

Abstract
A fragment-based drug discovery approach using a pyrazoloadenine fragment library was utilized to uncover new molecules that target the RET (REarranged during Transfection) oncoprotein, which is a driver oncoprotein in ~2% of non-small cell lung cancer. The fragment library was screened against the RET kinase and LC-2/ad (RET-driven), KM-12 (TRKA driven- matched control) and A549 (cytotoxic control) cells to identify selective scaffolds that could inhibit RET-driven growth. An unsubstituted pyrazoloadenine fragment was found active on RET in a biochemical assay but reduced cell viability in non-RET driven cell lines (EC50 = 1 μM and 3 μM, respectively). To increase selectivity for RET, the pyrazoloadenine was modeled in the RET active site, and two domains were identified that were probed with pyrazoloadenine fragment derivatives to improve RET affinity. Scaffolds at each domain were merged to generate a novel lead compound, 8p, which exhibited improved activity and selectivity for the RET oncoprotein (A549 EC50 = 5.92 μM, LC-2/ad EC50 = 0.016 μM, RET IC50 = 0.000326 μM).
Keywords: Pyrazoloadenines, Oncoprotein, Fragment-based drug discovery, RET inhibitor
Graphical Abstract

A fragment-based drug discovery approach was performed to target the RET lung cancer oncoprotein.
Introduction
The RET (REarranged during Transfection) proto-oncogene1 encodes a transmembrane receptor tyrosine kinase, which is responsible for activating numerous signalling cascades.2 Initially, RET was identified as a driver oncogene in thyroid cancers.3 RET mutations were subsequently identified in other cancers4 and were found to drive ~2% of non–small cell lung cancers (NSCLC). Several RET gene fusions have been identified in lung adenocarcinomas and inhibition of these fusions impair lung cancer growth.5 To target RET fusions, small molecule kinase inhibitors have been developed to obstruct RET signalling. Vandetanib and cabozantinib, with FDA approval for thyroid cancer, are in phase II clinical trials for RET-rearranged NSCLC (NCT01639508 for cabozantinib, NCT01823068 for vandetanib).6–9
Pyrazoloadenines, with appropriate structural attributes, inhibit a wide range of protein kinases such as BTK (Bruton’s tyrosine kinase), CDK1/2 (cyclin-dependent kinase 1/2), Src kinase, and RIPK1 (Receptor-Interacting Protein Kinase).10–13 Modification of pyrazoloadenine has generated RET inhibitors for papillary thyroid cancer (PTC) for example, PP1 and PP2 (Figure S1, see supporting information).14,15 Perceiving the plasticity of pyrazoloadenines, we sought to utilize a pyrazoloadenine fragment library to test a fragment-based discovery approach by screening the library against the RET kinase. With our prior interest in developing kinase inhibitors,16–19 a pyrazoloadenine-based fragment library was screened against RET in biochemical assays as well as cellular assays employing LC-2/ad, a RET-CCDC6 fusion-driven NSCLC cell line, and matched controls to determine oncoprotein specificity (TRK-driven, KM-12) and cytotoxicity (A549). This approach led to the discovery of 8p, a highly potent and selective pyrazoloadenine-based inhibitor of RET.
Chemistry
In Scheme S1 (see supporting information), pyrazoloadenine 1 was iodinated using N-iodosuccinimide in DMF heated to 85 oC for 18 hours to form 2. Iodinated intermediate 2 was then N-alkylated at the N1-pyrazole using different alkyl iodides or bromides in the presence of K2CO3 in DMF at 80 oC for 12 hours. Fragments 3b-d were synthesized by reacting 2 with the respective hydrochloride salt of the alkyl chloride using NaH in dry DMF under inert atmosphere and heated to 80 oC for 24–48 hours. Reacting intermediate 2 with 1-bromo-4-(methylsulfonyl)benzene under copper catalysis produced compound 3d.
C-3 substituted 1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-amines 4a-d were obtained by reacting 3a with the corresponding boronic acid using Pd(PPh3)CH2Cl2 and K3PO4 in Dioxane:H2O (4:1) under microwave irradiation for 1 hour at 100 oC (Scheme S2, see supporting information).
Suzuki coupling of 3a-k with ethyl 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetate provided fragments 6a-e. The ester was subjected to saponification by LiOH in THF:H2O (1:1) under microwave irradiation to generate intermediates 7a-c. The acid component was subsequently coupled to various heterocyclic amines to afford the pyrazolo[3,4-d]pyrimidin-3-yl)phenyl) acetamide derivatives (Scheme S3, see supporting information). Derivative 8s was synthesized according to the synthetic route depicted in Scheme S4 (see supporting information). Reaction of 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl) aniline with triphosgene in presence of triethylamine in THF at 60 oC for 6 hours provided the isocyanate 10. Subsequent addition of 5-(tert-butyl)isoxazol-3-amine to intermediate 10 provided 1-(5-(tert-butyl)isoxazol-3-yl)-3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)urea 11 which was subjected to Suzuki coupling to furnish the derivative 8s.
Results and Discussion
Inhibitor design via a fragment-based optimization approach
The pyrazoloadenine warhead (fragment 1) was subjected to a biochemical screen (RET and TRKA) and an oncogene-driven cellular screen (LC-2ad and KM-12). TRKA was employed as an oncoprotein control because of its similarity to the RET kinase domain;20 if compounds exhibit selectivity against TRKA, this suggests selectivity in the greater kinome. The biochemical screen indicated selectivity of fragment 1 for RET (RET IC50 = 9.20 μM) over TRK (TRKA IC50 = 57.07 μM). However, the fragment was found to reduce cell viability in both oncogene-driven cell lines (EC50 = 1.47 μM and 1.73 μM, respectively for LC-2ad and KM-12) along with A549 (EC50 = 3.02 μM; cytotoxic control). Despite exhibiting biochemical relevance, fragment 1 was found to be too cytotoxic by non-selectively reducing cell viability, suggesting the fragment did not possess oncoprotein selectivity. To remove the cytotoxicity profile, fragment 1 was visualized in the RET active site to identify ligand-protein interactions to improve affinity (Figure S2).
Fragment 1 was modelled in a RET DFG-out homology model since molecules that interact with the DFG-out fold of RET possess improved selectivity.21 By analyzing ligand-protein interactions, it was found that fragment 1 formed two hydrogen bonds with hinge residues Ala807 and Glu805 and π-π interactions with Phe893 at the DFG-motif (Figure S2, see supporting information). The modelling study suggested the possibility for fragment expansion at the solvent front (Domain II) and the allosteric pocket (Domain I) (Figure S2, see supporting information). Using modelling insight, fragment 1 underwent expansion at the R1 position. To probe interactions at the solvent front (Domain II), several pyrazoloadenines fragment derivatives were generated (Table S1, see supporting information). N-methyl substitution slightly improved potency against RET (RET IC50 = 3.3 μM; LC-2/ad EC50 = 1 μM) but was still cytotoxic (A549 EC50 = 1 μM). Introducing polar groups, such as morpholino ethyl 3b and N,N-dimethylethyl 3c, improved RET activity and lowered cytotoxicity but cell activity diminished likely from permeability issues. Substituting with aromatic groups such as 2-methyl pyridine 3e and 4-(methylsulfonyl)benzene 3d decreased potency while propanenitrile 3j and methoxyethane 3h drastically reduced activity (Table S1, see supporting information). On the other hand, N-substitution with terminal alcohol 3g, isopentane 3i, pyran 3k, and 3-methoxypropane 3f improved RET potency and selectivity against TRKA. Of these, 3-methoxypropane (3f; RET IC50 = 1.9±2.81 μM) was found to be the most potent in reducing cell viability with 50-fold selectivity over TRKA.
To probe interactions in the kinase back pocket of RET (Domain I), C-3 pyrazoloadenines fragment derivatives were generated (Table S2, see supporting information). All substitutions off the C-3 carbon were found selective for RET over TRKA (4a-d). Of these, phenyl 4a (RET IC50 = 6.82±2.22 μM) and 1-methyl-1H-pyrazole 4d (RET IC50 = 1.044±0.27 μM) were most potent. Analyzing 4a and 4d in the RET active site revealed that the phenyl ring of 4a accesses the RET back pocket through a π-π interaction with Phe893 of the DFG-out motif, while the pyrazole of 4d did not; hence 4a was considered for additional modification to better exploit Domain I (Figure S3, see supporting information). To further develop the pyrazoloadenine SAR, derivatives were synthesized to expand into the back pocket of RET. 6a and 6e were found to be most active against RET with TRKA selectivity, albeit without cell activity.
The pyrazoloadenine SAR was further developed by modifying substituents in the back pocket (Domain I) while simultaneously altering groups at the solvent front (6a-e, 7a-c) (Domain II) (Table S3, see supporting information). Among these 6a, its acidic derivative 7a, and the N-pyran substituted derivative 6e were most active. Extension of the aryl substituent at the C-3 position, via a carboxylic acid handle, facilitated the exploration of the RET allosteric pocket. A small library of pyrazoloadenine acetamides were synthesized using different aromatic and heterocyclic amines to investigate this pocket (Table 1, Table S4 see supporting information).
Table 1.
Inhibition values (IC50/EC50 in μM) and SAR for various N-functionalized substituted phenylacetamidesa
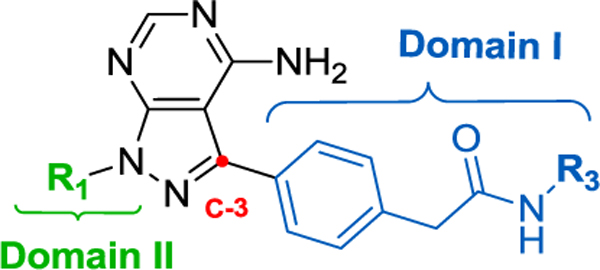
| |||||||
|---|---|---|---|---|---|---|---|
| Code | R1 | R3 | RET (IC50) | TRKA (IC50) | LC-2/ad (EC50) | KM-12 (EC50) | A549 (EC50) |
| 8a | methyl | 3-fluoro-aniline | 35.52±1.41 | 3.7±0.25 | 1.68±0.45 | 2.11±0.08 | 9.20±3.11 |
| 8b | methyl | 5-(tert-butyl)- isoxazol-3 | 0.00057±0.00005 | 0.20±0.74 | 0.024±0.02 | 0.71±0.07 | 20.35±0.51 |
| 8c | methyl | N-methyl-3-(trifluoromethyl)aniline | 0.0562±0.000004 | 4.03±0.93 | 0.37±0.04 | 2±0.03 | 56.3±5.91 |
| 8h | methyl | 3-(tert-butyl)-1-cyclohexyl-N-methyl-1H-pyrazol | 0.109±0.40 | 0.295±0.16 | 1.56±0.36 | 10.46±0.21 | > 20 |
| 8k | 2-ethylpyridyl | 3-(trifluoromethyl) aniline | 0.13±0.07 | 4.74±1.45 | 0.38±0.03 | 1.12±0.12 | > 20 |
| 8l | 2-ethylpyridyl | 3-(tert-butyl)-1-methyl-1H-pyrazol | 0.069±0.004 | 3.95±0.8 | 3.0±0.04 | 2.57±0.64 | > 20 |
| 8m | 2-ethylpyridyl | 3-cyclopropyl-1-methyl-1H-pyrazol-5-yl | 2.93±0.5 | 0.49±0.18 | > 20 | > 20 | > 20 |
| 8n | 4-ethylmorpholine | 5-(tert-butyl)- isoxazol | 0.08±0.02 | 0.12±0.06 | 7.83±0.80 | > 20 | > 20 |
| 8p | 1-methoxypropyl | 5-(tert-butyl)- isoxazol | 0.000326±0.00005 | 0.198±0.067 | 0.016 | 0.3±0.02 | 5.92±1.33 |
| 8q | 1-methoxypropyl | 3-fluoro-aniline | 0.25±0.07 | 1.74±0.43 | 2.41±0.16 | 4.38±0.30 | > 20 |
| 8r | 1-isobutyl | 3-fluoro-aniline | 3.48±0.42 | 2.48±0.83 | > 20 | > 20 | > 20 |
All data represents mean of at least n = 3 independent experiments.
In general, when R1 was methyl pyridine, modification with aryl and heteroaryl amines yielded compounds with weak activity (8i-j). However, when R3 was 3-cyclopropyl-N-methyl pyrazol-5-amine (8m), 3-(tert-butyl)-N-methyl pyrazol-5-amine (8l), or 3-(trifluoromethyl)aniline (8k) compounds exhibited improved cellular activities consistent with enzymatic activities. Solubilizing groups, such as morpholino ethyl (8n), exhibited decent RET inhibition but weak cellular activity, whereas N,N-dimethylethyl (8o) had diminished RET activity and very poor cellular activity.
A set of pyrazolo pyrimido phenylacetamides (8a-m) were synthesized to further define the SAR at the RET back pocket (Table 1, Table S4 see supporting information). Amide modifications with aromatic amines, such as 3-(fluoromethyl)aniline (8a), exhibited reduced RET inhibition (RET IC50 = 35.5 μM) in spite maintaining cellular activity (LC-2/ad EC50 = 1.68 μM), whereas 3-(trifluoromethyl)aniline (8c) exhibited RET inhibition (RET IC50 = 0.0562 μM) consistent with cellular activity (LC-2/ad EC50 = 0.37 μM). 5-membered heterocyclic amines, 3-(tert-butyl)pyrazol-5-amine (8d) and 3-(tert-butyl)-1-isopropyl pyrazol-5-amine (8f), exhibited RET inhibition consistent with cellular activity. While 3-cyclopropyl pyrazol-5-amine (8e) exhibited selectivity for RET over TRKA, the analogue had very poor cellular activity, whereas 3-(tert-butyl)-1-cyclohexyl pyrazol-5-amine (8h) exhibited inhibition of both RET and TRKA while maintaining cellular activity. When 5-(tert-butyl) isoxazol-3-amine (8b) was utilized as the amine input, the compound exhibited selectivity for RET over TRKA (RET IC50 = 0.00057 μM; TRKA IC50 = 0.202 μM). These results displayed that the phenyl isoxazole acetamide derivative 8b possessed notable potency against RET-driven lung cancer with a wide therapeutic window (A549 EC50 = 20.52 μM).
Our previous observations from Table S1 (see supporting information) suggested that N-substitutions with isopentane, pyran, and 3-methoxypropane provided improved RET potency and selectivity over TRKA; hence pyrazolo[3,4-d]pyrimidin-3-yl phenylacetamide derivatives with N-alkyl groups were synthesized and screened. 3-fluorophenyl acetamide (8r) with 1-isobutyl as the N-alkyl group exhibited greater TRKA inhibition over RET albeit with poor cellular activity.
On the other hand, 3-fluorophenylacetamide 8q with 3-methoxypropyl as the N-alkyl substituent inhibited RET without selectivity. Hence, after extensive SAR development from the initial fragment 1, it was identified that fragment 3f and compound 8b exhibited greatest RET selectivity with reduced cytotoxicity. Merging 3f and 8b into a single molecule generated 8p. The core of 8p was identified from the initial biochemical screen and interactions at both the solvent and back pocket of RET were optimized by expanding fragment 1. Merging 3f and 8b resulted in a 2-fold improvement in RET potency and cellular activity (LC-2/ad EC50 = 0.01 μM) (Figure 1).
Figure 1.

Scaffold development strategy that led to RET inhibitor 8p
Global kinase selectivity of 8p was assessed using a 97-kinase panel with three RET point mutations at a concentration of 20 nM. The results indicate that 8p exhibits excellent selectivity with a 0.10 S35 value defined as the percentage of the kinases showing >90% inhibition out of the total number of kinases profiled (see supporting information, Figure S4). Interestingly, 8p was also found to have high affinity for mutant forms of RET including RETM918T, RETV804L, and RETV804M.
To gain insight into the binding of 8p, in-silico docking studies were performed with the RET kinase DFG [Asp892, Phe893, Gly894]-out computational model (Figure 2). The binding of 8p to the RET DFG-out form of the kinase suggests 8p is a type-2 inhibitor.21 The pyrazolopyrimidine core of 8p hydrogen bonds with Ala807 and Glu805 at the hinge region. Pyrazoloadenine and the phenyl linker π-π stack with Phe893 of the DFG-motif. The 3-methoxypropane substituent orients towards the solvent front whereas the amide linker and oxygen of the isoxazole moiety form two hydrogen bonds with the backbone atoms of Asp892. This interaction permits access into the allosteric pocket, which is occupied by tert-butyl isoxazole.
Figure 2.

Computational modelling of 8p in a RET DFG-out homology model. Hydrogen bonds are illustrated in blue and π-π stacking interactions in brown.
Conclusion
This study utilized a fragment optimization approach, based on on both biochemical and cell based screens, along with computational studies to identify selective, pyazoloadenine-based RET inhibitors. Modelling uncovered two domains for fragment expansion to probe interactions within the RET active site. SAR studies led to the discovery of 8p, which is a selective RET inhibitor with sub-nanomolar IC50 values against RET (IC50 = 0.00032 μM) and high potency against LC-2/ad cells (EC50 = 0.01 μM). Kinome-wide selectivity profiling revealed that 8p exhibits excellent selectivity for RET and RET mutants over other similar kinases. In conclusion, this research illustrates the usefulness of an fragment-based optimization approach to rapidly probe an active site to refine a promiscuous molecule for a specific kinase oncoprotien.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of General Medical Sciences (P20 GM109005), a grant from the American Thyroid Association, a UAMS College of Pharmacy Seed grant, and a 2020 UAMS College of Pharmacy Summer Research Fellowship.
References
- 1.Attie-Bitach TA, Abitol M, Gerard M, Delezoide AL, Auge J, Pelet A, Amiel J, Pachnis V, Munnich A, Lyonnet S, Vekemans M, Am. J. Med. Genet. 1998, 80, 481–486. [DOI] [PubMed] [Google Scholar]
- 2.Arighi EB, Borrello MG, Sariola H, Cytokine Growth Factor Rev. 2005, 16, 441–467. [DOI] [PubMed] [Google Scholar]
- 3.Santoro M, Melillo RM, Carlomagno F, Vecchio G, Fusco A, Endocrinology, 2004, 145, 448–5451. [DOI] [PubMed] [Google Scholar]
- 4.Kohno T, Tabata J, Nakaoku T, Carcinogenesis, 2020, 41, 2, 123–129. [DOI] [PubMed] [Google Scholar]
- 5.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T, Nat. Med, 2012, 18, 375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choueiri TK, Pal SK, McDermott DF, Morrissey S, Ferguson KC, Holland J, Kaelin WG, Dutcher JP, Ann. Oncol, 2014, 25, 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi WX, Shen Z, Lin F, Sun YJ, Min DL, Tang LN, He AN, Yao Y, Br. J. Clin. Pharmacol. 2013, 75, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zang J, Wu S, Tang L, Xu X, Bai J, Ding C, Chang Y, Yue L, Kang E, He J, PLoS One, 2012, 7, e30352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang ZF, Wang T, Liu LH, Guo HQ, PLoS One, 2014, 9, e90135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rana F, Liub Y, Yua S, Guoa K, Tanga W, Chena X, Zhaoa G, Bioorg. Chem. 2020, 94, 103367. [DOI] [PubMed] [Google Scholar]
- 11.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ, Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherukupalli S, Chandrasekaran B, Kryštof V, Aleti RR, Sayyad N, Merugu SR, Kushwaha ND, Karpoormath R, Bioorg. Chem. 2018, 79, 46–59. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Xiong Y, Zhang G, Zhang L, Yang W, Yang J, Huang L, Qiao Z, Miao Z, Lin G, Sun Q, Niu T, Chen L, Niu D, Li L, Yang S, J. Med. Chem, 2018, 61, 11398–11414. [DOI] [PubMed] [Google Scholar]
- 14.Carlomagno F, Vitagliano D, Guida T, Napolitano M, Vecchio G, Fusco A, Gazit A, Levitzki A, Santoro M, Cancer Res. 2002, 62, 1077–1082. [PubMed] [Google Scholar]
- 15.Dinér P, Alao JP, Söderlund J, Sunnerhagen P, Grøtli M, J. Med. Chem. 2012, 55, 4872–4876. [DOI] [PubMed] [Google Scholar]
- 16.Saha D, Kharbanda A, Yan W, Lakkaniga NR, Frett B, Yu Li H, J. Med. Chem. 2020, 63, 441–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lakkaniga NR, Zhang L, Belachew B, Gunaganti N, Frett B, yu Li H, Med EJ Chem., 2020, 203, 112589. [DOI] [PubMed] [Google Scholar]
- 18.Bharate JB, McConnell N, Naresh G, Zhang L, Lakkaniga NR, Ding L, Shah NP, Frett B, Li H, Sci. Rep. 2018, 8, 3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saha D, Kharbanda A, Essien N, Zhang L, Cooper R, Basak D, Kendrick S, Brendan Frett H.-yu Li, Org. Chem. Front. 2019, 6, 2234. [Google Scholar]
- 20.Gudernova I, Balek L, Varecha M, Kucerova JF, Bosakova MK, Fafilek B, Palusova V, Uldrijan S, Trantirek L, Krejci P, Oncotarget, 2017, 65, 109319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frett B, Carlomagno F, Moccia ML, Brescia A, Federico G, Falco VD, Admire B, Chen Z, Qi W, Santoro M, Li HY, Angew. Chem. Int. Ed, 2015, 54, 8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.