

A machine learning model using multifeature fragmentome data from genome-wide cell-free DNA analyses detected hepatocellular carcinoma with high sensitivity and specificity in average and high-risk populations, including in early-stage disease.
Abstract
Liver cancer is a major cause of cancer mortality worldwide. Screening individuals at high risk, including those with cirrhosis and viral hepatitis, provides an avenue for improved survival, but current screening methods are inadequate. In this study, we used whole-genome cell-free DNA (cfDNA) fragmentome analyses to evaluate 724 individuals from the United States, the European Union, or Hong Kong with hepatocellular carcinoma (HCC) or who were at average or high-risk for HCC. Using a machine learning model that incorporated multifeature fragmentome data, the sensitivity for detecting cancer was 88% in an average-risk population at 98% specificity and 85% among high-risk individuals at 80% specificity. We validated these results in an independent population. cfDNA fragmentation changes reflected genomic and chromatin changes in liver cancer, including from transcription factor binding sites. These findings provide a biological basis for changes in cfDNA fragmentation in patients with liver cancer and provide an accessible approach for noninvasive cancer detection.
Significance:
There is a great need for accessible and sensitive screening approaches for HCC worldwide. We have developed an approach for examining genome-wide cfDNA fragmentation features to provide a high-performing and cost-effective approach for liver cancer detection.
See related commentary Rolfo and Russo, p. 532.
This article is highlighted in the In This Issue feature, p. 517
INTRODUCTION
Liver cancer causes a staggering amount of morbidity and mortality worldwide, with more than 900,000 newly diagnosed cases each year and more than 800,000 deaths (1). In the United States, liver cancer is one of the few cancers that has shown an increase in incidence and mortality over the last 20 years. Ninety percent of cases of liver cancer are hepatocellular carcinoma (HCC), and survival is highly dependent on the stage of the disease at diagnosis. The five-year survival rate is 34% when the cancer is localized (44% of patients), 12% when regional (27% of patients), and 3% when a distant disease is found (18% of patients; ref. 2). There is a large, well-defined population that is at significantly increased risk for HCC, including individuals with chronic hepatitis B (HBV) infection or with cirrhosis from various causes including hepatitis C (HCV; ref. 3), nonalcoholic fatty liver disease (NAFLD; ref. 4), heavy alcohol use (5), aflatoxin, and other conditions (6). Worldwide, there are 350 million individuals with chronic viral hepatitis infection and 50 million with cirrhosis (7). In the United States, 4.5 million individuals have chronic HCV and 29 million have been diagnosed with NAFLD. Up to one third of those with cirrhosis and between 25% and 40% with HBV will develop HCC over their lifetime, with an up to 8% annual risk for patients with cirrhosis (8). A growing group of individuals at risk for liver cancer, including 29 million in the United States, have NAFLD, and 20% of the HCC that develops in this population occurs without cirrhosis (9). Medical societies throughout the world recommend screening for the highest risk populations, currently with abdominal ultrasound imaging with or without alpha-fetoprotein (AFP). Overall adherence to international guidelines, however, remains low, with less than one in five eligible individuals worldwide receiving some level of surveillance and less than 2% following recommended screening (10–12). Many factors contribute to low adherence to screening guidelines, including the identification of high-risk individuals, the requirement of infrastructure, and personnel needed for imaging-based screening methods (11). Current screening tests that include ultrasound imaging, with or without AFP, have shown limited sensitivity, varying from 47% to 84% with specificities from 67% to over 90% (13). Additionally, the lack of noninvasive diagnostic approaches for NAFLD suggests that the population not currently covered by HCC screening recommendations is increasing. Therefore, there is a great need for the development of accessible and sensitive screening approaches for HCC worldwide.
One recent avenue for overcoming these challenges has been the development of novel blood-based cell-free DNA (cfDNA) biomarkers for the detection of cancer. Somatic mutation–based approaches have been used as biomarkers for liver cancer but are limited by the need for tissue-based mutation identification and by the few changes detectable in plasma (14). Methylation profiling, both at specific sites and throughout the whole genome, and copy-number changes have also provided feasible avenues for the detection of liver cancer, but their detection sensitivities in very early-stage disease remain suboptimal (15–20). Recently developed multicancer early detection tests appear useful for the detection of many cancers (including liver cancer) in an average-risk cohort (21), but there are no published reports of using these approaches in a population at high risk of HCC. Additionally, the cost of most cfDNA-based tests is much higher than estimates of what would be affordable for screening tests in the United States and worldwide (21). Combining these approaches with AFP has increased performance but requires two separate tests and still has limitations in early-stage disease (22). We have previously developed an approach called DNA evaluation of fragments for early interception (DELFI) that utilizes genome-wide fragmentation profiles to provide a high-performing and cost-effective approach to cancer detection (23, 24). Zhang and colleagues applied a variation of this approach to evaluate noninvasive detection of liver cancer in China, but the underlying source of fragmentation changes in these patients was not explored (25). Fragmentation and methylation information has also demonstrated the ability to differentiate patients with liver cancer from those without cancer (26), although such an approach requires two distinct methods of cfDNA library preparation and analysis. To date, no study has validated genome-wide approaches for detecting HCC in independent groups or across different high-risk populations.
Here we describe the development of a genome-wide fragmentome approach to detect individuals with liver cancer. We examine the molecular origins of cfDNA in these patients and identify genomic and chromatin features associated with fragmentation changes. Finally, we use this approach to detect liver cancer in the US population and validate this model in a separate Hong Kong cohort.
RESULTS
Clinical Cohorts and Genomic Analyses of cfDNA
We examined plasma samples from 501 individuals, including 75 individuals with HCC and 426 without cancer. Among individuals without cancer, 133 had conditions that increased HCC risk, including cirrhosis from all causes or viral hepatitis without cirrhosis. Blood samples were prospectively collected from patients with HCC at various cancer stages and from high-risk individuals at the Johns Hopkins Hospital, whereas the remaining samples were identified through screening efforts at other US or EU hospitals (US/EU cohort; Table 1; Supplementary Table S1). We isolated 0.5 to 5 mL of plasma from each of these individuals, generated genomic libraries, and sequenced the cfDNA fragments using low-coverage whole-genome sequencing (∼2.6× coverage) with an average of 49 million high-quality paired reads per sample comprising 9 Gb of sequence data (Supplementary Table S2; refs. 23, 24). In addition to the US/EU cohort, we examined as a validation cohort whole-genome sequence data from 223 patients from Hong Kong, including patients with resectable early-stage HCC (n = 90, stage A = 85, B = 5), HBV (n = 66), and HBV-related cirrhosis (n = 35), as well as healthy individuals without liver disease (n = 32; Hong Kong cohort; Table 1; Supplementary Table S3; refs. 15, 27).
Table 1.
Patient demographics and clinical information
| Patient characteristic | Noncancer individuals n = 426 | Cancer patients n = 75 | P valuea |
|---|---|---|---|
| Age | |||
| Mean | 57.5 | 64.5 | <0.001 |
| Range | 27–81 | 38–88 | |
| Sex | |||
| Male | 235 | 63 | <0.001 |
| Female | 191 | 12 | |
| Liver disease | |||
| None | 293 | ||
| Hepatitis B | 26 | 1 | |
| Hepatitis C | 29 | 1 | |
| Cirrhosis | 78 | 69 | <0.001 |
| HCV | 53 | 41 | |
| HBV | 2 | 4 | |
| EtOH | 13 | 12 | |
| NAFLD | 3 | 11 | |
| Child–Pugh stage | |||
| A | 20 | 49 | <0.001 |
| B | 10 | 21 | |
| C | 10 | 5 | |
| Unknown | 38 | ||
| BCLC stage | |||
| 0 | 7 | ||
| A | 17 | ||
| B | 30 | ||
| C | 21 | ||
| Previous treatment | |||
| Yes | 28 | ||
| No | 47 | ||
| Validation cohort (Hong Kong) b | 133 | 90 | |
| Liver disease | |||
| None | 32 | ||
| Cirrhosis (HBV) | 35 | 90 | |
| Active HBV | 66 | ||
| BCLC stage | |||
| A | 85 | ||
| B | 5 | ||
Abbreviations: BCLC, Barcelona Clinic Liver Cancer staging system; EtOH, alcohol associated.
a P values were calculated to compare data from individuals with and without liver cancer for the following variables: mean ages using Student unpaired two-tailed t tests, sex distribution, cirrhosis etiology, and Child–Pugh stage using a χ2 test.
bValidation cohort data were obtained from Jiang et al. (15).
Genome-wide cfDNA Fragmentation Profiles Informed by Underlying Chromatin Structure
We evaluated the fragmentome and generated fragmentation profiles across the genome in 473 nonoverlapping 5-Mb regions, each region comprising ∼80,000 fragments, and spanning approximately 2.4 Gb of the genome using the DELFI approach (23). The fragmentation profiles were consistent among individuals without cancer but highly variable among patients with HCC (Fig. 1A). Profiles of patients with cirrhosis were closer to noncancer individuals without cirrhosis than they were to those from patients with HCC (Fig. 1A). Likewise, patients with viral hepatitis had fragmentation profiles nearly identical to those of noncancer individuals without liver disease (Fig. 1A).
Figure 1.

Genome-wide fragmentation profiles reflect underlying chromatin structure. A, Fragmentation profiles of 501 individuals in 473 nonoverlapping 5-Mb genomic regions. Fragmentation profiles for individuals with cancer show marked heterogeneity as compared with noncancer individuals with and without liver disease. B, Comparison of plasma fragmentation features to reference A/B compartments. Track 1 shows A/B compartments extracted from liver cancer tissue (28). Track 2 shows a median liver cancer component extracted from the HCC plasma samples of 10 liver patients with high tumor fraction by ichorCNA (56). Track 3 shows the median fragmentation profile in the plasma for these 10 HCC samples, and track 4 shows the median profile for 10 healthy plasma samples. Track 5 shows A/B compartments for lymphoblast cells (28). These five tracks show chromosome 22 as an example, with darker shading indicating informative regions of the genome where the two reference tracks differ in domain (open/closed) or magnitude. C, Among these informative bins, for each chromosome, the log odds of the plasma component matching the HCC reference track in domain. Log odds greater than 1 indicate more similarity to the HCC reference track, whereas log odds less than 1 indicate more similarity to the lymphoblast reference track. The extracted HCC component has the greatest similarity to the HCC reference track, and the noncancer plasma has the greatest similarity to the lymphoblast reference track; the HCC plasma track is intermediate to the two.
To examine the origins of cfDNA fragmentation patterns, we compared genome-wide fragmentome profiles with high-throughput sequencing chromosome conformation capture (Hi-C) open (A) and closed (B) compartments. We found that cfDNA patterns of healthy individuals were highly correlated to those of lymphoblastoid cells (Fig. 1B). Analysis of cfDNA profiles from 10 HCC patients with high ctDNA levels revealed that their fragmentome reflected two components: one resembling the profile of individuals without cancer and a separate cfDNA component that had high similarity to A/B compartments previously estimated from liver cancers (Fig. 1B; ref. 28). Additionally, when these two components were estimated, the cfDNA profiles of the predicted liver component had high similarity to genome-wide A/B compartments of liver cancer, whereas the profiles of patients with HCC were intermediate in similarity to liver cancer (Fig. 1B and C). In contrast, the profiles of individuals without cancer were closer to A/B compartments of lymphoblastoid cells (Fig. 1B and C). These analyses suggested that cfDNA fragmentomes from individuals with HCC represent a mixture of cfDNA profiles of chromatin compartments of cells from peripheral blood as well as those from liver cancer.
Disease-Specific Transcription Factors Inferred from Genome-wide cfDNA Fragmentation
As chromatin organization reflects underlying cellular transcriptional programs (29–31), we examined whether cfDNA fragmentation characteristics might reflect changes derived from altered DNA binding of transcription factors (TF) in liver cancer. To identify DNA binding sites for all known TFs, we analyzed 5,620 chromatin immunoprecipitation sequencing (ChIP-seq) experiments from the ReMap 2020 database (32). For each TF, we calculated the aggregate cfDNA coverage across all binding sites identified (4,000–490,000 per sample) compared with the overall adjacent genomic coverage, producing a single metric for each TF in each sample. We compared these TFs in patients with and without HCC to identify those TFs with the largest and smallest differences in genome-wide binding site coverage in cfDNA (Fig. 2A and B). Gene set enrichment analyses using the DisGeNET database of gene–disease associations revealed that differences in cfDNA TF binding coverages between individuals with HCC and individuals without cancer were predicted to be related to liver and other cancers (Fig. 2C and D). Additionally, the top-scoring individual TFs represented those with known biological relevance to chromatin organization and liver cancer, whereas the low-scoring TFs did not (Table 2; Supplementary Table S4). These included members of the activator protein 1 (AP1) complex, including JUN, JUND, ATF2, and ATF7 genes, which integrate extracellular signals (33) and have been linked to liver tumorigenesis (34, 35); Transcriptional Enhancer Factor Domain Family member 4 (TEAD4), which has been shown to have oncogenic roles in HCC (36, 37); Poly(C)‑binding protein 2 (PCBP2) transcriptional coregulator, which when overexpressed is associated with a worse prognosis in patients with HCC (38); Prohibitin 2 (PHB), which promotes progression in HCC (39); and AT-rich interacting domain 3A (ARID3A), an oncogenic TF that when upregulated promotes liver cancer malignancy (40). A similar analysis of cfDNA fragmentation data from our recent study of patients in the LUCAS lung cancer diagnostic trial (23) revealed an enrichment of coverage differences in binding sites of TFs related to lung cancer (Fig. 2C and E). Altogether, these observations suggest that changes in cfDNA fragmentation in patients with liver and other cancers result from the multitude of altered transcriptional profiles present in the cancer cells.
Figure 2.

Fragmentation profiles in patients with HCC highlight liver-specific TFs. A, The coverage at and around the TF binding sites (TFBS) for the 9 TFs for which the relative coverage at the binding site had the highest separation of HCC from noncancer samples. The mean is plotted for each group, with ± 1 SD shown by shading. These confidence intervals (CI) show separation, highlighting that differences in coverage at a TFBS can provide information on cancer status. B, The coverage at and around the TFBS for the 9 TFs that had the lowest separation of HCC from noncancer samples in the US/EU cohort. These CIs are largely overlapping, reflecting their status as TFBS with poor discrimination.Gene set enrichment analysis of TFs analyzed in both HCC and lung adenocarcinoma showed TFs are selectively enriched in numerous pathways related to liver and lung cancer, respectively (C), including adult liver carcinoma and adenocarcinoma of the lung (D and E).
Table 2.
Top scoring TFs in US/EU cohort samples
| TF | Gene name | AUC | Cell type in ChIP-seq experiment | Gene function | Link to HCC |
|---|---|---|---|---|---|
| ZNF512 | Zinc Finger Protein 512 | 0.836 | K-562 | Unknown | Undescribed |
| ZNF184 | Zinc Finger Protein 184 | 0.826 | K-562 | Unknown | Undescribed |
| PCBP2 | Poly(C)-binding protein 2 | 0.791 | Hep-G2 | Transcriptional coregulator | Overexpression contributes to poor prognosis and enhanced cell growth in HCC (38) |
| JUN | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit | 0.785 | Hep-G2 | TF | Promotes HBV-related liver tumorigenesis (59) |
| PHB2 | Prohibitin 2 | 0.771 | K-562 | Transcriptional coregulator | Functions in mitophagy of HCC (39) |
| ATF2 | Activating Transcription Factor 2 | 0.770 | Hep-G2 | TF, HAT | Mediates suppression of liver tumor formation (34) |
| ATF7 | Activating Transcription Factor 7 | 0.765 | MCF-7 | TF | Regulates growth of liver cancer (60) |
| TEAD4 | TEA Domain Transcription Factor 4 | 0.760 | Hep-G2 | TF | Oncogenic role in HCC (36) |
| ARID3A | AT-rich interacting domain 3A | 0.756 | Hep-G2 | TF | Facilitates liver cancer malignancy (40) |
| JUND | JunD Proto-Oncogene, AP-1 Transcription Factor Subunit | 0.754 | HT29_DSMO | TF | Involved in PARγ signaling and NAFLD development (61) |
Abbrevation: HAT, histone acetyltransferase.
Genomic Changes in HCC Are Revealed from cfDNA Fragmentomes
As the cfDNA fragmentome may comprise changes related to large-scale genomic alterations released from cancer cells (23, 24), we also examined chromosomal gains and losses in the circulation of these patients. In addition to the genome-wide fragmentation profiles resulting from chromatin and TF changes observed in patients with liver cancer (Fig. 3A), our analyses revealed an altered representation of chromosomal arms matching those commonly gained or lost in liver cancer as reported in previous The Cancer Genome Atlas (TCGA) large-scale genomic studies of HCC (n = 372; Fig. 3B). These included increased cfDNA representation of 1q, 7p, 7q, and 8q and decreased levels of 4q, 8p, 9p, 13q, and 21q, all known to be gained or lost, respectively, in HCC (41, 42). Importantly, these alterations were observed in the patients with HCC but not in individuals without cancer, even if they had cirrhosis or chronic liver disease (Fig. 3B).
Figure 3.

High-dimensional fragmentation features reflect liver cancer biology and are incorporated in DELFI machine learning approaches. A, A heat map reflecting the complexity of genome-wide fragmentation and TF binding site (TFBS) features utilized in the DELFI machine learning approach. Each row represents a sample, whereas columns show individual genomic features.B, Analysis of copy-number changes in tissue from 372 TCGA liver cancers and plasma from 501 individuals reflects biological consistency. Copy-number changes that occur in TCGA (red = gains, blue = losses) were also found at the chromosomal arm level in HCC plasma, but not in individuals without cancer. CNV, copy-number variation. C, Heat map depicting the contributions of individual genomic regions to the final trained DELFI model. The fragmentation features were summarized as three principal components (PC) in the model, whereas aneuploidy was summarized as arm-level z-scores. The top, middle, and right depict the coefficients of fragmentation components, arm-level z-scores, and TFBS, respectively, in the model.
DELFI Model for HCC Detection
Given the direct connection between genomic and chromatin changes in liver cancer and cfDNA fragmentation, we used a machine learning approach to determine if changes in cfDNA fragmentomes could distinguish patients with HCC from those without cancer. We previously used this approach to develop a robust classifier for lung cancer detection that was externally validated in an independent population (23). We determined the performance of this classifier in the US/EU cohort by repeated 5-fold cross-validation, generating a score for each individual that is an average over 10 cross-validation repeats (DELFI score). The resulting model included a combination of regional and large-scale fragmentation characteristics that were optimal for identifying individuals with liver cancer (Supplementary Fig. S1; Fig. 3C). These features comprised the majority of the informative chromosomal, chromatin, and local changes identified above, comprising >90% of the variance of the fragmentation profiles across samples.
As clinical characteristics may affect tumor biomarkers, we investigated whether measures of liver dysfunction or demographic parameters such as age, sex, race, or weight were associated with DELFI scores in individuals without cancer where this information was available (Supplementary Table S1). We observed no association of DELFI scores with age (R = 0.18, P = 0.08, Spearman correlation; Supplementary Fig. S2A) and no difference in DELFI scores between males and females (P = 0.58, Wilcoxon test; Supplementary Fig. S2B). Asians and African Americans have been shown to have a higher incidence of liver cancer that is diagnosed at later stages (43), and we observed small differences in fragmentation scores among high-risk individuals without cancer across these or other racial or ethnic groups, although these analyses are limited by lack of information on clinical covariates in some of these cases (P = 0.037 in patients with viral hepatitis and P = 0.026 in patients with cirrhosis, Kruskal–Wallis test; Supplementary Fig. S3). Among individuals with cirrhosis, we observed a correlation between the degree of liver disease as measured by the Child–Pugh score and DELFI scores (R = 0.58 P = 8.6e−5, Spearman correlation; Supplementary Fig. S4). Increased body mass index (BMI), a risk factor for NAFLD and liver cancer, was not associated with changes in DELFI scores in patients with viral hepatitis (R = 0.027, P = 0.85, Spearman correlation); however, lower BMI in patients with cirrhosis was associated with higher DELFI scores, perhaps due to cachexia in patients with severe cirrhosis (R = −0.23, P = 0.043, Spearman correlation; Supplementary Fig. S5).
We next examined the relationship between DELFI scores and the presence and stage of liver cancer in a population at high risk for liver cancer. The DELFI scores for 133 individuals who were cancer-free were low, with median DELFI scores of 0.078 or 0.080 for those with viral hepatitis or cirrhosis, respectively. In contrast, the 75 patients with HCC had significantly higher median DELFI scores across all Barcelona Clinic Liver Cancer staging system (BCLC) stages, including stage 0 = 0.46, stage A = 0.61, stage B = 0.83, and stage C = 0.92 (P < 0.01 for stages 0, A, B, or C, Wilcoxon rank sum test; Fig. 4A). A receiver operator characteristic (ROC) curve of the DELFI approach to identify patients with HCC revealed an area under the curve (AUC) of 0.90 [95% confidence interval (CI), 0.86–0.94] among high-risk individuals (Fig. 4B). Performance remained robust for early-stage HCCs, with AUCs of 0.9 and 0.81 for BCLC stage 0 and A. Individuals with advanced-stage HCC (BCLC C) were almost perfectly detected among the individuals analyzed (AUC > 0.97; Fig. 4C).
Figure 4.

DELFI machine learning models detect liver cancer with high sensitivity and specificity. A, DELFI scores for the US/EU cohort across liver disease and cancer stage for the screening and surveillance models. Patients with cirrhosis have DELFI scores higher than individuals without cancer or with viral hepatitis on average, but lower than all stages of liver cancer. Patients with liver cancer across all stages have relatively high DELFI scores, with stage C individuals uniformly having the highest DELFI scores. B, ROC analyses of the US/EU general population cohort and the high-risk surveillance cohort. C, ROC analyses of the US/EU general population and surveillance cohorts separated by BCLC stage, showing high sensitivity and specificity across stages. D, ROC analyses for the fixed surveillance model applied to the Hong Kong cohort, which includes 90 individuals with HCC (85 with BCLC stage A cancer, and 5 with BCLC stage B cancer), 101 individuals with cirrhosis and viral hepatitis, and 32 individuals without cancer or liver disease.
To extend these analyses to individuals at low risk for developing liver cancer, we examined the ability of a DELFI model to distinguish between individuals with cancer and those from a general population (n = 293) without viral hepatitis or cirrhosis. In this larger cohort where additional features could be included in cross-validated training, we used the features of the model above and also included cfDNA coverage at ChIP-seq–derived TF binding sites from liver cell lines available in the ReMap database to create a DELFI model for a general population (Fig. 3C). This approach had high performance for cancer detection (AUC = 0.98) among these individuals. We evaluated the performance of this model at 98% specificity, a threshold appropriate for an average risk population (24), and observed an overall sensitivity of 88% in this setting (Fig. 4B), with sensitivity above 75% across all stages. Use of a model that did not incorporate TF binding sites led to a slightly reduced performance, and there was a high correlation among the rank-ordered scores using our DELFI models for high-risk and screening populations (R = 0.48, P = 2e−5; Supplementary Figs. S6A, S6B, and S7).
To examine the relationship between fragmentation profiles and liver cancer progression, we assessed whether the size, number, and characteristics of liver cancer lesions as well as the etiology of neoplasia were related to aberrant fragmentation profiles, where this information was available. We found that the tumor size and lesion number were positively correlated to DELFI scores (R = 0.42 and 0.31, P = 0.00026 and P = 0.0064, respectively, Spearman correlation; Supplementary Fig. S8A and S8B), consistent with the notion that the fragmentation profile was related to overall tumor burden. Among patients with liver cancer at resectable stages (0, A, and B), the cancer etiology, including viral hepatitis, or cirrhosis due to alcohol, NAFLD, or idiopathic sources, yielded similar DELFI scores (P = 0.43, Kruskal–Wallis test; Supplementary Fig. S9). These observations suggest that fragmentation profiles were a result of ongoing tumor-related cfDNA processes and were not affected by early events in tumorigenesis.
To examine the real-world impact of this method in the context of HCC detection, we compared the performance of the DELFI fragmentome with the current screening measurement of AFP levels. AFP levels were elevated above the recommended screening threshold of 20 ng/mL in 39 of 75 (52%) individuals with cancer, consistent with previous reports (44). Among individuals that had AFP levels below 20 ng/mL and who have been undetected by this approach, DELFI detected 30 of 36 (83%). The use of AFP measurements would have detected 8/24 (33%) stage 0/A patients, 17/30 (57%) stage B patients, and 14/21 (66%) stage C patients (Supplementary Fig. S10). In contrast, the DELFI approach detected 19/24 (79%) stage 0/A patients, 25/30 (83%) stage B patients, and 20/21 (95%) stage C patients. Overall, genome-wide cfDNA fragmentation analyses had improved performance compared with AFP detection of HCC, and the combination of DELFI and AFP may provide an improvement in detection over the DELFI approach alone, as we observed these to have a combined sensitivity of 92% at a combined specificity of 80%.
External Validation of DELFI Model in an East Asian Population with HCC
In addition to our cross-validated analysis of the US/EU cohort, we tested the fixed DELFI model in the 223 patients from the Hong Kong cohort. These included patients who had largely resectable early-stage HCC (n = 90, stage A = 85, B = 5) and 101 with cirrhosis or HBV infection. These samples were sequenced previously using a different sequencer (HiSeq 2000 vs. Novaseq; 76-bp vs. 100-bp read length), different library preparation, and a higher number of PCR cycles (14 vs. 4 cycles), but we observed similar genome-wide patterns to our earlier analyses (Supplementary Fig. S11). The fragmentation profiles of patients with viral hepatitis and cirrhosis, as well as healthy individuals, had highly consistent profiles throughout the genome, whereas those of patients with HCC were variable and disordered (Supplementary Fig. S12). Additionally, the chromosomal changes observed in plasma in the Hong Kong cohort were similar to those in the initial US/EU cohort, as well as the cancers from the TCGA (Supplementary Fig. S13). Overall, in this validation cohort, the DELFI model distinguished HCC patients with an AUC of 0.97 from those with high-risk disease (Fig. 4D). These observations suggest that the underlying characteristics of cfDNA fragmentation were similar in this cohort, and that DELFI is a robust method to detect HCC and is generalizable across different high-risk populations.
Simulation of DELFI Performance at Population Scale
To evaluate how our approach would perform for surveillance and detection in patients at high risk for liver cancer, we evaluated the DELFI model in a theoretical population of 100,000 high-risk individuals using Monte Carlo simulations. Given the importance of the detection of early-stage cancers, we focused our modeling on the detection of stage 0/A disease. We compared the DELFI approach with the current standard of care, concurrent ultrasound, and AFP and modeled the uncertainty of sensitivity and specificity of these surveillance modalities in this theoretical population through probability distributions centered at empirical estimates from our cohort or previous reports (ref. 11; see Methods). Despite surveillance recommendations, the adherence to HCC surveillance in the United States is low, with the most generous estimates suggesting 39% adherence (45), resulting in an average of 40,042 individuals tested in this theoretical population (95% CI, 21,320–61,890). As blood tests offer high accessibility and compliance, with adherence rates of 80% to 90% reported for blood-based biomarkers (46, 47), we conservatively assumed an average of 75% (95% CI, 60%–90%) of this population would be tested using the DELFI approach. As the prevalence of cirrhosis, viral hepatitis, and the co-occurrence of these comorbidities with HCC could vary by region, we used a prior probability distribution to reflect our uncertainty of the composition of these diseases and possible regional differences. Monte Carlo simulations from these probability distributions (Methods) revealed that ultrasound with AFP detected an average of 2,233 (95% CI, 1,088–3,699) individuals with liver cancer. Using DELFI, we would detect on average 2,794 additional liver cancer cases, or a 2.46-fold increase (95% CI, 1.25–4.57-fold increase), compared with ultrasound with AFP alone (Supplementary Fig. S14A and S14B). The DELFI approach would not only substantially improve the detection of liver cancer but would be expected to decrease the false-negative rate, or fraction of cancers missed at testing, from 38% for ultrasound with AFP (95% CI, 25%–51.5%) to 24% for DELFI (95% CI, 9%–42.6%). Additionally, the negative predictive value of the test (NPV) would be expected to increase from 95.7% for ultrasound with AFP (95% CI, 93.8%–97.3%) to 97.1% for DELFI (95% CI, 94.8%–99.0%; Supplementary Fig. S14C and S14D). These analyses suggest a significant population-wide benefit for using a high-specificity blood-based early detection test as a tool for the detection of liver cancer.
DISCUSSION
Overall, in this study, we demonstrate the use of genome-wide cfDNA fragmentome features to detect HCC with high sensitivity and specificity. Furthermore, we show that the fragmentation profiles capture genomic and chromatin characteristics, including alterations known to be important in HCC. Our cfDNA fragmentome approach has robust performance in detecting HCC, including very early-stage disease, independent of disease etiology. To our knowledge, this is the first genome-wide fragmentation analysis that has been independently validated in a separate high-risk population, with stable and robust performance across different racial and ethnic groups from the United States and Hong Kong.
Our results also revealed that disease-specific TF signatures can be obtained through analysis of genome-wide cfDNA fragmentation profiles. Although such analyses have been performed using specific TFs to distinguish small cell from non–small cell lung cancers (23), this study suggests that analyses of disease-specific transcriptional regulation using genome-wide cfDNA fragmentation may improve the detection and identification of the tissue of origin in patients with cancer. With sufficient numbers of patients, cfDNA transcriptional profiles could further improve machine learning algorithms to detect HCC and other cancers.
HCC is unique in comparison with other solid cancers in that there is a large, well-defined high-risk population with an average 3% to 4% annual risk of developing HCC (48) recommended to have routine cancer screening every 6 months. Unfortunately, currently available tests have limited diagnostic utility, especially for early-stage disease (13). In our study, AFP had 52% sensitivity in detecting HCC, consistent with the known performance of this biomarker (13). Ultrasound-based surveillance also has technical limitations with its operator dependency and lower sensitivity in patients with cirrhosis and obesity (49). Most importantly, ultrasound has low compliance to established guidelines, less than 20% worldwide (10, 11), compared with much higher adherence to blood tests for other conditions (46). Despite these challenges, HCC screening provides overall survival benefit in patients with HBV (50) and cirrhosis (10), highlighting the major need to improve current screening tests. The high performance of cfDNA fragmentome analyses in HCC detection, along with its cost-efficient characteristics, would allow DELFI to be an accessible screening test for HCC and to increase the screening rates beyond the currently dismal levels. An interesting aspect of cfDNA analysis specific to HCC is that transplantation is the most curative treatment for early to intermediate stage HCC, and HCC surveillance of posttransplantation patients with a liquid biopsy approach could have a dual role in tracking recurrence and rejection, as studies of cfDNA in posttransplant patients have shown promise (51).
Although this study represents a potential improvement in current screening approaches, there are some limitations. For example, this study included a relatively small sample size of individuals with HCC. Although the independent validation cohort was performed with preanalytical differences in laboratory and sequencing methods, the fact that the DELFI approach performed well in this population suggests that the method will ultimately be able to be utilized in a range of different diagnostic laboratories. Larger validation studies will be needed before this approach can be useful clinically. Nevertheless, the observations that scalable and cost-effective noninvasive cfDNA fragmentome analyses can detect patients with liver cancer may provide an opportunity to screen high-risk and general populations worldwide.
METHODS
Study Population
For the US/EU cohort, samples from 208 patients, including 75 with HCC and 133 high-risk patients without HCC, were collected prospectively as part of the HCC biomarker registry and the AIDS Linked to the IntraVenous Experience (ALIVE) study at the Johns Hopkins University School of Medicine under protocols approved by the Johns Hopkins Institutional Review Board. HCC was defined by histologic examination or the appropriate imaging characteristics as defined by accepted guidelines. Tumor staging was determined by the BCLC. Detailed clinical data were extracted from the electronic medical record. High-risk patients were defined as individuals with cirrhosis from any etiology and/or individuals with chronic HBV or HCV who were recommended for routine HCC screening by expert society guidelines (49). In addition, we included 38 patients with HBV or cirrhosis retrospectively collected by BioIVT. AFP levels were quantified by partnering centers in their clinical laboratories using FDA-approved AFP tests.
The US/EU cohort also included samples from 293 individuals without cancer that were previously analyzed (23), originally from two screening clinical trial cohorts for colorectal cancer in Denmark (Endoscopy III) and The Netherlands (COCOS, Netherlands Trial Register ID NTR182946). The protocol for the Endoscopy III Project was approved by the Regional Ethics Committee and the Danish Data Protection Agency; for the COCOS trial, ethical approval was obtained from the Dutch Health Council. The inclusion criteria for both the Dutch and the Danish cohorts were any individuals of age 50 to 75 eligible for colorectal cancer screening. All patients used had either a negative fecal immunochemical test result or a negative colonoscopy result.
For the Hong Kong cohort, all recruited subjects gave written informed consent, and the study was approved by the Joint Chinese University of Hong Kong and New Territories East Cluster Clinical Research Ethics Committee (15, 27).
Sample Collection and Preservation
The sample collection was performed as follows: Venous peripheral blood was collected in one K2-EDTA tube and two serum gel tubes. Within 2 hours from blood collection, tubes were centrifuged at 2330 × g at 4°C for 10 minutes, plasma was transferred to new tubes, and the samples were spun at 14,000 rpm (18,000 rcf) for 10 minutes at room temperature to pellet any remaining cellular debris. After centrifugation, EDTA plasma was aliquoted and stored at −80°C for cfDNA analyses.
Sequencing Library Preparation
Circulating cfDNA was isolated from 2 to 4 mL of plasma using the Qiagen QIAamp Circulating Nucleic Acids Kit (Qiagen GmbH), eluted in 52 μL of RNase-free water containing 0.04% sodium azide (Qiagen GmbH), and stored in LoBind tubes (Eppendorf AG) at −20°C. Concentration and quality of cfDNA were assessed using the Bioanalyzer 2100 (Agilent Technologies).
Next-generation sequencing cfDNA libraries were prepared for whole-genome sequencing using 15 ng cfDNA when available or entire purified amount when less than 15 ng (Supplementary Table S5). In brief, genomic libraries were prepared using the NEBNext DNA Library Prep Kit for Illumina (New England Biolab) with four main modifications to the manufacturer's guidelines: (i) the library purification steps followed the on-bead AMPure XP (Beckman Coulter) approach to minimize sample loss during elution and tube transfer steps; (ii) NEBNext End Repair, A-tailing, and adapter ligation enzyme and buffer volumes were adjusted as appropriate to accommodate on-bead AMPure XP purification; (iii) Illumina dual index adapters were used in the ligation reaction; and (iv) cfDNA libraries were amplified with Phusion Hot Start Polymerase. All samples underwent a 4-cycle PCR amplification after the DNA ligation step.
Low-Coverage Whole-Genome Sequencing and Alignment
Whole-genome libraries of patients with cancer and cancer-free individuals were prepared as in ref. 23 with the modification that they were sequenced using 100-bp paired-end runs (200 cycles) on the Illumina NovaSeq platform at 1 to 2× coverage per genome. Prior to alignment, adapter sequences were filtered from reads using the fastp software (52). Sequence reads were aligned against the hg19 human reference genome using Bowtie2 (53), and duplicate reads were removed using Sambamba (54). After alignment, each aligned pair was converted to a genomic interval representing the sequenced DNA fragment using bedtools (55). Only reads with a MAPQ score of at least 30 or greater were retained. Read pairs were further filtered if overlapping the Duke Excluded Regions blacklist (https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=wgEncodeMapability). To capture large-scale epigenetic differences in fragmentation across the genome estimable from low-coverage whole-genome sequencing, we tiled the hg19 reference genome into nonoverlapping 5-Mb bins. Bins with an average GC base content < 0.3 and an average mappability < 0.9 were excluded, leaving 473 bins spanning approximately 2.4 Gb of the genome. Following Mathios and colleagues (23), GC correction was performed independently for short (<150 bp) and long (≥150 bp) cfDNA fragments using an external panel of 20 individuals without cancer sequenced on a NovaSeq to generate a target distribution.
Fastq files for patients in the Hong Kong cohort were obtained from The Chinese University of Hong Kong (CUHK) Circulating Nucleic Acids Research Group, as reported (ref. 15; #1645) and processed as described above and in Mathios and colleagues, to generate the DELFI features. GC correction was performed by normalizing to the target distribution provided in https://github.com/cancer-genomics/PlasmaToolsNovaseq.hg19, the same target distribution used for GC correction in the US/EU cohort. The validation set consisted of libraries constructed with 14 cycles of PCR and sequenced on the HiSeq 2000. These libraries were normalized to the 4-cycle NovaSeq target distribution to facilitate comparisons between studies. One sample each from the cirrhotic and HBV groups were excluded, as they were identified to have an HCC diagnosis.
Chromatin Structure Analysis
A/B compartments for liver cancer tissue and lymphoblastoid cells were obtained from https://github.com/Jfortin1/TCGA_AB_Compartments as well as from https://github.com/Jfortin1/HiC_AB_Compartments as described previously (28). The two reference tracks were compared to identify informative 100-kb bins, defined as bins where the chromatin domain differed between the two reference tracks or the magnitude difference in eigenvalues corresponded to a z-score greater than 1.96 or less than −1.96 (P = 0.05) across all eigenvalue differences.
The median fragmentation profile for 10 liver samples with high estimated tumor fraction by ichorCNA (56) and 10 randomly selected individuals without cancer was calculated. This information was used to extract an estimated median liver component in the plasma weighted by the ichor score of the individual plasma samples.
Genome-wide TF Analyses
ChIP-seq peaks from 5,620 experiments were downloaded from the ReMap 2020 database (32). This set was filtered for experiments with more than 4,000 peaks, resulting in 4,293 experiments. For each peak in the autosomes, we defined the center of the peak as position 0.
The mean of the coverages at each position (−3,000 to +3,000 with respect to the center of each peak) was computed across all peaks for each sample. For the ROC curves, relative coverage was computed for each sample as the mean coverage in a ±100-bp window surrounding the center of the binding sites divided by the mean coverage in a ±250-bp window surrounding 2,750 bp upstream and downstream of the binding sites. The ROC curve was generated using pROC 1.16.2 (57). The AUC for each peak set was ranked. Each TF was matched with its NCBI ID, leaving 797 unique TFs ranked by AUC. This ranked list was the input for the gseDGN function from the DOSE package in R. The output from this was ranked by the normalized enrichment score.
Whole-Genome Fragment Features
Fragmentation features were calculated as in Mathios and colleagues (23). Briefly, the ratio of short to long fragments was calculated for 473 nonoverlapping 5-Mb bins across the genome, and z-scores representing arm gains/losses were calculated for autosomal chromosome arms. The principal components of the ratios representing greater than 90% of variance and the z-scores were used to train machine learning models.
Machine Learning and Cross-Validation Analyses
Two machine learning models were developed: one for high-risk populations (a Gradient Boosting Machine using the Mathios et al. features) and the second for average-risk general populations (a penalized logistic regression with the Mathios et al. features as well as coverage from TF binding sites). These models were trained on the US/EU cohort in Caret with 5-fold cross-validation with 10 repeats, and scores for each sample were calculated by the mean across repeats and evaluated using AUC-ROC as in Mathios and colleagues (23). The first model used the high-risk noncancer and HCC patients, whereas the second model used the noncancer individuals without liver pathology. The locked high-risk model trained on the US/EU cohort was applied to the Hong Kong cohort to generate cancer predictions on an external validation set.
TCGA Analysis
Copy-number data from the HCC cancer cohort in TCGA [liver hepatocellular carcinoma (LIHC) n = 372] were retrieved using the package RTCGA v1.16.0 and were analyzed to determine the frequency of copy-number gains and losses in the 473 5-mb bins for this cohort (23). The somatic copy-number alteration threshold used in Mathios and colleagues (23) was used to call gains and losses in the HCC cohorts (23, 58).
Association of Clinical Covariates with DELFI Score
Potential associations between clinical covariates (for those patients for whom this information was made available) and the DELFI score were assessed with Spearman rank correlation coefficient (continuous variables) and Kruskal–Wallis one-way analysis of variance (categorical variables).
Simulation
Monte Carlo simulations were used to compare the DELFI approach to ultrasound and AFP in a theoretical surveillance population. We used estimated 95% CIs of sensitivity and specificity for DELFI and published 95% CIs for ultrasound with AFP (13). The R package epiR was used to derive prior predictive probability distributions (beta distributions) from these CIs (R package version 2.47, epiR; RRID:SCR_021673). Zhao and colleagues (45) reported that adherence to ultrasound and AFP surveillance was 39% (95% CI, 21%–65%). As other noninvasive blood-based tests have a reported adherence of more than 75% (46, 47), we assumed that adherence to DELFI would be 60% or greater with a probability 0.975 or higher. Using these confidence estimates, epiR was used to derive beta prior predictive distributions for adherence. We simulated multinomial probabilities for the prevalence of HBV, cirrhosis, HBV + HCC, cirrhosis + HCC, and HBV + cirrhosis + HCC from a Dirichlet with parameters 230, 680, 60, 23, and 7, respectively. For a single Monte Carlo simulation for ultrasound with AFP testing, we
(i) sampled the probability of adherence (
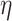 ) from the prior predictive distribution,
) from the prior predictive distribution,(ii) simulated the number of 100,000 individuals (S) who participated in surveillance (
 ),
),(iii) sampled probabilities of comorbidities [Dirichlet (230, 680, 60, 23, 7)],
(iv) computed the prevalence of HCC (
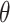 ),
),(v) simulated HCC cases
 and computed the number of individuals without cancer (
and computed the number of individuals without cancer ( ),
),(vi) sampled the sensitivity (
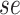 ) and specificity (
) and specificity (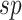 ) from the corresponding prior predictive distributions, and
) from the corresponding prior predictive distributions, and(vii) sampled the true positives (
 and false positives (
and false positives ( ).
).
Given TP and FP, we calculated the NPV as (true negatives)/(true negatives + false negatives), where true negatives =  and false negatives =
and false negatives =  . We repeated the above simulation 1,000 times, obtaining a distribution of TP, FP, and NPV. Using parameters for sensitivity, specificity, and adherence for the DELFI approach, we repeated the same Monte Carlo analysis to allow comparisons between these two surveillance methodologies.
. We repeated the above simulation 1,000 times, obtaining a distribution of TP, FP, and NPV. Using parameters for sensitivity, specificity, and adherence for the DELFI approach, we repeated the same Monte Carlo analysis to allow comparisons between these two surveillance methodologies.
Bioinformatic and Statistical Software
All statistical analyses were performed using R version 4.1.2. After trimming of adapter sequences using fastp (0.20.0), we used Bowtie2 (2.3.0) to align paired-end reads to the hg19 reference genome. PCR duplicates were removed using Sambamba (0.6.8), and the remaining aligned read pairs were converted to a bed format using Bedtools (2.29.0). We used the R package data.table (1.12.8) for manipulation of tabular data and binning fragments in 5-Mb windows along the genome. The R package Caret (6.0.84) was used to implement the classification by penalized logistic regression and resampling.
Data and Material Availability Statement
Sequence data and clinical variables used in this study are available at the European Genome-Phenome Archive (EGA) at accession EGAS00001005340, EGAD00001005093, and EGAS00001005340. Some data are not publicly available due to limitations in Institutional Review Board approval but are available upon reasonable request from the corresponding authors. The publicly available ChIP-seq data used in this study are available in the ReMap 2020 database (https://remap2020.univ-amu.fr/download_page). Segmented copy-number data, determined by analysis of the Affymetrix genome-wide human SNP array 6.0, were retrieved from the Broad Institute TCGA Genome Data Analysis Center (2016-01-28 release date, using RTCGA package, version 1.16.0). The remaining data are available within the article, Supplementary Information, or Source Data file. Computer code, software versions, and the computing environment for reproducing results from this study are available in the GitHub repository at https://github.com/cancer-genomics/reproduce_liver_final.
Supplementary Material
Supplementary Table S1 shows Clinical information of US/EU patients analyzed. Supplementary Table S2 is a summary of whole genome cfDNA analyses. Supplementary Table S3 shows clinical information of Hong Kong patients analyzed. Supplementary Table S4 shows the lowest scoring transcription factors in US/EU cohort samples. Supplementary Table S5 lists the plasma volume and cfDNA amounts used in genomic libraries.
Supplementary Figure S1 shows contributions of individual genomic regions to the final trained DELFI surveillance model. Supplementary Figure S2 shows DELFI scores in individuals without cancer by age and sex. Supplementary Figure S3 shows DELFI scores in high-risk individuals without cancer across racial or ethnic groups. Supplementary Figure S4 shows DELFI scores in cirrhotic patients in relation to severity. Supplementary Figure S5 shows DELFI scores in individuals in relation to BMI. Supplementary Figure S6 Shows Performance of alternative DELFI models. Supplementary Figure S7 shows Correlation between rank ordered DELFI. Supplementary Figure S8 shows DELFI scores in patients with HCC were related to tumor characteristics. Supplementary Figure S9 shows DELFI scores in by etiology of underlying liver disease. Supplementary Figure S10 shows the DELFI score in patients with HCC is correlated to AFP levels. Supplementary Figure S11 shows the correlation of fragmentation profiles. Supplementary Figure S12 shows Genome wide fragmentation profiles in the Hong Kong validation cohort Supplementary Figure S13 shows chromosomal copy number in Hong Kong cohort. Supplementary Figure S14 shows the implementation of DELFI in liver cancer screening.
Acknowledgments
This work was supported in part by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Stand Up To Cancer–Dutch Cancer Society International Translational Cancer Research Dream Team Grant (SU2C–AACR–DT1415), the Gray Foundation, the Commonwealth Foundation, Stand Up To Cancer–LUNGevity–American Lung Association Lung Cancer Interception Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT23-17), the Mark Foundation for Cancer Research, the Cole Foundation, a research grant from Delfi Diagnostics, NIH grants CA121113, CA006973, CA233259, GM136577, CA237624, CA062924, DA036297, and DA012568, and a Department of Defense CDMRP Award W81XWH-20-1-0605. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated SU2C grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. We thank individuals from our laboratories for critical review of this work. This study makes use of data from individuals without cancer collected through the Endoscopy III and the COCOS trials as previously reported (23). The results published here are in part based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and the Genotype-Tissue Expression (GTEx) Project. GTEx was supported by the Common Fund of the Office of the Director of the NIH, and by the NCI, the National Human Genome Research Institute, the National Heart, Lung, and Blood Institute, the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke. This study makes use of data generated by The Chinese University of Hong Kong (CUHK) Circulating Nucleic Acids Research Group, as reported by Peiyong Jiang and colleagues in Proc Natl Acad Sci U S A (27).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
Z.H. Foda reports a patent for detecting liver cancer using cfDNA fragmentation pending. A.V. Annapragada reports a patent for detecting liver cancer using cfDNA fragmentation pending. D.C. Bruhm reports a patent for 63/423,003 pending, a patent for 63/290,017 pending and licensed to Delfi Diagnostics, and a patent for PCT/US2021/0646 pending and licensed to Delfi Diagnostics. D. Mathios reports a patent for detection of lung cancer using cfDNA fragmentation pending. S. Cristiano reports other support from Delfi Diagnostics outside the submitted work. R.A. Anders reports grants and other support from Bristol Myers Squibb, other support from Merck SD, AstraZeneca, and GSK, and grants from RAPT Therapeutics outside the submitted work. D.L. Thomas reports personal fees from Merck, Excision Bio, and UpToDate outside the submitted work. G.D. Kirk reports grants from the NIH during the conduct of the study. V. Adleff reports personal fees from Delfi Diagnostics outside the submitted work, as well as a patent for cfDNA for assessing and/or treating cancer and related patents pending, issued, licensed, and with royalties paid from Delfi Diagnostics. J. Phallen reports other support from Delfi Diagnostics during the conduct of the study, as well as a patent for cfDNA for assessing and/or treating cancer pending, licensed, and with royalties paid from Delfi Diagnostics. R.B. Scharpf reports grants and personal fees from Delfi Diagnostics outside the submitted work; a patent for US-2022-0325343 licensed to Delfi Diagnostics; and is a founder of and holds equity in Delfi Diagnostics, and serves as the head of Data Science. This arrangement has been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies. A.K. Kim reports grants and personal fees from AstraZeneca and personal fees from Exelixis outside the submitted work. V.E. Velculescu reports grants, personal fees, and other support from Delfi Diagnostics during the conduct of the study; other support from Viron Therapeutics and Epitope outside the submitted work; patent applications related to early detection of cancer pending, issued, licensed, and with royalties paid from Delfi Diagnostics; and is a founder of Delfi Diagnostics, serves on the Board of Directors and as an officer of Delfi Diagnostics, and owns Delfi Diagnostics stock, which is subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics. V.E. Velculescu divested his equity in Personal Genome Diagnostics (PGDx) to LabCorp in February 2022. V.E. Velculescu is an inventor on patent applications submitted by Johns Hopkins University related to cancer genomic analyses and cfDNA for cancer detection that have been licensed to one or more entities, including Delfi Diagnostics, LabCorp, Qiagen, Sysmex, Agios, Genzyme, Esoterix, Ventana, and ManaT Bio. Under the terms of these license agreements, the university and inventors are entitled to fees and royalty distributions. V.E. Velculescu is an adviser to Viron Therapeutics and Epitope. These arrangements have been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies. No disclosures were reported by the other authors.
Authors’ Contributions
Z.H. Foda: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A.V. Annapragada: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. K. Boyapati: Formal analysis, investigation, methodology, writing–review and editing. D.C. Bruhm: Data curation, software, validation, investigation, methodology, writing–review and editing. N.A. Vulpescu: Software, validation, investigation, methodology, writing–review and editing. J.E. Medina: Validation, investigation, methodology, writing–review and editing. D. Mathios: Software, validation, investigation, methodology, writing–review and editing. S. Cristiano: Software, validation, methodology, writing–review and editing. N. Niknafs: Software, methodology, writing–review and editing. H.T. Luu: Resources, methodology, writing–review and editing. M.G. Goggins: Resources, writing–review and editing. R.A. Anders: Resources, writing–review and editing. J. Sun: Conceptualization, resources, investigation, writing–review and editing. S.H. Mehta: Resources, writing–review and editing. D.L. Thomas: Resources, writing–review and editing. G.D. Kirk: Conceptualization, resources, investigation, writing–review and editing. V. Adleff: Resources, data curation, methodology, writing–review and editing. J. Phallen: Software, validation, methodology, writing–review and editing. R.B. Scharpf: Resources, data curation, supervision, investigation, visualization, methodology, writing–review and editing. A.K. Kim: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing. V.E. Velculescu: Conceptualization, resources, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing.
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 3. Di Bisceglie AM. Hepatitis C and hepatocellular carcinoma. Hepatology 1997;26(3Suppl 1):34S–8S. [DOI] [PubMed] [Google Scholar]
- 4. Pinyopornpanish K, Khoudari G, Saleh MA, Angkurawaranon C, Pinyopornpanish K, Mansoor E, et al. Hepatocellular carcinoma in nonalcoholic fatty liver disease with or without cirrhosis: a population-based study. BMC gastroenterology 2021;21:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol 2002;155:323–31. [DOI] [PubMed] [Google Scholar]
- 6. Waly Raphael S, Yangde Z, Yuxiang C. Hepatocellular carcinoma: focus on different aspects of management. ISRN Oncol 2012;2012:421673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70:151–71. [DOI] [PubMed] [Google Scholar]
- 8. Frenette CT, Isaacson AJ, Bargellini I, Saab S, Singal AG. A practical guideline for hepatocellular carcinoma screening in patients at risk. Mayo Clin Proc Innov Qual Outcomes 2019;3:302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology 2019;157:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singal AG, Pillai A, Tiro J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta-analysis. PLoS Med 2014;11:e1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singal AG, Yopp A, Skinner CS, Packer M, Lee WM, Tiro JA. Utilization of hepatocellular carcinoma surveillance among American patients: a systematic review. J Gen Intern Med 2012;27:861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singal AG, Li X, Tiro J, Kandunoori P, Adams-Huet B, Nehra MS, et al. Racial, social, and clinical determinants of hepatocellular carcinoma surveillance. Am J Med 2015;128:90.e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, et al. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta-analysis. Gastroenterology 2018;154:1706–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benesova L, Belsanova B, Suchanek S, Kopeckova M, Minarikova P, Lipska L, et al. Mutation-based detection and monitoring of cell-free tumor DNA in peripheral blood of cancer patients. Anal Biochem 2013;433:227–34. [DOI] [PubMed] [Google Scholar]
- 15. Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A 2015;112:E1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cai J, Chen L, Zhang Z, Zhang X, Lu X, Liu W, et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut 2019;68:2195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu RH, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16:1155–61. [DOI] [PubMed] [Google Scholar]
- 18. Wang Y, Zhou K, Wang X, Liu Y, Guo D, Bian Z, et al. Multiple-level copy number variations in cell-free DNA for prognostic prediction of HCC with radical treatments. Cancer Sci 2021;112:4772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kisiel JB, Dukek BA, VSRK R, Ghoz HM, Yab TC, Berger CK, et al. Hepatocellular carcinoma detection by plasma methylated DNA: discovery, phase I pilot, and phase II clinical validation. Hepatology 2019;69:1180–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalasani NP, Ramasubramanian TS, Bhattacharya A, Olson MC, Edwards VD, Roberts LR, et al. A novel blood-based panel of methylated DNA and protein markers for detection of early-stage hepatocellular carcinoma. Clin Gastroenterol Hepatol 2021;19:2597–605. [DOI] [PubMed] [Google Scholar]
- 21. Klein E, Richards D, Cohn A, Tummala M, Lapham R, Cosgrove D, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol 2021;32:1167–77. [DOI] [PubMed] [Google Scholar]
- 22. Chalasani NP, Porter K, Bhattacharya A, Book AJ, Neis BM, Xiong KM, et al. Validation of a novel multitarget blood test shows high sensitivity to detect early stage hepatocellular carcinoma. Clin Gastroenterol H 2022;20:173–82. [DOI] [PubMed] [Google Scholar]
- 23. Mathios D, Johansen JS, Cristiano S, Medina JE, Phallen J, Larsen KR, et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun 2021;12:5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X, Wang Z, Tang W, Wang X, Liu R, Bao H, et al. Ultrasensitive and affordable assay for early detection of primary liver cancer using plasma cell-free DNA fragmentomics. Hepatology 2022;76:317–29. [DOI] [PubMed] [Google Scholar]
- 26. Chen L, Abou-Alfa GK, Zheng B, Liu JF, Bai J, Du LT, et al. Genome-scale profiling of circulating cell-free DNA signatures for early detection of hepatocellular carcinoma in cirrhotic patients. Cell Res 2021;31:589–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang P, Sun K, Tong YK, Cheng SH, Cheng THT, Heung MMS, et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci U S A 2018;115:E10925–E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol 2015;16:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi JK, Kim YJ. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat Genet 2009;41:498–503. [DOI] [PubMed] [Google Scholar]
- 30. Ulz P, Perakis S, Zhou Q, Moser T, Belic J, Lazzeri I, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 2019;10:4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016;164:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheneby J, Menetrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, et al. ReMap 2020: a database of regulatory regions from an integrative analysis of human and arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res 2020;48:D180–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bejjani F, Evanno E, Zibara K, Piechaczyk M, Jariel-Encontre I. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys Acta Rev Cancer 2019;1872:11–23. [DOI] [PubMed] [Google Scholar]
- 34. Gozdecka M, Lyons S, Kondo S, Taylor J, Li Y, Walczynski J, et al. JNK suppresses tumor formation via a gene-expression program mediated by ATF2. Cell Rep 2014;9:1361–74. [DOI] [PubMed] [Google Scholar]
- 35. Yan P, Zhou B, Ma Y, Wang A, Hu X, Luo Y, et al. Tracking the important role of JUNB in hepatocellular carcinoma by single‑cell sequencing analysis. Oncol Lett 2020;19:1478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coto-Llerena M, Tosti N, Taha-Mehlitz S, Kancherla V, Paradiso V, Gallon J, et al. Transcriptional enhancer factor domain family member 4 exerts an oncogenic role in hepatocellular carcinoma by hippo-independent regulation of heat shock protein 70 family members. Hepatol Commun 2021;5:661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Z, Fang X, Xie G, Zhu J. GATA3 is downregulated in HCC and accelerates HCC aggressiveness by transcriptionally inhibiting slug expression. Corrigendum in/10.3892/ol. 2021.12836. Oncol Lett 2021;21:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang X, Hua L, Yan D, Zhao F, Liu J, Zhou H, et al. Overexpression of PCBP2 contributes to poor prognosis and enhanced cell growth in human hepatocellular carcinoma. Oncol Rep 2016;36:3456–64. [DOI] [PubMed] [Google Scholar]
- 39. Xiang X, Fu Y, Zhao K, Miao R, Zhang X, Ma X, et al. Cellular senescence in hepatocellular carcinoma induced by a long non-coding RNA-encoded peptide PINT87aa by blocking FOXM1-mediated PHB2. Theranostics 2021;11:4929–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen M, Li S, Zhao Y, Liu Y, Liu Z, Huan L, et al. Hepatic ARID3A facilitates liver cancer malignancy by cooperating with CEP131 to regulate an embryonic stem cell-like gene signature. Cell Death Dis 2022;13:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marchio A, Pineau P, Meddeb M, Terris B, Tiollais P, Bernheim A, et al. Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients. Oncogene 2000;19:3733–8. [DOI] [PubMed] [Google Scholar]
- 42. Longerich T, Mueller MM, Breuhahn K, Schirmacher P, Benner A, Heiss C. Oncogenetic tree modeling of human hepatocarcinogenesis. Int J Cancer 2012;130:575–83. [DOI] [PubMed] [Google Scholar]
- 43. Stewart SL, Kwong SL, Bowlus CL, Nguyen TT, Maxwell AE, Bastani R, et al. Racial/ethnic disparities in hepatocellular carcinoma treatment and survival in California, 1988–2012. World J Gastroenterol 2016;22:8584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gupta S, Bent S, Kohlwes J. Test characteristics of alpha-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C. A systematic review and critical analysis. Ann Intern Med 2003;139:46–50. [DOI] [PubMed] [Google Scholar]
- 45. Zhao C, Jin M, Le RH, Le MH, Chen VL, Jin M, et al. Poor adherence to hepatocellular carcinoma surveillance: a systematic review and meta-analysis of a complex issue. Liver Int 2018;38:503–14. [DOI] [PubMed] [Google Scholar]
- 46. Bokhorst LP, Alberts AR, Rannikko A, Valdagni R, Pickles T, Kakehi Y, et al. Compliance rates with the Prostate Cancer Research International Active Surveillance (PRIAS) protocol and disease reclassification in noncompliers. Eur Urol 2015;68:814–21. [DOI] [PubMed] [Google Scholar]
- 47. Duffy MJ, van Rossum LG, van Turenhout ST, Malminiemi O, Sturgeon C, Lamerz R, et al. Use of faecal markers in screening for colorectal neoplasia: a European group on tumor markers position paper. Int J Cancer 2011;128:3–11. [DOI] [PubMed] [Google Scholar]
- 48. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J Hepatol 2020;72:250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358–80. [DOI] [PubMed] [Google Scholar]
- 50. Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004;130:417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goh SK, Do H, Testro A, Pavlovic J, Vago A, Lokan J, et al. The measurement of donor-specific cell-free DNA identifies recipients with biopsy-proven acute rejection requiring treatment after liver transplantation. Transplant Direct 2019;5:e462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–i90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31:2032–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017;8:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trierweiler C, Hockenjos B, Zatloukal K, Thimme R, Blum H, Wagner E, et al. The transcription factor c-JUN/AP-1 promotes HBV-related liver tumorigenesis in mice. Cell Death Differ 2016;23:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qu C, Wang Y, Wang P, Chen K, Wang M, Zeng H, et al. Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy. Proc Natl Acad Sci U S A 2019;116:6308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hasenfuss SC, Bakiri L, Thomsen MK, Hamacher R, Wagner EF. Activator protein 1 transcription factor Fos-related antigen 1 (Fra-1) is dispensable for murine liver fibrosis, but modulates xenobiotic metabolism. Hepatology 2014;59:261–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 shows Clinical information of US/EU patients analyzed. Supplementary Table S2 is a summary of whole genome cfDNA analyses. Supplementary Table S3 shows clinical information of Hong Kong patients analyzed. Supplementary Table S4 shows the lowest scoring transcription factors in US/EU cohort samples. Supplementary Table S5 lists the plasma volume and cfDNA amounts used in genomic libraries.
Supplementary Figure S1 shows contributions of individual genomic regions to the final trained DELFI surveillance model. Supplementary Figure S2 shows DELFI scores in individuals without cancer by age and sex. Supplementary Figure S3 shows DELFI scores in high-risk individuals without cancer across racial or ethnic groups. Supplementary Figure S4 shows DELFI scores in cirrhotic patients in relation to severity. Supplementary Figure S5 shows DELFI scores in individuals in relation to BMI. Supplementary Figure S6 Shows Performance of alternative DELFI models. Supplementary Figure S7 shows Correlation between rank ordered DELFI. Supplementary Figure S8 shows DELFI scores in patients with HCC were related to tumor characteristics. Supplementary Figure S9 shows DELFI scores in by etiology of underlying liver disease. Supplementary Figure S10 shows the DELFI score in patients with HCC is correlated to AFP levels. Supplementary Figure S11 shows the correlation of fragmentation profiles. Supplementary Figure S12 shows Genome wide fragmentation profiles in the Hong Kong validation cohort Supplementary Figure S13 shows chromosomal copy number in Hong Kong cohort. Supplementary Figure S14 shows the implementation of DELFI in liver cancer screening.