

Abstract
Background:
Parkinson’s disease (PD) is characterized by α-synuclein aggregation and loss of dopamine neurons. Risk of PD arises due to a combination of genetic and environmental factors, which may interact, termed gene-environment (GxE) interactions. An inverse association between smoking and risk of PD is well-established, and a previous genome-wide GxE interaction study identified genetic variation in the synaptic-vesicle glycoprotein 2C (SV2C) locus as an important mediator of the degree to which smoking is inversely associated with PD.
Objective:
We sought to determine the mechanism of the smoking-SV2C interaction in a Drosophila model of PD.
Methods:
Flies expressing human α-synuclein in all neurons develop the hallmarks of PD, including motor dysfunction, loss of DA neurons, and formation of α-synuclein inclusions. We assessed the effects of increasing doses of nicotine on these parameters of neurodegeneration, in the presence or absence of knockdown of two Drosophila orthologues of SV2, hereafter referred to as SV2L1 and SV2L2.
Results:
α-synuclein-expressing flies treated with nicotine had improved locomotion, DA neuron counts, and α-synuclein aggregation. However, in α-synuclein-expressing flies in which SV2L1 and SV2L2 were knocked down, nicotine failed to rescue neurodegeneration.
Conclusions:
This work confirms a GxE interaction between nicotine and SV2, defines a role for this interaction in α-synuclein proteostasis, and suggests that future clinical trials on nicotine should consider genetic variation in SV2C. Further, this provides proof of concept that our model can be used for mechanistic study of GxE, paving the way for investigation of additional GxE interactions or identification of novel GxE.
Graphical Abstract
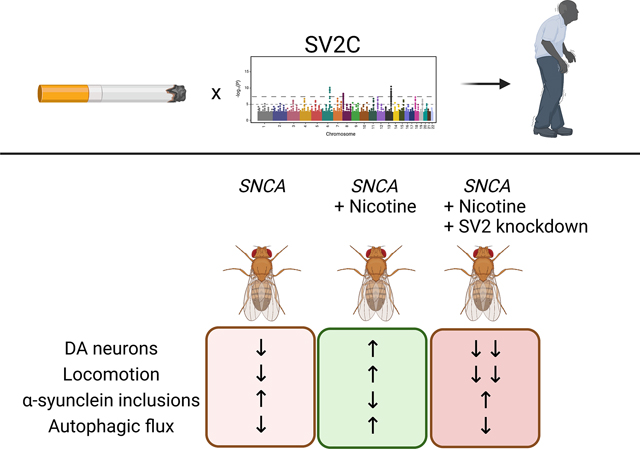
Previous work identified a gene-environment interaction between SV2C and smoking in Parkinson’s disease. We sought to determine the mechanism of interaction in a Drosophila model. Nicotine rescued neurodegeneration in flies expressing human alpha-synuclein but failed to do so when SV2 orthologs were knocked down, validating the interaction.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting 1% of the population over the age of 60. It arises due to a combination of genetic and environmental factors. How genes and environment interact together to influence risk remains incompletely understood. Traditional genome-wide association studies (GWAS) have been effective at identifying associated risk alleles, such as SNCA, LRRK2, and GBA, but can miss interactions with low effect sizes. Indeed, even the largest GWAS to date only accounts for a minority of heritable risk of PD1. Similarly, epidemiologic studies have identified numerous environmental factors that are positively or negatively associated with PD, but these studies can be challenging to reproduce, and causality difficult to determine. Thus, there is a need for mechanistic studies investigating gene-environment interactions (GxE).
By stratifying subjects into groups based on environmental exposures and performing a genome-wide association and interaction study (GWAIS), genes that exert their effects only in the context of certain environmental exposures can cross the threshold of significance. In a previous GWAIS, we identified an interaction between smoking and the synaptic vesicle protein SV2C in PD patients2. That is, the degree to which smoking was associated with PD was dependent on genetic variation in the SV2C locus2, ranging from strongly negatively associated to strongly positively associated, with an odds ratio (OR) of 0.44 – 3.23, depending on genotype at two independent SNPs.
SV2C belongs to a family of synaptic vesicle glycoproteins, along with SV2A and SV2B. The SV2 family proteins are transmembrane proteins found on secretory vesicles, including synaptic vesicles, and they have been reported to facilitate exocytosis of synaptic vesicles by rendering them responsive to calcium3,4. Expression of SV2C is primarily localized to the basal ganglia, with highest levels found in dopaminergic (DA) neurons of the substantia nigra (SNpc), ventral tegmental area (VTA), and in cholinergic interneurons of the striatum5. Certain SV2C variants in PD patients have been shown to predict sensitivity to levodopa6, and SV2C expression pattern is altered in PD patients7. Thus, SV2C and other SV2 family members represent credible targets as potential modifiers of PD.
Similarly, smoking has been inversely associated with PD in numerous epidemiologic studies. Although there are different hypotheses proposed to explain this relationship, including decreased sensation-seeking behavior in PD8, most evidence suggests that nicotine contributes significantly to neuroprotection against PD9. Nicotine has been shown to stimulate DA release in smokers10 and increase degradation of SIRT6, a pro-inflammatory protein11. Further, cholinergic deficits are also present in PD12, which could conceivably be alleviated by nicotine as a nicotinic acetylcholine receptor (nAChR) agonist. There is evidence for a protective effect of nicotine in vivo, with animal models of PD showing amelioration after nicotine exposure13,14. Results of nicotine in clinical trials have been mixed, however15–23. Further, even in trials demonstrating some benefit of nicotine, it is unknown whether this effect is merely symptomatic, alleviating symptoms, or neuroprotective, decreasing disease progression. Finally, it is possible that nicotine-free tobacco may also confer neuroprotection, suggesting a role for non-nicotinic mechanisms24. Thus, while nicotine is the most biologically plausible factor underlying the inverse relationship between smoking and PD, many questions regarding its mechanism remain.
In our prior work, in addition to identifying the smoking-SV2C interaction in PD patients, we also identified a gene-environment interaction between nicotine and a Drosophila ortholog of the SV2 family, in a paraquat-induced model of parkinsonism2. Here we sought to validate this interaction in an α-synuclein based Drosophila model of PD and to determine the mechanism underlying the interaction.
Methods
Drosophila genetics
All fly crosses and aging were performed at 25 °C. All experiments were performed at 10 days post-eclosion unless otherwise noted in the figure legends. All experiments include roughly equal numbers of male and female flies. While this study was not statistically powered to detect a difference between male and female flies in terms of response to nicotine, pilot studies did not suggest a sex-related differences. Human α-synuclein is expressed in neurons using the bipartite QUAS-QF2 expression system25 and the pan-neuronal driver neuronal-synaptobrevin (nSyb)-QF2. Additionally, genes of interest were knocked down using the UAS-GAL4 system, also driven by n-synaptobrevin. The following stocks were obtained from the Bloomington Drosophila Stock Center: 1) UAS-Atg8a-GFP (RRID:BDSC_51656, full genotype P{ry[+t7.2]=hsFLP}1, y[1] w[1118]; P{w[+mC]=UAS-Atg8a.GFP}3), 2) UAS-GFP-mCherry-Atg8a (RRID:BDSC_37749, full genotype y[1] w[1118]; P{w[+mC]=UASp-GFP-mCherry-Atg8a}2), 3) UAS-CG3168 JF02441 (RRID:BDSC_29301, full genotype y[1] v[1]; P{y[+t7.7] v[+t1.8]=TRiP.JF02441}attP2 (CG3168)) and 4) UAS-CG14691 JF03420 (RRID:BDSC_31986, full genotype y[1] v[1]; P{y[+t7.7] v[+t1.8]=TRiP.JF03420}attP2 (CG14691)). The transgenic RNAi stocks UAS-CG3168 48010 and UAS-CG14691 46365 were obtained from the Vienna Drosophila Resource Center. Flies lacking α-synuclein or the RNAi but containing both drivers were used as controls. The genotype for α-synuclein expressing flies was QUAS-Syn, nSyb-QF2, nSyb-GAL4/+ or QUAS-Syn, nSyb-QF2, nSyb-GAL4/UAS-RNAi, unless otherwise noted in the figure legend. These flies were compared to control flies expressing the same genetic drivers but lacking α-synuclein, that is, nSyb-QF2, nSyb-GAL4/+ or nSyb-QF2, nSyb-GAL4/UAS-RNAi.
Drug feeding and maintenance
Drosophila crosses were performed on standard fly food. At the time of eclosion flies were placed in vials containing a 1:1 ratio of instant medium (Carolina) to aqueous nicotine solution. Nicotine (Sigma) was diluted in 100% ethanol and then further diluted with distilled water to a final concentration of 0.1, 0.2, or 0.4 mg/ml in 4% ethanol. Control flies grown in the absence of drug were fed instant medium mixed with vehicle control (4% ethanol). dx.doi.org/10.17504/protocols.io.5jyl8j9n9g2w/v1
qRT-PCR
To confirm the knock down of selected genes, exon-spanning primers were selected from the DRSC FlyPrimerBank. 10 fly heads per condition were homogenized in Qiazol (Qiagen) and total RNA was isolated. Samples were treated with DNAse and cDNA prepared using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Amplification was reported by SYBR green in a QuantStudio 6 Flex System (ThermoFisher), and relative expression was determined using ΔΔCt method normalized to the RPL32 housekeeping gene. dx.doi.org/10.17504/protocols.io.q26g7y4r1gwz/v1
Locomotion assay
Flies were sorted into cohorts containing 9–14 flies per vial. At day 7 of exposure to either vehicle or drug, flies were transferred to a clean vial (without food) and allowed to acclimate for 1 min. The locomotion assay was performed as previously described26. Six technical replicates were averaged for each biological replicate, and there were six biological replicates for each condition. Significant difference between phenotypes at the same time-point was measured by two-way ANOVA with Tukey’s multiple comparison test. Two RNAi lines per gene were used for behavior studies (Figure 1A, Supplemental Figure 1), and a single RNAi was used for all subsequent confirmatory pathologic studies. The RNAi lines used in the confirmatory studies were the UAS-CG3168 JF02441 and UAS-CG14691 JF03420 lines. After identifying a dose response in locomotion to nicotine of 0.1–0.4 mg/ml (Figure 1A), the highest dose of 0.4 mg/ml was used for all subsequent confirmatory pathologic studies.
Figure 1: Rescue of α-synuclein toxicity by nicotine requires SV2.
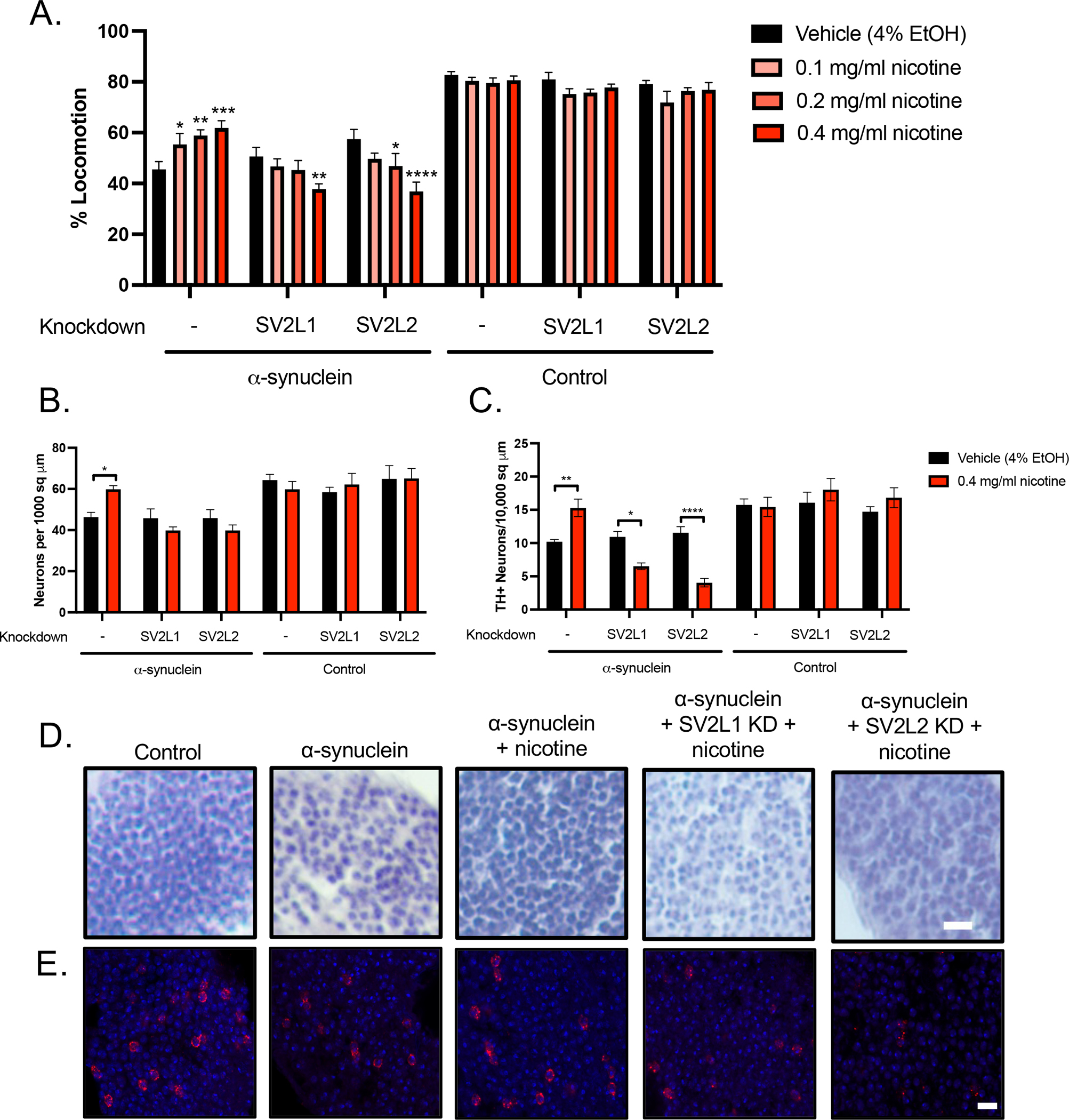
A. α-synuclein or control flies were fed media with 0.1–0.4 mg/ml nicotine or vehicle control from the time of eclosion up to 10 days of age. Locomotion was measured at 4, 7, and 10 days. The day 7 timepoint is shown; results at 4 and 10 days were similar. Nicotine improved locomotion in α-synuclein expressing flies (far left) in a dose-dependent manner. However, nicotine failed to rescue the locomotor deficit when the SV2 orthologs SV2L1 and SV2L2 were knocked down and in fact further worsened locomotion in this condition in a dose-dependent manner. N = minimum 6 biological replicates of 9–14 flies each per genotype. *p <0.05, **p < 0.01, *** p < 0.005, **** p < 0.001. Within each genotype, statistical significance was determined using two-way ANOVA with Dunnet’s multiple comparison test, comparing each dose of nicotine to vehicle control. B. Quantification of total neurons from hematoxylin-stained slides of anterior medulla, n = 6 replicates per genotype. * = p <0.05, ** = p <0.01, determined with one-way ANOVA with Dunnet’s test for multiple comparisons. C. Quantification of dopaminergic neurons. n = 6 replicates per genotype. * = p <0.05, ** = p <0.01, determined with one-way ANOVA with Dunnet’s test for multiple comparisons. D. Representative sections of the anterior medulla. Scale = 10 μm. E. Anterior medulla sections were stained with tyrosine hydroxylase (red) and mounted with media containing DAPI (blue). Scale = 5 μm.
Immunohistochemistry & Immunofluorescence
After 10 days of exposure to either vehicle or 0.4 mg/ml nicotine, flies were anesthetized, fixed in formalin overnight, and embedded in paraffin wax. A series of frontal sections of fly brain were cut either at 2 or 4 μm in thickness and processed through xylenes, ethanol, and water. For total neuron counts (2 μm), slides were stained with hematoxylin. For DA neuron counts (4 μm), immunohistochemistry was performed. Antigen retrieval for 30 min in 10 mM sodium citrate, pH 6.0, was performed in a pressure cooker. After cooling and depressurization, slides were blocked in 2% milk hydrated in PBS with 0.3% Triton for 1 hr and then incubated with primary diluted in the same blocking solution at room temperature overnight. Antibodies used include Atg8a/LC3 (clone EIJ4E, 1:2,000, rabbit, Cell Signaling, Cat# 13733, RRID:AB_2798306), and ref(2)P/p62 (1:5,000 rabbit). The next day slides were washed and incubated for 1 hr in a secondary biotin-conjugated anti-mouse antibody (1:200, goat anti mouse, SouthernBiotech, Birmingham, AL, Cat# 1010-08, RRID:AB_2794127) mixed in blocking solution. Following washing, avidin–biotin–peroxidase complex (Vectastain Elite) mixed in PBS was applied for 1 hr followed by development using diaminobenzidine (ImmPACT DAB, Vector, Torrance, CA).
For immunofluorescence, microwave antigen retrieval was conducted in 10 mM sodium citrate, and slides were blocked in 2% milk hydrated in PBS with 0.3% Triton for 1 hr. All primary antibodies were diluted in blocking solution. Primary antibodies used include tyrosine hydroxylase (1:200, mouse, Immunostar Cat#22941, RRID:AB_572268), α-synuclein 5G4 (1:50,000, mouse, Millipore Cat#MABN389, RRID:AB_2716647), and GFP (1:5, Developmental Studies Hybridoma Bank, Cat# N86/38, RRID:AB_2877499). Slides were incubated with fluorophore-conjugated secondary antibodies in blocking solution for 1 hr (1:200, Goat anti-mouse Alexa Fluor 555, Thermo Fisher Scientific, Cat#A21422, RRID:AB_141822) then mounted with DAPI-containing Fluoromount medium (SouthernBiotech). Tissue was excited and photographed using Zeiss LSM 800 confocal microscopy and analyzed with ImageJ.
Quantification of neuron counts
Images of the anterior medulla from hematoxylin-stained formalin-fixed paraffin-embedded tissue were captured at 40X magnification using brightfield microscopy. One image per fly and 6 flies per genotype were used for quantification. The number of nuclei in each tissue section was counted by a blinded reviewer and normalized to the area of the section. dx.doi.org/10.17504/protocols.io.4r3l24on4g1y/v1
Quantification of TH+ neuron counts and semi-quantitative analysis of tyrosine hydroxylase staining
Representative images of the anterior medulla from immunofluorescence-stained formalin-fixed paraffin-embedded tissue were captured on a confocal microscope at 63X magnification and are included in Figure 1E. For quantification of TH+ neurons, images of the anterior medulla from immunohistochemistry stained formalin-fixed paraffin-embedded tissue were captured at 20x magnification. One image per fly and 6 flies per genotype were used for quantification. The number of TH+ cells in each tissue section was counted by eye by a blinded reviewer and normalized to the area of the section.
Quantification of α-synuclein aggregates
Images of the anterior medulla from immunofluorescence-stained formalin-fixed paraffin-embedded tissue were captured on a confocal microscope at 63X magnification. One image per fly and 6 flies per genotype were used for quantification. The number α-synuclein aggregates in each tissue section was counted by a blinded reviewer and normalized to the area of the section.
α-synuclein monomer detection
At day 1 post-eclosion, one fly head per condition was homogenized in 2X Laemmli buffer, boiled for 10 min, and centrifuged. SDS-PAGE was performed (Lonza) followed by transfer to PVDF membrane (Bio-Rad) and microwave antigen retrieval in PBS. Membranes were blocked in 2% milk in PBS with 0.05% Tween 20 for 1 hour, then immunoblotted with primary antibody α-synuclein H3C (1:100,000, mouse, Developmental Studies Hybridoma Bank, Cat# H3C, RRID:AB_2618046) in 2% milk in PBS with 0.05% Tween-20 overnight at 4 °C. Membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (1:50,000, Goat anti-mouse IgG, Southern Biotechnology Associates, Cat# 1030-05, RRID: AB_2619742) in 2% milk in PBS with 0.05% Tween 20 for 3 hours. Signal was developed with enhanced chemiluminescence (Thermo Scientific). dx.doi.org/10.17504/protocols.io.8epv5j96jl1b/v1
α-synuclein oligomer detection
20 fly heads per genotype were homogenized in 20 ul TNE lysis buffer (10 mm Tris HCl, 150 mM NaCl, 5 mM EGTA, 0.5% NP40) supplemented with HALT protease and phosphatase inhibitor (Roche). The homogenate was briefly spun down to remove debris. The remaining supernatant was ultracentrifuged at 100,000 × g for 1 hour at 4 C. The supernatant was transferred to a new tube and combined with 2X Laemelli buffer at a 1:1 ratio. SDS-PAGE was then performed as above (dx.doi.org/10.17504/protocols.io.8epv5j96jl1b/v1) but without microwave antigen retrieval. Prior to incubating with primary antibody, the blot was cut to separate monomeric from oligomeric α-synuclein. Primary antibody was α-synuclein clone 42 (1:5,000 mouse, BD Bioscience, Cat# 610787, RRID:AB_398107).
Quantification of Atg8a and ref(2)P puncta
Images of the anterior medulla from immunohistochemistry-stained formalin-fixed paraffin-embedded tissue were captured at 40X magnification using brightfield microscopy. One image per fly and 6 flies per genotype were used for quantification. The number of large puncta (>0.25 μm2, measured in ImageJ (Version 1.52o, RRID:SCR_003070, https://imagej.net/) using the particle analysis function) in each tissue section were counted by a blinded reviewer and normalized to the area of the section.
Measurement of autophagic flux
Freshly dissected brains from 10-day-old animals were imaged at 63X on a Zeiss LSM 800 confocal microscopy. GFP-mCherry-Atg8a structures were analyzed as previously described27. For analysis of mCherry-GFP ratio across puncta, GFP-mCherry-Atg8a puncta within the field of view were automatically selected using h_watershed in Fiji (Version 2.3.0/1.53q, RRID:SCR_002285, http://fiji.sc) 28. For each punctum, intensity of fluorescence was measured for each channel, and the ratio of red to green fluorescence was calculated.
Data Sharing
The data that support the findings of this study are available from the corresponding author, upon reasonable request. The raw tabular data underlying each graph has been deposited in the data repository zenodo (https://doi.org/10.5281/zenodo.7023264).
Results
Nicotine rescues α-synuclein induced impaired locomotion in a dose-dependent manner
We have previously demonstrated that nicotine treatment improves lifespan in a dose-dependent manner in paraquat-induced parkinsonism in the fly2. To determine if nicotine has neuroprotective effects in an α-synuclein based model, we first measured the effects on locomotion using a previously published assay26. We assessed locomotion at day 7 post-eclosion and determined that nicotine rescues impaired locomotion in a dose-dependent manner (Figure 1A).
This rescue in locomotor activity due to nicotine is dependent on SV2. Drosophila have multiple SV2 family orthologs. We chose to examine two: CG3168, which is the most closely related to the human SV2 family as determined by Drosophila RNAi Screening Center Integrative Ortholog Predictor Tool (DIOPT), and CG14691, which was identified in our prior work as interacting with nicotine2. Because of their homology with the larger synaptic vesicle glycoprotein 2 (SV2) gene family, we have name these SV2L1 and SV2L2, respectively, for synaptic vesicle glycoprotein 2 like.
When SV2L1 or SV2L2 are knocked down (Figure 1A), nicotine no longer increases locomotion, and in fact, the highest dose of nicotine (0.4 mg/ml) significantly worsens locomotion in the α-synuclein flies. This dose of nicotine was used for all subsequent experiments. We confirmed the locomotion result with a second RNAi line (Supplemental Figure 1). Effective knockdown of SV2L1 and SV2L2 with each RNAi is demonstrated in Supplemental Figure 2. All subsequent experiments use RNA #1 for each gene.
Rescue of neurodegeneration by nicotine requires SV2
Compared to control flies, flies expressing α-synuclein in neurons demonstrate significant neurodegeneration, quantified by fewer cells per square micron in the densely cell-populated anterior medulla (Figure 1B, 1D). When α-synuclein flies were exposed to 0.4 mg/ml of nicotine, we observed a rescue of cell density. The effect of nicotine was abrogated with knockdown of SV2L1 and SV2L2. Similarly, nicotine rescued loss of dopaminergic neurons (Figure 1C, 1E), which also required SV2L1 and SV2L2 expression. Interestingly, with SV2L1 and SV2L2 knockdown, nicotine not only failed to rescue dopaminergic neuron loss but in fact further significantly exacerbated it, similar to the results seen with locomotion (Figure 1C, 1E). Collectively, these experiments confirm a gene-environment interaction between nicotine and SV2, which is statistically significant when quantified with a 2-way ANOVA (p <0.0001 for interaction).
Rescue of α-synuclein proteostasis by nicotine requires SV2
We next examined the effect of nicotine on α-synuclein proteostasis. Nicotine reduced the number of large α-synuclein aggregates (Figure 2A, 2C) as well as the number of high molecular weight oligomers (Figure 2B, 2D), but failed to reduce these α-synuclein species when SV2L1 and SV2L2 were knocked down. This experiment suggests that the neuroprotective effects of nicotine involve partial normalization of α-synuclein proteostasis, and that this requires SV2. Surprisingly, nicotine did not affect phosphorylation of α-synuclein at S129, however (Supplemental Figure 3).
Figure 2. High dose nicotine decreases α-synuclein aggregation and oligomerization.
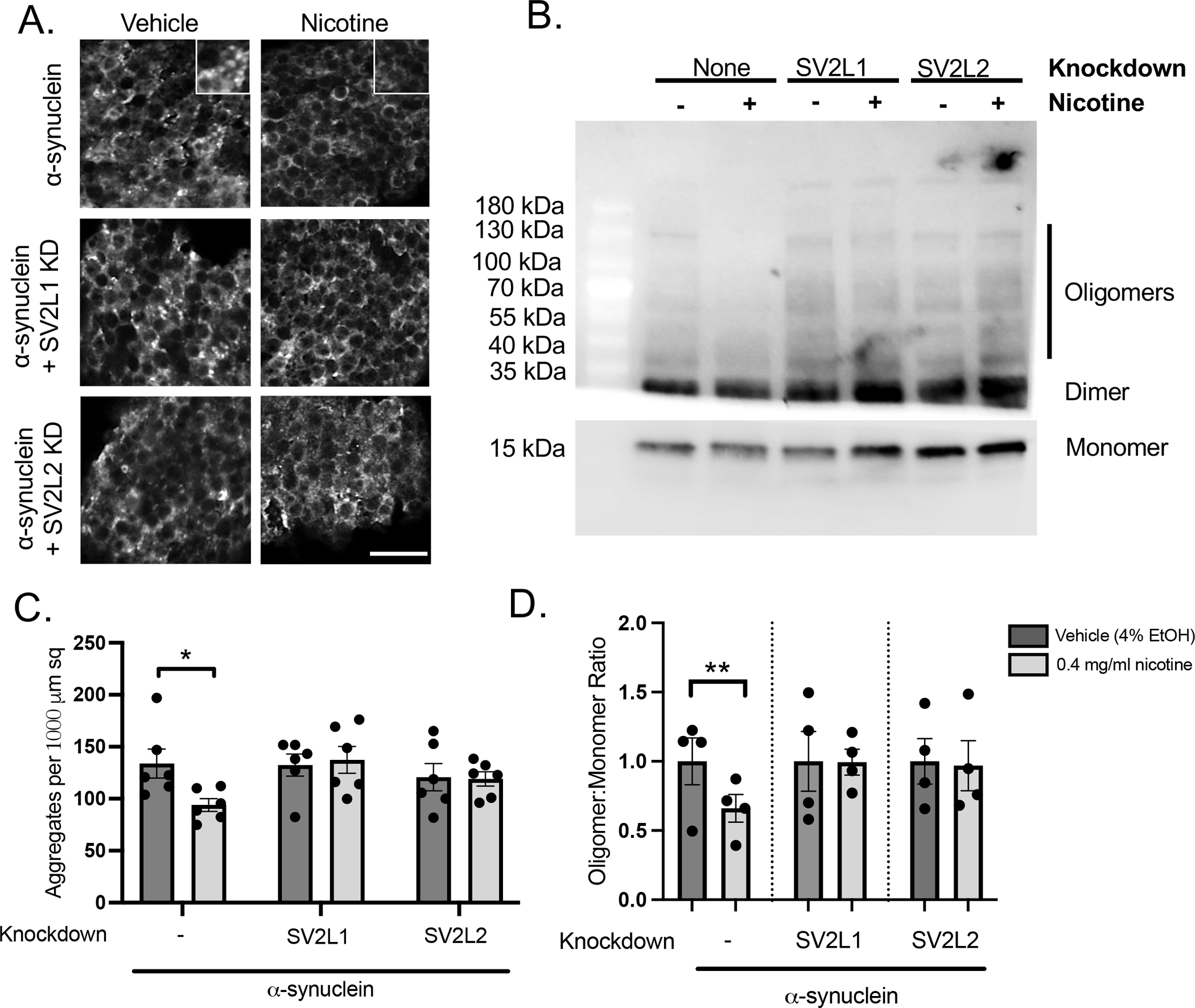
A. Slides were stained with anti-α-synuclein (clone 5G4, mouse, 1:50,000) and DAPI. Representative images are shown. B. Representative immunoblot demonstrating high molecular oligomers as well as monomeric α-synuclein. C. Quantification of α-synuclein aggregates. D. Quantification from 4 independent experiments. Each nicotine condition was normalized to its vehicle control, and statistical significance was determined using a 1 sample t-test.
Nicotine normalizes markers of autophagy
To further investigate the mechanism of the nicotine-SV2C interaction on proteostasis, we turned to the lysosomal autophagy system. We have previously investigated the relationship between α-synuclein and autophagy in detail in this model system, finding that α-synuclein expressing flies accumulate autophagosomes due to impaired autophagic flux27. Here we examined autophagy using two markers: ref(2)P (the Drosophila ortholog of p62) and Atg8a (the Drosophila ortholog of LC3). As we have demonstrated previously, α-synuclein transgenic flies demonstrated increased number of puncta staining for both of these markers, as compared to control flies (Figure 3). Further, high-dose nicotine partially rescued this, but failed to do so when SV2L1 and SV2L2 was knocked down. This suggests that the neuroprotective effects of nicotine involve the lysosomal autophagy system.
Figure 3: High dose nicotine reduces markers of pathologic autophagy.

Immunohistochemistry was performed for ref(2)P (p62), and Atg8a (LC3). Images were thresholded in ImageJ, and puncta >0.25 μm in area were counted using the Analyze Particles function. n = 6 flies per genotype. A. Quantification of Atg8a (LC3) puncta. B. Quantification of ref(2)P (p62) puncta. C. Representative images of Atg8a (LC3) puncta. D. Representative images of ref(2)P (p62) puncta. Scale = 10 μm. Knockdown of SV2 increases Atg8a puncta even in the absence of α-synuclein but has no effect of ref(2)P puncta. Statistical analysis was performed with a one-way ANOVA with Dunnet’s multiple comparison test. Nicotine and/or SV2 knockdown conditions were compared to α-synuclein or control flies treated with vehicle and with no RNAi. * = p <0.05, ** = p <0.01, *** = p <0.005, **** = p <0.001.
SV2C has independent effects on autophagy
Surprisingly, SV2L1 and SV2L2 knockdown independently increased accumulation of Atg8a+ puncta (Figure 3A, 3C), even in the absence of α-synuclein, though this was not the case for ref(2)P+ puncta (Figure 3B, 3D). To confirm the increase in Atg8a+ puncta, we expressed a GFP-tagged Atg8a. As expected, α-synuclein expressing flies demonstrated increased Atg8a-GFP puncta, which was rescued by high-dose nicotine (Supplemental Figure 4A–B). Further, SV2L1 and SV2L2 knockdown also increased the number of Atg8a-GFP puncta, even in the absence of α-synuclein (Supplemental Figure 4C–D). Thus, SV2C knockdown increases Atg8a+ puncta but not ref(2)P+ puncta.
Atg8a is the Drosophila ortholog of LC3, the canonical marker of autophagosomes, and is thus a general marker of autophagosomes. In contrast, ref(2)P, the Drosophila ortholog of p62, is found in protein aggregates in the setting of disrupted autophagy, as occurs in neurodegeneration. We therefore hypothesized that SV2C knockdown was increasing basal autophagy, independently of an effect on the pathologic autophagosomes that accumulate with α-synuclein. To investigate this hypothesis, we used a tandem tag reporter, GFP-mCherry-Atg8a. This reporter expresses both GFP and mCherry in newly formed autophagosomes, but the GFP fluorescence is quenched in the acidic autolysosome. Thus, a large mCherry to GFP ratio indicates preserved autophagic flux, whereas a small mCherry to GFP ratio indicates impaired autophagic flux.
We have previously reported that α-synuclein expressing flies have impaired autophagic flux using this reporter, as indicated by many dual GFP and mCherry positive puncta (Sarkar, 2021). Here we again demonstrate that α-synuclein expression leads to impaired autophagic flux, with more dual-positive puncta and a low mCherry to GFP ratio compared to control flies (Figure 4A–B, 4F). The mCherry to GFP ratio was normalized by either treatment with nicotine or by SV2C knockdown (Figure 4C–D, 4F). However, when both SV2C knockdown and nicotine treatment were present, autophagic flux was not normalized (Figure 4E–F). Thus, nicotine increases autophagic flux in α-synuclein expressing flies in an SV2C-dependent manner. In the absence of α-synuclein, SV2C knockdown led to an increase in the mCherry to GFP ratio (Figure 4G), supporting the idea that SV2C knockdown is increasing basal autophagy. Treatment with nicotine blunted this effect (Figure 4G).
Figure 4: SV2 knockdown increases physiologic autophagy.
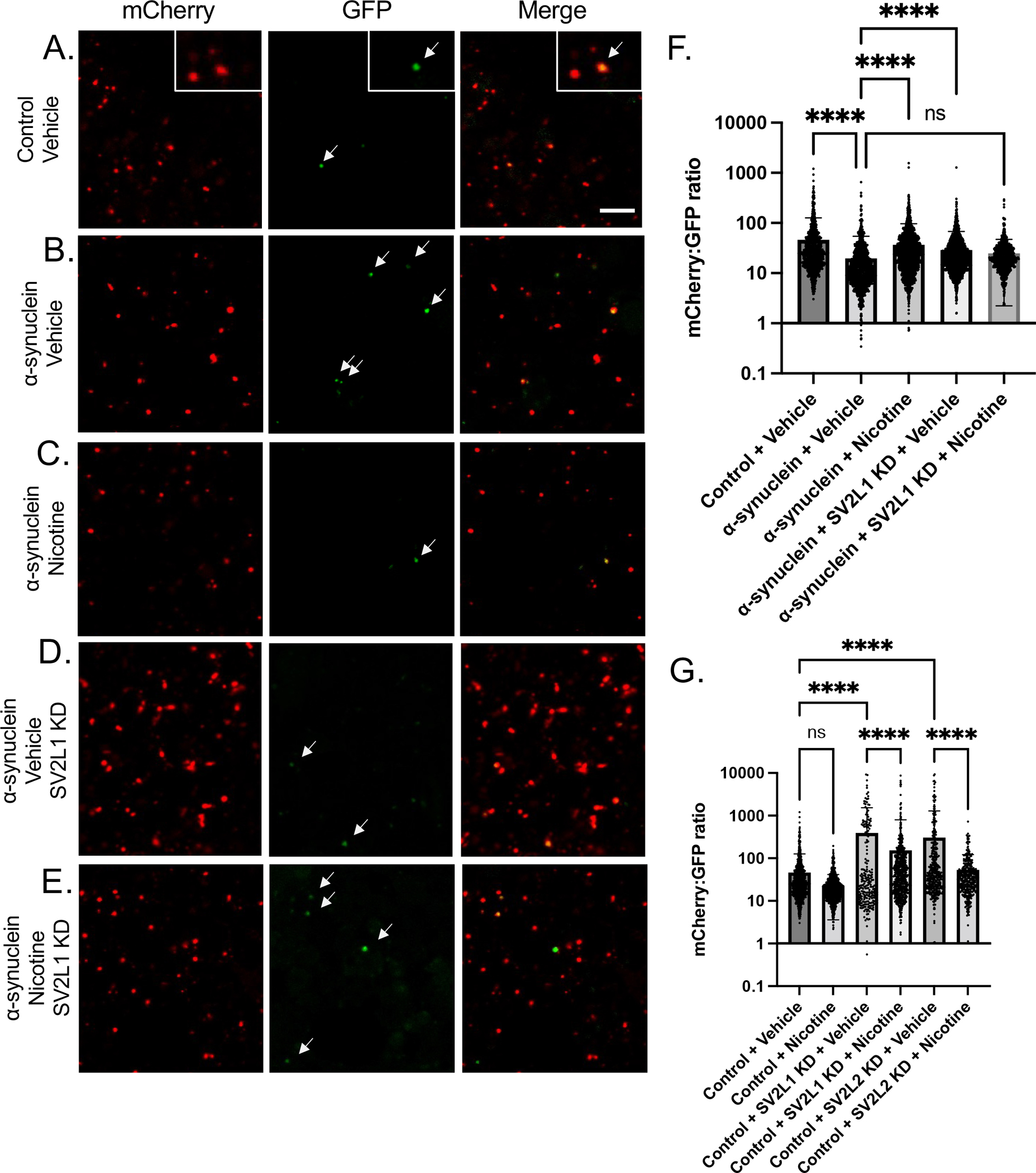
A. UAS-GFP-mCherry-Atg8a was expressed in control or α-synuclein flies, with or without SV2L2 knockdown, treated with nicotine or vehicle control. Brains were dissected in PBS and imaged immediately and sequentially in the red and green channels. A-E. Representative images. In control flies, numerous mCherry+ puncta are seen, with exceedingly rare dual positive puncta (see inset). Scale = 5 μm. F-G. Fluorescence intensity of mCherry:GFP was averaged across all puncta. n = 3 flies per condition. Genotype is UAS-GFP-mCherry-Atg8a/+; nSybQF2, Snyb-GAL4/UAS-CG14691 RNAi.
Discussion
Here, we demonstrate that nicotine rescues neurodegeneration in a genetic model of PD in which human α-synuclein is over-expressed in Drosophila neurons. While multiple previous investigations have identified a neuroprotective effect of nicotine in toxic models of PD, including MPTP1,11, 6-OHDA29, rotenone13, and paraquat2, studies involving genetic models are more limited. Nicotine has been examined in a parkin mutant Drosophila model14, as well as recent paper involving overexpression of Synphilin-1 in dopaminergic neurons, also in Drosophila30. Surprisingly, few studies have examined nicotine in α-synuclein based models of PD. Bono et. al found a protective effect of nicotine on α-synuclein accumulation in cultured mouse and human DA neurons31, and Kardani et. al found that nicotine slowed α-synuclein oligomerization in yeast32. Interestingly, Trinh et. al reported that nicotine-free tobacco, but not nicotine itself rescued dopaminergic neuron numbers in an α-synuclein based Drosophila PD model24. In comparison with our study, Trinh et. al used a lower dose of nicotine as well as a different promotor for α-synuclein, which may account for the differing results. Finally, to our knowledge, there is only one published study involving nicotine in α-synuclein-based PD model in a mammalian model in vivo: Subramaniam and colleagues investigated nicotine in α-synuclein over-expressing transgenic mice, finding improvement in cognitive performance but no improvement in neurodegeneration, α-synuclein aggregation, or motor impairment33.
Beyond investigating a role for nicotine in an α-synuclein-based model of PD, we sought to determine the mechanism of interaction between nicotine and SV2. Of interest, both SV2C and nicotine have been reported to affect release of DA. Using fast scan cycle voltammetry, Dunn et. al determined that nicotine increases DA release in response to high-intensity stimulation in the dorsal striatum of wild-type mice, but when tested in SV2C knockout animals, nicotine paired with high-intensity stimulation reduced DA release to less than half of the response at baseline7. Thus, SV2C levels determined the response of dopaminergic neurons to nicotine. Expanding on this, we found that nicotine rescued motor functioning and prevented DA neuron death in α-synuclein flies, but when SV2 orthologs were knocked down, nicotine not only failed to rescue but in fact significantly further decreased locomotion and DA neuron survival. Together, these results suggest that SV2 expression level mediates the effects of nicotine on DA neurons not only in normal DA neurons, as demonstrated in Dunn et. al, but also in parkinsonian animals. Dopamine homeostasis is a complex and tightly regulated process, because dopamine metabolism results in high basal levels of oxidative stress in DA neurons35, rendering them vulnerable to additional insults36. Further, dopaminergic dysfunction is present in PD long before the onset of motor symptoms and may underlie the pathology of some pre-motor symptoms34. Thus, abnormalities in dopamine homeostasis may be one mechanism underlying the nicotine x SV2C interaction.
Beyond the effects on DA neurons, we also found that the nicotine x SV2 interaction also influenced α-synuclein proteostasis. That is, nicotine treatment rescued α-synuclein oligomerization and aggregation in the presence of wildtype SV2L1 and SV2L2 expression, but it failed to do so in SV2L1 or SV2L2 knockdown conditions (Figure 2). Of interest, Dunn et al reported a direct protein-protein interaction between SV2C and α-synuclein, an increase in high molecular weight α-synuclein in SV2C knockout mice, and an abnormal SV2C expression pattern in an A53T α-synuclein mouse model and PD patient brains. How nicotine might further influence any interaction between α-synuclein and SV2C is unknown.
To further understand the effects of the nicotine x SV2 interaction on proteostasis, we examined autophagy. Autophagy is a highly regulated process – it is necessary for cellular homeostasis and response to stress, but when excessive, it causes apoptosis37. Further, there is a complex relation between autophagy and α-synuclein. Alterations in the lysosomal autophagy system increase α-synuclein accumulation, and excessive and/or abnormal α-synuclein then further impedes the lysosomal autophagy system in a vicious cycle38. Of note, nicotine has been shown to induce autophagy in several tissues outside of the brain, though the effects of this range from beneficial to harmful depending on the tissue39–46. Here we found that nicotine treatment rescued impaired autophagic flux in α-synuclein flies, and that this rescue was dependent on intact expression of SV2C (Figure 4–5).
Figure 5: Proposed model.
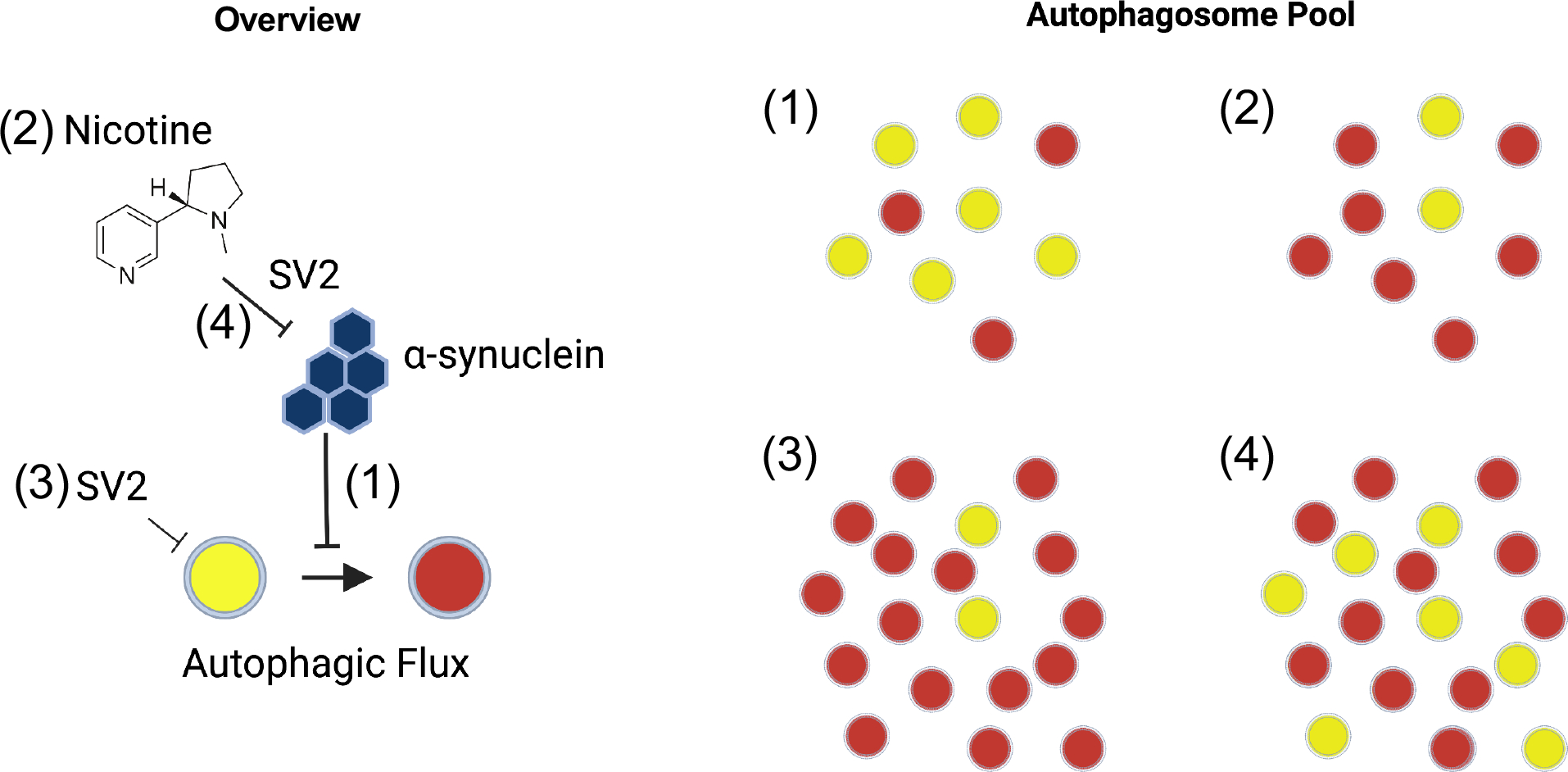
1. α-synuclein expressing flies have impaired autophagic flux. 2. Nicotine can restore autophagic flux in α-synuclein flies in an SV2-dependent manner. 3. SV2 knockdown increases basal autophagy, resulting in more autophagosomes but no delay in maturation. 4. In the absence of SV2C nicotine is unable to rescue autophagic flux, perhaps because the increase in basal autophagy resulting in more autophagosomes overwhelms the autophagy-lysosomal system.
Unexpectedly, we also found an effect of SV2C on autophagy that was independent of α-synuclein. That is, knockdown of SV2L1 or SV2L2 in either control or α-synuclein animals increased the number of autophagosomes, as indicated by the canonical autophagy marker Atg8a (LC3) (Figure 5). We believe that this is due most likely to an increase in basal autophagy levels, as ref(2)P (p62) levels were not increased by SV2L1 or SV2L2 knockdown (Figure 3B), and SV2L1 or SV2L2 knockdown led to an increase in autophagic flux (Figure 4–5). How SV2C may regulate autophagy is unknown, but it is worth mentioning that SV2 family proteins are transmembrane proteins that are thought to be present on all secretory vesicles, and they play a role in vesicular transport, trafficking, anchoring, and recycling47. If this membrane interaction affects other membranous organelles, like autophagosomes or lysosomes, this could explain the varied effects of SV2 observed in our study.
In summary, we show here that nicotine-mediated rescue of α-synuclein toxicity requires SV2. This work validates the gene-environment interaction between nicotine and SV2 and provides insight on the mechanism, finding an unexpected role for this interaction in α-synuclein proteostasis and autophagy. Further, the work provides proof of concept of the utility of this model for investigating additional GxE interactions. Understanding these GxE is critical for future clinical trial design and identifying which patients may be most likely to benefit from which therapies.
Supplementary Material
Supplemental Figure 1. Confirmation of locomotion rescue with a second RNAi line. α-synuclein or control flies with or without knockdown of SV2L1 and SV2L2 using VDRC lines. Flies were aged to 7 days post-eclosion on food containing 0.4 mg/ml nicotine or vehicle control. Symbols above the 0.4 mg/ml nicotine bar represent statistically significant difference compared to the vehicle control, respectively. *p <0.05, **p < 0.01, ***P<0.005, ns = p > 0.05. n = minimum of 60 flies per genotype (6 biological replicates of 10 flies each). Significance was determined by two-way ANOVA with Sidak’s test for multiple comparisons.
Supplemental Figure 2. Confirmation of knockdown. A. qRT-PCR for SV2L1 in control flies (no RNAi) or flies expressing one of two RNAi’s targeting SV2L1. RNAi #1 is the JF line. RNA #2 is the VDRC line. B. qRT-PCR for SV2L2 in control flies (no RNAi) or flies expressing one of two RNAi’s targeting SV2L2. RNAi #1 is the JF line. RNA #2 is the VDRC line.
Supplemental Figure 3. S129 phosphorylated α-synuclein. A. Representative immunoblot comparing S129 phosphorylated α-synuclein to total α-synuclein. B. Quantification of 4 independent experiments.
Supplemental Figure 4. SV2 knockdown increases Atg8a even in the absence of α-synuclein. A. Representative images. B. Quantification. Images were thresholded in ImageJ, and puncta > 0.12 μm in area were counted using the Analyze Particles function. Control flies expressing a GFP tagged Atg8a treated with ethanol vehicle or nicotine demonstrate sparse small puncta (small white arrows). Puncta are larger and more numerous with α-synuclein and large puncta are reduced in number with nicotine treatment (large yellow arrows). Scale = 5 μm. n = 6 flies per condition. Genotypes are nSyb-QF2, nSyb-GAL4/UAS-Atg8a-GFP or QUAS-Syn, nSyb-QF2, nSyb-GAL4/UAS-Atg8a-GFP for control and α-synuclein expressing flies, respectively. C. Representative images. D. Quantification of puncta was performed as in B. Genotypes are nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/+, nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/UAS-CG3168 RNAi or nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/UAS-CG14691 RNAi.
Acknowledgement
The HC3 α-synuclein antibody and GFP antibodies were provided by the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. Drosophila stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. We thank the Transgenic RNAi Project (TRiP) at the Harvard Medical School (NIH-NIGMS R01GM084947) for making transgenic RNAi stocks. Figure 5 and the table of contents imaged were created with Biorender.
Funding Sources
This work was funded by 1R21 NS0105151 (MBF), 1R01 NS098821 (MBF), 5K08-K08NS109344-03 (ALO), and W81XWH-18-1-0395 (ALO).
The study is funded by the joint efforts of The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and the Aligning Science Across Parkinson’s (ASAP) initiative. MJFF administers the grant [ASAP-000301] on behalf of ASAP and itself.
Financial Disclosures for all authors
MBF receives grant support from NIH-NINDS, NIH-NIA, and the Michael J Fox Association and Aligning Science Across Parkinson’s (ASAP) initiative. ALO receives grant support NIH-NINDS, the Department of Defense, and the American Parkinson’s Disease Association. SGC has no financial disclosures.
Footnotes
Financial Disclosures/COI concerning the current manuscript
None
References
- 1.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–1102. doi: 10.1016/S1474-4422(19)30320-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill-Burns EM, Singh N, Ganguly P, et al. A genetic basis for the variable effect of smoking/nicotine on Parkinson’s disease. Pharmacogenomics J. 2013;13(6):530–537. doi: 10.1038/tpj.2012.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang WP, Südhof TC. SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. J Neurosci. 2009;29(4):883–897. doi: 10.1523/JNEUROSCI.4521-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciruelas K, Marcotulli D, Bajjalieh SM. Synaptic vesicle protein 2: A multi-faceted regulator of secretion. Semin Cell Dev Biol. 2019;95:130–141. doi: 10.1016/j.semcdb.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dardou D, Dassesse D, Cuvelier L, Deprez T, De Ryck M, Schiffmann SN. Distribution of SV2C mRNA and protein expression in the mouse brain with a particular emphasis on the basal ganglia system. Brain Res. 2011;1367:130–145. doi: 10.1016/j.brainres.2010.09.063 [DOI] [PubMed] [Google Scholar]
- 6.Altmann V, Schumacher-Schuh AF, Rieck M, Callegari-Jacques SM, Rieder CRM, Hutz MH. Influence of genetic, biological and pharmacological factors on levodopa dose in Parkinson’s disease. Pharmacogenomics. 2016;17(5):481–488. doi: 10.2217/pgs.15.183 [DOI] [PubMed] [Google Scholar]
- 7.Dunn AR, Stout KA, Ozawa M, et al. Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci USA. 2017;114(11):E2253–E2262. doi: 10.1073/pnas.1616892114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans AH, Lawrence AD, Potts J, et al. Relationship between impulsive sensation seeking traits, smoking, alcohol and caffeine intake, and Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2006;77(3):317–321. doi: 10.1136/jnnp.2005.065417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quik M, Perez XA, Bordia T. Nicotine as a potential neuroprotective agent for Parkinson’s disease. Mov Disord. 2012;27(8):947–957. doi: 10.1002/mds.25028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi H, Fujimura Y, Hayashi M, et al. Enhanced dopamine release by nicotine in cigarette smokers: a double-blind, randomized, placebo-controlled pilot study. Int J Neuropsychopharmacol. 2008;11(3):413–417. doi: 10.1017/S1461145707008103 [DOI] [PubMed] [Google Scholar]
- 11.Nicholatos JW, Francisco AB, Bender CA, et al. Nicotine promotes neuron survival and partially protects from Parkinson’s disease by suppressing SIRT6. Acta Neuropathol Commun. 2018;6(1):120. doi: 10.1186/s40478-018-0625-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohnen NI, Yarnall AJ, Weil RS, et al. Cholinergic system changes in Parkinson’s disease: emerging therapeutic approaches. Lancet Neurol. 2022;21(4):381–392. doi: 10.1016/S1474-4422(21)00377-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mouhape C, Costa G, Ferreira M, Abin-Carriquiry JA, Dajas F, Prunell G. Nicotine-Induced Neuroprotection in Rotenone In Vivo and In Vitro Models of Parkinson’s Disease: Evidences for the Involvement of the Labile Iron Pool Level as the Underlying Mechanism. Neurotox Res. 2019;35(1):71–82. doi: 10.1007/s12640-018-9931-1 [DOI] [PubMed] [Google Scholar]
- 14.Chambers RP, Call GB, Meyer D, et al. Nicotine increases lifespan and rescues olfactory and motor deficits in a Drosophila model of Parkinson’s disease. Behav Brain Res. 2013;253:95–102. doi: 10.1016/j.bbr.2013.07.020 [DOI] [PubMed] [Google Scholar]
- 15.Villafane G, Thiriez C, Audureau E, et al. High-dose transdermal nicotine in Parkinson’s disease patients: a randomized, open-label, blinded-endpoint evaluation phase 2 study. Eur J Neurol. 2018;25(1):120–127. doi: 10.1111/ene.13474 [DOI] [PubMed] [Google Scholar]
- 16.Villafane G, Cesaro P, Rialland A, et al. Chronic high dose transdermal nicotine in Parkinson’s disease: an open trial. Eur J Neurol. 2007;14(12):1313–1316. doi: 10.1111/j.1468-1331.2007.01949.x [DOI] [PubMed] [Google Scholar]
- 17.Holmes AD, Copland DA, Silburn PA, Chenery HJ. Acute nicotine enhances strategy-based semantic processing in Parkinson’s disease. Int J Neuropsychopharmacol. 2011;14(7):877–885. doi: 10.1017/S1461145710001665 [DOI] [PubMed] [Google Scholar]
- 18.Lemay S, Chouinard S, Blanchet P, et al. Lack of efficacy of a nicotine transdermal treatment on motor and cognitive deficits in Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):31–39. doi: 10.1016/S0278-5846(03)00172-6 [DOI] [PubMed] [Google Scholar]
- 19.Clemens P, Baron JA, Coffey D, Reeves A. The short-term effect of nicotine chewing gum in patients with Parkinson’s disease. Psychopharmacology (Berl). 1995;117(2):253–256. doi: 10.1007/BF02245195 [DOI] [PubMed] [Google Scholar]
- 20.Lieberman A, Lockhart TE, Olson MC, et al. Nicotine Bitartrate Reduces Falls and Freezing of Gait in Parkinson Disease: A Reanalysis. Front Neurol. 2019;10:424. doi: 10.3389/fneur.2019.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebersbach G, Stöck M, Müller J, Wenning G, Wissel J, Poewe W. Worsening of motor performance in patients with Parkinson’s disease following transdermal nicotine administration. Mov Disord. 1999;14(6):1011–1013. doi: [DOI] [PubMed] [Google Scholar]
- 22.Fagerström KO, Pomerleau O, Giordani B, Stelson F. Nicotine may relieve symptoms of Parkinson’s disease. Psychopharmacology (Berl). 1994;116(1):117–119. doi: 10.1007/BF02244882 [DOI] [PubMed] [Google Scholar]
- 23.Vieregge A, Sieberer M, Jacobs H, Hagenah JM, Vieregge P. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology. 2001;57(6):1032–1035. doi: 10.1212/wnl.57.6.1032 [DOI] [PubMed] [Google Scholar]
- 24.Trinh K, Andrews L, Krause J, et al. Decaffeinated coffee and nicotine-free tobacco provide neuroprotection in Drosophila models of Parkinson’s disease through an NRF2-dependent mechanism. J Neurosci. 2010;30(16):5525–5532. doi: 10.1523/JNEUROSCI.4777-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter CJ, Tasic B, Russler EV, Liang L, Luo L. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 2010;141(3):536–548. doi: 10.1016/j.cell.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsen AL, Feany MB. Glial α-synuclein promotes neurodegeneration characterized by a distinct transcriptional program in vivo. Glia. 2019;67(10):1933–1957. doi: 10.1002/glia.23671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarkar S, Olsen AL, Sygnecka K, Lohr KM, Feany MB. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021;17(2):e1009359. doi: 10.1371/journal.pgen.1009359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brazill JM, Zhu Y, Li C, Zhai RG. Quantitative Cell Biology of Neurodegeneration in Drosophila Through Unbiased Analysis of Fluorescently Tagged Proteins Using ImageJ. J Vis Exp. 2018;(138). doi: 10.3791/58041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meshul CK, Kamel D, Moore C, Kay TS, Krentz L. Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp Neurol. 2002;175(1):257–274. doi: 10.1006/exnr.2002.7900 [DOI] [PubMed] [Google Scholar]
- 30.Carvajal-Oliveros A, Domínguez-Baleón C, Zárate RV, Campusano JM, Narváez-Padilla V, Reynaud E. Nicotine suppresses Parkinson’s disease like phenotypes induced by Synphilin-1 overexpression in Drosophila melanogaster by increasing tyrosine hydroxylase and dopamine levels. Sci Rep. 2021;11(1):9579. doi: 10.1038/s41598-021-88910-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bono F, Mutti V, Savoia P, et al. Nicotine prevents alpha-synuclein accumulation in mouse and human iPSC-derived dopaminergic neurons through activation of the dopamine D3- acetylcholine nicotinic receptor heteromer. Neurobiol Dis. 2019;129:1–12. doi: 10.1016/j.nbd.2019.04.017 [DOI] [PubMed] [Google Scholar]
- 32.Kardani J, Sethi R, Roy I. Nicotine slows down oligomerisation of α-synuclein and ameliorates cytotoxicity in a yeast model of Parkinson’s disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(6):1454–1463. doi: 10.1016/j.bbadis.2017.02.002 [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam SR, Magen I, Bove N, et al. Chronic nicotine improves cognitive and social impairment in mice overexpressing wild type α-synuclein. Neurobiol Dis. 2018;117:170–180. doi: 10.1016/j.nbd.2018.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bang Y, Lim J, Choi HJ. Recent advances in the pathology of prodromal non-motor symptoms olfactory deficit and depression in Parkinson’s disease: clues to early diagnosis and effective treatment. Arch Pharm Res. 2021;44(6):588–604. doi: 10.1007/s12272-021-01337-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signal. 2013;11(1):34. doi: 10.1186/1478-811X-11-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18(2):101–113. doi: 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo F, Sandhu AF, Rungratanawanich W, et al. Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Int J Mol Sci. 2020;21(19):E7174. doi: 10.3390/ijms21197174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xilouri M, Brekk OR, Stefanis L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov Disord. 2016;31(2):178–192. doi: 10.1002/mds.26477 [DOI] [PubMed] [Google Scholar]
- 39.Zhao X, Xu W, Wu J, et al. Nicotine induced autophagy of Leydig cells rather than apoptosis is the major reason of the decrease of serum testosterone. Int J Biochem Cell Biol. 2018;100:30–41. doi: 10.1016/j.biocel.2018.05.001 [DOI] [PubMed] [Google Scholar]
- 40.Xing R, Cheng X, Qi Y, et al. Low-dose nicotine promotes autophagy of cardiomyocytes by upregulating HO-1 expression. Biochem Biophys Res Commun. 2020;522(4):1015–1021. doi: 10.1016/j.bbrc.2019.11.086 [DOI] [PubMed] [Google Scholar]
- 41.Gao Q, Bi P, Luo D, et al. Nicotine-induced autophagy via AMPK/mTOR pathway exerts protective effect in colitis mouse model. Chem Biol Interact. 2020;317:108943. doi: 10.1016/j.cbi.2020.108943 [DOI] [PubMed] [Google Scholar]
- 42.Yan HY, Wen X, Chen LZ, et al. Augmented autophagy suppresses thymocytes development via Bcl10/p-p65 pathway in prenatal nicotine exposed fetal mice. Ecotoxicol Environ Saf. 2021;207:111272. doi: 10.1016/j.ecoenv.2020.111272 [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Liu B, Zhu J, Wang D, Wang Y. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway. Atherosclerosis. 2019;284:1–10. doi: 10.1016/j.atherosclerosis.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 44.Li Z, Zhang X, Jin T, Hao J. Nicotine promotes activation of human pancreatic stellate cells through inducing autophagy via α7nAChR-mediated JAK2/STAT3 signaling pathway. Life Sci. 2020;243:117301. doi: 10.1016/j.lfs.2020.117301 [DOI] [PubMed] [Google Scholar]
- 45.Feng Y, Xu F, Wang SM, et al. Melatonin attenuates nicotine-induced autophagy and neurological changes by decreasing the production of reactive oxygen species. Int J Neurosci. 2020;130(4):391–397. doi: 10.1080/00207454.2019.1692833 [DOI] [PubMed] [Google Scholar]
- 46.Pelissier-Rota MA, Pelosi L, Meresse P, Jacquier-Sarlin MR. Nicotine-induced cellular stresses and autophagy in human cancer colon cells: A supportive effect on cell homeostasis via up-regulation of Cox-2 and PGE(2) production. Int J Biochem Cell Biol. 2015;65:239–256. doi: 10.1016/j.biocel.2015.06.013 [DOI] [PubMed] [Google Scholar]
- 47.Stout KA, Dunn AR, Hoffman C, Miller GW. The Synaptic Vesicle Glycoprotein 2: Structure, Function, and Disease Relevance. ACS Chem Neurosci. 2019;10(9):3927–3938. doi: 10.1021/acschemneuro.9b00351 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Confirmation of locomotion rescue with a second RNAi line. α-synuclein or control flies with or without knockdown of SV2L1 and SV2L2 using VDRC lines. Flies were aged to 7 days post-eclosion on food containing 0.4 mg/ml nicotine or vehicle control. Symbols above the 0.4 mg/ml nicotine bar represent statistically significant difference compared to the vehicle control, respectively. *p <0.05, **p < 0.01, ***P<0.005, ns = p > 0.05. n = minimum of 60 flies per genotype (6 biological replicates of 10 flies each). Significance was determined by two-way ANOVA with Sidak’s test for multiple comparisons.
Supplemental Figure 2. Confirmation of knockdown. A. qRT-PCR for SV2L1 in control flies (no RNAi) or flies expressing one of two RNAi’s targeting SV2L1. RNAi #1 is the JF line. RNA #2 is the VDRC line. B. qRT-PCR for SV2L2 in control flies (no RNAi) or flies expressing one of two RNAi’s targeting SV2L2. RNAi #1 is the JF line. RNA #2 is the VDRC line.
Supplemental Figure 3. S129 phosphorylated α-synuclein. A. Representative immunoblot comparing S129 phosphorylated α-synuclein to total α-synuclein. B. Quantification of 4 independent experiments.
Supplemental Figure 4. SV2 knockdown increases Atg8a even in the absence of α-synuclein. A. Representative images. B. Quantification. Images were thresholded in ImageJ, and puncta > 0.12 μm in area were counted using the Analyze Particles function. Control flies expressing a GFP tagged Atg8a treated with ethanol vehicle or nicotine demonstrate sparse small puncta (small white arrows). Puncta are larger and more numerous with α-synuclein and large puncta are reduced in number with nicotine treatment (large yellow arrows). Scale = 5 μm. n = 6 flies per condition. Genotypes are nSyb-QF2, nSyb-GAL4/UAS-Atg8a-GFP or QUAS-Syn, nSyb-QF2, nSyb-GAL4/UAS-Atg8a-GFP for control and α-synuclein expressing flies, respectively. C. Representative images. D. Quantification of puncta was performed as in B. Genotypes are nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/+, nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/UAS-CG3168 RNAi or nSyb-QF2, nSyb-GAL4, UAS-Atg8a-GFP/UAS-CG14691 RNAi.