

Abstract
Hepatocellular carcinoma (HCC), the fourth leading cause of cancer mortality, develops almost exclusively in patients with chronic liver disease (CLD) and advanced fibrosis1,2. Here we interrogated functions of hepatic stellate cells (HSC), the main source of liver fibroblasts3, during hepatocarcinogenesis. Genetic depletion, activation or inhibition established HSC as tumour-promoting in mouse models of HCC. HSC were enriched in the preneoplastic environment, where they closely interacted with hepatocytes and modulated hepatocarcinogenesis by regulating hepatocyte proliferation and death. Analysis of mouse and human HSC subpopulations and their associated mediators by single cell RNA-sequencing in conjunction with genetic ablation revealed dual functions of HSC in hepatocarcinogenesis. Hepatocyte growth factor, enriched in quiescent and cytokine-producing HSC (cyHSC), protected from hepatocyte death and HCC development. In contrast, type I collagen, enriched in activated myofibroblastic HSC (myHSC), promoted proliferation and tumour development via increased stiffness and TAZ activation in pretumoural hepatocytes and via activation of discoidin domain receptor 1 in established tumours. An increasing HSC dysbalance between cyHSC and myHSC during liver disease progression was associated with elevated HCC risk in patients. In summary, the dynamic shift of HSC subpopulations and their mediators during CLD is associated with a switch from HCC protection to HCC promotion.
INTRODUCTION
Hepatocellular carcinoma (HCC) causes ≈800,000 deaths annually 1. In the US and other western countries, incidence has tripled in the past 30 years4 and is expected to further increase due to increasing rates of non-alcoholic fatty liver disease (NAFLD). Accordingly, while mortality has been decreasing for most solid cancers in the US, HCC mortality has increased1,5. Most HCCs are detected too late for curative approaches such as resection or liver transplantation. Despite an increased portfolio of FDA-approved therapies for advanced HCC6, the majority of patients still die within one year after diagnosis, making this an important unmet medical need. Furthermore, HCC represents the most common cause of death in patients with compensated liver cirrhosis and the lack of effective prevention strategies renders this precancerous condition a threat for millions of patients.
Chronic hepatocyte death, and the ensuing inflammation and fibrosis are sufficient to trigger HCC7, indicating that HCC is truly a “wound that never heals”. This is reflected by the clinical finding that >90% of HCCs develop in the presence of fibrosis or cirrhosis, including those associated with non-alcoholic steatohepatitis (NASH) despite the absence of overt cirrhosis in 30–50% of NASH-associated HCC2,8. Different from desmoplastic tumours such as pancreatic cancer or cholangiocarcinoma, where fibrosis in the tumour microenvironment (TME) follows tumour development, fibrosis precedes HCC development with tumors arising in a fibrotic premalignant environment (PME)2. Despite the strong association between cirrhosis and HCC and its high medical relevance, the causal relationship between fibrosis and HCC development remains poorly understood and therapeutically unexplored2. Virtually all in vivo evidence on the role of hepatic stellate cells (HSC), the primary fibrogenic cell of the liver3, in hepatocarcinogenesis to date is based on studies in GFAP-Cre transgenic mice9–11 and remains controversial, (i) as these mice do not efficiently delete genes in HSC3 and may also delete in the hepatic epithelial compartment3 including liver tumours9, and (ii) as some studies using GFAP-Cre mice reported reduced fibrosis being associated with reduced HCC9,11,12, while others found the same amount of HCC10. Mechanisms and mediators through which HSC affect HCC remain inconclusive, as it has been both suggested that HSC senescence restricts9 or promotes hepatocarcinogenesis12. In sum, the current gap of knowledge prevents rational development of novel therapeutic concepts for HCC prevention or treatment in patients with liver fibrosis and cirrhosis.
Here, we combined single cell RNA-sequencing (scRNAseq) analysis with genetic tools to activate, deplete, inhibit and manipulate HSC and their mediators during HCC development. Our preclinical and patient data suggest that there is an increased imbalance of HSC subpopulations during disease progression. Quiescent HSCs, which express protective factors, shift towards activated HSCs, which express tumour-promoting mediators, thereby setting the stage for HCC development in CLD.
RESULTS
Activated HSC in the fibrotic liver promote HCC development
In view of the poor HSC targeting by GFAP-Cre3 in previous studies on HSC in hepatocarcinogenesis9–12 and controversies on fibrosis10 and HSC senescence in hepatocarcinogenesis9,12, we revisited their role in hepatocarcinogenesis using LratCre-transgenic mice, a highly efficient HSC deleter strain3. For this, we first studied p53 and NF-κB/RelA, two key mediators of HSC senescence and the senescence-associated secretory phenotype (SASP)9,13,14. LratCre mice, crossed to Trp53fl/fl floxed (p53ΔHSC) or NF-κB subunit Relafl/fl (RelaΔHSC) mice, showed 93–99% reduction of Trp53 and Rela mRNA in HSC, but, surprisingly, there were no alterations in liver fibrosis, HSC activation or the development of DEN+CCl4-induced HCC, a well-established model of fibrosis-associated hepatocarcinogenesis, (Extended Data Fig. 1a–h). Together, these findings suggest that either p53 and NF-κB in HSC do not contribute to fibrosis-associated hepatocarcinogenesis, or that HSC are not functionally connected to hepatocarcinogenesis. As we, like others15, observed that cellular senescence occurred mostly in cell types other than HSC (Extended Data Fig. 1i–m), we shifted to interrogate more globally the role of HSC in hepatocarcinogenesis, independently of senescence. For this purpose, we developed genetic approaches, whereby we activated, inhibited or depleted HSC during HCC development, thus addressing potentially confounding limitations of these methods such as possible changes in immune cell infiltration caused by HSC killing in genetic depletion. HSC were genetically activated by the deletion of Lim Homeobox Domain 2 (Lhx2)16, a known repressor of HSC activation, by LratCre (Lhx2ΔHSC). Lhx2ΔHSC mice exhibited increased Col1a1 mRNA and hepatic collagen deposition (Fig. 1a, Extended Data Fig. 2a–c) and exacerbated tumourigenesis in DEN-induced HCC, as determined by the liver/body weight ratio, tumour number and size (Fig. 1b). Because of the moderate deletion efficiency in Lhx2ΔHSC mice (likely caused by escape for yet unknown reasons), we confirmed these findings by Mx1Cre-mediated Lhx2 deletion. Like Lhx2ΔHSC mice, Mx1Cre-mediated Lhx2 deletion increased HSC activation, fibrosis and DEN-induced HCC (Extended Data Fig. 2d–f). Complementing data from this HSC activation model, we observed that genetic inhibition of HSC activation and liver fibrosis by LratCre-mediated Yap1 deletion (YapΔHSC) (Fig. 1c, Extended Data Fig. 2g–h) decreased tumour burden in DEN+CCl4-induced HCC (Fig. 1d). Likewise, depletion of HSC in triple transgenic mice expressing LratCre, TdTomato (TdTom) and Cre-induced diphtheria toxin receptor (DTRpos) efficiently reduced TdTom+ HSC and HSC marker genes (Fig. 1e, Extended Data Fig. 2i), and strongly suppressed DEN+CCl4-induced HCC development in comparison to DT-treated LratCrepos, TdTompos iDTRneg littermates (DTRneg), as shown by decreased liver-body weight ratio, tumour number and tumour size (Fig. 1f). As an additional LratCre-independent approach, we employed αSMA-TK mice for the depletion of activated αSMA+ HSC, which also reduced HCC development (Extended Data Fig. 2j–k). These findings were confirmed in a spontaneous HCC model, induced by Mdr2 deficiency (Mdr2KO), in which LratCre-mediated deletion of Pdgfrb (PdgfrbΔHSC) inhibited HSC activation, liver fibrosis and tumourigenesis (Extended Data Fig. 2l–m). Lastly, HSC or fibroblast depletion via LratCre-induced DTR and αSMA-TK, respectively, significantly inhibited the development of NASH-HCC, the projected main cause for advanced liver disease and HCC in the near future17, in models induced either by (i) the combination of sleeping beauty-mediated overexpression of activated TAZ (S89A) and FPC NASH diet18, (ii) DEN and high fat choline-deficient amino acid-supplemented (HF-CDAA) diet, or (iii) NICD overexpression and HF-CDAA diet (Extended Data Fig. 2n–u). Of note, HSC depletion did not reduce liver cholesterol and triglyceride content (Extended Data Fig. 2q). Collectively, our data based on complementary genetic HSC activation, inhibition or depletion approaches in different models of spontaneous, carcinogen-, oncogene- and NASH-induced HCC demonstrate that HSC promote hepatocarcinogenesis.
Figure 1 |. Promotion of HCC development by hepatic stellate cells.

a-b, Induction of liver fibrosis, shown by Sirius Red staining, and Col1a1 by HSC-selective Lhx2 deletion via LratCre (Lhx2ΔHSC) compared to Lhx2f/f littermates (n=7–8 mice/group), (a) and increased DEN-induced HCC, shown by representative liver pictures, the liver-body ratio (LBR), tumour number and size (b). c-d, Decreased DEN+CCl4-induced HSC activation in YapΔHSC compared to Yapfl/fl mice (n=15–16/group), shown by αSMA IHC and Col1a1 qPCR (c), as well as reduced HCC burden (d). e-f, Efficient HSC depletion in non-tumour tissue in the DEN+CCl4 model in DTRpos compared DTRneg LratCrepos, TdTompos littermates (n=16–17/group), determined by TdTom fluorescence and Col1a1 qPCR (e) as well as reduced tumour development (f). Data are shown as mean ± SEM, each data point represents one individual, Tu: Tumour, HTVI: hydrodynamic tail vein injection, all scale bars: 200 μm. Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
HSC accumulate and regulate hepatocyte survival and proliferation in the PME
The majority of Col1a1-GFP+ myofibroblasts were derived from LratCre-labeled HSC in multiple HCC models (Fig. 2a, Extended Data Fig. 3a), both in tumour and non-tumour areas, suggesting that HSC could affect hepatocarcinogenesis in both compartments. We therefore compared fibroblast accumulation and fibrosis between tumours and surrounding fibrotic tissue. In contrast to intrahepatic cholangiocarcinoma (ICC), a desmoplastic liver malignancy, tumours from the DEN+CCl4, TAZ+FPC, Mdr2KO and HF-CDAA HCC models contained significantly less activated myofibroblasts and fibrosis than the surrounding liver (Fig. 2a, Extended Data Fig. 3b–c). Similar to our findings in mice, HCC lesions in a group of 45 patients with underlying fibrosis had overall significantly less fibrosis and αSMA+ myofibroblasts than tumour adjacent tissue or ICC tumours, which showed a pattern opposite to HCC (Fig. 2b). In the subset of patients without underlying fibrosis, there were, expectedly, more fibrosis and αSMA+ myofibroblasts in tumours than the adjacent non-fibrotic tissue (Extended Data Fig. 3d). Comparison of snRNA-seq data from non-tumour and tumour areas of patients with HCC also demonstrated more fibroblasts in adjacent non-tumour than in tumour tissue (Extended Data Fig. 3e and Supplementary Information.1a–b). In conjunction with previous studies showing a key role for an adjacent liver tissue signature and HSC signatures within non-tumour tissue in predicting HCC survival19,20, these data points towards a potential role of HSC within the PME in modulating HCC development. Accordingly, RNA-sequencing of tissue from the PME and TME of DEN+CCl4-treated Yap ΔHSC and Yapfl/fl mice, in which we had observed profound differences in HCC development, revealed only minimal differences between Yapfl/fl and YapΔHSC mice in the tumour tissue (reflecting the TME of established tumours), whereas there were substantial differences in the adjacent non-tumour tissue (reflecting the PME containing premalignant or early-stage lesions) (Extended Data Fig. 3f–g). In addition to ECM-related genes (Extended Data Fig. 3h), the most notable differences in gene set enrichment analysis (GSEA) were in proliferation, apoptosis and inflammation, which were downregulated in YapΔHSC mice (Extended Data Fig. 3i–j). Based on these data and the above-described accumulation of activated HSC in the PME and not the TME, we hypothesized that HSC may modulate HCC development by influencing hepatocyte responses in the injured liver. Of note, hepatocytes constitute the cellular source of HCC in most settings21, and hepatocyte proliferation and death as well as inflammation and immunity exert profound impact on hepatocarcinogenesis1,7. Consistent with our GSEA data and a previously published study22, ALT and TUNEL+ hepatocytes were reduced in the non-tumour tissue of YapΔHSC vs Yapfl/fl mice as well as after αSMA+ cell depletion (Extended Data Fig. 3k). Ki67+ hepatocytes were reduced after HSC or αSMA+ cell depletion but unchanged in YapΔHSC mice (Fig. 2c, Extended Data Fig. 3l). In contrast, we did not find changes in cell proliferation within tumors in YapΔHSC and HSC-depleted mice (Extended Data Fig. 3m–n). Consistent with these finding, predominant HSC/fibroblast depletion in established tumours via GCV injection into αSMA-TK mice after cessation of CCl4 treatment (Extended Data Fig. 4a) did not affect HCC development (Extended Data Fig. 4b). Furthermore, we did not observe any changes in tumour development in the non-fibrotic MET+CTNNB1-driven model of HCC when depleting HSC (Extended Data Fig. 4c–d). Together, these findings support our hypothesis that the predominant role of HSC-derived myofibroblasts, consistent with their anatomic location, lies outside established tumours. scRNA-seq-based ligand-receptor analysis revealed HSC as a hub of cell-cell communication in the liver, as previously shown by us and others 23–25. Our analysis of ligand-receptor interactions, focused on hepatocytes, the cellular source of HCC21, revealed that HSC were the cell population interacting most intensely with hepatocytes in comparison to all other hepatocyte-interacting cell populations (Fig. 2d). Similar findings were made when analysing hepatocyte interactions in patients (Extended Data Fig. 4e). Further analysis of hepatocyte subclusters revealed stronger interactions with proliferating hepatocytes as well as metabolically active and TGFβ-responsive hepatocytes (Extended Data Fig. 4f and Supplementary Table 1a–b). Genetic HSC depletion or inhibition did not lead to major changes in angiogenesis and immune cell infiltration, assessed by endomucin staining and qPCR for Pecam/Cd31 and Kdr transcripts, flow cytometric analysis of lymphocyte and myeloid populations and CD45 staining (Extended Data Fig. 4h–l and Supplementary Figure 2a–b). The only exception was an expected increase in myeloid subpopulations in non-tumour areas, consistent with their known role in clearing apoptotic fibroblasts, which typically increase during αSMA-TK depletion. Furthermore, we did not observe changes in tumour mutational profiles with similar rates of Egfr, Hras and Braf hotspot mutations in models with genetic HSC activation, inhibition and depletion as well as tumour grading and no differences in expression of immune, inflammatory and tumour genes (Extended Data Fig. 5a–e). Together, our data suggest that HSC predominantly promote tumour growth by modulating the PME and early stages of hepatocarcinogenesis rather than affecting established tumours or the immune landscape.
Figure 2 |. Hepatic stellate cell accumulation and activation occurs predominantly in the non-tumour environment.

a, HSC and collagen-expressing fibroblasts were visualized in non-tumour (NT) and tumour (Tu) areas of DEN+CCl4 and TAZ+FPC diet HCC models or the KRAS/sgp19 ICC model by LratCre-induced TdTom and Col1a1-GFP, respectively (n=2–3/group); liver fibrosis was determined by Sirius Red staining (n=3/group). b, myofibroblasts were visualized by αSMA IHC and liver fibrosis by Sirius Red staining in paired non-tumour and tumour sections from human HCC (n=45, displayed are HCCs arising in fibrotic livers) and from human ICC (n=4–5). c, proliferation determined by Ki67 IHC in non-tumour tissue from Yapfl/fl and YapΔHSC littermates or DTRpos and DTRneg mice in the DEN+CCl4 HCC model. d, analysis of cell-cell interactions by CellPhoneDB in scRNA-seq data from 8xCCl4-injured liver (n=3) revealed HSC as a major cell type interacting with hepatocytes. Data are shown as mean ± SEM, each data point (a,b,c) represents one individual; scale bars: 150 μm (a,b), 100 μm (c). Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
The fibrotic liver contains distinct HSC subpopulations
Based on above data suggesting ligand-based HSC-hepatocyte interactions in the PME, we next sought to identify mediators through which activated HSC may affect hepatocytes and hepatocarcinogenesis. To uncover functionally relevant candidate genes and mechanisms of HSC subpopulations and their potential effects on hepatocyte behaviour and hepatocarcinogenesis, we determined differential gene expression and associated pathways between the most and the least activated HSC clusters from different fibrosis models (Supplementary Figure 3a–d, Supplementary Tables 2–3). Signatures based on these differentially expressed genes divided mouse and human HSC reliably into more quiescent and activated subpopulations (Fig. 3a–b, Extended Data Fig. 6a, Supplementary Figure 1c). Pathway analysis revealed the expected enrichment of ECM-related molecules and pathways in highly activated HSC (Supplementary Table 4a), which were therefore termed myofibroblastic HSC (myHSC). Interestingly, quiescent to weakly activated HSC were enriched in genes and pathways related to cytokines and growth factors and were therefore termed cytokine- and growth factor-expressing HSC (cyHSC) (Supplementary Table 4b). We observed a continuous decrease in HSC with a high cyHSC signature, paralleled by a concomitant increase of HSC with a high myHSC signature in mice with liver fibrosis and in cirrhotic patients, as well as an inverse correlation of both (Fig. 3c, Extended Data Fig 6b). Furthermore, the shift of cyHSC-myHSC balance towards myHSC in the Lhx2del genetic HSC activation model, which increased HCC, and towards cyHSC in the YapΔHSC genetic inhibition model, which decreased HCC, suggested a functional contribution of this balance to hepatocarcinogenesis (Extended Data Fig. 6c). Trajectory and pseudotime analysis in fibrotic models revealed that myHSC were derived from cyHSC (Extended Data Fig. 6d). However, given that HSC may revert to quiescence, we cannot exclude that myHSC can revert to cyHSC or shuttle back and forth, especially when disease progression is not linear but undulating. Consistent with our pan-HSC CellPhoneDB analysis, we found that hepatocytes strongly interacted with cyHSC and myHSC subpopulations (Fig. 3d), suggesting that both populations may affect hepatocyte biology and tumourigenesis. After establishing methods to identify these subpopulations, we next focused on identifying functionally relevant mediators in cyHSC and myHSC. Of note, there are currently no tools to selectively deplete, inhibit or therapeutically target specifically the cyHSC and myHSC subpopulations in mice or patients, whereas relevant mediators enriched in these subpopulations can be genetically ablated in HSC and possibly also therapeutically targeted. Differential gene expression of secreted mediators between cyHSC and myHSC revealed type I collagen (Col I), the most abundant ECM component in the fibrotic liver, as a highly expressed myHSC-enriched mediator and HGF as a highly expressed cyHSC-enriched mediator (Supplementary Table 5a–b). An enrichment of Hgf in cyHSC and of Col1a1 in myHSC was found in all mouse models as well as in patients (Fig. 3e–f, Extended Data Fig. 6d). As additional in situ validation, we therefore determined the localization of Hgf mRNA, Col1a1-GFP and LratCre-induced TdTom in the liver. We detected almost mutually exclusive populations of Hgfhigh Col1a1-GFPlow TdTompos HSC, representing cyHSC and Hgflow Col1a1-GFPhigh TdTompos HSC, representing myHSC in CCl4, Mdr2KO and TAZ-FPC models (Fig. 3e, Extended Data Fig. 6e). Furthermore, Col1a1-GFPhigh HSC were enriched in the fibrotic septa in the CCl4 model, whereas Hgfhigh TdTompos HSC were more abundant in the surrounding non-fibrotic liver parenchyma as well as in the healthy liver (Fig. 3g). Similar to the dynamic shifts of cyHSC and myHSC, we observed a progressive decrease of Hgf mRNA in HSC with increased disease, paralleled by a progressive increase of Col1a1 mRNA, in both bulk RNA-seq data of isolated HSC and scRNA-seq (Extended Data Fig. 6f–g). Furthermore, activating or depleting HSC at different time revealed strong effects of HSC on hepatocarcinogenesis in later stages (but still preceding or concomitant with the development of macroscopically visible tumours) but not early stages (Extended Data Fig. 7a–d). Together, our findings suggest that the increasing number of myHSC or an impaired balance between cyHSC and myHSC in progressive liver disease may favor hepatocarcinogenesis. Next, we sought to unravel the contribution of cyHSC- and myHSC-expressed mediators to hepatocarcinogenesis.
Figure 3 |. Single cell RNA-seq analysis reveals subsets of HSC with differential expression of HGF and COL1A1 in the fibrotic liver.
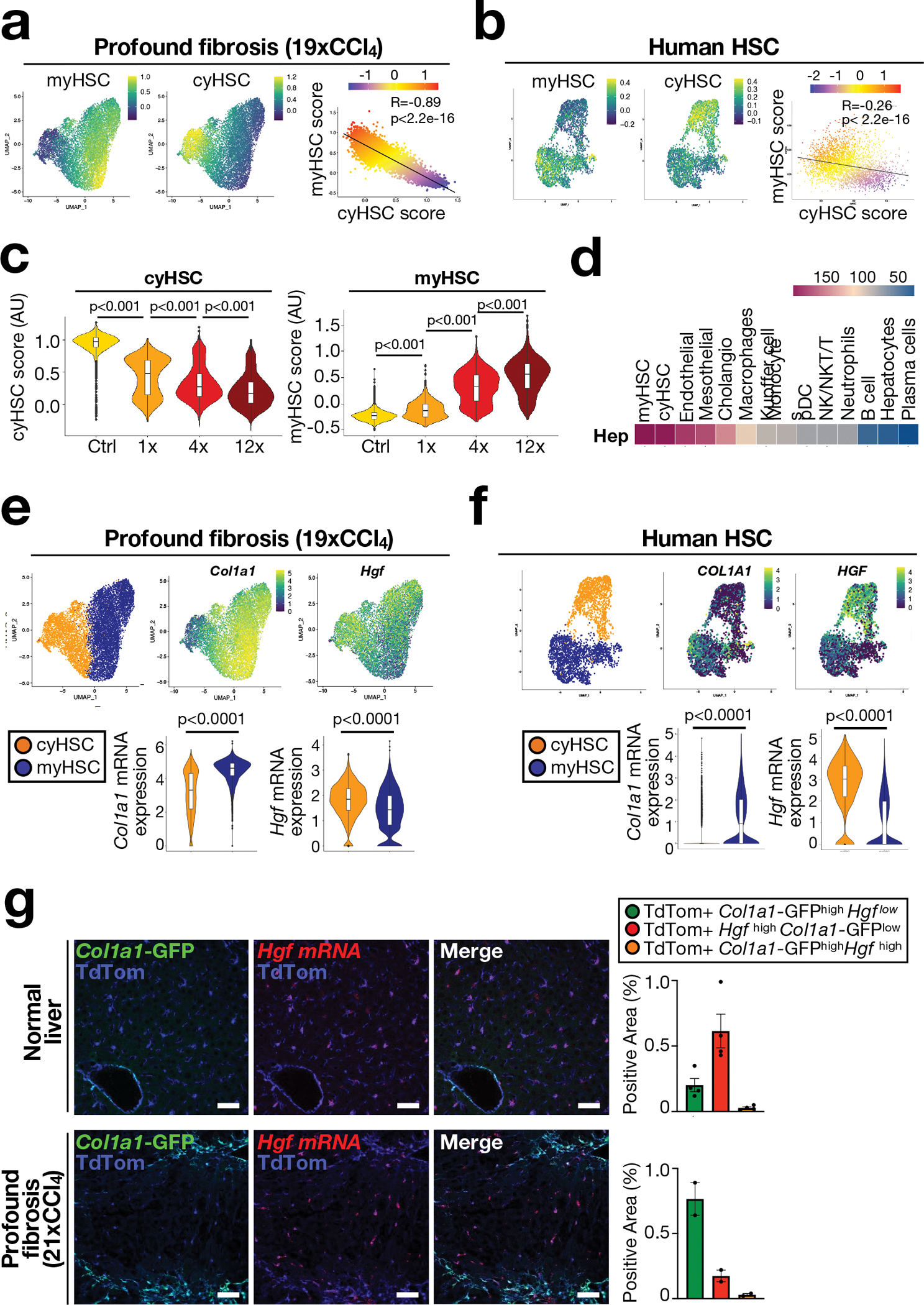
a-b, UMAP visualization of myHSC and cyHSC signatures from scRNAseq of HSC from 19xCCl4 fibrotic mouse liver (n=1) (a) or snRNAseq of HSC from normal and cirrhotic human livers (n=4 each) (b). c, Violin plots of scRNA-seq data show cyHSC and myHSC subsets in untreated (Ctrl) or injured (CCl4) livers (n=1 each). d, Cell-cell interaction analysis of hepatocytes with other liver cells by CellPhoneDB from 8xCCl4-injured liver (n=3). e-f, UMAP and violin plot aligning Col1a1 and Hgf mRNA expression with myHSC and cyHSC in scRNA-seq from 19xCCl4 treated liver (e, n=1) or snRNA-seq of HSC from human normal and cirrhotic livers (n=4 each) (f). g, in situ analysis of Hgf mRNA by RNAscope with Col1a1-GFP and LratCre-induced TdTom fluorescence, shows separate populations of Col1a1-GFPhigh Hgflow myHSC, and Col1a1-GFPlow Hgfhigh cyHSC in normal (n=1) and 21xCCl4-treated livers (n=1) (g). AU=arbitrary units. Data are shown as mean ± SEM. In c and e, box plots represent the interquartile range (IQR), Q1, median and Q3, whiskers (Q1−1.5×IQR and Q3+1.5×IQR) and outlier data as individual dots. each data point represents one cell (a,b,e,f), or one area (g). Scale bars: 50 μm. Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
Promotion of hepatocarcinogenesis by myHSC-enriched type I collagen
Type I collagen has been linked to stiffness-mediated tumour-promotion in several organs 26, but its role in hepatocarcinogenesis remains largely elusive. Col1a1, which is required for assembly of homo- and heterotrimeric type I collagen fibrils, was selectively expressed in HSC in human and mouse livers (Extended Data Fig. 8a–b) and strongly upregulated in CCl4-treated mouse liver and in patients with liver fibrosis (Extended Data Fig. 8c). Mx1Cre-mediated deletion of Col1a1 was highly efficient (Extended Data Fig. 8d). Reduced HCC development was observed in a profound fibrosis regimen but not in a model with once weekly injection of CCl4, which lacked strong fibrosis and stiffness and did not display reduced stiffness in Col1a1-deleted mice (Fig. 4a, Extended Data Fig. 8f–i). Thus, it appears that there is a threshold for fibrosis and/or stiffness for Col1a1-dependent tumour promotion, consistent with the increase risk of HCC development in patients with liver stiffness above 12 kPa 27–29. We confirmed our findings in the Mdr2KO model of spontaneously developing HCC, where we observed a significantly reduced tumour number and liver stiffness in Col1a1-deleted mice (Extended Data Fig. 8j–l). A similar reduction of hepatocarcinogenesis was seen in the DEN+CCl4 profound fibrosis model as well as the NASH-HCC model induced by 8 months of HF-CDAA diet when Col1a1 was selectively deleted in HSC by LratCre (Extended Data Fig. 8m–r). Col1a1-deletion was accompanied by decreased hepatocyte proliferation, which we observed in both the non-tumour and tumour compartments (Fig. 4b, Extended Data Fig. 8o), but without changes in hepatocyte death parameters (Extended Data Fig. 9a). Importantly, type I collagen deletion did not alter HSC activation or inflammation (Extended Data Fig. 9b–d). FACS-based analysis of immune cell subpopulations in Col1a1-deleted mice showed a trend toward increased CD45+ infiltration in tumour and non-tumour tissues, trends for increased CD4+, CD8+ and Ki67+ CD4+ T cells as well as accompanying increases Foxp3+ Treg and Natural Killer (NK) T cells in tumour tissue and CD11c+ MHCII+ dendritic cells in adjacent fibrotic liver, but no changes in granzyme B-positive T, NK and NKT cells (Extended Data Fig. 9e–g, Supplementary Information 4a–b). Together, these data suggest that Col I may also represent a barrier for immune cell influx and could affect HCC beyond the direct HSC-epithelial crosstalk, established here. Concomitant with reduced stiffness in Col1a1-deleted mice (Fig. 4c, Extended Data Fig. 8e), we observed significant decreases in the expression of YAP, TAZ and phosphorylated AKT, three mechanosensitive pathways that have been associated with tumour promotion, in the fibrotic liver (Fig. 4d). This finding was further confirmed by immunohistochemistry, revealing decreased nuclear translocation of TAZ (Fig. 4e) but not YAP (Extended Data Fig. 9h) in hepatocytes in the PME. To explore the relevance of these findings to hepatocarcinogenesis further, we determined the effect of YAP and TAZ deletion in our profound liver fibrosis HCC model. Hepatocyte-specific TAZ but not YAP deletion suppressed HCC development and hepatocyte proliferation in the non-tumour compartment but not in tumours (Fig. 4f–g, Extended Data Fig. 9i). Together with the reduced proliferation Col1a1-deleted mice, our findings suggest that the Col I-stiffness-TAZ pathway contributes to hepatocyte proliferation and a tumour-prone environment in the highly fibrotic and stiff liver. In addition to stiffness, Col I also interacts with cell surface receptors. We investigated the role of two receptors with known roles in carcinogenesis, integrin beta 1 (ITGB1) and discoidin domain receptor 1 (DDR1). Hepatocyte-specific deletion of Itgb1 exerted modest effects on DEN+CCl4-induced hepatocarcinogenesis, with significantly decreased tumour size but no change in tumour number or the liver-body weight ratio (Extended Data Fig. 9j). We observed an upregulation of DDR1 in non-tumour and tumour tissue in different mouse models of fibrosis-associated HCC as well as in patients (Fig. 5a–b, Extended Data Fig. 10a). Specifically, DDR1 increased both in hepatocytes and HCC tumor cells as shown by scRNA-seq, snRNA-seq and immunostaining (Fig. 5b, Extended Data Fig. 10a). Interestingly, DDR1 expression was induced in both hepatocytes and tumour cells by TGFβ (Extended Data Fig. 10b), suggesting that this profibrogenic master cytokine controls wound healing and ECM-mediated cell communication not only via upregulation of collagen production in HSC but also by increasing collagen receptor expression in the epithelial compartment. Importantly, hepatocyte-specific DDR1 deletion caused a significant reduction of DEN+CCl4-induced HCC (Fig. 5c, Extended Data Fig. 10c). Similar to our findings in our other models, i.e. Col1a1ΔHSC as well as HSC-depleted and Wwtr1ΔHep mice, we observed a decrease of proliferation in Ddr1ΔHep mice, but this was significant only in the tumour cell compartment, while there was only a trend towards reduced hepatocyte proliferation in the surrounding fibrotic liver (Fig. 5d). Our previous experiments showing a lower degree of fibrosis and fibroblast accumulation in tumours than in non-tumour liver contrasts the predominant role of DDR1 in the tumour compartment. We further characterized the profound fibrosis DEN+CCl4 model used for studying both Col I and DDR1 in hepatocarcinogenesis. We found significantly more fibrosis and fibroblast accumulation as well as a trend towards increased Ddr1 mRNA in tumors from the profound fibrosis than the regular fibrosis model (Extended Data Fig. 10d), also paralleling the high degree of fibrosis and αSMA+ fibroblasts seen in some HCC patients (Fig. 2b, Extended Data Figure 2d). It was previously reported that mice with non-degradable Col I, which have higher collagen levels, develop less HCC 30, which appears to contradict our findings. We therefore investigated if degradable and non-degradable Col I exert differential effects on DDR1 in HCC. Indeed, we found that ECM containing degradable Col I, but not ECM containing non-degradable type I R/R collagen, activated DDR1 and downstream tumour-promoting pathways such as AKT in tumour cells, which was blocked by DDR1 inhibitor 7rh (Fig. 5e–f, Extended Data Fig. 10e–g - see data in accompanying manuscript by Su et al. for mechanistic details). Consistent with the hypothesis that the collagen degradation status may affect DDR1 and thereby tumour development, we observed increased expression of collagen-degrading MMPs in tumour tissue and found that a signature of collagen-degrading MMPs correlated with poor survival in HCC patients (Extended Data Fig. 10h–i). Importantly, we did not observe decreased TAZ expression in DDR1-deleted livers (Extended Data Fig. 10j). Together, our data suggest that the Col I-stiffness-TAZ pathway, predominantly activated in severely fibrotic liver PME, and the Col I-DDR1 pathway, predominantly activated in the TME, represent two independent and spatially distinct pathways of Col I-driven hepatocarcinogenesis. Moreover, increases in immune cell infiltration, including CD3+ T cells in Col1a1ΔHSC but not in Ddr1ΔHep mice (Extended Data Fig. 9e–g and 10k), may provide an additional regulator of hepatocarcinogenesis at the level of anti-tumour immunity that is independent of epithelial TAZ- and DDR1 (currently under investigation). As HSC promoted HCC in moderate fibrosis settings but in a collagen-independent manner (Fig. 1a–f, Extended Data Fig. 8g–h), the tumour-promoting Col I -stiffness-TAZ and the Col I-DDR1 pathways appear to operate in parallel with other pathways through which HSC promote HCC (Fig. 1a–f). Accordingly, we observed a tumour-promoting role of a second ECM gene, Has2, encoding for hyaluronic acid (HA)-producing enzyme hyaluronan synthase 2. Has2 was enriched in myHSC, and mice with HSC-selective Has2 deletion (Has2 ΔHSC) displayed a small but significant reduction of HCC development (Extended Data Fig. 11a–d), consistent with reduced HCC formation in mice with global and hepatocyte-specific knockout of HA receptor CD44 reported by us previously31.
Figure 4 |. Promotion of HCC by type I collagen and stiffness-associated accumulation of TAZ in hepatocytes.
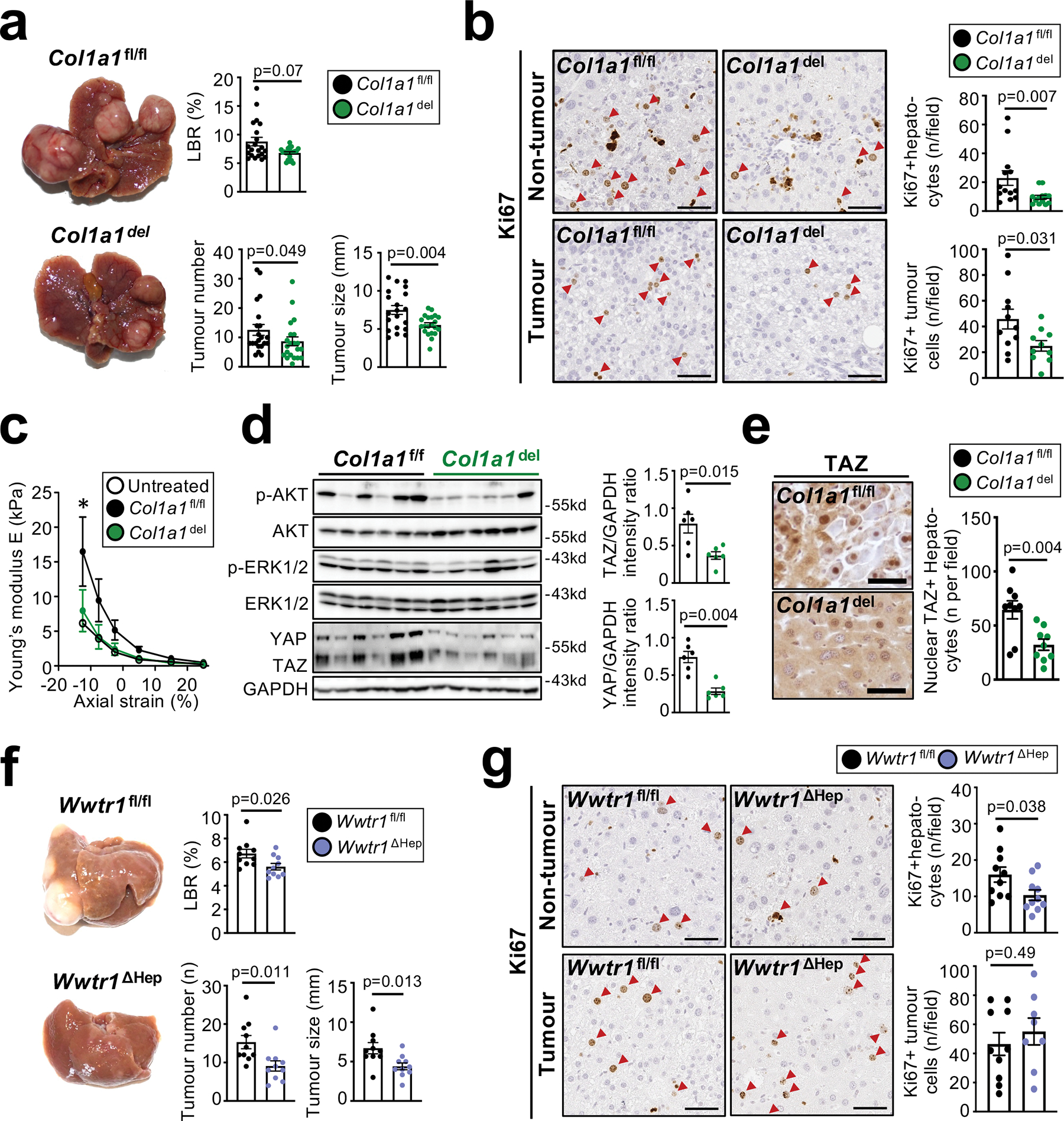
a, significantly reduced HCC, shown by representative images, liver body ratio (LBR), tumour size and number, in Mx1Crepos Col1a1f/f (Col1a1del) compared to Col1a1f/f littermates (n=21 per group) in a profound fibrosis DEN+44xCCl4 model. b, Ki67 staining in non-tumour and tumour tissue (n=10–13 mice per group) shows a reduction of hepatocyte proliferation and tumour cell proliferation in Col1a1del compared to Col1a1f/f mice. c, reduction of liver stiffness, shown by rheometry (untreated: n=1; DEN+19xCCl4: n=4/group). d-e, pAKT, pERK, YAP, TAZ and GAPDH, detected by western blot (d), and TAZ IHC and nuclear translocation (e), in livers from the DEN+19xCCl4 profound fibrosis model (macroscopic tumors not yet present) from Col1a1del and Col1a1f/f mice (n=6/group for d, n=9/group for e). f-g, HCC development (f) and hepatocyte proliferation (g) in mice with hepatocyte-specific TAZ deletion (Wwtr1ΔHep, n=8–10) compared to Wwtr1fl/fl littermates (n=10) in the profound fibrosis DEN+44xCCl4 HCC model. Using the same samples, immunoblots in d were done using one gel for YAP/TAZ and GAPDH and a second gel for pAKT, AKT, pERK1/2 and ERK. Data are shown as mean ± SEM, each data point represents one individual. Scale bars: 50 μm. Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
Figure 5 |. Promotion of hepatocarcinogenesis by DDR1.
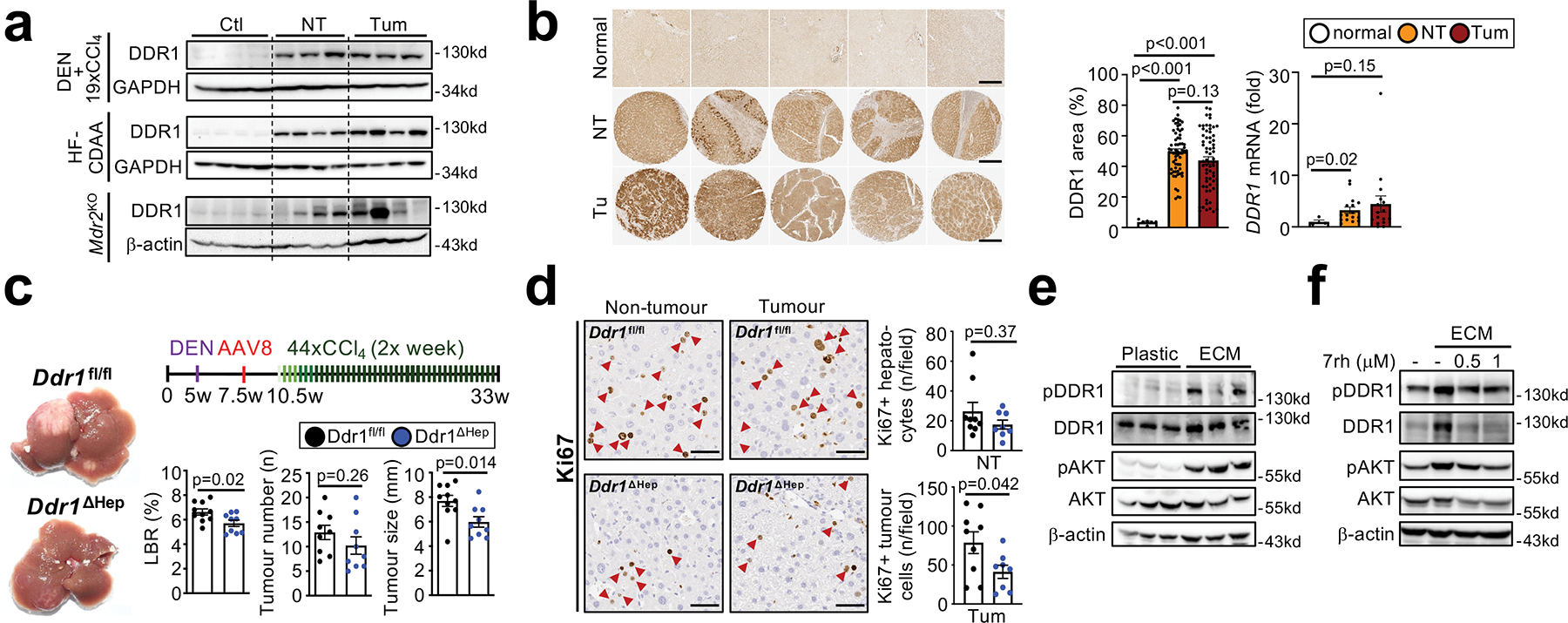
a, Western blot for DDR1 in healthy, non-tumour and tumour liver from different mouse models of HCC. b, DDR1 IHC in healthy human control livers (n=6) and in non-tumour and tumour areas from patients (TMA containing n=64 each), as well as DDR1 mRNA expression in healthy controls (n=4), amd non-tumor and tumor areas from HCC patients (n=16). c-d, HCC development (c) and Ki67+ hepaotcytes (d) were assessed in mice with hepatocyte-specific DDR1 deletion (Ddr1fl/fl, n=12 and Ddr1ΔHep, n=9) in the DEN+CCl4 profound fibrosis HCC model. e, Western blot for pDDR1, pAKT, DDR1, AKT and b-actin in HepG2 cells, plated on plastic or ECM. f, western blot for pDDR1, pAKT, DDR1, AKT and b-actin in Huh7 cells plated on plastic, or ECM and treated with or without DDR1 inhibitor 7rh. Data are shown as mean ± SEM, each datapoint represents one individual (a-d) or one well (e-f). Scale bars: 500 μm (b), 50 μm (d). Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
cyHSC-enriched HGF protects from hepatocarcinogenesis
After studies on other cyHSC-enriched factors such as hedgehog inhibitor HHIP, and chemokine CXCL12, did not reveal contributions of these mediators to hepatocarcinogenesis (Extended Data Fig. 11e–i), we next determined the role of HSC-expressed HGF. Via its receptor MET, HGF may exert dual roles in cancer, functioning as tumour promoter and therapeutic target in many cancers including HCC32 but also as mediator of protective effects in the precancerous state33–35. Hgf was predominantly found in HSC but also detected to a lesser degree in LSEC in both mice and human sc- and sn-RNA-seq datasets, whereas its receptor Met was primarily expressed in hepatocytes (Extended Data Fig. 12a–c). Deletion of Hgf in HSC via LratCre (HgfΔHSC) yielded, consistent with our scRNA-seq data, ≈70% reductions of Hgf mRNA and HGF protein levels in normal and fibrotic livers from HgfΔHSC (Extended Data Fig. 12d). Strikingly, Hgf deletion exerted effects opposite to Col1a1 deletion, with a drastic increase of HCC development (Fig. 6a). Hgf ΔHSC mice displayed increased hepatocyte injury, assessed by serum ALT and TUNEL staining and, most likely as consequence of increased injury, increased compensatory hepatocyte proliferation, fibrosis, inflammation and monocyte infiltration (Fig. 6b, Extended Data Fig. 12e–h, Supplementary Information 5a–b). A similar increase in hepatocyte injury was found in the dietary HF-CDAA high-fat NASH model (Fig. 6b). The cytoprotective functions of HGF were further confirmed by in vitro studies showing strong protection against Fas-mediated cell death by HGF in primary hepatocytes (Fig. 6c), consistent with previous reports 36. Importantly, HSC-selective Hgf deletion did not reduce hepatocyte proliferation after 70% partial hepatoectomy, suggesting the main effect of HSC-derived HGF is hepatoprotection rather than the promotion of hepatocyte proliferation (Extended Data Fig. 12i). Contrasting the above-described reduction of Hgf mRNA in HSC on a per cell base (Extended Fig. 6d), there was an increase of Hgf mRNA within the entire liver in mice and patients (Fig. 6d–e), suggesting that the downregulation of cyHSC mediators such as HGF within HSC is compensated by increased HSC numbers. While this compensation appears to be a protective mechanism in CLD, analysis in dysplastic nodules and HCC (Fig. 6d–e) indicate a failure of this compensation and increased risk for progression, as further suggested by the decreased survival of patients with low HGF expression (Fig. 6f). To test this concept of an impaired balance between protective cyHSC and tumour-promoting myHSC in clinical settings, we applied a scRNA-seq signature combining all myHSC and cyHSC genes encoding secreted mediators (Supplementary Table 6, Extended Data Fig. 13a) to a microarray dataset from patients with cirrhosis. This analysis revealed that an increased dysbalance between cyHSC and myHSC with progressive disease (Extended Data Fig. 13b) was associated with a significantly increased risk for HCC development (Extended Data Fig. 13c).
Figure 6 |. CyHSC-enriched HGF protects hepatocytes from death and reduces HCC development.
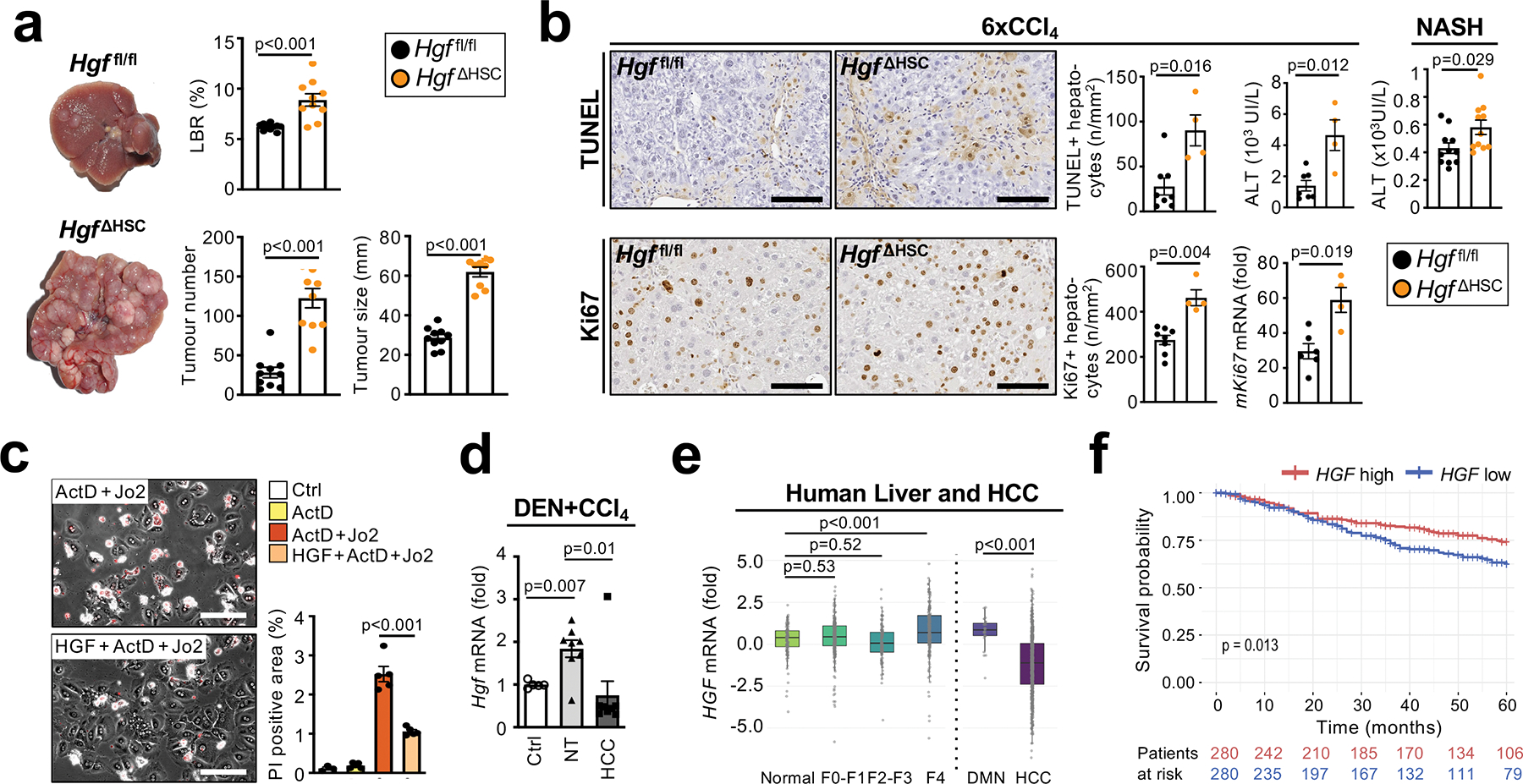
a, Hgf deletion in HSC promoted DEN+12xCCl4-induced HCC development (n=10/group). b, hepatocyte death assessed by TUNEL staining and ALT measurement as well as hepatocyte Ki67 IHC and mKi67 RNA in Hgf ΔHSC and to Hgf fl/fl mice in the 6xCCl4 model (n=4–8/group) and ALT measurement in mice on HF-CDAA-diet for 1 month (n=9/group). c, Propidium iodide staining (red) in cultured murine primary hepatocytes treated with ActD+Jo2-induced +/− murine recombinant HGF (50 ng/μL) for 6h (n=3–5 wells/group). d, Hgf mRNA expression in non-tumour (NT) and HCC tumour tissue (n=8 each) from mice after DEN+14xCCl4 treatment compared to untreated liver (Ctrl, n=5). e, HGF mRNA expression in normal liver (n=124), F0-F1 non-tumour (n=261), F2-F3 (n=115), F4 (n=182), dyplastic macronodules (DMN, n=31) and HCC (n=687) from patients. f, survival of HCC patients with low HGF (n=280) or high HGF (n=280) expression. Data are shown as mean ± SEM, box plots represent interquartile range (IQR), Q1, median and Q3, each data point represents one individual. Scale bars 100 μm. Details of sample sizes, biological replicates and statistical tests are given in the Additional Information and Source Data.
DISCUSSION
While our study uncovered an overall tumour-promoting role of HSC by genetic HSC depletion, activation and inhibition approaches in models of genotoxic, cholestatic and NASH-associated HCC, it also highlights unexpected tumour-suppressive functions of HSC. In this complex scenario, a dynamic shift between tumour-suppressive and tumour-promoting HSC subpopulations and their mediators during the course of CLD appears to determine cancer risk. Our finding that cyHSC-enriched HGF protects from HCC is consistent with the concept of normal fibroblasts suppressing tumours, referred to as “neighbour suppression” 37,38. The observed upregulation of HGF in fibrosis and cirrhosis, caused by increased numbers of HSC, likely constitutes a protective compensation mechanism, but appears to fail in advanced disease stages. A parallel increase of HCC-promoting myHSC-enriched mediator type I collagen contributes to the impaired balance between protective and disease-promoting factors in advanced liver disease. Deposition of type I collagen beyond a critical threshold led to the activation of tumour-promoting stiffness-TAZ pathway in the hepatic PME, reflecting the elevated HCC risk in patients with increased liver stiffness above 12 kPa27–29. Together, the dysbalance between cyHSC/myHSC and/or their mediators may contribute to increased hepatocyte proliferation and cell death, two well-established risk factors for HCC, and thereby contribute to an increased risk for HCC development Our finding that late but not early HSC depletion or HSC activation modulated hepatocarcinogenesis suggest the possibility that HSC exert distinct and possibly opposite functions at early stages (largely cyHSC) and late stages (largely myHSC). The lacking net effect of early HSC depletion, which reduces HSC early and late, would be consistent with such opposite functions. Depletion of αSMA+ cancer associated fibroblasts (CAF) in large established tumours did not affect tumour growth, suggesting that HSC mostly influence the development of HCC lesions rather than their progression, and is consistent with their low numbers in the tumour compartment. Indeed, our analysis in patients shows that an unfavorable ratio between myHSC and cyHSC at advanced stages confers an increased risk for HCC. It can be speculated that this tumour-promoting dysbalance between cyHSC and myHSC and/or their mediators is spatially confined to specific regions within the chronically injured liver, elevating risk for HCC development locally, which requires further investigation. Although complementary approaches for manipulating HSC functions and multiple tumor models revealed consistent findings on HSC functions, HSC subpopulations and their mediators, our study also has potential limitations: (i) our Cre-based approaches did not allow for selective deletion of Hgf and Col1a1 in cyHSC and myHSC subpopulations; (ii) depletion of HSC via LratCre-induced DTR and αSMA-TK cannot be selectively achieved in early stages as significant repopulation did not occur; (iii) we could only selectively deplete myHSC (via αSMA-TK) but not cyHSC; and (iv) killing of HSC in our depletion studies may have resulted in alterations of immunity beyond those investigated by our flow-cytometric analyses. Furthermore, it is likely that HSC increase HCC risk through mediators besides HGF and Col I. In this regard, we identified HA-producing enzyme Has2 as a tumour-promoting myHSC mediator, albeit with only modest effects and consistent with the known role of the HA receptor CD44 in HCC31. In addition to a tumour-promoting Col I-stiffness-TAZ pathway affecting premalignant hepatocytes or early-stage lesions in non-tumour areas, we also uncovered a tumour-promoting Col I-DDR1 pathway, that stimulated tumour cell proliferation and appears to be promoted by high expression of DDR1 on tumour cells and increased collagen degradation (see accompanying manuscript by Su et al for further mechanistic details). Different from breast cancer39, epithelial DDR1 did not promote immune cell infiltration in our study. Interestingly, the roles of HSC mediators HGF and type I collagen in HCC contrast their roles in other liver tumours 23,24 and pancreatic cancer 40: HSC-derived HGF, which suppresses HCC, promotes the growth of ICC and desmoplastic liver metastasis23,24. Conversely, type I collagen, which promotes HCC, reduces tumour growth in desmoplastic liver metastasis24 and pancreatic cancer 40. These differences may be explained by the accumulation of fibroblasts at disease-specific sites and the astounding regenerative capacity of hepatocytes, which is important for organismal survival but poses a unique risk for cancer development in the liver. In this context, PME-localized HSC and their mediators appear to mostly act on hepatocytes and/or early stages lesions in the chronically injured liver, thereby affecting cancer risk and development through mechanisms that are distinct from those of TME-localized CAF. In view of the lack of primary and secondary prevention strategies for HCC, one of the leading causes of cancer mortality1,5, and the alarming increase in NASH, our data suggest that restoring the balance between cyHSC and myHSC and/or their mediators may represent a novel approach to mitigate the risk for HCC development.
METHODS
Data availability
The microarray, RNA-seq, scRNA-seq and snRNAseq datasets reported in this study have been deposited in the Gene Expression Omnibus database (GEO) under the accession number GSE174748 and GSE212047. In addition, we analyzed previously published whole liver or isolated HSC scRNAseq dataset GSE172492 and GSE158183, normal human liver snRNAseq GSE185477 as well as the microarray dataset GSE15654, GSE49541, and GSE10140. Source data are provided with this paper.
Code availability
R markdown scripts enabling the main steps of the analysis have been deposited on Github (https://github.com/Schwabelab/HSC_in_HCC). Survival of HCC patients from the TCGA dataset was determined using http://gepia2.cancer-pku.cn/#survival.
Human specimens
For Sirius Red, αSMA and DDR1 quantification in human liver tissues as well as qPCR and single nucleus RNA sequencing of patients with HCC, written informed consent was obtained from each patient at the time of recruitment and samples were collected with approval from the Columbia University Medical Center Institutional Review Board (Protocol Number: IRB-AAAN2452). Patient records were anonymized and de-identified. Studies were conducted in accordance with National Institutes of Health and institutional guidelines for human subject research. Formalin-fixed paraffin-embedded (FFPE) tissue microarray (TMA) with paired non-tumour and hepatocellular carcinoma tissue (in triplicate) as well as FFPE slides from paired non-tumour and tumour intrahepatic cholangiocarcinoma cases were used to perform Sirius Red and ⍺SMA staining. Individuals were separated into two groups: HCC patients with or without underlying fibrosis based on Sirius Red and αSMA quantification (with cutoff threshold set 2.5-fold higher than non-tumour ICC tissue [considered normal]).
Local approval for procuring human liver tissue for single nucleus RNA-seq was also obtained from the NRS BioResource and Tissue Governance Unit at University of Edinburgh, Scotland (study number SR574), following review at the East of Scotland Research Ethics Service (reference 15/ES/0094). All subjects provided written informed consent. Healthy background non-lesional liver tissue was obtained intraoperatively from patients undergoing surgical liver resection for solitary colorectal metastasis at the Hepatobiliary and Pancreatic Unit, Department of Clinical Surgery, Royal Infirmary of Edinburgh. Patients with a known history of chronic liver disease, abnormal liver function tests or those who had received systemic chemotherapy within the last four months were excluded from this cohort. Cirrhotic liver tissue was obtained intraoperatively from patients undergoing orthotopic liver transplantation at the Scottish Liver Transplant Unit, Royal Infirmary of Edinburgh.
The study of COL1A1 and HGF mRNA in human liver tissues was approved by the local Ethics Committee (CCPPRB Paris Saint-Louis IRB00003835), and informed consent was obtained in accordance with French legislation for all patients. A total of 1400 fresh-frozen liver tissue samples, including 687 HCC, were collected and described previously41. Measurement of COL1A1 and HGF expression was performed by Fluidigm array with the specific Taqman probes COL1A1 (Thermofisher, Hs00164004_m1) and HGF (Thermofisher, Hs00300159_m1).
Animal studies
Animal care and experimental procedures were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” of the National Institutes of Health and were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC - protocols AC-AAAP8400, AC-AABM2555, AC-AAAZ8474, AC-AABP3560 and AC-AAAV2454). Mice were housed in the Irving Cancer Research Center (ICRC) at Columbia University fed with a standard mouse diet (ad libitum water and food access) with constant temperature of 21–24 °C, 45–65% humidity with a 12-h light–dark cycle. The following mice were obtained from the Jackson Laboratory: TdTomato (TdTom) Ai14 reporter (#007908); Rosa26-HBEGF (Cre-inducible diphtheria toxin receptor [DTR]) (#007900), Mx1-Cre (#003556); Trp53 floxed (#008462) mice; Cxcl12 floxed (#021773); Pdgfrb floxed (#010977). Ddr1tm1a (EM:09692) and Hhip floxed (EM:04541) mice were obtained from Infrafrontier. Wwtr1 floxed and Yap1 floxed mice were obtained from Dr. Eric Olson, University of Texas Southwestern. αSMA-TK mice were provided by Dr. Raghu Kalluri (available at Jackson # 031155). Col1a1-GFP reporter mice 42 and LratCre mice 3 were previously described and at least seven times backcrossed to C57Bl/6. Mdr2KO mice [in Friend Virus B (FVB) background] were provided by Pr. Detlef Schuppan (University of Mainz, Germany) and interbred with LratCrepos Pdgfrb floxed or Mx1-Crepos Col1a1 floxed mice (backcrossed three times). The following mice were previously described: Lhx2fl/fl mice 43, RelAfl/fl mice 44, Col1a1fl/fl mice 45, Hgf fl/fl mice 46,47, Has2fl/fl mice 48 and RosaNICD mice 49.
HSC depletion, fibrosis, regeneration and liver cancer models
For genetic HSC depletion, LratCrepos TdTompos DTRpos or LratCre pos TdTompos DTRneg controls were intraperitonially (i.p.) injected with diphtheria toxin (DT, Sigma #322326, 0.25 ng/g) once per week as indicated. αSMA-driven thymidine kinase (αSMA-TK50, backcrossed at least five times to C57Bl/6, were injected i.p. with ganciclovir (InvivoGen, i.p. 10 mg/kg) 3 times, once per day following the CCl4 injections or 2 to 3 times per week when fed with HF-CDAA-diet as described in figures. For Col1a1 depletion, Mx1Creneg Col1a1f/f and Mx1Crepos Col1a1f/f received polyI:C (GE Healthcare #27-432-01, i.p. 10 μg/g) 3 times every 3 days, 1 month after diethylnitrosamine (DEN) injection and 2 weeks before CCl4 administration. For deletion of Yap1, Wwtr1 or Ddr1 in hepatocytes and tumour compartments or induction of NICD1 overexpression, floxed mice were administrated i.v. with 1011 GC/mouse of AAV8-TBG-Cre (Addgene, #107787-AAV8) or AAV8-TBG-Null (Addgene, #105536-AAV8) diluted in saline solution at the indicated time. For all experiments, individuals from the same litter were allocated to different experimental groups on the basis of expression of the Cre recombinase. To induced hepatocyte gene deletion, half of each litter (Wwtr1f/f, YAPf/f, Ddr1f/f and Itgb1f/f mice) received AAV8-TBG-Null and the other half received a similar dose of AAV8-TBG-CRE. Groups were designed to have mice with similar weights in each group. When experiments were not based on CRE expression, C57Bl6/J mice were randomly allocated into different groups.
In order to induce HSC activation and fibrosis, CCl4 was injected (i.p. 0.5 ml/kg, diluted 1:4 in corn oil) every 3 days. Liver collection or HSC isolation was performed 2 days after the last injection of CCl4. NASH-induced fibrosis was performed using L-amino acid diet with 60 kcal% fat with 0.1% methionine and no added choline (HF-CDAA diet, Research Diet, A06071302) or with a high fat diet (FPC-NASH diet, Envigo, TD.160785) in combination with high-glucose (23 g/L) fructose (19 g/L) water for the time indicated. For bulk RNA-sequencing, quiescent HSC were isolated from mice fed with a low fat diet (Envigo, TD.08485).
Two-thirds partial hepatectomy was performed after subcutaneous injection of 5 mg/kg of carprofen and 0.15 mg/kg of buprenorphine, under sterile condition and anesthesia using isoflurane on 8 to 12 weeks old male mice as previously described 51. After skin closure, animals were monitored every day until day two when they were euthanized, and the liver was harvested and processed.
Mdr2KO-induced HCC was performed in female or male mice (as indicated) and all other HCC experiments were performed in male mice. For the induction of HCC in a moderate fibrosis setting, DEN (Sigma, N0258, i.p. 25 mg/kg) was administrated on day 15 postpartum to male mice as indicated, followed by CCl4 injections (i.p. 0.5 ml/kg, diluted 1:4 in corn oil) on a weekly basis as indicated. To induce HCC in a model with profound fibrosis, DEN (i.p. 80 mg/kg) was injected into 4.5 week-old mice in combination escalating doses of CCl4 over time (first week 1× 0.50 ml/kg diluted 1:8 in corn oil; second week: 3× 0.5 ml/kg, diluted 1:4 in corn oil; third week: 3× 1 ml/kg diluted 1:4 in corn oil; fourth week: 3× 1.5 ml/kg diluted 1:4 in corn oil and from the fifth to 20th weeks: 2× 1.5 ml/kg, diluted 1:4 in corn oil and is referred as “profound fibrosis regimen” when evaluated at different timepoints throughout the study). All analyses in the DEN+CCl4 once per week model were performed on livers harvested two days after the last CCl4 injection. Liver analysis from the DEN+CCl4 profound fibrosis regimen was performed on mice euthanatized one week after the last CCl4 injection with the exception of experiments on HSC activation and inflammation in Extended Data Fig 9b–d, which were performed in mice sacrificed two days after the last CCl4 injection. For hydrodynamic tail vein injection-induced HCC models, mice were injected with 20 μg of pT3-EF1α-TAZS89A52 kindly provided by Xin Chen (University of California, San Francisco); or 20 μg pT3-EF1a-cMet and 20 μg pT3-EF5a-CTNNB1-S45Y-Myc-tag53, both plasmids kindly provided by Dr. Paul Monga (University of Pittsburgh). All plasmids were injected in combination with sleeping beauty transposase (SB13) in a ratio of 4:1 at 7 to 9 weeks of age. One to four weeks after injection, mice were fed with a FPC-NASH diet in combination with high-glucose and fructose drinking water for the time indicated. For all samples, the following analysis was performed: tumour burden was evaluated by liver weight/body weight ratio (LBR), tumour number (tumours greater than 1 mm in the DEN+CCl4 [once per week] and Mdr2KO models or greater than 3 mm in all the other models) and tumour size (mean of the 3 largest tumours).
Kras/p19-induced ICC was generated in our previous study 23 by hydrodynamic tail vein injection of the pCaggs-KRAS G12D (human) transposon plasmid and CRISPR/Cas9 sg-p19 (pX330-sg-p19) in combination with SB13.
As stated in our IACUC protocols, all mice using above hepatocarcinogenesis methods were euthanized before developing tumours exceeding 2 cm in diameter.
Supplementary Table 7 and 8 summarize all animal studies of hepatocarcinogenesis.
Cell isolations and culture
Primary mouse HSC were isolated from normal or fibrotic livers (for the CCl4 model, all isolations from the CCl4 model were done 2 days after the last injection of CCl4) using pronase-collagenase perfusion as previously described 54,55. For scRNAseq, HSC and myofibroblasts were purified using a 34% Nycodenz gradient centrifugation followed by FACS-sorting on a BD Aria II Cell Sorter using the TdTomato or GFP reporters. For RNA and protein preparations, HSC were isolated using a 11% Nycodenz gradient and FACS-sorted on a BD Aria II Cell Sorter or Influx cell sorter using the endogenous retinoid fluorescence of HSC with or without the TdTomato fluorescence reporter. Primary hepatocytes were isolated from 10–12 week old untreated C57Bl6/J mice using collagenase type IV perfusion (Worthington #LS004188) followed by a Percoll (GE Healthcare #17-0891-01) gradient. In order to have a representation of all hepatic cell populations, mouse livers were perfused with collagenase type I (Worthington #LS004196), followed by separation of hepatocytes and non-parenchymal cells by two minutes centrifugation at 50g and loading onto a 35% Percoll gradient. Isolated non-parenchymal and parenchymal fractions were mixed at a 2.5:1 ratio.
Following isolation, primary hepatocytes were cultured with a seeding medium consisting of William’s medium (Gibco, #12551-032) supplemented with hepatocyte supplement (Gibco, #A13448), 10 μM dexamethasone (Gibco #A13449), 10 % fetal bovine serum (FBS), gentamicin and antibiotics. After 4h, the medium was changed to a maintenance medium (the seeding medium but without FBS and dexamethasone). For some experiments, hepatocytes were pretreated for 6h with 50 ng/ul recombinant murine HGF (R&D Systems #2207-HG) before cell death induction. For cell death induction, 0.3 μg/mL actinomycin D was added 30 min before treatment with an 0.5 μg/μL anti-Fas antibody (BD, clone Jo2, #554255). Cells were analyzed 15 h after Fas activation by propidium iodide (PI, diluted 1:100). Pictures of PI-positive cells were taken on an Olympus IX71S1F-3 microscope coupled to a QImaging Retiga camera and images were analyzed using Adobe Photoshop.
The HCC cell lines (Huh7, HepG2 and Hepa1-6) as well as the fibroblast cell line (MRC5) were obtained from ATCC and cultured in DMEM+10% FBS. To study the effect of ECM-induced DDR1 activation, MRC5 or WT and RR (MMP-resistant Col I) fibroblasts were cultured for 5 to 7 days in DMEM+10% FBS in the presence or absence of 100 μM of Vitamin C supplementation. Fibroblasts were removed using PBS with 0.5% (v/v) Triton X-100 and 20mM NH4OH and ECM was washed with PBS once before plating HCC cells or primary hepatocytes. For the experiment using WT and RR (MMP-resistant Col I) fibroblasts, medium was changed one day after plating to low glucose medium (glucose-free medium supplemented with 0.5 mM glucose and 10% dialyzed FBS) for 24 h. The DDR1 inhibitor 7rh (R&D, 5860/5) was used to block DDR1.
Induction of DDR1 was measured 24h after treatment using the following cytokines and growth factors: 2.5 ng/ml of rhTGFb1 (R&D Systems, #240-B), or 40 ng/ml of rmHGF (R&D Systems #2207-HG), 50 ng/ml of rmEGF (Invitrogen, PMG8043), 20 ng/ul of rhPDGF-BB (R&D Systems, #220-BB), 30 ng/ml of rmTNFa (R&D Systems, #410-MT), 5 ng/mL of rmIL-1b (R&D Systems, #401-ML).
Rheometry
Rheometry measurements were done at the University of Pennsylvania (UPENN) on liver samples shipped overnight in cold PBS. All measurements were done on the left lobe. Parallel plate shear rheometry was carried out using a Kinexus rheometer (Malvern Panalytical, Westborough, MA). Samples were attached to the top and bottom plates with fibrin glue made by mixing 10 μl of 5 mg/ml salmon fibrinogen and 10 μl of 150 U/ml salmon thrombin (Sea Run Holdings, Freeport, ME) for each side of the sample. The upper plate (8 mm diameter) was lowered until contact was made as determined by the application of 400 Pa normal stress, and the sample was allowed to rest for 5 min to ensure attachment to the metal plates. Shear storage modulus G’, loss modulus G”, and normal force were measured by applying a low oscillatory shear strain of 2% at a frequency of 1 rad/sec at room temperature. Simultaneously, samples were subjected to small stepwise axial strains in tension (0, 10, and 20 %) followed by compression (−10, −15, −20, and −25%), between which the samples were allowed to relax for 2 min. Samples were kept hydrated during experiments with PBS. The equilibrium G’ after 2 min of relaxation were plotted against axial strain. Young’s modulus was calculated as the slope of the axial stress versus strain curve at each tension or compression level.
Histological analysis
Liver tissues, at least one piece of each of the five main lobes, were fixed for 1 to 2 days with 10% formalin or 4% paraformaldehyde and embedded in paraffin or submerged in 30 % sucrose for at least 1 day before to be embedded in Tissue-Tek O.C.T. to prepare frozen blocks. Blocks were cut into 5 μm sections. Sections were subjected to Sirius Red staining for fibrosis detection. Fibrosis quantification was done using a polarized light filter. Pictures were acquired using QCapture Suite Plus (v3.1.3.10) software and quantification was performed using Adobe Photoshop (v11.0). 3,3’-diaminobenzidine (DAB)-based and fluorescent immunohistochemistry (IHC) were performed using standard protocols. Hyaluronan was detected using Biotin-labeled HA-binding protein (R&D Systems, rhAggrecan aa30-675/His [NSO/7], 4 μg/ml). Dead cells were stained using the ApopTag® Plus Peroxidase In Situ Apoptosis Kit (Sigma, S7101). For IHC, the following antibodies were used: anti-αSMA-FITC (Sigma, F3777, 1/2000); anti-p21 (Abcam, ab188224, 1/100); anti-Ki67 (Abcam, ab1667, 1/100), anti-CD45 (BD #550539, 1/500); anti-CD3 (Invitrogen, MA5-14524, 1/500); anti-FITC (Abcam, ab6655, 1/250); anti-RFP and anti-RFP-biotin (Rockland, 600-401-379S, 600-406-379, 1/500); anti-GFP (Abcam, ab6673, 1/200); anti-Endomucin (Santacruz, sc-65495, 1/50); anti-myc tag (Maine Medical Center, #Vli01, 1/2000); anti-macrophages (Abcam, RM0029-11H3, 1/500); anti-Cytokeratin 19 (Abcam, ab133496, 1/500) anti-TAZ (ABclonal, #A8202, 1/100) and anti-DDR1 (Novus Biological, NBP1-28861, 1/500). Detection was performed using either the Vectastatin Elite ABC-HRP kit (Vector Laboratories) with DAB Peroxidase Substrate kit (Vector Laboratories) or a fluorescent secondary antibody with various fluorescent conjugates (Alexa Fluor 488/594/647, 1/250, Life Technologies), followed by counterstaining with either hematoxylin or DAPI (Thermo Fisher Scientific). Non-fluorescence image acquisition was performed on a Leica SCN400 slide scanner with the Scanner Console (v102.0.7.5) or an Olympus IX71S1F-3 microscope coupled to a Qimaging Retiga camera and the QuantStudio Design And Analysis Software (v1.4.1). Quantification of the DAB positive area was performed using the LEICA Digital Image Hub 4.0 image server, using 10x or 20x magnification or the AperioImageScope software (v12.4.3.5008). TUNEL+ and Ki67+ hepatocytes were manually counted. All quantifications were done on 1–2 areas of 3–5 different lobes. Pictures and quantification showing colocalization of TdTom and Col1a1-GFP were acquired using a Nikon A1 confocal laser microscope (Nikon Instruments) and analyzed using ImageJ (v.1.53c) or Fiji (v.2.1.0) and Adobe Photoshop (v11.0).
Senescence associated-β-galactosidase staining.
Fixed-fresh frozen liver sections were re-hydrated with PBS and stained for pH-dependent β-galactosidase activity using senescence associated (SA)-β-galactosidase solution (pH 6.0) according to the manufacturer’s protocol (Cell Signaling, 9860S) for 12 h at 37 °C. Slides were washed and immunostaining was performed as mentioned above to mark different liver cell types.
RNAscope
The RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostic PN 310091) was used to detect RNAs by in situ hybridization of Hgf mRNA (probe from ACD; Cat No. 315631-C3) on 10 μm thick frozen liver sections. The manufacturer protocol was used and followed by immunofluorescence using an anti-RFP and anti-GFP (as described above) to detect the TdTomato and GFP reporters, respectively. After counterstaining with DAPI, pictures were captured using a Nikon A1 confocal laser microscope. Percentage of colocalization of Col1a1-GFP+/HSC-TdTom+ and mRNA_Hgf+/TdTom+ as well as percentage of Col1a1-GFP and mRNA_Hgf per area were determined using Fiji software.
RNA isolation and real-time qPCR
Total RNA from liver tissues was isolated using TRIZOL and the Tissue RNA isolation kit (Roche). RNA from HSC was extracted using the RNeasy Micro (Qiagen, #74004). RNA extraction from mouse HCC was done on single tumours. After RNA quantification with a spectrophotometer nanodrop ND-1000 using the software ND-1000 (v.3.3.1), cDNA was generated using TaqMan reverse transcription reagents (Applied Biosystems, # 4368813). Real-time qPCR was performed on an Applied Biosystems™ QuantStudio™ 5 Real-Time PCR System (Applied Biosystems) using PerfeCTa FastMix II buffer (Quantabio, #95120) and the following probes (Thermofisher): 18s (Hs99999901_s1); Acta2 (Mm01546133_m1); Afp (Mm00431715_m1); Ccl2 (Mm00441242_m1); Ccl5 (Mm01302427); Cd3e (Mm00599683_m1); Cd4 (Mm00442754_m1); Cd8a (Mm01182108_m1); Cd68 (Mm03047343_m1); Col1a1 (Mm00801666_g1); Col1a2 (Mm00483888_m1); Col3a1 (Mm00802300_m1); Cxcl5 (Mm00436451_g1); Cxcl12 (Mm00445553_m1); Des (Mm00802455_m1); Ddr1 (Mm01273496_m1, Mm00432256_m1 and Hs01058430); Epcam (Mm00493214_m1); Gli1 (Mm01160464_g1); Gpc3 (Mm00516722); Hgf (Mm01135184 and Mm01161535); Hhip (Mm00469580_m1); Il1b (Mm00434228_m1); Ifng (Mm01168134_m1); Krt19 (Mm00492980_m1); Lhx2 (Mm00839783_m1); Lox (Mm00495386_m1); Lrat (Mm00469972_m1),; Mki67 (Mm01278617_m1); Mmp1a (Mm00473485_m1); Mmp1b (Mm00473493_g1); Mmp2 (Mm00439498_m1); Mmp8 (Mm00439509_m1); Mmp9 (Mm00442991_m1); Mmp13 (Mm0043491_m1); Mmp14 (Mm00485054_m1); Prom1 (Mm01211402_m1); Ptprc (Mm01293577_m1); RelA (Mm00501346_m1); Sox9 (Mm00448840_m1); Tnfa (Mm00443258_m1); Trp53 (Mm01337166); Wwtr1 (Mm00513560_m1); Yap1 (Mm01143263_m1). Analysis was performed using QuantStudio Design And Analysis Software v1.4.1. Absolute values were quantified using relative standard curves, normalized to 18s expression and fold inductions were carried out with respect to livers of untreated mice (excepted for Figure 1a and Extended Data Figure 9h where fold induction was measured relative to floxed mice group) or normal livers from patients.
Tumour mutational analysis
After homogenisation of each single tumour, DNA was extracted using a PCR kit (Genesee scientific, # 17-207B). DNA sequences with hotspot mutation 56 were amplified by PCR for Hras (forward: 5’-atccatcagggtatgagaggtg; reverse: 5’-gcatgactgtgtccaggacatt), Egfr (forward: 5’-ggtgtttctgactatcctgg; reverse: 5’-tgaggactgttgggtgaaagg) and Braf (forward 5’-cagaggacatacgaatctctg; reverse: 5’-gcccttcagtgtatttctcgt). PCR products were sent to Genewiz for Sanger sequencing.
Immunoblotting
Proteins were extracted from liver tissue using RIPA buffer with anti-protease (Roche, cOmplete™) and anti-phosphatase (Roche, PhosSTOP™). Proteins were denaturated by adding Laemmli buffer, sonicated and boiled at 95°C. Samples were subjected to SDS-PAGE and transferred to a nitrocellulose membrane (Sigma) using a semi-dry blot system (BioRad). The following antibodies were used: anti-YAP/TAZ (Cell Signaling, #8418, 1/2000); anti-p-AKT (Cell Signaling #4060, 1/1000); anti-AKT (Cell Signaling #9272, 1/1000); anti-p-ERK1/2 (Cell Signaling #9106. 1/1000); anti-ERK1/2 (Cell Signaling, #4695, 1/1000); anti-p-DDR1 (pTyr513) (Sigma, SAB4504671, 1/1000),;anti-DDR1 (Cell Signaling #5583, 1/1000); anti-β-Actin (Sigma, A3854, 1/20000); anti-GAPDH (Sigma, G9295, 1/17500) and HRP-anti-Rabbit (Santa Cruz, sc-2004, 1/2000). Blots were visualized using ultra-sensitive enhanced chemiluminescent substrate (ThermoFisher Scientific, 34094) on a FluorChem M System instrument and quantified on ImageJ. Uncropped immunoblots are provided in Supplementary Information 6–10.
Enzyme-linked immunoassay (ELISA)
HGF protein levels were measured in PBS-homogenized mouse liver tissues by using the mouse HGF duoset ELISA (R&D, DY2207) according to the manufacturer’s instructions (R&D systems) and the use of a Synergy H1 Hybrid plate reader.
Immune cells analysis by flow cytometry
Non-tumour and tumour tissue (mix of 3 to 6 tumours/mouse) were mechanically homogenized followed by an enzymatic digestion with 1 mg/mL of collagenase A (Roche, #10103578001) and 0.5 μg/mL DNase I (Roche #10104159001) in isolation buffer (RPMI1640, 5% FBS, 1% L-glutamine, 1% penicillin–streptomycin and 10 mM HEPES) for 45 min at 150 rpm at 37°C. Cells were filtered through a 100 μm cell strainer, washed and separated in 2 parts to analyse the myeloid and the lymphocytes cell subsets. For the latter, cells were loaded onto a Percoll gradient (67% overlay with 40%) followed by red blood cells lysis using ammonium-chloride-potassium buffer and stained. Cells were incubated with Ghost Dye Red 780 (Tonbo Biosciences) to exclude dead cells and an anti-CD16/32 (Tonbo, 2.4G2, 1/500) before staining. Extracellular antibodies included: anti-CD45 (BD and Biolegend, clone 30-F11, 1/400); anti-CD19 (Tonbo, clone 1D3, 1/200); anti-CD3e (Tonbo, clone 145-2C11, 1/400); anti-CD4 (BD, clone RM4-5, 1/400); anti-CD8a (Tonbo, clone 53-6.7, 1/400); anti-NK1.1 (BD, clone PK136, 1/300); anti-CD11b (BD, clone M1/70, 1/500); anti-CD11c (BD, clone HL3,1/200); anti-F4/80 (Tonbo, clone BM8.1, 1/500); anti-Ly6C (Biolegend, clone HK1.4, 1/500); anti-Ly6G (Biolegend, clone 1A8, 1/500); anti-B220 (BD, RA3-6B2, 1/200); anti-CD279/PD1 (BioLegend, clone 29F.1A12, 1/400) and anti-MHC class II (Tonbo, clone M5/114.15.2, 1/400) antibodies. Intracellular antibodies included: anti-CD3e (BD, clone 145-2C11, 1/400); anti-TCRβ (BD, clone H57-597, 1/300); anti-FOXP3 (eBioscience, FJK-16s, 1/300); anti-Ki-67 (Thermo, clone SolA15, 1/200) and anti-Granzyme B (BioLegend, clone QA16A02, 1/200). Cells were fixed using the FOXP3/transcription factor staining buffer set (Tonbo) according to the manufacturer’s protocol. Samples were analysed using a BD LSR Fortessa cell analyser. Flow cytometry analysis was performed using FlowJo (v.10.7.2 and v.10.8.1).
Liver lipid measurements
Liver triglycerides and cholesterol were extracted from 45 to 55 mg of snap-frozen non-tumoural liver tissue using the Folch method. After homogenization with PBS, samples were added to a chloroform–methanol mixture and the aqueous layer evaporated under Nitrogen (N2) gas. Lipid content was measured with colorimetric assays (Thermo Fisher Scientific [TFS] Infinity Cholesterol Liquid Stable Reagent TR13421 and Infinity Triglycerides Liquid Stable Reagent TR22421).
Alanine transaminase (ALT) measurement
ALT was measured using a Dri-Chem and Element DC, veterinary chemistry Analyzer (Heska) in PBS-diluted serum samples.
Microarray and bulk RNA-sequencing
Microarray analysis was conducted on Lhx2f/f and Lhx2del HSC isolated 2 weeks after the last injection of polyI:C using the Affymetrix Mouse Gene 1.0 ST Array (Affymetrix, Santa Clara, CA). For bulk RNA sequencing, high-quality total RNA samples, with RIN > 8 (determined by Bioanalyzer 2100, Agilent Technologies) were processed by the Columbia Genome Center. For each sample, a minimum of 20 million 100-base-pair single-end reads were sequenced on the Illumina NovaSeq 6000. RTA (Illumina) was used for base calling and bcl2fastq2 (version 2.19 and 2.20) for converting BCL to fastq format, coupled with adaptor trimming. A pseudoalignment to a kallisto index was created from transcriptomes (Human: GRCh38; Mouse: GRCm38) using kallisto (0.44.0). To explore similarities and dissimilarities between samples, count data were normalized using the variance stabilizing transformation (VST) function from the DESeq2 package 57.
Gene set enrichment analysis (GSEA), heatmap and volcano plots
Enrichment analysis in Extended Data Figure 3h was determined using Metascape 58. GSEA heatmaps and volcano plots were performed on the normalized raw data after VST from bulk RNAseq or raw data from microarray. GSEA was performed using the GSEA_4.0.1 software (https://www.broadinstitute.org/gsea). Data were compared using signal to noise (Signal2Noise) calculation with 5,000 or 10,000 permutations on the Hallmark collection from the Molecular Signatures Database (MSigDB) or by running the myHSC and cyHSC signatures using the ‘text entry’ option. Heatmap in Extended Data Figure 3f was performed using the ComplexHeatmap R/bioconductor package v2.4.2 (42) in conjunction with the circlize R package. Volcano plots were generated after DEseq2 analysis on the non-tumour or tumour liver tissues from YAPf/f and YAPΔHSC mice treated with DEN+CCl4 (GSE206409) using R using and the ggplot2 library. HSC and hepatocyte genes were selected with a log2Foldchange >0.5 compared to other cell populations using the scRNA-seq dataset RS025 (whole liver 8xCCl4) and the function ‘FindAllMarkers’.
TCGA database survival analysis
The impact of an MMP signature (MMP1, MMP2, MMP8, MMP9, MMP13 and MMP14) on the survival of patients with HCC was determined in the TCGA liver cancer data set (n=364 patients) using the Gepia2 tool with a median group cutoff.
Single-cell RNA-sequencing
All cell samples were filtered and counted on a Countess II automated cell counter (ThermoFisher) before being loaded on a 10x Chromium instrument (10x Genomics). Single-cell RNA-sequencing libraries were prepared using the Chromium Single Cell 3′v2 or 3’v3 Reagent Kit (10x Genomics) according to the manufacturer’s instructions. 12 cycles of cDNA amplification and 12 cycles of library amplification were performed, and samples were sequenced on an Illumina NovaSeq 6000 Sequencing System at the JP Sulzberger Columbia Genome Center. The 10x Genomics Cell Ranger pipeline was used to process the data (v2.1.1 and v5.0.1). BCL files were demultiplexed with 10x Cell Ranger’s mkfastq command, and both analysis and alignment were performed using Cell Ranger’s count command with Cell Ranger’s reference mm10.
Single sample scRNAseq analysis
The scRNA-seq filtered raw counts matrices were analysed in Seurat (v4.0.5)59 and R (v4.0.2). During QC filtering, cells with a minimum of 200 genes and genes expressed in a minimum of 3 cells were retained for further analysis. In addition, cells with outlier values of mitochondrial gene expression, number of genes detected (nFeature_RNA), or number of unique mRNAs (nCount_RNA), when compared to other cells in the dataset, were removed. QC filtered expression data were normalized using ‘NormalizeData’ function with ‘LogNormalize’ and 1e4 ‘scale.factor’ parameters. Then, 2,000 or 3,000 most variable genes in the dataset were identified using ‘FindVariableFeatures’ function with ‘vst’ method. Normalized expression data was scaled and centred using ‘ScaleData’ function on the variable genes and by regressing out ‘nCount_RNA’ and ‘percent.mt’. Principal component analysis (PCA) was performed by ‘RunPCA’ function on the scaled and centred expression data. Top informative PCs were selected using ElbowPlot function which showed the PCs ranked by the percentage of variance and thus, identifying the ‘elbow’ region. ‘FindNeighbours’, ‘FindClusters’ and ‘RunUMAP’ functions were used to identify neighbours of cells, cluster the cells and visualize cell clustering, respectively, using top PCs. The resulting Seurat object was used for subsequent analysis of all single samples or for integration of multiple samples into one single Seurat object. The viridis palatte from ggplot2 (v3.3.6) was used for visualizing gene expression.
Multi-sample scRNA-seq analysis
Integration of multiple samples was performed using Seurat’s reciprocal PCA (RPCA) integration method. After processing each single-sample Seurat object, 3000 repeatedly variable genes were identified using ‘SelectIntegrationFeatures’ function. ‘FindIntegrationAnchors’ function with ‘rpca’ parameter, allowed to identify cell-pairs across datasets (cross-dataset pairs) that share the same biological state (called as ‘anchors’). ‘IntegrateData’ function uses the cross-dataset cell-pair anchors to remove technical differences (batch-effect correction) and integrate the datasets into a single Seurat object. Next, the default assay is set to ‘integrated’ and, as described above, ‘ScaleData’, ‘RunPCA’, and ‘ElbowPlot’ functions were used to identify top PCs. ‘FindNeighbours’, ‘FindClusters’ and ‘RunUMAP’ functions, on top PCs, were used to cluster and visualize the cells in the datasets. The cells that visually group separately in UMAP but shared the same cluster name were manually assigned separate clusters using ‘CellSelector’ function.
Nucleus isolation for snRNAseq
Human liver for snRNA-seq was processed as described previously60. In brief, frozen liver tissue was minced with scissors in 1 ml TST buffer in the well of a 6-well plate for 10 min on ice. The homogenised solution was then passed through a 40 μm cell strainer. An additional 1 mL of TST buffer and 3 ml of 1xST buffer were used to wash the well and passed through a 40 μm cell strainer. The resulting 5 ml of nuclei suspension was centrifuged for 5 min at 500 g at 4°C. The supernatant was discarded, and the pellet resuspended in 1 ml of 1xST buffer. The nuclei suspension was then passed through a 35 μm filter.
Droplet-based sn RNAseq
Samples from GSE174748 were prepared as follow: single nuclei were processed through the Chromium Single Cell Platform using the Chromium Single Cell 3′ Library and Gel Bead Kit v3 (10X Genomics, PN-1000075) and the Chromium Single Cell B Chip Kit (10X Genomics, PN-1000074) as per the manufacturer’s protocol. In brief, nuclei were counted using a Bio-Rad TC20 then loaded onto the 10X chip for a recovery target of 10,000. The nuclei were partitioned into Gel Beads in Emulsion in the Chromium instrument, in which bar-coded reverse transcription of RNA occurred. This was followed by amplification, fragmentation and 5′ adaptor and sample index attachment. Libraries were sequenced on an Illumina HiSeq 4000. Single nuclei to prepare the adjacent and HCC human livers (in GSE212046) were processed from OCT-embedded tissues using the 10x Genomics 5 prime v2 kit loading 14,000 nuclei per sample. We aligned to a modified version of the GRCh38 reference genome (counting intronic reads as well as those aligned to exons), and estimated cell-containing partitions and associated UMIs, using Cell Ranger v3.1.0 from 10X Genomics.
snRNA-seq analysis
There were a total of 10 human snRNA-seq datasets consisting of n=2 normal livers from GSE185477 61, n=2 normal and NAFLD cirrhotic samples each from GSE174748 and n=2; adjacent cirrhotic and HCC liver tissues each from GSE174748, n=2 and adjacent cirrhotic and HCC liver tissues: GSE212046 n=2 each). Technical artifacts such as ambient background RNA and empty droplets in these datasets were removed using ‘remove-background’ function in CellBender (v0.2.0)62. The unfiltered raw count matrix files in h5 or mtx format were used as input to the CellBender. The ‘epochs’ parameter was 80 and ‘expected-cells’ and ‘total-droplets-included’ parameters were selected based on the ‘UMI curve’ for each dataset, as suggested in CellBender. The output ‘raw_feature_bc_matrix_filtered.h5’ from CellBender for each sample were individually processed in Seurat according to the procedure in section ‘Single sample scRNA-seq analysis’. ‘FindVariableFeatures’ function was run with 3000 most variable genes parameter. Next, all the individual samples were batch-corrected and integrated as described above in section ‘Multi-sample scRNA-seq analysis’.
Cell type identification
For each dataset (single sample or multi-sample Seurat objects), the differentially expressed genes (DEGs) in every cell cluster were identified using ‘FindAllMarkers’ function in Seurat. In the human dataset, the ‘max.cells.per.ident’ parameter was set to 1000. The main cell types were identified either manually by checking if well-known markers-genes were present in the DEGs of a cluster or by checking the top DEGs in PanglaoDB 63. RColorBrewer v1.1-3 and ggsci v2.9 were used for assigning cell type colors in UMAP.
Trajectory analysis
The trajectory analysis of cyHSC to myHSC cell-state transition was performed with Monocle3 (v1.0.0)64 and Seurat. The Seurat object from each single sample was converted to a Monocle3 object using ‘as.cell_data_set’ function from SeuratWrappers (v0.3.0). Then, ‘cluster_cells’ function from Monocle3 was used to cluster and partition (higher-order superclusters) the cells. Next, the ‘learn_graph’ function was used to infer the cell transition pathways (principal graph). Then, the ‘order_cell’ function was used to interactively assign a root node or initial state to calculate the pseudotime of cell transition. The least-activated state of the HSC population was selected as the root node. Finally, ‘as.Seurat’ function from SeuratWrappers was used to convert the Monocle3 object with the identified cell transition pathways and pseudotime information back to a Seurat object.
Identification of cyHSC and myHSC signatures and cell populations
The differentially expressed genes between the most and least activated HSC clusters in samples RS039 (CCl4-induced profound fibrosis), RS042 (TAZ+FPC-NASH diet), RS043 (HF-CDAA-diet) and RS024 (Mdr2KO) were used to compute a common signature that can reliably determine the HSC activation status. First, cycling cells, portal fibroblast-like HSC, vascular smooth muscle-like HSC and non-HSC populations were excluded (Supplementary Information 3) and HSC were selected using the ‘subset’ function. Standard procedures for reclustering were performed, namely ‘ScaleData’, ‘FindVariableFeatures’, ‘RunPCA’, ‘FindNeighbours’, ‘FindClusters’ and ‘RunUMAP’ as described above. Next, we identified the most and least activated HSC clusters by computing a signature score comprised of activation markers genes Acta2, Lox, Col1a1 and Timp1 in all the cells using ‘AddModuleScore’ function and then averaging the score across each cell in a cluster. Next, clusters with the most and least average activation score were identified and DEGs both were computed using ‘FindMarker’ function in each sample individually. The highly expressed genes (DEG average log2FC >= 0.5) present in most-activated clusters in all the four samples were selected as the myHSC signature. The final average log2FC, percent of cells expressing the gene inside cluster (pct.1) and percent of cells expressing the gene outside cluster (pct.2) values were averaged across the four datasets. The adjusted p-values were converted to Z-scores, Stouffer-integrated across the four datasets and reconverted to obtain the final adjusted p-values. Similar steps were performed in least-activated clusters in the four samples to obtain cyHSC signature.
To identify the myHSC and cyHSC populations in mouse datasets, the myHSC score for every cell in each dataset was obtained by using ‘AddModuleScore’ function with the myHSC signature. myHSC score of each cluster in a dataset was obtained by averaging the myHSC score across all the cells in that cluster. The cyHSC score for each cluster was computed similarly. Clusters with a higher myHSC than cyHSC score were assigned as myHSC cell population and vice-versa for cyHSC. In humans, 1-to-1 homologs of mice genes in myHSC and cyHSC signatures were first obtained using mouse-human homolog genes from The Jackson Laboratory (http://www.informatics.jax.org/homology.shtml) downloaded on 2020-02-01. Then, identical steps to the mice datasets, were performed to identify the cyHSC and myHSC populations in human. The ggscatter function from ggpubr (v.0.4.0) was used for generating cy-myHSC correlation plots.
Impaired cyHSC-myHSC balance analysis in human livers
The impaired balance between HSC populations was measured using the signature described in Supplementary Table 5. Transcriptome profiles of NASH- and HCV-affected liver tissues were obtained from NCBI GEO (GSE 10140 and GSE49541). An impaired balance between myHSC and cyHSC was determined using Nearest Template Prediction (NTP) algorithm as previously described 65 and defined based on p <0.05. Using cohort GSE15654 of HCV infected patients with liver cirrhosis, the incidence of HCC was compared between patients with low and high cyHSC-myHSC dysbalance.
Pathway analysis
Pathway analysis using gene ontology (GO) enrichment was performed using enrichR66 (v 3.0) by selecting ‘GO_Biological_Process’ database and other default parameters. For the pathway analysis of the selected hepatocyte clusters (in Supplementary Table 1), first a Seurat object of only hepatocyte cells were created using ‘subset’ function. Then, the DEGs of the hepatocyte cell clusters were identified using ‘FindAllMarkers’ function. For each cluster, the DEGs with an average log2FC > 0.5 and up to a maximum of 100 genes were used as input gene list to enrichR. For the pathway analysis in Supplementary Table 4, the gene lists with all the genes from myHSC or cyHSC signatures were used as input to the enrichR for myHSC or cyHSC pathways, respectively. We manually curated relevant and representative pathways that showed significant adjusted p-value.
Cell-cell interaction analysis
Cell-cell interaction analysis was performed using CellPhoneDB67 (v2.0.0), running on Python, which is a curated database of ligand-receptor interactions along with subunit composition and monomeric or heteromeric complex details. This ligand-receptor database is integrated in a statistical framework to infer statistically significant ligand-receptor interactions between the different cell types in scRNA-seq data. After analysing and identifying different cell types, cell-cell interaction analysis was performed with a local implementation of CellPhoneDB v2.0.0, and following recommended procedures for data preparation 67,68. Briefly, QC filtered scRNA-seq gene expression raw counts for each cell were normalized to 1e4, mouse genes were replaced by 1-to-1 homologous human genes (as described above), and the metadata file was created from the cell-type annotations. CellPhoneDB analysis was performed with ‘cellphonedb method statistical analysis’ command with 0.01 ‘threshold’, a CellPhoneDB database with additional liver specific interactions 23,24 and other default parameters.
Cell-cell interaction analysis for the main cell types was performed using the CCl4-treated whole-liver dataset (Fig. 2d) and the human dataset with n=4 fibrotic samples (Extended Data Fig. 4e), for which we obtained cell type annotations as described above. Cell-cell interaction analysis in Fig. 3d used cyHSC and myHSC subtypes identified in HSC cell population, as mentioned previously, along with other cell-type annotations. R package pheatmap (v1.0.12) was used to generate cell-cell interaction heatmaps
Quantification and Statistical Analysis
Statistical significance was determined using GraphPad Prism v.9.0 or R (v 4.0.2). After assessing the normal distribution of the data using D’Agostino and Pearson omnibus normality tests and or Shapiro-Wilk normality test, p-values were calculated and all statistical tests used are described in the ‘Statistical tests’ section. Pearson’s correlation coefficient was used to measure the statistic relationship between myHSC and cyHSC signature using R and the library ggpubr. Survival curves were represented using the Kaplan-Meier method and compared with log-rank statistics. The median HGF expression levels on the total number of analyzed samples were used to determine to low- and high-expression groups. Exact values used for each graph are represented in the Source data file.
Extended Data
Extended Data Figure 1 |. Analysis of hepatic stellate cell senescence during liver fibrosis and HCC development.
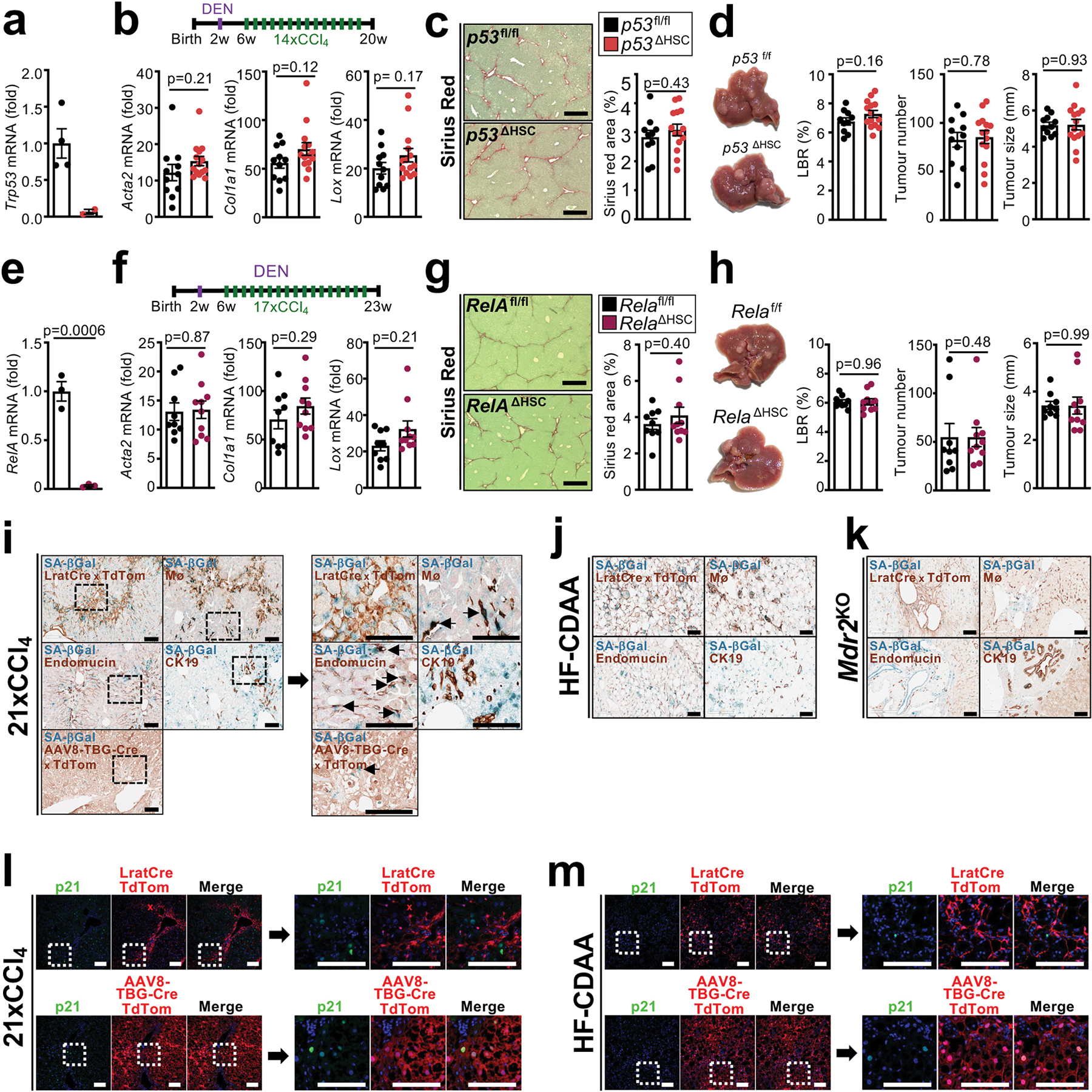
a, qPCR showing Trp53 mRNA in FACS-sorted HSC isolated from p53f/f (n=2 mice) and p53ΔHSC mice (n=4 mice). b-d, HCC was induced in p53f/f (n=11 mice) and p53ΔHSC (n=14 mice) by injection of DEN (i.p. 25 mg/kg at 2 weeks old) followed by 14 injections of CCl4 (i.p. 0.5 μL/g, 1x/week) starting one month after DEN. HSC activation and fibrogenesis were assessed in by qPCR for fibrogenic genes Acta2, Col1a1 and Lox in the liver (b). Fibrosis was evaluated by Sirius Red staining (c). HCC is shown by representative pictures and the tumour burden measured by liver/body weight ratio (LBR), tumour number and tumour size (d). e, qPCR showing Rela mRNA in FACS-sorted HSC from Relaf/f and RelaΔHSC mice (n=3 mice/group). f-h, HCC was induced in Relaf/f (n=9 mice) and RelaΔHSC (n=10 mice) mice by injection of DEN (i.p. 25 mg/kg at 2 weeks old) followed by 17 injections of CCl4 (i.p. 0.5 μL/g, 1x/week). HSC activation and fibrogenesis was assessed by qPCR for the fibrogenic genes Acta2, Col1a1 and Lox in the liver (f). Fibrosis was evaluated by Sirius Red staining (g). HCC is shown by representative pictures and tumour burden measured by LBR, tumour number and tumour size (h). i-k, representative images showing senescence in specific cell types by senescence associated beta-galactosidase (SA-bGal) staining and co-staining for markers or lineage tracers of HSC (LratCre × TdTom), macrophages (anti-macrophage antibody), endothelial cells (endomucin antibody), cholangiocytes (CK19 antibody) and hepatocytes (AAV8-TGB-Cre × TdTom) in the CCl4 (n=3 mice) (i), HF-CDAA diet (n=1 mouse) (j) and Mdr2KO (n=1 mouse) (k) mouse models of fibrosis. l-m, representative images showing senescence in specific cellular compartments by p21 IHC in combination with lineage markers for HSC (LratCre × TdTom) and hepatocytes (AAV8-TGB-Cre × TdTom) in the CCl4 (l) and HF-CDAA diet (m) mouse models of fibrosis (from n=1 mouse per model). Data are shown as mean ± SEM, each data point represents one individual. Scale bars: 400 μm (c,g) and 100 μm (i-m). LBR: liver/body weight ratio. Data in b, c, d, e, g, Acta2 mRNA and Col1a1 mRNA in f, and LBR and tumour size in h were analysed by two-tailed Student’s t-test. Lox mRNA in d and tumour number in h were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data.
Extended Data Figure 2 |. Genetic strategies to manipulate HSC during hepatocarcinogenesis.

a, Lhx2 mRNA in isolated HSC (n=5 mice), Kupffer cells (KC), endothelial cells (LSEC) and hepatocytes (n=3 mice each). b, Lhx2 mRNA in scRNAseq from normal mouse liver (n=1 mouse). c, qPCR showing deletion of Lhx2 by LratCre in whole liver: Lhx2f/f: n=8 mice, Lhx2ΔHSC: n=6 mice or FACS-sorted HSC (n=2 mice/group). d-f, deletion of Lhx2, achieved via Mx1Cre and poly I:C injection, increased liver fibrosis, shown by Sirius Red staining: Lhx2f/f: n=11 mice, Lhx2del: n=9 mice in non-tumour areas (d), HSC activation measured by qPCR: Lhx2f/f: n=11 mice, Lhx2del: n=8 mice (e); and promoted HCC development Lhx2f/f: n=11 mice, Lhx2del: n=9 mice (f) compared to Lhx2f/f littermates. g, LratCre-mediated Yap1 deletion (YapΔHSC) was confirmed in FACS-sorted HSC by qPCR: Yapf/f: n=2 mice , YapΔHSC: n=3 mice, and western blot (n=2 mice/group). h, YapΔHSC mice showed reduced fibrosis, evaluated by Sirius Red (n=15 mice/group) and HSC markers, measured by qPCR (Yapf/f: n=14 mice , Yap ΔHSC: n=15 mice), in non-tumour liver tissue from mice treated with DEN+CCl4. i, HSC depletion via LratCre-induced DTR significantly reduced Lrat mRNA in the DEN+CCl4 model: DTR neg: n=15 mice, DTR pos: n=16 mice. j-k, αSMA staining (n=13 mice/group) and qPCR for Acta2 and Col1a1 (n=12 mice/group) showed depletion of αSMA+ cells in non-tumour areas in αSMA-TKpos mice compared to αSMA-TKneg littermates after ganciclovir (GCV) injections in DEN+CCl4-induced HCC (j) and αSMA-TKpos mice developed fewer tumours (n=13 mice/group) (k). l, Liver fibrosis and deletion of Pdgfrb were determined by Sirius red staining and qPCR for Col1a1 and Pdgfrb in 4 month-old Mdr2KO PdgfrbΔHSC (n=13 mice) and Mdr2KO Pdgfrbfl/fl (n=13 mice) female mice. m, Tumour development was determined in 15 month-old Mdr2KO PdgfrbΔHSC (n=8 mice) and Mdr2KO Pdgfrbfl/fl (n=6 mice) female mice as described above. n, HCC development in mice overexpressing TAZS89A in hepatocytes receiving a NASH-FPC (n=13 mice) or chow diet (n=11 mice). o-q, DTRpos mice displayed efficient HSC depletion in the TAZ+FPC NASH-HCC model compared to DTRneg mice: DTRneg: n=10 mice, DTRpos: n=14 mice (o) as well as reduced tumour development: DTRneg: n=10 mice, DTRpos: n=14 mice (p), but no reduction of cholesterol and triglycerides measurement in non-tumour liver tissue (untreated: n=3 mice, TAZ+FPC in DTRneg: n=6 mice, TAZ+FPC in DTRpos: n=7 mice) (q). r-s, LratCre-positive DTRpos or DTRneg mice were subjected to DEN+HF-CDAA-induced spontaneous hepatocarcinogenesis, revealing efficient HSC depletion (n=4 mice/group) (r) as well as reduced tumour development in DTRpos mice (n=8 mice) compared to DTRneg mice (n=6 mice) (s). t-u, αSMA-TKpos or αSMA-TKneg mice were subjected to NICD+HF-CDAA-induced hepatocarcinogenesis, revealing efficient fibroblast depletion: αSMA-TKpos (n=8 mice) vs αSMA-TKneg mice (n=7 mice) (t) as well as reduced tumour development in αSMA-TKpos (n=8 mice) vs αSMA-TKneg mice (n=9 mice) (u). Data are shown as mean ± SEM, each data point represents one individual, all scale bars: 200 μm. GCV: Ganciclovir. Statistics: data in d, all data in e besides Lox mRNA, Sirius Red in h, i, tumour number and tumour size in k, data in l besides Col1a1 mRNA, o, p, r, s, t and data in u besides tumour number were analysed by two-tailed Student’s t-test. The following data: Lox mRNA in e, f, all data in h besides Sirius Red, j, LBR in k, Col1a1 mRNA in l, m, n, and tumour number in u were analysed by two-tailed Mann-Whitney test. Data in q were analysed by one-way ANOVA (p<0.001) followed by Tukey’s multiple comparison. Raw data are given in Source Data and uncropped western blots gels in Supplementary Information.
Extended Data Figure 3 |. Hepatic stellate cell accumulation occurs predominantly in the PME and affect genes and pathways relevant for tumourigenesis and fibrosis.
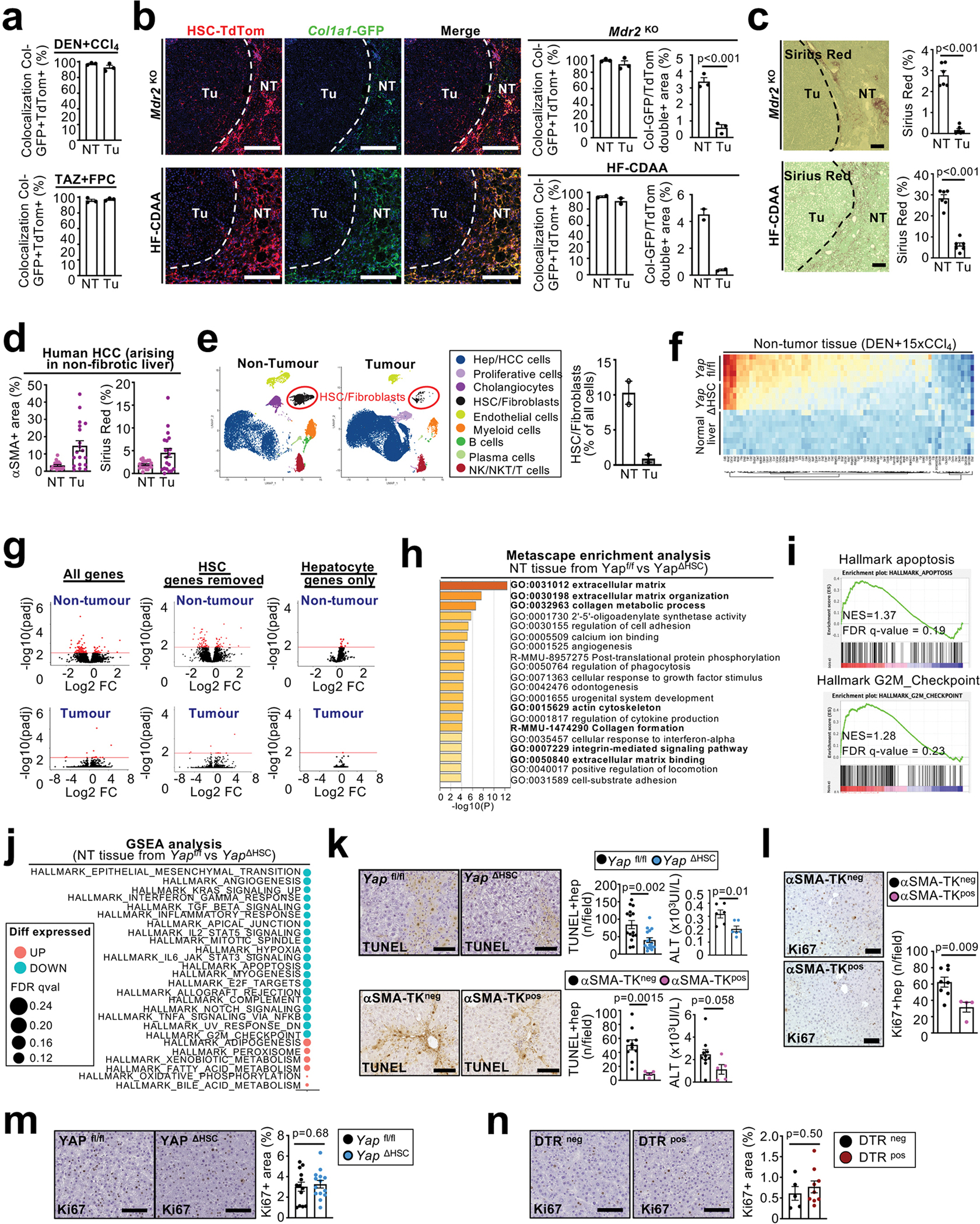
a, co-localization of Col1a1-GFP+ and LratCre-induced TdTom was quantified in non-tumour (NT) and tumour (Tu) areas of DEN+CCl4- and TAZ-FPC-induced HCC (n=3 mice/HCC model – data related to Figure 2a). b, LratCre-TdTom+ HSC and Col1a1-GFP+ fibroblasts were visualized, and co-localization of Col1a1-GFP+ and LratCre-induced TdTom and the Col1a1-GFP/TdTom double-positive area were quantified in 15 month-old Mdr2KO-induced (n=3 mice) and HF-CDAA-diet-induced (n=2 mice) HCC. c, Fibrosis was visualized and quantified by Sirius Red in 15 month-old Mdr2KO-induced and HF-CDAA-diet-induced HCC (n=6 mice/group/HCC model). d, αSMA and Sirius Red quantification of paired non-tumour and human HCC developing in non-fibrotic livers (n=20 cases - data related to Figure 2b). e. UMAP visualization of cell populations from snRNA-seq of matching human non-tumour cirrhotic liver (n=2) and tumour (n=2) liver tissue pairs as well as the proportion of HSC/fibroblasts in both compartments. f-j, Bulk RNAseq of liver tissue from DEN+CCl4-treated Yapf/f (n=5), YapΔHSC mice (n=5) and normal liver (n=8 mice). The heatmap displays up- and downregulated differentially expressed genes (DEG) in non-tumour tissue based on DESeq2 analysis from bulk RNAseq (compared to the normal liver, adjusted p-value <0.1 and log2FC>0.5 or < −0.5) (f). Comparison of genes expression in Yapfl/fl and YapΔHSC tumour and non-tumour areas in DEN+CCl4-induced HCC (n=5 mice/group) data are displayed as volcano plot before and after removal of HSC genes, identified by scRNA-seq analysis, or filtering on hepatocyte genes, identified by scRNA-seq analysis (n=5 mice/group) (g). Metascape enrichment analysis of down-regulated DEG genes in non-tumour tissues of YapΔHSC compared to Yapf/f, all the pathway related to fibrosis and HSC-activation are marked in bold (n=5 mice/group) (h). Gene set enrichment of RNA-seq data revealed apoptosis and G2/M checkpoint as enriched in Yapfl/fl vs YapΔHSC non-tumour tissue (i). Displayed is the gene set enrichment analysis of the collection “Hallmark gene set” from the MSigDB with a FDRqval <0.25 in non-tumour tissues of Yap ΔHSC compared to Yap f/f (n=5 mice/group) (j). k-l, hepatocyte death (k) and proliferation (l) were determined by TUNEL assay: YAP f/f: n=15 mice, YAPΔHSC: n=16 mice, αSMA-TKneg, n=10 mice, αSMA-TKpos: n=5 mice and serum ALT measurement: Yap f/f: n=6 mice, YapΔHSC: n=6 mice, αSMA-Tk neg, n=10 mice, αSMA-TKpos: n=5 mice (k) and Ki67 IHC: αSMA-TKneg, n=8 mice, αSMA-TKpos: n=4 mice (i) in non-tumoural tissues. m-n, proliferation was determined by Ki67 IHC in the tumor compartment of YapΔHSC (n=14) and Yapfl/fl (n=13) (m) as well as DTRpos (n=9) and DTRneg (n=5) mice (n). Data are shown as mean ± SEM, each data point represents one individual (e-g), all scale bars: 100 mm NT: Non-tumour, Tu: Tumour. Data are shown as mean ± SEM, each data point (a-d) as well as the HSC-fibroblast percentage quantification (e) represents one individual, in d each dot represent one gen, all scale bars: 100 mm. Statistics: all data displayed in graph dotplots besides ALT measured in αSMA-TKneg and αSMA-TKpos in k were analysed by two-tailed Student’s t-test. ALT in αSMA-TKneg and αSMA-TKpos in k were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data.
Extended Data Figure 4 |. Determination of the role of HSC/fibroblasts in hepatocarcinogenesis in established tumors and in a non-fibrotic HCC model and interaction with of regulation of hepatocyte proliferation, immune and endothelial cell compartments.

a-b, Ganciclovir injections into DEN+CCl4-treated αSMA-TKpos and αSMA-TKneg littermates at late time points, when large tumours were established and when CCl4 injections had ceased for 1.5 weeks, resulted in a strong reduction of αSMA+ cells in tumour but not in non-tumour areas (IHC: αSMA-TKneg, n=12 mice, αSMA-Tkpos: n=10 mice; Acta2 RNA: NT: αSMA-Tkneg, n=13 mice, αSMA-TKpos: n=9 mice; Tu: αSMA-TKneg, n=13 mice, αSMA-TKpos: n=11 mice) (a), and did not affect tumour progression (αSMA-TKneg, n=13 mice, αSMA-TKpos: n=11 mice) (b). c-d, HSC depletion via LratCre × DTR in a non-fibrotic HCC model, induced by hydrodynamic tail vein injection of sleeping beauty-mediated expression MET + CTNNB1-Myc-tag, was highly efficient, as determined by TdTom fluorescence (c) but did not affect liver/body weight ratio, tumour size, tumour number or tumour area assessed by Myc-tag staining (DTRneg: n=11 mice, DTRpos: n=7 mice) (d). e-f analysis of cell-cell interactions by CellPhoneDB in snRNA-seq data from cirrhotic liver patients (n=4 cases) revealed HSC as a major cell type interacting with hepatocytes (e) or between different hepatocytes clusters and liver cells populations in the mouse fibrotic liver induced by 8xCCl4 (n=3 mice) (f) as well as UMAP visualization of the proliferation marker Mki67 (n=3 mice) (f). g-h, FACS analysis of total CD45+ leukocytes (g), and lymphocytes and myeloid cells (h) in the PME and TME after αSMA+ cell depletion during HCC development induced by DEN+17xCCl4 (NT: n=8 mice per group, Tu: αSMA-TKneg, n=4 mice, αSMA-TKpos: n=5 mice - related to experiments in Extended Data Fig. 2 j–k) shows increased neutrophil and Ly6Chigh macrophage infiltration into non-tumour areas in αSMA-TK pos mice. i-j, CD45 IHC staining of the non-tumour and tumour tissue in DEN+CCl4 treated Yapfl/fl (n=10 mice) and YapΔHSC mice (n=12 mice) (i), and in HSC-depleted DTRpos mice compared to DTRneg mice (NT: DTR neg: n=10 mice, DTRpos: n=12 mice; Tu: DTR neg: n=9 mice, DT pos: n=11 mice) during HCC development induced by TAZ+FPC (j). k-l, endothelial cell analysis after HSC depletion induced by diphtheria toxin in DTRpos and DTRneg mice during HCC development induced by DEN+CCl4 or TAZ+FPC diet evaluated by endomucin staining (k) and qPCR for the endothelial cell markers Pecam1 (encoding for CD31) and Kdr (DEN+CCl4: NT: DTR neg: n=15 mice, DTR pos: n=14 mice; Tu: 16 mice per group; Kdr mRNA: NT: 14 mice per group; Tu: 16 mice per group; Pecam and Kdr mRNA: TAZ+FPC: NT: DTRneg: n=10 mice, DTRpos: n=14 mice; Tu: DTRneg: n=10 mice, DTRpos: n=13 mice) (l). Data are shown as mean ± SEM, each data point represents one individual (a-d and g-l), scale bars: 100 μm (a-j) or 200 μm (k). GCV: ganciclovir; LBR: liver/body weight ratio; NK: Natural Killer, KC: Kupffer Cell, DC: Dendritic cell, NT: Non-tumour, Tu: Tumour. Statistics: all data in a besides Acta2 in Tu, data in b besides LBR, d, g, all data in h besides B-cells, CD4+, CD8+, KC and Ly6ClowLy6Gneg in NT, CD45 in tumour in i, all data in l besides Kdr mRNA in DEN+CCl4 and TAZ FPC in tumour were analysed by two-tailed Student’s t-test. Data in a: Acta2 mRNA in Tu, LBR in b, data in c, B-cells, CD4+, CD8+, KC and Ly6ClowLy6Gneg in NT, CD45 in tumour in h, CD45 in NT in I, data in j and Kdr mRNA in tumour in l were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data and gating strategy for h in Supplementary Information 2.
Extended Data Figure 5|. Characterization of genetic mutations, inflammation, immune cell and tumor markers in mice with genetic HSC depletion, activation of inhibition.

a, Mutations detected by PCR and sequencing for Hras Q61K/R/L, Braf V584E and Egfr F254I in Lhx2 fl/fl (1 tumour/n; n=19 mice) vs Lhx2ΔHSC (1 tumour/n; n=7 mice) and Lhx2 del (1 tumour/n; n=19 mice) mice treatead with DEN; Yapfl/fl (2 tumours/n; n=14 mice) vs YapΔHSC (2 tumours/n; n=15 mice) mice and DTR neg (1 tumour/n; n=14 mice) vs DTR pos (1 tumour/n; n=15 mice) mice treated with DEN+CCl4. b, Tumour grading represents average of 3 tumours per mouse in Lhx2fl/fl (n=8 mice) and Lhx2ΔHSC (n=7 mice) mice subjected to the DEN model; in Yapfl/fl (n=14 mice) and YapΔHSC (n=16 mice) mice subjected to the DEN+CCl4 model and DTRneg (n=9 mice) and DTRpos (n=14 mice) mice subjected to the TAZ+FPC HCC model. c-e, Characterization of inflammation, immune cells and tumors severity by qPCR for T cell markers, inflammatory genes and tumor-relevant marker genes in tumors from Lhx2ΔHSC mice with HSC activation in comparison to mice to Lhx2 f/f littermates (n=7 mice per group) (c); YapΔHSC mice with HSC inhibition compared to Yapfl/fl mice (Cd3e, Cd4, Cd8, Ccl2, Ccl5, Tnfa, Ifng, Krt19, and Epcam mRNA: n=15 mice per group; Afp, Gpc3, Prom1, and Sox9 mRNA: YAP f/f: n=15 mice, YAPΔHSC: n=14 mice). (d); and DTRpos (n=16 mice) mice with HSC depletion compared to DTRneg (n=16 mice) mice (e). Data are shown as mean ± SEM, each data point represents one individual. Data are shown as mean ± SEM, each data point represents one individual. Statistics: a: Mutation frequency were analysed by Chi-square test. b: tumour grading data were analysed by two-tailed Student’s t-test. All data in c besides Afp and Sox9 mRNA, Afp, Gpc3 and Prom1 mRNA in d, and Prom1 and Sox9 mRNA in e were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data.
Extended Data Figure 6|. Analysis of the myHSC and cyHSC in mouse and human fibrotic livers.
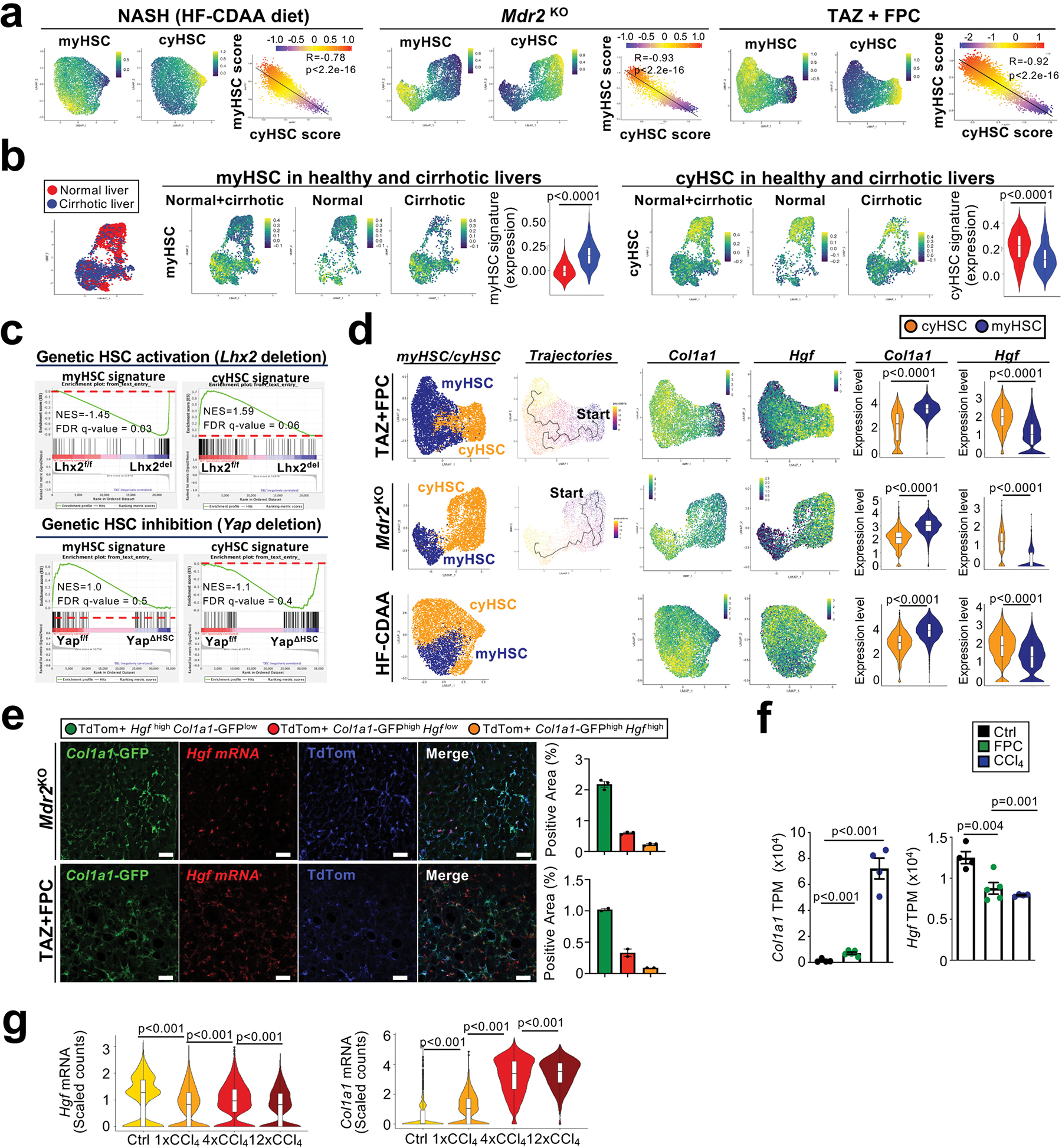
a, UMAPs of myHSC and cyHSC by scRNAseq, each visualized specific signatures as well as the correlation of cyHSC and myHSC signature score in HSC isolated from mice fed with HF-CDAA NASH diet for 12 weeks (n=1 mouse), from a 3 month old Mdr2KO mice (n=1 mouse) or a mouse treated with TAZ+FPC diet (n=1 mouse). b, visualization and quantification of myHSC and cyHSC populations in HSC from healthy (n=4 individuals) and cirrhotic (n=4 individuals) human livers by snRNAseq. c, analysis of microarray data from isolated HSC shows that genetic HSC activation via Mx1Cre-mediated Lhx2 deletion (n=4 Lhx2fl/fl, n=4 Lhx2del) resulted in enriched myHSC signature and decreased cyHSC signature; and that genetic inhibition induced by LratCre-mediated Yap1 deletion (n=5 Yapfl/fl, n=4 YapΔHSC) exerted the opposite effect with enriched cyHSC and decreased myHSC signature in isolated HSC after treatment with 6xCCl4. d, pseudotime analysis (of HSC from TAZ+FPC and Mdr2KO) as well as cyHSC, myHSC, Col1a1 and Hgf mRNA expression in HSC using the same dataset as in a. f, In situ analysis of Hgf mRNA, visualized by RNAscope, Col1a1-GFP and LratCre-induced TdTom and subsequent quantification shows separate populations of Col1a1-GFP high Hgf low myHSC, and Col1a1-GFPlow Hgfhigh cyHSC in Mdr2 KO (n=3 mice) and TAZ+FPC-treated livers (n=1 mouse). f-g, analysis of Hgf and Col1a1 expression by bulk RNA-seq (Ctrl and CCl4: n=4 mice, FPC: n=5 mice) (f) or scRNA-seq (n=1 per condition) (g) of FACS-sorted HSC from control mice (Ctrl), or from CCl4-treated or TAZ-FPC-treated mice. Dot plot bar graphsare shown as mean ± SEM. In the violin plots, box plots represent the interquartile range (IQR), Q1, median and Q3, whiskers as minimum (Q1−1.5×IQR) and maximum (Q3+1.5×IQR), and outlier data as individual dots. Each data point represents one cell (a,b,d) individual (f and Mdr2KO model in e) or one field of the same liver (TAZ+FPC model in e). Scale bar 50 μm. Statistics: a: P-values, coefficient of determination (R2) and statistical significance (P value) were determined using Pearson’s. Data in d were analysed by two-tailed Mann-Whitney test. Data in b, d and g were analysed by two-tailed Mann-Whitney test. Data in f were analysed by two-tailed Student’s t-test. Raw data are given in Source Data.
Extended Data Figure 7 |. Determination of the role of HSC in hepatocarcinogenesis at different time points.

a, The long-term effects of LratCre × DTR-mediated HSC depletion in the first half of DEN+CCl4-induced hepatocarcinogenesis was tested by sacrificing mice 1.5 days (n=3 mice per group), 28 days (DTRneg: n=5 mice, DTRpos: n=4 mice mice per group) and 42 days (DTRneg: n=6 mice, DTRpos: n=4 mice) after the last DT injection and determining LratCre-induced TdTom expression. b, LratCrepos DTRpos (n=9 mice) mice with HSC depletion in the first half of DEN+CCl4-induced hepatocarcinogenesis retained significant HSC depletion at time of sacrifice, as determined by qPCR for HSC markers Lrat and Des, but did not show significant changes in liver/body weight ratio (LBR), tumour number and tumour size compared to LratCrepos DTRneg littermates. c-d, Effects of poly I:C-induced Lhx2 deletion at early time points (c, n=9 mice per group) and late time points (d, (Lhx2f/f: n=10 mice, Lhx2del: n=12 mice) of DEN-induced HCC, as shown by representative images, liver/body ratio (LBR), tumour number and tumour size. Data are shown as mean ± SEM, each data point represents one individual, scale bars: 200 μm. Statistics: Data in a: one way ANOVA (p<0.001) followed by Bonferroni’s multiple comparison (comparison between samples sac at the same time point). Data in b, LBR in c, tumour number and tumour size in d, were analysed by two-tailed Student’s t-test. Tumour number and tumour size in c and LBR in d: were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data.
Extended Data Figure 8|. Collagen type 1 deletion reduces liver stiffness and HCC development in DEN+CCl4-, NASH diet- and Mdr2KO-induced HCC.

a-b, UMAP visualization showing that Col1a1 are mainly expressed in the HSC population in the 8xCCl4 injured mouse liver by scRNAseq (n=3 mice) (a) and in the HSC/fibroblast cluster in the human normal (n=4 individuals), cirrhotic (n=4 individuals) and HCC (n=2 individuals) livers by snRNAseq (b). c, Col1a1 mRNA expression, measured by qPCR, in untreated liver (n=5 mice, left panel) and the non-tumour tissue (n=8 mice) from DEN+14xCCl4 treated mice; COL1A1 mRNA expression in normal (n=84 individuals) and adjacent non-tumour tissue from patients with F0-F1 (n=143 individuals), F2-F3 (n=59 individuals) and F4 (n=76 individuals) fibrosis (right panel). d-e, Mx1Crepos Col1a1f/f (Col1a1del) showed significant reduction of fibrosis in non-tumour tissue, determined by Sirius Red staining and Col1a1 mRNA, compared to Col1a1f/f littermates in a profound fibrosis DEN+44xCCl4 model (Col1a1 f/f: n=13 mice, Col1a1 del: n=14 mice) (d), and reduction of liver stiffness, determined by rheometry (control: n=1 mouse; DEN+19xCCl4: n=4 mice/group) (e). f, Sirius red staining shows a strong increase of fibrosis, in the non-tumour tissue, induced by the profound fibrosis DEN+19xCCl4 regimen (0.5 to 1.5 μl of CCl4 per gram 2–3 injections/week; n=5 mice) compared to a moderate fibrosis model induced by DEN+10xCCl4 (0.5 μl of CCl4 per gram, 1 injection/week; n=5 mice; untreated controls n=3 mice). g, Col1a1del mice treated with the moderate fibrosis DEN+15xCCl4 (0.5 μl of CCl4 per gram, 1injection/week as in f) reduced liver fibrosis (Col1a1f/f: n=10 mice, Col1a1 del: n=14 mice) and Col1a1 mRNA (Col1a1f/f: n=11 mice, Col1a1del: n=8 mice) (g), but did not alter HCC development in comparison to Col1a1 fl/fl mice (Col1a1f/f: n=17 mice, Col1a1del: n=14 mice) (h) or i, liver stiffness assessed by rheometry in the non-tumour tissue from mice treated with DEN+10xCCl4 (one injection per week as in f-h, n=3 mice for untreated and n=5 mice/group for DEN+CCl4). j-k, Mx1Cre-mediated Col1a1 deletion in Mdr2KO female mice (Mdr2KO Col1a1 del, n=11 mice and n=7 mice for Col1a1 mRNA) efficiently reduced liver fibrosis (j) and HCC development (k), compared to Mx1Creneg Mdr2KO littermates (Mdr2KO Col1a1fl/fl, n=18 and n=7 mice for Col1a1 mRNA). l, Liver stiffness assessed by rheometry was reduced in 22 week old Mdr2 KO Col1a1del male mice (n=2 mice) compared to their. Col1a1fl/fl Mdr2KO (n=3 mice) littermates, livers from 8 week old untreated mice were used as control (n=3). m-o, HSC-selective ablation of Col1a1 in LratCrepos Col1a1f/f (Col1a1ΔHSC) efficiently reduced liver fibrosis (Sirius Red: Col1a1f/f: n=16 mice, Col1a1ΔHSC: n=14 mice; Col1a1 mRNA: Col1a1f/f: n=15 mice, Col1a1ΔHSC: n=13 mice) (m), HCC development (Col1a1f/f: n=16 mice, Col1a1ΔHSC: n=14 mice), (n) as well as hepatocyte proliferation in the non-tumour liver, as determined by Ki67 IHC and quantification of proliferating hepatocytes (Col1a1f/f: n=13 mice, Col1a1ΔHSC: n=14 mice; Ki67+ tumour cells: n=9 mice) (o), compared to Lratneg littermate controls (Col1a1fl/fl) in the DEN + CCl4 profound fibrosis model (as in d). p-r, Col1a1ΔHSC mice displayed strong reductions of liver fibrosis (p), HCC development (q), but not in liver triglycerides and cholesterol content (r) compared to Col1a1fl/fl control mice in a model of NASH-associated HCC induced by 8 months of HF-CDAA diet (Col1a1fl/fl: n=4 mice for Sirius red and triglycerides, n=11 mice for Col1a1 mRNA and HCC, Col1a1ΔHSC: n=7 mice for Sirius red and triglycerides, n=10 mice for Col1a1 mRNA and HCC). Data are shown as mean ± SEM, box plots represent the interquartile Range (IQR), Q1, median and Q3, each data point represents one individual (c-r), scale bars: 200 μm (d, f, g, j, m and p), 50 μm (o), Red arrows show the Ki67+ proliferating hepatocytes or tumour cells. Statistics: Col1a1 mRNA in mouse liver in c, data in d, f, g, LBR and tumour number in h, j, LBR and tumour size in k, m, LBR and tumour size in n, o, p, q and cholesterol in r were analysed by two-tailed Student’s t-test. Tumour size in h, tumour number in k and in n, and triglycerides in r were analysed by two-tailed Mann-Whitney test. # P<0.05, **,## P<0.001, ### P<0.001. Data in c and l: COL1A1 mRNA in human liver was analysed by one-way ANOVA (Kruskal-Wallis’s test) followed by Dunn’s multiple comparison test with normal liver group. Data in e: was analysed by two-way ANOVA followed by Tukey’s multiple comparison test. Raw data are given in Source Data.
Extended Data Figure 9|. Characterization of Col1a1 on hepatocyte death, HSC activation, inflammation and immune cell infiltration and YAP activation and effects of hepatocyte-specific Yap or Itgb1 deletion on hepatocarcinogenesis.

a-b, Col1a1f/f and Col1a1del mice were treated with the profound fibrosis DEN+44xCCl4 regimen. Hepatocyte death was determined by TUNEL staining (n=6 mice per group) in non-tumour tissue at the end of the DEN+44xCCl4 regimen 1 week after the last CCl4 injection, as well as by ALT measured 2 days after 20xCCl4 (n=6 per group) (a). HSC activation and inflammation were assessed by αSMA staining (n=8 per group) and qPCR for HSC activation and inflammatory markers in non-tumour tissue two days after 44xCCl4 (n=8 mice per group) (b). c-d, HSC activation and Il1b mRNA were assessed in Col1a1f/f (n=11 mice) and Col1a1ΔHSC (n=1 mice) mice fed eight months with HF-CDAA diet (c) and in 14 months-old Mdr2KO x Col1a1f/f and Mdr2KO x Col1a1del mice (n=7 mice per group) (d). e-g, Determination of CD45+ lymphocytes (e), myeloid subpopulations) (f) and lymphoid (g) subpopulations by FACS in tumour and non-tumour areas of LratCreneg Col1a1fl/fl mice (n=5 mice) and LratCrepos Col1a1fl/fl mice (Col1a1ΔHSC, n=5 mice) with the profound fibrosis DEN+44xCCl4 regimen. h, YAP staining in Col1a1fl/fl and Col1a1del mice treated with the profound fibrosis DEN+19xCCl4 showed YAP expression mostly in non-parenchymal cells rather than in hepatocytes. i, Yap was efficiently delete by injection of AAV8-TBG-Cre (YapΔHep, n=10 mice) compared to AAV-TGB-empty (Yapfl/fl, n=14 mice) but did not affect HCC development were in the DEN+CCl4 profound fibrosis HCC model determined by liver/body weight ratio (LBR), tumour number and tumour size. j, Itgb1fl/fl mice were injected with either AAV8-TGB-empty (Itgb1fl/fl, n=13) or AAV8-TBG-Cre (Itgb1ΔHep, n=11) and effects of hepatocyte-specific Itgb1 deletion on HCC development were determined in the DEN+CCl4 profound fibrosis HCC model by the liver/body weight ratio (LBR), tumour number and tumour size. Data are shown as mean ± SEM, each data point represents one individual. NT: non-tumour, Tum: tumour NK: Natural killer, Treg: T regulatory cells, GZB: Granzyme B, DC: Dendritic cells, KC: Kupffer cells, LBR: liver/body weight ratio. Scale bars: 100 μm (a and h), 200 μm (b). Statistics: TUNEL in a, data in b and d besides Il1b mRNA, Col1a2 mRNA in c, f, data in g besides B cells, NKT cells, CD8 and Tregs in non-tumour area, i, and data in j besides LBR were analysed by two-tailed Student’s t-test. ALT in a, Il1b mRNA in b and d, all data in c besides Col1a2, e, B cells, NKT cells, CD8 and Tregs in non-tumour area in g and LBR in j were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data and gating strategy for e-g in Supplementary Information 4.
Extended Data Figure 10 |. Regulation and role of DDR1 in the fibrotic liver and HCC development.

a, UMAPs of scRNA-seq data showing hepatocyte Ddr1 expression from normal (n=1 mouse) and 8xCCl4 fibrotic mouse livers (n=3 mice – top panel) and of snRNA-seq from patients showing DDR1 expression in hepatocytes HCC from healthy patients (n=4 individuals), non-tumor (n=2 individuals) and cirrhotic (n=2 individuals) or HCC tumour (n=2 individuals) liver tissues (bottom panel). b, Huh7, Hepa1–6, HepG2 HCC cells and mouse primary hepatocytes were treated with different cytokines for 24h, followed by immunoblotting for DDR1 and GAPDH (each n=1 well, n=1 experiment). c, Confirmation of Ddr1 deletion by pPCR in non-tumour liver and tumour liver from AAV8-TGB-empty treated Ddr1fl/fl and AAV8-TGB-Cre treated Ddr1ΔHep mice treated with DEN+44xCCl4 (in NT: Ddr1f/f: n=9 mice, Ddr1ΔHep n=8 mice; in Tu: Ddr1f/f: n=10 mice, Ddr1ΔHep n=8 mice - related to Fig. 5c). d, Fibrosis, αSMA+ myofibroblast accumulation and Ddr1 mRNA expression within tumours were quantified by Sirius Red staining, IHC and qPCR, respectively, in the ”moderate fibrosis” DEN + 14xCCl4 HCC model (αSMA and Sirius Red: n=10 mice; Ddr1 mRNA n=11 mice) and in the “profound fibrosis” DEN+44xCCl4 HCC model (αSMA and Sirius Red: n=9 mice; Ddr1 mRNA n=10 mice). e-f, Huh7 HCC cells (n=3 biological replicate; n=2 experiments) (e) and primary mouse hepatocyte (n=1 well; n=1 experiment) (f) were plated on either plastic or fibroblast ECM, samples were harvested at 24h or at the indicated times after plating, followed by western blot for pDDR1, DDR1, pAKT, AKT and b-actin or GAPDH. g, Huh7 cells were plated on ECM from wild-type fibroblasts or from MMP-resistant Col I (RR) fibroblasts, followed by western blot for pDDR1, DDR1, pAKT, AKT and b-actin (n=1 well, n=3 experiments). h, expression of collagen-degrading MMPs, determined by qPCR, in the profound fibrosis DEN+CCl4 model (n=4 normal liver controls [Ctrl], n=9 mice for non-tumour [NT] and tumour [Tu] samples). i, survival analysis in the TCGA dataset based on the expression of a collagen-degrading MMP signature (MMP1, MMP2, MMP8, MMP9, MMP13 and MMP14). j, YAP, TAZ, GAPDH, pAKT and AKT expression were determined by western blot in non-tumor tissue from Ddr1fl/fl and Ddr1ΔHep mice (n=6 mice per group). k, Infiltration of CD3+ T cells was determined by IHC and quantified in both non-tumor and tumor tissue (CD3 IHC, Cd3e and Ptprc mRNA in non-tumour area: Ddr1f/f: n=9 mice, Ddr1ΔHep n=8 mice; CD3 IHC, Cd3e and Ptprc mRNA in non-tumour area: Ddr1f/f: n=10 mice, Ddr1ΔHep n=8 mice); expression of Cd3e and Ptprc (encoding CD45) was determined by qPCR. Data are shown as mean ± SEM, each data point represents one well (b,e,f,g) or one individual (c,h,k); scale bars: 200 μm (d), 50 μm (k). nd: non-detected. Immunoblots in j were performed on two different gels using the same samples: one gel for YAP/TAZ and GAPDH and a second gel for pAKT and AKT. Data are shown as mean ± SEM. Statistics: Ddr1 mRNA in NT in c, data in d, CD3+ IHC and Cd3e mRNA in non-tumour and Ptprc mRNA in tumour in g were analysed by two-tailed Student’s t-test. CD3+ IHC and Cd3e mRNA in tumour and Ptprc mRNA in non-tumour in g were analysed by two-tailed Mann-Whitney test. Data in h were analysed by one way ANOVA (Kruskal Wallis) for Mmp2 (p<0.001), Mmp8 (p=0.004) and Mmp9 mRNA (p=0.004) followed by Dunn’s multiple comparison or for Mmp13 (p=0.001) and Mmp14 mRNA one-way one-way ANOVA (p<0.001) followed by Tukey’s multiple comparison. Raw data are given in Source Data and raw western blot gels in Supplementary Information.
Extended Data Figure 11|. Expression and role of myHSC-enriched hyaluronan and cyHSC-expressed HHIP and CXCL12 in hepatocarcinogenesis.
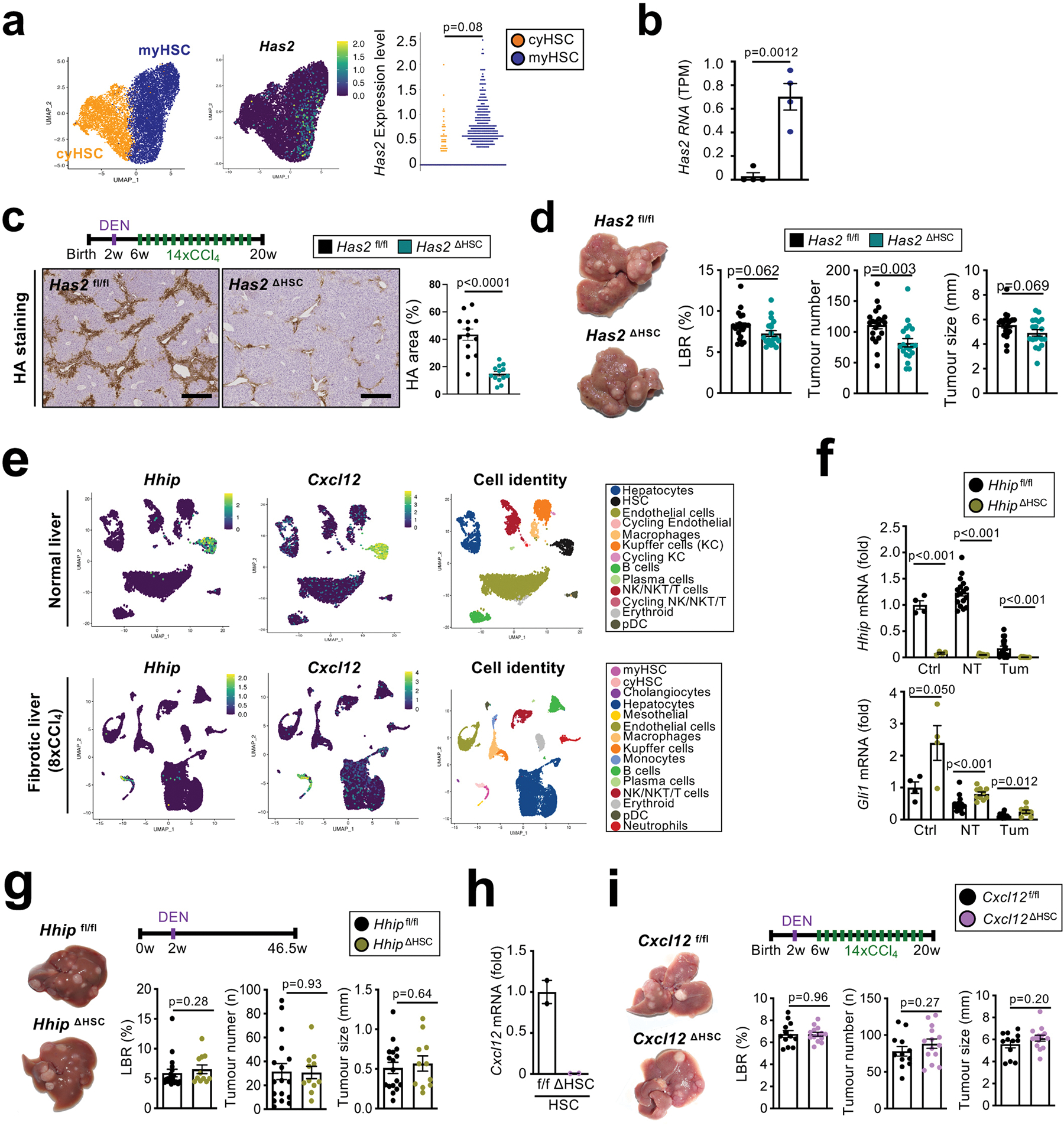
a, UMAP visualization and dotplot show predominant expression of Has2 in myHSC in the profound fibrosis regimen, induced by 19xCCl4 (n=1 mouse). b, bulk RNA sequencing in FACS-sorted HSC showed upregulation of Has2 in HSC from 12xCCl4-injured liver (n=4 mice) compared to quiescent HSC (n=4 mice). c-d, HCC was induced by DEN+14xCCl4 and hyaluronan (HA) staining showed strong reduction of HA deposition in Has2ΔHSC mice compared to Has2f/f littermates (n=13 mice per group) (c). HCC evaluation showed a slight reduction of HCC development in Has2ΔHSC (n=20 mice) mice compared to Has2f/f littermates (n=21 mice) (d). e, UMAPs of Hhip and Cxcl12 mRNA in normal (n=1 mouse) and in 8xCCl4-treated (n=3 mice) mouse livers. f, Mice with LratCre-mediated conditional deletion of Hhip (HhipΔHSC) show efficient deletion in healthy control liver (n=4 mice), non-tumor tissue and tumor tissue (Hhipf/f: n=17 mice, HhipΔHSC n=10 mice) as well as upregulation of Hedgehog target gene Gli1 in comparison to Hhip f/f controls. g, HhipΔHSC (n=11 mice) and Hhipfl/fl (n=17 mice) mice were subjected to DEN-induced hepatocarcinogenesis, followed by evaluation of the liver/body weight ratio (LBR), tumour size and tumour number. h, Deletion of Cxcl12 was determined by qPCR in HSC isolated from mice with LratCre-mediated deletion (Cxcl12ΔHSC) in comparison to Cxcl12fl/fl HSC (n=2 mice per group). i, Cxcl12ΔHSC (n=12 mice) and Cxcl12fl/fl (n=14 mice) mice were subjected to DEN+CCl4 induced HCC followed by evaluation of the liver/body weight ratio, tumour size and tumour number. Data are shown as mean ± SEM. In the dotplot graphs, each data point represent one cell (a) or one individual (b,c,d,g,h,i), scale bars: 400 μm. Statistics: Data in a, LBR in d, Gli1 mRNA in non-tumour in f, LBR in g and i were analysed by two-tailed Mann-Whitney test. Data in b, c, all data in d besides Gli1 mRNA, LBR in g and i, were analysed by two-tailed Student’s t-test. Raw data are given in Source Data and Supplementary Information.
Extended Data Figure 12|. HGF deletion in HSC promotes fibrosis and inflammation in CCl4-induced liver injury.

a-c, UMAP visualization of Hgf and/or Met in the 8xCCl4 injured (n=3 mice) (a) or normal mouse livers (n=1) by scRNAseq (c) and in the human normal (n=4 cases), cirrhotic (n=4 cases) and HCC (n=2 cases) livers by snRNAseq (b). d, significant reduction of Hgf mRNA in normal (n=4 mice/group) and CCl4 liver (UT: n=3 mice, Hgf fl/fl: n=8 mice, Hgf ΔHSC: n=7 mice) as well as of HGF protein in liver (UT: n=4 mice, CCl4: n=5 mice/group) e, Fibrosis was assessed by Sirius Red staining in the non-tumour tissue from Hgf ΔHSC mice (n=10 mice) compared to Hgf f/f littermates (n=11 mice) during HCC development induced by DEN+12xCCl4. f, HSC activation was determined by qPCR for the fibrogenic genes Col1a1 and Lox in livers from Hgf ΔHSC (n=4 mice) and Hgf f/f mice (n=7 mice) treated with injections of 6xCCl4. g, flow cytometric analysis of CD45+ cells as well as myeloid and lymphocyte populations in livers from Hgf ΔHSC (n=7 mice) compared to Hgf f/f mice (n=8 mice) treated with injections of 6xCCl4. h, qPCR for inflammatory genes from Hgf ΔHSC (n=7 mice) and Hgf f/f (n=8 mice) livers after treatment with 6xCCl4 injections. i, Liver/body ratio (LBR) in Hgf fl/fl (Ctrl: n=10 mice, pHx: n=6 mice) and Hgf ΔHSC (Ctrl: n=8 mice, pHx: n=7 mice) mice in untreated mice or after 48h partial hepatectomy (pHx) (left panel). Hepatocyte proliferation assessed by Ki67 staining 48h after PHx (right panel - Hgf f/f: n=6 mice, Hgf ΔHSC: n=7 mice). Data are shown as mean ± SEM, each data point represents one individual, scale bars: 200 μm. GZB, granzyme B. Statistics: Data in d-g and i were analysed by two-tailed Student’s t-test. Data in h were analysed by two-tailed Mann-Whitney test. Raw data are given in Source Data and gating strategy for g in Supplementary Information 5.
Extended Data Figure 13|. Dysbalance between myHSC and cyHSC occurs in advanced liver disease and elevates HCC risk in patients.
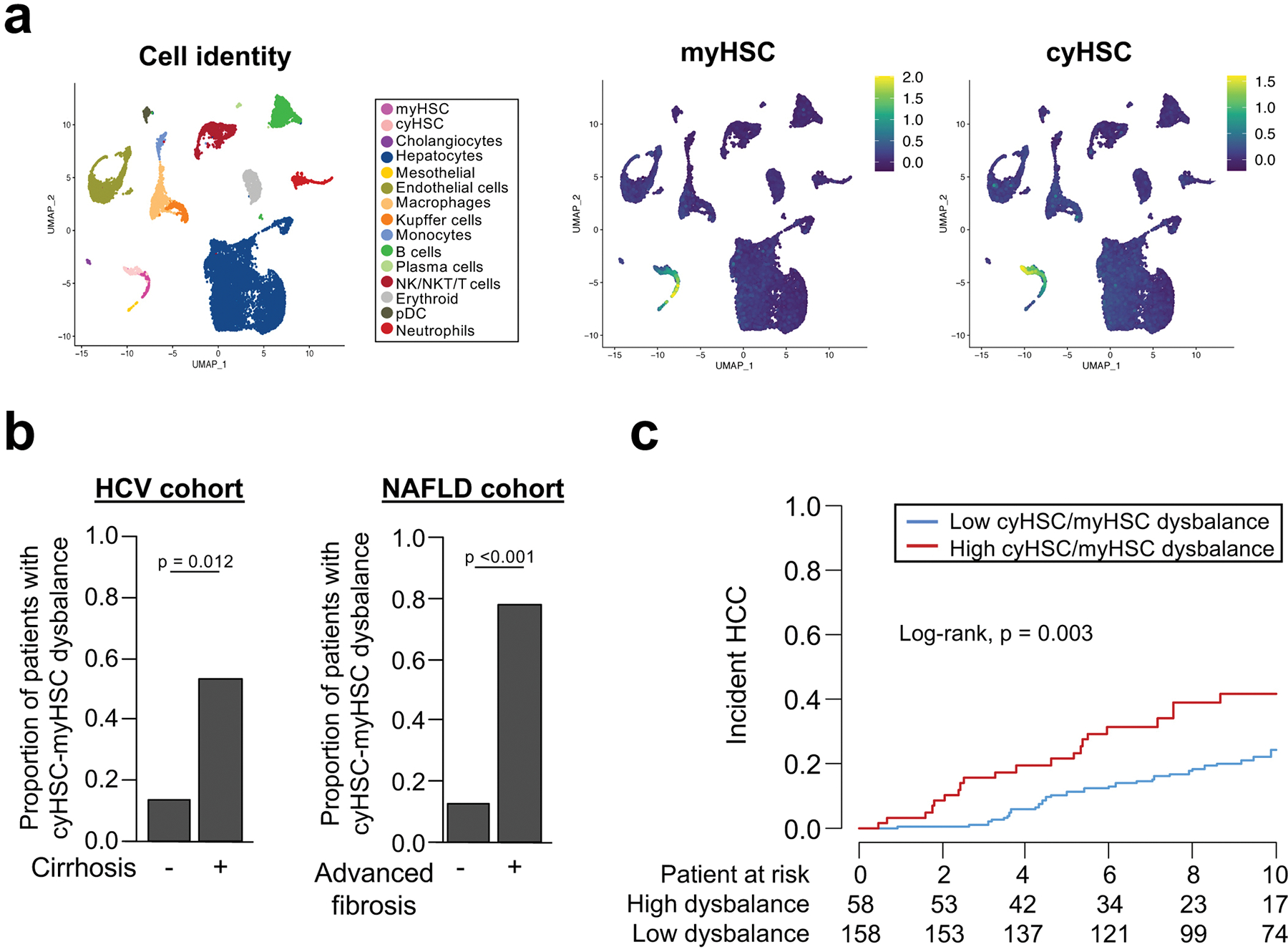
a, UMAP visualization showing that the genes from the myHSC and cyHSC signatures, encoding secreted mediators, are strongly enriched in myHSC or cyHSC, and weakly or not expressed in other liver cell populations in 8xCCl4 injured mouse liver (n=3 mice) analysed by scRNAseq. b, myHSC and cyHSC secreted gene signatures were applied to bulk RNA-seq data from cohorts of patients with HCV-induced liver disease (GSE10140) or NAFLD/NASH-induced liver disease (GSE49541), and the proportion of patients with high cyHSC/myHSC dysbalance was determined as described in the methods. c, HCC incidence was compared between patients with high cyHSC/myHSC dysbalance (i.e. a status with higher myHSC and lower cyHSC) and low cyHSC/myHSC dysbalance (i.e. a status lower myHSC and higher cyHSC) in a HCV-induced liver disease cohort (GSE15654). Statistics: data in b were analysed by two-sided Fisher’s exact test. Survival curves in c were represented using the Kaplan-Meier method and compared with log-rank statistics.
Supplementary Material
Acknowledgements
This work was supported by grants R01CA190844 and R01CA228483 (to RFS) and R01DK116620 (to RFS and IT) and the Columbia University Digestive and Liver Disease Research Center (1P30DK132710) and its Bioinformatics and Single Cell Analysis Core. J.Z-R was supported by Ligue Nationale contre le Cancer (Equipe Labellisée) and Labex OncoImmunology (Investissement d’avenir). N.C.H. is supported by a Wellcome Trust Senior Research Fellowship in Clinical Science (ref. 219542/Z/19/Z), Medical Research Council and a Chan Zuckerberg Initiative Seed Network Grant. Y.H. was supported by NIH grant CA233794 and Cancer Prevention and Research Institute of Texas grant RR180016; BI was supported by NIH grants R37CA258829 and R21CA263381. These studies used the resources of the Herbert Irving Comprehensive Cancer Center at Columbia University: The Flow Core Facility funded in part through Center Grant P30CA013696 and S10OD020056, the Molecular Pathology and the Confocal and Specialized Microscopy, supported by NIH grant #P30 CA013696. A.F. was funded by a Foundation pour la Recherche Medicale postdoctoral fellowship (SPE20170336778), an American Liver Foundation Postdoctoral Research Award, an International Liver Cancer Association’s Fellowship and the Mandl Connective Tissue Research Fellowship. Y.S by Uehara Memorial Foundation and Naomi Berrie Diabetes Center Russell Berrie Foundation. D.D by F31 DK091980, S.B. by Deutsche Forschungsgemeinschaft grant GZ:BH 155/1-1; S.A. was funded by an American Liver Foundation Postdoctoral Research Fellowship Award, a Cholangiocarcinoma Foundation’s Innovation Award, and a Research Scholar Award from the American Gastroenterological Association. We thank Dr. Edwin Monuki (University of California, Irvine) for the Lhx2 floxed mice and Dr. Matthias Mack (University of Regensburg, Germany) for the Col1a1 floxed mice, Dr. Raghu Kalluri for the αSMA-TK mice, and Dr. Yu Yamaguchi (Stamford Burnham Prebys Medical Discovery Institute, La Jolla) for the Has2 floxed mice. We thank Dr Ekihiro Seki (University of California, Los Angeles), Dr. Céline Hernandez (The University of Birmingham, United Kingdom) and Dr. Christian Kuntzen (Columbia University) for scientific support and discussions.
ADDITIONAL INFORMATION
Sample sizes, biological replicates and statistical tests
Additional information for Figure 1 | Promotion of HCC development by hepatic stellate cells. a: Sirius Red: Lhx2f/f: n=8 mice, Lhx2ΔHSC: n=7 mice; Col1a1 mRNA: Lhx2f/f: n=8 mice, Lhx2ΔHSC: n=6 mice. b: LBR, tumour number and tumour size: Lhx2f/f: n=8 mice, Lhx2ΔHSC: n=7 mice. c: αSMA area: YAPf/f: n=14 mice, YAPΔHSC: n=16 mice; Col1a1 mRNA: YAPf/f: n=15 mice, YAPΔHSC: n=14 mice. d: LBR, tumour number and tumour size: YAPf/f: n=15 mice, YAPΔHSC: n=16 mice. e: TdTomato: DTRneg: n=12 mice, DTRpos: n=7 mice; Col1a1 mRNA: DTRneg: n=15 mice, DTRpos: n=16 mice. f: LBR, tumour number and tumour size: DTRneg: n=15 mice, DTRpos: n=17 mice. Statistics: all data in a, b, d, e, αSMA in c and all data in f besides LBR were analysed by two-tailed Student’s t-test; Col1a1 in c and LBR in f were analysed by two-tailed Mann-Whitney test.
Additional information for Figure 2 | Hepatic stellate cell accumulation and activation occurs predominantly in the non-tumour environment. a: Col-GFP/TdTom double+ area: DEN+CCl4: n=3 mice per group, TAZ+FPC: n=2 mice per group, KRAS/sgp19: n=3 mice per group; Sirius Red: DEN+CCl4: n=3 mice per group, TAZ+FPC: n=3 mice per group, KRAS/sgp19: n=3 mice per group. b: αSMA: Human HCC: n=45 cases per group, ICC: n=5 cases per group, Sirius Red: Human HCC: n=45 cases per group, ICC: n=4 cases per group. c: Ki67+hepatocytes: YAPf/f: n=10 mice, YAPΔHSC: n=11 mice; DTRneg: n=10 mice, DTRpos: n=10 mice. d: CellPhoneDB in scRNAseq: n=3 mice. Statistics: all data in a and c were analysed by two-tailed Student’s t-test. Data in b were analysed by one-way ANOVA (Kruskal-Wallis’s test) followed by Dunn’s multiple comparison test.
Additional information for Figure 3 | Single cell RNA-seq analysis reveals subsets of HSC with differential expression of HGF and COL1A1 in the fibrotic liver. Fig a,e: scRNAseq on n=1 mouse, b,f: snRNAseq on normal and cirrhotic human livers n= 4 cases per group. c: scRNAseq on n=1 mouse per group. d: CellPhoneDB in scRNAseq: n=3 mice. g: normal liver: n=1 mouse and quantification on 4 areas per group; profound fibrosis: n=1 mouse and quantification on 4 areas per group. Statistics: P-values in a-b, coefficient of determination (R2) and statistical significance (P value) were determined using Pearson’s. Data in c were analysed by one-way ANOVA (Kruskal-Wallis test) followed by Dunn’s multiple comparison test. Data in e-f were analysed by two-tailed Mann-Whitney test.
Additional information for Figure 4 | Promotion of HCC by type I collagen and stiffness-associated accumulation of TAZ in hepatocytes. a: LBR and tumour number: n=21 mice per group; tumour size: Col1af/f: n=20 mice, Col1a1del: n=21 mice. b: Ki67+hepatocytes: n=13 mice per group; Ki67+tumour cells: Col1a1f/f: n=11 mice, Col1a1del: n=10 mice. c: Youngs modulus: untreated: 1 mouse, 19xCCl4: n=4 mice per group. d: TAZ/GAPDH and YAP/GAPDH: n=6 mice per group. e: nuclear TAZ+ hepatocytes: n= 9 mice per group. f: LBR, tumour number and tumour size: n= 10 mice per group. g: Ki67+hepatocytes: n=10 mice per group; Ki67+tumour cells: Wwtr1f/f: n=10 mice, Wwtr1ΔHep: n=8 mice. Statistics: tumour size in a, Ki67+ tumour cells in b, and all data in d,e,f and g were analysed by two-tailed Student’s t-test. LBR and tumour number in a and Ki67+ hepatocytes in b were analysed by two-tailed Mann-Whitney test. Data in e were analysed by two-way ANOVA followed by Tukey’s multiple comparison test. * Comparison between Col1a1f/f and Col1a1del mice, * p<0.05
Additional information for Figure 5 | Promotion of hepatocarcinogenesis by DDR1. b. DDR1 IHC: normal: n=6, non-tumour and tumour: n=64 per group; DDR1 mRNA: normal: n=4, non-tumour and tumour: n=16 per group. c: LBR and tumour size: Ddr1f/f: n=10 mice, Ddr1ΔHep n=9 mice, tumour number: n=9 mice per group. d: Ki67+ hepatocytes and Ki67+ tumours cells: Ddr1f/f: n=9 mice, Ddr1ΔHep n=8 mice. e: (n=3 biological replication/group; n=1 experiment). f, (n=1 biological replicate; n=2 experiments). Statistics: all data in b and Ki67+ tumour cells in d were analysed by two-tailed Mann-Whitney test. All data in c and Ki67+ hepatocytes in d were analysed by two-tailed Student’s t-test.
Additional information for Figure 6 | CyHSC-enriched HGF protects hepatocytes from death and reduces HCC development. a: LBR and tumour number: n= 10 mice per group; tumour size: Hgff/f: n=10 mice, HgfΔHSC: n=9 mice. b: TUNEL and Ki67+ hepatocytes: Hgff/f: n=8 mice, Hgf ΔHSC: n=4 mice; ALT (CCl4): Hgf f/f: n=7 mice, Hgf ΔHSC: n=4 mice; Mki67 mRNA: Hgf f/f: n=6 mice, Hgf ΔHSC: n=4 mice; ALT (NASH): n= 11 mice per group. c: PI+ area: control: n= 3 wells, treatment: n= 5 wells per group. d: Hgf mRNA: untreated: n= 5mice, NT and HCC: n= 8 mice per group. e: HGF mRNA: normal: n=124 cases, F0-F1 non-tumour: n=261 cases, F2-F3: n=115 cases, F4: n=182 cases, DMN: n=31 cases, HCC: n=687 cases. Statistics: all data in a, b, c and d were analysed by two-tailed Student’s t-test. Data in e between normal, F0-F1, F2-F3 and F4 were analysed by one-way ANOVA (Kruskal-Wallis’s test) followed by Dunn’s multiple comparison test with normal liver group. Dysplastic macronodules (DMN) and HCC comparison in e was analysed by two-tailed Mann-Whitney test. Kaplan-Meier survival curves in f were compared with log-rank statistics.
Footnotes
COMPETING INTEREST DECLARATION
The authors declare no conflict of interest beside:
B.I. has received honoraria from consulting with Merck, J&J/Janssen Pharmaceuticals, AstraZeneca and Volastra Therapeutics.
M.K. is a founder and SAB member of Elgia Pharma and received research support from Merck, Janssen Gossamer4 Bio.
REFERENCES
- 1.Villanueva A Hepatocellular Carcinoma. N Engl J Med 380, 1450–1462, doi: 10.1056/NEJMra1713263 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Affo S, Yu LX & Schwabe RF The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol 12, 153–186, doi: 10.1146/annurev-pathol-052016-100322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mederacke I et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 4, 2823, doi: 10.1038/ncomms3823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryerson AB et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 122, 1312–1337, doi: 10.1002/cncr.29936 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jemal A et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst 109, doi: 10.1093/jnci/djx030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol 19, 151–172, doi: 10.1038/s41571-021-00573-2 (2022). [DOI] [PubMed] [Google Scholar]
- 7.Schwabe RF & Luedde T Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 15, 738–752, doi: 10.1038/s41575-018-0065-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paradis V et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 49, 851–859, doi: 10.1002/hep.22734 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Lujambio A et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460, doi: 10.1016/j.cell.2013.03.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kocabayoglu P et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol 63, 141–147, doi: 10.1016/j.jhep.2015.01.036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duran A et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 30, 595–609, doi: 10.1016/j.ccell.2016.09.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimoto S et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101, doi: 10.1038/nature12347 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Krizhanovsky V et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667, doi: 10.1016/j.cell.2008.06.049 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loo TM et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov 7, 522–538, doi: 10.1158/2159-8290.CD-16-0932 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Omori S et al. Generation of a p16 Reporter Mouse and Its Use to Characterize and Target p16(high) Cells In Vivo. Cell Metab 32, 814–828 e816, doi: 10.1016/j.cmet.2020.09.006 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Wandzioch E, Kolterud A, Jacobsson M, Friedman SL & Carlsson L Lhx2−/− mice develop liver fibrosis. Proc Natl Acad Sci U S A 101, 16549–16554, doi: 10.1073/pnas.0404678101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anstee QM, Reeves HL, Kotsiliti E, Govaere O & Heikenwalder M From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16, 411–428, doi: 10.1038/s41575-019-0145-7 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Wang X et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 24, 848–862, doi: 10.1016/j.cmet.2016.09.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoshida Y et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 359, 1995–2004, doi: 10.1056/NEJMoa0804525 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji J et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 62, 481–495, doi: 10.1002/hep.27822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mu X et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest 125, 3891–3903, doi: 10.1172/JCI77995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puche JE et al. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 57, 339–350, doi: 10.1002/hep.26053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Affo S et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 39, 866–882 e811, doi: 10.1016/j.ccell.2021.03.012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharjee S et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 131, doi: 10.1172/JCI146987 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong X et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 75, 644–660 e645, doi: 10.1016/j.molcel.2019.07.028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayward MK, Muncie JM & Weaver VM Tissue mechanics in stem cell fate, development, and cancer. Dev Cell 56, 1833–1847, doi: 10.1016/j.devcel.2021.05.011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuzaki R et al. Prospective risk assessment for hepatocellular carcinoma development in patients with chronic hepatitis C by transient elastography. Hepatology 49, 1954–1961, doi: 10.1002/hep.22870 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Poynard T et al. Staging chronic hepatitis C in seven categories using fibrosis biomarker (FibroTest) and transient elastography (FibroScan(R)). J Hepatol 60, 706–714, doi: 10.1016/j.jhep.2013.11.016 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Wang HM et al. Liver stiffness measurement as an alternative to fibrotic stage in risk assessment of hepatocellular carcinoma incidence for chronic hepatitis C patients. Liver Int 33, 756–761, doi: 10.1111/liv.12118 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Baglieri J et al. Nondegradable Collagen Increases Liver Fibrosis but Not Hepatocellular Carcinoma in Mice. Am J Pathol 191, 1564–1579, doi: 10.1016/j.ajpath.2021.05.019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhar D et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 33, 1061–1077 e1066, doi: 10.1016/j.ccell.2018.05.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goyal L, Muzumdar MD & Zhu AX Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 2310–2318, doi: 10.1158/1078-0432.CCR-12-2791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takami T et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res 67, 9844–9851, doi: 10.1158/0008-5472.CAN-07-1905 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Ueki T et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat Med 5, 226–230, doi: 10.1038/5593 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Santoni-Rugiu E et al. Inhibition of neoplastic development in the liver by hepatocyte growth factor in a transgenic mouse model. Proc Natl Acad Sci U S A 93, 9577–9582, doi: 10.1073/pnas.93.18.9577 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki A et al. Hepatocyte growth factor promotes cell survival from fas-mediated cell death in hepatocellular carcinoma cells via Akt activation and Fas-death-inducing signaling complex suppression. Hepatology 32, 796–802, doi: 10.1053/jhep.2000.17738 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Stoker MG, Shearer M & O’Neill C Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci 1, 297–310 (1966). [DOI] [PubMed] [Google Scholar]
- 38.Alkasalias T et al. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc Natl Acad Sci U S A 111, 17188–17193, doi: 10.1073/pnas.1419554111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature 599, 673–678, doi: 10.1038/s41586-021-04057-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y et al. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39, 548–565 e546, doi: 10.1016/j.ccell.2021.02.007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
References (Methods)
- 41.Nault JC et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 71, 164–182, doi: 10.1002/hep.30811 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Krempen K et al. Far upstream regulatory elements enhance position-independent and uterus-specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr 8, 151–163 (1999). [PMC free article] [PubMed] [Google Scholar]
- 43.Mangale VS et al. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 319, 304–309, doi: 10.1126/science.1151695 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geisler F, Algul H, Paxian S & Schmid RM Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology 132, 2489–2503, doi: 10.1053/j.gastro.2007.03.033 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Buchtler S et al. Cellular Origin and Functional Relevance of Collagen I Production in the Kidney. J Am Soc Nephrol 29, 1859–1873, doi: 10.1681/ASN.2018020138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Affo S et al. Promotion of Cholangiocarcinoma Growth by Diverse Cancer-associated Fibroblast Subpopulations. Cancer Cell In Press (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phaneuf D, Moscioni AD, LeClair C, Raper SE & Wilson JM Generation of a mouse expressing a conditional knockout of the hepatocyte growth factor gene: demonstration of impaired liver regeneration. DNA Cell Biol 23, 592–603, doi: 10.1089/dna.2004.23.592 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto K et al. Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 136, 2825–2835, doi: 10.1242/dev.038505 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murtaugh LC, Stanger BZ, Kwan KM & Melton DA Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A 100, 14920–14925, doi: 10.1073/pnas.2436557100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ozdemir BC et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734, doi: 10.1016/j.ccr.2014.04.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitchell C & Willenbring H A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nature protocols 3, 1167–1170, doi: 10.1038/nprot.2008.80 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Zhang S et al. The Hippo Effector Transcriptional Coactivator with PDZ-Binding Motif Cooperates with Oncogenic beta-Catenin to Induce Hepatoblastoma Development in Mice and Humans. Am J Pathol 190, 1397–1413, doi: 10.1016/j.ajpath.2020.03.011 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao J et al. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology 64, 1587–1605, doi: 10.1002/hep.28601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mederacke I, Dapito DH, Affo S, Uchinami H & Schwabe RF High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nature protocols 10, 305–315, doi: 10.1038/nprot.2015.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pradere JP et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473, doi: 10.1002/hep.26429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung AS et al. Immune Checkpoint Inhibition is Safe and Effective for Liver Cancer Prevention in a Mouse Model of Hepatocellular Carcinoma. Cancer Prev Res (Phila) 13, 911–922, doi: 10.1158/1940-6207.CAPR-20-0200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523, doi: 10.1038/s41467-019-09234-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y et al. Integrated analysis of multimodal single-cell data. bioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slyper M et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med 26, 792–802, doi: 10.1038/s41591-020-0844-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andrews TS et al. Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol Commun 6, 821–840, doi: 10.1002/hep4.1854 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleming SJ, Marioni JC & Babadi M CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv doi: 10.1101/791699 (2019). [DOI] [Google Scholar]
- 63.Franzen O, Gan LM & Bjorkegren JLM PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019, doi: 10.1093/database/baz046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502, doi: 10.1038/s41586-019-0969-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoshida Y Nearest template prediction: a single-sample-based flexible class prediction with confidence assessment. PLoS One 5, e15543, doi: 10.1371/journal.pone.0015543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97, doi: 10.1093/nar/gkw377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vento-Tormo R et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353, doi: 10.1038/s41586-018-0698-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nature protocols 15, 1484–1506, doi: 10.1038/s41596-020-0292-x (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray, RNA-seq, scRNA-seq and snRNAseq datasets reported in this study have been deposited in the Gene Expression Omnibus database (GEO) under the accession number GSE174748 and GSE212047. In addition, we analyzed previously published whole liver or isolated HSC scRNAseq dataset GSE172492 and GSE158183, normal human liver snRNAseq GSE185477 as well as the microarray dataset GSE15654, GSE49541, and GSE10140. Source data are provided with this paper.