilitates the ECM overproduction by activating surrounding cells.7 Excess stiffness and overproduction of ECM deposits can further activate myofibroblasts. The fibroblast–myofibroblast transitions begin with the fibroblast being activated to a proto-myofibroblast phenotype.8 The proto-myofibroblasts contain collagen types I and III along with fibronectin EDA (Fibronectin extra domain A), an important protein used in wound healing closure.8 Mechanical tension, fibronectin EDA, and platelet-derived growth factor, help facilitate the transition of the fibroblast to the proto-myofibroblast.8 Increased cell motility, stress, and fiber formation are driven by PDGF. Once the fibroblast transitions to the proto-myofibroblast, the cell initiates expression of β- and γ-actin isoforms which are found in stress fibers along with N-cadherin.9 N-cadherin helps facilitate increased cell motility but does not have as high an adhesion force as OB-cadherin (Osteoblast-cadherin). OB-cadherin is expressed when the fibroblast fully transitions to the mature myofibroblast.10 This prolonged stress of the transition initiates the proto-myofibroblast to fully transition into a myofibroblast through the synthesis of α-SMA which gradually starts to form the stress fibers that phenotypically characterize myofibroblast cells.11 The presence of mature focal adhesions, expression of OB-cadherin and tensin, and lower motility/proliferation rate with enhanced contractility play the distinguishing factor in the full differentiation of fibroblast–myofibroblast transition (Fig. 2).10 The contractile nature expressed by these cells is shown as bundles of microfilaments rich with α-SMA ending with focal adhesions. The focal adhesions of myofibroblasts have been shown to be positive for integrins (α1, α3, α4, α5, αV, β1), vinculin, paxillin, talin, and tensin.Myofibroblasts are connected by gap junctions and have similar features to smooth muscle cells.12 Commonly expressed in mesenchymal cells, myofibroblasts express fibroblast surface protein (FSP) and vimentin.13
Fig. 2.
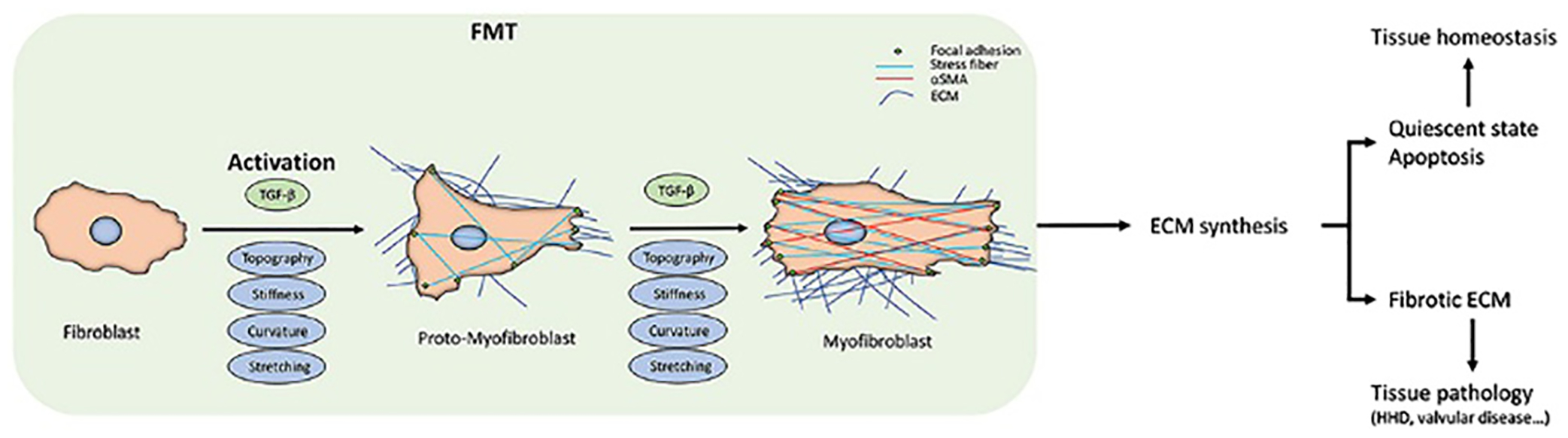
Fibroblast-to-myofibroblast transition (FMT). The scheme summarizes the FMT process, the corresponding changes in fibroblast behaviour, and the downstream effects at the tissue level. The transition starts from the fibroblast activation due to the different kinds of stimuli. The activation can sometimes be reversed or can proceed to the apoptosis of the myofibroblasts. When they escape these routes, due either to the persistent stimuli or to intracellular misregulations, FMT will lead to changes in the extracellular matrix (ECM) deposition and its architecture, driving the tissue into a pathological state. At the cellular level, FMT results are appreciable in the intracellular stress fibres and α-SMA expression. Reproduced with permission from ref. 15 Copyright, 2020 D’Urso and Kurniawam Frontier.
Since FMT is regulated by processes that are essential for normal body functions, using targeted treatment approaches is essential. FMT can be achieved through the development of targeted drug delivery systems. However, targeted drug delivery systems have limited progress due to various barriers. The cell-specific drug delivery systems can be more interested. Studies need to be conducted towards investigating the different types of materials and their specificity toward the cells and carbohydrates such as chitosan, polymers such as β-glucan, and inorganic materials such as boron nitrite sheets. With systems such as polymeric, inorganic, lipid, liposome, and dendrimers it is very important to explore the cell-specific uptake. Cell-specific targeted drug delivery enables prolongation of circulation time and reduces toxicity induced on cells and tissue. This system is novel to fight a range of disorders and we envision employing cell-specific targeting to develop treatments to target fibrosis. Specific biomarkers expressed on the cell membrane which are unique to fibrosis can be utilized to design the targeting therapeutics and delivery of the drug. Recently, the cell-specific targeting approach showed promising results compared to traditional approaches used for therapy.14 Working on the development of a targeted drug delivery system for cancer and diabetics has made great progress. Mainly, the focus is on the development of cell-specific drug delivery systems. Different types of materials and their specificity toward the cells were widely investigated. Currently, carbohydrates such as chitosan, polymers such as β-glucan, and inorganic materials such as boron nitrite sheets are the most interesting. Drug delivery systems such as polymeric, inorganic, lipid, liposome, and dendrimer ones are very important to explore cell-specific uptake. Cell-specific targeted drug delivery enables prolongation of circulation time and reduces toxicity induced on cells and tissue. This system is novel to fight a range of disorders.
Specific biomarkers expressed on the cell membrane which are unique to fibrosis can be utilized to design the targeting therapeutics and therefore the delivery of the drug. Our group has been researching cell-specific targeted drug delivery for over a decade and reported various unique approaches in cancer targeting for specific diagnosis and treatment. For instance, we have developed a quantum dot nanoparticle specific to the HER-2 receptor of breast cancer cells for targeted therapy and imaging.14 The studies conducted on a tumor-bearing mouse shows that the near-IR nanoparticle decorated with a biocompatible polymer and Herceptin monoclonal antibody significantly inhibit tumor growth within 30 days of intravenous administration. We have also developed a photo-sensitizer nanoparticle composed of heparin/gold,16 and a hybrid nanoparticle of iron oxide coated gold for target-specific photodynamic and photothermal therapy for cancer.17 Along the same line, we later reported the development of graphene derivatives for target-specific cancer imaging and phototherapy.18 Later, to improve the targetability of the graphene, we modified its surface with polydopamine to target the dopamine receptor,19 and with hyaluronic acid to target the CD44 receptor of specific cells.20 Our team has also been investigating oral delivery systems specific to bile acid transporters, and M-cell targeting, to improve the oral delivery of biologics, small molecules as well immunomodulators.21–24 Moreover, we have recently published a review article on the importance of cellular and sub-cellular specific targeting delivery systems and their mechanism.25 With our given expertise in cell-specific drug delivery and interest, we have further expanded our research program beyond cancer and are now vastly focused on myofibroblast specific targeting delivery of therapeutics to improve fibrosis treatment.
2. Identification of key cellular biomarkers and their targeting mechanism and method of integration
2.1. TGF-β as a catalyst for fibroblast–myofibroblast transition
Pathways that result in the initiation of fibrosis can be activated in various ways. TGF-β (transforming growth factor-β) has been shown to be a major driver of fibrosis progression. TGF-β operates by a canonical or Smad mediated pathway and a non-canonical or non-Smad mediated pathway.26 In the Smad mediated pathway TGF-β1 receptor I (TGF-β-RI), can also be referred to as activin-like kinases, where ALK-5 is the most common in terms of fibrogenesis.27 TGF-β receptors are regulated by phosphorylation of the heterotetrameric TGF-β latent complex.28 Phosphorylation activates the latent complex of TGF-β receptors which leads to the activation of Smad/non-Smad pathways.28 The activation forms a trimeric complex that eventually translocates across the nuclear membrane to aid transcription factors in transcribing specific target genes.13 In non-Smad signaling pathways which are independent of R-Smad, TGF-β fibrosis effects are activated by cell-specific reactions.26 TGF-β can activate PI3K (phosphoinositide 3-kinase) which activates p21-activated kinase/c-Abelson kinase along with Akt-mTORIC1 initiating a cascade of events that upregulate collagen production.29 JNK activation also leads to an increase in the highly potent transcription factor c-Jun. If TGF-β is phosphorylated on a tyrosine residue on TGF-β-RII this leads to the regulation of myofibroblast formation and ECM synthesis through the activation of ERK1/MAPK (Fig. 3).13
Fig. 3.
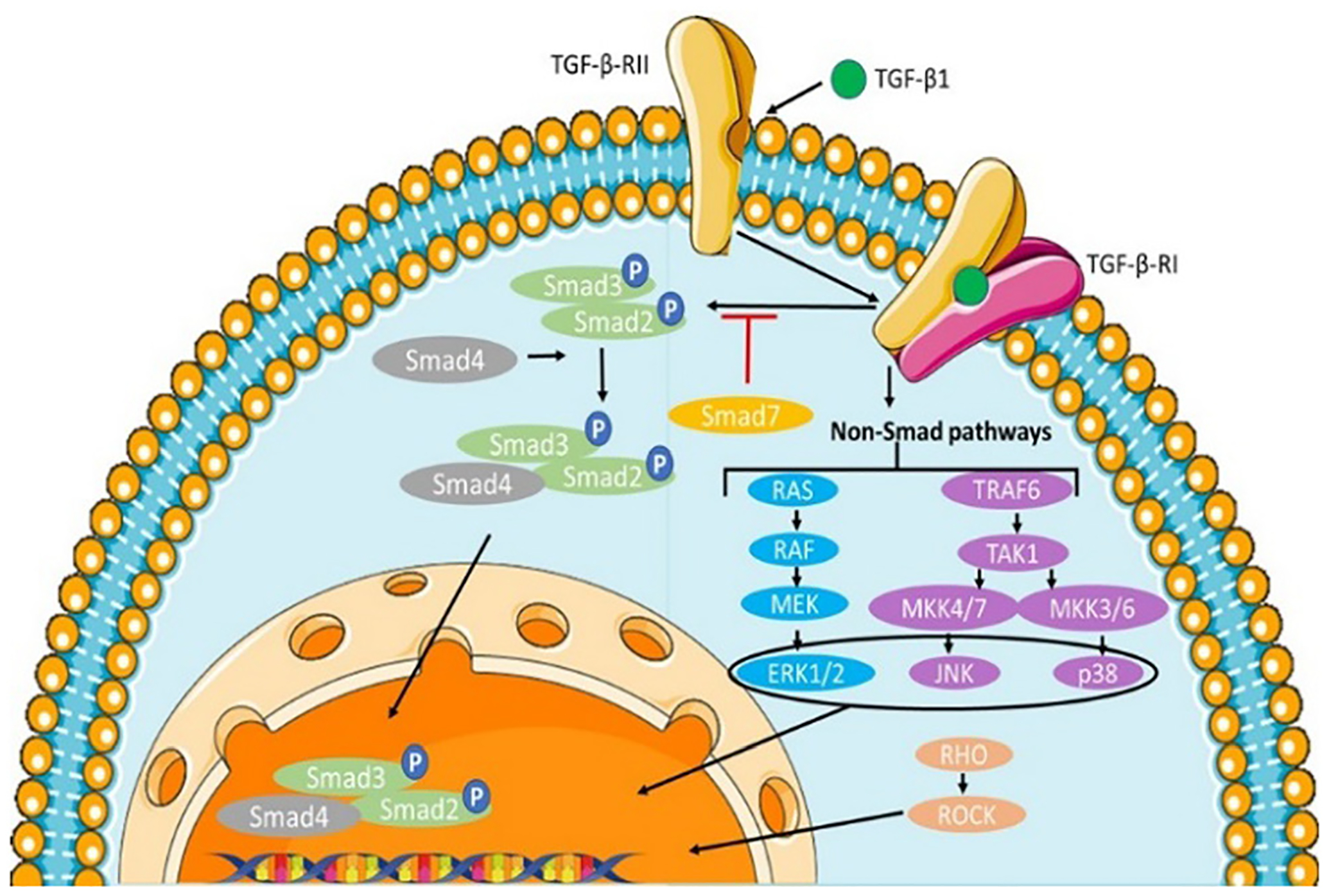
Canonical and noncanonical pathways of TGF-β signaling.
Activation of TGF-β also stimulates the other profibrotic mediators including connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF).30 Bone morphogenic growth factors are a member of the TGF-β family that also regulates fibrogenesis. However, they have also been shown to have anti-fibrotic effects specifically with BMP-6, BMP-7, and BMP-9. BMP signaling is like TGF-β pathways. BMPs bind to their specific cell surface receptors initiating a cascade of phosphorylation events that activate Smad1/5/8. The heteromeric complex binds with Smad4 for translocation to the cell nucleus inducing specific fibrogenic transcription factors.13
TGF-β is an essential molecule that regulates multiple systems in the body including cell proliferation and differentiation, wound healing, adhesion, and migration of immune cells.30 This molecule comes in three isoforms; TGF-β1 which is the most expressed isoform found throughout the body, TGF-β2 found in epithelial cells, and TGF-β3 found in mesenchymal cells.31 The TGF-β working mechanism is through the Smad mediated signaling cascade. It is a family of protein growth factors also called cytokines, that have a similar dimeric structure and have a “cysteine knot structural motif”.32 TGF-β is a known driver for fibrosis disease. TGF-β mediates diseases like lung, kidney, skin, and liver fibrosis not only by producing an excessive amount of ECM but by recruiting and activating monocytes and fibroblasts.33 The three isoforms, TGF-β1, TGF-β2, and TGF-β3 play an important role in various fibrotic diseases once they are activated. TGF-β1 has been demonstrated in lung, liver,15 and kidney fibrosis and is fundamental in regulating cellular processes and the production of ECM components such as collagen, fibronectin, and elastin.13 It binds to specific cell surface receptors TGF-β-RI and TGF-β-RII,33 that drive the cascade for EMT and fibrogenesis. The TGF-β mechanism of operation is a downstream regulatory process involving Smad2, Smad3, Smad4, and Smad7. Smad2 and Smad3 are the main vehicles of this process while Smad7 is shown to be a fibrosis antagonist that protects against fibroid tissue accumulation.13
The three isoforms of TGF-β are secreted by different cell types in latent complexes. These consist of a mature TGF-β dimer bound by a disulfide bond where hydrophobic interactions strengthen the bond.34 This pro-peptide dimer is noncovalently bonded to a latency-associated protein (LAP), which is linked to the latent TGF-β binding protein (LTBP). The noncovalent association of LAP creates the latency of the complex as well as the high binding affinity to TGF-β. LAP and LTBP are also bound through a disulfide bond.35 LTBP is made up of epidermal growth factor-like repeats and 8 cysteine residue domains; the disulfide bond to LAP can only happen on the third cysteine residue while the others help the LTBP localize in the ECM.35 The latent complex allows for storage of TGF-β to prevent unnecessary activation and signaling of the protein. However, an activation event is necessary to make the latent TGF-β biologically active.13 The activation process is usually dependent on the cell type and physiology of the surrounding area but in general, activation can occur through matrix metalloproteases (MMPs), plasmin, and thrombin. Regardless of the activation event, the activator targets the LTBP of the latent complex releasing TGF-β from the latent complex to allow binding to TGF-β receptors on the surface of the cell membrane.31
The canonical signaling pathway is shown in Fig. 3. In the canonical singling pathway, TGF-β binds to TGF-β receptor type II, which recruits and phosphorylates TGF-β receptor type I. The activated TGF-β-RI leads to Smad2/3 phosphorylation. The Smad2/3 complex then binds to Smad4 and translocates into the nucleus, where it induces gene transcription. The signaling can be inhibited by negative feedback of Smad7 on the TGF-β-R1.36
Activation of TGF-β begins by targeting LTBP for the release of TGF-β1 by proteolytic cleavage from the extracellular matrix,37 which then allows TGF-β1 to bind to integrins on the surface of the cell membrane. Plasmin, a serine protease, and metalloproteases (MMP-2 and MMP-9) are enzymes that degrade ECM to promote the activation of TGF-β. TGF-β1 can also be activated by αvβ6 integrin because TGF-β1 has an RGD integrin binding sequence.38 LTBP’s N-terminus is covalently linked to the ECM by an isopeptide bond.38 TGF-β1 binds to serine/threonine auto phosphorylated receptor protein TGF-β-RII to recruit and phosphorylate TGF-β-RI (ALK-5 expressed most in epithelial cells) into the receptor complex. Since TGF-β is a structure composed of two bound monomers, the resulting structure of the two activated receptors is a tetramer of two TGF-β-RII molecules and two TGF-β-RI molecules. Two molecules of TGF-β-RI stabilize TGF-β-RII dimers through ligand binding. Phosphorylation of the short juxta membrane glycine and serine-rich domains by TGF-β-RII kinases on TGF-β-RI serine and threonine induces a conformational change activating the TGF-β1 receptor kinase; the GS domain of the activated receptor gives the surface of interaction for MH2 domains of Smad2 and Smad3. At the same time, immunophilin inhibitor FKBP12 is released which helps maintain the TGF-β1 latent state of the unattached TGF-β receptor. Activated TGF-β1 phosphorylates two serine molecules on the C-terminus of the R-Smad allowing interaction that weakens the R-Smad binding to TGF-β1 to release the activated R-Smad from the receptor.39
The activated serine/threonine of TGF-β-RII activates TGF-β-RI receptors on the glycine and serine domain (GS domain) in the juxta membrane domain of the receptor.29,40,41 The activated TGF-β-RI receptor phosphorylates two members of the R-Smad family;40 Smad2 by ALK5 and Smad3 by ALK4 on a serine residue at the C-terminal.42 The Smad2/Smad3 complex detaches from the receptors into the cytoplasm of the cell to then join with a co-Smad, Smad4, together forming a hetero-trimeric complex.42 The trimeric Smad complex is translocated into the nucleus where it acts as a regulator for transcription by binding to DNA to target specific genes.43 The MH1 domain located at the C-terminal of the complex drives the translocation to the nucleus, while the MH2 domain of the N-terminal controls the binding affinity to DNA.44 CAGAC is a DNA sequence recognized by Smad proteins where the binding takes place, however, there is a low binding affinity, so Smad proteins associate with partners to bind to DNA.45 The heteromeric Smad complex translocates to the nucleus where binding to DNA proteins FAST1, FAST2, Fos/Jun, ATF2, TFE3, PECP2/CBF, and the Vitamin D receptor assists in specific genes such as integrins, E-cadherin, and collagen.46 Target genes AGAC or GTCT sequences are the most commonly efficient for TGF-β transcription that may also help regulate and restore Smad complexes.33
Besides the R-Smad (receptor-regulated Smad) pathway, TGF-β can also activate non-Smad signaling pathways. TGF-β has been shown to activate stress-induced kinases such as p38 and Jun N-terminal kinase (JNK) which can lead to EMT or apoptosis.47 Through activation of extracellular-signal-regulated kinases (ERK1 and ERK2) by mitogen-activated protein kinase (MAPK), initiated by TGF-β, it is also possible to induce EMT. Stimulated by TGF-β activation, activation of RhoA, Cdc42, and Rac (Rho GTPases) can lead to reorganization of the cytoskeleton, motility, and invasion of cells.48
While inhibiting TGF-β signaling has been challenging due to several automatic phosphorylation40 events designed to regulate TGF-β, there are some pathways that can stop Smad cascades. One example is with Smad7 which acts in a negative feedback loop to stop TGF-β signaling.41 Smad7 prevents formation of the Smad2 and Smad3 complex by preventing interaction with TGF-β-RI.41 Smad7 is also capable of seques-tering Smurf1/2 (ubiquitin ligases) and protein phosphatases to active TGF-β to stop its signals. Another means of TGF-β regulation is through clathrin-independent/dependent mechanisms of internalization by endocytic adapters to TGF-β receptors.49 Within the nucleus the Smad2/Smad3 complex can also be degraded by ubiquitin-proteasome mediation and dephosphorylated;50 in each case, the signaling process terminates. Epidermal growth factor and oncogenic Ras signaling can also negatively regulate nuclear translocated Smad signaling.
Intracellular kinase inhibitors such as MAPK kinases, calcium-calmodulin-dependent protein kinase II, cyclin-dependent kinase CDK2, casein kinase, protein kinase C, and G protein-coupled receptor kinase 2 (GRK2) can also stop Smad signal transduction through phosphorylation of the linker domain of R-Smads at serine and threonine locations.51
Regulation of TGF-β is modulated by various feedback mechanisms not only at the receptors but also through upstream and downstream mechanisms to terminate TGF-β signaling.52 One such mechanism is through Smad7 which happens at the receptors. Through TGF-β induction, Smad7 forms a complex with Smurf ligase and PP2C phosphatase to bind with TGF-β-RI; this mechanism promotes the ubiquitylation of the two proteins to initiate degradation and depho-sphorylation of the receptors which eventually leads to deactivation of the TGF-β-RI.40 While Smad7 can inhibit TGF-β type 1 receptors (ALK-5), Smad6 prefers binding to BMP type 1 receptors ALK-3 and ALK-6 to inhibit the signaling cascade.40
While R-Smad signaling plays a crucial role in the TGF-β signaling pathway mediating fibrosis, non-Smad signaling pathways can also drive TGF-β signaling through specific cell-mediated effects.53 One mechanism is through the activation of Erk1/2 MAP kinase by phosphorylated Shc tyrosine residues in TGF-β-RI.54 Ras nucleotide exchange protein Grb2 forms a complex with Sos1 and associates with the previously mentioned phosphorylated tyrosine residue activating the Ras protein to activate Erk1/2 MAP kinase.55
It has also been shown that ubiquitin ligase tumor necrosis factor receptor-associated factor 6 (TRAF6) plays an important role in several non-Smad pathways.56 Stimulation of TGF-β enhances TRAF6 binding to the juxta membrane part of TGF-β-RI promoting its activation.40 TAK1 is then activated through the ubiquitination of MAP-kinase by TRAF6.57 MAP kinase 3 or 6 is activated through phosphorylation by TAK1 which in turn activates p38 MAP kinase.57 P38 is an essential regulator of apoptosis and EMT through TGF-β induction.47 The three kinases come closer by binding to Smad7 and then to TGF-β-RI.40
P12 activated kinase (PAK2)-Abelson kinase (Abl) and Akt-mTOR1 are two profibrotic pathways that are activated by PI3K by the stimulation of TGF-β. C-Abl targets PKCδ/Fli-1, Egr, and Smad1 downstream fibrotic mediators.58 For Smad1 activation, activated c-Abl is required for the phosphorylation of protein kinase C-δ (PKC-δ). Since this process leads to the phosphorylation of the Fli-1 transcription factor which reverses its inhibition of collagen gene expression.58 Another profibrotic transcription factor is c-Jun which is activated through the activation of Jun N-terminal kinase (JNK).59 TGF-β-RI may also be phosphorylated on tyrosine residues which leads to Erk1/2 MAPK activation, important for myofibroblast regulation and the synthesis of matrix proteins.40 Connective tissue growth factor (CTGF/CCN2) is shown to play an important role in mediating tissue fibrotic responses, especially for SSc tissue fibrosis.60 In vitro studies have shown that overexpression of CTGF leads to fibrosis. TGF-β also promotes CTGF synthesis in multiple cells including fibroblasts, vascular smooth muscle cells, and endothelial cells, and may even act as a mediator to enhance TGF-β-activated fibrosis.61
The Wnt-signaling pathway has also been found in multiple fibrotic diseases such as pulmonary, renal, dermal, and liver.62 β-catenin is critical in canonical Wnt signaling. Proteins of Wnt signaling are ligands that send signals across the plasma membrane by interaction with Frizzled receptors and low-density lipoprotein.63 This signaling process disrupts the complex of adenomatosis polyposis coli (APC), axin, glycogen synthase kinase-3β (GSK-3β), and casein kinase, to stabilize β-catenin for transportation to the nucleus where it aids in the transcription of target genes.
Not only does TGF-β-RI phosphorylation contribute to activation of other signaling pathways, but TGF-β-RII phosphorylation can also activate different substrates.64–66 One example is through the interaction with Par6, the polarity complex protein. Par6 undergoes a phosphorylation process by TGF-β-RII after it interacts with TGF-β-RI who then presents it to RII.40 After this, Smurf1 is recruited and ubiquitylates RhoA, a small GTPase, for degradation.40 RhoA is an important protein for regulating actin filaments that are made by the accumulation of α-SMA found in myofibroblasts;67 degradation of RhoA interferes with maintaining tight junctions. For BMP, LIM kinase 1 is recruited by the BMP-RII C-terminal but remains inactive and therefore cannot phosphorylate cofilin.68 Cofilin inhibits the polymerization of actin, and since LIMK1 inactivates this protein BMP-RII aids in limiting actin polymerization.68
Pits in early endosomes coated in clathrin internalize the receptor complexes of TGF-β receptors, which is needed for the activation of Smad.69 Interaction with β2-adaptin of the clathrin-associated adapter of the AP2 complex is necessary for the internalization.69 Inside the endosome the receptors meet the Smad anchor for receptor activation (SARA) which helps facilitate the Smad2 presentation for TGF-β-R1.70 SARA works with two proteins, tumor suppressor protein cytoplasmic promyelocytic leukemia (cPML) and adaptor protein disabled-2 (DAB2), that stabilizes the complex. Internalization is a necessary step for Smad molecules because if the clathrin-dependent pathway interferes with R-Smad signaling and phosphorylation does not take place thereby making the Smad molecules incapable of binding to TGF-β receptors. Recycling of TGF-β receptors is also possible through adaptor CIN85 independent of Rab11 recycling vesicles for TGF-β-RI, and DAB2 adapter protein for TGF-β-RII where both can occur with or without ligand binding.71 Intracellularly there is a reservoir of TGF-β receptors that can be moved to the surface of the cell through phosphorylation of Rab-GTPase induced by Akt.26 PI3-kinase activates Akt which makes cells more susceptible to stimulation by TGF-β. The phosphorylation of β-glycan at the carboxyl tail by TGF-β-RII also promotes clathrin-dependent endocytosis.40 Independently of TGF-β, its receptor II can bind the retromer vacuolar protein-sorting 26 (Vps26).72
Besides clathrin-coated internalization, receptors can also be internalized by lipid rafts into caveolin-coated vesicles. This type of internalization promotes degradation of the TGF-β receptors. Degradation takes place in proteasomes and lysosomes after interaction with the ubiquitylation complex of Smad7/Smurf2.28 Clathrin-dependent or clathrin-independent internalization can also happen to β-glycan, the protein that anchors TGF-β receptors to the surface of the cell membrane. For successful activation of Smad2 and p38 map kinase β-glycan internalization is required.40 For β-glycan endocytosis promotion, β-glycan phosphorylation on Thr841 by TGF-β-RII fosters the interaction of β-arrestin which mediates flotillin of lipid rafts necessary for the endocytosis process. In competition with Smad7 binding which prevents receptor degradation, TGF-β-RII binds to transmembrane metalloproteinase ADAM12 to encourage the localization of receptor II to the early endosomes.40
The tissue repair response is a necessary and important function of the body to repair damaged tissues.73 This process is heavily regulated, however, when it becomes dysregulated it can have devastating effects on various organs in the body, especially in the case of chronic inflammatory disorders. When there is an injury in the body, the immune response is activated which promotes the production of extracellular matrix proteins such as collagen and fibronectin which is a normal function of tissue repair for bodily organs.73 Upon tissue injury, fibroblasts of the surrounding injured area become activated which increases mechanical functions such as cellular contractility which induces the secretion of inflammatory cytokines to begin the production of ECM proteins, initiating the body’s wound healing response.74 When the damage that occurs to a tissue is a minor injury or does not happen chronically the production of ECM deposits helps fix the damaged area and cease ECM component production.73 However, when there is chronic or repetitive injury to the tissue this produces a prolonged inflammatory response that dysregulates the normal wound repair response leading to excessive accumulation and production of ECM proteins making the surrounding tissue stiffer, adding to the synthesis of proteins such as collagens which eventually lead to organ failure.75 Recent studies have shown that inflammation could be a cause of fibrosis as embryonic tissue repair and tissue repair of immature tissues have the ability to heal without scarring.
Another pro-fibrotic trigger found in several tissues that activates the Smad 2/3 cascade is activins. Activins promote the proliferation of fibroblasts as well as their transition into myofibroblasts. In diseases like cystic fibrosis, chronic kidney disease, and heart failure activins have been found in increased concentrations.27 Activin is a ligand of TGF-β signaling through activating type 2 receptors known as Act-RII-A/Act-RII-B. A complex can be formed with four proteins, activin A, activin B, myostatin GDF-8/GDF-11, and Act-RII-A/Act-RII-B to initiate signaling by the Smad2/3 complex.76 Activin does not overlap with other ligands of TGF-β in terms of their bioactivities since activin is specific to the cell/tissue type. Activin has a specific preference for binding with type one receptors such as ALK4, ALK5, and ALK7.77 One distinct difference between activin is that unlike TGF-β isoforms, activin is released in an active form. Due to this, their activity has to be regulated to prevent excessive signaling.78 Activin is regulated by follistatin outside of the cell.79 Follistatin binds to receptor sites of activin ligands to limit their potential signaling.79 Despite this difference, like TGF-β isoforms, activin has an affinity for ECM binding proteins such as heparin-sulfated proteoglycans (HSPG).27 In fibrotic diseases such as human idiopathic lung fibrosis, Duchenne muscular dystrophy, liver disease, and kidney fibrosis, HSPGs are upregulated as well as heparinase which enhances the metabolization of the carbohydrate chains of such proteoglycans. Enhanced activity of heparinase drives the release and activation of activin and TGF-β. Due to activin’s high affinity for HSPG, there is a supplemented pool of growth factors in the ECM.27
3. Targeting fibroblast-derived proteins?
Collagen is the main protein of the ECM constituting between 30–70% of the total proteins present in all tissue types. Collagens are highly upregulated in fibrotic diseases and hence, the reduction of collagen biosynthesis, cross-linking and deposition is the primary target for new anti-fibrotic drugs to evaluate the fibrosis in vitro and in vivo.1,80,81 Ultimately, the drugs, which target the central pathway (TGF-b, CTGF and mTOR), affect the collagen synthesis and maturation but some drugs are not anti-collagenic due to the multiple phenotypic traits.81–83 Hence, the reduction in collagen biosynthesis and stopping the induction of its maturation has gained more attention as a therapeutic target during the last decade. However, collagen is an omnipresent protein in the body responsible for the important functions, such as a macromolecular scaffold, regulation of growth factors and receptor binding in each tissue. Hence, it is challenging to target the disease-specific collagen pathways (e.g. La-related protein 6 (LARP6); hsa-miR-29; Collagen Prolyl-4-Hydroxylase; FK506-binding protein 10 (FKBP10); heat shock protein 47 (HSP47); Transport and Golgi organization protein 1 (TANGO1); Lysyl Oxidases; Matrix metalloproteinase (MMPs)) without affecting right collagens.81
In all types of fibrosis such as lung, kidney, liver, and skin, the activated fibroblasts and their myofibroblasts play crucial roles in excessive essential structural productions of ECM proteins (majorly collagens I and III, and fibronectin),84 in all types of fibrosis.64–66 The fibroblasts and myofibroblasts are responsible for the production of large quantities of other proteins such as elastin, specialized adhesive proteins (e.g., laminin), or molecules dconstituting structural proteins with high water, ion, and growth factor binding capacity (space-filling hydrated gels of proteoglycans and glycosaminoglycans).7
The production of ECM components by fibroblasts is tissue-specific e.g., lung,64 skin,65 and liver.66 In addition, fibroblasts play a crucial role in the maintenance of the ECM and connective tissue, which is regulated by the secretion of proteolytic enzymes, mainly MMPs, as well as MMP inhibitors, largely tissue inhibitors of metalloproteinases (TIMP)-1.85
Amongst various collagens synthesized in the ECM, collagens type I (~80%), III (~15%), and V (<5%) are major products and the rate of estimated collagen molecule synthesis is 40 molecules/cells.86,87 The other types of collagens (type VI, XII, and XIV) are very limited in production but they are still considered modulators found on the surface (types XII and XIV) of the thick collagen fibrils (composites made up of collagens type I, III and V) which regulates the interaction with cells and other ECM components.88 The fibroblasts also produce fibrillar collagens (type I, III, and V), fibril-associated collagen (type XII or XIV), and interfibrillar micro filamentous collagen (type VI). The fibroblasts may produce α1(IV) and α2(IV) chains which are associated with the basement membrane collagen89–91 because it is reported that the fibroblast type IV collagen conditions their microenvironments and may reduce their migration.90
3.1. The role of collagens in organ-specific fibrosis
Activated fibroblasts (myofibroblasts) are primarily responsible for fibrotic ECM deposition in various organs such as the lung, liver, kidney, and skin.92 For example, the deposition of interstitial collagens (type I, III, and VI) are increased in the alveolar region when idiopathic pulmonary fibrosis (IPF) occurs.93 In the kidney, skin, and liver fibrosis, the deposition of the interstitial micro filamentous collagen (type VI) is increased with an increased risk of mortality.94
Collagens (type I and III) deposition are increased when the fibrosis progressed from mild to moderate to advanced (cirrhosis) which makes the ECM more dense and less flexible.94 The various types of collagens within the liver play distinct roles within the ECM and multicellular context. In addition, the collagens can be synthesized predominantly by non-fibroblastic cells both in rodents and in humans in vivo. For instance, the activated biliary cells and sinusoidal endothelia synthesize collagen type IV while type XVII are synthesized by hepatocytes and bile duct epithelial cells in advanced fibrosis stages.
The most relevant examples are the synthesis of collagen type IV and other basement membrane proteins by activated biliary cells and sinusoidal endothelia95 and collagen type XVIII by hepatocytes and bile duct epithelial cells, with an only modest up to 2-fold increase even in advanced fibrosis stages.96 Hence, the elevated level of type XVII collagen in liver fibrosis and cirrhosis is observed due to the high production of fibrillar collagens in hepatocytes and bile duct epithelial cells.
In addition, type IV collagen and other basement membrane components such as fibronectin are also increased and surrounded near myofibroblasts with the loss of a thin ECM network.97 Therefore, fibrosis is considered a complex disease that involves a change in the compositions of collagens including non-collagen ECM molecules with a relative loss of some types of collagens and the overexpression of others, or with collagens deposited “at the wrong place.”
Apart from the fibrillar collagens, other non-fibrillar collagens are accumulated forming a complex of branched networks by an association of larger collagen fibrils with each other.98 Thus, the normal ECM components are lost due to changes in collagen composition and location when liver fibrosis progresses to cirrhosis.99
3.2. Collagen binding mechanism in hemostasis, wound healing, and fibrosis
Collagens help in the initiation of coagulation and maintain the hemostatic environment by platelet activation and these platelets are separated from the ECM under normal physiological conditions.100 The basement membrane is destructed when the vascular and epithelial cells are damaged, and it can be extended towards the interstitial membrane if the damage is severe enough.101 This phenomenon exposes the collagens to platelets in the basement and interstitial matrix resulting in platelet adhesion, activation, and aggregation. This process is very important for clot formation and wound healing activity. Moreover, it induces the release of platelet-derived growth factor (PDGF) and TGF-β which are responsible for fibroblast activation.102
The collagens mostly bind with three main platelet receptors viz. integrin α2β1-, VWF-, and glycoprotein VI (GPVI)-receptors at the site of injury.103 The collagens type IV and VI directly interact with platelet receptors GPVI and α2β1 binding through a Gly-Pro-Hyp motif and a triple-helix motif (GFOGER) specific for type I and IV collagens.103
In addition to these receptors, the glycoprotein Von Willebrand Factor (VWF) also interacts with the collagen and platelet receptors GPIb and α2β3 indirectly and this interaction plays an important role in wound healing by facilitating the adhesion of platelets to collagens in the ECM, maintaining hemostasis and bleeding disorders such as Von Willebrand Disease (VWD).104 Collagens type I and III majorly interact with VWF via the A3 domain and play a crucial role in the binding with the activated platelets upon release of the agonists ADP and thromboxane A2.105
The coagulation and platelet activation together eventually forms the fibrin clots that heal the lesion, but they also act as a substrate and create the framework for tissue remodeling.106 If collagen remodeling is improper or the collagen turnover is uncontrolled, it can lead to malfunctioning of wound healing and the hemostasis process and generates bleeding disorders and fibrosis as well.101 The interstitial collagens, fibrillar collagens (type I and III), and microfibrillar type VI have crucial roles in the initiation of the fibrotic process. In addition, mutations of collagen type I and III are responsible for vascular and ECM related diseases such as Ehlers-Danlos syndrome.107,108
Scleroderma is the finest example for understanding collagen binding which involves excessive collagen fiber deposition in the skin layers leading to systemic sclerosis (SSc) with multiple organ manifestations such as the lung and gastro-intestinal tract in which vessel integrity and function is altered. Fibroblast activation and ECM production are primary hallmarks of SSc resulting in platelet activation by endothelial damage done by reactive oxygen species.109
Platelet activation is a central driving force of fibrosis, which was illustrated in a study of liver fibrogenesis, and it was determined that all PDGF-AB and -BB activity was due to platelet activation.110 It was concluded that anti-platelet therapy with aspirin was effective to reduce fibrosis progression and a potent PDGF-B neutralizing antibody. Moreover, activated platelets induce the secretions of other proinflammatory and fibrotic cytokines, chemokines, and TGF-β1 (most potent profibrogenic factor) when clot formation happens and make an important contribution to fibrosis in SSc and liver fibrosis.111
In addition, there are several reports which outline the agonistic activity of anti-PDGFR antibodies towards fibroblasts in vitro.112 Hence, platelets are a primary source of cytokines and chemokines which are responsible for fibrosis of SSc.113,114
3.3. Angiotensin
3.3.1. Angiotensin.
Angiotensin was isolated in the late 1930s and subsequently characterized and synthesized by groups at the Cleveland Clinic and Ciba laboratories.115 Angiotensin I is officially called proangiotensin. There are four types of angiotensin termed angiotensin I–IV. The major and active form of the hormone is angiotensin II. If the level of angiotensin is lower than normal it can cause hypotension, hyperkalemia, hyponatremia, and water loss through urine.116 When the blood pressure is low, kidneys release the enzyme renin into the bloodstream. Renin splits angiotensinogen, a protein made in the liver and releases pieces of it; one of the pieces is angiotensin I, an inactive form. Its further splits by angiotensin-converting enzyme in the lungs and kidneys.116 The other piece is angiotensin II, an active hormone. When the level of angiotensin is higher than the normal level, it can cause water retention and hypertension. Hence angiotensin is a target for fibrosis.
3.3.2. Role of angiotensin in fibrosis.
Angiotensin is linked to a group of hormones, enzymes, proteins, and reactions that help in blood pressure regulation. Angiotensin has a complex series of effects such as stimulating aldosterone release from adrenal glands and antidiuretic hormone from the pituitary gland, enhancing blood pressure by narrowing blood vessels, and triggering the thirst sensation.116,117 Aldosterone releases potassium through urine and enhances sodium in the blood-stream resulting in retention of water.116 Hence, blood pressure increases. The renin-angiotensin-aldosterone system (RAAS) is also activated by other hormones such as corticosteroids, estrogen, and thyroid hormones. If there is any issue among these it can affect blood pressure, sodium, and potassium levels.116
The renin-angiotensin-aldosterone system plays a crucial role in normal myocardial function. During connective tissue repair, activated fibroblasts relocate to the wound area and then synthesized the new extracellular matrix. Proteins such as angiotensin II contribute to myofibroblast differentiation and persistence. However, the elevation of angiotensin II (Ang II) is associated with hypertension, myocardial hypertrophy, fibrosis, and bad remodeling. A lack of treatment results in heart failure. Fibrotic diseases are major causes of mortality and morbidity globally. They are an economic burden to the healthcare system. This is mediated through Ang II receptor AT1. Signals from AT2 are opposed to AT1-dependent pro-inflammatory, pro hypertrophic and pro-fibrotic effects. Ang II upregulates matrix metalloproteinases (MMPs) which promote ECM degradation. This results in increased cardiac fibroblast migration and proliferation, ECM deposition, fibrosis, and adverse remodeling. The levels of MMPs are very low at normal heart function. Following injury, activation of MMP2, MMP9, MT1-MMP, and MMP7 promotes ECM breakdown and hence results in fibroblast invasion and proliferation, and fibrosis. Siddesha et al., studied fibroblast migration via differential regulation of matrixins and RECK. RECK is a unique membrane protein that regulates MMPs. Ang II infusion for 2 weeks induced cardiac fibrosis and enhanced levels of MMPs 2, 7, 9, and 14. But RECK expression was significantly reduced. These effects are inhibited by AT1 and losartan codelivery. They demonstrated that Ang II suppresses RECK and induces MMPs in vitro and in vivo. RECK overexpression blunts Ang II-induced MMP activation and cardiac fibroblast migration.118
3.3.3. Targeting angiotensin
3.3.3.1. Targeting RAAS in cardiac and renal fibrosis.
Recently cardiac fibroblasts have been found to possess a functional intracellular renin-angiotensin-aldosterone system (RAAS) that is particularly interesting for targeted treatment of cardiac fibrosis.119 The RAAS pathway regulates blood pressure. A schematic of RAAS is presented in Fig. 4. RAAS is an important pathway that contributes to cardiac fibrosis with TGF-β through downstream Ang II/AT1R to induce fibrosis in the heart. However, the full mechanism is not understood yet. The Ang II/AT1R/p38 MAPK pathway is activated in cardiac and renal fibrosis. Inhibition of renin is a promising strategy as it limits Ang II synthesis and hence combats cardiac and renal fibrosis. Fibroblast activation occurs in response to angiotensin II. Targeting fibroblasts is a great strategy which not only reduces collagen deposition but also limits macrophages in remodeling the heart. Clinical studies showed that renin-angiotensin system inhibition with ACE inhibitors or AT1 receptor antagonists reduced myocardial fibrosis. RAS inhibitors showed the ability to reduce fibrosis in human myocardium. The aldosterone antagonist spironolactone has also shown promising effects. Calcium channel blockers also showed promising pre-clinical performances. However, the exact mechanism of anti-hypertensive drugs against fibrosis is not fully understood yet. Ang II shows fibroblast activation, proliferation, and collagen production. Targeting the RAAS system may reverse cardiac fibrosis.119,120
Fig. 4.
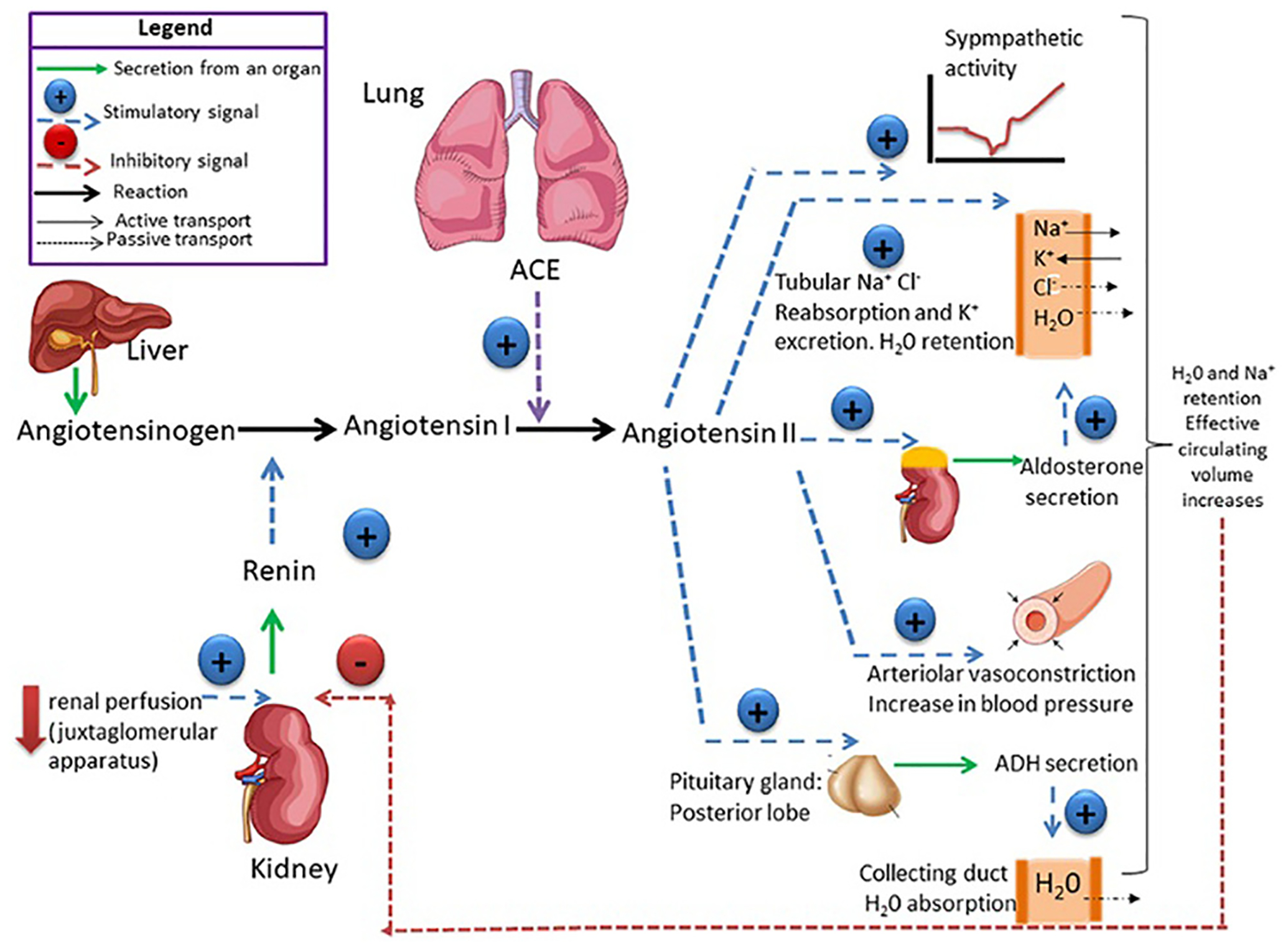
Schematic of the renin-angiotensin-aldosterone system (RAAS). Reproduced with permission from ref. 121, 2019 Frontiers, Open Access.
Studies found that angiotensin and TGF-β1 are integrated signaling networks, which promote fibrosis. Drugs that inhibit the angiotensin pathway are called angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists. These drugs are used for hypertension and cardiomyopathies.
Losartan, an angiotensin receptor inhibitor showed effectiveness in fibrosis treatment in a variety of animal models and humans.122 Compared to TGF-β signaling an angiotensin-converting enzyme inhibitor may be a potential approach.
The medication for high angiotensin levels is an angiotensin-converting enzyme inhibitor (e.g., enalapril) and angiotensin receptor blockers (e.g., losartan).116 One side effect of these drugs is an increase in potassium levels in the body. There are several angiotensin receptor blockers available such as Azilsartan (Edarbi), candesartan (atacand), Eprosartan, Irbesartan (Avapro), Losartan (Cozaar), Olmesartan (Benicar), Telmisartan (Macarids), and Valsartan (Diovan).116 Angiotensin II receptor blockers may also treat chronic kidney diseases, heart failure, and kidney failure in diabetes. The major side effects are dizziness, potassium level enhancement, and swelling of the skin.116 Several markers such as vimentin, fibronectin, periostin, and β1-integrin have been overexpressed in fibroblasts. None of them are fibroblast specific.119,120
Heart failure is usually caused by impaired cardiovascular reflexes and dysregulated neurohumoral activation mechanisms such as the renin angiotensin system, sympathetic nervous system, and cytokine system. The pathological activation of myocardial function and cardiovascular homeostasis causes heart failure.123 This also affects pulmonary and renal systems. Since the identification of the angiotensin-(I-7)- Mas receptor axis renin-angiotensin system it has become an important target in cardiovascular disease.
Aliskiren is a United States Food and Drug Administration (US-FDA) approved renin inhibitor for hypertension treatment. It attenuates collagen deposition, oxidative stress, and TGF-β synthesis in both normal and diabetic mice. It may affect anti-fibrotic activity independently of Ang II.124 Queisser et al. showed MR receptor blocking with spironolactone ameliorated heart fibrosis in hypertensive rats. At optimum doses, spironolactone prevented heart fibrosis. The advantage of RAAS inhibition in the reduction of reactive oxygen species in cardiac tissues.125 Lisinopril also showed collagen volume reduction in patients.126 So it can be a potential anti-fibrotic agent. Two weeks of enalapril treatment also showed great progress in cardiac fibroblast physiology which protects the heart.126 Pirfenidone is an anti-fibrotic agent which ameliorates fibrosis in different tissues. Recently, it was reported that pirfenidone converges the RAAS pathway in cardiac fibrosis. Pirfenidone balances the ACE/ACE2 ratio, inhibits AT1R/p38 MAPK, and controls RAAS balance. ARB losartan also inhibits AT1R/p38 MAPK and Liver X receptor and increases α (LXR α) expression which shows anti-fibrotic effects.125
However, extensive research specifically focused on fibrosis and targeting needs to be conducted including detailed clinical trials.
3.3.3.2. Targeting RAAS in liver fibrosis.
Hepatic fibrosis involves hepatic stellate cells (HSC) and Kupffer cells (KC), growth factors, chemokines, and cytokines. This results in disruption of homeostatic functions and organ failure. RAAS is overproduced at different stages of liver fibrosis. AT1R blockers (ARBs), ACE inhibitors, and selective aldosterone blockers have been reported to be effective for hepatic fibrosis suppression. ARB candesartan treatment for 6 months showed ameliorated liver fibrosis and reduced fibrotic area, scores, and markers of α-SMA and hydroxyproline levels in patients with alcoholic liver fibrosis. The clinical trials in patients with liver fibrosis were significantly reduced with RAAS inhibition. Ang (1–7) inhibits hepatic stellate cell activation which results in the reduction of fibrosis. The potential of the RAAS blockade for liver fibrosis therapy has been tested in several investigations. Randomized controlled clinical trials in patients with liver fibrosis showed that the RAAS inhibitor reduces liver fibrosis significantly. This data concluded that RAAS antagonism is a promising therapeutic approach for liver fibrosis. Altogether it can be concluded that RAAS antagonism is a promising approach for liver fibrosis treatment. However, further clinical trials need to be conducted.
3.3.3.3. Targeting RAAS in skin fibrosis.
RAAS regulates wound healing in the skin by stimulating synthesis and release of pro-fibrotic proteins. The skin expresses local RAAS components which are dysregulated during wound healing and result in hypertrophic scars and keloid formation. Blocking ACE/AT1R and activating ACE2/AT2R is a good choice of treatment.127 Fang et al. studied the potential of ACE inhibitors against scar formation. They used ACE inhibitors and studied the in vitro studies using NIH 3T3 fibroblasts and a rat scar model. Flow cytometry and western blotting analysis confirm the potential antifibrotic activity in the ramipril or losartan treated group compared to the control. This study concluded that ACE inhibitors inhibit scar formation by suppressing TGF-β/SMAD2/3 and TGF-β1/TAK1 pathways.127 The AT1R inhibitor losartan reduced scar formation significantly. ACE inhibitors decreased fibroblast proliferation, collagen, and TGF-β expression. In pilot clinical trials topical losartan showed a significant reduction in scar scores in patients with hypertrophic scars and keloids. Mouse of porcine with chronic diabetic wounds and aging diabetic pigs showed faster wound healing with good quality skin observed when treated with 1% losartan gel.128 However, the AT1R and AT2R skin healing is phase dependent and late treatment may take a longer time to heal. However, ACE inhibitors reduce Ang II production which results in a lack of stimulation to AT1R and AT2R, hence ACE inhibitors failed to produce beneficial effects. Losartan specifically blocks AT1R without affecting binding of AT2R.
Even through basic understanding of RAAS and other targeting pathways for treatment of fibrosis the exact mechanism needs to be investigated which may open a new window for the development of fibrosis therapy.
3.4. Cadherin
3.4.1. Fibroblasts – cadherin producers of the interstitial matrix.
The cells adhere to each other and interact with ECM through cellular adhesion molecule (CAM) proteins through the calcium-dependent cellular adhesion mechanism and this regulates many biological processes including ECM remodeling and inflammation.129 It has been reported that cadherins have a role in malignancy and tumor invasiveness.130–133 The epithelial cells become more invasive during the EMT and malignant transformation, which involves the down regulation of right cadherins (e.g. E-cadherin) and up-regulation of mesenchymal cadherins (e.g. N-cadherin and cadherin-11) responsible for fibrosis.131,133,134 It has been shown that cadherin-11 are overexpressed in fibrotic tissues and studied in the context of lung, liver, intestinal, cardiac, and skin fibrosis.135–139 In addition, it has been reported that cadherin-11 are overexpressed in fibroblasts within the synovial lining joints where they regulate the production of interleukin-6 and MMPs.140–142 Cadherin-11 regulates multiple cell populations such as macrophage, epithelial and fibroblast cells responsible for fibrosis. For instance, the fibrotic macrophages produce more TNFα than cadherin-11 deficient macrophages.135,142 Additionally, the mesenchymal gene expression and EMT were blocked when cadherin-11 was downregulated in lung epithelial cells.135 Furthermore, cadherin-11 regulates the migration and levels of β-catenin in dermal fibroblasts which is very important in the fibrosis development.139 All these reports suggest that cadherin-11 may be a therapeutic target for treating the fibrosis.
The biological process such as tissue remodeling and inflammation involves calcium-dependent cell-to-cell interaction in the extracellular matrix, and it determines the ability to adhere the cells with each other via cell adhesion molecules. These adhesion molecules are categorized into four families such as selectins, immunoglobulin, integrins, and cadherins according to their molecular structure.129 Amongst several molecules responsible for cellular adhesion, cadherins are important proteins responsible for the progression of fibrotic and cancer diseases.129 Cadherin is categorized into several subtypes: type I classical cadherins such as E-cadherin, N-cadherin, and P-cadherin; type II classical cadherins such as VE-cadherin (CDH5) and OB-cadherin (CDH11), the desmosomal cadherins; the seven-pass transmembrane cadherins, FAT and dachsous (DCHS) group cadherins, and protocadherins (PCDHs).143
Cadherins have a repeating amino acid sequence with about 110 residues, each representing an immunoglobulin-like fold structure called an “extracellular cadherin” or “EC” domain responsible for homotypic cadherin–cadherin interaction.144 The cadherin family are structurally diverse proteins including transmembrane cell adhesion molecules, cytoplasmic domain and G-protein coupled receptors (GPCR).145 The E-cadherins are expressed in epithelial cells while N-cadherin and cadherin-11 are expressed during mesenchymal cell transition in fibroblasts. The malignant cells are characterized by changes in the expression level of cadherins.146
3.4.2. The role of cadherins in organ fibrosis.
CDH11 expression is enhanced when the fibroblast to myofibroblasts differentiation occurs which further results in cell-to-cell adhesion and contractility.147 The expression of N-cadherin is increased in alveolar epithelial cells when the cells are in epithelial-mesenchymal transition.139 The significant role of cadherin-11 in dermal fibrosis has also been reported by several authors.148 There are enough reports available that suggest that cadherin-11 targeting could be a potential therapeutic strategy in fibrotic patients.
The mechanism by which cadherin-11 regulates fibrosis is still under investigation. It is demonstrated from the immuno-histochemistry that cadherin-11 is expressed in fibroblasts of fibrotic lungs, macrophages, and type II alveolar epithelial cells.135 The cadherin-11 modulates the fibrotic process through multiple steps. For instance, less TGF-β is produced by macrophages that are cadherin-11 deficient, but similar amounts of TNF-α are produced, compared to wild-type macrophages.135 Moreover, the mesenchymal gene expression and EMT are blocked in lung epithelial cells after cadherin-11 is downregulated.135 The migration of dermal fibroblast migration and of β-catenin levels are regulated by cadherin-11 in the development of fibrosis.139 Furthermore, the expression of the proinflammatory cytokine interleukin-6 (IL-6) and myofibroblast markers e.g., TGF-β1 is increased when the cadherin-11 is overexpressed in injured joints, lungs, and heart valves.135
3.4.3. Cadherins binding mechanism.
Besides, other ectodomains such as EC5, EC2, and EC4 would pair with EC1 domains when the cadherin-coated surfaces are “pulled apart” as shown by atomic force microscopy analysis.149 Chu et al. showed that cadherin binds between cells by association of their EC1 with EC5 domains measured by fluorescence resonance energy transfer (FRET) analysis (Table 1).149
Table 1.
Potential targets and approaches for various collagens and cadherins targeting
| Potential targets | Type of proteins targeted | Drug and delivery approaches | Ref. |
|---|---|---|---|
| HSP47 | Collagen-I, II, III, IV, V, XI | Liposomes, silica nanoparticles, small molecules inhibitors, siRNA | 81 and 150–154 |
| Lysyl oxidase | Collagen-I, II, III, IV, V, IX, XI | Antibody-siRNA, inhibitory monoclonal antibody, β-aminopropionitrile | 81 and 155–158 |
| TANGO1 | Collagen-I, II, III, IV, VII, IX | COPII and melanoma inhibitory activity member 3 carrier protein from the endoplasmic reticulum | 81, 159 and 160 |
| FKBP10 | Collagen-I, III, V, VI | siRNA, nintedanib, or pirfenidone | 81, 161 and 162 |
| LARP6 | Collagen-I, III | mRNA, small molecule inhibitors, TPI-2659, TPI-2435 | 163–165 |
| Hepatic stellate cells | Collagen-I, III, VI | Albumin nanocarriers, IFN-α1b liposomes, polyetherimine (RcP) nanoparticles, oligonucleotides delivery, RGD modified polymerosomes | 166–170 |
| Cadherins | Cadherin-11 | Anti-cadherin-11 antibody, repurposed celecoxib | 54, 171 and 172 |
| Cadherins | Cadherin-11, E-cadherin | siRNA, miR-199a-5p, miR-214–3p, miR-373, lipid nanoparticles | 173‒176 |
Moreover, the Ca2+ ion plays a major role in cell adhesion. Boggon et al. showed that the connection between multiple EC domains (EC1 and EC5) is regulated by the Ca2+ coordination and they are held at well-defined orientations which were observed from the crystal structure, electron micrographs, and EM tomograms of each ectodomain.177
Detailed information on type-I cadherins has been provided regarding its adhesive dimer interface.177 The amino-terminal β-strands of EC1 domains swap results in replacing the A strand of one monomer with the A strand of the other. The binding occurs through the insertion of the side chain from the A strand of the tryptophan-2 (Tryp-2) residue which is extended to the hydrophobic core. Any mutation in the Tryo-2 results in the loss of adhesive function of all cadherins (Fig. 5).177
Fig. 5.
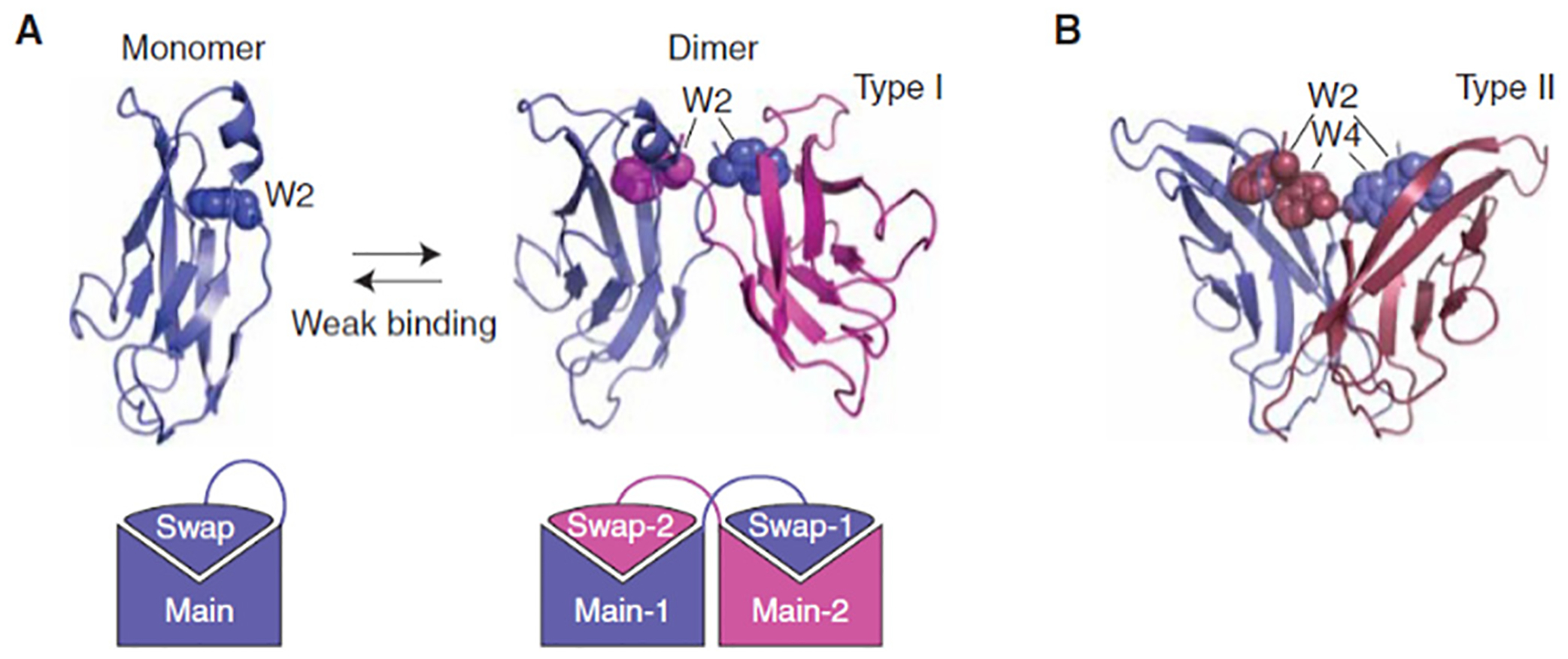
Strand-swap binding by classical cadherins. (A) 3D domain swapping by cadherin EC1 domains. 3D domain swapping provides a simple mechanism for constructing homophilic interfaces. A protein made up of a “main” domain and a “swapping” domain connected by a flexible region can form a “closed” monomer, or a multimer (a dimer in the case of cadherins). These two molecular configurations compete with one another, leading to weak binding affinities. (B) Comparison of strand-swap interfaces of type I and type II classical cadherins. Although the folding topology is identical, these two subfamilies have incompatible binding interfaces. Type II cadherins include two conserved Trp anchor residues, rather than one, and form a hydrophobic interface that runs the length of the EC1 domain. Reproduced with permission.149 Copyright 2009 Cold Spring Harbor Laboratory Press.
A similar type of strand-swap binding occurs in type II cadherin ectodomains observed by the crystal structures. In addition, Trp-4 also plays a role in binding with type II cadherins through the insertion into a hydrophobic pocket of the EC1 domain.178 In addition, the EC1–EC1 interface in type II cadherins offers a larger surface for interaction that runs along the entire length of the interface.
Kim and colleagues have reported another interaction mechanism between E-cadherin and α3β1 integrin, which was observed in an immunofluorescent study.179 E-cadherin also interacts with the α3β1 integrin and TGF-β receptor in alveolar epithelial cells.180 This trio of interaction results in the tyrosine phosphorylation of β-catenin at position Y654 (pY654). This pY654 binds with the pSmad2 (a TGFβ signaling molecule) and regulates the transcription by translocating into the nucleus. In addition, this interaction has been found in the myofibroblast of both mice and humans but not in the fibroblast.
4. Conclusions and future perspectives
Fibroblast targeting has not been studied extensively despite its great potential and clinical need. Among the several proteins, TGF-β, collagen, angiotensin, and cadherins are the key proteins that are closely associated with the development of fibrosis. Understanding the mechanism of proteins in fibroblast activation development and targeting these proteins can be an effective strategy for fibrosis therapy. Collagen research is still relatively unexplored. There is a need to develop remodeling strategies that remove the wrong collagen from the right place (fibrous tissue) but not the right collagen in the right place (normal interstitial ECM and bone). This could be feasible by developing activated (myo-) fibroblast targeted therapies.114 Another unexplored area is the replacement of a pathological ECM with the natural ECM and regeneration of the tissue by re-establishment of the spatial distribution of polarized cells on an ordered basement membrane. Moreover, it is necessary to think if a diseased ECM is removed/replaced by normal ECM, the epithelial cells undergoing a mesenchymal transition in fibrosis and cancer should be transitioned back to the normal condition.
In the past decade, significant progress has been made in understanding the mechanism of tissue fibrosis. In many ways, the many pathways are similar and interconnected in different tissue fibrosis such as lungs, skin, liver, and kidneys. So far very few drugs have been developed for treatment. Cadherin-11 plays an important role in tissue fibrosis as evidenced in several mouse models. Further investigation about how cadherins change the behaviour of fibroblasts and myofibroblasts responsible for the development of fibrosis is still needed. The targeting of cadherins, cadherin-11, appears to be a more targeted inhibition of key fibrotic pathways e.g., TGF-β and β-catenin. The clinical translation of these targets is still under investigation.
We have written this article after reviewing hundreds of excellent research publications from recent decades and highlighted several fibroblast activating pathways. This review provides insight into the different signalling pathways and discusses their chemical nature and their interference with fibrogenesis. We reviewed the key proteins that play crucial roles in fibrosis such as TGF-β, collagen, angiotensin, and cadherin, and their potential for developing myofibroblast-specific targeting therapy for an improved and efficient treatment approach.
This review provides insights into TGF-β and its importance in the regulation of multiple systems in the body including cell proliferation and differentiation, wound healing, adhesion, and migration of immune cells. TGF-β signalling is a well-established pathway of fibrosis. TGF-β is activated in fibrotic diseases and is sufficient and required for the induction of fibrosis. We comprehensively reviewed TGF-β driving canonical ALK5/Smad3 for fibrosis. The article provides TGF-β targeting strategies extensively. However, TGF-β plays a crucial role in tissue homeostasis, immunity, and cell proliferation. Hence blocking the TGF-β may lead to potential side effects. There are other strategies that have been developed but they have not shown promising antifibrotic potential in pre-clinical studies. However, combined therapies targeting TGF-β minimize the adverse effects even though there is a lack of clinical evidence.
We also briefly outline the various systems and conditions which enhance the production of collagens which lead to fibrosis. We extensively discussed strategies for the degradation of collagen to treat fibrosis including extracellular and intracellular pathways. Under homeostatic conditions, collagen is continuously produced and degraded, which prevents the formation of fibrosis. However, collagen degradation may be associated with the formation of permanent scar tissue. Moreover, collagen degradation reduces the pace of collagen production which results in extracellular accumulation of fibrillar collagen.
We also discussed the angiotensin II pathway toward fibrosis. We provided insights into the release of angiotensin and its mechanism which leads to fibrosis. Angiotensin II plays a role in increasing the blood pressure by narrowing the blood vessels. We have also provided a brief overview of the renal angiotensin aldosterone system. We discussed various strategies to target angiotensin II such as angiotensin II receptor blockers, angiotensin-converting enzyme inhibitors, and compounds that interfere with the binding of angiotensin II with a G-protein coupled receptor. However, the exact mechanism of angiotensin II has not been explored yet.
The last pathway we discussed in this article is based on cadherins. We provided insight into the potential pathways of cadherin II such as the regulation of fibroblast migration, alveolar macrophage production of TGF-β and mesenchymal gene expression, and possibly EMT of type II AECs. We also discussed the role of Cadherin-11 in tissue fibrosis including several pieces of evidence. The targeting of cadherins, cadherin-11, appears to be a more targeted inhibition of key fibrotic pathways e.g., TGF-β and β-catenin. The clinical translation of these targets is still under investigation.
There are a few drugs that have been developed to control fibroblast activation and have been approved by the FDA. These drugs include Trikafta to help treat cystic fibrosis, and nintedanib and pirfenidone for idiopathic pulmonary fibrosis. However, these drugs are limited to use due to adverse side effects. So, the control and targeted delivery systems need to be explored to minimize the side effects and improve the potential of drugs. We are currently developing various delivery systems to target specific myofibroblasts cells of the kidneys, lungs, and skin. We are using a novel targeting approach through engineering the nanocarriers loaded with payload. We have developed various myofibroblast targeting ligand functionalized nanoparticles. To target kidney myofibroblasts, we will use peptide ligands. To identify the potential peptide ligands, we synthesized a library of peptides. Targeted mRNA therapy can be a potential system still needing exploration. This article concludes that the fibroblast targeting system is a good choice for fibrosis therapy. However, the protein mechanism needs to be evaluated before clinical use.
The fibroblast protein targeting system may not be successful for fibrosis treatment due to unexpected barriers and a lack of ideal drugs and properties of drugs may also impact the delivery system. Identification of ideal carriers is also a big challenge. The relationship between carriers and drugs also has a critical role. However, recently, artificial intelligence and machine learning have received great interest. AI and ML play a crucial role in the identification and analysis of hypotheses using computers without the need for laboratory facilities and a work force. It is very clear that AI and ML are the most powerful techniques in science, technology, and medicine. However, the interface needs to be developed to make a connection between the computer and medicine. This is the major challenge we need to address in the future. We strongly believe that the advancement of technology may provide a solution through human body scanning. The technology may analyse the signals from the human body and provide point of care. The future of medicine could be ruled by human–computer–bio–nanotechnology.
Acknowledgements
We acknowledge funding by the Cancer Prevention Research Institute of Texas (CPRIT) through Texas Regional Excellence in Cancer Award (TREC) under Award No. PR210153, and the National Institutes of Health (NIH) under Award No. R03OD032624. The contents of this paper are solely the authors’ responsibility and do not necessarily represent the official views of NIH.
Biographies

Elfa Beaven
Elfa Beaven is currently pursuing a PhD in Biomedical Engineering at The University of Texas at El Paso which started in autumn 2020. She previously completed a bachelor’s degree in Biological Sciences from The University of Texas at El Paso in 2018. She is working under the mentorship of Dr Nurunnabi in the Department of Pharmaceutical Sciences where she is researching bioengineered therapeutics and working to develop targeted treatments for psoriasis and diabetes using ionic liquids.

Raj Kumar
Dr Raj Kumar is a Postdoctoral Research Fellow at the University of Texas at El Paso, USA. He completed a PhD in Chemistry (2017) at the Indian Institute of Technology (IIT) Mandi, India. He is currently exploring pharmaceutical nanotechnology, nanomedicine, and oral drug/gene delivery. He was awarded the UGC-JRF (2010, 11), SERBNPDF (2017), PBC Outstanding Postdoctoral Fellowship (2017–19), and Israel Government Scholarship (2017). He is serving as an Associate Editor for Frontiers in Nanotechnology and as a reviewer for more than 15 journals.

Himanshu N. Bhatt
Himanshu N. Bhatt is a postdoctoral fellow at the Department of Pharmaceutical Sciences, The University of Texas El Paso, USA. He was Postdoctoral Fellow at the University of Connecticut, Storrs, USA. He completed his PhD in Pharmaceutical Science from BITS, Pilani – Hyderabad Campus, India. He worked as a Formulation Scientist in Dr Reddy’s Laboratories Ltd, Hyderabad, India. His major research interest is improving the penetration of nanocarriers, continuous manufacturing of nanocarriers, and nanomedicine for multidrug resistance. He was awarded a Senior Research Fellowship from the CSIR, India, for pursuing doctoral studies, and a GPAT fellowship from the Government of India to pursue a master studies in Pharmaceutical Sciences.

Stephanie V. Esquivel
Stephanie Vargas Esquivel is an undergraduate student of Mechanical Engineering at the University of Texas at El Paso. She is carrying out a research project for the Space Health program in Dr Nurunnabi lab within the Aerospace Center (cSETR) at UTEP which consists of developing an oral drug delivery carrier that can target a specific region of interest and maintain their retention for a certain duration.

Md Nurunnabi
Dr Md Nurunnabi has been an assistant professor of Pharmaceutical Sciences and Biomedical Engineering at the University of Texas at El Paso (UTEP) since 2019. He is a core member of the Border Biomedical Research Center (BBRC), and the Lead Investigator for the Space Health program within the Aerospace Center (cSETR) at UTEP. His research interest lies between diseases detection and drug science using advanced bioinspired and bioengineering approaches. Currently he is serving as the Chair of the Bioengineering focus group of the Controlled Release Society. For details please go to https://www.utep.edu/NurunnabiLab.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Henderson NC, Rieder F and Wynn TA, Nature, 2020, 587, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotas ME and Medzhitov R, Cell, 2015, 160, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiskirchen R, Weiskirchen S and Tacke F, Mol. Aspects Med, 2019, 65, 2–15. [DOI] [PubMed] [Google Scholar]
- 4.Phan SH, Proc. Am. Thorac. Soc, 2008, 5, 334–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krieg T, Abraham D and Lafyatis R, Arthritis Res. Ther, 2007, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darby IA, Laverdet B, Bonté F and Desmoulière A, Clin. Cosmet. Investig. Dermatol, 2014, 7, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kendall RT and Feghali-Bostwick CA, Front. Pharmacol, 2014, 5, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai Y, Woods EL, Dally J, Kong D, Steadman R, Moseley R and Midgley AC, Biomolecules, 2021, 11, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinz B, Pittet P, Smith-Clerc J, Chaponnier C and Meister JJ, Mol. Biol. Cell, 2004, 15, 4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalik M, Wójcik-Pszczoła K, Paw M, Wnuk D, Koczurkiewicz P, Sanak M, Pękala E and Madeja Z, Cell. Mol. Life Sci, 2018, 75, 3943–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML and Gabbiani G, Am. J. Pathol, 2007, 170, 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou E, Zhang T, Bi C, Wang C and Zhang Z, Heart Vessels, 2020, 35, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbloom J, Macarak E, Piera-Velazquez S and Jimenez SA, Methods Mol. Biol, 2017, 1627, 1–23. [DOI] [PubMed] [Google Scholar]
- 14.Nurunnabi M, Cho KJ, Choi JS, Huh KM and Kyu Lee Y, Biomaterials, 2010, 31, 5436–5444. [DOI] [PubMed] [Google Scholar]
- 15.D’Urso M and Kurniawan NA, Front. Bioeng. Biotechnol, 2020, 8, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Nurunnabi M, Nafiujjaman M, Lee YK and Huh KM, J. Controlled Release, 2013, 171, 241–250. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Nurunnabi M, Nafiujjaman M, Jeong YY, Lee YK and Huh KM, J. Mater. Chem. B, 2014, 2, 2929–2937. [DOI] [PubMed] [Google Scholar]
- 18.Nurunnabi M, Khatun Z, Reeck GR, Lee DY and Lee YK, ACS Appl. Mater. Interfaces, 2014, 6, 12413–12421. [DOI] [PubMed] [Google Scholar]
- 19.Nurunnabi M, Khatun Z, Nafiujjaman M, Lee DG and Lee YK, ACS Appl. Mater. Interfaces, 2013, 5, 8246–8253. [DOI] [PubMed] [Google Scholar]
- 20.Khatun Z, Nurunnabi M, Nafiujjaman M, Reeck GR, Khan HA, Cho KJ and Lee YK, Nanoscale, 2015, 7, 10680–10689. [DOI] [PubMed] [Google Scholar]
- 21.Khatun Z, Nurunnabi M, Reeck GR, Cho KJ and Lee YK, J. Controlled Release, 2013, 170, 74–82. [DOI] [PubMed] [Google Scholar]
- 22.Khatun Z, Nurunnabi M, Cho KJ, Byun Y, Bae YH and Lee YK, J. Controlled Release, 2014, 177, 64–73. [DOI] [PubMed] [Google Scholar]
- 23.Nurunnabi M, Lee SA, Revuri V, Hwang YH, Kang SH, Lee M, Cho S, Cho KJ, Byun Y, Bae YH, Lee DY and Kyu Lee Y, J. Controlled Release, 2017, 268, 305–313. [DOI] [PubMed] [Google Scholar]
- 24.Lee DY, Nurunnabi M, Kang SH, Nafiujjaman M, Huh KM, Kyu Lee Y and Kim YC, Biomacromolecules, 2017, 18, 1172–1179. [DOI] [PubMed] [Google Scholar]
- 25.Nurunnabi M, Khatun Z, Badruddoza AZM, McCarthy JR, Lee YK and Huh KM, ACS Biomater. Sci. Eng, 2019, 5, 1645–1660. [Google Scholar]
- 26.Zhang YE, Cell Res, 2008, 19, 128–139, 2009 19:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walton KL, Johnson KE and Harrison CA, Front. Pharmacol, 2017, 8, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang F and Chen YG, Cell Biosci, 2012, 2, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biernacka A, Dobaczewski M and Frangogiannis NG, Growth Factors, 2011, 29, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.di Mola FF, Friess H, Martignoni ME, di Sebastiano P, Zimmermann A, Innocenti P, Graber H, Gold LI, Korc M and Büchler MW, Ann. Surg, 1999, 230, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X and Cao X, Bone Res, 2018, 6, 1–31, 2018 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu YL, Xu YR, Yang WX and Sun Y, Aging, 2018, 10, 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND and Zhao YY, Chem. – Biol. Interact, 2018, 292, 76–83. [DOI] [PubMed] [Google Scholar]
- 34.Hinck AP, Mueller TD and Springer TA, Cold Spring Harbor Perspect. Biol, 2016, 8, a022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K and Rifkin DB, Matrix Biol, 2015, 47, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung JYF, Chan MKK, Li JSF, Chan ASW, Tang PCT, Leung KT, To KF, Lan HY and Tang PMK, Int. J. Mol. Sci, 2021, 22, 7575.34299192 [Google Scholar]
- 37.Sun YBY, Qu X, Caruana G and Li J, Differentiation, 2016, 92, 102–107. [DOI] [PubMed] [Google Scholar]
- 38.Annes JP, Munger JS and Rifkin DB, J. Cell Sci, 2003, 116, 217–224. [DOI] [PubMed] [Google Scholar]
- 39.Xu P, Liu J and Derynck R, FEBS Lett, 2012, 586, 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heldin CH and Moustakas A, Cold Spring Harbor Perspect. Biol, 2016, 8, a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan X, Liu Z and Chen Y, Acta Biochim. Biophys. Sin, 2009, 41, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hata A and Chen YG, Cold Spring Harbor Perspect. Biol, 2016, 8, a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neel J-C, Humbert L and Lebrun J-J, ISRN Mol. Biol, 2012, 2012, 1–28. [Google Scholar]
- 44.Massagué J and Wotton D, EMBO J, 2000, 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin-Malpartida P, Batet M, Kaczmarska Z, Freier R, Gomes T, Aragón E, Zou Y, Wang Q, Xi Q, Ruiz L, Vea A, Márquez JA, Massagué J and Macias MJ, Nat. Commun, 2017, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katz LH, Likhter M, Jogunoori W, Belkin M, Ohshiro K and Mishra L, Cancer Lett, 2016, 379, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu L, Hébert MC and Zhang YE, EMBO J, 2002, 21, 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parri M and Chiarugi P, Cell Commun. SignaL, 2010, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen YG, Cell Res, 2008, 19, 58–702009 19:1. [Google Scholar]
- 50.Fukuchi M, Imamura T, Chiba T, Ebisawa T, Kawabata M, Tanaka K and Miyazono K, Mol. Biol. Cell, 2001, 12, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.García-Cárceles J, Caballero E, Gil C and Martínez A, J. Med. Chem, 2022, 65, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu T and Feng XH, Biochem. J, 2010, 430, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang YE, Cold Spring Harbor Perspect. Biol, 2017, 9, a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM and Derynck R, EMBO J, 2007, 26, 3957–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eblen ST, Adv. Cancer Res, 2018, 138, 99–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh MC, Lee J and Choi Y, Immunol. Rev, 2015, 266, 72–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kishida S, Sanjo H, Akira S, Matsumoto K and Ninomiya-Tsuji J, Genes Cells, 2005, 10, 447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bujor AM, Asano Y, Haines P, Lafyatis R and Trojanowska M, Arthritis Rheum, 2011, 63, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alcorn JF, van der Velden J, Brown AL, McElhinney B, Irvin CG and Janssen-Heininger YMW, Am. J. Respir. Cell Mol. Biol, 2009, 40, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mason RM, Int. J. Exp. Pathol, 2013, 94, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chambers RC, Leoni P, Kaminski N, Laurent GJ and Heller RA, Am. J. Pathol, 2003, 162, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G and Distler JHW, Nat. Comm, 2012, 3, 1–12, 2012 3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacDonald BT, Tamai K and He X, Dev. Cell, 2009, 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.v Pechkovsky D, Hackett TL, An SS, Shaheen F, Murray LA and Knight DA, Am. J. Respir. Cell Mol. Biol, 2012, 43, 641–651. [DOI] [PubMed] [Google Scholar]
- 65.Jelaska A and Korn JH, Arthritis Rheum, 2000, 43, 2230–2239. [DOI] [PubMed] [Google Scholar]
- 66.Popov Y and Schuppan D, Hepatology, 2009, 50, 1294–1306. [DOI] [PubMed] [Google Scholar]
- 67.Lamouille S, Xu J and Derynck R, Nature Rev. Mol. Cell Biol, 2014, 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foletta VC, Lim MA, Soosairaiah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massagué J and Bernard O, J. Cell Biol, 2003, 162, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang YE, Curr. Opin. Cell Biol, 2018, 51, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Runyan CE, Hayashida T, Hubchak S, Curley JF and Schnaper HW, J. Biol. Chem, 2009, 284, 25181–25189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Penheiter SG, Singh RD, Repellin CE, Wilkes MC, Edens M, Howe PH, Pagano RE and Leof EB, Mol. Bio.l Cell, 2010, 21, 4009–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin X, Murphy SJ, Wilkes MC, Ji Y and Leof EB, Mol. Biol. Cell, 2013, 24, 2285–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krafts KP, Tissue repair, Organogenesis, 2010, 6, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schultz GS, Chin GA, Moldawer L and Diegelmann RF, Diabetic Foot Problems, 2011, 395–402. [Google Scholar]
- 75.White ES and Mantovani AR, J. Pathol, 2013, 229, 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sako D, v Grinberg A, Liu J, v Davies M, Castonguay R, Maniatis S, Andreucci AJ, Pobre EG, Tomkinson KN, Monnell TE, Ucran JA, Martinez-Hackert E, Pearsall RS, Underwood KW, Seehra J and Kumar R, J. Biol. Chem, 2010, 285, 21037–21048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goebel EJ, Ongaro L, Kappes EC, Vestal K, Belcheva E, Castonguay R, Kumar R, Bernard DJ and Thompson TB, eLife, 2022, 11, e78197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Namwanje M and Brown CW, Cold Spring Harbor Perspect. Biol, 2016, 8, a021881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xia Y and Schneyer AL, J. Endocrinol, 2009, 202, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beachley VZ, Wolf MT, Sadtler K, Manda SS, Jacobs H, Blatchley MR, Bader JS, Pandey A, Pardoll D and Elisseeff JH, Nat. Methods, 2015, 12, 1197–1204, 2015 12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Staab-Weijnitz CA, Am. J. Respir. Cell Mol. Biol, 2022, 66, 363–381. [DOI] [PubMed] [Google Scholar]
- 82.Frangogiannis NG, J. Exp. Med, 2020, 217, e20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lipson KE, Wong C, Teng Y and Spong S, Fibrogenesis Tissue Repair, 2012, 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardie WD, Glasser SW and Hagood JS, Am. J. Pathol, 2009, 175, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McKleroy W, Lee TH and Atabai K, Am. J. Physiol.: Lung Cell. Mol. Physiol, 2013, 304, L709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.MacKenna D, Summerour SR and Villarreal FJ, Cardiovasc. Res, 2000, 46, 257–263. [DOI] [PubMed] [Google Scholar]
- 87.McAnulty RJ, Campa JS, Cambrey AD and Laurent GJ, Biochim. Biophys. Acta, Mol. Cell Res, 1991, 1091, 231–235. [DOI] [PubMed] [Google Scholar]
- 88.Stickel F, Urbaschek R, Schuppan D, Poeschl G, Oesterling C, Conradt C, McCuskey RS, Simanowski UA and Seitz HK, Dig. Dis. Sci, 2001, 46, 2025–2032. [DOI] [PubMed] [Google Scholar]
- 89.Cassiman D, Libbrecht L, Desmet V, Denef C and Roskams T, J. Hepatol, 2002, 36, 200–209. [DOI] [PubMed] [Google Scholar]
- 90.Urushiyama H, Terasaki Y, Nagasaka S, Terasaki M, Kunugi S, Nagase T, Fukuda Y and Shimizu A, Lab. Invest, 2015, 95, 872–885. [DOI] [PubMed] [Google Scholar]
- 91.Natori Y, O’Meara YM, Manning EC, Minto AWM, Levine JS, Weise WJ and Salant DJ, Am. J. Physiol. Renal Physiol, 1992, 262(1), F131. [DOI] [PubMed] [Google Scholar]
- 92.Klingberg F, Hinz B and White ES, J. Pathol, 2013, 229, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, Molyneaux PL, McKeever TM, Wells AU, Flynn A, Hubbard RB, Leeming DJ, Marshall RP, Karsdal MA, Lukey PT and Maher TM, Lancet Respir. Med, 2015, 3, 462–472. [DOI] [PubMed] [Google Scholar]
- 94.Fenton A, Jesky MD, Ferro CJ, Sørensen J, Karsdal MA, Cockwell P and Genovese F, PLoS One, 2017, 12, e0175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mak KM and Mei R, Anat. Rec, 2017, 300, 1371–1390. [DOI] [PubMed] [Google Scholar]
- 96.Duncan MB, Yang C, Tanjore H, Boyle PM, Keskin D, Sugimoto H, Zeisberg M, Olsen BR and Kalluri R, Dis. Model. Mech, 2013, 6, 942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cox TR and Erler JT, Dis. Model. Mech, 2011, 4, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holmes DF, Lu Y, Starborg T and Kadler KE, Curr. Top. Dev. Biol, 2018, 130, 107–142. [DOI] [PubMed] [Google Scholar]
- 99.Kaur J and Reinhardt DP, Stem Cell Biology and Tissue Engineering in Dental Sciences, 2015, pp. 25–45. [Google Scholar]
- 100.Weksler BB, Ley CW and Jaffe EA, J. Clin. Invest, 1978, 62, 923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Manon-Jensen T, Kjeld NG and Karsdal MA, J. Thromb. Hae-most, 2016, 14, 438–448. [DOI] [PubMed] [Google Scholar]
- 102.il Lee J, Wright JH, Johnson MM, Bauer RL, Sorg K, Yuen S, Hayes BJ, Nguyen L, Riehle KJ and Campbell JS, Am. J. Physiol.: Cell Physiol, 2016, 310, C436–C445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kehrel B, Wierwille S, Clemetson KJ, Anders O, Steiner M, Knight CG, Farndale RW, Okuma M and Barnes MJ, Blood, 1998, 91, 491–499. [PubMed] [Google Scholar]
- 104.Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, Gudmann NS, Rønnow S, Sand JM, Daniels SJ, Mortensen JH and Schuppan D, Adv. Drug Deliv. Rev, 2017, 121, 43–56. [DOI] [PubMed] [Google Scholar]
- 105.Bryckaert M, Rosa JP, Denis CV and Lenting PJ, Cell. Mol. Life Sci, 2015, 72, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ferreira Dos Santos G, Ferreira-Dos-Santos G, Friedrich Hurdle MB, Clendenen SR, Eldrige JS and Qu W, Pain Physician, 2022, 25, 15–27. [PubMed] [Google Scholar]
- 107.Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T and de Paepe A, Am. J. Hum. Genet, 2000, 66, 1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hamel BCJ, Pals G, Engels CHAM, van den Akker E, Boers GHJ, van Dongen PWJ and Steijlen PM, Clin. Genet, 1998, 53, 440–446. [DOI] [PubMed] [Google Scholar]
- 109.Silver RM, Ann. Rheum. Dis, 1991, 50, 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY, Miyamoto M, Kim YO, Ogawa S, Arch RH, Schuppan D and Popov Y, Gastroenterology, 2014, 147, 1378–1392. [DOI] [PubMed] [Google Scholar]
- 111.Trojanowska M, Rheumatology, 2008, 47, v2–v4. [DOI] [PubMed] [Google Scholar]
- 112.Iwayama T and Olson LE, Curr. Rheumatol. Rep, 2013, 15, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brown M and O’Reilly S, Clin. Exp. Immunol, 2019, 195, 310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Odagiri N, Matsubara T, Sato-Matsubara M, Fujii H, Enomoto M and Kawada N, Clin. Mol. Hepatol, 2021, 27, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saito S, J. Anesth, 2012, 26, 137–140. [DOI] [PubMed] [Google Scholar]
- 116.Mulrow PJ and Ganong WF, Yale J. Biol. Med, 1961, 33, 386. [PMC free article] [PubMed] [Google Scholar]
- 117.Hoffman WE, Phillips MI, Schmid PG, Falcon J and Weet JF, Neuropharmacology, 1977, 16, 463–472. [DOI] [PubMed] [Google Scholar]
- 118.Siddesha JM, Valente AJ, Sakamuri SSVP, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M and Chandrasekar B, J. Mol. Cell. Cardiol, 2013, 65, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schuetze KB, McKinsey TA and Long CS, J. Mol. Cell. Cardiol, 2014, 70, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hale TM, J. Mol. Cell. Cardiol, 2016, 93, 125–132. [DOI] [PubMed] [Google Scholar]
- 121.de Almeida LF and Coimbra TM, Front. Pediatr, 2019, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Couluris M, Kinder BW, Xu P, Gross-King M, Krischer J and Panos RJ, Lung, 2012, 190, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu B and Li H, Mol. Med. Rep, 2015, 12, 7823–7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Siragy H, Huang J and Lieb DC, Expt. Opin. Emerg. Drugs, 2008, 13, 417–430. [DOI] [PubMed] [Google Scholar]
- 125.Queisser N, Happ K, Link S, Jahn D, Zimnol A, Geier A and Schupp N, Toxicol. Appl. Pharmacol, 2014, 280, 399–407. [DOI] [PubMed] [Google Scholar]
- 126.AlQudah M, Hale TM and Czubryt MP, Matrix Biol, 2020, 91–92, 92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fang QQ, Wang XF, Zhao WY, DIng SL, Shi BH, Xia Y, Yang H, Wu LH, Li CY and Tan WQ, Sci. Rep, 2018, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hedayatyanfard K, Ziai SA, Niazi F, Habibi I, Habibi B and Moravvej H, Wound Repair Regen, 2018, 26, 340–343. [DOI] [PubMed] [Google Scholar]
- 129.Agarwal SK, Front. Pharmacol, 2014, 5, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shibata T, Ochiai A, Gotoh M, Machinami R and Hirohashi S, Cancer Lett, 1996, 99, 147–153. [DOI] [PubMed] [Google Scholar]
- 131.Hazan RB, Kang L, Whooley BP and Borgen PI, Cell Adhes. Commun, 1997, 4, 399–411. [DOI] [PubMed] [Google Scholar]
- 132.Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA and Byers SW, Cancer Res, 1999, 59, 947–952. [PubMed] [Google Scholar]
- 133.Wheelock MJ and Johnson KR, Ann. Rev. Cell Dev. Biol, 2003, 19, 207–235. [DOI] [PubMed] [Google Scholar]
- 134.Hazan RB, Phillips GR, Qiao RF, Norton L and Aaronson SA, J. Cell Biol, 2000, 148, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schneider DJ, Wu M, Le TT, Cho S-H, Brenner MB, Blackburn MR and Agarwal SK, FASEB J, 2012, 26, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Franzè E, Monteleone I, Laudisi F, Rizzo A, Dinallo V, di Fusco D, Colantoni A, Ortenzi A, Giuffrida P, di Carlo S, Sica GS, di Sabatino A and Monteleone G, J. Crohns. Colitis, 2020, 14, 406–417. [DOI] [PubMed] [Google Scholar]
- 137.Riley LA and Merryman WD, Cell Signal, 2021, 78, 109876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pedroza M, To S, Smith J and Agarwal SK, PLoS One, 2019, 14, e0218971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu M, Pedroza M, Lafyatis R, George AT, Mayes MD, Assassi S, Tan FK, Brenner MB and Agarwal SK, Arthritis Rheumatol, 2014, 66, 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GFM, Chisaka O, Takeichi M and Brenner MB, Science (1979), 2007, 315, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 141.Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM and Brenner MB, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 8402–8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Noss EH, Chang SK, Watts GFM and Brenner MB, Arthritis Rheum, 2011, 63, 3768–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oda H and Takeichi M, J. Cell Biol, 2011, 193, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Overduin M, Harvey TS, Bagby S, Tong KI, Yau P, Takeichi M and Ikura M, Science (1979), 1995, 267, 386–389. [DOI] [PubMed] [Google Scholar]
- 145.Sannigrahi MK, Srinivas S and Rakshit S, Adv. Exp. Med. Biol, 2018, 1112, 99–105. [DOI] [PubMed] [Google Scholar]
- 146.Nieman MT, Prudoff RS, Johnson KR and Wheelock MJ, J. Cell Biol, 1999, 147, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tabib T, Huang M, Morse N, Papazoglou A, Behera R, Jia M, Bulik M, Monier DE, Benos PV, Chen W, Domsic R and Lafyatis R, Nat. Commun, 2021, 12, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS and Lawson WE, Am. J. Respir. Crit. Care Med, 2009, 180, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shapiro L and Weis WI, Cold Spring Harbor Perspect. Biol, 2009, 1, a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yamakawa T, Ohigashi H, Hashimoto D, Hayase E, Takahashi S, Miyazaki M, Minomi K, Onozawa M, Niitsu Y and Teshima T, Blood, 2018, 131, 1476–1485. [DOI] [PubMed] [Google Scholar]
- 151.Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, Takimoto R, Takada K, Miyanishi K, Matsunaga T, Takayama T and Niitsu Y, Nat. Biotech, 2008, 26, 431–442. [DOI] [PubMed] [Google Scholar]
- 152.Bellaye PS, Burgy O, Bonniaud P and Kolb M, Expt. Opin. Therape. Targets, 2021, 16, 1239–1260. [DOI] [PubMed] [Google Scholar]
- 153.Morry J, Ngamcherdtrakul W, Gu S, Goodyear SM, Castro DJ, Reda MM, Sangvanich T and Yantasee W, Biomaterials, 2015, 66, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Natsume T, Koide T, Yokota SI, Hirayoshi K and Nagata K, J. Biol. Chem, 1994, 269, 31224–31228. [PubMed] [Google Scholar]
- 155.Vallet SD and Ricard-Blum S, Essays Biochem, 2019, 63, 349–364. [DOI] [PubMed] [Google Scholar]
- 156.Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, Barry V, Mikels-Vigdal A, Karpinski S, Kornyeyev D, Adamkewicz J, Feng X, Zhou Q, Shang C, Kumar P, Phan D, Kasner M, López B, Diez J, Wright KC, Kovacs RL, Chen PS, Quertermous T, Smith V, Yao L, Tschöpe C and Chang CP, Nat. Commun, 2016, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C, Garcia CA, Velayo AC, Jorgensen B, Biermann D, Tsai D, Green J, Zaffryar-Eilot S, Holzer A, Ogg S, Thai D, Neufeld G, van Vlasselaer P and Smith V, Nat. Med, 2010, 16, 1009–1017. [DOI] [PubMed] [Google Scholar]
- 158.Leiva O, Ng SK, Matsuura S, Chitalia V, Lucero H, Findlay A, Turner C, Jarolimek W and Ravid K, Int. J. Hematol, 2019, 110, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R and Malhotra V, Cell, 2009, 136, 891–902. [DOI] [PubMed] [Google Scholar]
- 160.Wilson DG, Phamluong K, Li L, Sun M, Cao TC, Liu PS, Modrusan Z, Sandoval WN, Rangell L, Carano RAD, Peterson AS and Solloway MJ, J. Cell Biol, 2011, 193, 935–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Knüppel L, Heinzelmann K, Lindner M, Hatz R, Behr J, Eickelberg O and Staab-Weijnitz CA, Respir. Res, 2018, 19, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Staab-Weijnitz CA, Fernandez IE, Knüppel L, Maul J, Heinzelmann K, Juan-Guardela BM, Hennen E, Preissler G, Winter H, Neurohr C, Hatz R, Lindner M, Behr J, Kaminski N and Eickelberg O, Am. J. Respir. Crit. Care Med, 2015, 192, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Parsons CJ, Stefanovic B, Seki E, Aoyama T, Latour AM, Marzluff WF, Rippe RA and Brenner DA, J. Biol. Chem, 2011, 286, 8609–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stefanovic B, Manojlovic Z, Vied C, Badger CD and Stefanovic L, Sci. Rep, 2019, 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stefanovic B, Michaels HA and Nefzi A, Med. Chem. Lett, 2021, 12, 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Beljaars L, Molema G, Schuppan D, Geerts A, de Bleser PJ, Weert B, Meijer DKF and Poelstra K, J. Biol. Chem, 2000, 275, 12743–12751. [DOI] [PubMed] [Google Scholar]
- 167.Du SL, Pan H, Lu WY, Wang J, Wu J and Wang JY, J. Pharmacol. Exp. Ther, 2007, 322, 560–568. [DOI] [PubMed] [Google Scholar]
- 168.Zhang Z, Wang C, Zha Y, Hu W, Gao Z, Zang Y, Chen J, Zhang J and Dong L, ACS Nano, 2015, 9, 2405–2419. [DOI] [PubMed] [Google Scholar]
- 169.Yang N, Ye Z, Li F and Mahato RI, Bioconjug. Chem, 2009, 20, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang J, Hou Y, Ji G, Song Z, Liu Y, Dai G, Zhang Y and Chen J, Eur. J. Pharm. Sci, 2014, 52, 180–190. [DOI] [PubMed] [Google Scholar]
- 171.Pedroza M, Welschhans RL and Agarwal SK, PLoS One, 2017, 12, e0187109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Assefnia S, Dakshanamurthy S, Auvil JMG, Hampel C, Anastasiadis PZ, Kallakury B, Uren A, Foley DW, Brown ML, Shapiro L, Brenner M, Haigh D, Byers SW, Assefnia S, Dakshanamurthy S, Guidry Auvil JM, Hampel C, Anastasiadis PZ, Kallakury B, Uren A, Foley DW, Brown ML, Shapiro L, Brenner M, Haigh D and Byers SW, Oncotarget, 2013, 5, 1458–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li J, Gordon I, Dejanovic D, Lin S, Wang J, Kurada S, Mao R, West G, Naydenov N, Lechuga S, Decaris M, Turner S, Fiocchi C, Ivanov A and Rieder F, Gastroenterology, 2021, 160, S46–S47. [Google Scholar]
- 174.Che M, Shi T, Feng S, Li H, Zhang X, Feng N, Lou W, Dou J, Tang G, Huang C, Xu G, Qian Q, Sun S, He L and Wang H, J. Am. Soc. Nephrol, 2017, 28, 2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang Q, Wang C, Miao S, Li C, Chen Z, Li F, Zhang Q, Wang C, Miao S, Li C, Chen Z and Li F, Oncotarget, 2017, 8, 93969–93983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xu M, Hu K, Liu Y, Huang Y, Liu S, Chen Y, Wang D, Zhou S, Zhang Q, Mei N, Lu H, Li F, Gao X and Chen J, Nat. Comm, 2021, 12, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM and Shapiro L, Science (1979), 2002, 296, 1308–1313. [DOI] [PubMed] [Google Scholar]
- 178.Patel SD, Ciatto C, Chen CP, Bahna F, Rajebhosale M, Arkus N, Schieren I, Jessell TM, Honig B, Price SR and Shapiro L, Cell, 2006, 124, 1255–1268. [DOI] [PubMed] [Google Scholar]
- 179.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA and Chapman HA, J. Clin. Invest, 2009, 119, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim Y, Kugler MC, Wei Y, Kim KK, Li X, Brumwell AN and Chapman HA, J. Cell Biol, 2009, 184, 309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]