

Abstract
Background and Aims
Peroxisome proliferator‐activated receptor α (PPARα) regulates fatty acid transport and catabolism in liver. However, the role of intestinal PPARα in lipid homeostasis is largely unknown. Here, intestinal PPARα was examined for its modulation of obesity and NASH.
Approach and Results
Intestinal PPARα was activated and fatty acid‐binding protein 1 (FABP1) up‐regulated in humans with obesity and high‐fat diet (HFD)–fed mice as revealed by using human intestine specimens or HFD/high‐fat, high‐cholesterol, and high‐fructose diet (HFCFD)‐fed C57BL/6N mice and PPARA‐humanized, peroxisome proliferator response element–luciferase mice. Intestine‐specific Ppara or Fabp1 disruption in mice fed a HFD or HFCFD decreased obesity‐associated metabolic disorders and NASH. Molecular analyses by luciferase reporter assays and chromatin immunoprecipitation assays in combination with fatty acid uptake assays in primary intestinal organoids revealed that intestinal PPARα induced the expression of FABP1 that in turn mediated the effects of intestinal PPARα in modulating fatty acid uptake. The PPARα antagonist GW6471 improved obesity and NASH, dependent on intestinal PPARα or FABP1. Double‐knockout (Ppara/Fabp1 ΔIE) mice demonstrated that intestinal Ppara disruption failed to further decrease obesity and NASH in the absence of intestinal FABP1. Translationally, GW6471 reduced human PPARA‐driven intestinal fatty acid uptake and improved obesity‐related metabolic dysfunctions in PPARA‐humanized, but not Ppara‐null, mice.
Conclusions
Intestinal PPARα signaling promotes NASH progression through regulating dietary fatty acid uptake through modulation of FABP1, which provides a compelling therapeutic target for NASH treatment.
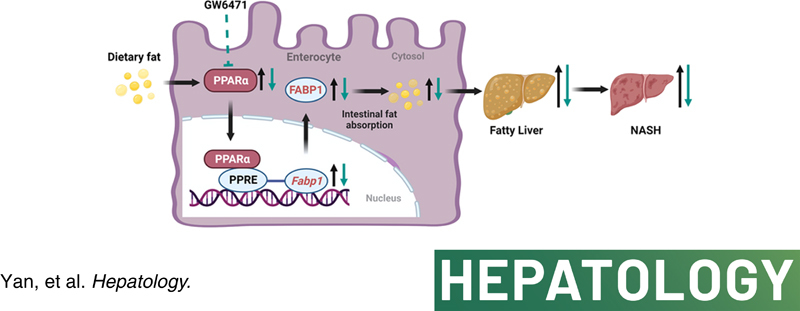
INTRODUCTION
NAFLD is the most common chronic liver disease globally.1 Persistent NAFLD could progress to NASH and increase the risk of end‐stage liver diseases such as cirrhosis and hepatocellular carcinoma.2 To date, no drug has been approved for the treatment of NASH.3 Bariatric surgery, an effective option for treating morbid obesity, was found to resolves NASH.4 However, surgical risk,nutrition and vitamin deficiencies, and other adverse health outcomes largely restrict its broad application for the treatment of NASH.5 Thus, pharmacological therapies for NASH treatment are warranted.
Peroxisome proliferator‐activated receptor α (PPARα) is a nuclear receptor that modulates hepatic lipid homeostasis and affects NASH progression.6 PPARα agonists such as fibrates are widely prescribed for the treatment of dyslipidemias as lipid‐lowering drugs in the clinic.7 However, their use in human NASH treatment has not been approved. Notably, global PPARα knockout mice have markedly enhanced NASH8 but are protected against insulin resistance,9 suggesting pleiotropic roles for PPARα. Hepatocyte‐specific PPARα knockout mice were found to only partially phenocopy the phenotype of global PPARα knockout mice in NAFLD and fasting‐induced hepatic steatosis.10,11 Understanding the tissue‐specific functions of extrahepatic PPARα may help guide drug discovery from PPARα modulators in the treatment of metabolic diseases.
Dietary fat is absorbed by the enterocytes in the form of free fatty acids and 2‐monoacylglycerols that are produced from dietary triglycerides (TG), whereas the absorbed fatty acids and monoacylglycerols are re‐esterified into TG and exported to the blood.12 Fatty acid‐binding protein 1 (FABP1) is known to facilitate the transport of fatty acids and other hydrophobic molecules in the liver,13 whereas the role of intestinal FABP1 in modulating dietary fat absorption and NASH is still unknown.
In the current study, the role of intestine PPARα‐FABP1 signaling in modulating NASH progression was studied using intestine‐specific PPARα and FABP1 knockout mice, intestine‐specific PPARα/FABP1 double‐knockout mice, PPARA‐humanized mice, peroxisome proliferator response element (PPRE)‐luciferase reporter (PPRE‐Luc) mice, primary intestinal organoids, and the PPARα‐specific antagonist GW6471 in combination with global transcriptome and molecular biological analyses. Correlative studies on PPARα/FABP1 signaling with obesity were carried out on human intestine samples.
PATIENTS AND METHODS
Human cohorts
Frozen biopsies were collected in the Second People’s Hospital of Lianyungang City (Lianyungang, Jiangsu, China; Ethics Approval Number: 2018‐017‐01) or the First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China; Ethics Approval Number: 20200949). All individuals gave written informed consent before participation. Paraffin‐embedded intestine tissues were obtained from the Cancer Hospital Chinese Academy of Medical Sciences and Beijing Friendship Hospital (Beijing, China). The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the respective institutional ethics committee. Ethics approval number 2018‐017‐01 was applied by the Ethics Committee of the Second People's Hospital of Lianyungang City and ethics approval number 2020949 was applied by the Ethics Committee of the First Affilliated Hospital of Anhui Medical University.
Animals
Villin‐cre and villin‐ERT2‐cre mice were obtained from Deborah L. Gumucio (University of Michigan)14 and Pierre Chambon (Institute of Genetics, Molecular and Cellular Biology, Illkirch, France),15 respectively. Ppara fl/fl and Ppara ∆IE mice on the C57BL/6N genetic background were described previously.16,17 Fabp1 fl/fl mice on a C57BL/6J background were provided by Nicholas O. Davidson (Washington University School of Medicine).18 PPRE‐Luc mice containing a transgene expressing a luciferase reporter gene under control of a PPRE was generated as described previously (Charles River Company).19,20 PPARA‐humanized mice with the complete human PPARA gene on the Ppara‐null background were described previously.21 Details about the animal studies and mouse strain breeding are listed in the Supporting Information. All animal studies were carried out in accordance with the Institute of Laboratory Animal Resources guidelines and all mice received humane care according to the criteria outlined in the NIH Guide for the Care and Use of Laboratory Animals. The animal protocols were approved by the respective Animal Care and Use Committee of the National Cancer Institute and the National Institute of Diabetes and Digestive and Kidney Diseases.
Statistical analysis
Statistical analysis was performed using Prism version 8.4.3 (GraphPad Software). Sample sizes were indicated in the figure legends. No statistical tool was used to predetermine sample sizes; rather, the availability of materials and estimates of variances based on previous studies determined the number of biological replicates that were used. Experimental values are presented as mean ± SEM. Statistical significance between two groups was determined using two‐tailed Student t‐test, whereas one‐way analysis of variance followed by Tukey’s post hoc correction was applied for multiple comparisons. Correlation analyses of human samples were assessed by nonparametric Pearson’s test. The p values were calculated with confidence intervals of 95%. A p value less than 0.05 was considered statistically significant.
For further details regarding the materials and other methods including immunofluorescence staining, animal treatments and metabolic studies, in vivo luminofluorescence imaging, quantitative polymerase chain reaction (qPCR), western blotting, histological analyses, biochemical analyses, luciferase reporter assays, chromatin immunoprecipitation (ChIP) assay, intestinal organoid studies, RNA sequencing (RNAseq) analyses, and mass spectrometry‐based analyses for fatty acids and bile acids, please refer to the Supporting Information. RNAseq data for mouse intestines can be found in the Gene Expression Omnibus under accession code GSE190140.
RESULTS
Intestinal PPARA signaling is induced in high‐fat diet–fed mice and obese humans
To investigate the effect of NAFLD on PPARα signaling, the messenger RNA (mRNA) levels of PPARA in the intestines of humans with and without obesity were analyzed and found to be induced by obesity and positively correlated with body mass index (BMI) and serum alanine aminotransferase (ALT) levels (Figure 1A). Similarly, high‐fat diet (HFD) treatment increased the intestinal Ppara and its target gene mRNAs in mice following an HFD feeding for 2 weeks and 15 weeks (Figure 1B for jejunum, Figure S1A,B for duodenum and ileum). Nuclear PPARα levels were substantially increased in the intestines from mice fed a 2‐week HFD, 15‐week HFD, or 21‐week high‐fat, high‐cholesterol, and high‐fructose diet (HFCFD) (Figure 1B). In the mouse PPARα‐expressing PPRE‐Luc mice, HFD significantly induced PPRE‐Luc activity in the small intestine both at 1 week and 10 weeks after HFD feeding (Figure 1C). Given that PPARα shows a species response difference between humans and rodents,22 human PPARA or PPARα knockout (Ppara −/−) PPRE‐Luc mice were generated (Figure S1C–E). Compared with chow diet, intestinal PPRE‐luciferase activity was enhanced by 1‐week HFD treatment in PPARA‐humanized PPRE‐Luc mice, and this induction was compromised in Ppara −/− PPRE‐Luc mice (Figure 1C). Luminescent imaging showed enhanced luciferase activity in the abdomens of HFD‐treated human PPARA‐expressing PPRE‐Luc mice, which was compromised in Ppara −/− PPRE‐Luc mice (Figure 1D). Enhanced abdominal luciferase activity of mice was derived from intestines, but not livers or adipose tissues, as revealed by tissue imaging (Figure 1D).
FIGURE 1.
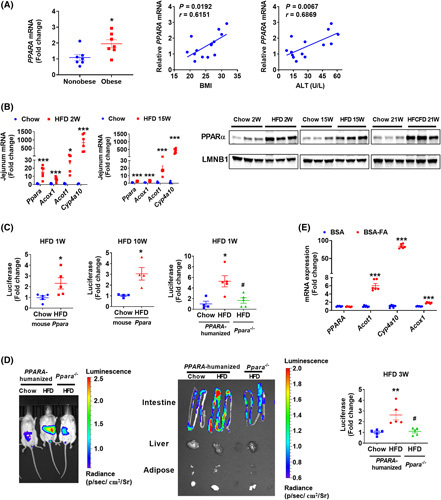
PPARα signaling was induced in the NASH progression. (A) Intestinal PPARA mRNA in biopsies from obese and nonobese humans (n = 7) and correlation analysis of intestinal PPARA mRNA expression with BMI and serum ALT levels (n = 14). (B) Jejunum mRNA levels of Ppara and its target genes in 2‐week and 15‐week HFD‐fed mice (n = 6) and intestinal nuclear PPARα protein from mice fed a HFD for 2 or 15 weeks or a HFCFD for 21 weeks. (C) Intestinal luciferase activities in 1‐week or 10‐week HFD‐fed mouse PPARα‐expressing PPRE‐Luc mice or 1‐week HFD‐fed PPARA‐humanized or Ppara −/− PPRE‐Luc mice (n = 4–5). (D) Representative luminofluorescence imaging of mouse, tissues, and quantitation of intestinal luminofluorescence of 3‐week HFD‐fed mice (n = 5). (E) qPCR analyses of fatty acids‐treated intestinal organoids isolated from PPARA‐humanized mice (n = 6). BSA‐FA, bovine serum albumin (BSA)‐conjugated fatty acids (0.4 mM of palmitic acid plus 0.8 mM of oleic acid). *p < 0.05, **p < 0.01, ***p < 0.001 compared with control. # p < 0.05 compared with HFD‐fed PPARA‐humanized mice
Multiple HFD‐derived fatty acids act as the ligands of PPARα.23 Thus, the direct effect of fatty acid treatment on intestinal PPARα signaling was studied in primary intestinal organoids isolated from PPARA‐humanized mice in vitro. Although PPARA mRNA (Figure 1E) and PPARα protein (Figure S1F) were not changed, the mRNA levels of PPARα target genes were highly induced by palmitic acid/oleic acid treatment (Figure 1E), indicative of human PPARα activation in intestine by fatty acids.
Intestine‐specific PPARA disruption attenuates obesity and NASH
To explore the role of intestinal PPARα in the development of metabolic disorders, control (Ppara fl/fl) and intestine‐specific Ppara‐null (Ppara ∆IE) mice were fed a HFD for 12 weeks. Compared with the Ppara fl/fl mice, Ppara ∆IE mice had less body weight gain with comparable food intake, improved insulin sensitivity, and shortened length of small intestines without measurable changes in intestinal morphology (Figure S2A). Test of energy expenditure of HFD‐fed Ppara fl/fl and Ppara ∆IE mice showed no significant change during which food intake and total activity were comparable (Figure S2B). Under chow diet, Ppara ∆IE mice showed no difference in body weight gain, food intake, liver weight, serum ALT, hepatic total cholesterol (TC) and TG, or serum TC/TG/nonesterified fatty acid (NEFA) levels, and liver histology, whereas significantly improved insulin resistance was shown (Figure S2C). Hematoxylin and eosin (H&E) and Oil Red O staining showed a reduction of hepatic lipid droplets in Ppara ∆IE mice (Figure 2A), consistent with decreased liver weights/indexes, hepatic TC/TG, serum TC/ALT/NEFA levels, with a tendency toward a decrease in serum TG levels (Figure 2B).
FIGURE 2.
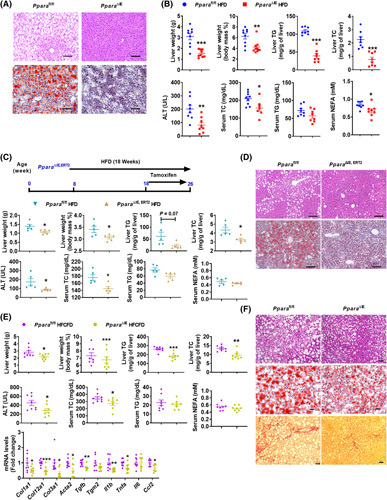
Intestine‐specific Ppara disruption attenuated obesity and NASH. (A,B) Ppara ∆IE and Ppara fl/fl mice were fed an HFD for 12 weeks (n = 8). (A) Representative H&E staining and Oil Red O staining; scale bar 100 µm. (B) Liver weight, liver index, hepatic TG and TC, serum ALT, TC, TG, and NEFA. (C,D) Ppara ∆IE,ERT2 and Ppara fl/fl mice were fed an HFD for 18 weeks and injected with tamoxifen for the last 8 weeks (n = 5). (C) Experimental scheme, liver weight and index, hepatic TG and TC, and serum ALT, TC, TG, and NEFA. (D) H&E and Oil Red O staining; scale bar 100 µm. (E,F) Ppara ∆IE and Ppara fl/fl mice were fed a HFCFD for 21 weeks (n = 8). (E) Liver weight and index; hepatic TG and TC; serum ALT, TC, TG, and NEFA; and hepatic mRNA levels of fibrogenesis‐ and inflammation‐related genes. (F) Representative H&E, Oil Red O, and Sirius Red staining; scale bar 50 µm. *p < 0.05, **p < 0.01, ***p < 0.001
To further test whether the intestinal Ppara disruption had a therapeutic effect in metabolic disorders, villin‐ERT2‐cre Ppara fl/fl (Ppara ∆IE,ERT2) mice were established. Intestinal Ppara disruption induced by tamoxifen did not affect body weight, liver weight, hepatic TC/TG, or serum ALT/TG/TC levels under chow diet (Figure S2D). Then, Ppara ∆IE,ERT2 mice were fed a HFD for 18 weeks with tamoxifen treatment for the last 8 weeks (Figure 2C). Ppara ∆ IE,ERT2 mice showed less body weight gain, liver weights/indexes, hepatic TG/TC, and serum ALT/TC levels; reduced hepatic lipid droplets; and a tendency towards decrease of serum TG/NEFA, accompanied by improved insulin resistance (Figures 2C,D and S2E).
To further explore the effect of intestinal Ppara disruption in the NASH progression, mice were fed a HFCFD to induce NASH characterized with liver inflammation and fibrosis. In HFCFD‐fed mice, Ppara ∆IE mice developed less body weight gain and fat mass without a change in food intake (Figure S2F). The liver weights/indexes, hepatic TG/TC, serum ALT/TC levels, hepatic fibrosis, and inflammation were significantly reduced, whereas serum TG/NEFA tended to be decreased in Ppara ∆IE mice compared with Ppara fl/fl mice (Figure 2E,F).
Compared with Ppara fl/fl mice, the expression of mRNAs encoded by genes involved in lipid synthesis, transport and β‐oxidation, glycolysis and gluconeogenesis, and PPARα signaling were markedly decreased in the livers of 12‐week HFD‐fed Ppara ∆IE mice (Figure S3A), whereas most mRNAs, except diacylglycerol O‐acyltransferase 1( Dgat1), Dgat2, and Gck (glucokinase), remained unchanged in the liver of 21‐week HFCFD‐fed Ppara ∆IE mice (Figure S3B). In 10‐day HFD‐fed mice, the mRNAs involved in hepatic lipid modulation and PPARα signaling remained unchanged, whereas hepatic TG and TC contents were already slightly but significantly decreased in Ppara ∆IE mice compared with Ppara fl/fl mice, with similar body weight and serum ALT/TG/TC/NEFA levels (Figure S3C,D). These data suggest that hepatic lipid modulation pathways and PPARα pathways were changed in an experimental context‐dependent manner and may be a secondary effect of the systemic metabolic changes.
Intestine FABP1 is encoded by a PPARA target gene
To clarify the mechanism underlying the above phenotypes, RNAseq was carried out with RNAs from the intestines of 10‐day chow/HFD‐fed Ppara ∆IE and Ppara fl/fl mice. Principle component analysis distinguished different expression profiles among the four groups (Figure S4A). Compared with chow diet, 430 genes were significantly up‐regulated and 250 genes down‐regulated in the intestines of Ppara fl/fl mice fed a HFD compared with chow diet, whereas 21 genes were up‐regulated and 25 genes down‐regulated in HFD‐fed Ppara ∆IE mice compared with HFD‐fed Ppara fl/fl mice (fold change > 2, P‐adj < 0.05) (Figure S4A). The Venn diagram demonstrates that 15 HFD‐up‐regulated genes were decreased and 4 HFD‐down‐regulated genes were increased by intestinal Ppara disruption (Figure 3A, left), respectively, among which Fabp1, encoding FABP1, ranked the highest HFD‐induced gene (Figure 3A, right). The changes in Fabp1 mRNA were further validated by qPCR analyses (Figure S4A).
FIGURE 3.
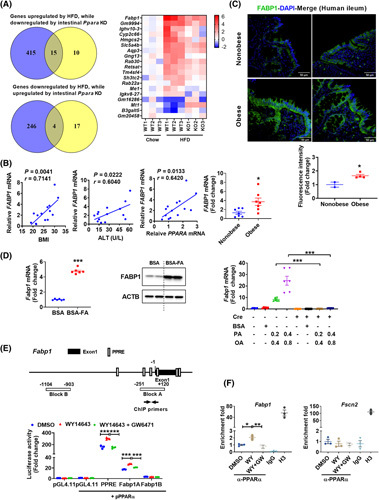
FABP1 is a target of intestinal PPARα. (A) Venn diagram and heat map. (B) correlation analysis of intestinal FABP1 mRNA expression with BMI, ALT, and PPARA mRNA levels (n = 14); mRNA levels of intestinal FABP1 in nonobese or obese humans (n = 7). (C) Representative immunofluorescence staining of FABP1 in human ileum (upper) and statistics (bottom); scale bar 50 µm. (D) Fabp1 mRNA (left) (n = 6) and FABP1 protein (middle) in fatty acid (0.4 mM palmitic acid plus 0.8 mM oleic acid)–treated PPARA‐humanized intestinal organoids, and Fabp1 mRNA (right) (n = 6) in primary Ppara fl/fl and Ppara ∆IE organoids treated with fatty acids for 24 h. (E) Schematic diagram of the mouse Fabp1 promoter illustrating the PPREs (upper) and luciferase reporter assay (bottom) (n = 3). (F) ChIP assay (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. BSA‐FA, BSA‐conjugated fatty acids
Human FABP1 mRNA levels positively correlated with BMI, serum ALT, and intestinal PPARA mRNA levels and were significantly increased by obesity (Figure 3B), along with elevated FABP1 protein in the duodenum (Figure S4B) and terminal ileum (Figure 3B,C) of small intestines collected from humans with obesity relative to nonobese controls. Fabp1 mRNA and FABP1 protein were also significantly increased in fatty acid–treated intestinal organoids isolated from PPARA‐humanized mice (Figure 3D, left and middle). Similarly, intestinal Fabp1 mRNA and FABP1 protein levels were markedly increased in mice fed a 2‐week HFD, 15‐week HFD, or 21‐week HFCFD (Figure S4C). A decrease in Fabp1 mRNA expression was observed in 12‐week HFD‐fed Ppara ∆IE mice, 18‐week HFD‐fed Ppara ∆IE,ERT2 mice treated with tamoxifen for the last 8 weeks, and 21‐week HFCFD‐fed Ppara ∆IE mice (Figure S4D). Moreover, fatty acids sharply induced Fabp1 mRNA expression in organoids isolated from Ppara fl/fl mice, an effect diminished in similarly treated Ppara ∆IE organoids (Figure 3D, right), whereas the Ppara target gene Cyp4a10 served as a positive control (Figure S4E).
Several PPREs are located within 1.5 kb upstream and 0.5 kb downstream of the Fabp1 transcription start site as identified by the Genomatix MatInspector (Figure 3E, upper). To verify if Fabp1 is an intestinal PPARα target gene, luciferase reporter assays and ChIP assays were performed. The luciferase activity of pGL4.11‐Fabp1‐A, which contains PPREs, was induced by the PPARα agonist WY14643 and repressed by the PPARα antagonist GW6471 (Figure 3E, bottom). A validated PPRE reporter vector from Addgene24 served as a positive control and pGL4.11‐Fabp1‐B that contains no PPRE was used as a negative control. ChIP assays revealed enhanced PPARα binding on the Fabp1 promoter after WY14643 treatment, an effect diminished by GW6471. Fscn2, which is not a PPARα target gene, served as a negative control (Figure 3F). These data demonstrate that Fabp1 is a direct PPARα target gene in the intestine.
FABP1 is known to facilitate hepatic fatty acid uptake.13 Thus, the role of intestinal Ppara in the absorption of dietary fatty acids was investigated. Ppara ∆IE mice had a significantly higher level of fecal NEFA compared with Ppara fl/fl mice both at 2 weeks and 12 weeks after HFD feeding as well as at 2 weeks following HFCFD treatment (Figure S4F). Serum postprandial TG levels were also significantly decreased in Ppara ∆IE mice (Figure S4F), suggesting less dietary fatty acid absorption in vivo.
Given that the absorbed fatty acids and monoacylglycerols are re‐esterified into TG and exported to the blood12 and FABP1 mainly facilitates the transport and absorption of long‐chain fatty acids (LCFA),13 serum total LCFA profiles were analyzed. Intestinal PPARα deficiency significantly decreased serum total LCFA in 10‐day HFD, 12‐week HFD, and 21‐week HFCFD‐fed mice (Figure S5A). The expression of some common intestinal lipid transporter mRNAs other than Fabp1 mRNA showed minor or no change in 10‐day or 12‐week HFD‐fed Ppara ∆IE mice, whereas several other mRNAs involved in lipid transport including Fabp2, Cd36, fatty acid transport protein 2 (Fatp2), and microsomal triglyceride transfer protein (Mttp) were decreased in 21‐week HFCFD‐fed Ppara ∆IE mice (Figure S5B).
Bile acids are known to facilitate fat absorption, and thus the FXR and TGR5 pathways controlling bile acid signaling were examined and bile acid profiling performed. Intestinal bile acid levels were not significantly changed in 10‐day HFD, 12‐week HFD, and 21‐week HFCFD‐fed mice with comparable mRNA levels of genes involved in FXR and TGR5 pathways (Figure S5C–E). Because decreased fat absorption may affect the absorption of fat‐soluble vitamins, serum levels of vitamin A, E, and D3 were measured. The levels of serum vitamin A were significantly decreased and serum vitamin E and D3 remained unchanged in 12‐week HFD and 21‐week HFCFD‐fed mice, whereas all three vitamins remain unchanged in 10‐day HFD‐fed mice between the two genotypes (Figure S5F).
A PPARA antagonist reduces intestinal FABP1 expression and NASH, dependent on intestinal PPARA
Next, the effect of a PPARα‐specific antagonist GW6471 in treating obesity and NASH was examined. Pharmacokinetic analysis showed that GW6471 accumulated in the small intestine to much higher levels than in liver (Figure S6A, left and middle). In HFD‐fed PPRE‐Luc mice, 1 week of GW6471 gavage significantly decreased intestinal PPRE‐luciferase activity (Figure S6A, right), supporting the efficacy of GW6471 in inhibiting intestinal PPARα activation. Under HFD, GW6471 reduced HFD‐induced body weight and liver weight gain, hepatic steatosis, and insulin resistance along with lower serum ALT/TC/NEFA, hepatic TG/TC, reduced intestinal FABP1 expression levels, and reduced postprandial serum TG levels in HFD‐fed Ppara fl/fl mice, but not in Ppara ∆IE mice, without significant changes in food intake and serum TG (Figures 4A–C and S6B). Under HFCFD, Ppara ∆IE mice and GW6471‐treated mice displayed less body weight and liver weight gain, hepatic TC/TG, lower serum ALT/TC/TG levels, improved hepatic steatosis, reduced inflammation, reversed fibrosis, and decreased intestinal FABP1 expression as compared with vehicle‐treated Ppara fl/fl mice, whereas these effects of GW6471 were lost in Ppara ∆IE mice (Figures S6C and 4D–F). Fatty acid uptake assays using 12‐N‐methyl‐(7‐nitrobenz‐2‐oxa‐1,3‐diazo) aminostearic acid (NBD‐stearate) and 4,4‐difluoro‐5‐methyl‐4‐bora‐3a,4a‐diaza‐s‐indacene‐3‐dodecanoic acid (BODIPY‐ C12) as substrates, demonstrated that GW6471 decreased fatty acid uptake in intestinal organoids isolated from Ppara fl/fl but not Ppara ∆IE mice (Figure S6D). In C57BL/6N mice fed a chow diet for 12 weeks, GW6471 had no significant effect on body weight, liver weight/index, serum ALT, insulin sensitivity, or hepatic and intestinal histology (Figure S6E). Thus, GW6471 decreases intestinal fatty acid uptake and improves obesity and NASH dependent on the presence of intestinal PPARα, without causing hepatotoxicity.
FIGURE 4.
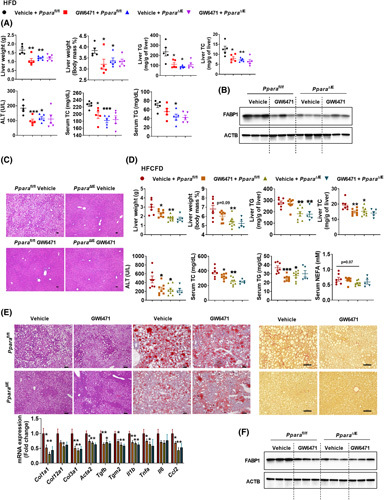
GW6471 reduced obesity and NASH dependent on intestinal PPARα. (A–C) The therapeutic effect of GW6471 in HFD‐induced obesity and fatty liver was examined (n = 5). (A) Liver weight and index, hepatic TG and TC, and serum ALT, TC, and TG. (B) Intestinal FABP1 protein in HFD‐fed mice. (C) Representative H&E staining; scale bar 50 µm. (D–F) The therapeutic effect of GW6471 in HFCFD‐induced NASH were examined (n = 5–8). (D) Liver weight and index, hepatic TG and TC, and serum ALT, TC, TG, and NEFA. (E) Representative H&E, Oil Red O, and Sirius Red staining (scale bar, 100 µm) and mRNA levels of hepatic fibrogenesis and inflammation‐related genes (n = 5–8). (F) Intestinal FABP1 protein in HFCFD‐fed mice. *p < 0.05, **p < 0.01, ***p < 0.001
Intestine‐specific FABPI disruption attenuates obesity and NASH
The role of intestinal FABP1 in modulating obesity and NASH is unknown. Therefore, intestine‐specific FABP1 knockout mice (Fabp1 ∆IE) were generated and studied. Fabp1 mRNA expression was lost in the small intestine but not in extraintestinal tissues (Figure S7A). FABP1 protein was absent in the small intestines of Fabp1 ∆IE mice (Figure 5A). Fabp1 ∆IE and Fabp1 fl/fl mice were fed a HFD for 15 weeks. Body weight gain, fat mass, insulin resistance and small intestine length were markedly reduced in Fabp1 ∆IE mice without change in the food intake and intestinal morphology compared with Fabp1 fl/fl mice (Figure S7A,B, upper). Compared with Fabp1 fl/fl mice, Fabp1 ∆IE mice had a marked reduction of hepatic lipid droplets, lower liver/indexes, hepatic TC/TG, and serum ALT/TC/NEFA levels without changing serum TG (Figure 5B,C). Fecal NEFA levels were substantially higher in Fabp1 ∆IE mice relative to Fabp1 fl/fl mice (Figure 5C).
FIGURE 5.
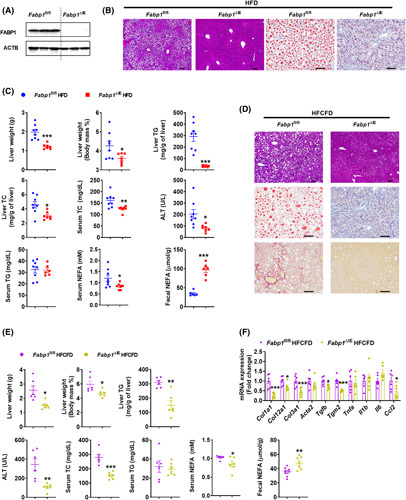
Intestine‐specific Fabp1 disruption attenuated obesity and NASH. (A) Western blot analysis of intestinal FABP1 protein. (B) Representative H&E and Oil Red O staining; scale bar 100 µm. (C) Liver weight and index, hepatic TC and TG, and serum ALT, TC, TG, and NEFA of 15‐week HFD‐fed mice (n = 7–8) and fecal NEFA levels at 3–5 days after HFD feeding (n = 6). (D) Representative H&E, Oil Red O, and Sirius Red staining of 21‐week HFCFD‐fed mice; scale bar 100 µm. (E) Liver weight and liver index, hepatic TG, and serum ALT, TC, TG and NEFA levels of 21‐week HFCFD‐fed mice (n = 6) and fecal NEFA levels of 3–5‐day HFCFD‐fed mice (n = 7). (F) mRNA levels of hepatic fibrosis and inflammation‐related genes (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001
To further evaluate the effect of intestinal FABP1 disruption on NASH, Fabp1 ∆IE and Fabp1 fl/fl mice were fed a HFCFD diet for 21 weeks. HFCFD‐fed Fabp1 ∆IE mice showed decreased body weight gain, fat mass, and insulin resistance without changes in food intake and intestinal histology compared with Fabp1 fl/fl mice (Figure S7C,B, bottom). Liver weights/indexes, hepatic TG and serum ALT/TC/NEFA levels, hepatic lipid accumulation, liver inflammation, and fibrosis were markedly reduced in Fabp1 ∆IE mice, whereas serum TG remained unchanged and fecal NEFA levels increased relative to Fabp1 fl/fl mice (Figure 5D–F). Under a chow diet, albeit to a lesser extent compared with HFD or HFCFD, Fabp1 ∆IE mice developed less body weight gain, liver weights, insulin resistance, and serum/hepatic TG levels, whereas small intestine length, hepatic TC levels, and serum TC/NEFA levels tended to be decreased, without a change in serum ALT levels (Figure S7D), suggesting a role for intestinal FABP1 disruption in decreasing adult‐onset obesity even under chow diet.
Intestinal PPARA antagonism decreases obesity‐associated metabolic disorders dependent on intestinal FABP1
To determine whether the metabolic effect of antagonized PPARα signaling is mediated by FABP1, Fabp1 ∆IE and Fabp1 fl/fl mice were treated with an HFD for 16 weeks and with GW6471 treatment for the last 8 weeks. GW6471 decreased body weight, adipose weight, and insulin resistance in Fabp1 fl/fl but not Fabp1 ∆IE mice (Figure S8A). Hepatic lipid droplets, liver weights/indexes, hepatic TC/TG, serum ALT/TC/NEFA levels, and postprandial serum TG levels were markedly reduced, accompanied with an increase of fecal NEFA levels and a tendency towards decreased serum TG levels in GW6471‐treated Fabp1 fl/fl but not Fabp1 ∆IE mice (Figure 6A,B). GW6471 administration led to decreased small intestine length and FABP1 expression both at the mRNA and protein levels in Fabp1 fl/fl but not in Fabp1 ∆IE mice (Figures 6C and S8A,B). Furthermore, both GW6471 and intestinal Fabp1 disruption markedly decreased fatty acid uptake in primary intestinal organoids, whereas GW6471 treatment did not further affect the fatty acid uptake in Fabp1 ∆IE organoids (Figures 6D and S8C). Furthermore, rescue experiments for fatty acid uptake assays in primary intestinal organoids were performed by overexpressing FABP1 in the primary organoids isolated from Ppara ∆IE mice. The efficacy of FABP1 overexpression was validated (Figure S8D). Ppara gene disruption resulted in significantly less fatty acid uptake compared with Ppara fl/fl organoids, whereas lentivirus‐mediated FABP1 overexpression rescued the decreased fatty acid uptake in Ppara ∆IE organoids (Figures 6E and S8E), supporting the view that down‐regulation of FABP1 at least partially contributed to the intestinal PPARα deficiency‐mediated decrease of fatty acid uptake.
FIGURE 6.
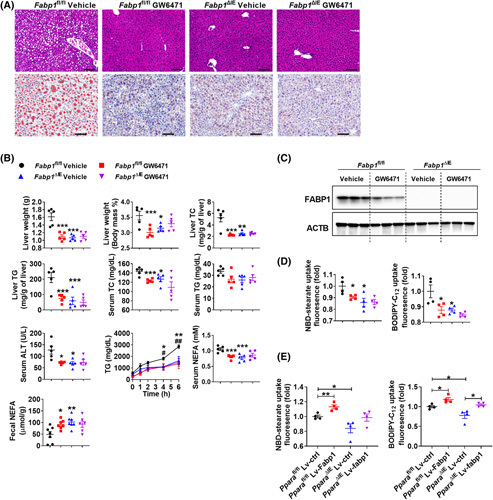
GW6471 decreased intestinal fatty acid uptake and improved obesity and NASH depending on the presence of intestinal FABP1. (A,B) The anti‐NAFLD effect of GW6471 in Fabp1 ∆IE and Fabp1 fl/fl mice. (A) Representative H&E and Oil Red O staining; scale bar 100 µm. (B) Liver weight and liver ratio, hepatic TC and TG, and serum ALT, TC, TG, and NEFA of 16‐week‐HFD‐fed mice (n = 5), postprandial serum TG levels (n = 5) and fecal NEFA levels (n = 7) at 1 week after GW6471 dosing. (C) Intestinal FABP1 protein levels. (D) NBD‐stearate (left) or BODIPY‐C12 (right) uptake in intestinal organoids (n = 4). (E) NBD‐stearate (left) or BODIPY‐C12 (right) uptake in intestinal organoids infected with Lv‐ctrl or Lv‐Fabp1 (n = 4). *p < 0.05; **p < 0.01, ***p < 0.001. (B) Postprandial serum TG, # p < 0.05, ## p < 0.01, Fabp1 ∆IE vehicle group versus Fabp1 fl/fl vehicle group; *p < 0.05, **p < 0.01, ***p < 0.001, Fabp1 fl/fl vehicle group versus Fabp1 fl/fl GW6471 group. Lv‐Ctrl, control lentivirus; Lv‐Fabp1, lentivirus that carries the mouse Fabp1 cDNA
To further assess the contribution of intestinal FABP1 to the effect of intestinal Ppara disruption on metabolic syndrome, an intestine‐specific PPARα/FABP1 double‐knockout (Ppara/Fabp1 ∆IE) mouse line was generated. Ppara and Fabp1 mRNA levels were both diminished specifically in the small intestines but not in the livers of Ppara/Fabp1 ∆IE mice (Figure S9A). Western blot analyses confirmed the intestinal depletion of PPARα and FABP1 protein in Ppara/Fabp1 ∆IE mice (Figure 7A). Fabp1 fl/fl, Fabp1 ∆IE, and Ppara/Fabp1 ∆IE mice were fed a HFD for 15 weeks. Both Fabp1 ∆IE and Ppara/Fabp1 ∆IE mice showed less body weight/fat mass, small intestine length, improved insulin resistance with no change in intestine histology compared with the Fabp1 fl/fl controls, whereas no significant change found between the Fabp1 ∆IE and Ppara/Fabp1 ∆IE groups (Figure S9B–D). Liver weights/indexes, hepatic TC/TG, serum TC/ALT/NEFA levels, and hepatic lipid droplets were reduced, whereas serum TG levels remained unchanged in Fabp1 ∆IE and Ppara/Fabp1 ∆IE mice compared with Fabp1 fl/fl mice, and no change was found between Fabp1 ∆IE and Ppara/Fabp1 ∆IE mice (Figure 7B,C). After feeding a NASH‐promoting HFCFD for 21 weeks, Fabp1 ∆IE and Ppara/Fabp1 ∆IE mice showed less body weight/fat mass, insulin resistance, liver weights/indexes, hepatic TC/TG, serum TC/TG/ALT/NEFA levels, hepatic lipid droplets, postprandial serum TG, hepatic fibrosis and inflammation, and serum LCFA levels, whereas fecal NEFAs increased, compared with Fabp1 fl/fl controls, and no significant change was found between Fabp1 ∆IE and Ppara/Fabp1 ∆IE mice (Figures S9E,F and 7D–F). Thus, intestinal Ppara gene disruption improves obesity and NASH depending on the presence of intestinal FABP1.
FIGURE 7.
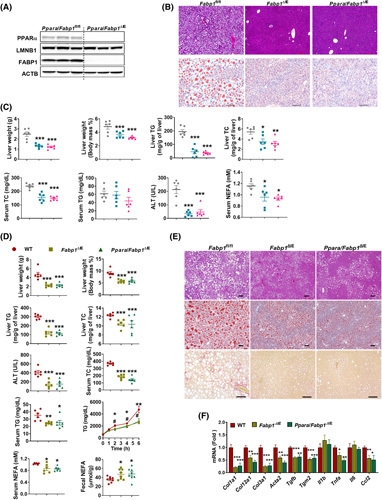
Intestinal Ppara gene disruption improved metabolic disorders depending on the presence of intestinal FABP1. (A) Intestinal FABP1 and PPARα protein levels in Ppara/Fabp1 ∆IE mice. (B,C) Fabp1/Ppara ∆IE mice, Fabp1 ∆IE mice, and Fabp1 fl/fl mice were fed a HFD for 15 weeks (n = 6). (B) H&E, Oil Red O staining; scale bar 100 µm. (C) Liver weight, liver index, liver TG and TC (upper), and serum TC, TG, ALT, and NEFA (bottom). (D–F) Fabp1/Ppara ∆IE mice, Fabp1 ∆IE mice, and Fabp1 fl/fl mice were fed a HFCFD for 21 weeks (n = 6). (D) Liver weight; liver ratio; liver TG and TC; serum ALT, TC, TG, and NEFA levels of 21‐week‐HFCFD‐fed mice (n = 6); and postprandial serum TG levels (n = 5) and fecal NEFA levels of 1‐week HFD‐fed mice (n = 8). (E) H&E, Oil Red O staining, and Sirius Red staining of livers; scale bar 100 µm. (F) mRNA levels of hepatic fibrosis and inflammation‐related genes (n = 6). *p < 0.05; **p < 0.01, ***p < 0.001 compared with the control group. # p < 0.05, ## p < 0.01 indicate comparison between WT and Ppara/Fabp1 ∆IE groups. WT, wild type
Intestinal PPARA antagonist decreases metabolic disorders in PPARA‐humanized mice
Global PPARA‐humanized mice and their matched global Ppara‐null (Ppara −/−) mice were employed to determine whether intestinal human PPARα antagonism decreased NAFLD. To assess the preventive effect, both PPARA‐humanized and Ppara −/− mice were fed an HFD and treated with GW6471 or vehicle for 12 weeks. GW6471 decreased hepatic lipid accumulation and reduced body weight, liver weight, hepatic TG/TC, serum TC/TG/ALT/NEFA levels, and insulin resistance in HFD‐fed PPARA‐humanized mice but not in Ppara −/− mice (Figure S10A). GW6471 also decreased the expression of the PPARα target gene Fabp1 mRNA and FABP1 protein levels in the small intestines of PPARA‐humanized mice but not in Ppara −/− mice (Figure S10B,C).
Next, both global PPARA‐humanized and Ppara −/− mice were fed a HFD for 16 weeks and treated with GW6471 for the last 8 weeks to examine the therapeutic effect of GW6471. Under HFD, GW6471 decreased body weight and insulin resistance in PPARA‐humanized mice, but not in Ppara −/− mice (Figure S10D). GW6471 significantly reduced liver weights, hepatic TC/TG, serum ALT/TG/NEFA levels, and intestinal FABP1 protein levels, whereas serum TC levels tended to decrease in HFD‐fed PPARA‐humanized but not Ppara −/− mice (Figures 8A–C and S10D). Notably, compared with global PPARA‐humanized mice, Ppara −/− mice developed reduced insulin resistance but enhanced NAFLD as evidenced by increased liver weights, hepatic lipid levels, and serum ALT/NEFA levels both during testing the preventive effects and therapeutic effects (Figures S10A,D and 8A,B), which was consistent with earlier studies8,9,25 supporting the view that global mouse PPARα knockout enhanced NAFLD and improved diabetes. To further determine if human PPARα activity regulates intestinal fatty acid uptake, primary intestinal organoids isolated from PPARA‐humanized mice were treated with the PPARα agonist WY14643 or together with the PPARα antagonist GW6471. WY14643 markedly induced the expression of PPARα target genes including Fabp1, an effect diminished by GW6471 treatment (Figures 8D and S10E). Activation of human PPARα by WY14643 increased fatty acid uptake, which was reversed by GW6471, supporting a direct role for human PPARα in facilitating intestinal fatty acid uptake (Figure 8E).
FIGURE 8.
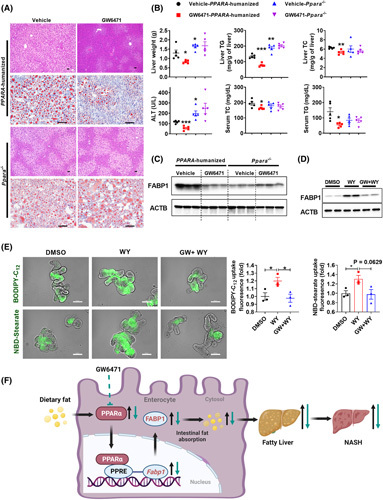
GW6471 decreased fatty acid uptake and fatty liver in PPARA‐humanized mice. (A–C) The anti‐NAFLD effect of GW6471 in PPARA‐humanized mice and Ppara −/− mice, n = 5. (A) H&E (scale bar, 50 µm) and Oil Red O staining (scale bar 100 µm). (B) Liver weight, TG, and TC (upper) and serum ALT, TG, and TC (bottom) (n = 5). (C) Western blot analyses of intestinal FABP1 protein. (D,E) Intestinal organoids isolated from PPARA‐humanized mice were treated with WY14643 (100 µM) or together with GW6471 (6 µM) for 24 h. (D) Western blot analyses of FABP1 protein. (E) BODIPY‐C12 and NBD‐stearate uptake in organoids (n = 3) and representative immunofluorescence images; scale bar, 100 µm. (F) A schematic diagram depicting the role of intestinal PPARα‐FABP1 axis in NASH progression. *p < 0.05, **p < 0.01, ***p < 0.001. WY, WY14643. GW, GW6471
DISCUSSION
The risks of surgery procedures and side effects render bariatric surgery the last, but also almost the only, choice to treat morbid obesity and NASH.4,5 When voluntary lifestyle and dietary strategies fail, alternative pharmacological therapeutics are urgently warranted. Here, we show that intestinal PPARα signaling is highly induced in HFD‐fed mice and humans with obesity. Intestine‐specific PPARα deficiency or chemical inhibition of intestinal PPARα improves obesity‐associated metabolic disorders and NASH. Mechanistically, intestinal FABP1, encoded by a PPARα target gene Fabp1, determines the effect of PPARα on intestinal fatty acid uptake. Intestinal Fabp1 disruption reduces obesity and NASH, whereas intestinal PPARα functional loss reduces NASH depending on the presence of intestinal FABP1. Intestinal PPARα antagonism improves NAFLD in PPARA‐humanized mice depending on the presence of human PPARα. In summary, a PPARα/FABP1 axis in the small intestine is shown that modulates dietary fatty acid uptake, thus providing compelling therapeutic strategies for treating NASH (Figure 8F).
Global PPARα knockout mice have increased fatty liver and NASH while conversely improving insulin resistance.8,25,26 Hepatocyte‐specific PPARα knockout mice just partially reflected the phenotypes of global PPARα knockout mice.10,11 In contrast, Ppara ∆IE mice were protected from obesity‐associated metabolic disorders and NASH. Tissue‐specific and distinct roles of hepatic and intestinal PPARα may at least partially explain the pleotropic roles of global PPARα in glycolipid homeostasis, although the possibility cannot be ruled out that extrahepatic and extraintestinal PPARα also play important roles in NASH development. Great efforts have been invested in the discovery of PPARα agonists based on the understanding of global or hepatic PPARα activation in NAFLD development.27,28 However, the PPARα agonists fibrates as lipid‐lowering drugs have not been approved for NASH treatment despite a history of use for decades,7 and there are no compelling experimental data to support how tissue‐specific roles of PPARα may explain their poor efficacy in treating NASH. Here, we suggest that systematic PPARα agonists may not work in the treatment of NAFLD due to intestinal PPARα activation compromising its anti‐NAFLD effect resulting from hepatic PPARα activation. Thus, demonstrating a role for intestinal PPARα distinct from hepatic PPARα in modulating NASH is of great importance to explain the failure of fibrates in treating NASH and address the potential for drug discovery from intestinal PPARα antagonists for treating NASH.
The causal relationship for the contribution of intestinal FABP1 down‐regulation by intestinal PPARα deficiency to the anti‐NASH effect of intestinal PPARα functional loss was further validated by using GW6471‐treated Fabp1 ∆IE mice or intestine‐specific Ppara/Fabp1 ∆IE mice. Adult‐onset fatty liver and dietary NASH were decreased in global Fabp1 −/− mice,29–33 and hepatocyte‐specific FABP1 knockout mice were protected from fasting‐induced hepatic steatosis and dietary fibrosis, phenocopying the global Fabp1 −/− mice, whereas hepatic stellate cell loss of FABP1 had no effect in modulating fibrosis.18 In contrast, another study showed that global Fabp1 −/− mice had enhanced HFD‐induced obesity compared with the wild‐type controls.34 Thus, the current study phenocopies the antiobesity effect of the global FABP1 knockout and demonstrates that intestine‐specific FABP1 deficiency improves NASH by decreasing dietary fatty acid uptake, which at least partially contributes to the phenotype of the intestinal PPARα knockout mouse. In line with this finding, FABP1 knockdown in the human enterocyte cell line Caco‐2 decreased cell proliferation accompanied by lower fatty acid uptake.35 A recent study also demonstrated that intestinal PPARα was necessary to control the overeating‐induced enhancement of gut surface size and absorptive capacity,36 which, consistent with the present study, supports a positive role for intestinal PPARα in regulating dietary lipid absorption. The present data supports the view that intestinal FABP1 at least partially contributes to the improved metabolic syndrome by intestinal PPARα depletion or inhbition, albeit the contribution of some other genes involved in intestinal lipid transport could not be excluded. In addition to FABP1, FABP2 was suggested to play a major role in the intestinal fatty acid transport under basal conditions,34,37 and global Fabp2‐null mice showed decreased obesity.34 However, HFD feeding only slightly altered Fabp2 expression, whereas it highly induced Fabp1 expression. Thus, intestinal FABP1 may have a more important role in controlling diet‐induced obesity‐related metabolic disorders.
Adverse effects have been found to be associated with decreasing fat absorption, including steatodiarrhea, deficiency of fat‐soluble vitamins, and enhanced hepatic de novo fatty acid synthesis.12 However, no sign of steatodiarrhea was observed in the current HFD or HFCFD‐fed Ppara ∆IE mice, whereas intestinal PPARα deficiency had minor effects on serum levels of fat‐soluble vitamins in the current study. Intestinal PPARα deficiency does not enhance, but actually compromises hepatic lipogenesis in long‐term models, whereas it shows no significant effects on hepatic lipogenesis gene expression in short‐term models. Thus, it is less likely that intestinal PPARα deficiency would cause these fat malabsorption‐accompanied adverse effects, albeit the minor but significant reduction in serum vitamin A levels deserves further attention. Although hepatic PPARα is known to play a role in modulating bile acid homeostasis,38 no significant change in intestinal bile acids was found between wild‐type and intestinal Ppara‐deficient mice in the current study, suggesting that bile acids may not play a crucial role in the metabolic improvement in the current models.
In summary, the present work reveals an intestinal PPARα‐FABP1 axis that modulates obesity and NASH throught the control of dietary fatty acid uptake. This study offers insights into gut‐liver cross‐talk, and suggests the potential for drug discovery of intestinal PPARα and FABP1 inhibitors for use in the treatment of NASH.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Buckley, Chad N. Brocker, Cen Xie, Qiao Wang, Jie Zhao, Jiang Yue, and Dasheng Lu for expert advice and help with animal dissection. We thank Linda G. Byrd for submitting animal protocols and particularly thank Nicholas O. Davidson for providing the Fabp1 fl/fl mice.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Conceptualization and design: Tingting Yan, Yuhong Luo, and Frank J. Gonzalez. Methodology: Tingting Yan, Yuhong Luo, Oksana Gavrilova, Aijuan Qu, and Weiwei Liu. Formal analysis: Tingting Yan, Yuhong Luo, Aijuan Qu, Xuan Wu, and Oksana Gavrilova. Investigation: Tingting Yan, Yuhong Luo, Nana Yan, Aijuan Qu, Weiwei Liu, Changdong Zhao, Dan Qi, Nan Zhao, Xiaoting Yu, Xuan Wu, Keisuke Hamada, Yangliu Xia, Ping Wang, Shoumei Yang, Qiong Wang, Lulu Sun, Jie Cai, Kristopher W. Krausz, Xuan Wu, and Daxesh P. Patel. Writing—original draft, review, and editing: Tingting Yan, Yuhong Luo, Frank J. Gonzalez, Aijuan Qu, and Oksana Gavrilova. Supervision: Frank J. Gonzalez, Aijuan Qu, Weiwei Liu, Haiping Hao, and Changtao Jiang. Funding acquisition: Frank J. Gonzalez, Aijuan Qu, and Weiwei Liu.
Footnotes
Abbreviations: ALT, alanine aminotransferase; BMI, body mass index; BODIPY‐ C12, 4,4‐difluoro‐5,7‐dimethyl‐4‐bora‐3a,4a‐diaza‐s‐Indacene‐3‐dodecanoic acid; ChIP, chromatin immuinoprecipitation; FABP1, fatty acid‐binding protein 1; H&E, hematoxylin and eosin; HFD, high‐fat diet; HFCFD, high‐fat, high‐cholesterol, and high‐fructose diet; LCFA, long‐chain fatty acids; mRNA, messenger RNA; NBD‐stearate, 12‐N‐methyl‐(7‐nitrobenz‐2‐oxa‐1,3‐diazo)aminostearic acid; NEFA, nonesterified fatty acids; PPAR, peroxisome proliferator‐activated receptor; PPRE, peroxisome proliferator response element; PPRE‐Luc, PPRE‐luciferase reporter; qPCR, quantitative polymerase chain reaction; RNAseq, RNA sequencing; TC, total cholesterol; TG, triglycerides.
Funding informationThis work was funded by the National Cancer Institute Intramural Research Program and grants from the National Natural Science Foundation of China (82070474), Key Science and Technology Project of Beijing Municipal Institutions (KZ202010025032), and Special Clinical Research Project of Shanghai Municipal Health Commission (202140147).
Tingting Yan and Yuhong Luo contributed equally.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
Contributor Information
Weiwei Liu, Email: huashanvivian@126.com.
Aijuan Qu, Email: aijuanqu@ccmu.edu.cn.
Frank J. Gonzalez, Email: gonzalef@mail.nih.gov.
REFERENCES
- 1.Younossi ZM. Non‐alcoholic fatty liver disease − a global public health perspective. J Hepatol. 2019;70(3):531–44. [DOI] [PubMed] [Google Scholar]
- 2.Huang DQ, El‐Serag HB, Loomba R. Global epidemiology of NAFLD‐related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non‐alcoholic fatty liver disease. Gut. 2017;66(1):180–90. [DOI] [PubMed] [Google Scholar]
- 4.Lassailly G, Caiazzo R, Ntandja‐Wandji LC, Gnemmi V, Baud G, Verkindt H, et al. Bariatric surgery provides long‐term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology. 2020;159(4):1290–301.e5. [DOI] [PubMed] [Google Scholar]
- 5.Arterburn DE, Telem DA, Kushner RF, Courcoulas AP. Benefits and risks of bariatric surgery in adults: a review. JAMA. 2020;324(9):879–887. [DOI] [PubMed] [Google Scholar]
- 6.Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity‐induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13(1):36–49. [DOI] [PubMed] [Google Scholar]
- 7.Goldfine AB, Kaul S, Hiatt WR. Fibrates in the treatment of dyslipidemias ‐ time for a reassessment. N Engl J Med. 2011;365(6):481–44. [DOI] [PubMed] [Google Scholar]
- 8.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARα‐dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123–32. [DOI] [PubMed] [Google Scholar]
- 9.Guerre‐Millo M, Rouault C, Poulain P, André J, Poitout V, Peters JM, et al. PPAR‐alpha‐null mice are protected from high‐fat diet‐induced insulin resistance. Diabetes. 2001;50(12):2809–14. [DOI] [PubMed] [Google Scholar]
- 10.Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, et al. Liver PPARα is crucial for whole‐body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65(7):1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brocker CN, Patel DP, Velenosi TJ, Kim D, Yan T, Yue J, et al. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting‐induced hepatosteatosis in mice. J Lipid Res. 2018;59(11):2140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol. 2020;17(3):169–83. [DOI] [PubMed] [Google Scholar]
- 13.Wang GQ, Bonkovsky HL, de Lemos A, Burczynski FJ. Recent insights into the biological functions of liver fatty acid binding protein 1. J Lipid Res. 2015;56(12):2238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277(36):33275–83. [DOI] [PubMed] [Google Scholar]
- 15.El Marjou F, Janssen KP, Hung‐Junn Chang B, Li M, Hindie V, Chan L, et al. Tissue‐specific and inducible Cre‐mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–93. [DOI] [PubMed] [Google Scholar]
- 16.Luo Y, Xie C, Brocker CN, Fan J, Wu X, Feng L, et al. Intestinal PPARα protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology. 2019;157(3):744–59.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brocker CN, Yue J, Kim D, Qu A, Bonzo JA, Gonzalez FJ. Hepatocyte‐specific PPARA expression exclusively promotes agonist‐induced cell proliferation without influence from nonparenchymal cells. Am J Physiol Gastrointest Liver Physiol. 2017;312(3):G283–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newberry EP, Xie Y, Lodeiro C, Solis R, Moritz W, Kennedy S, et al. Hepatocyte and stellate cell deletion of liver fatty acid binding protein reveals distinct roles in fibrogenic injury. FASEB J. 2019;33(3):4610–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El‐Jamal N, Dubuquoy L, Auwerx J, Bertin B, Desreumaux P. In vivo imaging reveals selective PPAR activity in the skin of peroxisome proliferator‐activated receptor responsive element‐luciferase reporter mice. Exp Dermatol. 2013;22(2):137–40. [DOI] [PubMed] [Google Scholar]
- 20.Xie G, Yin S, Zhang Z, Qi D, Wang X, Kim D, et al. Hepatocyte peroxisome proliferator‐activated receptor α enhances liver regeneration after partial hepatectomy in mice. Am J Pathol. 2019;189(2):272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Q, Nagano T, Shah Y, Cheung C, Ito S, Gonzalez FJ. The PPARα‐humanized mouse: a model to investigate species differences in liver toxicity mediated by PPARα. Toxicol Sci. 2008;101(1):132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts RA. Peroxisome proliferators: mechanisms of adverse effects in rodents and molecular basis for species differences. Arch Toxicol. 1999;73(8–9):413–8. [DOI] [PubMed] [Google Scholar]
- 23.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator‐activated receptors α and δ. Proc Natl Acad Sci U S A. 1997;94(9):4312–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARγ through the production of endogenous ligand. Proc Natl Acad Sci USA. 1998;95(8):4333–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costet P, Legendre C, More J, Edgar A, Galtier P, Pineau T. Peroxisome proliferator‐activated receptor α‐isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J Biol Chem. 1998;273(45):29577–85. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator‐activated receptor α‐inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275(37):28918–228. [DOI] [PubMed] [Google Scholar]
- 27.Ferri N, Corsini A, Sirtori C, Ruscica M. PPARα agonists are still on the rise: an update on clinical and experimental findings. Expert Opin Investig Drugs. 2017;26(5):593–602. [DOI] [PubMed] [Google Scholar]
- 28.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol. 2015;62(3):720–33. [DOI] [PubMed] [Google Scholar]
- 29.Newberry EP, Xie Y, Kennedy S, Han X, Buhman KK, Luo J, et al. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid‐binding protein gene. J Biol Chem. 2003;278(51):51664–72. [DOI] [PubMed] [Google Scholar]
- 30.Newberry EP, Kennedy SM, Xie Y, Luo J, Crooke RM, Graham MJ, et al. Decreased body weight and hepatic steatosis with altered fatty acid ethanolamide metabolism in aged L‐Fabp−/− mice. J Lipid Res. 2012;53(4):744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newberry EP, Kennedy SM, Xie Y, Sternard BT, Luo J, Davidson NO. Diet‐induced obesity and hepatic steatosis in L‐Fabp−/− mice is abrogated with SF, but not PUFA, feeding and attenuated after cholesterol supplementation. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G307–G314. [DOI] [PubMed] [Google Scholar]
- 32.Newberry EP, Xie Y, Kennedy SM, Luo J, Davidson NO. Protection against Western diet‐induced obesity and hepatic steatosis in liver fatty acid‐binding protein knockout mice. Hepatology. 2006;44(5):1191–205. [DOI] [PubMed] [Google Scholar]
- 33.Chen A, Tang Y, Davis V, Hsu FF, Kennedy SM, Song H, et al. Liver fatty acid binding protein (L‐Fabp) modulates murine stellate cell activation and diet‐induced nonalcoholic fatty liver disease. Hepatology. 2013;57(6):2202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gajda AM, Zhou YX, Agellon LB, Fried SK, Kodukula S, Fortson W, et al. Direct comparison of mice null for liver or intestinal fatty acid‐binding proteins reveals highly divergent phenotypic responses to high fat feeding. J Biol Chem. 2013;288(42):30330–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez Sawicki L, Bottasso Arias NM, Scaglia N, Falomir Lockhart LJ, Franchini GR, Storch J, et al. FABP1 knockdown in human enterocytes impairs proliferation and alters lipid metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(12):1587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stojanović O, Altirriba J, Rigo D, Spiljar M, Evrard E, Roska B, et al. Dietary excess regulates absorption and surface of gut epithelium through intestinal PPARα. Nat Commun. 2021;12(1):7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu KT, Storch J. Fatty acid transfer from liver and intestinal fatty acid‐binding proteins to membranes occurs by different mechanisms. J Biol Chem. 1996;271(23):13317–23. [DOI] [PubMed] [Google Scholar]
- 38.Xie C, Takahashi S, Brocker CN, He S, Chen L, Xie G, et al. Hepatocyte peroxisome proliferator‐activated receptor α regulates bile acid synthesis and transport. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(10):1396–411. [DOI] [PMC free article] [PubMed] [Google Scholar]