

Abstract
Purpose:
Proton craniospinal irradiation (pCSI) is a promising treatment for patients with solid tumor leptomeningeal metastasis (LM). We hypothesize that genetic characteristics before and changes resulting after pCSI will reflect clinical response to pCSI. We analyzed the cerebrospinal fluid (CSF) circulating tumor DNA (ctDNA) from patients receiving pCSI for LM and explored genetic variations associated with response.
Experimental Design:
We subjected CSF from 14 patients with LM before and after pCSI to cell-free DNA sequencing using a targeted-sequencing panel. In parallel, plasma ctDNA and primary tumors were subjected to targeted-sequencing. Variant allele frequency (VAF) and cancer cell fraction (CCF) were calculated; clonality of observed mutations was determined. Kaplan Meier analysis was used to associate genomic changes with survival.
Results:
The median overall survival (OS) for the cohort was 9 months (IQR: 5 – 21 months). We showed clonal evolution between tumor and ctDNA of the CSF and plasma with unique mutations identified by compartment. Higher CSF ctDNA mean VAF before pCSI (VAFpre) had worse OS (6 months for VAFpre≥0.32 vs. 9 months for VAFpre<0.32, p=0.05). Similarly, increased VAF after pCSI portended worse survival (6 vs 18 months, p=0.008). Higher mean CCF of subclonal mutations appearing after pCSI was associated with worse OS (8 vs 17 months, p=0.05).
Conclusions:
In solid tumor LM patients undergoing pCSI, we found unique genomic profiles associated with pCSI through CSF ctDNA analyses. Patients with reduced genomic diversity within the leptomeningeal compartment demonstrated improved OS after pCSI suggesting that CSF ctDNA analysis may have use in predicting pCSI response.
Keywords: proton therapy, leptomeningeal metastases, ctDNA, palliative, genomics
Introduction
Leptomeningeal metastasis (LM) is a dreaded complication of solid tumor malignancy with poor prognosis and limited treatment options. Conventional treatments result in median survival of 3-6 months, depending on the histology, functional status, and response to systemic therapy1. Recently, proton craniospinal irradiation (pCSI) has emerged as a potentially efficacious treatment for patients with solid tumor LM with promising survival outcomes2,3.
Irradiation of mammalian cells results in direct and indirect DNA damage that can lead to genetic alterations, including mutations, insertions, deletions, and copy number aberrations (CNA)4. Because tumor cells often carry highly abnormal and heterogeneous genomes, the genetic effects of ionizing radiation on tumor cells may differ greatly, even among cells of the same histology5. Thus, the efficacy of radiation therapy may depend in part on the complement of genetic alterations present in tumor cells before and after treatment. Analysis of detectable genetic aberration in LM can reveal clonal evolution of metastatic cancer cells, and the emergence of specific clones after pCSI may predict specific responses to radiotherapy6.
Though there are currently no established biomarkers of response to radiotherapy, liquid biopsies are an emerging non-invasive method to diagnose tumors7, characterize their natural history8, and understand their response to therapy9. Cerebrospinal fluid (CSF) can be obtained through lumbar puncture, a minimally invasive procedure with a low complication rate10. We have previously shown that baseline levels and dynamic changes in the number of CSF circulating tumor cells (CTCs) in patients with LM could be used to stratify response to pCSI11. Like CTCs, circulating tumor DNA (ctDNA) in the CSF12,13 and plasma14 of LM patients have been shown to reflect tumor evolution and are of potential use as a biomarker of treatment response. When compared with plasma ctDNA, CSF ctDNA demonstrates higher sensitivity to detect LM compared to traditional cytology15. Mutational analysis of ctDNA of the CSF reveals clinically relevant genomic alterations in patients with primary central nervous system (CNS) tumors and CNS metastases with higher allelic fractions compared to plasma-detected mutations16,17.
Even when detecting genetic variation within a tumor or ctDNA, most detected variants have unclear significance and clinical applications pose a significant challenge18,19. Several methods have been proposed to derive clinically relevant genetic alteration summary scores, with varying application. In patients with primary brain tumors or CNS metastases, variant allele frequencies (VAFs) have been shown to correlate with tumor burden and decrease with therapy (i.e. surgery and systemic therapy)12. However, the predictive value of ctDNA alterations and mutational summary measures in LM patients treated with pCSI remains unknown.
We analyzed the ctDNA in patients from our phase I trial of pCSI for solid tumor LM (NCT03520504). We assessed the changes in the mutational landscape that are specific to the selective pressure of pCSI. We also explored whether ctDNA summary measures, like mean VAF and mean CCF, which incorporates the inferred clonal architecture, can predict clinical outcomes in LM patients treated with pCSI.
Methods
For the patients on protocol receiving pCSI for LM, ctDNA from the CSF and plasma were collected at baseline, and at 1-month, 3-months and 6-months post-pCSI 2. Fourteen patients had both baseline CSF ctDNA measured and at least one follow-up timepoint measurement. Systemic therapy was held 7 days prior to initiation and during pCSI, with the exception of endocrine therapy and trastuzumab for breast cancer patients, and patients resumed their previous systemic therapy or initiated a new systemic therapy 4 weeks after completing pCSI. Per study exclusion criteria, patients with extensive systemic disease without reasonable systemic therapy options and with multiple, serious neurologic deficits were excluded. Patients were followed until death or censoring on December 31, 2020.
For all ctDNA samples, we performed targeted exome sequencing using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), an in-house panel assaying 505 genes20. All samples were compared to their germline tissue to determine somatic mutations, except for one patient who was missing germline tissue and was compared to a pooled normal tissue reference. The sequencing depth for each sample is reported in Supplementary Table 1. Mutations were independently called without any prior knowledge using total depth ≥20, variant allele depth ≥8 and variant allele frequency (VAF) ≥2% for known mutational hotspots and VAF ≥5% for novel mutations6 (Supplemental Table 2). On manual verification of all called variants, 14 variants were included that had a total read depth <20 but ≥10, because they occurred in multiple samples from the same patient or had a VAF >25%. All variants had at least 3 supporting reads which is the absolute minimum recommended allele depth to be considered a mutation21. For all mutations called in either the plasma or the CSF, a secondary analysis was performed for all other samples collected from the same patient, in which less stringent criteria were applied to detect the full compilation of alterations (called as present if variant allele depth ≥1). If these criteria were not met, a mutation was marked as not present and considered to be limited to the plasma or CSF. For this secondary mutation analysis, we report only the frequency of mutations that are common and unique to plasma and CSF as well as the overall agreement between the plasma and CSF pre- and post-pCSI using Cohen’s kappa statistic. Mutations pre- and post-pCSI were compared using a paired Wilcoxon signed rank test.
The mean VAF was calculated as the average proportion of sequencing reads showing any alteration22 the mean VAF pre-CSI (VAFpre) and post-CSI (VAFpost) were determined for all patients. The mean change in VAF pre- and post-pCSI (ΔVAF) was derived by calculating the average difference in VAF for each variant and using a value of zero for undetected variants (i.e. a mutation that is not detectable or was only detectable post-pCSI).
Allele-specific copy number variation and the clonality of mutations were determined using FACETS (version 0.3.9)23. A two-pass implementation with a low-sensitivity run (critical value = 100) was used to determine the purity and sample ploidy, and a high sensitivity run (critical value = 50) was used to determine gene-level segmentation and integer copy number calls. A threshold of +/−0.5 was used to call an amplification or deletion of a chromosomal segment. Samples with a purity that could not be estimated (typically ≤0.30) were excluded from the clonality analysis. Estimated purity of plasma and CSF ctDNA samples was compared using an unpaired Wilcoxon signed rank test. For 11 analyzable samples, pre-existing MSK-IMPACT mutation calls using an automated clinical pipeline were obtained for either their primary tumor or a metastatic tumor from prior biopsies. The clonal evolution that took place from initial biopsy (i.e. either primary tumor or metastasis) to ctDNA before pCSI, and ctDNA after pCSI was estimated using cancer cell fraction (CCF). The FACETS algorithm is an exploratory approach and with the default settings, mutations with a high estimated CCF are determined to be clonal whereas lower CCF estimates (i.e. ≤80%) indicate a subclonal mutation. For new subclonal mutations that were only detected after pCSI, we calculated the mean CCF (CCFSUBCLONAL). CCFSUBCLONAL was only calculated for all samples with a high enough purity to determine clonality using the default FACETS algorithm settings.
We separately analyzed all genes with alterations present in tumor and metastases, genes with alterations occurring in ctDNA of the CSF and plasma, genes with alterations that are unique to ctDNA of the CSF and plasma before pCSI and genes with alterations in ctDNA after pCSI. For these gene lists, we used Gene Set Enrichment Analysis (GSEA) to interrogate any significant overlap with Reactome pathways genes. We report all pathways with significant overlap at a false discovery rate (FDR) q value ≤0.05.
We identified an independent cohort of patients with CNS metastases, including brain metastases or LM, who had CSF ctDNA MSK-IMPACT testing at the time of diagnosis. Genomic variants had already been determined according to a clinically validated pipeline20. These patients were further stratified into those who had CNS-directed radiotherapy, defined as whole brain radiotherapy, spine radiotherapy, or both, as a component of their initial CNS metastatic disease treatment but did not have pCSI, and those who did not have radiotherapy. For these samples, the mean VAF at the time of diagnosis prior to intervention was calculated as specified previously.
We estimated overall survival (OS), defined as the time from the start of pCSI to death or last follow-up using the Kaplan Meier method. Analysis was performed to find associations between VAF, CCF and OS. CNS response and progression of disease was recorded per protocol using MRI and clinical assessment, and we also analyzed CNS progression free survival (PFS), defined at the time from start of pCSI to CNS progression, death or last follow-up2. Variables were either dichotomized by the median value or, for measures of change pre- and post-pCSI, a value close to zero was chosen to include patients with decreased and essentially no change. For the comparison cohort, OS was calculated as the time from initial therapy after diagnosis to death or last follow-up. Survival estimates are shown with 95% confidence intervals (CI). Unless otherwise specified, significance was assessed an alpha = 0.05.
Written informed consent to use patient samples was obtained from all participants in adherence with the MSKCC IRB. This study was conducted in accordance with the ethical guidelines outlined in the Belmont Report.
Data Availability
Raw data for this study were generated at the Memorial Sloan Kettering Cancer Center Integrated Genomic Operation. Derived data supporting the findings of this study are available from the corresponding author upon request. Other data generated in this study are available within the article and its supplementary data files.
Results
Study cohort characteristics
The patient characteristics are shown in Table 1. The median patient age was 53 years. The majority of patients (64%) had NSCLC. All patients received at least 1 systemic therapy since cancer diagnosis, and most patients were heavily pre-treated with the median number of lines of pre-CSI chemotherapy as 4 (range 1-9). At the study end, 79% (n=11) of patients had died and 79% (n=11) had CNS progression of disease. No patients had CNS progression within 3 months of pCSI, and though 6 patients had systemic progression before CNS progression, only 3 were within 3 months of the pCSI. The CNS and systemic disease status is shown in Supplemental Table 3, and most patients had stable systemic disease limited to bone metastases, pulmonary nodules, or no detectable lesions. The median OS for the cohort was 9 months (IQR: 5 – 21 months, Supplement Figure 1).
Table 1.
Patient characteristics prior to proton craniospinal irradiation (pCSI).
| Patient Characteristics | N=14 | ||
|---|---|---|---|
| Age | median (range) | 53 (30 - 62) | |
|
| |||
| Sex | Female | n (%) | 10 (71%) |
| Male | n (%) | 4 (29%) | |
|
| |||
| KPS | 70 | n (%) | 4 (29%) |
| ≥80 | n (%) | 10 (71%) | |
|
| |||
| Histology | NSCLC | n (%) | 9 (64%) |
| Breast | n (%) | 3 (21%) | |
| Esophageal | n (%) | 1 (7%) | |
| Rectal | n (%) | 1 (7%) | |
|
| |||
| Months from LM dx to pCSI | median (range) | 1 (0 - 24) | |
|
| |||
| Prior Lines of Chemotherapy | median (range) | 4 (1- 9) | |
Abbreviation: Karnofksy performance status (KPS), diagnosis (dx), leptomeningeal metastasis (LM)
Specific genetic alterations observed before and after pCSI
Of the 14 patients with pre-pCSI CSF ctDNA measured, 13 had a post-CSI CSF ctDNA measurement taken at 1-month post-pCSI and 1 patient had a measurement taken 3 months post- pCSI. We observed changes in genetic aberrations occurring in ctDNA on the CSF after pCSI compared to the baseline (Figure 1A), but there was no significant change in the overall number of observed mutations after pCSI (paired Wilcoxon signed rank p = 0.66, Figure 1B). A non-significant increase in the number of patients with altered GNAS, PIK3C2G, and NOTCH2 was observed after pCSI (Figure 1C). In patients with available data, we observed variants in ctDNA pre- and post-pCSI that can be classified as those that were: 1) present in the primary tumor or metastatic tumor (Supplemental Figure 2A), 2) present in ctDNA but were not observed in the primary or metastases (Supplemental Figure 2B), 3) present before pCSI but not detectable afterwards (Supplemental Figure 2C), or 4) only detectable after pCSI (Supplemental Figure 2D). Using GSEA analysis of ctDNA, we found that many altered genes were involved in transcription regulation such as the RUNX1 pathway, DNA damage repair, and cell cycle regulation, and there appears to be enrichment for altered transcription pre- and post-pCSI (Supplemental Table 4). GNAS, encoding the G-protein alpha subunit, was the only recurrently mutated gene after pCSI occurring in 2 patients.
Figure 1.

Mutational analysis CSF ctDNA from patients pre and post pCSI. A) An oncoplot of called mutations for CSF samples pre- and post-pCSI. Alteration type is identified by color. Each vertical column represents a patient; horizontal bars indicate primary cancer histology (NSCLC, breast or other) and the overall survival divided into less than 6 months, 6 months to 1 year and more than 1 year. The number of mutations called per sample is demonstrated in histogram. (n=14 patients). B) Genomic alterations detected in CSF pre- and post-pCSI (n=134 alterations pre, 172 alterations post; Wilcoxon signed-rank test p = 0.68) C) For the top recurrently mutated genes in CSF ctDNA, the number of patients with the listed alterations is shown pre- and post-pCSI.
Comparisons of plasma and CSF ctDNA alterations
We next compared mutations detected in the plasma and CSF ctDNA in pre-pCSI and post-pCSI samples. The purity of the ctDNA samples, determined using FACETS, from the plasma was significantly different than that of the CSF and typically lower (Wilcoxon rank sum p=0.001) (Supplemental Figure 3). The kappa statistic pre-pCSI comparing mutations found in the CSF and plasma was 0.255 (p<0.001) indicating fair agreement. Conversely, the kappa statistic post-pCSI comparing mutations found in the CSF and plasma was 0.005 (p=0.92) indicating no agreement. There were no differences in the distribution of clonal and subclonal alterations in CSF ctDNA pre and post-pCSI, Wilcoxon paired signed rank tests for clonal mutations, p=0.432, indeterminate mutations, p=0.779, and subclonal, p=0.922 (Supplemental Figure 4). While some patients showed a large number of shared mutations, there were a considerable number of mutations that were unique to the CSF ctDNA (Supplemental Figure 5). An increased number of mutations unique to the CSF at 1-month post-pCSI was associated with worse OS (HR: 1.24 per mutation [95% CI:1.05 - 1.45], p=0.009).
Variant allele fraction and association with survival
We then examined whether the mean VAF before and after pCSI was associated with survival outcomes. Having a higher VAFpre when dichotomized by the median (VAFpre=0.32) was associated with worse OS (median 6 months (95% CI: 5 months – not estimated (NE)) for VAFpre ≥0.32 vs 9 months (95% CI:9 months – NE) for VAFpre<0.32, p=0.05, Figure 2A). However, VAFpost (when dichotomized by the median, VAFpost=0.35) was not associated with differences in OS (median 8 months (95% CI: 5 months - NE) for VAFpost ≥0.35 vs 9 months (95% CI: 6 months – NE) for VAFpost <0.35, p=0.9). We instead considered whether ΔVAF, which increased for 5 patients and decreased or remained stable for 9 patients (Figure 2B) could predict survival outcomes. Patients with an increased ΔVAF after pCSI had significantly worse OS (median 6 months (95% CI: 5 months – NE) for ΔVAF ≥0.01 vs 18 months (95% CI: 9 months- NE) for ΔVAF <0.01, p=0.008, Figure 2C). Similarly, allele fractions incorporating clonality also predicted response to pCSI in LM patients: CCFSUBCLONAL greater than the median was associated with worse OS (median 8 months (95% CI: 5 months – NE) for CCFSUBCLONAL ≥0.61 vs. 17 months (95% CI: 6 months – NE) for CCFSUBCLONAL <0.61, p=0.05) (Figure 2D). We did not find significant associations with CNS PFS.
Figure 2.

Variant allele fraction and cancer cell fraction associations with overall survival (OS). A) pre-pCSI VAF was dichotomized, (median VAF=0.32) and associated with OS using Kaplan-Meir analysis and a log-rank test. p=0.05. B) Change in VAF pre and post pCSI. n=13 patients. C) The change in VAF pre- and post-pCSI was dichotomized using a threshold of 0.01 to associate having stable or decreased VAF (n=9) compared to increased VAF (n=5) with OS using Kaplan Meier analysis and log-rank test (p=0.008). D) The mean CCF was dichotomized by the median (median CCF=0.61) and having a mean CCF above the median (n=5) compared to below the median (n=6) was associated with OS using Kaplan Meier analysis and log-rank test. (p=0.05).
Clonal evolution for select patients
We present the putative framework for clonal evolution observed in ctDNA (Figure 3A) and for three patients, clonal evolution from a solid tumor to leptomeningeal metastasis is shown along with treatment history in Figure 3B–D. For patient 10, who had a relatively poor survival (OS=8.5 months), we detected the emergence of multiple alterations after pCSI (Figure 3B). In contrast, for patient 5, who had extended survival (OS=28 months), we did not appreciate a substantial increase in detected alterations after pCSI (Figure 3C). For patients with analyzable ctDNA in both the plasma and the CSF, we examined clonal evolution and show an example of a patient with poor survival (OS=3.6 months) in Figure 3D, to demonstrate that alterations can be shared between ctDNA in the plasma and CSF or unique to each compartment. Through this approach, it is possible to test whether pCSI provides a selection pressure for clonal evolution. While there are some alterations unique to each compartment, we observe that only the CSF demonstrated an increase in observed alterations, some of which are clonal mutations (SESN3, BRD4, CCNE1, GNAS) and CNA with loss of EGFR and FGFR.
Figure 3.
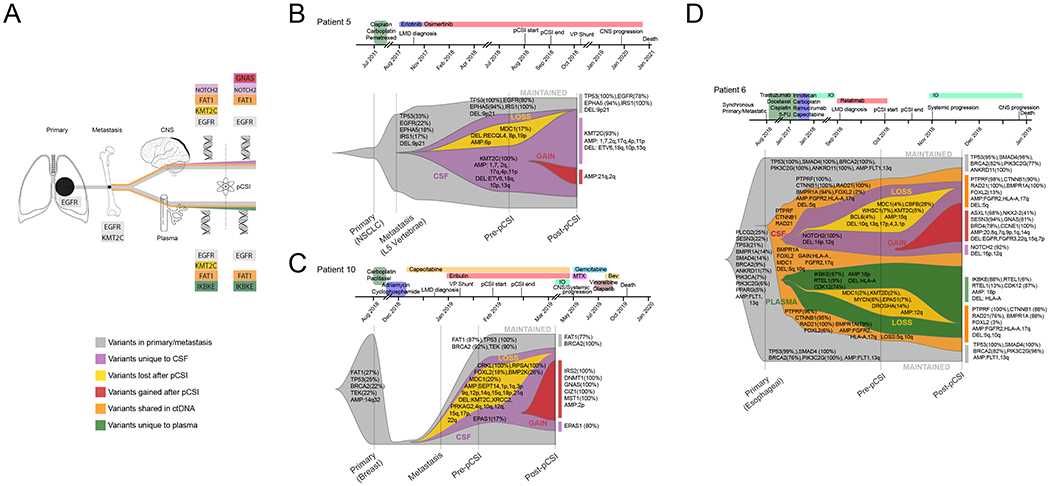
Clonal evolution from primary tumor to leptomeningeal metastasis. A schematic explaining the information conveyed in the fish plots in shown (A). Example genes are shown that are altered in the original tissue(s) (gray), altered in ctDNA that are not observed in preceding tissue (orange), altered in CSF ctDNA occurring both pre- and post-pCSI (purple), altered in plasma ctDNA occurring both pre- and post-pCSI (green), lost after pCSI (gold) and gained after pCSI (red). For some altered genes, multiple and overlapping colored rectangles are shown to demonstrate that a gene can be altered in different contexts (e.g. unique to CSF ctDNA and gained after pCSI). Clonal evolution is shown for studied patients without plasma ctDNA (B and C) and with plasma ctDNA (D). The clinical history is indicated above each patient. Cancer cell source is indicated along X axis. The left-most alterations indicate those present in a primary or a solid tumor metastasis with VAF in parentheses. At each subsequent stage, the altered genes with CCF are in parenthesis are shown classified by category of mutation (e.g lost after pCSI). For CNV, a threshold of +/−0.5 was used to define an amplification or deletion. D) Clonal evolution for a patient including information about plasma ctDNA is shown which allows an additional category of mutations shared by plasma and CSF ctDNA (orange) to be considered.
VAF in a comparison cohort of LM patients who did not receive pCSI
We identified 50 patients with CSF MSK-IMPACT performed at CNS metastasis diagnosis. Within this cohort, 60% (n=30) received non-pCSI CNS radiotherapy; 40% (n=20) did not receive radiotherapy. Like our study patient population, the comparison cohort consisted primarily of lung (42%, n=21) and breast cancers (42%, n=21). There was no difference in median OS for the patients who received radiotherapy vs. those who did not (p=0.70). Among patients who received radiotherapy, lower CSF VAF at baseline was associated with improved OS (median 8 months (95% CI: 4 months – 15 months) vs. 9 months (95% CI- 7 months- NE) in with lower CSF VAF at baseline, p=0.05), while no difference in OS by CSF VAF was .govn in patients who did not receive RT (p=0.10) (Figure 4). Therefore, pre-treatment CSF VAF may be a predictor of survival in patients with solid tumor CNS metastases who receive radiotherapy.
Figure 4.

Comparison cohort of 50 patients with CNS metastases treated without RT (n=20) or with CNS-directed non-pCSI RT (n=30). Increased VAF prior to CNS-directed treatment is significantly associated with worse OS for patients that had RT (p=0.05), right panel, but not for those without RT (p=0.10), left panel.
Discussion
In this study, we examined the genetic variation of ctDNA before and after pCSI in patients with LM in a subset of a patients who were treated on a phase I trial of pCSI for solid tumor LM. We found that the pre- and post-pCSI VAF and the change in VAF may have utility as a clinical biomarker. We also provided evidence that pCSI may exert a selective pressure in the CSF compartment potentially inducing genomic changes that are specific to LM treated with pCSI. To our knowledge, this is the first study to evaluate CSF ctDNA alterations associated with pCSI for LM. Though we detected associations with CSF ctDNA VAFpre,, ΔVAF, CCFSUBCLONAL, and OS, we did not find any association between these measures and CNS PFS. We have previously shown that pCSI is associated with improved CNS PFS and OS in LM patients3, and we are actively pursuing biomarkers that may be associated with both outcomes11. It is possible that evaluating genetic changes detected in the CSF at 1 month for most patients was too early for detectable associations with CNS PFS, and a larger study with additional, regularly-obtained ctDNA measurements may be needed.
For patients with CNS metastases, it is not currently standard of care to incorporate detectable genetic aberration, especially for variants of unknown significance, into clinical decision making for CNS-directed radiotherapy. In this study, we showed that the VAF is a simple measurement that can be applied clinically to stratify patient response to pCSI and target high-risk patients for earlier intervention after pCSI. The ability to calculate VAF likely reflects both the tumor burden and the degree of tumor heterogeneity, and we showed the higher pre-pCSI VAF as well as an increasing change in VAF after pCSI are associated with worse OS. Our results are consistent with reports demonstrating that a higher tumor burden in the CNS is associated with worse outcomes in LM24.
Because the VAF does not take into account the clonality of mutations in ctDNA or the purity of the cfDNA sample, we also demonstrated that CCF calculated only using subclonal mutations is a significant predictor of survival after pCSI for LM in a cohort of patients with multiple types of cancer, supporting the robustness of ctDNA mutational analysis in this context. Given our limited sample size and heterogenous cohort, we were not powered to discriminate between small differences in the CCF of clonal mutations, which generally only had values greater than 80%. Future studies will likely be able to determine whether there is biologic significance to altered clonal mutations before and after pCSI, and it is possible that this effect will be different by histology, requiring a stratified analysis. In addition, we compared our findings with a cohort of patients with CNS metastases who did not receive pCSI and found that VAF of CSF ctDNA was associated with OS after radiotherapy, indicating potential broad application for response in patients with CNS metastasis undergoing radiotherapy, although a larger validation cohorts with statistical analysis controlling for potential confounders are needed to understand the relationship between CSF ctDNA VAF and OS after radiotherapy for CNS metastasis.
Targeted exome sequencing revealed low tumor purity and clonal architecture that could not be assessed in plasma ctDNA samples compared with CSF ctDNA. Although alternative methods have been demonstrated to have better efficacy in sequencing low purity plasma ctDNA using a hybridization capture of a smaller panel of genes and deep sequencing utilizing techniques to suppress background sequencing errors25, the high purity of ctDNA in the CSF may overcome the known limitations of bulk sequencing of heterogeneous solid tumor or competing cfDNA in the plasma16. In a cohort of patients with CNS tumors of which 3 had LM, CSF ctDNA was shown to have a greater mutational diversity compared to plasma ctDNA26. Similarly, in patients with brain metastases, mutational diversity of CSF ctDNA was observed to be distinct from tumor primaries and plasma ctDNA12,25,27. In this context, CSF ctDNA may be an ideal, highly-sensitive method to detect subclonal mutations. We show that using the exploratory CCF estimates of the subclonal mutations is also associated with OS.
We detected substantial genetic mutational and copy number diversity in the CSF of LM patients, even in a limited cohort. By tracing the presence of mutations from primary/metastatic tissue to ctDNA (Figure 3A), we found that many of the observed mutations in ctDNA were present in preceding solid tumor tissue, and we showed that the presence of more alterations before pCSI was associated with worse OS. Interestingly, while we observe that some variants are not detectable after pCSI, some patients have an increased genetic diversity after treatment, and larger increases in detected alterations are associated with poorer survival after pCSI (Figure 3B–C). We also demonstrated that there are genetic variants that are unique to the CSF ctDNA, consistent with prior findings in LM patients showing CNS-limited single nucleotide variants16,28 (Figure 3D). Overall, our findings suggest that there could be mutations that occur in response to pCSI, and radiation is known to lead to genetic variation through several mechanisms29. While alterations to ctDNA after pCSI are expected, it remains unclear why some patients would develop new clonal and subclonal mutations and others would have decreased alterations after pCSI. This difference may be related to defective DNA damage response (DDR) pathways in some tumors, which may also be a risk factor for poor patient survival. Many of the detected mutations after pCSI involve genes implicated in DDR, cell cycle control, and radiosensitivity, such as ARID1B30, EPHB231, BRD432, CCNE133, NKX2-234, RTEL135, and CDKN2A36.
There are several possible sources of observed genetic variation that may correlate with outcomes independently of pCSI. Because patients in this study, and LM patients in general, have often had multiple systemic therapies, it is likely that the observed mutations reflect prior selective pressures. While the baseline ctDNA would reflect the selective pressures of prior treatment, which we would also expect to vary by histology, this cohort represents a heterogenous sample of leptomeningeal metastasis patients. A larger, stratified cohort could help distinguish the contribution of prior systemic therapy from the unique metastatic and leptomeningeal genetic signals. Given that the bulk of our VAF measurements were pre-pCSI compared to 1-month post-pCSI, the changes have a higher probability of being related to the pCSI than to the ongoing systemic therapy, which was held until 1month post-pCSI. In addition, the observed ctDNA in the plasma and CSF may reflect systemic and CNS disease, respectively, but the possibility of ctDNA crossover remains. Though the blood brain barrier is thought to minimize plasma ctDNA transfer to the CSF37, CNS confined disease was demonstrated to have some ctDNA circulating in the periphery at relatively low levels38. In the present study, only 3 patients had systemic disease progression before CNS progression within 3 months of pCSI; 2 had a decrease in CNS ΔVAF and 1 showed an increase, indicating that the presence of systemic disease is not likely biasing our findings.
There are several limitations to this study. This study is exploratory in nature with a small cohort, so we did not perform correction for multiple comparisons; thus, all specific cut points, thresholds, and biomarkers may be unique to this cohort and, therefore, require further study. Though only a subset of the trial patients underwent both pre- and post-pCSI ctDNA measurements, the median OS was similar between this cohort and the full phase I trial cohort2, and a larger cohort8 of patients treated with pCSI for LM. The sample set was too small to detect many common mutations between samples; we overcame this limitation by focusing on summary measures and pathway level findings. We also chose mutation calling parameters that were more permissive, while still requiring sufficient evidence for mutation calls, to increase our mutation calling sensitivity. This allowed us to broaden our analysis of a limited number of patients at the expense of specificity, and we performed manual validation of called variants to increase our confidence in mutation calls. However, as a result, the presence or absence of an alteration cannot exclude false positives or lack of coverage, respectively. Lastly, we reported the CCF as determined using FACETS, but CCF is still an exploratory method, and the clinical utility and interpretation of CCF requires additional study.
Conclusion
Proton CSI is an emerging and promising therapy for patients with LM. Analysis of ctDNA from LM patients before and after pCSI shows that pCSI efficacy may depend on genomic characteristics before treatment and may exert genomic selection after treatment. We found that patients with improved outcomes are associated with lower frequency of mutations, and in particular, a lower mean VAF. We showed that the quantification of these mutations has important implications for patient stratification in CNS-directed radiotherapy.
Supplementary Material
Translational Relevance.
Proton craniospinal irradiation (pCSI) is a promising treatment for patients with leptomeningeal metastasis (LM). Circulating tumor DNA (ctDNA) in the CSF should provide insight into the dynamic processes occurring during LM development and pCSI treatment. By quantifying the mean variant allele fraction (VAF) of gene alterations, we demonstrate that genetic changes in CSF ctDNA resulting from pCSI are associated with clinical response to pCSI. Like VAF, we also provide evidence that the cancer cell fraction, which adjusts the VAF for cancer cell clonality, can be analyzed to understand the dynamic response to pCSI. These findings indicate that the genetic signals in CSF ctDNA can be used in the clinic to guide LM treatment choices, such as patient selection for pCSI and dose escalation for patients at higher risk of disease progression after pCSI. Furthermore, we show that quantifying VAF may predict response to all central nervous system metastasis irradiation.
Financial support:
This work was supported by grants from Cycle for Survival Equinox Innovation Initiative Award (to Yang) and the National Institutes of Health/National Cancer Institute (P30-CA008748 and R01 CA245499 (to Boire))
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.Chowdhary S, Chamberlain M. Leptomeningeal metastases: current concepts and management guidelines. Journal of the National Comprehensive Cancer Network. 2005. Sep 1;3(5):693–703. [DOI] [PubMed] [Google Scholar]
- 2.Yang TJ, Wijetunga NA, Yamada J, Wolden S, Mehallow M, Goldman DA, et al. Clinical trial of proton craniospinal irradiation for leptomeningeal metastases. Neuro-oncology. 2021. Jan 30;23(1):134–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang JT, Wijetunga NA, Pentsova E, Wolden S, Young RJ, Correa D, et al. Randomized Phase II Trial of Proton Craniospinal Irradiation Versus Photon Involved-Field Radiotherapy for Patients With Solid Tumor Leptomeningeal Metastasis. Journal of Clinical Oncology. 2022. Jul:JCO–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Progress in nucleic acid research and molecular biology. 1988. Jan 1;35:95–125. [DOI] [PubMed] [Google Scholar]
- 5.Gao Y, Duan Q, Wu N, Xu B. A heterogeneous cellular response to ionizing radiation revealed by single cell transcriptome sequencing. American journal of cancer research. 2021;11(2):513. [PMC free article] [PubMed] [Google Scholar]
- 6.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019. Jan;565(7741):654–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straathof CS, de Bruin HG, Dippel DW, Vecht CJ. The diagnostic accuracy of magnetic resonance imaging and cerebrospinal fluid cytology in leptomeningeal metastasis. Journal of neurology. 1999. Sep;246(9):810–4. [DOI] [PubMed] [Google Scholar]
- 8.Zheng MM, Li YS, Jiang BY, Tu HY, Tang WF, Yang JJ, et al. Clinical utility of cerebrospinal fluid cell-free DNA as liquid biopsy for leptomeningeal metastases in ALK-rearranged NSCLC. Journal of Thoracic Oncology. 2019. May 1;14(5):924–32. [DOI] [PubMed] [Google Scholar]
- 9.Malani R, Fleisher M, Kumthekar P, Lin X, Omuro A, Groves MD, et al. Cerebrospinal fluid circulating tumor cells as a quantifiable measurement of leptomeningeal metastases in patients with HER2 positive cancer. Journal of neuro-oncology. 2020. Jul;148(3):599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doherty CM, Forbes RB. Diagnostic lumbar puncture. The Ulster medical journal. 2014. May;83(2):93. [PMC free article] [PubMed] [Google Scholar]
- 11.Wijetunga NA, Boire A, Young RJ, Yamada Y, Wolden S, Yu H, et al. Quantitative cerebrospinal fluid circulating tumor cells are a potential biomarker of response for proton craniospinal irradiation for leptomeningeal metastasis. Neuro-oncology advances. 2021. Jan;3(1):vdab181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattos-Arruda D, Mayor R, Ng CK, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nature communications. 2015. Nov 10;6(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan C, Diplas BH, Chen X, Wu Y, Xiao X, Jiang L, et al. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta neuropathologica. 2019. Feb;137(2):297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, Barkhoudarian G, et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS oncology. 2019. Jun 1;8(2):CNS34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White MD, Klein RH, Shaw B, Kim A, Subramanian M, Mora JL, t al. Detection of leptomeningeal disease using cell-free DNA from cerebrospinal fluid. JAMA Network Open. 2021. Aug 2;4(8):e2120040-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YS, Jiang BY, Yang JJ, Zhang XC, Zhang Z, Ye JY, et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Annals of Oncology. 2018. Apr 1;29(4):945–52. [DOI] [PubMed] [Google Scholar]
- 17.Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. Journal of clinical oncology. 2016. Jul 7;34(20):2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. BMC medicine. 2012. Dec;10(1):1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Febbo PG, Ladanyi M, Aldape KD, De Marzo AM, Hammond ME, Hayes DF, et al. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. Journal of the National Comprehensive Cancer Network. 2011. Nov 1;9(Suppl_5):S–1. [DOI] [PubMed] [Google Scholar]
- 20.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. The Journal of molecular diagnostics. 2015. May 1;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kassahn KS, Holmes O, Nones K, Patch AM, Miller DK, Christ AN, et al. Somatic point mutation calling in low cellularity tumors. PloS one. 2013. Nov 8;8(11):e74380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Luo J, Wu S, Si H, Gao C, Xu W, et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint BlockadectDNA Dynamics as a Biomarker of Response to Immunotherapies. Cancer discovery. 2020. Dec 1;10(12):1842–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic acids research. 2016. Sep 19;44(16):e131-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nevel KS, DiStefano N, Lin X, Skakodub A, Ogilvie SQ, Reiner AS, et al. A retrospective, quantitative assessment of disease burden in patients with leptomeningeal metastases from non-small-cell lung cancer. Neuro-oncology. 2020. May 15;22(5):675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nature Communications. 2021. Jun 18;12(1):1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan W, Gu W, Nagpal S, Gephart MH, Quake SR. Brain tumor mutations detected in cerebral spinal fluid. Clinical chemistry. 2015. Mar 1;61(3):514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mouliere F, Mair R, Chandrananda D, Marass F, Smith CG, Su J, et al. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO molecular medicine. 2018. Dec;10(12):e9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge M, Zhan Q, Zhang Z, Ji X, Zhou X, Huang R, et al. Different next-generation sequencing pipelines based detection of tumor DNA in cerebrospinal fluid of lung adenocarcinoma cancer patients with leptomeningeal metastases. BMC cancer. 2019. Dec;19(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cannan WJ, Pederson DS. Mechanisms and consequences of double-strand DNA break formation in chromatin. Journal of cellular physiology. 2016. Jan;231(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, Stuschke M. Targeting ARID1A-mutant colorectal cancer: depletion of ARID1B increases radiosensitivity and modulates DNA damage response. Scientific Reports. 2019. Dec 3;9(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatia S, Hirsch K, Bukkapatnam S, Baig NA, Oweida A, Griego A, et al. Combined EphB2 receptor knockdown with radiation decreases cell viability and invasion in medulloblastoma. Cancer cell international. 2017. Dec;17(1):1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013. Jun;498(7453):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caldon CE, Musgrove EA. Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell division. 2010. Dec;5(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Zheng J, Zhang A, You Z. A six-gene-based signature for breast cancer radiotherapy sensitivity estimation. Bioscience reports. 2020. Dec 23;40(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O’Neil NJ, Petalcorin MI, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008. Oct 17;135(2):261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (p16) gene and human cancer. Molecular medicine. 1997. Jan;3(1):5–20. [PMC free article] [PubMed] [Google Scholar]
- 37.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N,et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci Transl Med. 2014; 6 (224): 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izquierdo E, Proszek P, Pericoli G, Temelso S, Clarke M, Carvalho DM, et al. Droplet digital PCR-based detection of circulating tumor DNA from pediatric high grade and diffuse midline glioma patients. Neuro-oncology advances. 2021. Jan;3(1):vdab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for this study were generated at the Memorial Sloan Kettering Cancer Center Integrated Genomic Operation. Derived data supporting the findings of this study are available from the corresponding author upon request. Other data generated in this study are available within the article and its supplementary data files.