

Abstract
Asymmetric ring-opening of 7-oxabenzonorbornadienes is achieved via Co-catalyzed indole C–H functionalization. The utilization of chiral Co-catalyst consisting of a binaphthyl-derived trisubstituted cyclopentadienyl ligand resulted in high yields (up to 99%) and excellent enantioselectivity (>99% ee) for the target products with tolerance for diverse functional groups. Opposite diastereoselectivities are obtained with chiral Co-catalyst or Cp*CoI2CO. Combined experimental and computational studies suggest β-oxygen elimination being the selectivity-determining step of the reaction. Meanwhile, the reactions of 7-azabenzonorbornadiene could also be executed in a diastereodivergent manner.
Subject terms: Synthetic chemistry methodology, Asymmetric catalysis
Asymmetric ring-opening (ARO) reactions, which can help quickly build molecular complexity, have not been extensively shown via β-oxygen elimination. Here, the authors report a cobalt(III)-catalyzed ARO reaction, providing mechanistic insights and the origins of the diastereo- and enantioselectivity.
Introduction
Tremendous developments have been achieved in the past decades for transition-metal-catalyzed C–H functionalization reactions1–8. Recently, exploring 3d metal complexes in C–H functionalization has attracted considerable attention because of the earth-abundance and low toxicity of these metals compared with the 4d and 5d counterparts9–12. Pioneered by the groups of Matsunaga and Kanai13,14, Ackermann15, Ellman16, Glorius17, and Chang18, cyclopentadienyl (Cp)-Co complexes have been identified as competent catalysts for various C–H functionalization reactions, which often exhibit complementary reactivity and selectivity to those with Cp-Rh catalysts14,18–26. Particularly, the Cp-Co complexes derived from a chiral Cp ligand27–29 or achiral Cp ligand in combination with chiral counteranions30–37 were demonstrated suitable for asymmetric C–H functionalization reactions (Fig. 1a)12. In this regard, the groups of Ackermann30, Matsunaga31–34, Cramer27–29, and Shi35–37 independently reported Co-catalyzed asymmetric aryl C–H functionalization with unactivated olefins, maleimides or dioxazolones. Previously, the modifications of the Cp ring have resulted in an extensive array of Cp-metal catalysts38–44. However, whether the use of chiral Cp ligands in Co-catalyzed C–H functionalization reactions can influence the reaction outcomes of beyond enantioselectivity remains underdeveloped13,27–29,45,46.
Fig. 1. Asymmetric ring-opening of bicyclic olefins via Co-catalyzed C–H functionalization.
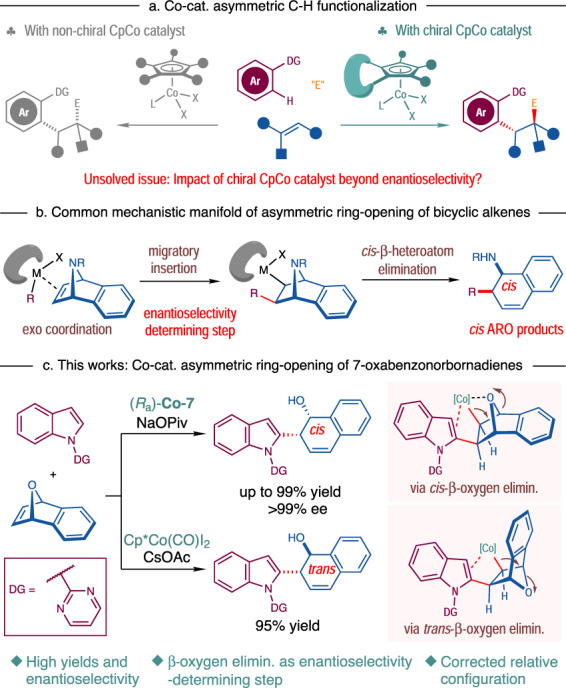
a Co catalyzed asymmetric C–H functionalization. b Common mechanistic manifold of asymmetric ring-opening of bicyclic alkenes. c Co-catalyzed asymmetric ring-opening of 7-oxabenzonorbornadienes (this work).
Asymmetric ring-opening (ARO) of strained bicyclic olefins is an important platform for building-up molecular complexity (Fig. 1b)47–49. Merging ARO reactions with transition-metal-catalyzed C–H activation has become an area of recent interest50–60. In this regard, the groups of Zheng and Li53, and Cramer54 independently reported the ARO reactions of 7-azabenzonorbornadienes and other aza-bicyclic olefins under Rh-catalysis, in which β-nitrogen elimination was a key step. However, to the best of our knowledge, the C–H activation-induced ARO reactions of 7-oxabenzonorbornadienes via β-oxygen elimination remained elusive, probably due to the competitive dehydration61,62 or direct reductive elimination63,64 as a side reaction pathway. Mechanistically, it was regarded that the ARO reactions proceeded via the exo-coordination of a carbon–metal species to the olefin moiety, migratory insertion and cis-β-heteroatom elimination47,53–60. And the migratory insertion was speculated as the enantioselectivity-determining step in the ARO reactions (Fig. 1b)47,53,54. In line with our continuous interest in developing transition-metal-catalyzed asymmetric C–H functionalization reactions65–71, we envisioned that asymmetric indole C–H functionalization with 7-oxabenzonorbornadienes might be accomplished by using a chiral Cp-Co catalyst56 (Fig. 1c).
In this work, high yields and excellent stereoselectivity were obtained for a wide range of cis-ring-opening products under the optimal conditions. More interestingly, the utilization of Cp*Co(CO)I2 as the catalyst led to reversed diastereoselectivities favoring the trans-ring-opening products. The misassigned relative configuration of the products in the previous literature was corrected accordingly56. Combined experimental and computational studies suggested the mechanistic pictures of the CpCo-catalyzed ARO reactions where cis- and trans-β-oxygen elimination work as the selectivity-determining step for enantioselective and racemic reactions, respectively. Such results are different from the previous cognition47,53–64. As an extension, 7-azabenzonorbornadiene could also be applied in the similar ARO reactions in a diastereodivergent manner. Herein, we report the results from this study.
Results
Reaction development
Our study commenced with the optimization of the reaction conditions using N-pyrimidinylindole (1a, 0.05 mmol) and 7-oxabenzonorbornadiene (2a, 1.5 equiv) as model substrates (Table 1). When achiral catalyst Cp*CoI2CO (Co-1, 5 mol%) and CsOAc (30 mol%) were utilized, the reaction proceeded smoothly in trifluoroethanol (TFE, 0.3 mL) at 50 °C, leading to the designed ring-opening product 3aa as a mixture of cis and trans isomers (10:90 dr) in a combined yield of 95% (entry 1). Variating the substituent of the Cp ring of the catalyst (Co-2 and Co-3, R1 = H and Cy) did not provide better yields or ratio of cis-3aa/trans-3aa (entries 2 and 3). On the contrary, when the chiral Co-complexes consisting of a chiral binaphthyl-derived Cp ligand [(Ra)-Co-4–(Ra)-Co-7] were used, the diastereoselectivity of the reaction was reversed, with cis-3aa being the major product (entries 4–7). As the substituent of the Cp ring became bulkier (R2 = H, Me, i-Pr and t-Bu), both the ratio of cis-3aa/trans-3aa (80:20–95:5 dr) and the ee of cis-3aa (69 to >99%) increased steadily albeit with moderate combined yield of 3aa (10–25%). It was found that the reaction efficiency could be significantly influenced by the additive and solvent. Replacing CsOAc to CsOPiv, NaOPiv or HOPiv resulted in the elevated combined yields of cis-3aa (39–60%), high diastereoselectivity (up to 96:4 dr) and superior enantioselectivity (>99% ee), with NaOPiv as the best choice among tested (entries 8–10). Switching the solvent to MeOH, DCM or toluene totally shutdown the reaction. However, the utilization of hexafluoroisopropanol (HFIP) led to nearly quantitative formation of cis-3aa (98% yield) but with slightly decreased dr (93:7) and ee (98%) (entry 11). The stereochemical control could be restored to excellent level by employing mixed solvents of TFE and HFIP (entries 12 and 13). Overall, the optimal reaction outcomes (98% yield, 95:5 dr and >99% ee for cis-3aa) were obtained when the ratio of TFE and HFIP (v/v) was set to 3:1 (entry 13). In addition, the reactions between 1a and 2a were conducted under the (Ra)-Rh-1 catalysis (entry 14, also see the Supplementary Information for details)53, but both cis- and trans-3aa were not detected, while 1a and 2a were recovered. These results demonstrated that the Co-catalysts exhibit unique reactivity for the ARO reaction of 7-oxabenzonorbornadiene.
Table 1.
Optimization of reaction conditionsa
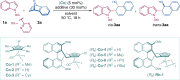
| entry | [Co] | solvent | additive | yield (%)b | dr (cis-3aa/trans-3aa)c | ee of cis-3aa (%)c |
|---|---|---|---|---|---|---|
| 1 | Co-1 | TFE | CsOAc | 95 | 10:90 | – |
| 2 | Co-2 | TFE | CsOAc | 67 | 43:57 | – |
| 3 | Co-3 | TFE | CsOAc | 94 | 18:82 | – |
| 4d | (Ra)-Co-4 | TFE | CsOAc | 10 | 80:20 | 69 |
| 5 | (Ra)-Co-5 | TFE | CsOAc | 12 | 90:10 | >99 |
| 6 | (Ra)-Co-6 | TFE | CsOAc | 19 | 93:7 | >99 |
| 7 | (Ra)-Co-7 | TFE | CsOAc | 25 | 95:5 | >99 |
| 8 | (Ra)-Co-7 | TFE | CsOPiv | 39 | 96:4 | >99 |
| 9 | (Ra)-Co-7 | TFE | NaOPiv | 60 | 96:4 | >99 |
| 10 | (Ra)-Co-7 | TFE | HOPiv | 59 | 92:8 | >99 |
| 11 | (Ra)-Co-7 | HFIP | NaOPiv | 98 | 93:7 | 98 |
| 12 | (Ra)-Co-7 | TFE/HFIP (v/v = 1:1) | NaOPiv | 98 | 94:6 | 99 |
| 13 | (Ra)-Co-7 | TFE/HFIP (v/v = 3:1)e | NaOPiv | 98 | 95:5 | >99 |
| 14 | (Ra)-Rh-1 | TFE/HFIP (v/v = 3:1)e | NaOPiv | ND | – | – |
aReaction conditions: 1a (0.05 mmol), 2a (0.075 mmol), [Co] (5 mol%), additive (30 mol%) in solvent (0.3 mL) at 50 °C for 18 h.
bCombined yield of cis-3aa and trans-3aa.
cThe ratio of cis-3aa/trans-3aa and the ee value of cis-3aa were determined by HPLC with a chiral stationary phase.
dAt 80 °C for 10 h.
eTFE (0.3 mL) and HFIP (0.1 mL) were used as the solvent. ND = not detected.
With the optimal conditions in hands, the scope of substituted indoles was next investigated (Fig. 2). Indoles bearing a methyl group at the C4-, C5- or C6-position were well tolerated. The corresponding products cis-3ba–3da were afforded in high yields (77–99%) with good to excellent diastereoselectivity and excellent enantioselectivity (90:10 to >95:5 dr and 92 to >99% ee). However, the reaction of C7-methyl substituted indole became sluggish where the yield and ee value of cis-3ea were decreased to 78% and 74%, respectively. The influences of other electron-donating groups including methoxy and benzyloxy at the C4-, C5- or C6-position were also investigated. The desired products cis-3fa–3 ha were all obtained efficiently (98–99% yields, 93:7 to >95:5 dr, 99 to >99% ee). Furthermore, a variety of electron-deficient indoles bearing halogen substituents (fluoride, chloride and bromide) or an ester group (CO2Me) at these positions were well tolerated. In general, good yields (70–88%) and excellent stereochemical control (90:10 to >95:5 dr and 97 to >99% ee) were achieved for the formation of cis-3ia, 3ja, 3la–3oa. Notably, introducing a bromide group at the C7-position of the indole ring largely retarded the reaction, leading to cis-3ka in moderate yield (55%) with lowered stereoselectivity (87:13 dr and 83% ee). The reaction could further accommodate [1,3]dioxolo-substituted indole substrate and variations of the electronic properties of the N-pyrimidinyl directing group (cis-3pa–3ra, 58–93% yields, 93:7 to >95:5 dr and 90 to >99% ee). Pyrrole derivatives also well participated in the reaction and the C2-substituents (methyl or phenyl) did not lead to deleterious effects on the reaction outcomes (cis-3sa–3ua, 57–95% yields, >95:5 dr and 69–81% ee). Moreover, thiophene and benzene derivatives can also be applied in the reactions with 7-oxabenzonorbornadiene, albeit with diminished yields of cis-3va and cis-3wa (25–32% yields, >95:5 dr and 53–70% ee) mainly due to recovered substrates 1v and 1w. Samples of cis-3da (enantiopure) and trans-3da (racemic) were subjected to X-ray crystallographic analyses. Their structures, relative configurations, and the absolute configuration of cis-3da (1S,2R) were assigned unambiguously.
Fig. 2. Substrate scope: Indoles and other arenes.

Reaction conditions: 1 (0.10 mmol) 2a (0.15 mmol), (Ra)-Co-7 (5 mol%), NaOPiv (30 mol%) in TFE/HFIP (0.4 mL, v/v = 3:1) at 50 °C for 18 h. Note: (a) In TFE/HFIP/DCM (0.5 mL, v/v/v = 3:1:1) at 50 °C for 18 h. (b) (Ra)-Co-7 (10 mol%), Zn(OAc)2 (30 mol%) instead of NaOPiv (30 mol%) was used, in TFE/HFIP (0.4 mL, v/v = 3:1) at 60 °C for 24 h. (c) (Ra)-Co-7 (10 mol%), Zn(OAc)2 (30 mol%) instead of NaOPiv (30 mol%) was used, in TFE/HFIP (0.4 mL, v/v = 3:1) at 70 °C for 24 h.
The reactions of various 7-oxabenzonorbornadiene-derivatives were also considered (Fig. 3). Symmetrically disubstituted 7-oxabenzonorbornadienes (2,3-dimethyl, 2,3-dimethoxy, 2,3-difluoro, 2,3-benzo, 1,4-dimethoxy and 1,4-dimethyl) participated in the desired reactions smoothly, giving rise to cis-3ab, 3ad–3ag in reasonable yields (50–81%) with good to excellent stereoselectivity (87:13 to >95:5 dr and 94 to >99% ee). To be noted, cis-3ac was afforded in only 35% yield, but with >95:5 dr and 95% ee. It was postulated that the electron-rich nature of cis-3ac caused the side reaction of dehydration61. Methyl-substituent at two bridge-head positions led to moderate yield (62%) and enantiopurity (52% ee) of cis-3ah. The reaction of unsymmetrically disubstituted (1,2-dimethyl) substrate generated a pair of regioisomers cis-3ai (43% yield) and cis-3ai′ (32% yield), both of which were obtained with excellent diastereo- and enantioselectivity (up to >95:5 dr and >99% ee).
Fig. 3. Substrate scope: 7-Oxabenzonorbornadienes.
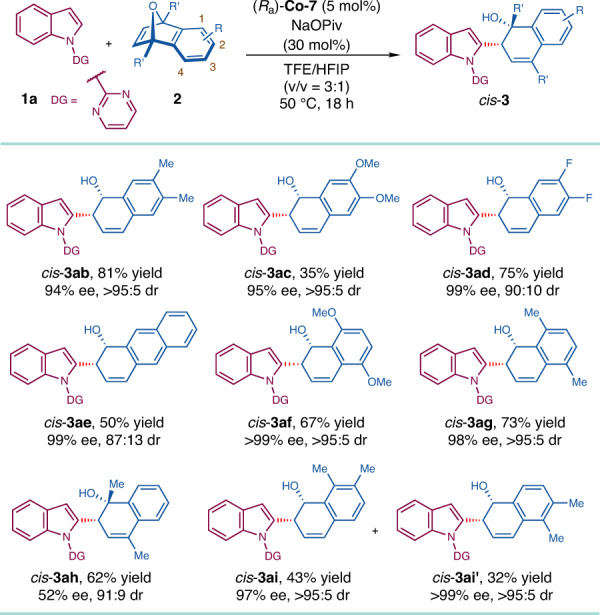
Reaction conditions: 1a (0.10 mmol), 2 (0.15 mmol), (Ra)-Co-7 (5 mol%), NaOPiv (30 mol%) in TFE/HFIP (0.4 mL, v/v = 3:1) at 50 °C for 18 h.
Mechanistic studies
In order to understand the origins for the opposite diastereoselectivity with Co-1 (Conditions A) or (Ra)-Co-7 (Conditions B) and for the enantioselectivity in the latter case, a series of mechanistic studies was performed. In a simplified catalytic cycle (Fig. 4a), the reaction starts with the C–H activation of indole substrate 1a by a Cp-Co complex I (X = OAc or OPiv, L = CO or solvent), which leads to a cyclometallated Co complex II. Deuterium-labeling reactions of 1a were conducted under both conditions using TFE-D3 or mixed solvents of TFE-D3 and HFIP-D2 (v/v = 3:1) (Fig. 4b). High deuteration ratio at the C2 position of the indole ring was observed in both cases (>95% for Conditions A and 83% for Conditions B), which implied the indole C–H activation step should be reversible. Meanwhile, H/D kinetic isotope effect (KIE) experiments using 1a and 1a-2-D revealed small kH/kD values of 1.1 for Conditions A and 1.3 for Conditions B, respectively (Fig. 4c). These results suggested that the indole C–H activation was not involved in the turn-over limiting step of the catalytic cycle. Next, Co complex II undergoes ligand exchange with 7-oxabenzonorbornadiene 2a, which generates an olefin-coordinated Co complex III. Starting from this intermediate, there exist two competing reaction pathways, exo- or endo-migratory insertion, leading to cis-IV and trans-IV, respectively. The subsequent C–O bond-cleavage proceeds via the cis- or trans-β-oxygen elimination, which could be assisted by acetic acid generated in the C–H activation step. The catalytic cycle is finally closed by releasing the products cis-3aa or trans-3aa with the concomitant regeneration of Co complex I. In addition, 12C/13C KIE at natural 13C abundance was determined by quantitative 13C NMR experiments for the reaction of 1a with 2a catalyzed by (Ra)-Co-7 (3 mmol scale)72,73. Significant 12C/13C KIE values were observed for both carbon atoms [1.025(3) and 1.012(3)] of the newly formed C=C double bond of cis-3aa, suggesting the β-oxygen elimination being the rate-determining step (Fig. 4d).
Fig. 4. Proposed catalytic cycle and deuterium-labeling and kinetic isotope effect experiments.

a Proposed catalytic cycle. b Deuterium-labeling reactions of 1a. c H/D Kinetic isotope effect (KIE) experiments. (d) 12C/13C KIE experiments.
DFT calculations were performed for the reactions between 1a and 2a with either Co-1 or (Ra)-Co-7 as the catalyst (Fig. 5, also see the Supplementary Information and Supplementary Data 1 for details). The calculated energy profiles indicated that the β-oxygen elimination was the rate- and stereoselectivity-determining step in both cases, and thus all the precedent steps were reversible. In the reaction promoted by Co-1, the energy of the transition state for trans-β-elimination TS-trans-rac was lower than that for the cis-β-elimination transition state TS-cis-rac by 3.2 kcal/mol, which qualitatively reproduced the preferential formation of trans-3a over cis-3a. It should be noted that the cis-β-elimination relied on a temporary Co–O coordination bond [B(Co–O) = 2.02 Å] in a cationic transition state TS-cis-rac, while in TS-trans-rac such interaction did not exist and the oxygen atom was located in the opposite side of the original 7-oxabenzonorbornadiene ring. Thus, an additional acetate anion was allowed to coordinate to the Co center in TS-trans-rac, forging a highly charge-separate but overall neutral transition state. The variance on the coordination mode of the Co center in TS-cis-rac and TS-trans-rac was believed to contribute to the calculated energy difference between these two transition states. The situation was changed significantly when (Ra)-Co-7 was employed as the catalyst. The steric congestion around the Co center could not accommodate the coordination of an additional anion to the Co center as in TS-trans-rac, which raised the energetic barrier of the trans-β-oxygen elimination, and thus made the cis-β-oxygen elimination the dominant reaction pathway. In order to shed light on the origin of the asymmetric induction with (Ra)-Co-7, the key transition states of cis-β-oxygen elimination leading to (1S,2R)-3aa (TS-cis-SR, 0.0 kcal/mol) and (1R,2S)-3aa (TS-cis-RS, 8.1 kcal/mol), respectively, were located. The large energy difference between these two transition states was in well accordance with the exceedingly high enantioselectivity (>99% ee) observed in the most cases experimentally. The specific substrate orientation in TS-cis-RS induced severe destabilizing structural features including (1) weak coordination of the C2=C3 bond of the indole ring to the Co center [B(Co–C2) = 2.26 Å and B(Co–C3) = 2.62 Å] resulted from the geometric distortion of the substrates compared with that in TS-cis-SR [B(Co–C2) = 2.35 Å and B(Co–C3) = 2.33 Å], and (2) steric repulsion between the substrates and the tert-butyl substituent on the cyclopentadienyl ring in TS-cis-RS, which was exemplified by the short distances between several pairs of hydrogen atoms in the two structural components [B(Ha–Hd) = 2.08 Å, B(Hb–He) = 2.06 Å and B(Hc–He) = 2.29 Å]. To be noted, some minor effects which cause diminished enantioselectivity for the substrates with certain substitution patterns might not be included in the computational model.
Fig. 5. Optimized structures of the transition states for the β-oxygen elimination step.

Calculated at the PWPB95-D3(BJ)/def2-TZVPP (SMD, TFE)//B3LYP-D3(BJ)/def2-SVP (gas) level of theory. The relative Gibbs free energies (ΔGsol) are in kcal/mol. The distances of forming/cleaving bonds are in Å.
Synthetic Applications
As an extension, N-Boc 7-azabenzonorbornadiene 6 was also investigated in this chiral Cp-Co catalyzed ARO reactions (Fig. 6, see the Supplementary Information for details). In the presence of (Sa)-Co-4 as the catalyst, AgSbF6, Zn(OAc)2, as the additives, and TFE as the solvent, the cis-ring-opening products (cis-7aa–7ae) were delivered in morderate yields (55-63%) with excellent stereoselectivity (92:8 to 95:5 dr, 94–96% ee). More interestingly, when the solvent was changed to toluene, the ring-opening products with the opposite relative configuration (trans-7aa–7ae) were obtained in promising results (51–57% yields, 93:7 to >95:5 dr, 63–86% ee). Besides, the reactions of N-tosyl 7-azabenzonorbornadiene could also be executed in a diastereodivergent manner, giving the cis-7af/trans-7af in morderate yields (44–50%) with moderate to good stereoselectivity (95:5 to >95:5 dr, 63–91% ee). Unfortunatly, when 7-oxabenzonorbornadiene 2a was subjected to the conditions for the synthesis of trans-7, only the dehydration product was delivered in almost quantitative yield61,62.
Fig. 6. ARO reactions of 7-azabenzonorbornadiene.

Reaction conditions A: 1 (0.15 mmol), 6 (0.10 mmol), (Sa)-Co-4 (10 mol%), AgSbF6 (20 mol%), Zn(OAc)2 (50 mol %) in TFE (1.0 mL) at 80 °C for 48 h. Reaction conditions B: 1 (0.15 mmol), 6 (0.10 mmol), (Sa)-Co-4 (10 mol%), AgNTf2 (20 mol%), Fe(OAc)2 (50 mol%) in toluene (1.0 mL) at 70 °C for 48 h.
The Co-catalyzed indole C–H functionalization/asymmetric ring-opening of 7-oxabenzonorbornadienes could be performed on a gram-scale. The reaction between 1a (3.0 mmol) and 2a (4.5 mmol) proceeded well in the presence of a lowered loading of (Ra)-Co-7 (2.5 mol%), delivering cis-3aa in 97% yield (989.4 mg) with 95:5 dr and 98% ee after 48 h (Fig. 7a). Besides, cis-3aa and cis-3ia underwent hydrogenation and Suzuki–Miyaura cross-coupling reactions, respectively, generating the corresponding products 4 and 5 in good yields (95% and 85%) without the erosion of enantiopurity (Figs. 7b, 7c). In addition, the N-Boc protecting group of trans-7ab could be readily removed in the presence of hydrogen chloride (Fig. 7d), and the further transformation to trans-7ab′ unambiguously assigned their absolute configuration (Fig. 7e).
Fig. 7. Gram-scale reaction and product transformations.
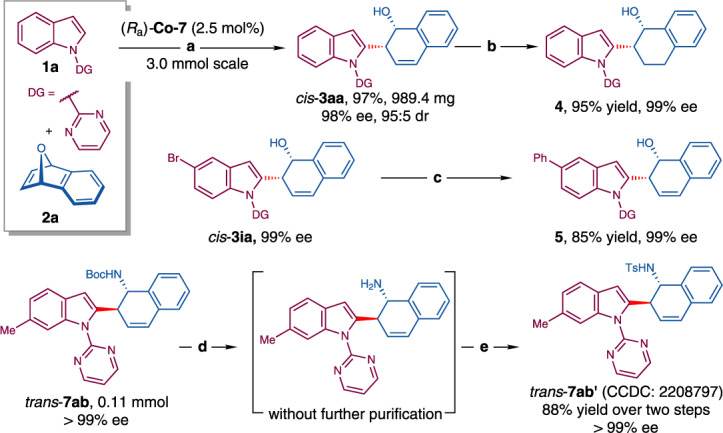
Reaction conditions: (a) 1a (3.0 mmol), 2a (4.5 mmol), NaOPiv · H2O (0.9 mmol), (Ra)-Co-7 (0.075 mmol), TFE/HFIP (12.0 mL, v/v = 3:1), 50 °C, 48 h. (b) Pd/C (0.1 equiv), H2 (1 atm), EtOAc (0.1 M), rt., 24 h. (c) PhB(OH)2 (2.0 equiv), Pd2dba3 (2.5 mol%), SPhos (10 mol%), Na2CO3 (0.5 equiv), toluene/EtOH/H2O (1.25 mL, v/v/v = 3:1:1). (d) HCl (2.0 mL, 4 M in dioxane), rt., overnight. (e) DMAP (1.0 equiv), TsCl (1.2 equiv), Et3N (6.0 equiv), DCM (2.0 mL), rt., 12 h.
Discussion
In conclusion, we have developed Co-catalyzed ARO reactions of 7-oxabenzonorbornadienes via indole C–H activation. The reactions demonstrated opposite diastereoselectivity with binaphthyl-derived chiral cyclopentadienyl-derived cobalt catalysts when compared with that using Cp*Co(CO)I2. Good yields and excellent enantioselectivity were obtained for a wide range of substrates. Combined experimental and computational studies offered deep mechanistic insights into the reaction profile and the origins of the diastereo- and enantioselectivity. Moreover, the Co-catalyzed ARO reactions of 7-azabenzonorbornadiene were also realized in a diastereodivergent manner.
Methods
General procedure for the enantioselective synthesis of cis-3
A sealed tube with a magnetic stir bar was charged with (Ra)-Co-7 (4.0 mg, 0.005 mmol), NaOPiv · H2O (4.4 mg, 0.03 mmol), 1 (0.1 mmol), 2 (0.15 mmol), TFE (0.3 mL) and HFIP (0.1 mL) under argon atmosphere. The mixture was stirred at 50 °C for 18 h. Afterwards, the mixture was cooled to room temperature and quenched by saturated NH4Cl aqueous solution (20 mL). The resulted mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over anhydrous Na2SO4 and filtered. All the volatiles were evaporated under reduced pressure. The remaining residue was purified by column chromatography on silica gel (hexanes/ethyl acetate = 8:1 to 4:1) to afford cis-3.
General procedure for the enantioselective synthesis of cis-7
A sealed tube with a magnetic stir bar was charged with (Sa)-Co-4 (7.6 mg, 0.010 mmol), AgSbF6 (6.8 mg, 0.020 mmol), Zn(OAc)2 (9.2 mg, 0.050 mmol), 1 (0.15 mmol), 6 (24.3 mg, 0.10 mmol) and TFE (1.0 mL) under argon atmosphere. The mixture was stirred at 80 °C for 48 h. Then, the mixture was cooled to room temperature and quenched by saturated NH4Cl aqueous solution (20 mL). The resulted mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over anhydrous Na2SO4 and filtered. All the volatiles were evaporated under reduced pressure. The remaining residue was purified by column chromatography on silica gel (hexanes/ethyl acetate = 10:1 to 6:1) to afford cis-7.
General procedure for the enantioselective synthesis of trans-7
A sealed tube with a magnetic stir bar was charged with (Sa)-Co-4 (7.6 mg, 0.010 mmol), AgNTf2 (7.8 mg, 0.020 mmol), Fe(OAc)2 (8.7 mg, 0.050 mmol), 1 (0.15 mmol), 6 (24.3 mg, 0.10 mmol) and toluene (1.0 mL) under argon atmosphere. The mixture was stirred at 70 °C for 48 h. Afterwards, the mixture was cooled to room temperature and quenched by saturated NH4Cl aqueous solution (20 mL). The resulting mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over anhydrous Na2SO4 and filtered. All the volatiles were evaporated under reduced pressure. The remaining residue was purified by column chromatography on silica gel (hexanes/ethyl acetate = 10:1 to 6:1) to afford trans-7.
Supplementary information
Description of Additional Supplementary File
Acknowledgements
The authors thank National Key R&D Program of China (2021YFA1500100), NSFC (21821002, 22171282, and 92256302), Youth Innovation Promotion Association of Chinese Academy of Sciences (Y2021075), and Science and Technology Commission of Shanghai Municipality (21520780100) for generous financial support. Y.Z. thanks the China Postdoctoral Science Foundation (2021M693279).
Author contributions
S.-L.Y. conceived the project. Y.Z. performed the experiments and analyzed the data. W.-Y.Z. and C.Z. performed the DFT calculations. Y.Z., W.-Y.Z., Q.G., C.Z., and S.-L.Y. cowrote the paper.
Peer review
Peer review information
Nature Communications thanks Baomin Fan, and the other, anonymous, reviewers for their contribution to the peer review of this work.
Data availability
The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC 2164323 (cis-3da), 2164324 (trans-3da) and CCDC 2208797 (trans-7ab′) (www.ccdc.cam.ac.uk/data_request/cif). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chao Zheng, Email: zhengchao@sioc.ac.cn.
Shu-Li You, Email: slyou@sioc.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-36723-6.
References
- 1.Park Y, Kim Y, Chang S. Transition metal-catalyzed C–H amination: scope, mechanism, and applications. Chem. Rev. 2017;117:9247–9301. doi: 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- 2.Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Mild metal-catalyzed C–H activation: examples and concepts. Chem. Soc. Rev. 2016;45:2900–2936. doi: 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]
- 3.Wang F, Yu S, Li X. Transition metal-catalysed couplings between arenes and strained or reactive rings: combination of C–H activation and ring scission. Chem. Soc. Rev. 2016;45:6462–6477. doi: 10.1039/C6CS00371K. [DOI] [PubMed] [Google Scholar]
- 4.He J, Wasa M, Chan KSL, Shao Q, Yu J-Q. Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev. 2017;117:8754–8786. doi: 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Z, Ren Z, Thompson SJ, Xu Y, Dong G. Transition-metal-catalyzed C–H alkylation using alkenes. Chem. Rev. 2017;117:9333–9403. doi: 10.1021/acs.chemrev.6b00574. [DOI] [PubMed] [Google Scholar]
- 6.Wozniak L, et al. Catalytic enantioselective functionalizations of C–H bonds by chiral iridium complexes. Chem. Rev. 2020;120:10516–10543. doi: 10.1021/acs.chemrev.0c00559. [DOI] [PubMed] [Google Scholar]
- 7.Liu C-X, Zhang W-W, Yin S-Y, Gu Q, You S-L. Synthesis of atropisomers by transition-metal-catalyzed asymmetric C-H functionalization reactions. J. Am. Chem. Soc. 2021;143:14025–14040. doi: 10.1021/jacs.1c07635. [DOI] [PubMed] [Google Scholar]
- 8.Yu X, Zhang Z-Z, Niu J-L, Shi B-F. Coordination-assisted, transition-metal-catalyzed enantioselective desymmetric C–H functionalization. Org. Chem. Front. 2022;9:1458–1484. doi: 10.1039/D1QO01884A. [DOI] [Google Scholar]
- 9.Gandeepan P, et al. 3d Transition metals for C−H activation. Chem. Rev. 2019;119:2192–2452. doi: 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]
- 10.Loup J, Dhawa U, Pesciaioli F, Wencel-Delord J, Ackermann L. Enantioselective C−H activation with earth-abundant 3d transition metals. Angew. Chem. Int. Ed. 2019;58:12803–12818. doi: 10.1002/anie.201904214. [DOI] [PubMed] [Google Scholar]
- 11.Woźniak L, Cramer N. Enantioselective C−H bond functionalizations by 3d transition-metal catalysts. Trend Chem. 2019;1:471–484. doi: 10.1016/j.trechm.2019.03.013. [DOI] [Google Scholar]
- 12.Zheng Y, Zheng C, Gu Q, You S-L. Enantioselective C−H functionalization reactions enabled by cobalt catalysis. Chem. Catal. 2022;2:2965–2985. doi: 10.1016/j.checat.2022.08.020. [DOI] [Google Scholar]
- 13.Yoshino T, Ikemoto H, Matsunaga S, Kanai M. A cationic high-valent Cp*CoIII complex for the catalytic generation of nucleophilic organometallic species: directed C−H bond activation. Angew. Chem. Int. Ed. 2013;52:2207–2211. doi: 10.1002/anie.201209226. [DOI] [PubMed] [Google Scholar]
- 14.Ikemoto H, Yoshino T, Sakata K, Matsunaga S, Kanai M. Pyrroloindolone synthesis via a Cp*CoIII-catalyzed redox-neutral directed C−H alkenylation/annulation sequence. J. Am. Chem. Soc. 2014;136:5424–5431. doi: 10.1021/ja5008432. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Ackermann L. Cobalt-catalyzed C−H cyanation of arenes and heteroarenes. Angew. Chem. Int. Ed. 2015;54:3635–3638. doi: 10.1002/anie.201409247. [DOI] [PubMed] [Google Scholar]
- 16.Hummel JR, Ellman JA. Cobalt(III)-catalyzed synthesis of indazoles and furans by C–H bond functionalization/addition/cyclization cascades. J. Am. Chem. Soc. 2015;137:490–498. doi: 10.1021/ja5116452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu D-G, Gensch T, de Azambuja F, Vásquez-Céspedes S, Glorius F. Co(III)-catalyzed C–H activation/formal SN-type reactions: selective and efficient cyanation, halogenation, and allylation. J. Am. Chem. Soc. 2014;136:17722–17725. doi: 10.1021/ja511011m. [DOI] [PubMed] [Google Scholar]
- 18.Park J, Chang S. Comparative catalytic activity of group 9 [Cp*MIII] complexes: cobalt-catalyzed C–H amidation of arenes with dioxazolones as amidating reagents. Angew. Chem. Int. Ed. 2015;54:14103–14107. doi: 10.1002/anie.201505820. [DOI] [PubMed] [Google Scholar]
- 19.Sun B, Yoshino T, Kanai M, Matsunaga S. Cp*CoIII catalyzed site-selective C–H activation of unsymmetrical O-acyl oximes: synthesis of multisubstituted isoquinolines from terminal and internal alkynes. Angew. Chem. Int. Ed. 2015;54:12968–12972. doi: 10.1002/anie.201507744. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki Y, et al. Dehydrative direct C–H allylation with allylic alcohols under [Cp*CoIII] catalysis. Angew. Chem. Int. Ed. 2015;54:9944–9947. doi: 10.1002/anie.201503704. [DOI] [PubMed] [Google Scholar]
- 21.Lerchen A, Knecht T, Daniliuc CG, Glorius F. Unnatural amino acid synthesis enabled by the regioselective cobalt(III)-catalyzed intermolecular carboamination of alkenes. Angew. Chem. Int. Ed. 2016;55:15166–15170. doi: 10.1002/anie.201608729. [DOI] [PubMed] [Google Scholar]
- 22.Kalsi D, Laskar RA, Barsu N, Premkumar JR, Sundararaju B. C-8-selective allylation of quinoline: a case study of β-hydride vs β-hydroxy elimination. Org. Lett. 2016;18:4198–4201. doi: 10.1021/acs.orglett.6b01845. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Lerchen A, Glorius F. A comparative investigation: group 9 Cp*M(III)-catalyzed formal [4 + 2] cycloaddition as an atom-economic approach to quinazolines. Org. Lett. 2016;18:2090–2093. doi: 10.1021/acs.orglett.6b00716. [DOI] [PubMed] [Google Scholar]
- 24.Zhou X, et al. Cp*CoIII-catalyzed branch-selective hydroarylation of alkynes via C–H activation: efficient access to α-gem-vinylindoles. ACS Catal. 2017;7:7296–7304. doi: 10.1021/acscatal.7b02248. [DOI] [Google Scholar]
- 25.Zhou X, Pan Y, Li X. Catalyst-controlled regiodivergent alkyne insertion in the context of C−H activation and Diels–Alder reactions: synthesis of fused and bridged cycles. Angew. Chem. Int. Ed. 2017;56:8163–8167. doi: 10.1002/anie.201704036. [DOI] [PubMed] [Google Scholar]
- 26.Bera SS, Sk MR, Maji MS. Weakly coordinating, ketone-directed (η5-pentamethylcyclopentadienyl)-cobalt(III)- and (η5-pentamethylcyclopentadienyl)-rhodium(III)-catalyzed C−H amidation of arenes: a route to acridone alkaloids. Chem. Eur. J. 2019;25:1806–1811. doi: 10.1002/chem.201805376. [DOI] [PubMed] [Google Scholar]
- 27.Ozols K, Jang YS, Cramer N. Chiral cyclopentadienyl cobalt(III) complexes enable highly enantioselective 3d-metal-catalyzed C–H functionalizations. J. Am. Chem. Soc. 2019;141:5675–5680. doi: 10.1021/jacs.9b02569. [DOI] [PubMed] [Google Scholar]
- 28.Ozols K, Onodera S, Wozniak L, Cramer N. Cobalt(III)-catalyzed enantioselective intermolecular carboamination by C–H functionalization. Angew. Chem. Int. Ed. 2021;60:655–659. doi: 10.1002/anie.202011140. [DOI] [PubMed] [Google Scholar]
- 29.Herraiz AG, Cramer N. Cobalt(III)-catalyzed diastereo- and enantioselective three-component C–H functionalization. ACS Catal. 2021;11:11938–11944. doi: 10.1021/acscatal.1c03153. [DOI] [Google Scholar]
- 30.Pesciaioli F, et al. Enantioselective cobalt(III)-catalyzed C−H activation enabled by chiral carboxylic acid cooperation. Angew. Chem. Int. Ed. 2018;57:15425–15429. doi: 10.1002/anie.201808595. [DOI] [PubMed] [Google Scholar]
- 31.Fukagawa S, et al. Enantioselective C(sp3)−H amidation of thioamides catalyzed by a cobalt(III)/chiral carboxylic acid hybrid system. Angew. Chem. Int. Ed. 2019;58:1153–1157. doi: 10.1002/anie.201812215. [DOI] [PubMed] [Google Scholar]
- 32.Kurihara T, Kojima M, Yoshino T, Matsunaga S. Cp*CoIII/chiral carboxylic acid-catalyzed enantioselective 1,4-addition reactions of indoles to maleimides. Asian J. Org. Chem. 2019;9:368–371. doi: 10.1002/ajoc.201900565. [DOI] [Google Scholar]
- 33.Sekine D, et al. Chiral 2-aryl ferrocene carboxylic acids for the catalytic asymmetric C(sp3)–H activation of thioamides. Organometallics. 2019;38:3921–3926. doi: 10.1021/acs.organomet.9b00407. [DOI] [Google Scholar]
- 34.Hirata Y, et al. Cobalt(III)/chiral carboxylic acid-catalyzed enantioselective synthesis of benzothiadiazine-1-oxides via C−H activation. Angew. Chem. Int. Ed. 2022;61:e202205341. doi: 10.1002/anie.202205341. [DOI] [PubMed] [Google Scholar]
- 35.Liu YH, et al. Cp*Co(III)/MPAA-catalyzed enantioselective amidation of ferrocenes directed by thioamides under mild conditions. Org. Lett. 2019;21:1895–1899. doi: 10.1021/acs.orglett.9b00511. [DOI] [PubMed] [Google Scholar]
- 36.Liu YH, et al. Cp*Co(III)-catalyzed enantioselective hydroarylation of unactivated terminal alkenes via C-H activation. J. Am. Chem. Soc. 2021;143:19112–19120. doi: 10.1021/jacs.1c08562. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Y-B, Zhou T, Qian P-F, Li J-Y, Shi B-F. Synthesis of sulfur-stereogenic sulfoximines via Co(III)/chiral carboxylic acid-catalyzed enantioselective C–H amidation. ACS Catal. 2022;12:9806–9811. doi: 10.1021/acscatal.2c02691. [DOI] [Google Scholar]
- 38.Piou T, Rovis T. Electronic and steric tuning of a prototypical piano stool complex: Rh(III) catalysis for C–H functionalization. Acc. Chem. Res. 2017;51:170–180. doi: 10.1021/acs.accounts.7b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S, Semakul N, Rovis T. Direct regio- and diastereoselective synthesis of δ-lactams from acrylamides and unactivated alkenes initiated by RhIII-catalyzed C−H activation. Angew. Chem. Int. Ed. 2020;59:4965–4969. doi: 10.1002/anie.201916332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piou T, Rovis T. Rhodium-catalysed syn-carboamination of alkenes via a transient directing group. Nature. 2015;527:86–90. doi: 10.1038/nature15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka J, Nagashima Y, Dias AJA, Tanaka K. Photo-induced ortho-C–H borylation of arenes through in situ generation of rhodium(II) ate complexes. J. Am. Chem. Soc. 2021;143:11325–11331. doi: 10.1021/jacs.1c05859. [DOI] [PubMed] [Google Scholar]
- 42.Shibata Y, Tanaka K. Catalytic [2+2+1] cross-cyclotrimerization of silylacetylenes and two alkynyl esters to produce substituted silylfulvenes. Angew. Chem. Int. Ed. 2011;50:10917–10921. doi: 10.1002/anie.201105517. [DOI] [PubMed] [Google Scholar]
- 43.Wodrich MD, Ye B, Gonthier JF, Corminboeuf C, Cramer N. Ligand-controlled regiodivergent pathways of rhodium(III)-catalyzed dihydroisoquinolone synthesis: experimental and computational studies of different cyclopentadienyl ligands. Chem. Eur. J. 2014;20:15409–15418. doi: 10.1002/chem.201404515. [DOI] [PubMed] [Google Scholar]
- 44.Tomita E, et al. Iridium(III) catalysts with an amide-pendant cyclopentadienyl ligand: double aromatic homologation reactions of benzamides by fourfold C−H activation. Angew. Chem. Int. Ed. 2020;59:10474–10478. doi: 10.1002/anie.202003009. [DOI] [PubMed] [Google Scholar]
- 45.Zell D, Bu Q, Feldt M, Ackermann L. Mild C−H/C−C activation by Z-selective cobalt catalysis. Angew. Chem. Int. Ed. 2016;55:7408–7412. doi: 10.1002/anie.201601778. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka R, Tanimoto I, Kojima M, Yoshino T, Matsunaga S. Imidate as the intact directing group for the cobalt-catalyzed C–H allylation. J. Org. Chem. 2019;84:13203–13210. doi: 10.1021/acs.joc.9b01972. [DOI] [PubMed] [Google Scholar]
- 47.Kumar SV, Yen A, Lautens M, Guiry PJ. Catalytic asymmetric transformations of oxa- and azabicyclic alkenes. Chem. Soc. Rev. 2021;50:3013–3093. doi: 10.1039/D0CS00702A. [DOI] [PubMed] [Google Scholar]
- 48.Boutin R, Koh S, Tam W. Recent advances in transition metal-catalyzed reactions of oxabenzonorbornadiene. Curr. Org. Synth. 2019;16:460–484. doi: 10.2174/1570179416666181122094643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rayabarapu DK, Cheng C-H. New catalytic reactions of oxa- and azabicyclic alkenes. Acc. Chem. Res. 2007;40:971–983. doi: 10.1021/ar600021z. [DOI] [PubMed] [Google Scholar]
- 50.Newton CG, Wang S-G, Oliveira CC, Cramer N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem. Rev. 2017;117:8908–8976. doi: 10.1021/acs.chemrev.6b00692. [DOI] [PubMed] [Google Scholar]
- 51.Rej S, Chatani N. Rhodium-catalyzed C(sp2)- or C(sp3)−H bond functionalization assisted by removable directing groups. Angew. Chem. Int. Ed. 2019;58:8304–8329. doi: 10.1002/anie.201808159. [DOI] [PubMed] [Google Scholar]
- 52.Kumar SV, Banerjeea S, Punniyamurthy T. Transition metal-catalyzed coupling of heterocyclic alkenes via C–H functionalization: recent trends and applications. Org. Chem. Front. 2020;7:1527–1569. doi: 10.1039/D0QO00279H. [DOI] [Google Scholar]
- 53.Yang X, Zheng G, Li X. Rhodium(III)-catalyzed enantioselective coupling of indoles and 7-azabenzonorbornadienes by C–H activation/desymmetrization. Angew. Chem. Int. Ed. 2019;58:322–326. doi: 10.1002/anie.201811998. [DOI] [PubMed] [Google Scholar]
- 54.Wang S-G, Cramer N. Enantioselective CpRhx(III)-catalyzed C–H functionalization/ring-opening route to chiral cyclopentenylamines. Angew. Chem. Int. Ed. 2019;58:2514–2518. doi: 10.1002/anie.201813953. [DOI] [PubMed] [Google Scholar]
- 55.Mi R, Zheng G, Qi Z, Li X. Rhodium-catalyzed enantioselective oxidative [3+2] annulation of arenes and azabicyclic olefins through twofold C–H activation. Angew. Chem. Int. Ed. 2019;58:17666–17670. doi: 10.1002/anie.201911086. [DOI] [PubMed] [Google Scholar]
- 56.Muralirajan K, Prakash S, Cheng C-H. Cobalt-catalyzed mild ring-opening addition of arenes C−H bond to 7-oxabicyclic alkenes. Adv. Synth. Catal. 2017;359:513–518. doi: 10.1002/adsc.201601026. [DOI] [Google Scholar]
- 57.Tan H, et al. Cobalt-catalyzed ring-opening addition of azabenzonorbornadienes via C(sp3)–H bond activation of 8-methylquinoline. Chem. Commun. 2020;56:12570–12573. doi: 10.1039/D0CC05374K. [DOI] [PubMed] [Google Scholar]
- 58.Vinayagam V, Mariappan A, Jana M, Jeganmohan M. Rhodium(III)-catalyzed diastereoselective ring-opening of 7-azabenzonorbornadienes with aromatic ketoximes: synthesis of benzophenanthridine derivatives. J. Org. Chem. 2019;84:15590–15604. doi: 10.1021/acs.joc.9b02582. [DOI] [PubMed] [Google Scholar]
- 59.Aravindan N, Jeganmohan M. A short total synthesis of benzophenanthridine alkaloids via a rhodium(III)-catalyzed C–H ring-opening reaction. J. Org. Chem. 2021;86:14826–14843. doi: 10.1021/acs.joc.1c01612. [DOI] [PubMed] [Google Scholar]
- 60.Aravindan N, Vinayagam V, Jeganmohan M. A ruthenium-catalyzed cyclization to dihydrobenzo[c]phenanthridinone from 7-azabenzonorbornadienes with aryl amides. Org. Lett. 2022;24:5260–5265. doi: 10.1021/acs.orglett.2c01734. [DOI] [PubMed] [Google Scholar]
- 61.Kong L, et al. Cobalt(III)-catalyzed C–C coupling of arenes with 7-oxabenzonorbornadiene and 2-vinyloxirane via C–H activation. Org. Lett. 2016;18:3802–3805. doi: 10.1021/acs.orglett.6b01806. [DOI] [PubMed] [Google Scholar]
- 62.Liao G, et al. Synthesis of chiral aldehyde catalysts by Pd-catalyzed atroposelective C−H naphthylation. Angew. Chem. Int. Ed. 2019;58:11464–11468. doi: 10.1002/anie.201906700. [DOI] [PubMed] [Google Scholar]
- 63.Brandes DS, Sirvent A, Mercado BQ, Ellman JA. Three-component 1,2-carboamidation of bridged bicyclic alkenes via RhIII-catalyzed addition of C–H bonds and amidating reagents. Org. Lett. 2021;23:2836–2840. doi: 10.1021/acs.orglett.1c00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trifonova EA, et al. A planar-chiral rhodium(III) catalyst with a sterically demanding cyclopentadienyl ligand and its application in the enantioselective synthesis of dihydroisoquinolones. Angew. Chem. Int. Ed. 2018;57:7714–7718. doi: 10.1002/anie.201801703. [DOI] [PubMed] [Google Scholar]
- 65.Pan C, Yin S-Y, Wang S-B, Gu Q, You S-L. Oxygen-linked cyclopentadienyl rhodium(III) complexes-catalyzed asymmetric C−H arylation of benzo[h]quinolines with 1-diazonaphthoquinones. Angew. Chem. Int. Ed. 2021;60:15510–15516. doi: 10.1002/anie.202103638. [DOI] [PubMed] [Google Scholar]
- 66.Wang Q, et al. Rhodium(III)-catalyzed enantioselective C–H activation/annulation of ferrocenecarboxamides with internal alkynes. ACS Catal. 2022;12:3083–3093. doi: 10.1021/acscatal.2c00083. [DOI] [Google Scholar]
- 67.Wang Q, Zhang W-W, Zheng C, Gu Q, You S-L. Enantioselective synthesis of azoniahelicenes by Rh-catalyzed C–H annulation with alkynes. J. Am. Chem. Soc. 2021;143:114–120. doi: 10.1021/jacs.0c11735. [DOI] [PubMed] [Google Scholar]
- 68.Cui W-J, Wu Z-J, Gu Q, You S-L. Divergent synthesis of tunable cyclopentadienyl ligands and their application in Rh-catalyzed enantioselective synthesis of isoindolinone. J. Am. Chem. Soc. 2020;142:7379–7385. doi: 10.1021/jacs.0c02813. [DOI] [PubMed] [Google Scholar]
- 69.Wang Q, et al. Rhodium-catalyzed atroposelective oxidative C–H/C–H cross coupling reaction of 1-aryl isoquinoline derivatives with electron-rich heteroarenes. J. Am. Chem. Soc. 2020;142:15678–15685. doi: 10.1021/jacs.0c08205. [DOI] [PubMed] [Google Scholar]
- 70.Zheng J, Wang S-B, Zheng C, You S-L. Asymmetric synthesis of spiropyrazolones via Rh-catalyzed C(sp2)-H functionalization/annulation reactions. Angew. Chem. Int. Ed. 2017;56:4540–4544. doi: 10.1002/anie.201700021. [DOI] [PubMed] [Google Scholar]
- 71.Zheng J, Cui W-J, Zheng C, You S-L. Synthesis and application of chiral spiro Cp ligands in rhodium-catalyzed asymmetric oxidative coupling of biaryl compounds with alkenes. J. Am. Chem. Soc. 2016;138:5242–5245. doi: 10.1021/jacs.6b02302. [DOI] [PubMed] [Google Scholar]
- 72.van Dijk L, et al. Mechanistic investigation of Rh(I)-catalysed asymmetric Suzuki–Miyaura coupling with racemic allyl halides. Nat. Catal. 2021;4:284–292. doi: 10.1038/s41929-021-00589-y. [DOI] [Google Scholar]
- 73.Rathbun CM, Johnson JB. Rhodium-catalyzed acylation with quinolinyl ketones: carbon−carbon single bond activation as the turnover-limiting step of catalysis. J. Am. Chem. Soc. 2011;133:2031–2033. doi: 10.1021/ja109686v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary File
Data Availability Statement
The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC 2164323 (cis-3da), 2164324 (trans-3da) and CCDC 2208797 (trans-7ab′) (www.ccdc.cam.ac.uk/data_request/cif). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon request.