

Summary
Long-read sequencing (LRS) facilitates both the genome assembly and the discovery of structural variants (SVs). Here, we built a graph-based pig pangenome by incorporating 11 LRS genomes with an average of 94.01% BUSCO completeness score, revealing 206-Mb novel sequences. We discovered 183,352 nonredundant SVs (63% novel), representing 12.12% of the reference genome. By genotyping SVs in an additional 196 short-read sequencing samples, we identified thousands of population stratified SVs. Particularly, we detected 7,568 Tibetan specific SVs, some of which demonstrate significant population differentiation between Tibetan and low-altitude pigs, which might be associated with the high-altitude hypoxia adaptation in Tibetan pigs. Further integrating functional genomic data, the most promising candidate genes within the SVs that might contribute to the high-altitude hypoxia adaptation were discovered. Overall, our study generates a benchmark pangenome resource for illustrating the important roles of SVs in adaptive evolution, domestication, and genetic improvement of agronomic traits in pigs.
Subject areas: Animals, Genomics, Phylogenetics
Graphical abstract
Highlights
-
•
De novo genome assemblies for 10 pigs using long-read sequencing
-
•
Built a graph-based pig pangenome revealing 206-mb novel sequences
-
•
Discovered 183,352 nonredundant structural variation with 63% novel
-
•
Tibetan specific structural variations associated with the high-altitude adaptation
Animals; Genomics; Phylogenetics
Introduction
Pigs (Sus scrofa) are not only one of the most important livestock species because of their enormous food supply but also important model animals in many areas of biomedical research because of their high anatomical, pathological, and physiological similarity to humans.1,2 Long-term local adaptation and artificial selection have resulted in a variety of breeds with abundant phenotypes in pigs around the world, e.g., body size, fertility, growth, resistance to disease, high-altitude hypoxia adaptation, and cold or hot climate adaptation. This provides a valuable resource for understanding the genetic basis of complex phenotypes, domestication and adaptive evolution in animals as well as in humans.
A single reference genome from one individual is not enough to represent the genomic diversity of all breeds within a species.3,4 Hence, the concept of the pangenome is proposed, which incorporates the nonredundant collection of DNA sequences in a species.5,6 Although the current pig reference genome (Sscrofa11.1) has greatly facilitated genetic and genomic studies in pigs,7 it was highly limited because it is a single Duroc individual, which might result in the inability to identify many important variants because of the distinct genetic backgrounds of different breeds, such as Chinese and European pigs.8 Pangenomic analysis has been proposed to improve the mapping rate of sequences, variant discovery, and genetic associations of both disease and economic traits.4,9,10 In plants, pangenomic analysis has greatly facilitated improvements in various crops, including rice, tomato and soybean, by uncovering numerous associations between agronomic traits and presence-absence variations (PAVs) in specific genes.11,12 In goats, compared to the ARS1 reference genomes,13 a total of 38.3 Mb of new sequences were detected by de novo assembly of nine genomes using short-read sequencing (SRS) technology. In pigs, a previous pangenome analysis based on 12 SRS de novo assemblies revealed a total of 72.5 Mb nonredundant sequences that were missing in the Sscrofa11.1 reference genome.10 However, these pangenomes in animals were highly limited in terms of both small population size and sequence technology. In addition, structural variants (SVs), as an important part of the pansequence,14 were not fully investigated, partly because of the limitation of the SRS technology, hindering their further applications in association with complex traits and adaptive evolution.
SVs are defined as genomic variants over 50 bp in length,15 including deletions (DELs), duplications (DUPs), insertions (INSs), inversions (INVs), translocations and complexes or nested variants thereof.16 They often contribute to greater nucleotide variation than single nucleotide variants (SNVs).17 Numerous studies have implicated SVs in human diseases,18,19 e.g., autism, cancer, and high-altitude hypoxia adaptation in the Tibetan population and body weight adaptation.20,21,22 A comprehensive set of SVs has been identified to be associated with key phenotypic traits in diverse plants and animals, including kernel weight in maize,23 fruit shape, fruit flavor, flowering, and fertility in tomato,24 production in soybean,9 fruit maturity and shape in peach,25 the dominance of a white or patch coat in European domestic pigs,26 white coat color in sheep27 and water buffalo,28 color sidedness in cattle.29 However, these studies have only deciphered a small set of SVs that contribute to complex traits in plants and animals because SVs have not been fully and accurately discovered, particularly in animals, owing to the limitations of BeadChip or SRS.30 Compared to SRS, long-read sequencing (LRS) allows us to detect SVs more accurately and comprehensively, particularly for complex SVs, and has the advantage of directly sequencing naked DNA with read lengths averaging approximately 10 kb and up to more than 1 Mb.31
In this study, by combining LRS and SRS technologies, we sequenced whole-genomes for eight Chinese domestic pigs, one European domestic pig and one European wild pig. First, we constructed a pig pangenome reference using the ten de novo assemblies together with a published Bama miniature (Bamamini) genome assembly. Second, we built an atlas of SVs in pigs and genotyped additional 196 pigs with SRS data to explore the SV spectrum on a population scale. Finally, to explore the contribution of SVs to complex phenotypic traits, we detected candidate SVs that are associated with high-altitude adaptation in Tibetan pigs. In summary, we offered the community a valuable pangenome resource of genomic variants in pigs, particularly for SVs, and provided novel biological insights into the roles of these variants in phenotypes and adaptation.
Results
De novo assembly of ten representative pig genomes
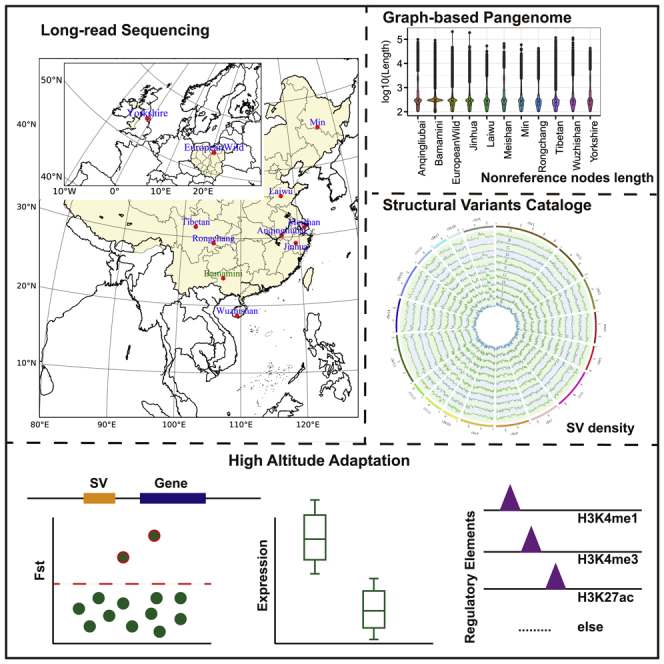
Local Chinese pigs can be divided into six categories, including the North China Type, South China Type, Central China Type, Lower Changjiang River Basin Type, Southwest Type, and PlateauType.32 At least one sample is selected for all these types. Moreover, one European commercial pig and a European wild boar were also collected. Finally, we sequenced the genomes of 10 distinct pig breeds, including eight geographically distributed local breeds in China (Anqingliubai, Jinhua, Laiwu, Meishan, Min, Rongchang, Tibetan, and Wuzhishan) and two originating from Europe (European wild and Yorkshire) using Nanopore PromethION sequencing technology (Figures 1A and 1B). On average, we obtained 7.95 M reads with a median read length of 28.08 kb (mean = 20.96 kb) and a coverage of 55.22-fold per sample (Figure 1B; Table 1). After error correction and genome assembly, we generated 910 contigs with an average contig N50 length of 32.48 mega-base pairs (Mb) per sample, resulting in an average genome size of 2.40 giga-base pairs (Gbp) (Figure 1B; Table 2). These assembled genomes attained an average of 94.01% (ranging from 92% to 94.7%) BUSCO completeness score (Figure S1), suggesting that they are potentially in high quality.
Figure 1.

Overview of samples and datasets
(A) Geographic distribution of the samples used in our study across Eurasia. The sequence data of Bama miniature were downloaded from public databases.
(B) Left panel: SNP-based phylogenetic tree based on Illumina sequencing of the 10 sequenced pig genomes. Middle panel: Sequence N50 of the ONT reads for each sample. Right panel: Contig N50 for each sample.
(C) The pangenome size changes with the added assembly.
(D) The length of nonreference nodes for each assembly added.
(E) The length of core, private and variable nodes for all pangenome nodes and nonreference nodes. The Wilcoxon rank-sum test was used for significance test, ∗∗∗ indicates p-value ≤0.001 and ∗∗∗∗ indicates p value ≤ 0.0001.
(F) Subgraphs of gap18 (Chr3:59,594,615-59,594,715, 100 bp) and gap4 (Chr1:214,753,360-214,753,460, 100 bp) in the reference genome Sscrofa11.1. Reference nodes are colored in green, whereas the nonreference nodes are colored in blue. Gap18 and gap4 are represented in s81414 and s37294 respectively.
(G) Improvement in mappability using the pangenome. Comparison of the mapping ratio of 24 resequencing datasets using the pangenome constructed in this study by LRS (panONT) and the pig reference genome Sscrofa11.1 (Sus11). ∗∗∗∗ indicates p value ≤ 0.0001 with two-tailed paired t test.
Table 1.
Long-read sample information for SV discovery
| Sample | Source | Sex | Seq Num | N50 Len | Mean Len | Mean Depth | Coverage (%) | Mapping Ratio (%) |
|---|---|---|---|---|---|---|---|---|
| Anqingliubai | This study | M | 6,416,566 | 36,465 | 27,120 | 62.83 | 98.35 | 97.34 |
| EuropeanWild | This study | M | 10,831,444 | 20,186 | 14,086 | 53.62 | 98.43 | 95.38 |
| Jinhua | This study | F | 5,698,043 | 36,517 | 26,539 | 54.14 | 97.83 | 97.06 |
| Laiwu | This study | M | 6,939,893 | 27,189 | 21,857 | 52.62 | 98.29 | 93.01 |
| Meishan | This study | M | 6,335,778 | 29,151 | 24,693 | 57.03 | 98.16 | 97.89 |
| Min | This study | M | 7,385,966 | 25,296 | 20,479 | 55.55 | 98.23 | 97.76 |
| Rongchang | This study | M | 6,370,478 | 30,677 | 24,116 | 55.2 | 98.14 | 96.99 |
| Tibetan | This study | M | 13,021,069 | 24,159 | 11,574 | 51.16 | 98.22 | 89.42 |
| Wuzhishan | This study | M | 10,443,802 | 23,255 | 15,731 | 58.34 | 98.22 | 96.75 |
| Yorkshire | This study | M | 6,049,845 | 27,896 | 23,441 | 51.66 | 98.44 | 98.00 |
| Bamamini | SRR9851973 | M | 1,871,364 | 25,057 | 20,288 | 13.85 | 96.61 | 97.35 |
A summary of the samples used in this study. “Source” denotes whether the genome was generated in this study or by other studies. “N50 Len” represents the N50 length.
Table 2.
Basic statistics of the assembly
| Assembly | Contig Number | Contig N50 | Genome Size (bp) |
|---|---|---|---|
| Anqingliubai | 923 | 41,403,868 | 2,431,005,403 |
| EuropeanWild | 1,023 | 28,389,353 | 2,405,396,245 |
| Jinhua | 831 | 35,656,463 | 2,397,491,931 |
| Laiwu | 959 | 31,833,351 | 2,347,999,432 |
| Meishan | 889 | 29,048,242 | 2,375,004,806 |
| Min | 1,028 | 35,544,885 | 2,411,955,771 |
| Rongchang | 979 | 35,927,340 | 2,410,787,528 |
| Tibetan | 467 | 28,944,720 | 2,447,452,149 |
| Yorkshire | 946 | 29,608,198 | 2,391,390,410 |
| Wuzhishan | 1,057 | 28,456,181 | 2,388,962,303 |
Pangenome of eleven de novo pig genome assemblies
By integrating our 10 newly generated de novo pig assemblies and an published assembly from Bama miniature breed using the minigraph toolkit,33 we constructed a pig pangenome graph, which consisted of 552,018 segments (referred to as nodes) and 664,789 links (referred to as edges), containing 2,705,225,506 total bases (Figure 1C). With the incremental integration of Anqingliubai, Bamamini, European wild, Jinhua, Laiwu, Meishan, Min, Rongchang, Tibetan, Wuzhishan and Yorkshire assemblies, we added 44,814, 19,059, 15,428, 21,820, 20,316, 17,624, 16,311, 15,806, 14,224, 21,489, and 14,192 nonreference nodes (221,083 in total), respectively, spanning 43.19, 17.76, 12.07, 19.50, 18.75, 18.92, 14.43, 16.11, 9.20, 21.03, and 14.49 Mb (206 Mb in total), respectively. Tibetan nonreference nodes were short on average compared with other assemblies (Figure 1D). Following a previous study,34 we colored nodes of the pangenome graph and obtained 99.99% accuracy of mapping as indicated by an F1 score. Approximately 77.29% of the pangenome sequence (157,534 nodes with a cumulative length of 2.09 Gb) was core (present in all assemblies), 15.77% of the sequence (241,658 nodes with a cumulative length of 426.71 Mb) was variable (present in more than one assembly but not all assemblies), and the remaining 6.94% of the sequence (152,825 nodes with a cumulative length of 187.71 Mb) was private (present in only one assembly) (Figure 1C). Most of the nonreference nodes and sequences were private to a single assembly (73.51% of all the nonreference sequence lengths, 151.79 Mb), followed by the sequence shared across all assemblies (0.95%, 1.97 Mb) (Figure S2). The mean length of the nodes of the core sequence (21.07 kb and 13.27 kb for nonreference nodes and all nodes, respectively) was significantly longer than that of the codes in the private (1.03 kb and 1.23 kb for nonreference nodes and all nodes, respectively) or variable sequences (0.74 kb) whether for nonreference nodes or all pangenome nodes (Figure 1E). A total of 241,263 bubbles were identified in the graph-base pig pangenome. There were 728 nonreference nodes in the bubbles across the fixed gaps in the current pig reference genome (Sscrofa11.1). For example, the gap18 presented in s81414 node across with the nonreference node s445965 (a simple bubble), and the gap4 presented in s37294 node across with the nonreference nodes of s473270 and s555154 (Figure 1F) (a more complex bubble). Moreover, we found a total of 395 gaps (76%) in the reference genome that can be partially or fully closed (Table S1).
We compared the mappability between Sscrofa11.1 and the new reference pangenome based on publicly available deep SRS genomes (Table S2). A total of 24 samples were randomly selected and separately mapped to Sscrofa11.1 and the new reference pangenome for comparison. The average mapping rate of the 24 samples to the new reference pangenome was increased by 0.17% compared that to Sscrofa11.1 (99.65% versus 99.48%, respectively; p <0.0001, two-tailed paired t test) (Figure 1G), which showed that the reference pangenome has higher power in subsequent variant calling analysis.
Discovery of SVs
According to a previous study,35 we employed a read-mapping-based approach to call SVs from Oxford Nanopore Technologies (ONT) sequencing long reads, yielding an average mapping rate of 96.09% to the pig reference genome Sscrofa11.1 (from 89.42% to 98.00% across individuals) using NGMLR (Table 1). For each LRS dataset, we detected SVs with lengths greater than 50 bp, including INSs, DELs, INVs and DUPs, using Sniffles.35 On average, we observed 67,225 SVs (ranging from 32,893 to 83,489) in each genome, covering 96.83 Mb (ranging from 26.43 Mb to 120.35 Mb). DELs and INSs accounted for most of the SVs, and each sample contained on average 48.64% DELs, 1.79% DUPs, 48.37% INSs and 1.21% INVs (Table S3). With respect to the SV length, each sample contained an average of 43.21%, 26.95%, 15.17% and 14.67% for DELs, DUPs, INSs and INVs, respectively, of the total base pairs (bp) of SVs (Table S3). Although the number of INVs and DUPs was low, these SVs contributed substantially to the total length of SVs because of their large size.
As illustrated in Figure 2A, we classified all identified SVs of the 11 de novo assembled pig genomes into three categories: shared SVs (present in all samples), polymorphic SVs (present in more than one sample but not all samples) or singleton SVs (breed-specific SVs, only present in one sample) (Figures 2A–2C). Among these individuals, relatively few SVs were detected in Bamamini, Yorkshire, and European wild pigs. The low number of SVs detected in Yorkshire and European wild pigs was likely because of their close relationship with the reference genome Sscrofa11.1 that was derived from a Duroc pig. The low sequencing depth of 13X for Bamamini led to fewer detected SVs. In addition, Figure 2A further demonstrates that polymorphic SVs dominated all the variants, whereas 5,780 shared SVs only contributed a low proportion (3.15%) of all identified SVs. Singleton SVs also had a low contribution in each breed. We obtained 5,075 (6.80%), 1,255 (3.82%), 6,070 (12.64%), 5,253 (6.90%), 4,393 (6.29%), 4,810 (6.45%), 4,453 (6.35%), 6,298 (7.84%), 7,032 (8.90%), 8,876 (10.63%) and 4,171 (8.26%) singleton SVs for Anqingliubai, Bamamini, European wild, Jinhua, Laiwu, Meishan, Min, Rongchang, Tibetan, Wuzhishan, and Yorkshire pigs, respectively (Figure 2A).
Figure 2.

SV characterization of the eleven samples
(A) The number of SVs for each category per sample. Shared SVs were identified in all samples, and singleton SVs were identified in only one sample. The remaining SVs identified in more than one but not all samples were polymorphic.
(B) Proportion of SV classes in each type.
(C) The number of nonredundant SVs changes with the sample added.
(D) Length distribution of the SVs of each type.
(E) Proportion of SVs for each type and functional SV after VEP annotation. The inner ring shows the percentages of merged SVs for each SV type. INSs, DELs, DUPs and INVs are colored red, blue, green and purple, respectively. The outer ring shows the proportion of SVs inducing potential effects after VEP annotation.
(F) Fold enrichment for structural variants intersected with the chromatin state.
A total of 183,352 nonredundant SVs were obtained after merging SVs across the 11 individuals, including 76,792 DELs (41.88%), 5,236 DUPs (2.86%), 98,480 INSs (53.71%), and 2,844 INVs (1.55%) (Figures 2D, 2E, and S3). Nearly 38% of the SVs were singletons, among which DUPs had a higher proportion than other types (Figure 2B). The frequencies of these four SV types decreased sharply with the increase of their length. Median lengths of 265 bp and 285 bp were observed for INSs and DELs, respectively, which were significantly shorter than DUPs and INVs with median lengths of 2,774 bp and 4,394 bp, respectively (Figure 2D). The results clearly showed two peaks at sizes of ∼300 bp and ∼8 kilobases (kb) for both INSs and DELs (Figures 2D and S4). Further results showed that most SVs of ∼300 bp were short interspersed nuclear elements (SINEs) for DELs, and a high number of SVs of ∼300 bp were SINEs or long interspersed nuclear elements (LINEs) for INSs. The peak at approximately 8 kb corresponded to LINEs, particularly for DELs. Similar results have been reported in humans, with peaks at ∼300 bp, corresponding to SINEs and LINEs, and 6 kb, corresponding to LINEs.4,21 In total, nonredundant SVs covered 299.56 Mb, representing approximately 12.12% of the pig reference genome (Sscrofa11.1), and these included 116.09 Mb, 94.31 Mb, 49.98 Mb and 39.19 Mb of DELs, DUPs, INSs, and INVs, respectively (Figures 2C and S3).
Here, we compared our long-read SV discovery callsets to previously reported SV callsets based on Illumina SRS data obtained from 11 previous studies (Figure S5; Table S4). We found that only 14.7% of base pairs overlapped between these two datasets, whereas 80.66% of SV base pairs (63% of SV numbers) discovered in this study were novel, highlighting the increased sensitivity of LRS for SV detection compared to SRS. To better understand and investigate the potential functions of these SVs, we conducted gene structure and epigenetic annotations. Nearly 53.25% of the SVs overlapped with repetitive elements (required at least half of an SV overlapped with a repetitive element), and most of the SV regions (55.27%) contained repetitive elements (Figure S6A). Most of these elements were LINEs (30.65%), followed by SINEs (15.41%) (Table S5). The transposable elements (TEs) characteristics of INS sequence were focused because the sequence of INSs was novel for reference genome. We found that 57.94% bases of INS sequence were masked, and LINEs sequences contributed the most (96.11%). Moreover, the divergence analysis for all classified TEs families was performed. The recently active TEs were mainly concerned with LINE/L1, LTR/REVL, LTR/REVK, LTR/REV1, DNA/hAT-Tip100 families (Figures S6B and S6C). Next, we systematically evaluated the effect of SVs on the function of genomic features. The results showed that most of the SVs were located in intergenic (40.63%) or intronic (41.49%) regions (Figure 2E). Only 0.96% of SVs were coding sequence variants, and 8.68% of SVs spanned multiple genes (Figure 2E). Finally, to explore the relationship between these SVs and regulatory elements, we used chromatin states predicted by ChromHMM.36 In general, we observed that SVs were enriched in repressed regions and depleted from active chromatin regions (Figure 2F).
Population structure of Chinese and European pigs based on SVs
DELs or INSs can be used as genetic markers to explore the population structure. As shown in Figure 3A, PC1 of DELs or INSs showed clear separation between Chinese pigs and European pigs, whereas PC2 showed separation between breeds, in agreement with the clustering result from SNPs (Figure S8). The breed clustering based on DUPs and INVs was not as clear as DELs or INSs, perhaps because of their small numbers. In admixture analysis, Chinese and European pigs showed two distant ancestral lineages when K = 2, and gene flows existed between the two groups. Next, Meishan pigs separated a lineage represented by Eastern Chinese (ECN) pigs when K = 3. Anqingliubai pigs were a severe mixture of Chinese and European lineages (Figures 3B and S7).
Figure 3.

Population structure of the 196 samples
(A) PCA structure for each type of SV. The color and shape of each point represent the breed and regional information per sample. See Table S2 for more details.
(B) Admixture analysis based on DELs.
Based on singleton SVs identified in ONT samples, we explored breed-specific SVs and observed several outliers of interest (Figure 4A). Specifically, Meishan pigs are known for their excellent reproduction and disease resistance ability, we found some SVs with high frequencies in Meishan pigs might be related to their high reproduction trait. For example, we found an intronic deletion in TEX11, a gene belongs testis expressed (TEX) gene family, which plays an important role in spermatogenesis and male fertility,37 with a frequency of 0.64 in Meishan pigs (Figure 4A). Other genes such as TBC1D8 and TPST1 related to fertility were also with a frequency of 0.75 and 0.21, respectively, in Meishan pigs.38,39 In addition, there was a high-frequency deletion (0.73) in the intron of IL18RAP in Meishan, which mediates high-affinity IL-18 binding and plays an important role in immunity by regulating the function of neutrophils.40 These SVs may be important candidate variants for high reproductive performance and disease resistance in Meishan pigs by regulating their target genes.
Figure 4.

Breed and region-specific variation
(A) Left panel: The breed-specific SVs for each population. Right panel: The region-specific SVs for each population. ECN: Eastern Chinese, EUD: European domestic, EW: European wild, NCN: Northern Chinese, SCN: Southern Chinese, SWCN: South West Chinese. See Table S2 for more details.
(B) The genome-wide SV frequency distribution in 1-Mbp non-overlapping windows for the 11 pigs. Circos from the outside (from A to K) to the inside present Anqingliubai, Bamamini, European wild, Jinhua, Laiwu, Meishan, Min, Rongchang, Tibetan, Wuzhishan and Yorkshire, respectively.
(C) The density of SVs per Mb on the X chromosome (averaged by the total number of SVs on the X chromosome) for each sample. The top and right panels reveal the number of SVs of each type. Nonredundant SVs represent a collection of merged SVs across all individuals. Chinese-specific SVs (CSSVs), defined as SVs, appear only in the Chinese pig group.
(D) The frequency of Southern Chinese specific (SCN-specific) and Chinese-specific SVs on the X chromosome.
For the LRS samples, the numbers of SVs in the nonoverlapping 1-Mb window were counted across the entire genome for each individual and then plotted in Figure 4B. The results clearly showed that SVs tend to be enriched at the start and end of chromosomes, consistent with findings in previous studies.4,24 Of interest, a 43-Mb SV hotspot on the X chromosome (44 Mb–87 Mb) was detected in most of Chinese pigs (Figure 4C). Therefore, we further performed a comprehensive comparison of SVs between European and Chinese pigs. In total, 113,058 SVs were only present in the Chinese pig population (refer to as Chinese-specific SVs, CSSVs) (Figure S3), including the 43-Mb SV hotspot on the X chromosome. Of interest, we found that this hotspot could be divided into two clusters. The first was a 13-Mb region in ChrX: 44–57 Mb that include 852 SVs and covering 53 genes mostly present in the Bamamini and Wuzhishan genomes. The second cluster a 30-Mb region in ChrX: 57–87 Mb that include 2,405 SVs and 84 genes present in Chinese pigs except for the Laiwu pigs (Figure 4B). However, no significant pathways were enriched for these genes. By genotyping additional 196 samples, we found a high frequency SV region on ChrX:44-57 Mb in Southern Chinese (SCN) pigs. The high-frequency Chinese-specific SVs were at ChrX:57-87 Mb (Figure 4D). PheWAS analysis revealed that most of the genes affected by the SCN-specific SVs were significantly associated with male baldness in humans (Table S6), suggesting that these genes may be related to the specific hair growth of southern Chinese pigs to adapt to the local hot environments in South China.
Tibetan pig specific SVs associated with high-altitude adaptation
We identified a total of 7,568 SVs specific to Tibetan pigs (TPSSVs) using LRS data (Table S7). The analysis of TPSSVs with previously reported quantitative trait loci (QTLs, www.qtldb.org) showed that these SVs were significantly enriched for traits related to fatness and blood parameters, such as the creatinine level, haptoglobin concentration, creatine kinase level and HDL cholesterol (Figures 5A and 5B; Table S8). In addition, other terms including hemoglobin, red blood cell count, hematocrit and mean corpuscular hemoglobin concentration, which were reported to be closely related to high-altitude adaptation,41,42 were also enriched, even insignificant (p-value threshold: 0.05) after accounting for multiple false positive correction (Table S8). Furthermore, 2,508 genes associated with TPSSVs (TPSSVGs) were significantly enriched in eight pathways, including neuron functions (‘axon guidance’ and ‘long-term depression’), disease and immune response (‘arrhythmogenic right ventricular cardiomyopathy’ and ‘human papillomavirus infection’), and tissue and organ morphogenesis and angiogenesis (‘focal adhesion’, ‘ECM-receptor interaction’, ‘regulation of actin cytoskeleton’, and ‘PI3K-Akt signaling pathway’) (Figure 5C; Table S9). As reported previously, angiogenesis involved coordinated changes in endothelial cells (ECs) and the actin cytoskeleton.43,44 Focal adhesion is essential to heart valve morphogenesis and blood vessel morphogenesis.45 Extracellular matrix (ECM) scaffolding influences angiogenesis and capillary integrity, and specific endothelial cell ECM receptors are critical for vascular morphogenetic changes during wound repair and high-altitude adaptation.46 Notably, the PI3K-Akt signaling pathway is reportedly associated with cardiovascular function and plays a vital role in erythropoiesis,47 which can regulate the expression of hypoxia-induced factor-1α and then further regulate the expression of downstream proteins related to angiogenesis, including erythropoietin and vascular endothelial growth factor.48 In addition, the GO enrichment analysis revealed that these genes were enriched in GTPase binding and small GTPase-mediated signal transduction. GTPase activity has been observed in Tibetan-specific SVs in humans,20 which is needed for the activation of hypoxia-inducible factor 149. We further collected 868 previously reported candidate genes (pHAAGs) for high-altitude adaptation in pigs (Table S10) and 2,260 differentially expressed genes (DEGs) between high-altitude and low-altitude pigs in a previous RNA-seq study50 (Table S10). We found that 127 and 298 of the 2,508 TPSSVGs overlapped with pHAAGs and DEGs, respectively, among which 28 genes were covered by both TPSSVGs and pHAAGs (Figure 5D).
Figure 5.

Candidate SVs of high-altitude adaptation
(A) QTL enrichment analysis of Tibetan-specific SVs (TPSSVs).
(B) Significantly enriched QTLs for blood parameters.
(C) KEGG pathway enrichment analysis of 2,508 genes located within 5 kb upstream or downstream of the TPSSVs.
(D) Overlap among three gene sets. TPSSVGs refer to Tibetan-specific SV-associated genes, pHAAGs are candidate genes for high-altitude adaptation reported in the literature, and DEGs are the differentially expressed genes identified by Tang.
(E) Manhattan plot of the Fst statistics (Tibetan versus low-altitude pigs) of 3,141 SVs for TPSSVs in the NGS population supported by at least one sample. The dotted line indicates the threshold defining the top 5% (Vst = 0.299).
Furthermore, we analyzed 120 publicly available SRS datasets, consisting of 22 Tibetan pigs and 98 Chinese lowland pigs (Table S2), to identify SVs and perform selective sweeps. Our results showed that 3,141 out of the 7,568 TPSSVs, including 1,907 DELs, 1,202 INSs, 7 DUPs and 25 INVs, could be supported by at least one NGS sample (Tables S6 and S10). Among the Tibetan-specific SVs identified from SRS data, 157 SVs (top 5% of the 3,141 SVs) with the highest divergence between the highland and lowland populations were associated with 72 genes (Figure 5E; Table S11), and two of these genes (SEMA5A and SOD1) overlapped with both TPSSVGs and pHAAGs. In addition, a total of 12 candidate genes (ADAMTS12, ATP6V0A1, EPHA2, HIPK2, NAV2, PDGFRA, REV1, SEMA5A, SGCD, SLC44A5, SOD1 and TRPC5) were identified according to the function of genes and relevant literature reports (Table S12).
A 124-bp DEL on Chromosome 13 (13:195,324,807-195,324,931, Fst: 0.391) in the upstream of the antioxidant superoxide dismutase 1 (SOD1) gene exhibited SV allele frequencies of 22.7% in Tibetan but null in low-altitude pigs. This DEL overlapped with the H3K27ac peak regions (13:195,322,800-195,325,600), an indicator of the chromatin state of an active enhancer in the liver51 (Figure 6A). In addition, positive selection signature was also observed in SNPs surrounding this SV (Figure 6A), which confirmed that SOD1 was under positive selection for high-altitude adaptation.52,53 The SOD1 gene encodes Fancc and Cu/Zn superoxide dismutase, which plays a vital role in antioxidant and anti-radiation mechanisms.54 It is relevant to the erythrocyte cell number and hemoglobin content in mice, peripheral blood erythrocytes are decreased in Fancc−/−Sod1−/−mice.55 Superoxide dismutase enzymes play a very important role in NO bioavailability, and NO-induced vascular relaxation is related to the regulation of blood pressure.56 The content of SOD1 is higher in organs with high metabolic activity, such as the liver and kidney, and in erythrocytes.57,58 The overexpression of SOD1 could decrease oxidative stress and protect against vascular dysfunction.59,60 Compared with low-altitude pigs, high expression of SOD1 was observed in the liver and spleen of Tibetan pigs (Figure 6B), which suggested that SOD1 is an important candidate gene related to high-altitude hypoxia adaptation. Further pheWAS based on the GWAS atlas (https://atlas.ctglab.nl/PheWAS) showed that SOD1 was significantly associated with the mean corpuscular hemoglobin concentration (p value: 0.00157) (Figure 6C and Table S13). Another divergent SV was a 1477-bp DEL on Chromosome 16 (16:72,750,546-72,752,023, Fst: 0.377) in the intronic region of semaphorin 5A (SEMA5A), which exhibited allele frequencies of 0.523 and 0.115 in Tibetan pigs and low-altitude pigs, respectively. This DEL overlapped with the ATAC-seq and H3K27me3 peak regions,51 suggest that it may play a regulatory role. In addition, a significant selection signal was also found in the SNPs surrounding this DEL (Figure S8A). The SEMA5A gene promotes angiogenesis by increasing endothelial cell proliferation and migration and decreasing apoptosis.61 Lower expression of SEMA5A in the kidneys of Tibetan pigs could reduce the incidence of cancer.62 In addition, we also take the other two of the 12 candidate gene as examples for detailed description (Table S12). A 371-bp DEL on Chromosome 3 (3: 54,164,916-54,165,287) in the intron region of REV1 showed allele frequencies of 0.273 and 0.031 in Tibetan and low-altitude pigs, respectively (Figure S8B). REV1 responds to ultraviolet (UV) light and is indispensable for translesion synthesis.63 A selective sweep of this DEL was also observed between high- and low-altitude pigs (Fst: 0.318). Furthermore, this SV was overlapped with the peak regions of ATAC-seq, H3K4me1 and H3K27ac and chromatin states of an active enhancer,51which suggest that this SV may play an important role in resistance to UV radiation in high-altitude Tibetan pigs. A 115-bp DEL on Chromosome 16 (16:66,780,487-66,780,602, Fst: 0.564) in the intronic region of sarcoglycan delta (SGCD) showed allele frequencies of 0.386 and 0.011 in Tibetan and low-altitude pigs, respectively (Figure S8C). Knockout of SGCD gene in domestic pigs will lead to the myocardial tissue degeneration, systolic dysfunction, and sudden death,64 which indicated that SGCD gene plays vital role in cardiovascular system and is an important candidate gene for high altitude adaptation.
Figure 6.

SVs of SOD1
(A) A 124-bp DEL upstream of SOD1. The upper panel shows the Hi-C map surrounding SOD1. The black arrow indicates the position of the DEL. The bottom distribution shows the 5-kb regions upstream and downstream of this SV and the Fst statistic (Tibetan versus low-altitude pigs) of SNPs in this region, as demonstrated in the left (gray) and right coordinate axes (blue), respectively.
(B) The gene expression value of SOD1 in corresponding tissues.
(C) pheWAS plot for SOD1.
(D) Analysis of the DEL using gel electrophoresis.
Discussion
Over the last few years, the reference genome has shown marked improvements in terms of the completeness and annotation of the genome with the rapid development of sequencing technology, e.g., LRS technology, Iso-Seq technology and Hi-C technology, which has resulted in fewer gaps in the reference genome and more novel genes or transcripts for genome annotation.65,66,67 Furthermore, we can obtain a telomere-to-telomere (T2T) assembly without a gap in the chromosome or even the whole genome using PacBio high-fidelity (HiFi) LRS technology.68,69,70 For pangenome analysis, three general approaches have been developed, including comparative de novo assembly, iterative assembly and graph-based pangenome assembly.12 The comparative de novo assembly was using the whole genome comparison of de novo assembling genomes with annotations, which required the individual genome must be assembled. The iterative assembly was to extract the unmapped reads for assembly and then updating the reference genome which can incorporate large samples. Graph-based pangenome assembly is the most recently developed approach and has shown rapid development,33,71 using a graph representing all of the genomic diversity and will facilitate population comparison and annotation without liftover between different breed reference genomes. Here, we used the graph-based assembly to generate a pangenome of 11 pig breeds. We explored novel sequences with a total length of 206-Mb in the pangenome which are absent in the pig reference genome Sscrofa11.1, and the total length of the novel sequences is markedly longer than that (72.5 Mb) reported in the pig pangenome study of Tian et al.,10 which may be mainly because of the different assembly methods, sequencing technologies and breeds involved in the studies. Not only were high quality assembled genomes base on LRS sequencing used, but also the most recently developed approach of graph-based pangenome was used in this study. The graphic pangenomes could represent all of the genomic diversity and display genome variation through the bubble. However, only SRS-based and an iterative assembly approach were used in the study of Tian et al.,10 which is a linear pangenome and is not a good indicator of the variants in a species, especially in complex regions of the genome. Moreover, the differences in number of breeds, pangenome construction strategies and criteria for filtering in pangenome analysis could lead to differences in pangenome sequence even for the same species. In different human pangenome studies, the total length of novel sequences varied from 0.16 Mb to 14.2 Mb per individual and from 0.33 Mb to 296 Mb per study.72
For novel SV detection, LRS also outperformed SRS in terms of sensitivity and accuracy, particularly for the detection of large and complex SVs and non-CNV SVs.4,31,66,73 Here, we found that LRS yielded more novel SVs: SVs with 331-Mb novel base pairs were identified in 11 LRS genomes in this study, whereas only 129-Mb novel SVs were identified by SRS in even more samples in published datasets (Figure S5). These results indicate that more novel SVs can be identified with LRS technology. There are three methods for SVs detection of LRS, including the approach of de novo assembly detection, graph-pan based and reads mapping. The de novo assembly approach of SVs detection requires the assembled genomes with high quality to reduce the errors in variants discovery. The graph-based approach of SVs detection is developed with the rise of graphs based pangenome, which is in rapid development, and has not been thoroughly evaluated in real-world data at large scale.74 It is worth mentioning that the genotype of variants (heterozygous or homozygous) cannot be determined using either the pangenome graph or the de novo assembly because the genomes we assembled were not haplotype-resolved. In addition, the graph-based and de novo assembly approaches are more intended to resolve complex SVs. Here, we focus on simple SVs (DELs, DUPs, INSs, and INVs), so the reads mapping method was applied as used in other studies.22,75
Furthermore, following in other studies,75,76,77 we applied a graph-based SV genotyper Paragraph78 to genotype long-read SV callsets in a large population of samples with SRS data. Although Paragraph achieves higher accuracy than other genotyping methods, it only works in INSs, DELs and INVs. For DUPs, it did not work well (Figure S9). Here, we also found that there was no significant correlation between sequencing depth and the missing rate of genotyping (p value = 0.2414). After filtering, only 61.9% of SVs were successfully genotyped, of which 79.22% were DELs, 51.19% were INSs, 5.63% were DUPs and 23.14% were INVs were retained, which may lead to the preference of subsequent population analysis. Therefore, better genotyping tools need to be developed. SVs were enriched at both ends of a chromosome, which is consistent with other studies4 (Figure 4B) because telomeres and subtelomeric regions are prone to mutation.79 In particular, an SV hotspot region (44 Mb–87 Mb, Sscrofa11.1) was identified on the X chromosome all Chinese pigs except the Laiwu pig, as reported by Zhao et al.,80 who identified a 35-Mb (65–100 Mb, Sscrofa10.2) SV hotspot on the X chromosome among pigs of Chinese origin. Of interest, this hotpot region was also located in the low-recombination region reported by Ai et al.8 and the surrounding centromeric region after alignment to the same Sscrofa11.1 reference genome. Centromeric DNA comprises satellite repeat DNAs and transposable elements,81 which makes this region an easy hotspot of SVs.79 Large introgressions could also result in SV hotspots as reported in tomato genomes.24 The absence of the hotpot on the X chromosome of the Laiwu pig may be because this region of this Laiwu pig was introgressed from European pigs, since it was close to other Laiwu pigs in the autosome-based phylogenetic trees but separated from other Laiwu pigs and closed to European pigs in the Chromosome X based phylogenetic tree (Figure S10).
Taking high-altitude hypoxia adaptation as an example, we investigated the roles of SVs underlying this phenotype. High-altitude hypoxia adaptation is a complex process under long-term selection involving many types of genomic variants, including SNPs and SVs.20 Many candidate genes related to high-altitude adaptation have been identified, and two of the most famous genes are EPAS1 and EGLN1 in humans.42,82 In addition, a Tibetan-enriched SV with a 3.4-kb DEL located 80 kb downstream of EPAS1 has been reported.83 In this study, 7,568 Tibetan pig-specific SVs were identified, which are associated with 2,508 genes enriched in eight KEGG pathways that play a vital role in high-altitude hypoxia adaptation. By combining multiomics data, LRS for SV identification, SRS for population validation, RNA-seq for differential expression analysis and published functional annotation of pig genome, several key candidate SVs affecting the SOD1, SEMA5A, REV1 and SGCD genes related to high-altitude adaptation were successfully identified in this study. We speculate these SVs alter the corresponding gene expression by affecting regulatory elements.21,84 However, more research is needed to examine the function and mechanism of these SVs in pigs.
Limitations of the study
We sequenced and assembled the genomes of ten diverse pigs using nanopore sequencing technology. However, they were not assembled to the chromosome level or T2T level, there may be some errors in the complex region of the genome, and SVs in a long length might be missed. In addition, part of the SVs (especially for DUPs and INVs) will be filtered when using Paragraph for the genotyping of SVs in the NGS population and these filtered SVs cannot be analyzed in the population scale. Additionally, it is a heavy work to verify the causal relationship between the candidate SVs and phenotypes as transgenic animals (CRISPR/Cas-based protocol85) are required. In addition to genomic variation, epigenetic variation is also one of the reasons for high-altitude hypoxic adaptation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Bama miniature assembly | Zhang et al., 201986 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/007/644/095/GCA_007644095.1_ASM764409v1 |
| Bama miniature sequencing reads | Zhang et al., 201986 | ftp.sra.ebi.ac.uk/vol1/fastq |
| 10 ONT sequencing reads | This paper | Genome Sequence Archive (https://ngdc.cncb.ac.cn/): PRJCA005901 |
| Software and algorithms | ||
| Bcftools v1.10.2 | Danecek et al., 202187 | https://github.com/samtools/bcftools |
| Bedtools v2.30.0 | Quinlan and Hall, 201088 | https://github.com/arq5x/bedtools2 |
| BUSCO v3.1.0 | Seppey et al., 201989 | https://busco.ezlab.org/ |
| BWA v0.7.17-r1188 | Li and Durbin, 200990 | https://github.com/lh3/bwa |
| Canu v1.5 | Koren et al., 201791 | https://github.com/marbl/canu |
| ClusterProfiler | Yu et al., 201292 | https://github.com/GuangchuangYu/clusterProfiler |
| g:Profile | Raudvere et al., 201993 | https://biit.cs.ut.ee/gprofiler/gost |
| GATK v3.7 | McKenna et al., 201094 | https://github.com/broadinstitute/gatk |
| gfatools v0.4-r165 | – | https://github.com/lh3/gfatools |
| GfaViz v1.0.0 | Gonnella et al., 201995 | https://github.com/ggonnella/gfaviz |
| Guppy v1.5 | Nanopore | https://community.nanoporetech.com/ |
| Minigraph toolkit v0.10-r356 | Li et al., 202033 | https://github.com/lh3/minigraph |
| NGMLR v0.2.7 | Sedlazeck et al., 201835 | https://github.com/philres/ngmlr |
| Paragraph v2.4a | Chen et al., 201978 | https://github.com/Illumina/paragraph |
| Pilon v1.22 | Walker et al., 201496 | https://github.com/broadinstitute/pilon |
| Racon v1.4.6 | Vaser et al., 201797 | https://github.com/lbcb-sci/racon |
| RepeatMasker v4.1.4 | Tempel, 201298 | https://www.repeatmasker.org/ |
| SAMTools v1.9 | Li et al., 200999 | https://github.com/samtools/samtools |
| Sniffles v1.0.11 | Sedlazeck et al., 201835 | https://github.com/fritzsedlazeck/Sniffles |
| SURVIVOR v1.0.6 | Jeffares et al., 2017100 | https://github.com/fritzsedlazeck/SURVIVOR |
| VCFtools v0.1.16 | Danecek et al., 2011101 | https://github.com/vcftools/vcftools |
| VEP release 100 | McLaren et al., 2016102 | https://github.com/Ensembl/ensembl-vep |
| wtdbg2 v2.5 | Ruan and Li, 2020103 | https://github.com/ruanjue/wtdbg2 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xiang-Dong Ding (xding@cau.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
All animal work (Sus scrofa, pig) was approved by the Institutional Animal Care and Use Committee (IACUC), China Agricultural University. One female and nine male individuals from ten diverse breeds (two originating in Europe and eight originating in China) (Figure 1A) were chosen for sequencing in this study (Anqingliubai, European wild, Jinhua, Laiwu, Meishan, Min, Rongchang, Tibetan, Wuzhishan and Yorkshire) (Table 1). In addition, one Bama miniature (Bamamini) individual was obtained from a published source.86
Method details
Data generation
To obtain LRS data, high-quality genomic DNA was isolated from peripheral blood samples obtained from the pigs. The libraries were constructed and then sequenced on the Nanopore PromethION platform following Oxford Nanopore Technologies (ONT) standard operating procedures. Each individual was sequenced to a coverage depth of 50X, and base calling was performed using Guppy (version: 1.5) with the default parameters.
For short-read DNA sequencing, genomic DNA was prepared using the Illumina Nextera DNA Library Preparation Kit, and the DNA samples were sequenced on the Illumina NovaSeq platform with an insert size of 350 bp, which resulted in 150-bp paired-end reads with an average coverage of 50X for each sample. In addition, we aggregated public short-read whole-genome sequencing (WGS) datasets of 196 pigs obtained in previous studies, which included 120 Chinese pigs and 76 non-Chinese pigs and exhibited an average depth of 14X (ranging from 4X to 25X).
Pangenome construction
The de novo genomes were assembled using the following steps: 1) Correcting and trimming the raw sequencing reads using Canu (version: 1.5).91 First, longer seed reads were selected with the settings ‘genomeSize = 2500000000’ and ‘corOutCoverage = 60’, and overlapping raw reads were then detected using a highly sensitive overlapper MHAP algorithm. Second, error correction was performed using the option ‘correctedErrorRate = 0.1’. Third, using the default parameters, the error-corrected reads were trimmed with unsupported bases and hairpin adapters to obtain their longest supported range. 2) Construction of a draft assembly using wtdbg2 (version 2.5).103 First, this program generates a draft assembly using the command ‘wtdbg2-i Longest∗.correctedReads.fasta-t 30-fo wtdbg-p 19-AS 2-L 5000’. A consensus assembly was then obtained with the command ‘wtpoa-cns-t 30-i wtdbg.ctg.lay.gz-fo wtdbg.ctg.lay.fa’. 3) Polishing the draft assembly to obtain the final assembly using Racon and Pilon. The first round of polishing was performed using the Racon (version: 1.4.6)97 algorithm with ONT data. The second polishing was conducted with a Pilon algorithm (version: 1.22)96 using Illumina data with the parameters ‘--mindepth 10--changes--threads 4--fix bases’.
Subsequently, BUSCO (version 3.1.0)89 was used to evaluate the gene completion of the assembly using the odb9 Mammalia ortholog dataset, which benchmarked 4104 universal single-copy orthologous genes.
The Minigraph toolkit (version: 0.10-r356)33 was then used to construct the Sus scrofa reference pangenome by integrating the Sscrofa11.1 pig reference genome, ten de novo genomes obtained in this study and one Bamamini de novo genome described by Zhang.86 The pangenome was visualized with GfaViz (version: 1.0.0).95 And the bubbles were called using gfatools bubble (version 0.4-r165). All the nodes that were not in the Duroc-based pig reference genome (Sscrofa11.1) are referred to nonreference nodes. We colored the nodes that aligned each assembly back to the pangenome graph separately to determine the support of each node according to Crysnanto et al.’s research.34 The output pangenome of the rGFA format was then converted to a FASTA file using gfatools (version 0.4-r165).
Gap-filling of the reference genome
A GFA procedure was used for closing gaps in the pig reference genome (Sscrofa11.1) following the previous study.104 A region consisting of continuous N in the Sscrofa11.1 was defined as a gap and merging gaps that are less than 500 bp apart, there are 519 gaps existed on the Sscrofa11.1 reference genome. The gfa.pl script (https://github.com/WGLab/uniline) was used to estimate the gap-filling ability of each assembly.
Reads mapping and SV detection
For long-read ONT sequencing data, SVs were identified based on read-mapping SV detection methods. First, the mapping software NGMLR (version: 0.2.7)35 with the parameter “--presets ont” was used to align the reads to the pig reference genome Sscrofa11.17. Subsequently, Sniffles (version 1.0.11)35 was implemented to call SVs from the bam file of each individual with the parameter “-s 10-L 50”,which required each variant to have at least ten reads of support and a length of at least 50 bp. Then, SVs with lengths greater than 2 Mb for DUPs and INVs, with lengths of more than 1 Mb for INSs and DELs, or located on the Y chromosome, mitochondria or nonchromosome were removed. Finally, individual SVs were merged using SURVIVOR (version 1.0.6).100
For the NGS short-read Illumina data, the reads were mapped to the pig reference genome Sscrofa11.17 using BWA (Burrows-Wheeler Aligner, version: 0.7.17-r1188)90 with the command ‘bwa men-M-R’. The SAM file was then converted to a bam file using SAMtools (version 1.9).99 The sorting and marking of potential PCR duplicates were then performed with Picard SortSam and Picard MarkDuplicates (http://broadinstitute.github.io/picard/). Indel realignment was performed using GATK (version 3.7)94 with the variants from the dbSNP database (Build ID: 150). Then, Paragraph (v2.4a)78 was used to genotype the SVs in these samples with the maximum permitted read count set to 20 times the mean sample depth. Bcftools (v1.10.2)87 was used to combine the genotypes of all samples. All the missing genotypes were replaced by (./.). We removed variants with a missing rate ≥50% of samples to obtain high-quality sets.
Comparison with published SV callsets
We collected SV callsets from eleven published studies identified from SRS data, including hundreds of pig genomes across diverse breeds26,80,105,106,107,108,109,110,111,112,113 (Table S3). Their bed files were then liftover into Sscrofa11.1 if they were called against Sscrofa10.2. Subsequently, a nonredundant SV callset was generated using bedtools merge (version: 2.30.0).88 Compared with these published sets, the coincident base pair was counted.
Annotation of SVs and gene enrichment analysis
For repeat annotation, repeats were identified by incorporating the repeat database of the Ssrofa11.1 reference genome (ftp://hgdownload.soe.ucsc.edu/goldenPath/susScr11/database/rmsk.txt.gz) and were summarized into ten categories: SINEs, LINEs, long terminal repeat (LTR) elements, rolling circles (RCs), DNA repeat elements, RNA repeats, satellite repeats, simple repeats, low-complexity repeats and unknown repeats. The novel sequence of INSs were masked by RepeatMasker (version: 4.1.4)98 with the known Dfam (version: 3.6) and Repbase (version: 10/26/2018) library. The Kimura 2-parameter divergence value of each families or subfamily of TEs was calculated using the calcDivergenceFromAlign.pl utility packaged in RepeatMasker. The ages of each families were then calculated according to the formula T = K/2r114, of which r is the average nucleotide substitution. Here, we assumed a substitution rate of 2.2 × 10−9 substitutions/site/year for pigs.115,116
For the functional annotation, Variant Effect Predictor102 (VEP; http://asia.ensembl.org/info/docs/tools/vep/index.html; ensembl release 100) was used, and the upstream and downstream distances were both 5 kbp.
For the chromatin state annotation, the regioneR package of R was used to perform overlap enrichment analysis of SVs versus chromatin states36 with 1000 permutation tests.
Functional enrichment analysis and pathway analysis of the specific related genes were performed using g:Profile.93
Overlap and enrichment of quantitative trait loci in specific SVs
The pig QTL database was downloaded from the Animal QTL database (release 40; https://www.animalgenome.org/cgi-bin/QTLdb/SS/index), which includes 30,170 pig QTLs for 688 different pig traits. These 688 traits were divided into 30 types, including meat and carcass traits, health traits, external traits, production traits and reproduction traits. ClusterProfiler92 was used for enrichment analysis of quantitative trait loci overlapping with specific SVs. Here, a base pair overlap of more than 1 bp was considered to indicate overlap. The statistics were significant if the Benjamini-Hochberg-corrected p value <0.05.
Natural selective sweep of SNPs around the focus SVs
After generating the bam file using the same protocol for data processing as that described for NGS data, GATK HaplotypeCaller was used for SNP calling to generate the gVCF file for each sample, and CombineGVCFs and GenotypeGVCFs were then used for genotyping the SNPs. After filtering using VariantFiltration with “QD < 2.0 || MQ < 40.0 || FS > 60.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0 || SOR >3.0” and then removing the SNPs with MAF<0.01 or missing rate greater than 0.2, a total of 36,561,094 autosomal SNPs were retained for the following analysis.
To further detect selective sweeps around the candidate SVs in Tibetan pigs, Weir and Cockerham’s Fst was calculated based on the SNP data of the NGS population comprising 22 high-altitude Tibetan pigs and 98 low-altitude pigs using VCFtools (version 0.1.16).101
SV validation by polymerase chain reaction (PCR)
Three important candidate SVs for high-altitude adaptation were selected for PCR validation. Primer Premier5 was used for primer design, and a 100-bp sequence at the SV breakpoints was extended as the PCR target. Agarose gel electrophoresis was used to visualize the PCR products. The primers used in this study are listed in Table S14.
Quantification and statistical analysis
Details of the statistical tests applied, including the statistical methods, number of replicates, mean and error bar details and significances are indicated in the relevant figure legends.
Additional resources
This study does not include additional resources.
Acknowledgments
This work was supported by grants for Key R&D Program of Shandong Province (2022LZGC003); China Agriculture Research System of MOF and MARA; the National Key Research and Development Project (2019YFE0106800); the 2020 Research Program of Sanya Yazhou Bay Science and Technology City (SKJC-2020-02-007); the National Natural Science Foundation of China (32070568, 31671327 and 31872327); Anhui Academy of Agricultural Sciences Key Laboratory Project (2019YL021); Hefei City Postdoctoral Science Foundation (2018PD22); China Postdoctoral Science Foundation (2020M681977); Key R&D Projects in Zhejiang Province (2021C02007) and Public Projects of Zhejiang Province (LGN19C170005).
Author contributions
X.D.D., D.D.W., Q.Z., C.L.W., and R.H.X. designed and supervised the project. W.W.W., Y.J., L.J., L.H.D., J.R.W., X.H.C., and Y.Q.Z. prepared the DNA sampling and experiments; Y.F.J., S.W., C.L.W., and R.H.X. performed the majority of the analysis with contributions from M.S.W.; Y.F.J., X.D.D., and S.W. prepared the manuscript under the revision of D.D.W., L.Z.F., and M.S.W. J.Y. performed PCR validation experiments. All authors approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106119.
Supplemental information
Data and code availability
-
•
All raw sequencing data generated in this study have been submitted to the Genome Sequence Archive (https://ngdc.cncb.ac.cn/bioproject/) under accession number PRJCA005901.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Andersson L. Domestic animals as models for biomedical research. Ups. J. Med. Sci. 2016;121:1–11. doi: 10.3109/03009734.2015.1091522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perleberg C., Kind A., Schnieke A. Genetically engineered pigs as models for human disease. Dis. Model. Mech. 2018;11:dmm030783. doi: 10.1242/dmm.030783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li R., Li Y., Zheng H., Luo R., Zhu H., Li Q., Qian W., Ren Y., Tian G., Li J., et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2010;28:57–63. doi: 10.1038/nbt.1596. [DOI] [PubMed] [Google Scholar]
- 4.Audano P.A., Sulovari A., Graves-Lindsay T.A., Cantsilieris S., Sorensen M., Welch A.E., Dougherty M.L., Nelson B.J., Shah A., Dutcher S.K., et al. Characterizing the major structural variant alleles of the human genome. Cell. 2019;176:663–675.e19. doi: 10.1016/j.cell.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eizenga J.M., Novak A.M., Sibbesen J.A., Heumos S., Ghaffaari A., Hickey G., Chang X., Seaman J.D., Rounthwaite R., Ebler J., et al. Pangenome graphs. Annu. Rev. Genomics Hum. Genet. 2020;21:139–162. doi: 10.1146/annurev-genom-120219-080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lei L., Goltsman E., Goodstein D., Wu G.A., Rokhsar D.S., Vogel J.P. Plant pan-genomics comes of age. Annu. Rev. Plant Biol. 2021;72:411–435. doi: 10.1146/annurev-arplant-080720-105454. [DOI] [PubMed] [Google Scholar]
- 7.Warr A., Affara N., Aken B., Beiki H., Bickhart D.M., Billis K., Chow W., Eory L., Finlayson H.A., Flicek P., et al. An improved pig reference genome sequence to enable pig genetics and genomics research. GigaScience. 2020;9 doi: 10.1093/gigascience/giaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ai H., Fang X., Yang B., Huang Z., Chen H., Mao L., Zhang F., Zhang L., Cui L., He W., et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015;47:217–225. doi: 10.1038/ng.3199. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y., Du H., Li P., Shen Y., Peng H., Liu S., Zhou G.A., Zhang H., Liu Z., Shi M., et al. Pan-genome of wild and cultivated soybeans. Cell. 2020;182:162–176.e13. doi: 10.1016/j.cell.2020.05.023. [DOI] [PubMed] [Google Scholar]
- 10.Tian X., Li R., Fu W., Li Y., Wang X., Li M., Du D., Tang Q., Cai Y., Long Y., et al. Building a sequence map of the pig pan-genome from multiple de novo assemblies and Hi-C data. Sci. China Life Sci. 2020;63:750–763. doi: 10.1007/s11427-019-9551-7. [DOI] [PubMed] [Google Scholar]
- 11.Tao Y., Zhao X., Mace E., Henry R., Jordan D. Exploring and exploiting pan-genomics for crop improvement. Mol. Plant. 2019;12:156–169. doi: 10.1016/j.molp.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Bayer P.E., Golicz A.A., Scheben A., Batley J., Edwards D. Plant pan-genomes are the new reference. Nat. Plants. 2020;6:914–920. doi: 10.1038/s41477-020-0733-0. [DOI] [PubMed] [Google Scholar]
- 13.Li R., Fu W., Su R., Tian X., Du D., Zhao Y., Zheng Z., Chen Q., Gao S., Cai Y., et al. Towards the complete goat pan-genome by recovering missing genomic segments from the reference genome. Front. Genet. 2019;10:1169. doi: 10.3389/fgene.2019.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X., Liu X., Ge S., Jensen J.D., Hu F., Li X., Dong Y., Gutenkunst R.N., Fang L., Huang L., et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2011;30:105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- 15.Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.Y., et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H., et al. A structural variation reference for medical and population genetics. Nature. 2020;581:444–451. doi: 10.1038/s41586-020-2287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huddleston J., Chaisson M.J.P., Steinberg K.M., Warren W., Hoekzema K., Gordon D., Graves-Lindsay T.A., Munson K.M., Kronenberg Z.N., Vives L., et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017;27:677–685. doi: 10.1101/gr.214007.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weischenfeldt J., Symmons O., Spitz F., Korbel J.O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 2013;14:125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 19.Eichler E.E. Genetic variation, comparative genomics, and the diagnosis of disease. N. Engl. J. Med. 2019;381:64–74. doi: 10.1056/NEJMra1809315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouzhuluobu, He Y., Lou H., Cui C., Deng L., Gao Y., Zheng W., Guo Y., Wang X., Ning Z., et al. De novo assembly of a Tibetan genome and identification of novel structural variants associated with high-altitude adaptation. Natl. Sci. Rev. 2020;7:391–402. doi: 10.1093/nsr/nwz160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quan C., Li Y., Wang Y., Ping J., Lu Y., Zhou G. Characterization of structural variation in tibetans reveals new evidence of high-altitude adaptation and introgression. bioRxiv. 2020 doi: 10.1101/2020.12.01.401174. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Z., Jiang Z., Li T., Xie C., Zhao L., Yang J., Ouyang S., Liu Y., Li T., Xie Z. Structural variants in chinese population and their impact on phenotypes, diseases and population adaptation. bioRxiv. 2021 doi: 10.1101/2021.02.09.430378. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang N., Liu J., Gao Q., Gui S., Chen L., Yang L., Huang J., Deng T., Luo J., He L., et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat. Genet. 2019;51:1052–1059. doi: 10.1038/s41588-019-0427-6. [DOI] [PubMed] [Google Scholar]
- 24.Alonge M., Wang X., Benoit M., Soyk S., Pereira L., Zhang L., Suresh H., Ramakrishnan S., Maumus F., Ciren D., et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell. 2020;182:145–161.e23. doi: 10.1016/j.cell.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo J., Cao K., Deng C., Li Y., Zhu G., Fang W., Chen C., Wang X., Wu J., Guan L., et al. An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biol. 2020;21:258. doi: 10.1186/s13059-020-02169-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubin C.J., Megens H.J., Martinez Barrio A., Maqbool K., Sayyab S., Schwochow D., Wang C., Carlborg Ö., Jern P., Jørgensen C.B., et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA. 2012;109:19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norris B.J., Whan V.A. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008;18:1282–1293. doi: 10.1101/gr.072090.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang D., Zhao P., Si J., Fang L., Pairo-Castineira E., Hu X., Xu Q., Hou Y., Gong Y., Liang Z., et al. Genomic analysis revealed a convergent evolution of LINE-1 in coat color: a case study in water buffaloes (Bubalus bubalis) Mol. Biol. Evol. 2021;38:1122–1136. doi: 10.1093/molbev/msaa279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durkin K., Coppieters W., Drögemüller C., Ahariz N., Cambisano N., Druet T., Fasquelle C., Haile A., Horin P., Huang L., et al. Serial translocation by means of circular intermediates underlies colour sidedness in cattle. Nature. 2012;482:81–84. doi: 10.1038/nature10757. [DOI] [PubMed] [Google Scholar]
- 30.Alkan C., Coe B.P., Eichler E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho S.S., Urban A.E., Mills R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2020;21:171–189. doi: 10.1038/s41576-019-0180-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang S.L., Wang Z.G., Liu B., Zhang G.X., Zhao S.H., Yu M., Fan B., Li M.H., Xiong T.A., Li K. Genetic variation and relationships of eighteen Chinese indigenous pig breeds. Genet. Sel. Evol. 2003;35:657–671. doi: 10.1186/1297-9686-35-7-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Feng X., Chu C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020;21:265. doi: 10.1186/s13059-020-02168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crysnanto D., Leonard A.S., Fang Z.H., Pausch H. Novel functional sequences uncovered through a bovine multiassembly graph. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2101056118. e2101056118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sedlazeck F.J., Rescheneder P., Smolka M., Fang H., Nattestad M., von Haeseler A., Schatz M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods. 2018;15:461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan Z., Yao Y., Yin H., Cai Z., Wang Y., Bai L., Kern C., Halstead M., Chanthavixay G., Trakooljul N., et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021;12:5848. doi: 10.1038/s41467-021-26153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellil H., Ghieh F., Hermel E., Mandon-Pepin B., Vialard F. Human testis-expressed (TEX) genes: a review focused on spermatogenesis and male fertility. Basic Clin. Androl. 2021;31:9. doi: 10.1186/s12610-021-00127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcello M.R., Jia W., Leary J.A., Moore K.L., Evans J.P. Lack of tyrosylprotein sulfotransferase-2 activity results in altered sperm-egg interactions and loss of ADAM3 and ADAM6 in epididymal sperm. J. Biol. Chem. 2011;286:13060–13070. doi: 10.1074/jbc.M110.175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borghei A., Ouyang Y.B., Westmuckett A.D., Marcello M.R., Landel C.P., Evans J.P., Moore K.L. Targeted disruption of tyrosylprotein sulfotransferase-2, an enzyme that catalyzes post-translational protein tyrosine O-sulfation, causes male infertility. J. Biol. Chem. 2006;281:9423–9431. doi: 10.1074/jbc.M513768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J., Lam I.K.Y., Lau C.S., Chan V.S.F. Elevated interleukin-18 receptor accessory protein mediates enhancement in reactive oxygen species production in neutrophils of systemic lupus erythematosus patients. Cells. 2021;10 doi: 10.3390/cells10050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beall C.M. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. USA. 2007;104:8655–8660. doi: 10.1073/pnas.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beall C.M., Cavalleri G.L., Deng L., Elston R.C., Gao Y., Knight J., Li C., Li J.C., Liang Y., McCormack M., et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA. 2010;107:11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eelen G., Treps L., Li X., Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ. Res. 2020;127:310–329. doi: 10.1161/circresaha.120.316851. [DOI] [PubMed] [Google Scholar]
- 44.Bayless K.J., Johnson G.A. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J. Vasc. Res. 2011;48:369–385. doi: 10.1159/000324751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer R.S., Lam P.Y., Huttenlocher A., Waterman C.M. Filopodia and focal adhesions: an integrated system driving branching morphogenesis in neuronal pathfinding and angiogenesis. Dev. Biol. 2019;451:86–95. doi: 10.1016/j.ydbio.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchand M., Monnot C., Muller L., Germain S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin. Cell Dev. Biol. 2019;89:147–156. doi: 10.1016/j.semcdb.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Jafari M., Ghadami E., Dadkhah T., Akhavan-Niaki H. PI3k/AKT signaling pathway: erythropoiesis and beyond. J. Cell. Physiol. 2019;234:2373–2385. doi: 10.1002/jcp.27262. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Z., Yao L., Yang J., Wang Z., Du G. PI3K/Akt and HIF1 signaling pathway in hypoxiaischemia (Review) Mol. Med. Rep. 2018;18:3547–3554. doi: 10.3892/mmr.2018.9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirota K., Semenza G.L. Rac1 activity is required for the activation of hypoxia-inducible factor 1. J. Biol. Chem. 2001;276:21166–21172. doi: 10.1074/jbc.M100677200. [DOI] [PubMed] [Google Scholar]
- 50.Tang Q., Gu Y., Zhou X., Jin L., Guan J., Liu R., Li J., Long K., Tian S., Che T., et al. Comparative transcriptomics of 5 high-altitude vertebrates and their low- altitude relatives. GigaScience. 2017;6:1–9. doi: 10.1093/gigascience/gix105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kern C., Wang Y., Xu X., Pan Z., Halstead M., Chanthavixay G., Saelao P., Waters S., Xiang R., Chamberlain A., et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021;12:1821. doi: 10.1038/s41467-021-22100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li M., Tian S., Jin L., Zhou G., Li Y., Zhang Y., Wang T., Yeung C.K.L., Chen L., Ma J., et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013;45:1431–1438. doi: 10.1038/ng.2811. [DOI] [PubMed] [Google Scholar]
- 53.Wei C., Wang H., Liu G., Zhao F., Kijas J.W., Ma Y., Lu J., Zhang L., Cao J., Wu M., et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016;6:26770. doi: 10.1038/srep26770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hadjur S., Ung K., Wadsworth L., Dimmick J., Rajcan-Separovic E., Scott R.W., Buchwald M., Jirik F.R. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood. 2001;98:1003–1011. doi: 10.1182/blood.v98.4.1003. [DOI] [PubMed] [Google Scholar]
- 56.Carlström M., Lai E.Y., Ma Z., Steege A., Patzak A., Eriksson U.J., Lundberg J.O., Wilcox C.S., Persson A.E.G. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension. 2010;56:907–913. doi: 10.1161/hypertensionaha.110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Torsdottir G., Kristinsson J., Snaedal J., Sveinbjörnsdóttir S., Gudmundsson G., Hreidarsson S., Jóhannesson T. Case-control studies on ceruloplasmin and superoxide dismutase (SOD1) in neurodegenerative diseases: a short review. J. Neurol. Sci. 2010;299:51–54. doi: 10.1016/j.jns.2010.08.047. [DOI] [PubMed] [Google Scholar]
- 58.Okado-Matsumoto A., Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 59.Eskandari-Nasab E., Kharazi-Nejad E., Nakhaee A., Afzali M., Tabatabaei S.P., Tirgar-Fakheri K., Hashemi M. 50-bp Ins/Del polymorphism of SOD1 is associated with increased risk of cardiovascular disease. Acta Med. Iran. 2014;52:591–595. [PubMed] [Google Scholar]
- 60.Wakisaka Y., Chu Y., Miller J.D., Rosenberg G.A., Heistad D.D. Critical role for copper/zinc-superoxide dismutase in preventing spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. Stroke. 2010;41:790–797. doi: 10.1161/strokeaha.109.569616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neufeld G., Sabag A.D., Rabinovicz N., Kessler O. Semaphorins in angiogenesis and tumor progression. Cold Spring Harb. Perspect. Med. 2012;2:a006718. doi: 10.1101/cshperspect.a006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sadanandam A., Sidhu S.S., Wullschleger S., Singh S., Varney M.L., Yang C.S., Ashour A.E., Batra S.K., Singh R.K. Secreted semaphorin 5A suppressed pancreatic tumour burden but increased metastasis and endothelial cell proliferation. Br. J. Cancer. 2012;107:501–507. doi: 10.1038/bjc.2012.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoon J.H., Park J., Conde J., Wakamiya M., Prakash L., Prakash S. Rev1 promotes replication through UV lesions in conjunction with DNA polymerases eta, iota, and kappa but not DNA polymerase zeta. Genes Dev. 2015;29:2588–2602. doi: 10.1101/gad.272229.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsunari H., Honda M., Watanabe M., Fukushima S., Suzuki K., Miyagawa S., Nakano K., Umeyama K., Uchikura A., Okamoto K., et al. Pigs with delta-sarcoglycan deficiency exhibit traits of genetic cardiomyopathy. Lab. Invest. 2020;100:887–899. doi: 10.1038/s41374-020-0406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beiki H., Liu H., Huang J., Manchanda N., Nonneman D., Smith T.P.L., Reecy J.M., Tuggle C.K. Improved annotation of the domestic pig genome through integration of Iso-Seq and RNA-seq data. BMC Genom. 2019;20:344. doi: 10.1186/s12864-019-5709-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He Y., Luo X., Zhou B., Hu T., Meng X., Audano P.A., Kronenberg Z.N., Eichler E.E., Jin J., Guo Y., et al. Long-read assembly of the Chinese rhesus macaque genome and identification of ape-specific structural variants. Nat. Commun. 2019;10:4233. doi: 10.1038/s41467-019-12174-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Low W.Y., Tearle R., Bickhart D.M., Rosen B.D., Kingan S.B., Swale T., Thibaud-Nissen F., Murphy T.D., Young R., Lefevre L., et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 2019;10:260. doi: 10.1038/s41467-018-08260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miga K.H., Koren S., Rhie A., Vollger M.R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G.A., et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020;585:79–84. doi: 10.1038/s41586-020-2547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Logsdon G.A., Vollger M.R., Hsieh P., Mao Y., Liskovykh M.A., Koren S., Nurk S., Mercuri L., Dishuck P.C., Rhie A., et al. The structure, function and evolution of a complete human chromosome 8. Nature. 2021;593:101–107. doi: 10.1038/s41586-021-03420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. bioRxiv. 2021 doi: 10.1101/2021.05.26.445798. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tao Y., Jordan D.R., Mace E.S. A graph-based pan-genome guides biological discovery. Mol. Plant. 2020;13:1247–1249. doi: 10.1016/j.molp.2020.07.020. [DOI] [PubMed] [Google Scholar]
- 72.Sherman R.M., Salzberg S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020;21:243–254. doi: 10.1038/s41576-020-0210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sedlazeck F.J., Lee H., Darby C.A., Schatz M.C. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018;19:329–346. doi: 10.1038/s41576-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 74.Wang T., Antonacci-Fulton L., Howe K., Lawson H.A., Lucas J.K., Phillippy A.M., Popejoy A.B., Asri M., Carson C., Chaisson M.J.P., et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature. 2022;604:437–446. doi: 10.1038/s41586-022-04601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quan C., Li Y., Liu X., Wang Y., Ping J., Lu Y., Zhou G. Characterization of structural variation in Tibetans reveals new evidence of high-altitude adaptation and introgression. Genome Biol. 2021;22:159. doi: 10.1186/s13059-021-02382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li R., Gong M., Zhang X., Wang F., Liu Z., Zhang L., Xu M., Zhang Y., Dai X., Zhang Z., et al. The first sheep graph pan-genome reveals the spectrum of structural variations and their effects on different tail phenotypes. bioRxiv. 2021 doi: 10.1101/2021.12.22.472709. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan S.M., Sherman R.M., Taylor D.J., Nair D.R., Bortvin A.N., Schatz M.C., McCoy R.C. Local adaptation and archaic introgression shape global diversity at human structural variant loci. Elife. 2021;10:e67615. doi: 10.7554/eLife.67615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen S., Krusche P., Dolzhenko E., Sherman R.M., Petrovski R., Schlesinger F., Kirsche M., Bentley D.R., Schatz M.C., Sedlazeck F.J., Eberle M.A. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019;20:291. doi: 10.1186/s13059-019-1909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nesta A.V., Tafur D., Beck C.R. Hotspots of human mutation. Trends Genet. 2020 doi: 10.1016/j.tig.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao P., Li J., Kang H., Wang H., Fan Z., Yin Z., Wang J., Zhang Q., Wang Z., Liu J.F. Structural variant detection by large-scale sequencing reveals new evolutionary evidence on breed divergence between Chinese and European pigs. Sci. Rep. 2016;6:18501. doi: 10.1038/srep18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Warburton P.E., Waye J.S., Willard H.F. Nonrandom localization of recombination events in human alpha satellite repeat unit variants: implications for higher-order structural characteristics within centromeric heterochromatin. Mol. Cell Biol. 1993;13:6520–6529. doi: 10.1128/mcb.13.10.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simonson T.S., Yang Y., Huff C.D., Yun H., Qin G., Witherspoon D.J., Bai Z., Lorenzo F.R., Xing J., Jorde L.B., et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–75. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 83.Lou H., Lu Y., Lu D., Fu R., Wang X., Feng Q., Wu S., Yang Y., Li S., Kang L., et al. A 3.4-kb copy-number deletion near EPAS1 is significantly enriched in high-altitude Tibetans but absent from the denisovan sequence. Am. J. Hum. Genet. 2015;97:54–66. doi: 10.1016/j.ajhg.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shanta O., Noor A., Human Genome Structural Variation Consortium HGSVC. Sebat J. The effects of common structural variants on 3D chromatin structure. BMC Genom. 2020;21:95. doi: 10.1186/s12864-020-6516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kraft K., Geuer S., Will A.J., Chan W.L., Paliou C., Borschiwer M., Harabula I., Wittler L., Franke M., Ibrahim D.M., et al. Deletions, inversions, duplications: engineering of structural variants using CRISPR/cas in mice. Cell Rep. 2015;10:833–839. doi: 10.1016/j.celrep.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 86.Zhang L., Huang Y., Wang M., Guo Y., Liang J., Yang X., Qi W., Wu Y., Si J., Zhu S., et al. Development and genome sequencing of a laboratory-inbred miniature pig facilitates study of human diabetic disease. iScience. 2019;19:162–176. doi: 10.1016/j.isci.2019.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seppey M., Manni M., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019;1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 90.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gkz369. W191-w198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next- generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonnella G., Niehus N., Kurtz S. GfaViz: flexible and interactive visualization of GFA sequence graphs. Bioinformatics. 2019;35:2853–2855. doi: 10.1093/bioinformatics/bty1046. [DOI] [PubMed] [Google Scholar]
- 96.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vaser R., Sović I., Nagarajan N., Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tempel S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012;859:29–51. doi: 10.1007/978-1-61779-603-6_2. [DOI] [PubMed] [Google Scholar]
- 99.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jeffares D.C., Jolly C., Hoti M., Speed D., Shaw L., Rallis C., Balloux F., Dessimoz C., Bähler J., Sedlazeck F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017;8:14061. doi: 10.1038/ncomms14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruan J., Li H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shi L., Guo Y., Dong C., Huddleston J., Yang H., Han X., Fu A., Li Q., Li N., Gong S., et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016;7:12065. doi: 10.1038/ncomms12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paudel Y., Madsen O., Megens H.J., Frantz L.A.F., Bosse M., Bastiaansen J.W.M., Crooijmans R.P.M.A., Groenen M.A.M. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom. 2013;14:449. doi: 10.1186/1471-2164-14-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paudel Y., Madsen O., Megens H.J., Frantz L.A.F., Bosse M., Crooijmans R.P.M.A., Groenen M.A.M. Copy number variation in the speciation of pigs: a possible prominent role for olfactory receptors. BMC Genom. 2015;16:330. doi: 10.1186/s12864-015-1449-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang L., Xu L., Liu X., Zhang T., Li N., Hay E.H., Zhang Y., Yan H., Zhao K., Liu G.E., et al. Copy number variation-based genome wide association study reveals additional variants contributing to meat quality in Swine. Sci. Rep. 2015;5:12535. doi: 10.1038/srep12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang R., Fang S., Wang J., Zhang C., Zhang R., Liu D., Zhao Y., Hu X., Li N. Genome-wide analysis of structural variants reveals genetic differences in Chinese pigs. PLoS One. 2017;12:e0186721. doi: 10.1371/journal.pone.0186721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Revilla M., Puig-Oliveras A., Castelló A., Crespo-Piazuelo D., Paludo E., Fernández A.I., Ballester M., Folch J.M. A global analysis of CNVs in swine using whole genome sequence data and association analysis with fatty acid composition and growth traits. PLoS One. 2017;12:e0177014. doi: 10.1371/journal.pone.0177014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li M., Chen L., Tian S., Lin Y., Tang Q., Zhou X., Li D., Yeung C.K.L., Che T., Jin L., et al. Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Res. 2017;27:865–874. doi: 10.1101/gr.207456.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keel B.N., Nonneman D.J., Lindholm-Perry A.K., Oliver W.T., Rohrer G.A. A survey of copy number variation in the porcine genome detected from whole-genome sequence. Front. Genet. 2019;10:737. doi: 10.3389/fgene.2019.00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zheng X., Zhao P., Yang K., Ning C., Wang H., Zhou L., Liu J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 2020;11:42. doi: 10.1186/s40104-020-00442-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang H., Wang C., Yang K., Liu J., Zhang Y., Wang Y., Xu X., Michal J.J., Jiang Z., Liu B. Genome wide distributions and functional characterization of copy number variations between Chinese and western pigs. PLoS One. 2015;10:e0131522. doi: 10.1371/journal.pone.0131522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;16:111–120. doi: 10.1007/bf01731581. [DOI] [PubMed] [Google Scholar]
- 115.Chen C., Wang W., Wang X., Shen D., Wang S., Wang Y., Gao B., Wimmers K., Mao J., Li K., Song C. Retrotransposons evolution and impact on lncRNA and protein coding genes in pigs. Mob. DNA. 2019;10:19. doi: 10.1186/s13100-019-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen C., D'Alessandro E., Murani E., Zheng Y., Giosa D., Yang N., Wang X., Gao B., Li K., Wimmers K., Song C. SINE jumping contributes to large-scale polymorphisms in the pig genomes. Mob. DNA. 2021;12:17. doi: 10.1186/s13100-021-00246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw sequencing data generated in this study have been submitted to the Genome Sequence Archive (https://ngdc.cncb.ac.cn/bioproject/) under accession number PRJCA005901.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.