

Summary
T cell exhaustion limits anti-tumor immunity, but the molecular determinants of this process remain poorly understood. Using a chronic stimulation assay, we performed genome-wide CRISPR-Cas9 screens to systematically discover regulators of T cell exhaustion, which identified an enrichment of epigenetic factors. In vivo CRISPR screens in murine and human tumor models demonstrated that perturbation of the INO80 and BAF chromatin remodeling complexes improved T cell persistence in tumors. In vivo Perturb-seq revealed distinct transcriptional roles of each complex and that depletion of canonical BAF complex members, including Arid1a, resulted in the maintenance of an effector program and downregulation of exhaustion-related genes in tumor-infiltrating T cells. Finally, Arid1a-depletion limited the acquisition of exhaustion-associated chromatin accessibility and led to improved anti-tumor immunity. In summary, we provide an atlas of the genetic regulators of T cell exhaustion and demonstrate that modulation of epigenetic state can improve T cell responses in cancer immunotherapy.
In Brief
Belk et al. systematically dissect the genetic regulators of T cell exhaustion with a series of in vitro and in vivo CRISPR-Cas9 screens. Inhibition of chromatin remodeling factors - in particular Arid1a - improves T cell function and reduces the transcriptional and epigenetic hallmarks of exhaustion.
Graphical Abstract
Introduction
T cell exhaustion is a process that is driven by chronic T cell receptor (TCR) stimulation and induces the stable expression of inhibitory surface receptors, poor response to tumor antigens, and low cell proliferation and persistence in vivo (Wherry and Kurachi, 2015; Collier et al., 2021). Originally identified in the setting of chronic viral infection (Zajac et al., 1998; Barber et al., 2006), T cell exhaustion is now appreciated to occur in diverse disease settings, including cancer and autoimmune disease (McKinney et al., 2015; McLane et al., 2019). Importantly, studies have demonstrated that T cell exhaustion represents a major barrier for the efficacy of both checkpoint blockade and chimeric antigen receptor T (CAR-T) cell immunotherapies, and that manipulating this process may lead to improved efficacy of T cell responses in cancer (Sakuishi et al., 2010; Long et al., 2015; Fraietta et al., 2018a, 2018b; Ribas and Wolchok, 2018; Lynn et al., 2019; Yost et al., 2019; Weber et al., 2021).
Recent genomic studies in murine models of chronic infection and cancer have demonstrated that T cell exhaustion is associated with a broad remodeling of the transcriptional and epigenomic landscape, which is conserved across disease settings (Pauken et al., 2016; Sen et al., 2016; Philip et al., 2017; Scott-Browne et al., 2016; Pritykin et al., 2021). This unique epigenetic state is primarily driven by chronic antigen and TCR signaling, and results in a stable cellular phenotype that is not changed by anti-PD-1 treatment (Pauken et al., 2016; Pritykin et al., 2021; Schietinger et al., 2016; Belk et al., 2022). Indeed, in cancer patients receiving PD-1 blockade, exhausted T cells display a distinct differentiation trajectory and end-stage chromatin profile, compared to functional effector T cells, and clonal tracing of exhausted T cells demonstrated that these cells are limited in their capacity to proliferate and perform effector functions in response to immunotherapy (Yost et al., 2019; Philip et al., 2017; Satpathy et al., 2019).
CRISPR-Cas9 screening has emerged as a powerful discovery tool for the molecular determinants of immune cell differentiation and function (Parnas et al., 2015; Shalem et al., 2014; Shifrut et al., 2018; Wang et al., 2014). For example, prior CRISPR-Cas9 screens in T cells have been used to identify transcription factors and metabolic regulators of T cell fate in vivo, as well as therapeutic targets (Chen et al., 2021; Dong et al., 2019; Huang et al., 2021; LaFleur et al., 2019; Wei et al., 2019). However, inherent limitations in scaling these in vivo assays have constrained library diversity of these screens, largely preventing genome-wide analysis and an unbiased discovery of novel regulators of T cell phenotypes. Furthermore, assays that simultaneously screen for multiple functions of T cells—for example, tissue localization, infiltration, and differentiation in tumors—have also made it challenging to interrogate the impact of a particular gene perturbation on a single aspect of T cell function and phenotype, such as exhaustion.
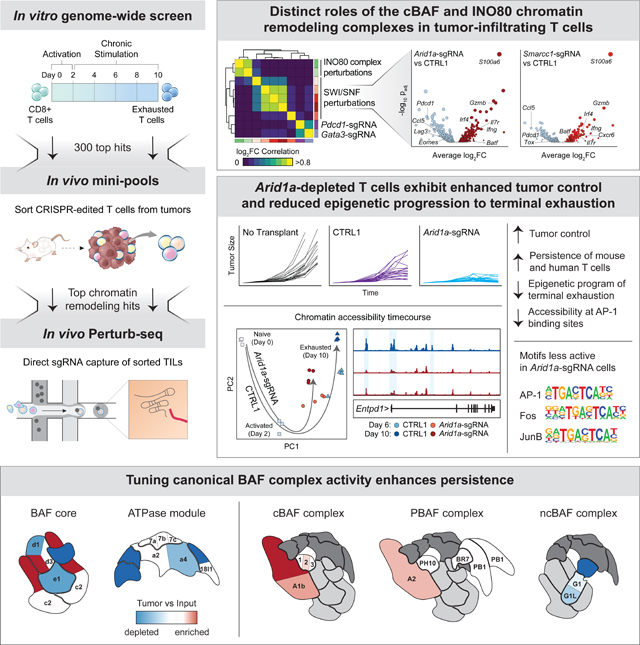
Here, we developed an in vitro model of CD8+ T cell exhaustion, which recapitulates the epigenomic features of exhaustion that are observed in vivo and is scalable for genome-wide CRISPR-Cas9 screens. Using this model, we provide a comprehensive view of the genetic regulators of T cell exhaustion. Strikingly, these factors are enriched for chromatin remodeling proteins, including subunits of the INO80 (inositol requiring mutant 80) nucleosome positioning complex and the SWI/SNF (switch/sucrose non-fermentable) chromatin remodeling complex. Depletion of INO80 and canonical BRG1 or BRM-associated factor (cBAF; SWI/SNF family) complex members—in particular, Arid1a—led to increased persistence of T cells in vivo, and Perturb-seq analysis revealed distinct transcriptional programs controlled by each complex in tumor-infiltrating T cells. Epigenomic profiling of Arid1a-depleted T cells demonstrated that Arid1a was required for the acquisition of exhaustion-associated chromatin remodeling that occurs during chronic antigen stimulation. Finally, Arid1a-depleted cells exhibited improved tumor control, suggesting that modulation of the epigenetic state of T cell exhaustion via chromatin remodeling factors may be an effective path to improve T cell responses in cancer immunotherapy.
Results
An in vitro chronic stimulation assay recapitulates the epigenetic program of terminal T cell exhaustion.
To develop an assay that is amenable to genome-wide CRISPR-Cas9 screening of T cell exhaustion, we adapted our previously described approach, which used anti-CD3 antibodies to enforce clustering of the T cell co-receptor, CD3, and thereby induce chronic TCR signaling in an antigen-independent manner (Figure 1A) (Vardhana et al., 2020). Compared to in vivo assays, this model isolates the core determinant of T cell exhaustion—chronic stimulation through the TCR complex—and removes T cell localization and trafficking effects, as well as immunosuppressive factors in the tumor microenvironment (TME). Importantly, this assay is scalable; we were able to culture upwards of 108 cells, enabling coverage of genome-wide CRISPR sgRNA libraries. Over the course of 8 days of anti-CD3 stimulation (after 2 days of antiCD3/CD28 activation), we confirmed a progressive upregulation of the inhibitory receptors, PD-1 and TIM3, and a growth defect in the chronically-stimulated T cells, compared to cells passaged without further TCR stimulation after initial activation (acute stimulation; p < 0.0001, unpaired t-test; Figure 1B–C, S1A). Chronically stimulated T cells exhibited defects in the secretion of IFNɣ and TNFɑ after restimulation with phorbol myristate acetate and ionomycin (PMA/IO), compared to acutely stimulated cells, and defects in tumor killing in vitro and in vivo (Figure S1B–D).
Figure 1: In vitro chronic antigen stimulation assay recapitulates the epigenetic hallmarks of T cell exhaustion.

(A) Diagram of in vitro exhaustion assay. (B) Surface phenotype of CD8+ T cells at day 0 and day 10 of the T cell exhaustion assay, gated on live cells. (C) Expansion of chronically stimulated and acutely stimulated T cells in vitro. Statistical significance was assessed by Student’s t-test, n=3. (D) Principal component analysis of ATAC-seq profiles of CD8+ T cells throughout the course of chronic stimulation, n=3. (E) ATAC-seq signal tracks in the Pdcd1 and Entpd1 gene loci at each time point in the in vitro exhaustion assay, as well as previously published reference ATAC-seq profiles from T cells in tumors or LCMV (Miller et al., 2019). (F) Heatmap showing ATAC-seq coverage of each peak in the “Terminal Exhaustion peak set” for each time point in the in vitro exhaustion assay. Reference data from TILs is also included. Selected nearest genes are indicated on the right. (G) chromVAR motif accessibility heatmap for each ATAC-seq sample. Selected TF motifs are indicated on the right. Top 100 most variable motifs are shown. See also Figure S1.
We next asked whether the in vitro exhaustion assay recapitulated epigenetic hallmarks of T cell exhaustion in vivo (Pauken et al., 2016; Sen et al., 2016; Satpathy et al., 2019). We performed the assay for transposase-accessible chromatin with sequencing (ATAC-seq) over the course of chronic stimulation and analyzed chromatin accessibility profiles. Principal component analysis (PCA) of ATAC-seq profiles showed that PC1 separated naïve cells (Day 0) from all other samples, while PC2 captured a progressive epigenetic differentiation of the T cells during chronic stimulation (Figure 1D). Analysis of individual gene loci, including Pdcd1 and Entpd1, demonstrated an increase in accessibility at known exhaustion-specific regulatory elements (Figure 1E) (Miller et al., 2019). We evaluated the global epigenetic similarity of in vitro stimulated cells to reference T cell exhaustion data from tumors and chronic infection (Miller et al., 2019). We defined a terminal exhausted T cell (TEX) peak set as ATAC-seq peaks that are specifically active in terminally exhausted T cells, compared to progenitor exhausted T cells, and we identified 3,537 terminal exhaustion ATAC-seq peaks in the B16 melanoma tumor model and 2,346 peaks in the lymphocytic choriomeningitis virus (LCMV) chronic infection model (Log2 FC ≥ 1; FDR ≤ 0.05; Figure 1F, S1E–G). The in vitro assay recapitulated global epigenomic changes observed in Terminal TEX cells in vivo in both systems: 88.6% of ATAC-seq peaks in tumors and 70.1% of ATAC-seq peaks in chronic infection showed a shared increase in accessibility in the in vitro model at Day 10 (FDR ≤ 0.05; Figure 1F, S1F–G). By contrast, analysis of the 2,926 Progenitor TEX peaks identified in TILs demonstrated that these sites showed decreased accessibility with repeated stimulation (Figure S1E–G). Finally, we analyzed chromatin accessibility at transcription factor (TF) binding sites using chromVAR (Schep et al., 2017), which showed that TF motifs previously associated with terminal exhaustion, including Batf, Fos, Jun, and Nr4a motifs were highly accessible in vitro at day 10. Moreover, we observed progressive loss of accessibility at naive and progenitor exhaustion-associated Lef1 and Tcf7 motifs, early increased accessibility of NF-κB and Nfat motifs, and later increased accessibility of AP-1 and Nr4a motifs, mirroring the progression of TF activity observed in T cell exhaustion in vivo (Figure 1G) (Lynn et al., 2019; Miller et al., 2019; Beltra et al., 2020; Daniel et al., 2021). In summary, these results demonstrate that the in vitro T cell exhaustion assay displayed hallmark functional and genomic features of in vivo T cell exhaustion.
Genome-wide CRISPR screens identify genetic regulators of T cell exhaustion.
We next adapted the in vitro exhaustion assay to be compatible with CRISPR screening (Figure 2A). We isolated CD8+ T cells from Rosa26-Cas9 knockin mice, transduced the cells with a genome-wide retroviral sgRNA library containing 90,230 sgRNAs, split the cells into acute (IL-2 only) and chronic (anti-CD3 and IL-2) stimulation conditions on Day 4 and sequenced both pools on Day 10 (Figure 2A) (Platt et al., 2014; Henriksson et al., 2019). We introduced a 48-hour delay after activation to allow time for efficient gene editing and puromycin selection of transduced cells and validated that this modified protocol caused similar defects in cytokine production after re-stimulation (Figure S2A–B). We performed replicate screens and confirmed a low multiplicity of infection, upregulation of inhibitory receptors on Day 10 of the chronic culture, and high coverage of the sgRNA library in each condition (Figure S2C–F, Table S1). Positive controls for the screen are components of the TCR signaling pathway, since depletion of these factors impairs antigen-driven (or anti-CD3-driven) signaling. Accordingly, we first analyzed the enrichments of the CD3 receptor subunits (Cd3e, Cd3d, Cd3g, Cd247; Figure S2G in red) and observed a robust enrichment of guides targeting these genes in both replicates. Merging the replicates yielded an overall z-score and ranking for each gene (Figure 2B–C, Table S1) (Flynn et al., 2021). We validated this analysis approach by comparing screen hits obtained from two additional CRISPR sgRNA enrichment analytical methods and two normalization strategies (Figure S3A–E, Table S2, Methods) (Li et al., 2014; Morgens et al., 2016; Gilbert et al., 2014).
Figure 2: Genome-wide functional interrogation of T cell exhaustion.

(A) Diagram of genome-wide T cell exhaustion screen. (B) Correlation of replicate screens (n=2) with selected functional categories of genes colored as indicated. Gene sets were based on GO Terms and were supplemented with manual annotations. (C) casTLE volcano plot of the Chronic vs Acute stimulation screen comparison, with top hits labeled. (D) Individual sgRNA z-scores for top hits in “integrin signaling” or “TCR signaling” functional categories. (E) GO Term analysis of the top 100 positive hits. (F) Individual sgRNA z-scores for genes in different functional categories: chromatin (left), selected receptors and transcription factors (center), or other (right). In (D) and (F) n=10 sgRNA-replicates per gene are shown. 1,000 randomly selected guides are shown in the background of each row in grey, for visual reference. (G) Correlation of Acute vs Chronic z-scores in the mini-pool versus the genome-wide screen. (H) Correlation of the mini-pool Chronic vs Acute z-scores against Acute vs Input (left) or Chronic vs Input (right). Genes in (G) and (H) are colored by functional category: TCR signaling (red), integrin signaling (orange), chromatin (blue), or other (grey). Colored boxes in (H, left) denote enhanced (purple), similar (yellow), or reduced (green) expansion after acute stimulation. See also Figures S2–5 and Tables S1–3.
In addition to Cd3e, Cd3d, and Cd3g, top enriched genes in the screen included other known components of the TCR signaling pathway, such as Zap70, Lcp2, Lat, and Lck, as well as cell adhesion and integrin-related genes Fermt3, Tln1, Itgav, and Itgb3 (Figure 2B–D). GO Term analysis of the top 100 positive regulators of exhaustion confirmed that the “T cell receptor signaling pathway” term was highly enriched (padj = 5.44 × 10−3; Figure 2E). Surprisingly, in addition to TCR-related GO terms, the other top terms were related to epigenetics, including “chromatin remodeling” (padj = 1.74 × 10−6) and “nucleosome disassembly” (padj = 1.90 × 10−5; Figure 2E). Indeed, analysis of additional highly enriched genes identified a number of chromatin-related factors, including Arid1a, Smarcc1, Smarcd2, Ino80, Actr8, and Actr5 (Figure 2F, left). Of note, the co-stimulatory and inhibitory receptors Icos, Pdcd1, Ctla4, Cd28, Havcr2, Lag3, and Tigit were not significantly enriched by the screen (Figure 2F, center). Among TFs, Irf4, Junb, Eomes, and Batf3 were depleted, while Tbx21 and Nr4a3 were modestly enriched, supporting previous demonstrations of their roles in exhaustion (Figure 2F, center) (Ataide et al., 2020; Chen et al., 2019; Paley et al., 2012; Seo et al., 2021). In contrast, Tox and Tox2, which are critical for the development of exhaustion, were not hits, supporting previous studies demonstrating that deletion of these factors may not improve T cell persistence in vivo (Figure 2F, center) (Alfei et al., 2019; Khan et al., 2019; Scott et al., 2019). Similarly, Jun and Batf were not hits, suggesting that while overexpression of these factors improves T cell persistence, deletion has no significant effect (Lynn et al., 2019; Seo et al., 2021).
We next used Cytoscape to visualize the protein-protein interaction network of top enriched and depleted genes (Figure S4A) (Shannon et al., 2003). This analysis confirmed the highly interconnected and enriched network of factors that directly associate with the TCR complex and downstream signaling components, as well as several other protein complexes and functional categories. These included the INO80 complex (hits included Ino80, Ino80b, Ino80c, Actr5, and Actr8) and the BAF complex (hits included Arid1a, Smarcb1, Smarcd2, Smarca4, and Smarcc1) - both ATP-dependent chromatin remodeling complexes that are essential in many aspects of development (Figure S4A; Hargreaves and Crabtree, 2011). Finally, analysis of single-cell RNA-seq data from chronic viral infection showed that nearly all (98/100) top-ranked hits were expressed in exhausted T cells in vivo (Figure S4B–D) (Raju et al., 2021). In summary, the in vitro genome-wide CRISPR-Cas9 exhaustion screen provides a comprehensive catalog of genetic factors that govern the process of chronic antigen-induced T cell exhaustion and identifies chromatin remodeling factors as potential targets for improving T cell persistence.
Chromatin remodeling factors limit T cell persistence in vitro and in vivo.
To further characterize the top 300 ranked genome-wide screen hits, we created a custom sgRNA mini-pool (Table S2). We repeated the in vitro stimulation screen and collected acute and chronic samples, as well as input samples on day 4 (Table S3, Figure S5A). We observed high concordance between biological replicates and therefore merged the replicates (Figure S5B–E, Table S3). We first considered the chronic vs acute enrichments, which served as validation of the original genome-wide screen. Of the 88 genes in the pool that were statistically significant positive hits in the genome wide screen, 52 (59.1%) were validated in the mini-pool (FDR < 0.05; Figure 2G, S5C). Next, we compared the chronic vs acute gene enrichments to acute vs input or chronic vs input enrichments, which measured the fitness advantage or disadvantage of each gene knockdown relative to the initial pool (Figure 2H, S5E). Most genes displayed either similar (233/300; 77.7%) or reduced (64/300; 21.3%) enrichments in acute stimulation compared to input, enabling the identification of sgRNAs that specifically improve T cell persistence in the presence of chronic antigen, rather than T cell proliferation in general, and that maintain proliferative capacity after acute stimulation (Similar: −3.5 ≤ z ≤ 3, reduced: z < −3.5, improved: z > 3.5; Figure 2H, left, S5E).
To characterize the in vivo function of the top hits, we next screened the sgRNA mini-pool in two murine tumor models. On day 0, we bilaterally injected MC-38 colon adenocarcinoma or B16 melanoma tumors that ectopically expressed ovalbumin into Rag1−/− mice and isolated CD8+ T cells from Cas9/OT-1 mice. On day 1, we transduced the T cells with the custom sgRNA mini-pool (Figure 3A). We transplanted 1×106 T cells per mouse 6 days after tumor inoculation, harvested the tumors and spleens of mice on day 15, sorted T cells from each organ, and sequenced the bulk sgRNA content in these cells (Figure S6A, Table S3). We computed sgRNA enrichments as described above and merged the results from all mice to create an aggregate tumor LFC z-score and spleen LFC z-score for each gene in each tumor model, relative to the control distribution (Figure 3B–E, Table S3).
Figure 3: Targeted in vivo screening identifies subunits of the INO80 and BAF complexes that limit T cell persistence.

(A) Diagram of in vivo pooled CRISPR screening. (B) Correlation of tumor LFC z-scores to spleen LFC z-scores, colored by functional category. (C) Correlation of in vivo z-score and in vitro z-scores for genes in the CRISPR mini-pool. (D) Correlation of in vivo MC-38 and B16 tumor z-scores for genes in the CRISPR mini-pool. (B-D) Results shown are merged from 3 mice per tumor model (n=6 tumors, n=3 spleens). (E) Cytoscape protein-protein interaction network colored by z-score in MC-38 tumors. (F) Top: Boxplot of MC-38 tumor versus input log fold change for each sgRNA targeting the indicated gene, with the mean control log fold change subtracted. Bottom: heatmaps showing the sgRNA average of the indicated in vivo or in vitro screen for the same hits. Box plots show 25th, 50th (median), and 75th percentiles with outliers shown as dots. Each dot represents one sgRNA-replicate, n=36 per target gene. (G) Individual sgRNA-replicate z-scores for six top hits showing the MC-38 Tumor vs Input comparison (left, n=36), MC-38 Spleen vs Input (center, n=18), and in vitro Chronic vs Acute (right, n=12). See also Figure S6 and Tables S2–3.
Cells containing TCR complex/signaling sgRNAs should have an impaired ability to recognize antigen and thus, in contrast to the in vitro screen, were depleted in vivo, except for Itk (Figure S6B). However, a select group of in vitro hits were highly enriched in tumors and spleens in both tumor models, including Arid1a, Itk, Smarcd2, B4galnt1, Gata3, Gpr137c, Trp53, and Vstm4 (Figure 3B–D). Visualizing the tumor enrichments of each gene in the context of the Cytoscape network revealed that many of the positive hits in vivo were epigenetic factors, including subunits of the INO80 complex, (Ino80c and Actr5) and the BAF complex (Arid1a, Smarcd2, and Smarcc1; Figure 3E). The top ranked gene knockdowns improved T cell accumulation in tumors by up to 3.4-fold. For comparison, T cells lacking Cd3d were depleted 6.7-fold and T cells lacking Cd3e were depleted 3.3-fold, demonstrating that targeting the top hits substantially improved T cell persistence in tumors (Figure 3F, S6C). These results validate the genome-wide screen, identify perturbations that improve T cell persistence only in the setting of chronic antigen stimulation, rather than improving general T cell fitness, and nominate the BAF and INO80 complexes for further investigation (Figure 3F–G, S6C).
Tuning cBAF activity enhances T cell persistence and improves tumor control.
We next validated the persistence advantage of Arid1a-sgRNA cells (the top epigenetic hit in the screen). We used a cell competition assay where cells were transduced with either a single-targeting control (CTRL1) sgRNA or an Arid1a-targeting sgRNA with different fluorescent reporters (Methods), mixed, and then put into the in vitro chronic stimulation assay (Figure 4A) or the in vivo MC-38 tumor model (Figure 4B). The activity of both Arid1a-targeting sgRNAs was confirmed at the DNA and protein level (Figure S6D–F). In vitro and in vivo, Arid1a-sgRNA cells demonstrated significantly enhanced persistence, compared to control cells, confirming the results of the pooled screens (Figure 4A–B). Moreover, Arid1a-sgRNA cells exhibited lower levels of PD-1 and Tim3 after chronic stimulation in vitro (Figure 4A). Finally, we evaluated whether the observed enhanced persistence of Arid1a-sgRNA cells resulted in improved anti-tumor responses in vivo. We inoculated Rag1−/− mice with MC-38 tumors as previously described, and on day 6, transplanted 5×105 Cas9/OT-1 CD8+ T cells transduced with either CTRL1 retrovirus or Arid1a-sgRNA retrovirus and monitored tumor growth (Figure 4C). By day 15, transfer of Arid1a-sgRNA cells significantly improved tumor clearance, compared to transfer of control cells (Arid1a-sgRNA vs CTRL1 tumor size, Day 15: p = 5 × 10−8; Welch Two Sample t-test). Importantly, survival of mice receiving Arid1a-sgRNA T cells was significantly extended, compared to mice receiving CTRL1 T cells (median survival = 12 days (no transplant), 15 days (CTRL1), 25 days (Arid1a-sgRNA); Arid1a-sgRNA vs CTRL1: p =1.20 × 10−8; Figure 4D).
Figure 4: SWI/SNF mini-pool CRISPR screens and functional studies demonstrate that tuning cBAF activity can enhance anti-tumor immunity.

(A) In vitro competition assay of Arid1a-sgRNA versus CTRL1 T cells. Left: cells were mixed on Day 4 at the indicated ratios and passaged in the chronic stimulation assay for 6 days. On Day 10, proliferation relative to CTRL1 T cells and surface phenotype were assessed by flow cytometry, n=3 or 4 as indicated. (B) In vivo competition assay of Arid1a-sgRNA versus CTRL1 T cells. Cells were mixed on Day 6 (input) and then transplanted into tumor bearing mice. On Day 15, relative proliferation in the tumor was assessed by flow cytometry, n=6 or 10 as indicated. (A-B) Error bars denote mean ± SD and significance was assessed by Welch Two Sample t-test. (C) Tumor sizes for each cohort. Statistical significance was assessed at Day 15 by Wilcoxon rank sum exact test, n=20 tumors per group. (D) Survival curves of tumor-bearing mice in each treatment group. Statistical significance was assessed by log-rank test, n=10 mice per group. (E) Correlation of SWI/SNF CRISPR mini-pool tumor enrichments in MC-38 versus B16 tumor models. Results shown are merged from 4 mice for MC-38 (n=8 tumors, n=4 spleens) or 2 mice for B16 tumors (n=4 tumors, n=2 spleens). (F) Cartoons of the three BAF complexes colored by z-score from SWI/SNF CRISPR mini-pool experiments in MC-38 tumors. BAF complex cartoons adapted from (Mashtalir et al., 2018). * p < 0.05, *** p < 0.001. See also Figure S6 and Table S4.
To provide deeper mechanistic insight into the role of BAF complex factors in T cell exhaustion, we performed an additional CRISPR mini-pool screen targeting each of the 29 SWI/SNF complex subunit genes in the B16 and MC-38 tumor models and interpreted these results in the structural context of SWI/SNF complex assembly (Mashtalir et al., 2018) (Table S2). As observed in the prior in vivo screen, the three most significant hits were in the cBAF complex (Arid1a, Smarcc1, and Smarcd2) and notably were in positions of the complex that can be substituted by paralogs in other forms of the complex (Figure 4E–F; Table S4) (Mashtalir et al., 2018). In contrast, perturbation of irreplaceable subunits of the BAF core (e.g. Smarce1, Smarcb1) or ATPase module components was deleterious and led to depletion of these sgRNAs. Therefore, we propose a model in which tuning (reducing) the presence of cBAF on chromatin is beneficial for T cell persistence. This concept is supported by prior mechanistic studies demonstrating that ARID1A-deficient tumors exhibit reduced (but not ablated) levels of cBAF complex on chromatin, which results in decreased access of key transcription factors (including AP-1 factors) (Mathur et al., 2017; Xu et al., 2020). In addition to cBAF, we also observed positive enrichments of sgRNAs targeting the PBAF complex member, Arid2, and strong depletion of sgRNAs targeting the ncBAF complex members, Bicral, Bicra, and Brd9 (Figure 4E–F; Table S4). In summary, these results demonstrate that perturbation of cBAF complex subunit genes can improve T cell persistence and anti-tumor immunity in vivo.
Perturbation of ARID1A improves T cell persistence in primary human T cells.
We asked whether perturbation of cBAF subunits could also improve the persistence of primary human T cells in an in vitro chronic stimulation assay (Figure 5A). We introduced CRISPR-Cas9 sgRNA ribonucleoproteins (RNPs) targeting ARID1A (two independent sgRNAs) or a control RNP into primary human T cells. We split the cells into acute and chronic cultures, and the chronic condition was stimulated for 6 days with anti-CD3 coated plates. In acutely stimulated cultures, we observed no difference between the genotypes for proliferation or viability. However, in chronically stimulated cultures, ARID1A-sgRNA cells proliferated significantly more and maintained higher viability than CTRL T cells (ARID1A-sgRNA vs CTRL1 cells: mean increase of 5.25-fold expansion, p = 0.013; Figure 5A).
Figure 5: Conserved function of ARID1A in human T cells in vitro and in vivo.

(A) Proliferation and viability of primary human T cells after electroporation of the indicated RNP. Left: Acutely stimulated T cells. Right: Chronically stimulated T cells using anti-CD3-coated plates. Data shown is representative of 3 independent experiments and 3 donors. Error bars denote mean ± SD and significance was assessed by Student’s t-test, n=2 replicates per sgRNA. (B) Schematic of CRISPR mini-pool screen in primary human CD8+ T cells transduced with the NY-ESO-1-specific TCR, 1G4. (C) Results of the human CRISPR mini-pool screen aggregated by gene. (D) Results of the human CRISPR mini-pool screen with individual sgRNA replicates shown as dots. Genes are ordered from highest to lowest average LFC. Box plots show 25th, 50th (median), and 75th percentiles. Results shown in (C-D) are combined from 2 independent donors, 2 mice per donor, and 2 sgRNAs per target gene (n=8 sgRNA replicates per target). In (C-D), orange indicates inhibitory receptors, red indicates TCR signaling pathway genes, blue indicates chromatin remodelers and grey indicates negative controls. See also Table S5.
We next validated the persistence advantage of ARID1A-sgRNA T cells in vivo. We designed a CRISPR mini-pool for in vivo human T cell experiments, which encompassed 48 sgRNAs targeting 20 genes and included 8 negative control guides (Table S5). We included sgRNAs targeting ARID1A, as well as the inhibitory receptors, PDCD1, LAG3, and HAVCR2, and other top-ranked genes from our prior screens, such as TMEM222, CBLB, TCEB2, and SOCS1 (Shifrut et al., 2018). We performed the screen in the A375 human melanoma xenograft model, which expresses the NY-ESO-1 antigen. We introduced the cognate 1G4 TCR into primary human T cells on day 1 along with the sgRNAs and transplanted T cells into NOD-SCID-IL2Rγ-null (NSG) tumor-bearing mice on day 14 (Figure 5B). 7 days later, we sorted T cells from the tumors and spleens, sequenced sgRNAs present in each organ, and compared their abundance to input samples prior to transplant. As expected, we did not observe enrichments in control sgRNAs or sgRNAs targeting inhibitory receptors but did observe depletion of sgRNAs targeting CD3D (Figure 5C–D). In contrast, sgRNAs targeting ARID1A were significantly enriched in tumors compared to input samples in both donors, demonstrating that the function of cBAF in limiting T cell persistence is conserved in human T cells (ARID1A-sgRNA versus CTRL LFC p = 0.0010 by Wilcoxon test; Figure 5C–D).
In vivo Perturb-seq reveals distinct transcriptional effects of chromatin remodeling complexes in TILs.
To understand the molecular mechanisms driving improved T cell function in hits identified by the CRISPR screens, we performed Perturb-seq, which simultaneously captures CRISPR sgRNAs and the transcriptome in single cells (Adamson et al., 2016; Dixit et al., 2016; Replogle et al., 2020). We designed a third custom sgRNA pool (micro-pool) targeting the INO80 and BAF complexes. For SWI/SNF genes, we targeted Arid1a, Smarcc1, and Smarcd2 (top hits identified in vitro and in vivo), as well as Arid2 and Arid1b, which were enriched in the SWI/SNF-specific mini-pool screen. From the INO80 complex, we selected Actr5 and Ino80c, which were enriched in both the in vitro and in vivo screens. Finally, we included positive controls, Pdcd1 and Gata3, as well as 12 single targeting negative controls for a total of 48 sgRNAs targeting 9 genes (Table S2). We performed a similar in vivo T cell protocol as described above for the larger CRISPR screen, including collecting an input sample to evaluate the persistence phenotype of each sgRNA. Nine days after T cell transplantation, we harvested tumors, isolated TILs, and used direct-capture Perturb-seq to simultaneously read out sgRNA identity and scRNA-seq profiles (Figure 6A, S7A–B) (Replogle et al., 2020).
Figure 6: In vivo Perturb-seq reveals distinct transcriptional roles of the cBAF and INO80 complexes in TILs.

(A) Diagram of direct-capture Perturb-seq of sorted TILs. (B) scRNA-seq profiles of TILs colored by cluster assignment. (C) scRNA-seq profiles of cells colored by the perturbation detected in each cell. Cells where no guide, or multiple guides, were detected are shown in grey. (D) Expression of selected marker genes in each single cell. (E) Analysis of LCMV signature gene sets for each cluster. Gene set enrichment scores were calculated for each single cell, cell values were averaged by cluster and z-scored. (F) Histogram of Pearson correlation of gene expression differences of pairs of sgRNAs. Top: Pairs targeting the same gene are shown in blue (n=120), other pairs are shown in gray (n=1,008). Bottom: Pairs targeting the same protein complex are shown in red (n=96), other pairs are shown in gray (n=912). Complexes considered in the analysis are cBAF (Arid1a, Arid1b, Smarcd2, and Smarcc1) and INO80 (Ino80c and Actr5) and pairs of sgRNAs that target the same gene are excluded. (G) Left: Heatmap of the correlation of gene expression differences of each pair of sgRNAs. Center (from left to right): Representation of each sgRNA in the pre-transplant sample, cell count of each sgRNA in the Perturb-seq dataset, and estimated fold change of each sgRNA relative to controls. Right: Proportion of cells in each cluster for each sgRNA. See also Figure S7.
After quality control filtering, we obtained high-quality single cell RNA sequencing (scRNA-seq) profiles from 70,646 cells and identified 6 clusters (Figure 6B). We determined a high-confidence sgRNA identity for 52,607 cells (74.4%; Figure 6C; Methods). Cell type clusters expressed varied levels of inhibitory receptors, effector cytokines, and key transcription factors, indicating that they represented a mix of exhausted and effector T cells (Figure 6D, S7C–D). Cluster 1 expressed high levels of Klf2 and S1pr1 (T effector memory; TEM), Cluster 2 expressed high levels of interferon stimulated genes (ISGs) such as Mx1 (TISG), Cluster 3 expressed high levels of Tnfrsf9 (encoding 41BB) and Cd160 (T-41BB), Cluster 4 expressed high levels of progenitor exhaustion genes including Pdcd1, Tcf7 and Slamf6 (TEXProg), Cluster 5 expressed the highest levels of inhibitory receptors Pdcd1, Lag3, and Havcr2 (TEXTerm), and Cluster 6 consisted primary of cycling cells, marked by Mki67 and confirmed by cell cycle analysis (T-Cycling; Figure S7C–D). To confirm cluster identities, we generated gene signatures from previously published CD8+ T cell types present in acute or chronic LCMV infection in vivo (Figure S7E–F; Methods) (Daniel et al., 2021). We used the top 100 marker genes for each LCMV T cell cluster to score each single cell in our Perturb-seq dataset according to the average expression of these signature gene sets. Visualizing the enrichment of these LCMV signatures in each cluster demonstrated transcriptional similarity of several clusters to cell types in the reference dataset (Figure 6E). For example, Cluster 1 was enriched for the effector memory-related genes (TEM signature), Cluster 2 was similar to the TEXISG signature, and the progenitor and terminally exhausted clusters (Clusters 4 and 5) enriched the corresponding LCMV signatures (Figure 6E).
We performed sgRNA-level quality controls to assess the reproducibility of effects of independent sgRNAs (Figure 6F–G). We first computed gene expression differences between each sgRNA and all other cells in the dataset. Independent sgRNAs targeting the same gene had highly correlated gene expression changes, relative to pairs of sgRNAs targeting different genes (Figure 6F–G). Interestingly, pairs of sgRNAs targeting the same complex, (grouping together sgRNAs targeting cBAF genes or sgRNAs targeting INO80 genes) also induced highly correlated changes, indicating common transcriptional effects of targeting distinct subunits within the same complex (Figure 6F–G). Arid2 clustered separately from the rest of the BAF targeted sgRNAs, suggesting distinct roles for the cBAF and PBAF complexes (Figure 6G). We next used the input representation of each sgRNA to estimate the T cell accumulation advantage of each sgRNA relative to controls, which demonstrated that the majority of sgRNAs enhanced T cell accumulation in the tumor, relative to control sgRNAs, in line with the in vivo screen results (Figure 6G). In particular, Arid1a-sgRNA cells were enriched 2.74-fold on average relative to CTRL1 cells (Figure 6G). Finally, we examined the cell type cluster composition of cells containing each sgRNA (Figure 6G, far right). All perturbations contained cells from each cluster with similar proportions, suggesting that depletion of each target gene may not impact wholescale changes in cell type composition, but rather modulates gene expression in one or more clusters.
To further investigate this possibility, we aggregated cells that contained sgRNAs targeting the same gene and computed differential gene expression for each perturbation, compared to CTRL1 cells (Figure 7A–F; Table S6). Targeting cBAF subunits Arid1a, Smarcd2, or Smarcc1 induced shared global changes in the transcriptional program of T cells, including the upregulation of effector molecules, Gzmb and Ifng, cell surface receptors, Cxcr6 and Il7r, and transcription factors, Irf4 and Batf. Meanwhile, Pdcd1, Lag3, and Ccl5 were consistently downregulated by cBAF perturbation (Figure 7A–F). In contrast, Arid2 perturbation induced a distinct gene expression program, albeit with some similarities, including the downregulation of Pdcd1 and Lag3. Perturbation of Gata3 and Pdcd1 induced distinct gene expression changes from either cBAF or Arid2 perturbation; for example, the most upregulated gene after Pdcd1 depletion was Tox, perhaps consistent with the proposed impact of PD-1 deletion on accelerating differentiation to terminal exhaustion (Figure 7A–F) (Odorizzi et al., 2015). Finally, when gene expression changes were analyzed within each cluster, we found that each perturbation induced highly concordant changes in gene expression regardless of the T cell subtype (Figure S7G). GO Term analysis of the cBAF upregulated gene set enriched effector terms, including T cell activation, cell adhesion, cytokine production, and T cell proliferation (Figure 7H). In contrast, INO80 perturbation substantially modulated metabolism related genes (Figure 7E, 7H). Projection of genes upregulated by cBAF depletion onto canonical T cell states identified in chronic LCMV infection showed an enrichment in effector T cell clusters, while projection of downregulated genes showed an enrichment in terminally exhausted T cell clusters (Figure 7G, S8B). In summary, these data demonstrate that subunits of the cBAF and INO80 chromatin remodeling complexes have distinct roles in T cell exhaustion that are largely conserved within the same complex, with cBAF primarily regulating effector- and exhaustion-related genes and INO80 regulating metabolism. Furthermore, the transcriptional impact of targeting chromatin remodeling factors minimally overlaps with the impact of previously known targets, Pdcd1 and Gata3, suggesting the potential to synergistically target multiple pathways to improve T cell function (Figure 7F, S8A).
Figure 7: cBAF-depleted T cells exhibit enhanced effector gene signatures and reduced terminal exhaustion.

(A) Volcano plots comparing aggregated cells with the indicated perturbation versus CTRL1 cells. FDR values were calculated via Wilcoxon Rank Sum test, as implemented in Seurat. Sample size: n=4,668 (Arid1a-sgRNA), n=5,891 (Smarcc1-sgRNA), n=1,448 (Smarcd2-sgRNA), n=3,712 (Arid2-sgRNA), n=2,625 (Gata3-sgRNA), n=6,465 (Pdcd1-sgRNA), n=18,569 (CTRL1). (B) Pairwise correlations of gene expression differences induced by each perturbation. (C) Heatmap of all upregulated (up) or downregulated (down) genes in at least one perturbation, grouped by which perturbation has the strongest effect on expression. Selected genes in each block are labeled. (D) Comparison of upregulated or downregulated gene sets by perturbation of cBAF subunits, Arid1a, Smarcd2, or Smarcc1. (E) Comparison of gene sets up- or downregulated by perturbation of INO80 subunits Actr5, or Ino80c. (F) Comparison of gene sets upregulated by perturbation of cBAF subunits, INO80 subunits, or Pdcd1, Gata3, or Arid2. (G) Enrichments of upregulated and downregulated gene sets in LCMV expression data (Daniel et al., 2021). Module scores of each gene set were computed for each single cell in the LCMV dataset, averaged by cluster, and then z-scored to obtain the indicated enrichment z-scores. (H) Selected GO Terms of indicated gene sets. See also Figure S8 and Table S7.
Arid1a perturbation limits the acquisition of terminal exhaustion-associated chromatin accessibility.
We next asked how perturbation of Arid1a impacted the epigenetic landscape of T cell exhaustion. We performed a competition assay as described above, wherein CTRL1 and Arid1a-sgRNA cells were mixed and subjected to in vitro exhaustion. At Day 6 and Day 10, we isolated CTRL1 and Arid1a-sgRNA cells from the same culture and performed ATAC-seq on each population. To analyze these results in the context of our initial assay characterization (Figure 1), we included the profiles of naïve (Day 0) and activated (Day 2) WT T cells (Figure 8A). The chromatin state progression in CTRL1 cells proceeded similarly to that observed previously in unperturbed cells; however, Arid1a-sgRNA cells proceeded down a distinct trajectory, remaining closer to naïve and activated samples than the CTRL1 cells at both time points (Figure 8A).
Figure 8: Arid1a is required for the acquisition of the exhausted T cell chromatin state.

(A) Principal component analysis of ATAC-seq profiles of Arid1a-sgRNA and CTRL1 cells in the in vitro exhaustion competition assay (n=3 or 4 as indicated). Unperturbed naïve and activated samples (Day 0 and 2) are included for reference (n=3). (B) Comparison of ‘opened’ and ‘closed’ ATAC-seq peak sets from Day 6 to Day 10 for each genotype. (C) Visualization of ‘opened’ and ‘closed’ ATAC-seq peak sets, with selected nearest genes labeled. (D) ATAC-seq signal tracks of selected gene loci. Representative replicates are shown for each condition. (E) Heatmap showing ATAC-seq coverage of each peak in the “Terminal Exhaustion peak set” for Arid1a-sgRNA and CTRL1 cells at Day 6 and Day 10 in the in vitro exhaustion assay. Reference data from TILs is also included, as well as reference naïve and activated cell profiles. (F) chromVAR motif accessibility heatmap for Arid1a-sgRNA and CTRL1 ATAC-seq samples. Selected motifs are indicated on the right. Top 100 most variable motifs are shown. See also Figure S8.
We defined regulatory elements as ‘opened’ peaks if we observed increased accessibility at Day 10, compared to Day 6, and as ‘closed’ peaks if we observed decreased accessibility at Day 10, compared to Day 6 (padj < 0.05, Log2 FC > 1). Analysis of these peak sets demonstrated substantially different chromatin remodeling changes in Arid1a-sgRNA T cells, compared to CTRL1 T cells (Figure 8B–C). First, Arid1a-sgRNA cells exhibited a marked global decrease in the number of opened peaks, likely representing a relative inability of cBAF-depleted cells to establish accessible chromatin (Figure 8B). Second, while Arid1a-sgRNA cells and CTRL1 cells closed chromatin to a similar extent, the majority of these regions were non-overlapping (Figure 8B). Analysis of individual exhaustion-associated regulatory elements, including those in Pdcd1, Lag3, Entpd1, and Ifng gene loci, revealed a substantial loss of accessibility in Arid1a-sgRNA cells, compared to CTRL1 cells (Figure 8D). Analysis of the terminal TEX-specific peak set (defined in Figure 1) showed that these sites were significantly less accessible in Arid1a-sgRNA cells than in CTRL1 cells at both time points (Figure 8E, S8C–D). We next analyzed chromatin accessibility at TF binding sites, which showed that terminal exhaustion-associated TF motifs, including Fos, Jun, and AP-1 motifs were significantly less accessible in Arid1a-sgRNA cells, compared to CTRL1 cells (Figure 8F). Conversely, several TF motifs associated with effector T cell function, including Ets, Klf, and Irf motifs, showed increased accessibility in Arid1a-sgRNA cells. Finally, ATAC-seq analysis of chronically stimulated ARID1A-sgRNA human T cells demonstrated a similar loss of global chromatin accessibility at AP-1 motifs, compared to control T cells, supporting the conserved epigenetic function of ARID1A in human T cells (Figure S8E–G). In summary, these results suggest that depletion of cBAF subunits, including Arid1a, may improve T cell function by restricting the access of AP-1 TFs to chromatin and thereby preventing the acquisition of the terminal exhaustion-associated chromatin state.
Discussion
In this study, we performed genome-wide CRISPR screens in chronically stimulated T cells, which provide a comprehensive atlas of genes that regulate T cell exhaustion. We used a complementary in vitro and in vivo screening strategy: (1) the development of an in vitro exhaustion assay that is compatible with genome-wide CRISPR screening enabled us to scale the number of cells and sgRNA library coverage compared to prior screens, providing an unbiased discovery tool, and (2) in vivo follow-up screens identified perturbations that significantly improved T cell persistence in immunotherapy-relevant tumor models. Importantly, this strategy recovered known regulators of exhaustion, including Gata3, which has been demonstrated to limit T cell function in tumor models (Singer et al., 2016). However, these screens also uncovered new genes, with a surprising enrichment of epigenetic factors involved in chromatin and nucleosome remodeling, including the cBAF and INO80 complexes. In vivo Perturb-seq experiments revealed that depletion of cBAF and INO80 complex subunits impacted distinct gene programs: cBAF perturbation led to the upregulation of an effector program and downregulation of terminal exhaustion genes, while INO80 perturbation primarily impacted gene expression related to metabolic function. Finally, depletion of the cBAF complex subunit, Arid1a, improved T cell persistence in in vitro and in vivo competition assays and improved anti-tumor immunity after adoptive T cell transfer.
Our strategy was to isolate a key determinant of T cell dysfunction in cancer — chronic stimulation through the TCR — from the multifactorial process involving tumor localization, trafficking, and immunosuppressive effects in the TME. The advantage of this strategy is its specificity; sgRNA abundance is impacted by a single selection factor, and therefore, we provide a precise conceptual picture of the molecular drivers of T cell exhaustion. For example, T cell inhibitory receptors such as PD-1 and CTLA-4 were not hits in our screen, supporting the notion that checkpoint blockade does not work by reversing or preventing the process of T cell exhaustion, but rather by recruiting new functional T cell clones to enter the TME (Yost et al., 2019; Pauken et al., 2016; Spitzer et al., 2017; Yost et al., 2021). However, we wish to acknowledge that this strategy does not account for additional dysfunction pathways in T cells that may be mediated by other external stimuli, for example TGFβ-mediated suppression, or metabolic or nutrient stressors (Mariathasan et al., 2018; DePeaux and Delgoffe, 2021). Similarly, our follow-up in vivo screen selected for one functional aspect of exhaustion — T cell persistence in tumors — but did not account for additional aspects, such as cytokine secretion, and thus, additional genes identified in the in vitro screen may be uncovered as important regulators of other facets of T cell exhaustion in future studies.
The enrichment of chromatin remodeling factors as hits in both in vitro and in vivo screens provides a complementary message to previous epigenomic profiling studies in T cell exhaustion (Pauken et al., 2016; Sen et al., 2016; Philip et al., 2017; Scott-Browne et al., 2016). Namely, these prior studies demonstrated that exhaustion is mediated by global chromatin remodeling, which maintains a stable dysfunctional cellular phenotype that is not altered by anti-PD-1 treatment. We now show that targeting nucleosome remodeling complexes may be sufficient to prevent the acquisition of features of this exhaustion-associated chromatin state, and thereby improve T cell persistence and maintenance of an effector-like state. It is possible that deletion of these factors may ‘dampen’ the downstream epigenetic impact of chronic TCR signaling and extend the window in which T cells can engage antigens without accumulating exhaustion-associated epigenetic changes. Recent studies in fibroblasts have demonstrated that AP-1 family TFs may have an essential role in signal-dependent enhancer selection by collaboratively binding to nucleosomal enhancers and recruiting the BAF complex to establish accessible chromatin (Vierbuchen et al., 2017). Indeed, analysis of Arid1a-sgRNA T cells revealed a dramatic loss of accessibility at exhaustion-induced AP-1 motif-containing regulatory elements, suggesting a similar mechanism in T cells. We envision that future work will build upon these findings to pursue novel avenues to effectively improve T cell function in the context of cancer immunotherapy.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ansuman Satpathy (satpathy@stanford.edu).
Materials availability
CRISPR sgRNA mini-pool plasmids generated in this study are available from the lead contact upon request.
Data and code availability
CRISPR screen counts tables and z-score tables are available with the manuscript as supplemental data. ATAC-seq and Perturb-seq data are available on GEO under accession GSE203593. Scripts used to analyze CRISPR screen data have been previously open-sourced and are available at: https://github.com/juliabelk/sarscov2_chirp_ms
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild type mice were C57BL/6J mice (JAX: 000664). Rosa26-Cas9 knockin mice were bred in house (JAX: 026179). OT-1 mice (JAX: 003831) were crossed with Cas9 mice and then bred in-house. Rag1−/− mice were bred in-house (JAX: 002216). C57BL/6 Scid mice (JAX: 001913) and NSG mice (JAX: 005557) were procured from JAX. All animal studies were performed in accordance with the Stanford University Institutional Animal Care and Use Committee under protocol APLAC-33814. All studies were performed in animals between 8 to 12 weeks of age.
METHOD DETAILS
Primary murine T cell isolation and culture
Spleens were collected and mashed through a 70 μM filter. Red blood cells were lysed with ACK lysis buffer (Gibco) and incubated for 6 mins before washing with PBS. Cells were counted and then resuspended in MACS buffer (PBS + 0.5% BSA + 2 μM EDTA). CD8 T cells were enriched using the mouse CD8+ T cells isolation kit from Miltenyi (Miltenyi Cat# 130–104-075) and then resuspended in RPMI with 10% FBS, 1 % Sodium pyruvate, 1% Non-essential amino-acids, 100U Pen/Strep, 50 nM of B-mercaptoethanol (cRPMI) and supplemented with 10 ng/ml of mouse IL-2. Cells were seeded at a concentration of 1 million cells/ml on plates coated with 5 ug/ml of anti-CD3 and 2 μg/ml of anti-CD28. Cells were kept on these activation plates for 48 hours at the beginning of all experiments. CD8+ T cell purity was verified via flow cytometry. Cells were passaged every two days and maintained at 1 million cells/mL.
In vitro T cell exhaustion assay
To induce T cell exhaustion, chronic stimulation was performed using plates coated with anti-CD3 at 5 μg/mL (in the continued presence of 10 ng/ml IL-2). Cells were passaged onto a fresh coated plate every two days and analyzed on Day 6, 8, or 10 as described in the Results. In contrast, acutely stimulated cells were maintained in 10 ng/ml IL-2 alone, passaged every two days, and analyzed on Day 6, 8, or 10, as described in the Results.
Measurement of cytokine production
T cells were re-stimulated with phorbol myristate acetate (Sigma, 50 ng/mL) and ionomycin (Sigma, 500 ng/mL) or plate bound anti-CD3 at 3 μg/ml. After 90 minutes, cells were treated with brefeldin A to block cytokine secretion. Then, 3 hours later, cells were stained for surface markers and simultaneously labeled with Live/Dead Blue Viability Dye (Thermo Fisher) for 20 min at 4 °C. Cells were washed twice and fixed overnight using a FoxP3 Fixation/Permeabilization Kit (Thermo Fisher). The next day, cells were washed and stained for intracellular cytokines for 1 hour at room temperature. They were then washed three times and analyzed using an LSR Fortessa machine (Beckman Dickinson). FlowJo v.10.0 was used for data analysis. All experiments were performed with at least two biological replicates. Antibodies used (at 1:100 unless otherwise noted) were TNF-PE (BioLegend, MP6-XT22, 506306), PD-1-PECy7 (BioLegend, RMP1–30, 109110) IFN-γ-FITC (BioLegend, XMG1.2, 505806), CD4-BV711 (BioLegend, RM4–5, 100550), and CD8α-BV786 (BioLegend, 53–6.7, 100750).
Growth curves
After activation (described above), T cells were plated in 24-well plates at 5 × 105 cells in 1 mL of RPMI-1640 medium containing 10% FBS, 2 mM l-glutamine, 5 μM β-ME and 10 ng/mL IL-2, and with (chronic) or without (acute) plate-bound anti-CD3. Every 2 days throughout the experiment, cells were collected and counted using a Beckmann Coulter Counter with a cell volume gate of 75–4,000 femtoliters. Then, 50% of the cells were re-plated in 1 mL of fresh T cell medium. All experiments were performed at least two independent times.
In vitro killing assay
B16 cells expressing a Luciferase reporter were pulsed with SIINFEKL peptide (Invivogen) at the concentrations noted in Figure S1 for 4 hours at 37C. They were then washed twice and plated at 4 × 104 cells per well along with 1 × 105 OT-1 transgenic T cells that had been acutely or chronically stimulated for 8 days as previously described. After 24 hours of co-culture, cells were lysed and luciferase activity was measured using a Luciferase Assay Kit (Promega), following manufacturer’s instructions. Luciferase activity was normalized to cells cultured in the absence of T cells.
B16-ovalbumin in vivo tumor models
C57BL/6 scid (Jackson 001913) mice were injected subcutaneously with 2 × 105 B16-OVA cells in a 1:1 mix of PBS and Matrigel (Corning). 5 days later, 2 × 106 OT-1 T cells that had been acutely or chronically stimulated as described previously were adoptively transferred to mice via retro-orbital injection. Mice were monitored once per day and were euthanized for signs of morbidity.
ATAC-seq sample processing
ATAC-seq was performed using the Omni-ATAC protocol (Corces et al., 2017). Briefly, 50,000 live cells were purified by flow cytometry immediately prior to ATAC-seq. Lysis, nuclei isolation, and transposition were performed according to the Omni-ATAC protocol. Libraries were prepared for sequencing and sequenced in 2×75 dual-indexed format on an Illumina NovaSeq.
Genome-wide sgRNA library
Retroviral Mouse Genome-wide CRISPR Knockout Library was a gift from Sarah Teichmann (Addgene #104861). The library was amplified via electroporation and confirmed by sequencing.
sgRNA pool design and cloning
The sgRNA mini-pool was designed using our previously developed protocol for cloning into a lentiviral backbone and then subcloned into retroviral construct pMSCV (Flynn et al., 2021). The lentiCRISPR-v2 was a gift from Feng Zhang (Addgene plasmid #52961). pMSCV-U6sgRNA(BbsI)-PGKpuro2ABFP was a gift from Sarah Teichmann (Addgene plasmid #102796).
The mini-pool targeting 300 top hits included 2,000 sgRNAs, with 6 sgRNAs per gene as well as 100 non-targeting and 100 single-targeting controls. Briefly, six 20bp variable sgRNA sequences per target gene were obtained from the Broad Genetic Perturbation Platform (GPP) genome wide designs: sgRNA_design_10090_GRCm38_SpyoCas9_CRISPRko_NCBI_20200317.txt.gz, available online at https://portals.broadinstitute.org/gpp/public/dir?dirpath=sgrna_design. 100 non-targeting and 100 single-targeting negative control guides designed for the mouse genome, also from the Broad GPP web portal, were included. The single-targeting sequences are designed to match exactly one intergenic location in the reference genome. A “G” was added to the start of each 20bp sequence. This 21bp sequence was flanked by BsmBI-v2 enzyme sites and then two nested PCR handles. Pooled oligos were synthesized by Twist Bioscience. Oligos were amplified by two rounds of PCR and the lentiCRISPR-v2 backbone was digested overnight with Esp3I. One step digestion/ligation of amplified oligos into lentiCRISPR-v2 was performed at 37C for 1 hour in a 20 uL reaction with 1 uL T4 ligase, 1 uL Esp3I, 2 uL T4 ligase buffer, 200 ng digested backbone, and 50 ng amplified insert. Reaction was heat inactivated for 15 minutes at 65C and then 1 uL was electroporated using 25 uL Lucigen Endura electrocompetent cells and a BioRad MicroPulser with 0.1 cm gap cuvettes. After 1 hour recovery in SOC, a 1000x dilution was plated onto an agar plate to confirm library coverage. The remainder was cultured overnight in a 150 mL liquid culture and then purified by maxiprep. Finally, the pool was subcloned into pMSCV by Gibson Assembly of the sgRNA variable region amplified via PCR and pMSCV backbone pre-digested with BbsI. Electroporation was repeated as described above. Guide representation was confirmed by sequencing.
The sgRNA SWI/SNF mini-pool and micro-pool for Perturb-seq were designed with 4 guides per gene, as described above for the mini-pool using the Broad GPP mouse genome-wide designs. The SWI/SNF mini-pool contained 50 single-targeting controls and Perturb-seq micro-pool contained 12 single-targeting controls. Two primers were ordered per designed guide, for cloning via annealing. The pMSCV vector was digested with BbsI. All primer pairs were annealed separately. Annealed products were pooled equally, diluted, and then ligated into pMSCV. Amplification was performed using Stbl3 Chemically Competent cells (ThermoFisher C737303) and library coverage was confirmed via colony counting and then sequencing.
Retrovirus production and transduction
The pMSCV plasmid was transfected into GP2–293 cells (Takara, RetroPack™ PT67 Cell Line) or 293T HEK cells at roughly 80% confluency in 15 cm tissue culture plates coated with poly-d-lysine. Viral supernatant was collected at 48h and 72h post-transfection, filtered via a 0.45 μm filtration unit (Millipore). Filtered virus was concentrated using the LentiX concentrator (Takara) at 1500 × g for 45 minutes. The concentrated supernatant was subsequently aliquoted, flash frozen, and stored in −80°C until use.
CD8+ T cells were transduced with concentrated retrovirus 24 hours after isolation. 4 μg/mL of polybrene was added to each well. Plates were sealed and then spun at 1100x g at 32 C for 90 minutes. 24 hours after spinfection (ie, starting on day 2) cells were checked for fluorescence via flow cytometry and 2 μg/mL puromycin was added to the media.
sgRNA library preparation and sequencing
For samples from in vitro chronic culture, live cells were first isolated via FACS. gDNA was extracted using a commercially available kit (Zymo Cat# D3025). sgRNA libraries were prepared for sequencing as previously described (Flynn et al., 2021). Briefly, a standard three-step amplification protocol was used. First, sgRNAs were amplified off of gDNA using primers specific to the pMSCV vector for 22 cycles of PCR. 100 uL reactions with up to 4 μg of gDNA per reaction were used, and the number of reactions was scaled up until all gDNA was used. For sequencing of plasmid pools, this first PCR was skipped. For the second PCR, a 0–7bp offset was added to the front of the library using 8 pooled stagger primers to increase the diversity of the library. PCR2 primer target sites were nested inside those of PCR1 to improve the specificity of the product. Finally, in PCR3, index sequences were added. Libraries were sequenced in dual-indexed 1×75 bp or 1×150 bp format on either an Illumina NextSeq or NovaSeq.
Tumor inoculation and T cell adoptive transfer for in vivo CRISPR experiments
MC-38 or B16 cells ectopically expressing an mCherry-ovalbumin fusion construct were prepared for injection by resuspending in a 1:1 mixture of matrigel and PBS. 106 cells per tumor were injected subcutaneously into the flanks of Rag1−/− mice (two tumors per mouse). Tumors were measured every three days. Cas9/OT-1 CD8+ T cells were transduced with sgRNA pools or individual sgRNAs and selected with puromycin for 4 days, as described above. T cells were then intravenously injected into tumor-bearing mice on day 6. For in vivo competition assays, cells were mixed immediately prior to injection. 9 days after T cell injection, the spleen and tumors were harvested from each mouse.
Tissue processing and isolation of tumor infiltrating lymphocytes
Tumors were weighed and then minced into small pieces. The tumors were transferred to a gentleMACS C tube and digested in the protocol-recommended enzyme mix with a gentleMACS octo dissociator using the soft/medium tumor program. Tumor suspensions were then filtered with a 70 μM filter and then subject to RBC lysis. Spleens were mashed and filtered through a 70 μM strainer, then treated with RBC lysis buffer. For bulk sgRNA sequencing and Perturb-seq, T cells were isolated from the tumors and/or spleens by FACS. Samples were washed twice with MACS buffer and stained for 30 minutes on ice. CD8+ BFP+ cells were isolated via flow cytometry.
Competition assay for validation of individual sgRNA proliferation
The pMSCV retroviral vector was modified to replace the BFP-puromycin fusion with a VEX-puromycin fusion (pMSCV-VEX). CTRL1 sgRNA was cloned into pMSCV-VEX, while two Arid1a-sgRNA sgRNAs (Arid1a-1 and Arid1a-2) were cloned into pMSCV. Cells were separately transduced with either vector, selected with puromycin to enrich for transduced cells, mixed together, and then subjected to either the in vitro exhaustion assay or injected into tumor bearing mice. Individual guides were cloned by annealing pairs of primers, as described above. The Arid1a-1 sgRNA sequence used was GCAGCTGCGAAGATATCGGG and the Arid1a-2 sequence used was CAGCAGAACTCGCACGACCA. The CTRL sgRNA sequence used was CTTACTCGACGAATGAGCCC. Tumor processing was performed as described above for the in vivo validation.
Validation of Arid1a-targeting sgRNAs
Tracking of indels by decomposition (TIDE): Genomic DNA was isolated from transduced cells using a commercially available kit (Zymo Cat# D3025). PCR reactions were performed with primers surrounding the expected edit site and 50 ng of input DNA. PCR conditions were 30 seconds at 98C, followed by 10 seconds at 98 C, 10 seconds annealing at 60C, 25 seconds at 72C for 35 cycles, then 2 minutes at 72C. The PCR amplicons were purified with a commercially available Zymo DNA clean up kit and sanger sequenced. Quantification of edits was performed using the online tool https://tide.nki.nl/.
Western blot: Protein lysates were prepared from mouse T cells transduced with the indicated sgRNA using a radioimmunoprecipitation assay (RIPA) buffer system (Santa Cruz, sc-24948). Protein concentrations were quantified using the bicinchoninic Acid (BCA) assay (Pierce, ThermoFisher 23225). 20 μg of protein per sample was loaded and run on a 4–12% Bis-Tris PAGE gel (NuPAGE 4–12% Bis-Tris Protein Gel, Invitrogen) and transferred onto a polyvinylidene fluoride (PVDF) membrane (Immobilon-FL, EMD Millipore). After blocking membranes for 1 hour with 5% milk in PBST at room temperature (RT), membranes were incubated overnight at 4 °C with primary antibodies against Arid1a (rabbit, 1:1000, Cell Signaling, 12354S: Lot 4), Arid1b (mouse, 1:1000, Abcam, ab57461: Lot GR3345290–4), Smarca4 (rabbit, 1:1000, Cell Signaling, 49360S: Lot 3) and Tbp (mouse, Abcam, ab51841: Lot GR3313213–3). The next day, PBST was used to wash membranes three times. Next, membranes were incubated for 1 hour at RT with species-specific secondary antibodies conjugated to near-infrared fluorophores: Goat Anti-Mouse IgG Polyclonal Antibody (IRDye 680RD, 1:10,000, LI-COR Biosciences, 926–68070) or Goat Anti-Rabbit IgG Polyclonal Antibody (IRDye 800CW, 1:10,000, LI-COR Biosciences, 926–32211). After secondary antibody application, PBST was used to wash membranes three times, and then membranes were imaged using a LI-COR Odyssey CLx imaging system (LI-COR). Protein band intensities were quantified using Image Studio Lite (LI-COR) with built-in background correction and normalization to Tbp controls. Statistical analysis comparing Arid1a levels normalized to Tbp was performed using Dunnett’s multiple comparisons test on Prism (v9.2.0).
In vitro experiments in primary human T cells
T cell expansion and viability assays: T cells were activated for 4 days at a 1:3 ratio of T cells to anti-CD3/28 Dynabeads (Invitrogen). T cell expansion assays were performed with IL-2 in the culture medium at 10 ng/mL. Cell counts and viability measurements were obtained using the Cellaca Mx Automated Cell Counter (Nexcelom). Cells were stained with acridine orange and propidium iodide to assess viability.
Targeted CRISPR gene editing: Ribonucleoprotein (RNP) was preparing using synthetic sgRNA with 2’-O-methyl phosphorothioate modification (Synthego) diluted in TE buffer at 100 μM. 5 μl sgRNA was incubated with 2.5 μl Duplex Buffer (IDT) and 2.5 μg Alt-R S.p. Cas9 Nuclease V3 (IDT) for 30 minutes at room temperature. 100 μl reactions were assembled with 10 million T cells, 90 μl P3 buffer (Lonza), and 10 μl RNP. Cell were pulsed with protocol EO115 using the P3 Primary Cell 4D-Nucleofector Kit and 4D Nucleofector System (Lonza). Cells were recovered immediately with warm media for 6 hours. Guide sequences: AAVS1-sg1 5’ GGGGCCACUAGGGACAGGAU 3’, ARID1A-sg58 5’ CCUGUUGACCAUACCCGCUG 3’, ARID1A-sg60 5’ UGUGGCUGCUGCUGAUACGA 3’.
Assessment of targeted CRISPR gene editing: 4–7 days after editing, genomic DNA was extracted with QuickExtract DNA Extraction Solution (Lucigen) and ~500 bp regions flanking the cut site were amplified with Phusion Hot Start Flex 2X Master Mix (New England Biolabs) according to manufacturer’s instructions. Sanger sequencing traces were analyzed by Inference of CRISPR Editing (ICE).
Pooled CRISPR screen in primary human T cells in vivo
Activated human T cells from two donors were transduced by lentivirus to express the NY-ESO specific TCR, in parallel to lentiviral transduction of a sgRNA library with 2 sgRNAs per target gene and 8 negative controls. 24 hours after transduction, cells were electroporated with Cas9 Protein, as previously described (Shifrut et al., 2018). After electroporation, T cells were expanded in complete X-vivo 15 medium and split every two days, supplementing IL-2 at 50 U/ml. On Day 7, 2 NSG mice per donor were injected subcutaneously with 1 × 106 A375 cells, as previously described (Roth et al, Cell 2020). 1 × 106 TCR-positive T cells were transferred to mice 7 days later via retro-orbital injection. Tumors and spleens were collected 7 days after T cell transfer and processed to single cell suspension, as described previously (Roth et al., 2020). T cells were sorted by CD45 staining and gDNA was extracted using commercial kits. Library preparation, next generation sequencing and analysis was performed as previously described (Shifrut et al., 2018). The guide abundance in the spleen and tumor of each mouse was used to calculate log fold change of each guide, and MAGeCK scores were calculated with default parameters.
Direct-capture Perturb-seq
For Perturb-seq experiments, we used direct-capture Perturb-seq because it does not require a vector with a barcode sequence separate from the sgRNA, or other modifications to standard sgRNA vectors, and thus was immediately compatible with our retroviral reagents (Replogle et al., 2020). We adapted the 10x Chromium Next GEM Single Cell V(D)J Reagent Kits v1.1 5’ scRNA with Feature Barcoding reagents and protocol to be compatible with direct capture of sgRNAs in single cells. Our procedure is conceptually similar to that of Replogle et al and the modifications to the 10X genomics protocol are summarized here. For Step 1, GEM Generation and Barcoding, 5 pmol of primer KP_bead_sgRNA_RT was spiked into the reaction, enabling capture of sgRNAs in droplets and then reverse transcription of sgRNAs. Step 3.2B, Supernatant Cleanup for Cell Surface Protein Library was performed to isolate the sgRNA library. Finally, 2 uL of the product of Step 3.2B was amplified and indexed using 3 rounds of PCR. The 250bp library was purified via agarose gel and sequenced together with the gene expression (GEX) library in 26×91 format, according to 10X protocol guidelines. For Perturb-seq replicate samples shown in Figure S7B, each replicate represents either an individual tumor or two tumors from the same mouse combined into one sample. Tumors from the same mouse were combined if the cell yield was well under 10X guidelines for targeted recovery of 10,000 cells per capture. If cell yield was well over the amount needed for recovery of 10,000 cells, in certain cases samples were split across multiple 10X captures to maximize cell yield. Samples split across multiple captures were computationally merged and not counted as separate replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis summary
Statistical analysis and all software used is detailed in the below sub-sections. Statistical details for experiments, including the statistical test used, the value of n, and what n represents, can be found in the figure legends. Statistical significance was determined as p < 0.05 (or FDR < 0.05, where appropriate) unless otherwise specified.
ATAC-seq analysis
Fastq files were trimmed using fastp (Chen et al., 2018) and aligned to the mm10 genome using hisat2 (Kim et al., 2019, p. 2). Reads were deduplicated and a bed file for each sample containing filtered, deduplicated ATAC-seq fragments was created. Peaks for each sample were called individually using MACS2 (Zhang et al., 2008) and then filtered into reproducible peaks based on peaks present in the majority of replicates for that sample. A union peak set for all samples was constructed by merging reproducible peaks for each sample into a set of high-confidence non-overlapping fixed width (500bp) peaks, which was used to create a peak by sample matrix used in downstream analysis. Differential peaks were determined using DESeq2 (Love et al., 2014, p. 2). Principal component analysis was performed on the peak matrix by first normalizing using `DESeq2::varianceStabilizingTransformation` and then `stats::prcomp`. Genome track files were created by loading the fragments for each sample into R, and exporting bigwig files normalized by reads in transcription start sites using `rtracklayer::export`. Coverage files were visualized using the Integrative Genomics Viewer. For analysis of previously published ATAC-seq data (Miller et al., 2019), fastq files were downloaded from accession GSE123236 and re-processed using our pipeline for consistency. Terminal and Progeintor TEX ATAC-seq peaks were computed using DESeq2 with cutoffs of Log2 FC ≥ 1 and FDR ≤ 0.05 when comparing Terminal versus Progenitor TEX samples (either TIL samples or LCMV samples, as indicated). For quantification of overlapping peaks between published data and in vitro assay data, a union peak set was created encompassing all samples and re-analyzed. For HOMER motif enrichment analysis shown in Figure S8G, the HOMER findMotifsGenome command line utility was used to identify motifs present in peaks in the indicated peak set relative to a background peak set. For the background peak set, we used the union peak set of the considered samples, and as a result the enriched motifs correspond to motifs enriched in the differential peak set relative to our samples in aggregate, rather than motifs enriched in human T cells relative to random genomic regions.
Bulk sgRNA screening data analysis
sgRNA sequencing data was analyzed using our previously published pipelines (Flynn et al., 2021). Briefly, fastq files were trimmed using `fastp -f 10 --max_len1=50`. Trimmed reads were aligned to a custom fasta file of the relevant pool (either the genome wide pool or the mini-pool) which was constructed by taking the sgRNA variable sequences and flanking them with the adjacent sequences in the pMSCV vector backbone. Alignment was performed using hisat2 with the --no-spliced-alignment option. Bam files were imported into R and converted into counts per guide using `Rsamtools::scanBam`. A table of guides per sample was constructed in R and normalized by multiplying each count by 1e6, dividing by the total counts in that sample, adding 1, and then log2 normalizing. Log fold changes between two conditions (chronic vs acute or tumor vs input) were computed and then z-scored by subtracting the reference LFC average and dividing by the reference LFC standard deviation. For genome-wide screens, all guides were used as the reference and for mini-pool screens the control guides were used as the reference. P-values were computed from z-scores using the normal distribution and then FDR was computed by correcting for multiple hypothesis testing using `p.adjust` in R. For the Gini index analysis shown in Figure S2, the ìneq` R package was used.
Comparison of CRISPR screen analytical methods
To validate our analysis strategy, we also analyzed the genome-wide screen results with two widely used methods, MAGeCK and casTLE, which showed a high correlation between effect size estimates (casTLE effect size correlation: R = 0.66; MAGeCK log fold change correlation: R = 0.77; Figure S3A–D, Table S2) (Li et al., 2014; Morgens et al., 2016). A comparison of the genes classified as hits using each method revealed that the largest group of hits were shared by all three methods (“hit” corresponds to FDR < 0.05 for our pipeline and MAGeCK or casTLE score > 10; Figure S3B). Finally, we sought to ensure that the identification of screen hits was robust to the choice of reference sgRNAs. We compared our normalization strategy (relative to all sgRNAs in the pool) to a strategy that utilizes a set of sgRNAs targeting olfactory receptors that are not expressed or predicted to function in T cells (Gilbert et al., 2014). We found that normalizing sgRNA enrichments to the olfactory receptor sgRNA set modestly boosted the statistical power of the screen results but otherwise had a minimal impact on the results (Figure S3E).
GO Term analysis
For gene categorizations shown in Figure 2B and elsewhere, gene sets were defined as: TCR - KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY, Chromatin - GOCC_CHROMATIN, Integrin - GOBP_INTEGRIN_ACTIVATION, Inhibitory receptor - GOBP_NEGATIVE_REGULATION_OF_LYMPHOCYTE_ACTIVATION. Gene lists were manually supplemented with the following genes: Chromatin - ZFP219, TBX21, KDM6A, ELMSAN1, DNTTIP1, SETD1B, TADA2B, ZFP217, EOMES. Integrins - ITGB3, APBB1IP, ITGAV. Inhibitory receptors - PDCD1. For the gene set enrichment analysis shown in Figure 2D and elsewhere, the indicated gene list was uploaded to the online gProfiler tool (available at https://biit.cs.ut.ee/gprofiler/gost).
Cytoscape interaction network
100 top enriched genes and 20 top depleted genes were imported into Cytoscape (Shannon et al., 2003). Edges were created by using the stringApp Cytoscape plugin to import known protein-protein interactions curated from string-db (Szklarczyk et al., 2019). A cutoff of stringdb score ≥ 0.75 was used to filter these protein protein interactions, which represents a conservative cutoff for identifying only high confidence interactions. Nodes were grouped based on GO Term analysis, subcellular localization, and/or manual curation. A small number of poorly characterized and/or disconnected nodes were removed from the visualization.
Direct-capture Perturb-seq analysis
Fastq files were processed using the 10X cellranger count pipeline with feature barcode analysis enabled to process the GEX library and sgRNA library together. The mm10 reference transcriptome was used for the GEX library. For the sgRNA library, a feature reference spreadsheet was constructed which contained the variable sequence of each guide (reverse complemented since it was sequenced as part of read 2), guide ID, and target gene. The filtered matrices for both `Gene Expression` and `CRISPR Guide Capturè were loaded into Seurat for downstream analysis (Hao et al., 2021). The Seurat ÌntegrateDatà utility was used to merge the samples from the two independent experiments.
To assign sgRNAs to cells, we computed row z-scores for the `CRISPR Guide Capturè matrix. We computed z-scores quantifying how enriched each sgRNA was relative to other sgRNAs detected in the same cell. We also computed the difference in z-scores between the most-enriched and second-most enriched sgRNA. Cells which had a maximum sgRNA z-score ≥ 5 and a z-score difference ≥ 2 was determined to contain the guide with maximum z-score, while cells with no sgRNA counts were assigned as “no guide,” and other cells were assigned “multi guide”. With this strategy, cells with multiple enriched sgRNAs due to retroviral infection doublets, single-cell capture doublets, and/or background reads were removed from further analysis. The guide assignments were added to the Seurat metadata for downstream processing. Seurat cell cycle scoring was used to predict the cell cycle phase of each single cell. All differential gene comparisons were performed using Seurat FindMarkers using the Wilcoxon test (the default statistical test). For volcano plot analysis, significantly differential genes were identified as FDR < 0.05. For comparisons of different gene sets across perturbations, an addition fold change cutoff was applied of average log2 FC > 0.1 or average log2 FC < −0.1. For categorization of shared ‘up’ and ‘down’ gene sets within the cBAF and INO80 complexes (analysis shown in Figure 7D–E), the union set of significantly differential genes within each complex was aggregated, and then ‘up’ and ‘down’ genes for each subunit were defined simply as LFC > 0 or LFC < 0. This strategy was chosen to compare gene sets despite the different amounts of cells collected for each perturbation and resulting difference in statistical power to reach the FDR < 0.05 threshold.
For comparisons to T cell signatures from acute and chronic LCMV infections (for example, in Figure 6E and Figure 7G), we analyzed a previously described scRNA-seq dataset (Daniel et al., 2021). Briefly, the TEFF, TMEM, and TEM clusters primarily contain T cells from the acute LCMV infection samples, and TEXeeff, TEXKLR, TEXISG, TEXTerm, TEXInt, and TEXProg clusters primarily contain cells from the chronic LCMV infection samples. Clonal analysis supports a differentiation trajectory wherein early effector TEX cells (TEXeeff) give rise to TEXProg (progenitor exhausted) and TEXInt (intermediate exhausted) subtypes. These cells differentiate into terminally exhausted cells (TEXTerm) or effector-like KLR-expressing exhausted cells (TEXKLR). Seurat gene module scoring was used to convert the LCMV gene sets (consisting of the top 100 marker genes per LCMV cluster) into a gene module score for each cell in the Perturb-seq dataset. Gene module scoring was also used to convert the upregulated and downregulated gene sets into module scores for each cell in the expanded LCMV data set, as shown in Figure S8.
Supplementary Material
Table S1. Genome-wide screen data, related to Figure 2. Results from the genome-wide screen, including sgRNA counts for both replicates and gene-level results merged across sgRNAs and replicates. Outputs are included for our pipeline, MAGeCK, and casTLE.
Table S2. sgRNA pool designs, related to Figures 2–4, 6, 7. sgRNA sequences for custom CRISPR mini-pools, including the top hit mini-pool, SWI/SNF mini-pool, and perturb-seq micro-pool.
Table S3.Top hit mini-pool screen data, related to Figures 2 and 3. Results from the top hit mini-pool screens, including sgRNA counts for all samples in the in vitro and in vivo mini-pool screens and z-score and FDR values for each gene, merged across sgRNAs and replicates.
Table S4. SWI/SNF mini-pool screen data, related to Figure 4. Results from the in vivo SWI/SNF mini-pool screens, including sgRNA counts for all samples and z-score and FDR values for each gene in, merged across sgRNAs and replicates.
Table S5. Human mini-pool sgRNA designs and results, related to Figure 5. sgRNA sequences and counts for all samples in the in vivo human mini-pool screen.
Table S6. In vivo Perturb-seq differential genes, related to Figure 7. Differential genes for each perturbation versus CTRL1 cells in the in vivo Perturb-seq dataset. Genes are included if they are significant in at least one comparison.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF Purified anti-mouse CD3ε Antibody (clone: 145–2C11) | Biolegend | Cat#100340; RRID:AB_11149115 |
| Ultra-LEAF(TM) Purified anti-mouse CD28 Antibody (clone: 37.51) | Biolegend | Cat#102116; RRID:AB_11147170 |
| PerCP Cy5.5 anti-mouse CD8 Antibody (clone: 53–6.7) | Biolegend | Cat#100734; RRID:AB_2075239 |
| PE/Cyanine7 anti-mouse CD279 (PD-1) antibody (clone: RMP1–30) | Biolegend | Cat# 109110, RRID:AB_572017 |
| PE anti-mouse CD366 (Tim-3) antibody (clone: RMT3–23) | Biolegend | Cat# 119704, RRID:AB_345378 |
| PE/Cyanine7 anti-mouse CD3 antibody (clone: 17A2) | Biolegend | Cat# 100220, RRID:AB_1732057 |
| APC/Cyanine7 anti-mouse CD45 antibody (clone: 30-F11) | Biolegend | Cat# 103116, RRID:AB_312981 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse IL-2 Protein | R&D system | Cat# 402-ML-020 |
| RPMI 1640 Medium | GIBCO | Cat#11995073 |
| Penicillin-Streptomycin (10,000 U/mL) | GIBCO | Cat#15140122 |
| 2-Mercaptoethanol | Sigma Aldrich | Cat#M6250–10ML |
| LIVE/DEAD™ Fixable Green Dead Cell Stain Kit, for 488 nm excitation | Invitrogen | Cat#L23101 |
| Critical commercial assays | ||
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi | Cat# 130–104-075 |
| Mitenyi tumor Dissociation Kit | Miltenyi | Cat# 130–096-730 |
| EasySep™ Mouse CD8a Positive Selection Kit II | Stemcell | Cat# 18953 |
| Deposited data | ||
| Mouse and human ATAC-seq | This paper | GEO: GSE203591 |
| In vivo Perturb-seq | This paper | GEO: GSE203592 |
| Experimental models: Cell lines | ||
| B16-F10 | ATCC | Cat# CRL-6475 |
| A-375 | ATCC | Cat# CRL-1619 |
| MC-38 | Stanford Tumor Core | N/A |
| Experimental models: Organisms/strains | ||
| Rosa26-Cas9 knockin mice | Jackson Labs | Cat# 026179 |
| OT-1 mice | Jackson Labs | Cat# 003831 |
| Rag1−/− mice | Jackson Labs | Cat# 002216 |
| C57BL/6J mice | Jackson Labs | Cat# 000664 |
| Recombinant DNA | ||
| pMSCV-U6sgRNA(BbsI)-PGKpuro2ABFP | Addgene | Cat# 102796 |
| Teichmann Retroviral Mouse Genome-wide CRISPR Knockout Library | Addgene | Cat# 104861 |
| Custom sgRNA mini-pools detailed in Table S2. | N/A | N/A |
| Software and algorithms | ||
| Seurat | ||
| HISAT2 | (Kim et al., 2019) | http://daehwankimlab.github.io/hisat2/ |
| cellranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count |
| Seurat | (Hao et al., 2021) | https://satijalab.org/seurat/index.html |
Highlights.
In vitro T cell exhaustion assay enables genome wide CRISPR-Cas9 screening
In vitro and in vivo genetic screens converge on cBAF and INO80 complex subunits
Arid1a-sgRNA T cells improve tumor control and enhance persistence of human T cells
Arid1a is required for acquisition of the epigenetic state of terminal exhaustion
Acknowledgements
We thank SciStories LLC for illustrations. This work was supported by the National Institutes of Health grant U01CA260852 (A.T.S.), the Burroughs Wellcome Fund Career Award for Medical Scientists (A.T.S. and S.A.V.), the Parker Institute for Cancer Immunotherapy (A.T.S.), a Pew-Stewart Scholars for Cancer Research Award (A.T.S.), a Cancer Research Institute Technology Impact Award (A.T.S.), a Baxter Foundation Faculty Scholar Award, and the Stanford Innovative Medicine Accelerator and Stanford ChEM-H (A.T.S). A.M. has received the Burroughs Wellcome Fund Career Award for Medical Scientists and The Cancer Research Institute (CRI) Lloyd J. Old STAR award. The Marson lab has received support from the Parker Institute for Cancer Immunotherapy (PICI), the Innovative Genomics Institute (IGI), and the Chan Zuckerberg Biohub. J.A.B was supported by a Stanford Graduate Fellowship and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518. S.A.V. was supported by a Special Fellow Award from the Parker Institute for Cancer Immunotherapy and a Mentored Clinical Scientist Career Development Award from NCI/NIH (K08 CA237731).
Footnotes
Declaration of Interests
A.T.S. is a scientific founder of Immunai and founder of Cartography Biosciences and receives research funding from Arsenal Biosciences, Allogene Therapeutics, and Merck Research Laboratories. J.A.B. is a consultant to Immunai. S.A.V. is an advisor to Immunai. K.E.Y. is a consultant to Cartography Biosciences. C.L.M. is a co-founder of Lyell Immunopharma and Syncopation Life Sciences, and consults for Lyell, Syncopation, NeoImmune Tech, Apricity, Nektar, Immatics, Mammoth, and Ensoma. A.A. is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation, and Kytarro; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, GenVivo, Ambagon, Phoenix Molecular Designs and GSK; a member of the SAB of Genentech, GLAdiator, Circle and Cambridge Science Corporation; receives research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca. A.M. is a co-founder of Spotlight Therapeutics, Arsenal Biosciences, and Survey Genomics. A.M. is a member of the scientific advisory board of NewLimit. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma, and Merck. A.M. has received fees from 23andMe, PACT Pharma, Juno Therapeutics, Trizell, Vertex, Merck, Amgen, Genentech, AlphaSights, Rupert Case Management, Bernstein, and ALDA. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem. K.A.F., E.S., J.C., A.A., A.M., and C.L.M. hold patents in the arena of CAR T cell therapeutics. J.A.B. and A.T.S. have filed a patent related to the contents of this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS, 2016. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. 10.1016/j.cell.2016.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D, Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, Zehn D, 2019. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269. 10.1038/s41586-019-1326-9 [DOI] [PubMed] [Google Scholar]
- Ataide MA, Komander K, Knöpper K, Peters AE, Wu H, Eickhoff S, Gogishvili T, Weber J, Grafen A, Kallies A, Garbi N, Einsele H, Hudecek M, Gasteiger G, Hölzel M, Vaeth M, Kastenmüller W, 2020. BATF3 programs CD8+ T cell memory. Nat Immunol 21, 1397–1407. 10.1038/s41590-020-0786-2 [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R, 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. 10.1038/nature04444 [DOI] [PubMed] [Google Scholar]
- Belk JA, Daniel B, Satpathy AT, 2022. Epigenetic regulation of T cell exhaustion. Nat Immunol 1–13. 10.1038/s41590-022-01224-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra J-C, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, Yan P, Nzingha K, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Huang AC, Wherry EJ, 2020. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841.e8. 10.1016/j.immuni.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, López-Moyado IF, Seo H, Lio C-WJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, Rao A, 2019. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. 10.1038/s41586-019-0985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J, 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, Huang H, Manne S, Cao Z, Baxter AE, Cai Z, Freilich E, Ali MA, Giles JR, Wu JE, Greenplate AR, Hakeem MA, Chen Q, Kurachi M, Nzingha K, Ekshyyan V, Mathew D, Wen Z, Speck NA, Battle A, Berger SL, Wherry EJ, Shi J, 2021. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280.e22. 10.1016/j.cell.2021.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier JL, Weiss SA, Pauken KE, Sen DR, Sharpe AH, 2021. Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat Immunol 22, 809–819. 10.1038/s41590-021-00949-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ, Chang HY, 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. 10.1038/nmeth.4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel B, Yost KE, Sandor K, Xia Y, Qi Y, Hiam-Galvez KJ, Meier SL, Belk JA, Giles JR, Wherry EJ, Chang HY, Egawa T, Satpathy AT, 2021. Divergent clonal differentiation trajectories of T cell exhaustion. 10.1101/2021.12.16.472900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePeaux K, Delgoffe GM, 2021. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol. 10.1038/s41577-021-00541-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, Regev A, 2016. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, Park JJ, Kim HR, Errami Y, Guzman CD, Zhou X, Chen KY, Renauer PA, Du Y, Shen J, Lam SZ, Zhou JJ, Lannin DR, Herbst RS, Chen S, 2019. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204.e23. 10.1016/j.cell.2019.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Belk JA, Qi Y, Yasumoto Y, Wei J, Alfajaro MM, Shi Q, Mumbach MR, Limaye A, DeWeirdt PC, Schmitz CO, Parker KR, Woo E, Chang HY, Horvath TL, Carette JE, Bertozzi CR, Wilen CB, Satpathy AT, 2021. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 184, 2394–2411.e16. 10.1016/j.cell.2021.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang W-T, Pequignot E, Ambrose DE, Zhang C, Wilcox N, Bedoya F, Dorfmeier C, Chen F, Tian L, Parakandi H, Gupta M, Young RM, Johnson FB, Kulikovskaya I, Liu L, Xu J, Kassim SH, Davis MM, Levine BL, Frey NV, Siegel DL, Huang AC, Wherry EJ, Bitter H, Brogdon JL, Porter DL, June CH, Melenhorst JJ, 2018a. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571. 10.1038/s41591-018-0010-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, Hwang Y, Gohil M, Kulikovskaya I, Nazimuddin F, Gupta M, Chen F, Everett JK, Alexander KA, Lin-Shiao E, Gee MH, Liu X, Young RM, Ambrose D, Wang Y, Xu J, Jordan MS, Marcucci KT, Levine BL, Garcia KC, Zhao Y, Kalos M, Porter DL, Kohli RM, Lacey SF, Berger SL, Bushman FD, June CH, Melenhorst JJ, 2018b. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312. 10.1038/s41586-018-0178-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, 2014. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, 2021. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR, 2011. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21, 396–420. 10.1038/cr.2011.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J, Chen X, Gomes T, Ullah U, Meyer KB, Miragaia R, Duddy G, Pramanik J, Yusa K, Lahesmaa R, Teichmann SA, 2019. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell 176, 882–896.e18. 10.1016/j.cell.2018.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhou P, Wei J, Long L, Shi H, Dhungana Y, Chapman NM, Fu G, Saravia J, Raynor JL, Liu S, Palacios G, Wang Y-D, Qian C, Yu J, Chi H, 2021. In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8+ T cell fate decisions. Cell 184, 1245–1261.e21. 10.1016/j.cell.2021.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, 2019. TOX transcriptionally and epigenetically programs CD8 + T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL, 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, Brown FD, Gillis JE, Coxe DJ, Doench JG, Haining WN, Sharpe AH, 2019. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nature Communications 10, 1668. 10.1038/s41467-019-09656-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS, 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biology 15, 554. 10.1186/s13059-014-0554-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 21, 581–590. 10.1038/nm.3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, Nagaraja S, Granja J, de Bourcy CFA, Majzner R, Satpathy AT, Quake SR, Monje M, Chang HY, Mackall CL, 2019. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. 10.1038/s41586-019-1805-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel Iii EE, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Şenbabaoğlu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Höglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, Powles T, 2018. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. 10.1038/nature25501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St. Pierre R, Valencia AM, Poynter SJ, Cassel SH, Ranish JA, Kadoch C, 2018. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20. 10.1016/j.cell.2018.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, Roberts CWM, 2017. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet 49, 296–302. 10.1038/ng.3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC, 2015. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616. 10.1038/nature14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, Wherry EJ, 2019. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annual Review of Immunology 37, 457–495. 10.1146/annurev-immunol-041015-055318 [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, 2019. Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nature Immunology 20, 326–336. 10.1038/s41590-019-0312-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgens DW, Deans RM, Li A, Bassik MC, 2016. Systematic comparison of CRISPR-Cas9 and RNAi screens for essential genes. Nat Biotechnol 34, 634–636. 10.1038/nbt.3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ, 2015. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med 212, 1125–1137. 10.1084/jem.20142237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ, 2012. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 338, 1220–1225. 10.1126/science.1229620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, Shalem O, Satija R, Raychowdhury R, Mertins P, Carr SA, Zhang F, Hacohen N, Regev A, 2015. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686. 10.1016/j.cell.2015.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ, 2016. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. 10.1126/science.aaf2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A, 2017. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. 10.1038/nature22367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F, 2014. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 159, 440–455. 10.1016/j.cell.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritykin Y, van der Veeken J, Pine AR, Zhong Y, Sahin M, Mazutis L, Pe’er D, Rudensky AY, Leslie CS, 2021. A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol Cell 81, 2477–2493.e10. 10.1016/j.molcel.2021.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju S, Xia Y, Daniel B, Yost KE, Bradshaw E, Tonc E, Verbaro DJ, Kometani K, Yokoyama WM, Kurosaki T, Satpathy AT, Egawa T, 2021. Identification of a T-bethi Quiescent Exhausted CD8 T Cell Subpopulation That Can Differentiate into TIM3+CX3CR1+ Effectors and Memory-like Cells. The Journal of Immunology. 10.4049/jimmunol.2001348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ, Meer EJ, Terry JM, Riordan DP, Srinivas N, Fiddes IT, Arthur JG, Alvarado LJ, Pfeiffer KA, Mikkelsen TS, Weissman JS, Adamson B, 2020. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat Biotechnol 38, 954–961. 10.1038/s41587-020-0470-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Wolchok JD, 2018. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, Yu R, Nguyen MLT, Lee Y, Truong A, Hiatt J, Wu D, Nguyen DN, Goodman D, Bluestone JA, Ye CJ, Roybal K, Shifrut E, Marson A, 2020. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 181, 728–744.e21. 10.1016/j.cell.2020.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, 2010. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207, 2187–2194. 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, Shah P, Bell JC, Jhutty D, Nemec CM, Wang J, Wang L, Yin Y, Giresi PG, Chang ALS, Zheng GXY, Greenleaf WJ, Chang HY, 2019. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936. 10.1038/s41587-019-0206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, Greenleaf WJ, 2017. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. 10.1038/nmeth.4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, Schell TD, Garbi N, Greenberg PD, 2016. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 45, 389–401. 10.1016/j.immuni.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, Schietinger A, 2019. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, Pereira RM, 2016. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 45, 1327–1340. 10.1016/j.immuni.2016.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao H-W, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, Haining WN, 2016. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. 10.1126/science.aae0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C-WJ, Rao A, Hogan PG, 2021. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol 1–13. 10.1038/s41590-021-00964-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, Marson A, 2018. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e15. 10.1016/j.cell.2018.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, Xia J, Kwon JYH, Nevin J, Herbst RH, Yanai I, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC, 2016. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511.e9. 10.1016/j.cell.2016.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, Fong L, Nolan GP, Engleman EG, 2017. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502.e15. 10.1016/j.cell.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering C von, 2019. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613. 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, van den Brink MRM, Cross JR, Thompson CB, 2020. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nature Immunology 21, 1022–1033. 10.1038/s41590-020-0725-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME, 2017. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082.e12. 10.1016/j.molcel.2017.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES, 2014. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 343, 80–84. 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, Malipatlolla M, Xu P, Bashti M, Heitzeneder S, Labanieh L, Vandris P, Majzner RG, Qi Y, Sandor K, Chen L-C, Prabhu S, Gentles AJ, Wandless TJ, Satpathy AT, Chang HY, Mackall CL, 2021. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372. 10.1126/science.aba1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang Y-D, Qian C, Xu B, Kc A, Saravia J, Huang H, Yu J, Doench JG, Geiger TL, Chi H, 2019. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476. 10.1038/s41586-019-1821-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M, 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. 10.1038/nri3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, Ferrando L, Selenica P, Ladewig E, Chan C, Da Cruz Paula A, Witkin M, Cheng Y, Park J, SernaTamayo C, Zhao H, Wu F, Sallaku M, Qu X, Zhao A, Collings CK, D’Avino AR, Jhaveri K, Koche R, Levine RL, Reis-Filho JS, Kadoch C, Scaltriti M, Leslie CS, Baselga J, Toska E, 2020. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet 52, 198–207. 10.1038/s41588-019-0554-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Chang HY, Satpathy AT, 2021. Recruiting T cells in cancer immunotherapy. Science 372, 130–131. 10.1126/science.abd1329 [DOI] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, Gupta RK, Curtis C, Bucktrout SL, Davis MM, Chang ALS, Chang HY, 2019. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259. 10.1038/s41591-019-0522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R, 1998. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213. 10.1084/jem.188.12.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome-wide screen data, related to Figure 2. Results from the genome-wide screen, including sgRNA counts for both replicates and gene-level results merged across sgRNAs and replicates. Outputs are included for our pipeline, MAGeCK, and casTLE.
Table S2. sgRNA pool designs, related to Figures 2–4, 6, 7. sgRNA sequences for custom CRISPR mini-pools, including the top hit mini-pool, SWI/SNF mini-pool, and perturb-seq micro-pool.
Table S3.Top hit mini-pool screen data, related to Figures 2 and 3. Results from the top hit mini-pool screens, including sgRNA counts for all samples in the in vitro and in vivo mini-pool screens and z-score and FDR values for each gene, merged across sgRNAs and replicates.
Table S4. SWI/SNF mini-pool screen data, related to Figure 4. Results from the in vivo SWI/SNF mini-pool screens, including sgRNA counts for all samples and z-score and FDR values for each gene in, merged across sgRNAs and replicates.
Table S5. Human mini-pool sgRNA designs and results, related to Figure 5. sgRNA sequences and counts for all samples in the in vivo human mini-pool screen.
Table S6. In vivo Perturb-seq differential genes, related to Figure 7. Differential genes for each perturbation versus CTRL1 cells in the in vivo Perturb-seq dataset. Genes are included if they are significant in at least one comparison.
Data Availability Statement
CRISPR screen counts tables and z-score tables are available with the manuscript as supplemental data. ATAC-seq and Perturb-seq data are available on GEO under accession GSE203593. Scripts used to analyze CRISPR screen data have been previously open-sourced and are available at: https://github.com/juliabelk/sarscov2_chirp_ms