

Abstract

The asymmetric Michael reaction of methyl alkynyl ketone and α,β-unsaturated aldehyde catalyzed by diphenylprolinol silyl ether was developed. Although methyl alkynyl ketone is a good Michael acceptor, it also acts as a Michael donor to afford the synthetically important δ-oxo aldehydes with excellent enantioselectivity. The products possessing several functional groups, such as alkyne, ketone, and aldehyde moieties, are useful chiral building blocks for further synthesis. Using this reaction as a key step, a side chain of atorvastatin (Lipitor), an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, was synthesized in a two-pot sequence with excellent diastereo- and enantioselectivities.
Keywords: organocatalyst, Michael reaction, diphenylprolinol silyl ether, iminium ion, Lipitor, methyl alkynyl ketone, HMG-CoA reductase
Alkynyl ketones (conjugated ynones) are versatile building blocks in synthetic organic chemistry1,2 because, in addition to acting as good Michael acceptors, the ketone moiety can be converted into several functional groups. Moreover, the alkynyl moiety can be utilized in metal-mediated [2 + 2 + 2] cycloadditions,3 reductive coupling reactions,4 and alkyne–alkene and alkene–alkyne coupling reactions.5 For the derivatization of alkynyl ketones, the addition reaction of alkynyl ketones is a useful method. In the case of asymmetric catalytic addition reactions of alkynyl ketones as a nucleophile, Trost6,7 and Shibasaki8 reported the use of chiral Zn9,10 and a Lewis acid–Brønsted base two-center catalyst,11 respectively, to promote aldol reactions. Trost also reported the Mannich reaction of alkynyl ketones using the same chiral catalyst and demonstrated the synthetic utility of an alkynyl ketone moiety.12,13
On the other hand, the Michael reaction is an important carbon–carbon bond-forming reaction.14−16 However, an asymmetric catalytic Michael reaction of alkynyl ketones as a Michael donor has been regarded as a difficult reaction because it also acts as a good Michael acceptor. To our knowledge, there are no reports on the asymmetric metal-catalyzed reaction with alkynyl ketones as a nucleophile.
The field of organocatalysis has developed very rapidly17,18 and many organocatalyst-mediated Michael reactions have been reported.19 Even in the field of organocatalysis, the direct use of alkynyl ketone as a Michael donor is difficult. Ramachary reported the reaction of methyl alkynyl ketone with α,α-doubly activated alkene to afford a domino Michael/Michael product without isolation of a Michael product (eq 1).20−22 Instead of methyl alkynyl ketones, activated methyl alkynyl ketones such as alkoxycarbonyl, aryl, vinyl, or alkoxy-substituted methyl alkynyl ketones23−25 are reported to react with nitroalkene, catalyzed by bifunctional Brønsted base/H-bond catalyst, to afford the Michael products with excellent enantioselectivity (eq 2 and 3). There are no reports of the asymmetric Michael reaction of methyl alkynyl ketone itself as a nucleophile, even in the field of organocatalysis.
 |
1 |
Recently, we described a diphenylprolinol silyl ether26,27-mediated Michael reaction of ketone,28 in which an enolate is a reactive species.29 We thought that methyl alkynyl ketones would act as good Michael donors to generate the corresponding enolate in the presence of diphenylprolinol silyl ether to react with α,β-unsaturated aldehyde to afford synthetically useful δ-oxo aldehydes (eq 4). Here, we describe the first asymmetric catalytic Michael reaction of methyl alkynyl ketones and its application to the pot-economical synthesis of a key intermediate of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase.
First, we examined the reaction of 4-(p-nitrophenyl)-but-3-yn-2-one and cinnamaldehyde, catalyzed by diphenylprolinol silyl ether. Although we investigated the reaction under several reaction conditions, the starting aldehyde remained unchanged in most cases. When an equimolar amount of alkynyl ketone and a catalyst was used, the Michael adduct of the catalyst and alkynyl ketone was isolated (eq 5). This result indicates that alkynyl ketone is a good Michael acceptor, which reacts with the amine catalyst. With an expectation that the bulky substituent at the 4-position of but-3-en-2-one would suppress this Michael reaction, we examined the reaction of 4-triisopropylsilyl-but-3-yn-2-one (1) in the presence of several acids (Table 1). Using strong acids such as trifluoroacetic acid, the reaction did not proceed (entry 1). The desired product 3a was isolated in a 10% yield when benzoic acid was employed, and two side products, 4 and 5, were also obtained (entry 2). Compound 4 was an aldol (1,2-addition) condensation product, whereas 5 was derived from the double Michael reaction of 1 and two molecules of 2a, followed by intramolecular aldol condensation. Better yields were obtained when acetic acid and pivalic acid were used (entries 3 and 4). In the case of p-nitrophenol, the amount of acid was important. In the presence of 100 mol % p-nitrophenol, the desired product 3a was obtained in a 22% yield, along with the side product 5 (entry 7); in the presence of 10 mol % p-nitrophenol, however, 3a was obtained in the highest yield (69%) with excellent enantioselectivity (entry 5).
Table 1. Effects of Acids and Their Amounts in the Michael Reaction of 1 and 2aa.

| yield
(%)b |
|||||
|---|---|---|---|---|---|
| entry | acid (x mol %) | time (h) | 3a | 4 | 5 |
| 1 | CF3CO2H (10) | 24 | 0 | 0 | 0 |
| 2 | PhCO2H (10) | 12 | 10 | 15 | 20 |
| 3 | CH3CO2H (10) | 8 | 23 | 11 | 15 |
| 4 | t-BuCO2H (10) | 7 | 39 | 10 | 9 |
| 5 | p-nitrophenol (10) | 3 | 69c | 0 | 0 |
| 6 | p-nitrophenol (50) | 2.5 | 42 | 8 | 8 |
| 7 | p-nitrophenol (100) | 2 | 22 | 13 | 18 |
Unless otherwise shown, reactions were performed by employing cinnamaldehyde 2a (0.18 mmol), ketone 1 (0.15 mmol), organocatalyst (0.030 mmol), water (0.45 mmol), and acid (indicated amount) in EtOH (0.60 mL) at room temperature for the indicated time.
Isolated yield.
Enantiomeric excess is 95%.
The reaction mechanism would be similar to that of the Michael reaction of cyclohexanone:29 An iminium ion, which was generated from 2a and diphenylprolinol silyl ether, would react with an enolate of methyl ketone 1 to afford Michael product 3a.
 |
5 |
Having established the best reaction conditions, the generality of the reaction was then investigated (Table 2). Substrates with electron-rich aryl groups such as p-methoxyphenyl at the 3-position of propenal were suitable, affording good yield and excellent enantioselectivity, although the reaction was slow (entry 2). Electron-deficient aryl groups such as p-bromo, m-bromo-, and o-bromo phenyls were also suitable substituents, and the reaction was completed within 2 h in good yield with excellent enantioselectivity (entries 3–5). Heteroaromatics such as furyl afforded good results (entry 6), as did silyl-substituted propenal. In the latter case, the C–Si bond could be converted into the corresponding C–OH bond (see below)30,31 to provide the Michael product with 98% ee (entry 7). The Michael reaction also proceeded in the case of crotonaldehyde, for which moderate enantioselectivity was obtained (76% ee, entry 8).
Table 2. Generality of the Michael Reaction of 1 and 2a.

| entry | R | time (h) | yieldb (%) | eec (%) |
|---|---|---|---|---|
| 1 | Phenyl (2a) | 3 | 69 | 95 |
| 2 | p-MeO-C6H4 (2b) | 24 | 50 | 95 |
| 3 | p-Br-C6H4 (2c) | 2 | 71 | 94 |
| 4 | m-Br-C6H4 (2d) | 2 | 75 | 95 |
| 5 | o-Br-C6H4 (2e) | 2 | 78 | 95 |
| 6 | 2-furyl (2f) | 30 | 76 | 94 |
| 7 | SiMe2Ph (2g) | 12 | 69 | 98 |
| 8 | Me (2h) | 24 | 55 | 76 |
Reactions were performed by employing α,β-unsaturated aldehyde 2 (0.18 mmol), ketone 1 (0.15 mmol), organocatalyst (0.030 mmol), water (0.45 mmol), and p-nitrophenol (0.015 mmol) in EtOH (0.60 mL) at room temperature for the indicated time.
Isolated yield.
Enantiomeric excess (ee) of the products, as determined by HPLC analysis over a chiral solid phase after conversion to α,β-unsaturated ester by the treatment with Ph3P = CHCO2Et.
Inhibitors of HMG-CoA reductase are important medicines,32,33 and several synthetic statins such as atorvastatin (Lipitor),34−36 rosuvastatin,37 and fluvastatin38 are commonly used (Figure 1). The key common unit in the side chain of these statins is β,δ-syn-dihydroxy carboxylic acid, and a useful synthetic intermediate of these medicines is protected δ-alkynyl β,δ-syn-dihydroxy ester 6.39,40
Figure 1.

Synthetic statins and key intermediate 6.
One-pot operations are an effective method for carrying out several transformations and forming several bonds in a single pot, while simultaneously eliminating several purification steps, minimizing chemical waste generation, and saving time.41−43 Based on this concept, we examined the pot-economical synthesis of 6 (Scheme 1).
Scheme 1. Two-pot Synthesis of Key Intermediate 6 of Inhibitors of HMG-CoA Reductase.
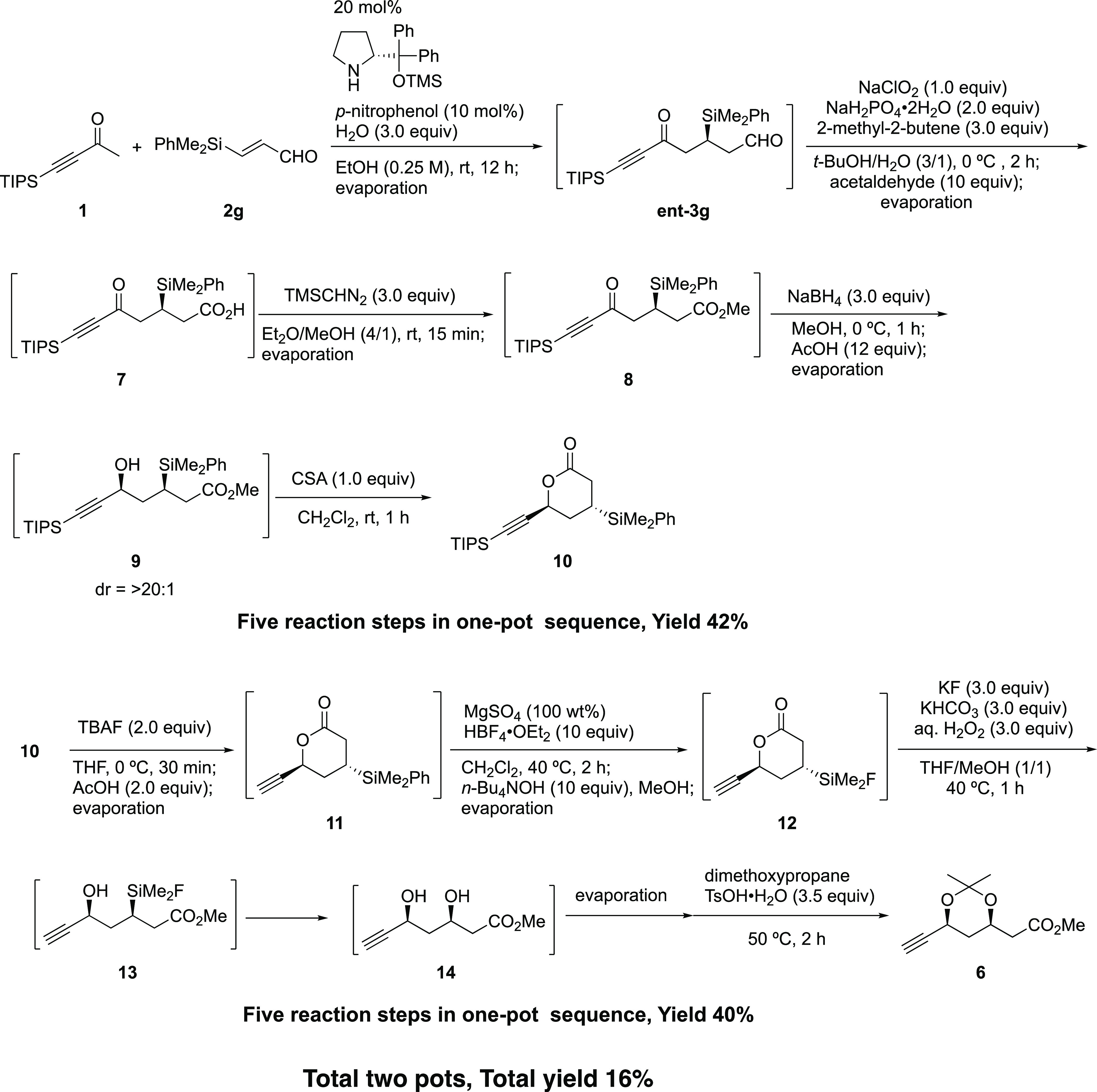
The first pot reaction started from the asymmetric Michael reaction of 1 and silylated propenal 2g using an (R)-diphenylprolinol silyl ether catalyst. After the removal of the solvent under reduced pressure and addition of t-BuOH and water in the same reaction vessel, the Michael adduct ent-3g was converted into carboxylic acid 7 by Pinnick–Kraus oxidation.44,45 Remaining oxidants were reduced upon the addition of acetaldehyde,46,47 which was converted into acetic acid. Generated acetic acid and excess acetaldehyde were removed under reduced pressure. The treatment of 7 with TMSCHN2 afforded methyl ester 8, which was reduced with NaBH4 to afford syn alcohol 9 as a single isomer with excellent diastereoselectivity (see below). Acetic acid was added, which reacted with the remaining NaBH4, and the solvent was changed from MeOH to CH2Cl2. Addition of CSA in the same reaction vessel promoted the lactonization to afford lactone 10, which was purified and isolated in a 42% yield from the first Michael reaction. Five reaction steps, (1) Michael reaction, (2) oxidation, (3) esterification, (4) reduction, and (5) lactonization, were conducted in a single reaction vessel. It should be noted that the one-pot reaction proceeds efficiently by suitable quenching of the reagents in situ,42 even though both oxidation and reduction steps are involved.
The second pot sequence started with the removal of the TIPS group from 10 by the addition of n-Bu4NF (TBAF) to afford 11. After neutralization of the reaction mixture by the addition of acetic acid, the addition of HBF4·OEt2 and MgSO4 converted the Si–Ph bond into a Si–F bond,30,31 in which MgSO4 suppressed the side reaction of alkyne and water. After neutralization of the reaction mixture with n-Bu4NOH, addition of H2O2, KF, and KHCO3 in a mixed solvent of THF and MeOH30,31 afforded the dihydroxy methyl ester 14 by opening lactone to hydroxy ester 13, followed by oxidative conversion of the C–Si bond into the C–OH bond. Protection of diol moiety 14 with dimethoxypropane gave acetal 6. The latter was obtained in a 40% yield from lactone 10 in a single reaction vessel. Five reaction steps, (1) the removal of the TIPS group, (2) conversion of the Si–Ph bond into a Si–F bond, (3) conversion of the C–Si bond into a C–OH bond, (4) opening of lactone to hydroxy ester, and (5) acetal protection of 1,3-diol, were conducted in a single reaction vessel.
It should be mentioned that the position of the Tamao–Fleming reaction in the sequence is important. The Tamao–Fleming reaction was successful from 11 to 14, but it did not proceed well for compounds 8 and 9.
The useful key intermediate 6 was prepared from the Michael reaction using only two reaction vessels with a total yield of 16%. This is a highly diastereo- and enantioselective synthesis. Compound 6 was further transformed into vinyl derivative 15,39,40,48 which is also a useful chiral building block for the synthesis of inhibitors of HMG-CoA reductase (eq 6).
 |
6 |
The diastereoselectivity of the reduction of 8 was notable. When the diastereoselective nucleophilic addition of β-silyl-substituted ketone was systematically investigated by Fleming,49 moderate selectivity was obtained in most cases. In the present reduction, excellent diastereoselectivity was observed, which is very rare. We investigated the use of several reducing reagents with isolated 8 (Table 3). Chiral reducing reagents such as (−)-B-chlorodiisopinocamphenylborane ((−)-DIP-Cl)50,51 gave excellent diastereoselectivity. Bulky reducing reagents such as l-selectride also gave excellent selectivity. On the other hand, a 1:1 mixture was obtained in the case of BH3·(t-BuNH2). When NaBH4 was employed in MeOH, both an excellent yield and diastereoselectivity were observed; however, for similar substrate 16, without a TIPS moiety, low diastereoselectivity was observed (the relative configuration was determined after conversion into the corresponding lactone; eq 7). Thus, the use of a TIPS protecting group for the alkyne is essential for the high selectivity, even though it is far from the reacting carbonyl group. This selectivity would be explained by the Felkin–Anh model, as proposed by Fleming in a similar system (Figure 2). The bulky PhMe2Si and alkynyl ketone adopt an antiperiplanar conformation. As conformer B is unfavorable because of the repulsion caused by the bulky TIPS group, the reaction would proceed from conformer A to afford the excellent diastereoselectivity observed.
 |
7 |
Table 3. Effect of Reducing Reagents in the Reduction of 8a.

| entry | reductant | solvent | temp (°C) | yield (%)b | syn:anti |
|---|---|---|---|---|---|
| 1 | (−)-DIP-Clc | THF | –20 | 62 | >20:1 |
| 2 | l-selectride | THF | –78 | 58 | >20:1 |
| 3 | BH3·(t-BuNH2) | CH2Cl2 | 0 | 81 | 1:1 |
| 4 | NaBH4d | MeOH | 0 | 88 | >20:1 |
Unless otherwise shown, reactions were performed by employing ketone 8 (0.10 mmol) and reductant (0.10 mmol) in the indicated solvent (0.30 mL) at the indicated temperature.
Isolated yield.
Amount of the reductant is 0.20 mmol.
Amount of the reductant is 0.30 mmol.
Figure 2.

Stable conformation of 8.
In summary, we developed the first asymmetric Michael reaction of methyl alkynyl ketone and α,β-unsaturated aldehyde, which was catalyzed by diphenylprolinol silyl ether to afford the synthetically important δ-oxo aldehydes bearing an alkyne moiety, with excellent enantioselectivity. Although 4-triisopropylsilyl-but-3-yn-2-one was the only methyl alkynyl ketone used in the present study,52 the silyl substituent can be converted into many substituents and functional groups. Because the products possess several functional groups such as alkyne, ketone, and aldehyde moieties, and are generated with excellent enantioselectivity, they would be useful chiral building intermediates. The synthetic utility of the alkynyl ketone moiety has already been well established by Trost’s group.12,13 By the application of this Michael reaction, chiral building block 6 of the inhibitors of HMG-CoA reductase can be synthesized in two pots with excellent diastereo- and enantioselectivities.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number JP20H04801 in Hybrid Catalysis for Enabling Molecular Synthesis on Demand and JP19H05630.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.1c00054.
Experimental procedures and analytical data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nájera C.; Sydnes L. S.; Yus M. Conjugated Ynones in Organic Synthesis. Chem. Rev. 2019, 119, 11110–11244. 10.1021/acs.chemrev.9b00277. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhu H.; Peng T.; Yang D. Conjugated ynones in catalytic enantioselective reactions. Org. Biomol. Chem. 2021, 19, 2110–2145. 10.1039/D0OB02521F. [DOI] [PubMed] [Google Scholar]
- Saito S.; Yamamoto Y. Recent Advances in the Transition- Metal-Catalyzed Regioselective Approaches to Polysubstituted Benzene Derivatives. Chem. Rev. 2000, 100, 2901–2916. 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- Jang H.-Y.; Krische M. J. Catalytic C-C Bond Formation via Capture of Hydrogenation Intermediates. Acc. Chem. Res. 2004, 37, 653–661. 10.1021/ar020108e. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Toste F. D.; Pinkerton A. B. Non-Metathesis Ruthenium-Catalyzed C–C Bond Formation. Chem. Rev. 2001, 101, 2067–2096. 10.1021/cr000666b. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Fettes A.; Shireman B. T. Direct Catalytic Asymmetric Aldol Additions of Methyl Ynones. Spontaneous Reversal in the Sense of Enantioinduction. J. Am. Chem. Soc. 2004, 126, 2660–2661. 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Stivala C. E.; Hull K. L.; Huang A.; Fandrick D. R. A concise synthesis of (-)-lasonolide A. J. Am. Chem. Soc. 2014, 136, 88–91. 10.1021/ja411270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K.; Maki K.; Kanai M.; Shibasaki M. Formal Catalytic Asymmetric Total Synthesis of Fostriecin. Org. Lett. 2003, 5, 733–736. 10.1021/ol027528o. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hung C.-I.; Mata G. Dinuclear Metal- ProPhenol Catalysts: Development and Synthetic Applications. Angew. Chem., Int. Ed. 2020, 59, 4240–4261. 10.1002/anie.201909692. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Bartlett M. J. ProPhenol-Catalyzed Asymmetric Additions by Spontaneously Assembled Dinuclear Main Group Metal Complexes. Acc. Chem. Res. 2015, 48, 688–701. 10.1021/ar500374r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa N.; Yamada Y. M. A.; Das J.; Sasai H.; Shibasaki M. Direct Catalytic Asymmetric Aldol Reaction. J. Am. Chem. Soc. 1999, 121, 4168–4178. 10.1021/ja990031y. [DOI] [Google Scholar]
- Trost B. M.; Hung C. I. Broad Spectrum Enolate Equivalent for Catalytic Chemo-, Diastereo-, and Enantioselective Addition to N-Boc Imines. J. Am. Chem. Soc. 2015, 137, 15940–15946. 10.1021/jacs.5b11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Hung C. I. J.; Gnanamani E. ACS Catal. 2019, 9, 1549–1557. 10.1021/acscatal.8b04685. [DOI] [Google Scholar]
- Carbon-Carbon Bond Formation. In Comprehensive Organic Synthesis, 2nd ed.; Knochel P.; Molander G. A., Eds.; Elsevier: Amsterdam, 2014; Vol. 3. [Google Scholar]
- Alexakis A.; Krause N.; Woodward S.. Copper-catalyzed Asymmertric Conjugate Addition. In Copper-Catalyzed Asymmetric Synthesis; Alexakis A.; Krause N.; Woodward S., Eds.; Wiley-VCH: Weinheim, 2014; pp 33–68. [Google Scholar]
- Non-Noble Metal Catalysis; Gebbink R. J. M. K.; Moret M., Eds.; Wiley-VCH: Weinheim, 2019; 191–208. [Google Scholar]
- Enantioselective Organocatalysis: Reactions and Experimental Procedures; Dalko P. I., Eds.; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- Asymmetric Organocatalysis 1: Lewis Base and Acid Catalysts; List B., Ed.; Georg Thieme Verlag KG: New York, 2012. [Google Scholar]
- Vicario J. L.; Reyes E.; Carrillo L.; Uria U.. Organocatalytic Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, 2014; Vol. 4, pp 119–188. [Google Scholar]
- Ramachary D. B.; Venkaiah Ch.; Krishna P. M. Discovery of 2-aminobuta-1,3-enynes in asymmetric organocascade catalysis: construction of drug-like spirocyclic cyclohexanes having five to six contiguous stereocenters. Chem. Commun. 2012, 48, 2252–2254. 10.1039/c2cc17219d. [DOI] [PubMed] [Google Scholar]
- Ramachary D. B.; Venkaiah C.; Madhavachary R. Asymmetric Synthesis of Druglike Six-Membered Spirooxindoles through an Amino Enyne Catalysis. Org. Lett. 2013, 15, 3042–3045. 10.1021/ol401227q. [DOI] [PubMed] [Google Scholar]
- Qi S.-S.; Yin H.; Wang Y.-F.; Wang C.-J.; Han H.-T.; Man T.-T.; Xu D.-Q. Catalytic Asymmetric Conjugate Addition/Hydro- alkoxylation Sequence: Expeditious Access to Enantioenriched Eight- Membered Cyclic Ether Derivatives. Org. Lett. 2021, 23, 2471–2476. 10.1021/acs.orglett.1c00392. [DOI] [PubMed] [Google Scholar]
- Liu W.; Zou L.; Fu B.; Wang X.; Wang K.; Sun Z.; Peng F.; Wang W.; Shao Z. A Multifaceted Directing Group Switching Ynones as Michael Donors in Chemo-, Enantio-, and γ-Selective 1,4-Conjugate Additions with Nitroolefins. J. Org. Chem. 2016, 81, 8296–8305. 10.1021/acs.joc.6b01425. [DOI] [PubMed] [Google Scholar]
- Iriarte I.; Olaizola O.; Vera S.; Gamboa I.; Oiarbide M.; Palomo C. Controlling the α/γ-Reactivity of Vinylogous Ketone Enolates in Organocatalytic Enantioselective Michael Reactions. Angew. Chem., Int. Ed. 2017, 56, 8860–8864. 10.1002/anie.201703764. [DOI] [PubMed] [Google Scholar]
- Campano T. E.; Iriarte I.; Olaizola O.; Etxabe J.; Mielgo A.; Ganboa I.; Odriozola J. M.; García J. M.; Oiarbide M.; Palomo C. Enantioselective Addition of Alkynyl Ketones to Nitroolefins Assisted by Brønsted Base/H-Bonding Catalysis. Chem. - Eur. J. 2019, 25, 4390–4397. 10.1002/chem.201805542. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Gotoh H.; Hayashi T.; Shoji M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]
- Marigo M.; Wabnitz T. C.; Fielenbach D.; Jørgensen K. A. Enantioselective organocatalyzed alpha sulfenylation of aldehydes. Angew. Chem., Int. Ed. 2005, 44, 794–797. 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Umekubo N. Direct Asymmetric Michael Reaction of α,β-Unsaturated Aldehydes and Ketones Catalyzed by Two Secondary Amine Catalysts. Angew., Chem., Int. Ed. 2018, 57, 1958–1962. 10.1002/anie.201710085. [DOI] [PubMed] [Google Scholar]
- Umekubo N.; Terunuma T.; Kwon E.; Hayashi Y. Evidence for an enolate mechanism in the asymmetric Michael reaction of α,β- unsaturated aldehydes and ketones via a hybrid system of two secondary amine catalysts. Chem. Sci. 2020, 11, 11293–11297. 10.1039/D0SC03359F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamao K.; Ishida N.; Tanaka T.; Kumada M. Hydrogen Peroxide Oxidation of the Silicon-Carbon Bond in Organoalkox- ysilanes. Organometallics 1983, 2, 1694–1696. 10.1021/om50005a041. [DOI] [Google Scholar]
- Fleming I.; Henning R.; Plaut H. The Phenyldimethylsilyl Group as a Masked Form of the Hydroxy Group. J. Chem. Soc., Chem. Commun. 1984, 29–31. 10.1039/c39840000029. [DOI] [Google Scholar]
- Goldstein J. L.; Brown M. S. Regulation of the mevalonate pathway. Nature. 1990, 343, 425–430. 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Maron D. J.; Fazio S.; Linton M.-R. F. Current Perspectives on Statins. Circulation 2000, 101, 207–213. 10.1161/01.CIR.101.2.207. [DOI] [PubMed] [Google Scholar]
- Roth B. D.; Mich A. A.. Trans-6-[2-(3- or 4-carboxamido-substituted pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of Cholesterol Synthesis. U.S. Patent US46818931987.
- Roth B. D.; Blankley C. J.; Chucholowski A. W.; Ferguson E.; Hoefle M. L.; Ortwine D. F.; Newton R. S.; Sekerke C. S.; Sliskovic D. R.; Wilson M. W. Inhibitors of Cholesterol Biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-l-yl)ethyl]-2H-pyran-2-one Inhibitors of HMG-CoA Reductase. 2. Effects of Introducing Substituents at Positions Three and Four of the Pyrrole Nucleus. J. Med. Chem. 1991, 34, 357–366. 10.1021/jm00105a056. [DOI] [PubMed] [Google Scholar]
- Roth B. D.; Mich A.. A. [R-(R*R*)]-2-(4-fluorophenyl)-β,δ-dihydroxy-5-(1-methylethyl-3-phenyl-4-[(phenylamino) carbonyl]-1H-pyrrole-1-heptanoic Acid, Its Lactone Form and Salts Thereof. U.S. Patent US52739951991.
- McTaggart F.; Buckett L.; Davidson R.; Holdgate G.; McCormick A.; Schneck D.; Smith G.; Warwick M. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am. J. Cardiol. 2001, 87, 28–32. 10.1016/S0002-9149(01)01454-0. [DOI] [PubMed] [Google Scholar]
- Hayashidani S.; Tsutsui H.; Shiomi T.; Suematsu N.; Kinugawa S.; Ide T.; Wen J.; Takeshita A. Fluvastatin, a 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitor, Attenuates Left Ventricular Remodeling and Failure After Experimental Myocardial Infarction. Circulation 2002, 105, 868–873. 10.1161/hc0702.104164. [DOI] [PubMed] [Google Scholar]
- Griffin J.; Lanza G.; Yu J.. Compositions and Treatments for Inhibiting Kinase and/or HMG-CoA Reductase. U.S. Patent US20050261354 A12005.
- Griffin J.; Lanza G.; Yu J.. Compositions and Treatments for Inhibiting Kinase and/or HMG-COA Reductase. U.S. Patent US20060084695 A12006.
- Hayashi Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y. Time Economy in Total Synthesis. J. Org. Chem. 2021, 86, 1–23. 10.1021/acs.joc.0c01581. [DOI] [PubMed] [Google Scholar]
- Hayashi Y. Time and Pot Economy in Total Synthesis. Acc. Chem. Res. 2021, 54, 1385–1398. 10.1021/acs.accounts.0c00803. [DOI] [PubMed] [Google Scholar]
- Kraus G. A.; Taschner M. J. Model studies for the synthesis of quassinoids. 1. Construction of the BCE ring system. J. Org. Chem. 1980, 45, 1175–1176. 10.1021/jo01294a058. [DOI] [Google Scholar]
- Bal B. S.; Childers W. E. Jr; Pinnick H. W. Oxidation of α,β-unsaturated aldehydes. Tetrahedron 1981, 37, 2091–2096. 10.1016/S0040-4020(01)97963-3. [DOI] [Google Scholar]
- Hayashi Y.; Koshino S.; Ojima K.; Kwon E. Pot Economy in the Total Synthesis of Estradiol Methyl Ether by Using an Organocatalyst. Angew. Chem., Int. Ed. 2017, 56, 11812–11815. 10.1002/anie.201706046. [DOI] [PubMed] [Google Scholar]
- Koshino S.; Kwon E.; Hayashi Y. Total Synthesis of Estradiol Methyl Ether and Its Five-Pot Synthesis with an Organocatalyst. Eur. J. Org. Chem. 2018, 2018, 5629–5638. 10.1002/ejoc.201800910. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Snider B. B. Synthesis of ent-Haterumalide NA (ent-Oocydin A) Methyl Ester. Org. Lett. 2003, 5, 4385–4388. 10.1021/ol0356789. [DOI] [PubMed] [Google Scholar]
- Barbero A.; Blakemore D. C.; Fleming I.; Wesley R. N. In search of open-chain 1,3-stereocontrol. J. Chem. Soc., Perkin Trans. 1 1997, 1329. 10.1039/a607545b. [DOI] [Google Scholar]
- Chandrasekharan J.; Ramachandran P. V.; Brown H. C. Diisopinocampheylchloroborane, a remarkably efficient chiral reducing agent for aromatic prochiral ketones. J. Org. Chem. 1985, 50, 5446–5448. 10.1021/jo00225a109. [DOI] [Google Scholar]
- Ohta C.; Kuwabe S.; Shiraishi T.; Shinohara I.; Araki H.; Sakuyama S.; Makihara T.; Kawanaka Y.; Ohuchida S.; Seko T. An Improved Synthesis of the Selective EP4 Receptor Agonist ONO-4819. J. Org. Chem. 2009, 74, 8298–8308. 10.1021/jo901497u. [DOI] [PubMed] [Google Scholar]
- The reaction was not completed with low yield in the cases of 3-(2-nitrophenyl)- but-3-yn-2-one and 3-(2-triethylsilyl-phenyl)- but-3-yn-2-one.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.