


Abstract
In Escherichia coli, replication of both strands of genomic DNA is carried out by a single replicase—DNA polymerase III holoenzyme (pol III HE). However, in certain genetic backgrounds, the low-fidelity TLS polymerase, DNA polymerase V (pol V) gains access to undamaged genomic DNA where it promotes elevated levels of spontaneous mutagenesis preferentially on the lagging strand. We employed active site mutants of pol III (pol IIIα_S759N) and pol V (pol V_Y11A) to analyze ribonucleotide incorporation and removal from the E. coli chromosome on a genome-wide scale under conditions of normal replication, as well as SOS induction. Using a variety of methods tuned to the specific properties of these polymerases (analysis of lacI mutational spectra, lacZ reversion assay, HydEn-seq, alkaline gel electrophoresis), we present evidence that repair of ribonucleotides from both DNA strands in E. coli is unequal. While RNase HII plays a primary role in leading-strand Ribonucleotide Excision Repair (RER), the lagging strand is subject to other repair systems (RNase HI and under conditions of SOS activation also Nucleotide Excision Repair). Importantly, we suggest that RNase HI activity can also influence the repair of single ribonucleotides incorporated by the replicase pol III HE into the lagging strand.
INTRODUCTION
Escherichia coli DNA replication is carried out by the single replicase, DNA polymerase III holoenzyme (pol III HE), which is a multi-subunit complex responsible for simultaneous and coordinated replication of the leading and lagging DNA strands (1,2). pol III HE contains 2–3 catalytic cores, and each core comprises three proteins: α polymerizing subunit (encoded by the dnaE gene), ϵ 3′→5′ exonucleolytic proofreading subunit (dnaQ), and θ stabilizing subunit (holE) (1,2). While the leading strand is believed to be replicated in a nearly continuous manner, the lagging strand is synthesized as ∼1000 nucleotide (nt) Okazaki fragments, each starting with a ∼10 nt RNA primer (3). Okazaki fragment maturation is performed by a repair polymerase, DNA polymerase I (pol I). RNase HI was suggested to play an auxiliary role during Okazaki primer processing (4,5). There are three other DNA polymerases in E. coli which play accessory roles: DNA polymerase II (pol II) is a high-fidelity polymerase that serves as a backup replicase, while DNA polymerases IV and V (pol IV, pol V) are low fidelity polymerases mainly responsible for translesion synthesis (TLS) (6).
Despite the fact that both DNA strands in E. coli are replicated by the same replicase, the rates of mutations on both DNA strands in wild-type E. coli cells are unequal, with the lagging strand being synthesized with higher fidelity (7). Recent studies demonstrated that the frequency of dissociation of the major replicase from the DNA primer terminus (8,9) is an important factor responsible for the observed strand bias. Higher frequency of dissociation from the free primer terminus on the lagging strand enables additional options for error removal (e.g. by pol III’s innate proofreading activity) (6,8). On the other hand, the accessibility of 3′ primer termini on the lagging strand to other polymerases provides them an opportunity to replicate regions of undamaged DNA and thereby leading to a significant change in spontaneous mutagenesis (10–14).
Apart from ensuring correct base-pairing during DNA synthesis, DNA polymerases must also select nucleotides with the right sugar (15). Most DNA polymerases are particularly adept at excluding ribonucleotides from being incorporated into DNA. However, due to their vast intracellular abundance over deoxynucleotide pools, ribonucleotides (rNMPs) misincorporated by DNA polymerases during replication are believed to be the most common endogenous lesions in DNA, which if not removed, can lead to severe consequences in all organisms (16,17). On the other hand, ribonucleotides misincorporated by DNA polymerases can serve as excellent markers to detect the participation of particular polymerases and/or repair enzymes on a genome-wide scale (18).
In many polymerases, the efficacy of ribonucleotide discrimination relies on a single amino acid residue in the active site of the DNA polymerase (termed the ‘steric gate’), whose side chain clashes with the 2′-OH of the incoming ribonucleotide, limiting the rate of its incorporation into DNA (15,19). In E. coli replicase's polymerizing α subunit this activity is provided by Histidine 760 (20,21), while in pol V, which is comprised of UmuD'2C, the steric gate residue is Tyrosine 11 in the catalytic UmuC subunit (22). Mutant DNA polymerases with single amino acid substitutions in either the steric gate (pol V_Y11A) or an adjacent amino acid (pol IIIα_S759N) can readily incorporate ribonucleotides into DNA both in vitro and in vivo (21,22). The usage of such mutant variants helped uncover specific cellular pathway responsible for ribonucleotide recognition and removal from DNA, called ribonucleotide excision repair (RER). This system was shown to be necessary to maintain genome stability and proper functioning of both prokaryotic and eukaryotic cells (23,24).
The primary enzyme involved in RER is ribonucleotide-specific endonuclease, ribonuclease HII (RNase HII), encoded by the rnhB gene in procaryotes (25,26). Unlike its eukaryotic counterpart, which is capable of recognizing all kinds of RNA/DNA hybrids, bacterial RNase HII requires the presence of an RNA-DNA junction and cleaves at the 5′-side of the rNMP (27). The lack of RNase HII has no phenotypic effect in otherwise wild-type bacterial cells, unlike in eukaryotic cells, which exhibit multiple defects resulting from increased genetic instability (16). Another enzyme capable of detecting and removing rNMPs from DNA in bacterial cells is RNase HI (encoded by the rnhA gene), however its role in RER is limited due to substrate specificity. It can recognize DNA/RNA hybrids as well as chimeric DNA containing at least 3–4 consecutive rNMPs, cleaving within the RNA segment in a distributive manner at the 3′-side of the rNMP (27,28). It is believed that this enzyme does not recognize single rNs, but one in vitro study suggests that in collaboration with RNase HII it can be involved in excision of single ribonucleotides (29). Additionally, in the presence of a 3′ overhang in the opposite strand, RNase HI can work as a processive exoribonuclease, which might help with its role in Okazaki primer processing (30). However, the major cellular function of RNase HI in vivo is the removal of R-loops formed during rehybridization of RNA transcripts to DNA template (31,32). Additionally, in bacteria, RNase HI is involved in replication of ColE1-type plasmids as well as replication termination (33,34). In eukaryotic cells, it participates in telomere replication, while in mitochondria, it plays a role during the removal of DNA replication primers as well as separation of mtDNA (35,36). The lack of RNase HI leads to severe phenotypic changes in E. coli, e.g., slower growth or unscheduled initiation of replication, called constitutive stable DNA replication (cSDR) (37,38). E. coli mutants deficient in both RNase HI and HII exhibit stronger negative effects related to accumulation of DNA lesions containing rNMPs and replication stress (38,39).
Our understanding of the molecular mechanisms of RER in E. coli cells was expanded due to the usage of pol V_Y11A, as well as the steric gate variant of pol V’s homolog, pol VR391 (Y13A) (22,40). These studies revealed that RNase HII plays a key role in RER, with pol I being responsible for gap-filling synthesis during the repair process (41). When RNase HII-dependent RER is inactive, RNase HI-RER and NER can serve as back-up repair systems (17,41).
In this study, using active site variants of two differentially acting DNA polymerases; pol IIIα_S759N variant of the major replicase (21) and the Y11A steric gate variant error-prone pol V (10,21,22,42–48), we aimed to uncover whether the efficiency of the known RER pathways (RNase HII, RNase HI, and NER) differs between the leading and lagging DNA strands during genome duplication in E. coli. We provide evidence that there is a RER strand bias in E. coli which is related to the different efficiency of ribonucleotide repair systems on the leading and lagging strand that persists from normal replication to TLS.
MATERIALS AND METHODS
Escherichia coli strains used in the study
The E. coli strains used in this study are listed in Table 1.
Table 1.
Escherichia coli K-12 strains used in this study
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| BW7261 | Hfr(PO2A) leu-63::Tn10 fhuA22 Δ(argF-lac)169 ompF627 relA1 spoT1 | CGSC#6787 |
| RW1448a | rnhB + rnhA + uvrA + | RW698c x BW7261 |
| RW1450a | ΔrnhB782 | RW970c x BW7261 |
| RW1632a | rnhA339::cat | RW1044c x BW7261 |
| EC10713a | ΔuvrA753::Kan | RW1448 x RW1634c |
| EC10714a | ΔrnhB782 ΔuvrA753::Kan | RW1450 x RW1634c |
| RW1636a | ΔrnhB782::Kan rnhA339::cat | RW1092c x BW7261 |
| EC10459 | as RW1448 but galK+ | RW1448 x MC4100d |
| EC10460 | as RW1450 but galK+ | RW1450 x MC4100d |
| EC10534 | as EC10459 but attB::lacIZYA(L) | EC10459 x EC9429d |
| EC10535 | as EC10459 but attB::lacIZYA(R) | EC10459 x EC9428d |
| EC10536 | as EC10460 but attB::lacIZYA(L) | EC10460 x EC9429d |
| EC10537 | as EC10460 but attB::lacIZYA(R) | EC10460 x EC9428d |
| EC9882 | as RW1448 but attB::lacIZYA(CC105)L | RW1448 x EC3138e |
| EC9883 | as RW1448 but attB::lacIZYA(CC105)R | RW1448 x EC3144e |
| EC9886 | as RW1450 but attB::lacIZYA(CC105)L | RW1450 x EC3138e |
| EC9887 | as RW1450 but attB::lacIZYA(CC105)R | RW1450 x EC3144e |
| EC10212 | as RW1632 but attB::lacIZYA(CC105)L | RW1632 x EC3138e |
| EC10213 | as RW1632 but attB::lacIZYA(CC105)R | RW1632 x EC3144e |
| EC10522 | as EC10713 but attB::lacIZYA(CC105)L | EC10713 x EC3138e |
| EC10523 | as EC10713 but attB::lacIZYA(CC105)R | EC10713 x EC3144e |
| EC10524 | as EC10714 but attB::lacIZYA(CC105)L | EC10714 x EC3138e |
| EC10525 | as EC10714 but attB::lacIZYA(CC105)R | EC10714 x EC3144e |
| EC10216 | as RW1636 but attB::lacIZYA(CC105)L | RW1636 x EC3138e |
| EC10217 | as RW1636 but attB::lacIZYA(CC105)R | RW1636 x EC3144e |
| RW1628b | dnaE + | (21) |
| RW1612b | dnaE_S759N | (21) |
| RW1630b | dnaE + ΔrnhB782::Kan | (21) |
| RW1718b | dnaE_S759N ΔrnhB782::Kan yafC502::Tn10 | (21) |
| RW1624b | dnaE_S759T ΔrnhB782 ΔyafC727::Kan | (21) |
| RW1736b | dnaE_S759C ΔrnhB782::Kan yafC502::Tn10 | (21) |
| RW1620b | dnaE + ΔrnhB782 dnaQ920 ΔyafC727::Kan | (21) |
| EC10545b | dnaE_S759N ΔrnhB782::Kan dnaQ920 yafC502::Tn10 | (21) |
| EC10426b | dnaE + ΔrnhB782::Kan rnhA339::cat | RW1630 x RW1632 |
| EC10427b | dnaE_S759N ΔrnhB782::Kan yafC502::Tn10 rnhA339::cat | RW1630 x RW1632 |
a thr-1 araD139 Δ(argF-lac)169 tsx-33 supE44 galK2 hisG4 rpsL31 xyl-5 mtl-1 argE3 thi-1 sulA211 lexA51(Def) recA730 Δ(umuDC)596::ermGT ΔdinB61::ble.
b thr-1 Δ(argF-lac)169 tsx-33 supE44 galK2 hisG4 rpsL31 xyl-5 mtl-1 argE3 thi-1 sulA211 Δ(umuDC)596::ermGT ΔdinB61::ble ΔaraD-polB::ΩSpc.
cReferenced in (41).
dReferenced in (8).
eReferenced in (7).
The strains for lacI and lacZ assays using pol V and pol V_Y11A mutant
The strains used were derived from isogenic recA730 lexA51(Def) ΔumuDC596::ermGT RER-deficient strains previously described (22). The full genotype of the RER-proficient ‘wild-type’ strain is: recA730 lexA51(Def) ΔumuDC596::ermGT thr-1 araD139 Δ(gpt-proA)62 lacY1 tsx-33 glnV44 galK2 hisG4 rpsL31 xyl-5 mtl-1 argE3 thi-1 sulA211. Note that these strains carry the lacY1 allele and should therefore be phenotypically Lac−. However, the lacY1 allele is leaky, and bacteria have a red color appearance when grown on MacConkey lactose plates (unpublished observations). The parental strains were therefore rendered Lac– by interrupted bacterial mating of recipient strains with the E. coli strain BW7261 (proA+ Δ(argF-lac)169; CGSC#6787 (49)). Sexductants were selected on Zeocin containing minimal agar plates lacking proline. Strains were confirmed to be Lac– by their light pink color when grown on MacConkey lactose plates.
For the lacI spectrum analysis, the synthetic carbon source phenyl-β-D-galactopyranoside is broken down by β-galactosidase into phenol and galactose. In order to be able to utilize galactose, the strains were made galK+ by P1virA-mediated transduction using MC4100 as a donor. We subsequently constructed pairs of strains via transduction, using a donor strain containing the lacIqZYA operon inserted into the phage λ attachment site in the two orientations (Left and Right) with respect to the origin of replication (8). The isolates were then transduced with plasmids pGB2-lac-kan, pRW134-lac-kan, or pJM963-lac-kan (see below).
For the lacZ reversion assay, we used a similar approach, but transduced the galK2 strains with a donor phage lysate containing lacZ alleles in two orientations (7). For the analysis of the leading and lagging strand mutagenesis described here, we utilized a lacZ missense allele that allows scoring of mutagenesis via reversion to Lac+ by an A·T→T·A transversion (50). The final step of strain construction was the transformation of recipient strains with low-copy-number plasmids expressing either wild-type pol V (pRW134), or the pol V_Y11A variant (pJM963), or the control vector pGB2.
Strains for HydEn-seq and alkali gel electrophoresis assays using dnaE active site mutants
The strains are derivatives of RW732 and share a common genotype: thr-1 Δ(argF-lac)169 tsx-33 glnV44 galK2 hisG4 rpsL31 xyl-5 mtl-1 argE3 thi-1 sulA211 ΔumuDC596::ermGT ΔdinB::ble Δ(araD-polB)::ΩSpc ΔrnhB782.
Media for bacterial culture cultivation
Solid and liquid media were prepared as described in (51). Minimal medium plates contained 0.5% glucose, 0.5% lactose, or 750 μg/ml p-gal as the carbon source and were supplemented with 200 μg/ml casamino acids, 25 μg/ml thiamine and 100 μg/ml amino acids (arginine, threonine, histidine, leucine, isoleucine, valine), if necessary. Antibiotics, when required during strain construction, were added at the following concentrations: ampicillin (50 μg/ml); kanamycin (50 μg/ml or 75 μg/ml, as stated); chloramphenicol (25 μg/ml); tetracycline (12.5 μg/ml); zeocin (25 μg/ml) or spectinomycin (50 μg/ml).
Construction of plasmids expressing a kanamycin resistance gene under the lac promoter
Expression of the lac operon genes is controlled by the presence of LacI repressor which binds to the lacO operator sequence that overlaps the lac promoter, blocking its transcription in the absence of lactose. lacO contains two strong mutational hot-spots at positions +5 and +6 (G→A and T→C, respectively) (52). Preliminary sequencing revealed that these mutations constitute ∼50% of all mutations occurring (data not shown).
To select against irrelevant lacO mutations, we introduced a kanamycin resistance gene under the lac promoter (plac-kan) along with the lacO operator sequence on a low-copy-number vector and then screened for kanamycin resistance (52). In this instance, when p-gal+ mutants arise due to mutation in the chromosomal lacO sequence, the LacI repressor remains wild-type and can bind to the plasmid copy of lacO, preventing the expression of the kanamycin resistance gene, and rendering the strains phenotypically KanS. In contrast, when a p-gal+ mutation occurs in the lacI gene, the mutant LacI protein binds neither chromosomal, nor plasmid copy of lacO, and the strains are KanR.
We designed a nucleic acid sequence containing the 165 nt long region upstream of lacZ and the Kanamycin resistance gene from pKD13 (with a point mutation removing the PstI restriction site) along with a 50 nt long downstream fragment. The fragment upstream of lacZ contains lacO1, lacP and lacO3 loci. PstI restriction sites were added on both sides and the whole fragment was synthesized as a custom service (Genscript). The plac-kan sequence was then sub-cloned into pGB2, pRW134 and pJM963. The correct orientation of the insert, such that transcription of plac-kan occurs in the opposite direction to the transcription of the Spectinomycin resistance gene which creates a head-on collision between the two transcription machineries, was verified by digestion with NcoI alone or in combination with PvuI. The new plasmids were named pGB2-lac-kan, pRW134-lac-kan and pJM963-lac-kan.
lacI mutagenesis and mutational spectra analysis
Fluctuation assay for mutation rate estimation
36 parallel cultures of the appropriate galK+ lacL/lacR strains were grown in 2 ml fresh LB with Spectinomycin overnight. 100 μl of –1 dilution was spread on MM + p-gal selective plates and 100 μl of –6 dilution was spread on MM + glc non-selective plates. The colonies were grown for 2–3 days at 37°C. Mutation rates were calculated using maximum likelihood estimation method and 95% confidence intervals using inverted likelihood ratio test with the R statistical package rSalvador (53,54).
Analysis of mutational spectra
We used the method described in (8) with modifications. 6–7 independent colonies for each strain were inoculated in 2 ml fresh LB with Spectinomycin and incubated overnight with shaking at 37°C. The next day, the cultures were diluted 104-fold, and 10 μl portions of the dilutions (∼103 cells) were re-inoculated into 400 μl fresh LB with Spectinomycin in 96-deep-well plates. The plates were incubated overnight with shaking at 37°C. 10–20 μl of the cell cultures was spread on the quarter-sectors of minimal plates supplemented with phenyl-β-D-galactopyranoside and kanamycin (75 μg/ml) using a sterile disposable 10 μl inoculating loop and incubated for 3 days at 37°C. After that time, single colonies closest to the center of the plate were re-streaked onto fresh minimal plates supplemented with p-gal and kanamycin (75 μg/ml) and grown for another 3 days at 37°C.
Single colonies from the latter plates were tooth-picked and used to PCR amplify the ∼1800 bp fragment comprising the lacI coding sequence using primers lacI133F (TGGGATCAGGAGGAGAAG) and lacO249R (ATGGGATAGGTCACGTTGG) (8). The PCR reaction protocol was as follows: 1 cycle of 95°C for 5 min; 30 cycles of: 95°C for 30 s, 64°C for 30 s, 72°C for 1 min; 1 cycle of 72°C for 7 min. DNA sequencing was performed using primers lacI1F (CATCTTCCGGCGCTACAACG) and lacI3R (AAACGACGGCCAGTGAATCC) as a custom service (TacGen). The contig assembly and analysis were performed using Sequencher 5.4.6 (GeneCodes). The results of the analyses are included in Supplementary Files S2 and S3.
lacZ reversion assay
lacZ mutation rates for each strain were determined as follows: two to three independent lacZ isolates for each orientation carrying the low-copy plasmids expressing either wild-type pol V (pRW134), or steric gate mutant pol V_Y11A (pJM963), or control plasmid pGB2 were used to assess the Lac+ revertant frequencies. Mutant frequencies for each strain were determined by growing 10–40 cultures from single colonies overnight at 37°C (in 2 ml of LB medium supplemented with Spectinomycin). 100 μl portions of the cultures were plated on MM + lac plates to determine the number of Lac+ mutants and 100 μl of –6 dilution was spread on MM + glc non-selective plates to determine the total cell count. The plates were incubated for 2 days at 37°C. Mutation rates were calculated using Maximum Likelihood Estimation method and 95% confidence intervals using inverted Likelihood Ratio Test with the R statistical package rSalvador (53,54). 95% confidence intervals for the relative mutagenesis levels were determined using profile likelihood method (55). The results for strains harboring control vector pGB2 were excluded from further analyzes due to negligible mutability in all tested genetic backgrounds.
HydEn-seq method
Cells were thawed from frozen stocks and streaked out on LB agar plates containing 50 μg/ml kanamycin, 25 μg/ml spectinomycin and 25 μg/ml zeocin. The next day, 5–6 colonies were picked and diluted in 2–3 ml LB medium supplemented with the aforementioned antibiotics and incubated at 37°C overnight. The culture was diluted to an OD600 = 0.05 in 25 ml LB containing kanamycin, spectinomycin and zeocin. After 2–3 hours, the cells reached exponential growth (OD600 = 0.4 – 0.6), at which time they were centrifuged to pellet and used as the starting material for the HydEn-seq method (18). Briefly, DNA extraction was performed using MasterPure™ Complete DNA and RNA Purification Kit (Lucigen, Epicentre, MC85200). After measuring the DNA concentration using the Qubit® dsDNA BR Assay Kit (Life Technologies, molecular probes, Q32853), 1 μg DNA was hydrolyzed using 0.3 M KOH by incubation at 55°C for 2 h. Ethanol-precipitated DNA was denatured by 3 min incubation at 85°C. The single strand DNA containing 5′-OH ends was phosphorylated using 3′-phosphatase minus T4 polynucleotide kinase (New England Biolabs, M0236L), incubated at 65°C for 20 min, washed with beads (GC biotech, CPCR-0050), denatured, ligated to ARC140 adaptor using T4 RNA Ligase 1 (New England Biolabs, M0204S), denatured, polymerized using T7 DNA Polymerase (New England Biolabs, M0274L) and ARC76/77 oligos, and bead-washed. KAPA HiFi hot-start ready-mix (2X) kit (KAPA biosystems, 07958935001) was used to amplify the libraries. Paired-end sequencing was performed using Illumina NextSeq 500 system.
Sequence data analysis
Trimming for quality and adaptor sequence of all reads was performed with cutadapt 1.12 (56) Pairs containing one, or both reads, shorter than 15 nt were discarded. Bowtie 1.2 (57) was used to align Mate 1 of all remaining pairs to the list of index primers used to prepare the libraries; all matching pairs were discarded. All remaining pairs were aligned with bowtie (-v2 –X2000-best) to the E. coli K12, DH10B reference genome build 2008-03-17 from NCBI. Single-end alignments were then performed for mate 1 of all unaligned pairs (-m1, -v2). The count of 5′-ends of all unique paired-end and single-end alignments was determined for all samples and shifted one base upstream to the location of the hydrolyzed ribonucleotide.
For visual comparison of individual libraries, end counts were normalized to counts per million uniquely mapped reads (divided by the total of uniquely mapped ends and multiplied by 106). To remove reads stemming from free 5′-ends, scaled end counts from strains with wild-type polymerases were subtracted from the steric gate mutants. For each position where both the forward and reverse strands presented numeric values, the forward strand value was divided by the sum of the forward and reverse strand values. If no reads were encountered in both the forward and reverse reads, a no value was returned. If the forward read was numeric, but the reverse read was not numeric, then 1 was returned, For the inverse scenario where the reverse read was numeric, but the forward read not, then a 0 was returned.
End-mapped reads were sorted into bins of 10 000 bp after background normalization using the wild-type strain. The average fraction of ends mapping to the top strand for positions 1 690 000 to 3 920 000—corresponding to the approximate region between TerB (1 684 227 to 1 684 247) and oriC (3 925 744 to 3 925 975) in MG1655, was calculated for three biological replicates and referred to as ‘TerB to oriC’. The average of all other positions (0 to 1 680 000 and 3 930 000 to 4 670 000) was also calculated and referred to as ‘oriC to TerB’. The results of the analysis are included in Supplementary File S4.
High-molecular-weight genomic DNA isolation
Genomic DNA for RNase cleavage assay was isolated using MasterPure™ Complete DNA and RNA Purification Kit (Lucigen, Epicentre, MC85200) according to a modified manufacturer's protocol. In brief, 300 μl overnight bacterial culture was centrifuged and suspended in 300 μl Tissue and Cell Lysis Solution with 1 μl manufacturer-supplemented Proteinase K and incubated at RT for 30 min. After that time the lysates were incubated for 10 min on ice, 150 μl MPC Protein Precipitation Reagent was added followed by another 10 min incubation on ice. The samples were centrifuged at 4°C for 20 min and the supernatant was transferred to a clean tube using a wide-bore pipette tip. 500 μl isopropanol was added, and the samples were centrifuged at 4°C for 25 min and washed with 70% ethanol. The dried pellet was re-suspended in 35 μl TE. DNA concentration was measured using the Quant-iT® dsDNA BR Assay Kit (Life Technologies, Q33130) in a Varioskan™ LUX microplate reader (Thermo Scientific).
Genomic DNA treatment
500 ng DNA was incubated in a 50 μl reaction volume for 3 h at 37°C with different enzymes: 2.5U RNase HII (NEB, M0288L) + 5 μg RNase A (Thermo Fisher, EN0531) in ThermoPol® Reaction Buffer (NEB) for estimating the total number of ribonucleotides; 2.5 U RNase H (NEB, M0297L) in RNase H Buffer (NEB) for RNase HI cleavage assay; 5 μg RNase A (Thermo Fisher, EN0531) in TE or TE with 300 mM NaCl) for RNase A cleavage assay. After incubation, the salt concentration was increased to 500 mM and DNA was precipitated using 50 μl isopropanol, followed by centrifugation at 4°C for 25 min. DNA was washed with 70% ethanol and the dried pellet was re-suspended in 20 μl TE.
Alkaline gel electrophoresis and determination of the number of genome-embedded ribonucleotides
Rehydrated DNA samples were mixed with 4 μl Alkaline Loading Dye (Alfa Aesar, J62157) and loaded into 0.7% agarose gel prepared in alkaline buffer (50 mM NaOH, 1 mM EDTA). Alkaline gel electrophoresis was run in alkaline buffer (50 mM NaOH, 1 mM EDTA) for 22 h at 1.5 V/cm. After separation, the gel was washed twice in neutralization buffer (1.5 M NaCl, 0.7 M Tris–Cl, pH 8.0) for 40 min and then stained for 30 min with SYBR™ Gold Nucleic Acid Gel Stain (Invitrogen, S11494). Image acquisition was done using FluorChem® Q MultiImage III Gel Imaging System (Alpha Innotech). Image analysis was done using ImageQuant™ TL 10.1 (Cytiva).
Determination of the number of ribonucleotides per chromosome was performed as described previously (21). To minimize the impact of variability in technical parameters (such as differences in voltage, mobility, buffer temperature, ladder separation, background levels in image acquisition, vignetting close to the edges of the image, etc.) on the estimates of the average number of ribonucleotides between different experiments, a paired t-test was used for the analysis of statistical significance.
RESULTS
The approach used to investigate strand-specific efficiency of ribonucleotide excision repair in Escherichia coli
In the present study, we aimed to determine the activity of RER pathways during two major replicative events: normal replication carried out by DNA polymerase III (using an active site mutant of E. coli replicase's polymerase subunit, pol IIIα_S759N), and pol V-dependent SOS mutator activity (using the steric gate mutant of E. coli major TLS polymerase, pol V_Y11A). We started with the analysis of the mutational activity of pol V and pol V_Y11A, which are error-prone, poorly processive polymerases with limited access to DNA replication (6,22,58). Because pol V replicates only short patches of DNA, we assume that its effect on the overall ribonucleotide content in the whole genome is rather small in comparison to the replicase, but at the same time, all of its errors (both ribonucleotides and mismatched deoxyribonucleotides) must be concentrated in these small patches of DNA. We can therefore expect that ribonucleotide excision repair will result in removal of not only ribonucleotides, but also neighboring mismatched nucleotides. Thus, we investigated ribonucleotide removal indirectly, via its effect on the repair of adjacent base mispairs in the reporter genes, as we have done previously (23,40,41). To this end, we analyzed pol V- and pol V_Y11A-dependent forward and reverse mutation rates in lacI and lacZ, respectively, inserted into the chromosome in two orientations, such that leading- and lagging-strand mutagenesis can be scored separately.
This assumption does not apply to the pol III mutant. pol IIIα_S759N incorporates roughly 1 mutation per 108 nucleotides in a proofreading-defective background and 1 ribonucleotide per 2.4 × 103 nucleotides (21). Thus, in the case of pol IIIα_S759N, ribonucleotides are unlikely to be present near mispairs. On the other hand, because pol III replicates the whole genome, ribonucleotides in DNA can be directly investigated on a genome-wide scale: we employed techniques allowing us to study the amount (RNase HII cleavage assay with alkaline gel electrophoresis), as well as the strand-specific distribution (HydEn-seq), of the ribonucleotides across the whole chromosome. The usage of techniques suited for the specific properties of pol III and pol V allowed us to therefore perform a comprehensive analysis of the efficiency of RER on both DNA strands.
Leading- and lagging-strand mutational spectra promoted by pol V and pol V_Y11A steric gate mutant
E. coli pol V is subject to multiple levels of regulation, such that its activity on undamaged DNA is normally kept to a minimum (58,59). However, much of this regulation is circumvented in strains harboring the recA730 allele, which encodes a mutant RecA protein (E38K), considered to be in a constitutively activated (RecA*) state required for pol V mutasome assembly (48). As a consequence, recA730 strains exhibit high levels of spontaneous mutagenesis that is further elevated by up to ∼50% when combined with a lexA51(Def) allele, encoding an inactive form of the SOS repressor LexA (45). Using the pol V_Y11A mutant, it was shown that bacterial RER can significantly influence the fidelity of DNA replication by removing not only ribonucleotides incorporated by the steric gate variant of pol V, but also mispaired deoxyribonucleotides inserted in their vicinity (22).
As uncovered by the analysis of the mutational spectra promoted in rpoB, as well as the whole E. coli chromosome, pol V replication is characterized by a high incidence of A·T→T·A transversions (44,45). Such events also dominate the rpoB spectrum of RER-deficient strains expressing pol V_Y11A steric gate mutant (23). As DNA mutations occur through the easier to extend Pyr·Pyr mismatches, pol V creates A·T→T·A transversions predominantly via T·T mispairs rather than A·A mispairs (7,10,60,61). This observation allows us to discriminate between leading- and lagging-strand synthesis in various genetic studies.
Here, we employed a recently developed genetic system that allows us to analyze the specificity of mutagenesis by scoring for forward mutations in the lacI reporter gene inserted into the chromosome in two orientations with regard to oriC (8,62) (Supplementary Figure S1A). Depending on its orientation, the lacI coding sequence is replicated as a leading strand in one strain and as a lagging strand in the other strain (Supplementary Figure S1B). Mutations spanning the ∼1,100 bp target that deactivate the LacI repressor allow for constitutive expression of β-galactosidase, and therefore also for growth on a synthetic carbon source phenyl-β-D-galactopyranoside (p-gal) (Supplementary Figure S1D, E).
For this purpose, we used AB1157 derivatives that: (a) carry chromosomal deletions of lac and umuDC operons, (b) carry recA730 lexA51(Def) alleles to ensure robust pol V activation and (c) were additionally made galK+ such that galactose, a product of p-gal metabolism, can be utilized as a carbon source. We subsequently introduced the lac operon into the lambda phage attachment site (attB) in one of two orientations, and then transformed strains with plasmids pGB2-lac-kan (vector), pRW134-lac-kan (wild-type pol V), or pJM963-lac-kan (Y11A steric gate mutant of pol V) (see Materials and Methods). Because constitutive expression of β-galactosidase can result from mutations in either lacI or lacO, the plasmids additionally carried Kanamycin resistance gene under the lac promoter, which ensured that we only scored lacI mutations (Supplementary Figure S1F) (52).
We analyzed mutation rates, as well as nucleotide changes in the lacI reporter gene in strains producing either pol V or pol V_Y11A by sequencing ∼2300 mutant lacI clones carrying the previously mentioned plasmids, with an active (rnhB+) or inactive (ΔrnhB) RNase HII-dependent RER pathway. Our study focused on A·T→T·A transversions, which are particularly responsive to SOS induction and characteristic for error-prone DNA replication by pol V (10,44,63). There are 68 A·T→T·A positions with 22 unique 3-nucleotide sequence contexts (Supplementary File S2), which we assume to be representative of the whole chromosome. The rate of lacI mutations in pol V-deficient E. coli is approximately ∼5 × 10–8 per locus per cell division (Table 2). Expression of pol V from a low-copy-number vector increases the mutagenesis 8- to 11–fold up to ∼4–6 × 10–7, regardless of the presence (rnhB+), or absence of RNase HII (ΔrnhB). This is consistent with the previously reported lack of phenotype caused by an RNase HII deficiency. Of all pol V mutations, ∼30% are A·T→T·A transversions, characteristic of the pol V-dependent spontaneous mutator effect (Figure 1, Supplementary File S2). In contrast, in strains expressing the pol V_Y11A steric gate mutant, the level of mutagenesis is ∼20% of that of wild-type pol V at ∼1 × 10–8 (Table 2), and the share of pol V-induced A·T→T·A mutations decreases to < 10%, along with other types of base-pair substitutions which now constitute less than 50% of all mutations (Figure 1). This reiterates previous observations in hisG4 and lacZ105 reversion assays, as well as in the rpoB mutational spectra showing that the steric gate mutations of pol V and its homolog, pol VR391 have an anti-mutagenic effect (22,40,41). The ‘anti-mutator’ phenotype has been attributed to stimulation of RER pathways which, during repair of a target ribonucleotide, also remove adjacent mismatched nucleotides (Supplementary Figure S1G) (41). This effect is specific to pol V, which is a highly mutagenic but poorly processive, distributive polymerase, meaning that its errors are concentrated in small fragments of DNA, and mismatched nucleotides located in the vicinity of ribonucleotides can be concomitantly proofread by pol I resynthesizing a portion of DNA after RNase HII-dependent incision of an rNMP-containing DNA. Indeed, the lack of RNase HII in strains expressing pol V_Y11A mutant not only increases the mutagenesis to levels comparable or slightly above that of wild-type pol V (120–150%, Table 2), but also restores the wild-type-like share of A·T→T·A transversions (Figure 1), all consistent with the lack of concomitant repair of mismatches by RER.
Table 2.
Rates of spontaneous lacI mutations in recA730 lexA51(Def) strains carrying plasmid-expressed genes encoding wild-type pol V or pol V_Y11A mutant
| Genotype | lacI orientation | Vector | wt pol V | pol V_Y11A |
|---|---|---|---|---|
| rnhB + | lacL | 3.77 (3.03–4.59) | 38.1 (34.2–42.0) | 11.3 (9.6–13.2) |
| lacR | 4.78 (3.86–5.78) | 56.4 (50.8–61.9) | 13.0 (10.9–15.2) | |
| ΔrnhB | lacL | 4.18 (3.40–5.03) | 38.2 (33.8–42.5) | 55.2 (48.9–61.3) |
| lacR | 5.22 (4.33–6.19) | 47.0 (40.8–53.2) | 54.4 (48.7–60.0) |
Mutation rate (×108) per lacI locus per cell division along with 95% confidence intervals in brackets was calculated as described in Materials and Methods using n = 36 cultures per strain.
Figure 1.
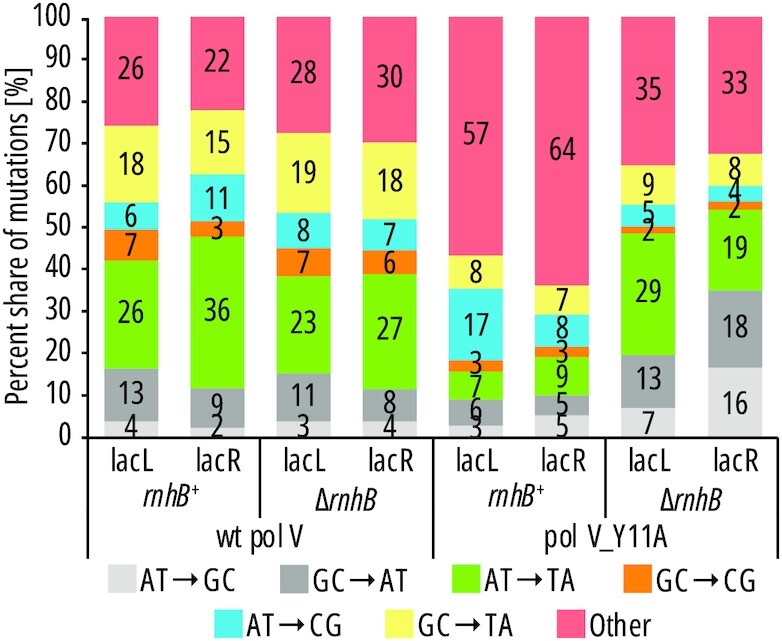
Share of different types of mutations in mutational spectra of pol V and pol V_Y11A. Base substitutions: transition mutations are in grey while transversions are in various colors. Other mutations: indels, complex mutations and IS element insertions.
Next, we were interested if the repair of ribonucleotides (observed indirectly by the levels of mutagenesis) incorporated by pol V_Y11A differs between the two DNA strands. We used the rationale described in (8) but focused on the most typical A·T→T·A transversions. In brief, pairs of strains denoted as lacL or lacR contain the lac operon inserted into the chromosome in either of the two orientations such that the coding sequence of lacI is replicated as a leading strand in the lacR strain and as a lagging strand the lacL strain (Supplementary Figure S1B). While the observed rate of A·T→T·A transversions is a sum of T·T or A·A events, based on the biochemical data described above, we can assume that these mutations occurred mainly via T·T mispairs. By analyzing on which strand the more frequent T·T mispair occurs, we could ascribe mutational events to either the leading- or lagging-strand. For example, if the lacI coding sequence contained T in a mutational hot spot, we assume that the mutation in this locus occurred during leading strand replication in the lacL strain and during lagging strand replication in the lacR strain, while the opposite is true for A in the coding sequence (Supplementary Figure S1C).
For the control strains with the wild-type pol V, the rate of T→A events (originating from a T·T mispair) is higher on the lagging than on the leading strand (Figure 2, first graph, 22.9 × 10–8 in lacR vs. 5.4 × 10–8 in lacL strains) and the same strand bias is observed in both RNase HII proficient (rnhB+) and deficient (ΔrnhB) backgrounds. The observed mutagenic effect of pol V on the lagging strand is consistent with previous observations in a recA730 background, where DNA pol V actively participates in preferential replication of the lagging strand (8,10). In rnhB+ strains expressing the steric gate variant pol V_Y11A, mutagenesis significantly decreases on both strands (Figure 2, third graph, to 0.6 × 10–8 and 1.0 × 10–8 in lacL and lacR strains, respectively). A more striking effect is observed on the lagging strand which can be explained by stronger activation of RER due to a higher content of ribonucleotides inserted by pol V on this strand. The lack of RNase HII (ΔrnhB) in Y11A strains resulted in an increase in mutagenesis on both DNA strands, but more so on the leading than on the lagging strand (Figure 2, fourth graph, 16.9 × 10–8 in lacL vs. 6.0 × 10–8 in lacR strains). The strand bias is reversed in comparison to that observed for the wild-type pol V and now the level of Y11A-dependent mutagenesis is higher (and above that of pol V) on the leading strand than on the lagging strand, where it is only partially restored. Overall, these results show that repair of ribonucleotides inserted by pol V_Y11A into both strands can be successfully tracked by analyzing its impact on the levels of mutagenesis promoted by this polymerase in SOS-induced E. coli cells. Furthermore, the differences in A·T→T·A leading- and lagging-strand mutation rates between pol V- and Y11A-producing strains suggests that the repair of ribonucleotides inserted into both DNA strands by pol V_Y11A might not be equal.
Figure 2.
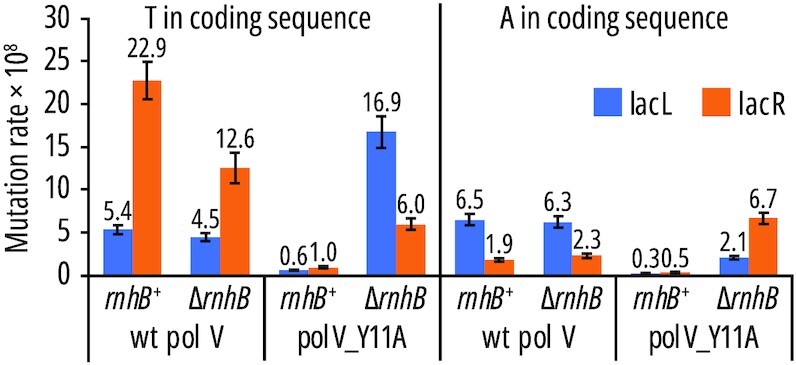
lacI A·T→T·A mutagenesis promoted by pol V and pol V_Y11A in lacL and lacR strains. Partial mutation rates were calculated using overall mutation rates from Table 1 and the frequencies of transversion events (separated into these occurring in lacI positions with either A or T in the coding sequence) from Supplementary File S2. Error bars represent 95% confidence intervals.
Leading- and lagging-strand mutagenesis promoted by pol V_Y11A steric gate mutant in various RER-deficient backgrounds
For a more precise analysis of how effective the particular RER pathways (RNase HII- and RNase HI-dependent RER, or NER) are in removing genome-embedded ribonucleotides from both DNA strands we employed a simpler lacZ assay that enables us to specifically measure the levels of the most characteristic pol V-dependent A·T→T·A transversions on both DNA strands by scoring for reversion mutations in the lacZ reporter gene inserted into the chromosome in two orientations with regard to oriC (7). We constructed pairs of recA730 lexA(Def) strains carrying a lacZ allele which reverts through the defined transversion and compared the mutation rates promoted by pol V and pol V_Y11A on the leading and lagging DNA strands in various RER-deficient backgrounds (Figure 3). Our analysis focused on the relative levels of pol V_Y11A mutagenesis, which is pol V_Y11A-dependent mutagenesis expressed as a percent of wild-type pol V-dependent mutagenesis in the isogenic strain. By always relating pol V_Y11A mutagenesis to wild-type pol V mutagenesis in an isogenic strain, we can disregard all factors except increased ribonucleotide incorporation that could potentially influence mutation rates in each genetic background under study (41). Therefore, we assume that any difference in mutation rates between pol V and pol V_Y11A can only be attributed to the repair of ribonucleotides, as well as adjacent mismatched nucleotides (Supplementary Figure S1G), or a lack thereof. Since pol V_Y11A is as error-prone, if not more so, than wild-type pol V (22), we assumed that any pol V_Y11A-dependent mutagenesis that is lower than that of wild-type pol V is indicative of active RER-dependent processes in vivo. In contrast, relative levels of pol V_Y11A mutagenesis higher than wild-type pol V would indicate that RER has been inactivated. This is because when there is no RER activity, neither ribonucleotides nor adjacent mispairs can be repaired, and the true extent of pol V_Y11A mutagenesis becomes exposed.
Figure 3.

Absolute and relative A·T→T·A mutation rates promoted by pol V and pol V_Y11A in various RER-deficient backgrounds. Table: A·T→T·A mutation rates (×109) per lacZ locus per cell division is shown in bold typeface with 95% confidence intervals in curly brackets. Chart: relative mutagenesis calculated by dividing pol V_Y11A mutagenesis by wild-type pol V mutagenesis in each isogenic strain. Error bars represent 95% confidence intervals.
As expected, in an rnhB+ background the presence of the pol V_Y11A significantly decreases mutagenesis on both DNA strands (Figure 3). This result is in agreement with previously reported phenotype of pol V_Y11A in a His+ reversion assay (41) and corroborates the lacI mutagenesis results described above, showing that the impact of pol V_Y11A on the fidelity of DNA replication is independent of the size of the mutational target. The extent of pol V_Y11A mutagenesis on the leading strand is 45% and on the lagging strand it is just 8% of that observed for wild-type pol V, indicating that at least 92% of the pol V_Y11A-dependent mutations are repaired in the lagging strand. The lower levels of mutagenesis promoted by the pol V_Y11A mutant on the lagging strand may be interpreted as the result of the greater accumulation of errant rNMPs incorporated by pol V_Y11A into the lagging strand and the subsequent extensive activation of RER pathways on this strand.
In RNase HII-deficient (ΔrnhB) strains, mutagenesis promoted by pol V_Y11A is completely restored on the leading strand and reaches ∼180% of wild-type pol V mutagenesis whereas on the lagging strand it increases only up to 20% of the values observed for wild-type pol V (Figure 3). Inactivation of either RNase HI-dependent RER (ΔrnhA) or NER (ΔuvrA) in rnhB+ strains does not significantly increase the mutagenesis on either DNA strand (Supplementary Table S1). These observations are in agreement with the hypothesis that the first line of defense against misincorporated ribonucleotides is RNase HII-mediated RER, and our current studies suggest that this occurs primarily on the leading strand, while repair on the lagging DNA strand remains efficient even upon RNase HII deletion (80% repair).
Inactivation of both RNases HII and HI further increases pol V_Y11A-dependent mutagenesis on both DNA strands, with a stronger effect on the lagging strand (from ∼176% to ∼434% on the leading strand and from ∼20% to ∼114% on the lagging strand). Thus, when RNase HII is inactivated, RNase HI promotes RER on both the leading and lagging DNA strands, with a slight preference for the lagging strand. Similarly, simultaneous defects in RNase HII and NER significantly increase the relative levels of pol V_Y11A-dependent mutagenesis on both DNA strands, to ∼365% mutagenesis on the leading strand and ∼92% mutagenesis on the lagging strand (Figure 3). Importantly, in both cases, lagging-strand relative mutagenesis is restored to levels comparable to wild-type pol V when the RNase HII deletion is combined with other RER pathway deficiencies. We conclude that when RNase HII is inactivated, RNase HI and NER promote ribonucleotide repair on both DNA strands, with a preference for the lagging strand, suggesting their overlapping roles in RER.
To summarize, our results from the lacZ system suggest that RNase HII primarily, but not exclusively, operates on the leading strand while the role of other repair systems (RNase HI-RER) and (NER-RER) is more important on the lagging DNA strand.
Tracking DNA synthesis by pol IIIα_S759N using HydEn-seq: strand specificity, origin of replication, and termination region identification
The above results, that suggest split roles of RNase HII and backup repair systems on both DNA strands during RER under conditions of constitutive SOS induction, are based on the usage of a pol V variant with a limited, distributive role in DNA replication and preferential access to the lagging DNA strand. It was therefore possible that at least a part of the observed effects resulted from our experimental setup in which we track ribonucleotide repair under specific conditions and only via their effect on leading- vs. lagging-strand mutagenesis, which depends on DNA polymerase selectivity. A question therefore arises whether strand-specific RER happens also during normal replication? To analyze ribonucleotide incorporation and removal from the E. coli chromosome under conditions of normal replication, we extended our analyses by employing an active site variant of the major replicase responsible for the replication of both DNA strands; pol IIIα_S759N, which was recently shown to incorporate ∼8× more ribonucleotides than the wild-type replicase (21). As pol III replicates the whole genome, we performed our analyses on a genome-wide scale.
HydEn-seq has been previously used to track the replication enzymology of variants of yeast replicases (pol α, δ and ϵ) that have an increased propensity to incorporate ribonucleotides into the S. cerevisiae genome (18). The technique allows for the identification of embedded ribonucleotides in DNA isolated from strains with defective RER. Using such an approach, pol ϵ was confirmed to be the main leading strand polymerase, with pol α and δ mainly contributing to lagging strand synthesis during S. cerevisiae genome duplication (18,64,65).
In this study, we used the same methodology to track pol IIIα enzymology of the E. coli replicase, pol III. Unlike eukaryotes, which have multiple origins of replication, E. coli replication is initiated at only one site, oriC, at an approximate position of 3926 kb (66) (Figure 4A). DNA replication occurs in a bidirectional manner in E. coli, and clockwise/counterclockwise forks meet each other ∼50 centisomes from oriC in a region called the terminus. Unlike the single ori site, several different replication terminators have been described in the terminus region. TerB, which is one of the most well-defined terminators in E. coli (67), is located at nucleotides 1 684 227–1 684 247 in MG1655 (Figure 4A).
Figure 4.

Genome-wide mapping of pol III strand-specific ribonucleotide incorporation. (A) A schematic representation of E. coli chromosome replication. oriC, the replication origin, as well as the termination region containing TerA, TerB and TerC are marked. (B, C) Results for pol IIIα_S759N active site mutant strains are shown with the fraction of end-mapped reads in bins of 10 000 bp after background normalization using the wild-type strain. The numbers depicted on the right side of the graphs show vertical viewing range. Average fraction of ends mapping to the top strand ± SD between oriC and TerB (green), or TerB and oriC (orange), is presented next to the figure. Results for two other dnaE mutants (dnaE_S759C and dnaE_S759T) are shown in Supplementary Figure S4A. (B) The dnaE_S759N mutant shows a strong strand switch that correlates to the positions of oriC and TerB. The average difference between the leading and lagging strands is 0.0207. Three biological replicates for each dnaE allele are presented in Supplementary Figure S3A. (C) The strand bias in the strain expressing dnaE_S759N mutant slightly decreases when proofreading is impaired (dnaQ920) to 0.0154 and is essentially abolished (0.0040) when in combination with an inactive RNase HI (rnhA339::cat). Three biological replicates of each strain are presented in Supplementary Figure S3B, C.
To track the activity of the replicase, we analyzed ribonucleotide incorporation of the dnaE_S759N variant, which was previously shown to incorporate a high number of ribonucleotides into the E. coli genome, as well as two other mutants, dnaE_S759C and dnaE_S759T, with higher sugar selectivity as controls (21). Interestingly, HydEn-seq analysis of the S759N mutant shows a clear strand bias with more ribonucleotides in the leading strand (Figure 4B; Table 3; Supplementary File S4). This was unexpected, since pol III replicates both strands equally (1,68–73).
Table 3.
Average number of ribonucleotides in the chromosomes of ΔrnhB strains carrying wild-type dnaE or dnaE_S759N allele and defects in proofreading (dnaQ920) or RNase HI (rnhA339::cat)
| dnaE + | S759N | dnaQ920 | dnaQ920 S759N | rnhA339 | rnhA339 S759N | |
|---|---|---|---|---|---|---|
| Exp. 1 | 729 | 3819 | 832 | 4638 | 668 | 4459 |
| Exp. 2 | 576 | 2853 | 443 | 3357 | 434 | 3924 |
| Exp. 3 | 1130 | 5652 | 929 | 4801 | 1222 | 7265 |
| Exp. 4 | 636 | 4383 | 979 | 4647 | 781 | 4833 |
| Exp. 5 | 388 | 3887 | 632 | 4024 | 497 | 4404 |
| Average | 692 ± 341 | 4119 ± 1276 | 763 ± 277 | 4293 ± 748 | 721 ± 387 | 4977 ± 1638 |
| P vs S759N | 0.569 | 0.017 |
Average number was calculated based on the total number of nucleotides in E. coli genome (9.28 Mb). A paired two-sided t-test was used to calculate P values.
In contrast, neither dnaE_S759C nor dnaE_S759T mutants gave a signal that was sufficient to assign any strand bias using HydEn-seq (Supplementary Figure S4), consistent with much lower levels of ribonucleotides incorporated by these variants than the dnaE_S759N mutant (21).
Investigation of the factors influencing ribonucleotide strand bias observed in a pol IIIα_S759N strain deficient in RNase HII
The ribonucleotide strand bias revealed by HydEn-seq analysis of the ΔrnhB dnaE_S759N strain is puzzling, as there is no reason to assume that pol III HE is intrinsically more prone to misinsert rNTPs into one strand, or the other. Consequently, we expected that the number of ribonucleotides residing in both DNA strands after replication would be similar. On the other hand, the ribonucleotide strand bias corroborates our findings from the pol V_Y11A studies, which suggested that the lagging strand could be subject to additional ribonucleotide repair in the absence of RNase HII.
One immediate possibility is the engagement of backup RER pathways on the lagging strand (as described in previous sections), which would be manifested by the greater number of ribonucleotides detected on the leading strand than on the lagging strand in the ΔrnhB dnaE_S759N strain. Our primary candidate is RNase HI-dependent RER, as this enzyme is not SOS-regulated and therefore should be present at constant levels during both normal replication and TLS.
Proofreading activity of DNA replicative polymerase could be another important factor that influences ribonucleotide levels in genomic DNA. It has been shown that newly incorporated ribonucleotides can be proofread by the yeast replicases, pol ϵ (74) and pol δ (75). The mechanism of preferential proofreading of lagging-strand ribonucleotides would be the same as the one causing mutator strand bias in wild-type E. coli, which is the discontinuous mechanism of the lagging strand replication described in the Introduction. Since the dnaE_S759N variant was shown to be compromised for both base and sugar discrimination (21) it was interesting to check the possible role of pol III proofreading (provided by the DnaQ protein) in removal of ribonucleotides from the E. coli genome.
To test both hypotheses, we first compared the ribonucleotide distribution on both DNA strands (using HydEn-seq) in ΔrnhB dnaE_S759N strains carrying defects in two additional repair pathways: impaired pol III proofreading activity, or RNase HI deficiency (Figure 4C). We observed a 26% difference in the leading vs. lagging ribonucleotide incorporation pattern between strains with active (dnaQ+) (0.0207; Figure 4B; Supplementary Figure S2) or defective (dnaQ920) proofreading activity of pol III (0.0154; Figure 4C; Supplementary Figure S2). In contrast, the lack of RNase HI activity (rnhA339) completely abolishes the leading-strand bias observed in the dnaE_S759N strain (0.0040; Figure 4C; Supplementary Figure S2).
Next, we performed alkaline gel electrophoresis of RNase HII-treated genomic DNA (Figure 5) and compared the number of ribonucleotides embedded into genomic DNAs of strains proficient, or deficient in RNase HI activity (rnhA+ vs. rnhA339, Table 3). The presence of the dnaE_S759N allele in cells deficient in RNase HII leads to increased fragmentation of chromosomal DNA (Figure 5) and increased ribonucleotide load in the chromosome compared to dnaE+ (Table 3), which is consistent with our previous observations (21). The lack of RNase HI further increases genomic DNA fragmentation (Figure 5) and the number of embedded ribonucleotides (Table 3). The average number of ribonucleotides incorporated into DNA isolated from the ΔrnhB rnhA339 dnaE_S759N strain (∼5 × 103, Table 3, column 6) is significantly higher than the number observed in the ΔrnhB dnaE_S759N background (∼4.2 × 103, Table 3, column 2). In contrast, no change in DNA fragmentation was observed in the ΔrnhB dnaE_S759N strain with defective proofreading activity of pol III (dnaQ920) compared to the ΔrnhB dnaE_S759N mutant (Figure 5). Consequently, the number of ribonucleotides incorporated into genomic DNA does not significantly differ between the ΔrnhB dnaE_ S759N and the ΔrnhB dnaQ920 dnaE_S759N strains (∼4.2 × 103 and ∼4.3 × 103, respectively, Table 3).
Figure 5.

Increased ribonucleotide incorporation in dnaE_S759N strains due to an RNase HI deficiency. Genomic DNAs from E. coli strains expressing wild-type dnaE (marked as ‘S’ in the figure) or dnaE_S759N allele (‘N’) in different combinations with RNase HII– (ΔrnhB, ‘–’), HI–deficiency (rnhA339), or impaired proofreading of the replicase (dnaQ920), were isolated, treated with purified RNase HII, and separated in alkaline agarose gel as described in Materials and Methods. 500 ng DNA was loaded per lane. Electrophoresis migration patterns (A) were converted to densitometry curves (B) as described in Materials & Methods. Untreated controls separated in non-denaturing (1× TAE) buffer are shown in Supplementary Figure S7.
Based on these results, we conclude that the strand bias observed in the ΔrnhB dnaE_S759N strain is due to enhanced activation of the RNase HI-dependent repair pathway on the lagging strand. These observations are consistent with the pol V_Y11A results showing engagement of RNase HI during lagging strand RER and confirm a back-up repair role for RNase HI on the lagging strand in an ΔrnhB dnaE_S759N background and show that the division of labor between RNase HI and HII during RER is not specific to conditions of SOS induction, but rather a more general mechanism. On the other hand, the role of proofreading activity of pol III in ribonucleotide removal appears to be limited.
DISCUSSION
Recent studies have demonstrated that ribonucleotides are the most common non-canonical nucleotides in DNA (76). Stretches of RNA can be incorporated into DNA when RNase HI fails to remove transcripts invading the DNA duplex, but the major source of ribonucleotides in the chromosome is misincorporation by replicative polymerases (16). The deleterious consequences of lack of RNase HII-dependent ribonucleotide repair particularly in unicellular and higher eukaryotes are not observed in Escherichia coli, which might be related to the existence of backup RER pathways that rely on the activities of RNase HI and Nucleotide Excision Repair (NER) (17).
In the current study, we were interested in exploring to what extent particular RER pathways in bacterial cells are involved in ribonucleotide removal from both DNA strands. The usage of a steric gate variant of a distributive but highly mutagenic TLS polymerase pol V, as well as an active site mutant of the major replicase pol III responsible for replication of both DNA strands, allowed us to indirectly (by comparing mutational spectra and the levels of mutagenesis) or directly (by measuring the number and distribution of ribonucleotides in DNA with HydEn-seq and RNase HII cleavage assay combined with alkaline gel electrophoresis) test the efficiency of particular RER pathways in ribonucleotide removal from both DNA strands.
Pivotal role of RNase HII in leading strand RER
Here, we present evidence that RNase HII primarily, but not exclusively, operates on the leading strand during RER.
Comparison of pol V_Y11A-dependent rates of lacI mutations in rnhB+ to ΔrnhB strains revealed significant increase in Y11A-dependent mutagenesis (Table 2), observed preferentially on the leading strand upon RNase HII-RER knockout (Figure 2). Analysis of the mutational spectra promoted by pol V_Y11A within the lacI target sequence confirmed an increase in the share of pol V-specific A·T→T·A mutations when RNase HII is absent, as participation of A·T→T·A mutations in the mutational spectra is now comparable to those promoted by wild-type pol V (Figure 1). These observations are based on the analysis of the level and specificity of mutations arising at >500 detectably mutable sites throughout a large mutational target (∼1100-bp lacI, Supplementary Files S2 and S3) (8). Introduction of the pol V_Y11A steric gate variant resulted in a considerable decline of pol V-specific transversions on both strands in an rnhB-proficient background (Figure 2), with a particularly significant (e.g. 23-fold for T→A in coding sequence, Figure 2) reduction observed on the lagging strand.
Comparison of the relative extent of the leading strand pol V_Y11A mutagenesis in rnhB+ strains to ΔrnhB, calculated from A·T→T·A mutation rates in the lacZ reporter gene (Figure 3), resulted in a 3.9-fold increase in the amount of pol V_Y11A mutagenesis relative to wild-type pol V on the leading strand, and a 2.6-fold increase on the lagging strand in ΔrnhB strains, recapitulating the lacI spectra results (Supplementary Table S1).
The data presented in these studies suggest that leading strand RER is strongly dependent upon the activity of RNase HII, as a rnhB deletion elevates the relative pol V_Y11A lacZ reversion rate to ∼180% on this strand (Figure 3). On the contrary, the lack of RNase HII has only modest impact on pol V_Y11A-dependent mutagenesis on the lagging strand (increase of mutagenesis to 20% of that of wild-type, Figure 3), indicating that the lagging strand is subject to the action of other ribonucleotide repair pathways. Due to the accumulation of a significant number of rNMPs in the lagging strand in strains expressing the pol V_Y11A mutant, one may expect RER on this strand to be particularly robust. Indeed, we observe that the relative level of mutagenesis promoted by pol V_Y11A in an RER-proficient background is lower on the lagging (8%) than on the leading strand (45%) (Figure 3). The abundance of ribonucleotides on the lagging strand could be a signal for the recruitment of NER and RNase HI during RER, resulting in increased removal of ribonucleotides, as well as misincorporated deoxyribonucleotides. Indeed, when either both RNases H (I and II), or RNase HII and NER, are inactive, the relative levels of pol V_Y11A lacZ mutation rates significantly increased on both DNA strands, to levels comparable to wild type pol V (Figure 3). The stronger effects of back-up repair pathway deactivation are observed on the lagging strand (e.g. 2.5 vs. 5.7-fold effects on the leading and lagging strand, respectively, when comparing ΔrnhB rnhA339 to ΔrnhB strains, Supplementary Table S1).
We are aware that the analysis of the activity ribonucleotide repair mechanisms in the genetic tests used here may be affected by factors such as polymerase mismatch preference, sequence context, or position of mismatched nucleotides in relation to ribonucleotides, which affects the likelihood of their ‘accidental’ repair. The analysis of pol V mutagenesis presented above is based on the assumption that both the wild-type and the steric gate mutant of pol V have the same nucleotide misincorporation specificity and T·T mispairs are made more frequently. We cannot fully confirm this using in vitro assays as the pol V_Y11A steric gate mutant exhibits almost no sugar selectivity in vitro (22). The rate of T·T mismatch formation is virtually impossible to assess as pol V_Y11A will correctly pair dT with ATP, which is added to the reaction as a pre-requisite for mutasome assembly (22). Thus, it would be possible to consider a situation in which it is the A·A mispair that arises more frequently as a consequence of pol V_Y11A mutator activity in vivo. Were that the case, lacI and lacZ mutation rates for the leading and lagging strand in pol V_Y11A strains would need to be reversed. This would have a small impact on our conclusions from the lacZ reversion assay as the absolute values of pol V_Y11A mutation rates are similar in both lacZ orientations. On the other hand, the pol V_Y11A-dependent relative mutagenesis levels in the lacI reporter gene would be 100–130% on both DNA strands (for example in ΔrnhB strains, for T in coding sequence: 4.5 × 10–8 for wild-type pol V and 6.0 × 10–8 for pol V_Y11A on the leading strand; 12.6 × 10–8 for wild-type pol V and 16.9 × 10–8 for pol V_Y11A on the lagging strand, Figure 2), which would suggest a bigger role of RNase HII in the removal of lagging-strand ribonucleotides in the lacI assay.
However, another confirmation for a pivotal role of RNase HII in the leading comes from genome-wide mapping of ribonucleotide incorporation in strains carrying pol III variant (dnaE_S759N). The observed strand bias in dnaE_S759N ΔrnhB strains (Figure 4B), with more ribonucleotides observed on the leading strand, that is abolished when RNase HI is absent (Figure 4C), strongly favors the hypothesis that RNase HII plays an important role during RER on the leading strand, while the lagging strand is subject to additional ribonucleotide removal pathway (RNase HI-dependent RER) not only during TLS, but also during normal replication by the replicase. In the latter case, however, increased participation of RNase HI during RER on the lagging strand cannot be explained by asymmetric distribution of ribonucleotides in the chromosome, as pol III is the sole replicase responsible for replication of both DNA strands. Possible scenario will be discussed below.
Role of RNase HI in lagging-strand RER
While RNase HII-dependent RER is the primary pathway responsible for ribonucleotide repair in E. coli, the work presented in this manuscript suggests that it plays a more significant role on the leading DNA strand. Repair of lagging-strand rNMPs is, in part, dependent on the activity of backup RER systems, prominently RNase HI-dependent RER. This result was somewhat surprising as RNase HI can only recognize substrates containing multiple consecutive ribonucleotides, which were not expected to be present in pol IIIα_S759N-replicated genome given a lack of apparent stress phenotype in dnaE_S759N ΔrnhB strains (unpublished observations), as well as the fact that expression of the dnaE_S759N mutation increases the ribonucleotide load by only 6- to 8-fold (Table 3, (21)). Nevertheless, to explore the possibility that pol IIIα_S759N inserts polyribonucleotide tracts into the chromosome, we compared the fragmentation patterns of genomic DNAs isolated from rnhA339 ΔrnhB dnaE_S759N with rnhA339 ΔrnhB controls digested with RNase HI or RNase A under either low salt (RNase A|LS), or high-salt (RNase A|HS) conditions. These enzymes have different specificities: RNase HI can cleave only RNA patches opposite DNA, RNase A|HS recognizes single stranded RNA, such as RNA tracts within DNA where there is a DNA gap on the opposite strand, while RNase A|LS can recognize both these substrates, as well as single ribonucleotides, albeit with only partial activity compared to RNase HII (38). Analysis of the distribution of products of RNase HI and RNase A cleavage shows no apparent sensitivity of genomic DNAs isolated from dnaE_S759N ΔrnhB and dnaE_S759N ΔrnhB rnhA339 strains to either RNase HI or RNase A|HS treatment, and a partial sensitivity to RNase A|LS (Supplementary Figures S5 and S8). RNase HI activity in the assay was confirmed by an in vitro cleavage of an oligonucleotide substrate followed by polyacrylamide gel electrophoresis (Supplementary Figure S6). This suggests that polyribonucleotide tracts, if any, must be sporadic and cannot explain a ∼20% increase in the number of ribonucleotides in dnaE_S759N ΔrnhB upon RNase HI deletion (Figure 5; Table 3). This raises the question of how to explain the involvement of RNase HI on the lagging strand during RER. RNase HI was suggested to be involved in the removal of single ribonucleotides in collaboration with RNase HII (29), which, however, is not present in the tested strains; this prompted us to consider non-biochemical factors that can be at play. One such mechanistic factor is the discontinuous manner of replication of the lagging strand which is known to be responsible for higher fidelity of its replication (6). Novel discoveries strengthen the hypothesis that RNase HI is involved in the removal of RNA primers during Okazaki fragment processing, which has been suggested as early as 1984 (4,30). Even though RNase HI is not essential in the cell, a polyribonucleotide primer bound to DNA template is a substrate recognized by RNase HI (30), and recent evidence shows that RNase HI can interact with SSB protein which is abundant on the lagging strand (77) and the interaction with SSB allows RNase HI to localize at the replication forks (78). On the other hand, in the same study, the authors observed that the lack of an RNase HI-SSB interaction did not exacerbate the phenotype of a polA12(Ts) allele, which encodes a mutant pol I with poor polymerase activity and no proofreading activities, concluding that this interaction is not required for processing RNA primers by RNase HI (78). Nevertheless, it is possible that the presence of short RNA patches every ∼1000 nt may attract RNase HI to the lagging strand, and in turn, its presence stimulates removal of single ribonucleotides embedded by the replicase.
Other explanations for HydEn-seq bias observed in dnaE_S759N ΔrnhB strain
Participation of other DNA polymerases in genome replication could influence the ribonucleotide strand bias as pol IIIα is the only mutated polymerase with increased ribonucleotide incorporation rate in the tested strains. Our previous reports showed increased access of accessory DNA polymerases to the lagging strand replication (10,12,14,79). Here we used strains deficient in pol II (ΔpolB), pol IV (ΔdinB) and pol V (ΔumuDC), so we may assume that the leading strand is replicated mainly by the pol III variant, while the lagging strand is replicated by pol III and partially by pol I which is responsible for replication of ∼1% of the lagging strand during maturation of Okazaki fragments (14). The disappearance of the HydEn-seq strand bias upon RNase HI removal (Figure 4; Supplementary Figure S2) could, in principle, be caused by changes in the mechanism of DNA replication: either accumulation of R-loops and initiation of unscheduled replication (called constitutive stable DNA replication – cSDR) away from the origin of replication region (80), or direct re-priming of the leading strand using R-loops, making its replication more discontinuous and lagging-strand-like, for what we have preliminary genetic evidence (data not shown; see also (81)). In the former case fragments of DNA normally replicated as a lagging strand could be replicated as a leading strand and vice versa (37,82) such that pol I replication during Okazaki fragment maturation could be more evenly spread throughout the whole genome and both DNA strands. In the latter case pol I synthesis would increase on the leading strand, on top of its participation in lagging strand synthesis. However, the fact that upon rnhA knockout we observe a further increase (by about 1000 rNMPs) in the total number of ribonucleotides in DNA (Table 3, columns 2 and 6) would require pol I to replicate much less genomic DNA in dnaE_S759N ΔrnhB rnhA339 strains, which is the opposite of what we would expect if the lack of ribonucleotide strand bias in the rnhA339 background was caused by cSDR, or re-priming. It is also unlikely that replication of ∼1% of the lagging strand during Okazaki fragment maturation by pol I could result in a 20% difference in the observed number of genome-embedded ribonucleotides between rnhA+ and rnhA339 backgrounds. Therefore, even if pol I-associated replication of the lagging strand contributes, to some extent, to the ribonucleotide strand bias in rnhA339 strains, we believe that the results of RNase cleavage assay with alkaline gel electrophoresis suggest that the major driving force behind HydEn-seq ribonucleotide strand bias is not pol I-dependent replication of the lagging strand fragments during Okazaki fragment maturation, but rather the engagement of RNase HI in ribonucleotide repair (in which pol I also participates).
Role of cSDR in shaping HydEn-seq bias in rnhA339 strains
It has been shown that cSDR affects the replication profile in strains lacking RNase HI activity due to initiation from non-canonical ori sites (37,82). A major alternative ori has been found in the rrn operon region close to oriC, which encodes ribosomal RNAs, and is actively transcribed (83). On the other hand, the sharp peak near the termination region is associated with RNase HI’s role in replication completion by processing overreplicated regions of DNA (34). Interestingly, even upon cSDR induction, oriC seems to have a dominant effect as the replication profile is not completely flat in rnhA339 strains (37,82). A randomness in replication initiation requires blocking oriC initiation site with the usage of a dnaA mutant allele (37,82). Our HydEn-seq data show that in the dnaE_S759N ΔrnhB rnhA339 strain the ribonucleotide strand bias disappears such that it is no longer possible to locate oriC and ter region, even though we did not introduce any mutations that would affect replication initiation from oriC (Figure 4). Based on these data we believe that the disappearance of the ribonucleotide strand bias in dnaE_S759N ΔrnhB rnhA339 strain cannot be attributed solely to cSDR, although it may be responsible for part of the effect. In any case, both the increase in the number of ribonucleotides by ∼1000 upon RNase HI deletion in strain producing pol IIIα_S759N, but not in strain with wild-type pol IIIα, as well as the existence of a ribonucleotide strand bias in dnaE_S759N ΔrnhB strains in the first place, suggest additional RER activity on the lagging strand regardless of cSDR.
Proposed model of RER in E. coli
Based upon the data presented here, we propose the following strand-dependent model of RER in E. coli (Figure 6). In an RER-proficient background, RNase HII-mediated RER appears to play a crucial role in the removal of leading-strand ribonucleotides and a lesser role on the lagging strand. In the absence of RNase HII, RNase HI and the NER proteins facilitate RER on both DNA strands, with a preference for the lagging strand. While the role of NER is more likely to be limited to SOS induced conditions, RNase HI can also participate in RER during normal DNA replication. The exact mechanism of RNase HI involvement in the removal of single ribonucleotides incorporated by the replicase on the lagging strand is currently unknown, but is more likely to be indirect, for example as a byproduct of cleavage of Okazaki primers. The role of 3′→5′ exonucleolytic proofreading in reducing the number of genome-embedded ribonucleotides seems to be limited, but cannot be fully excluded.
Figure 6.
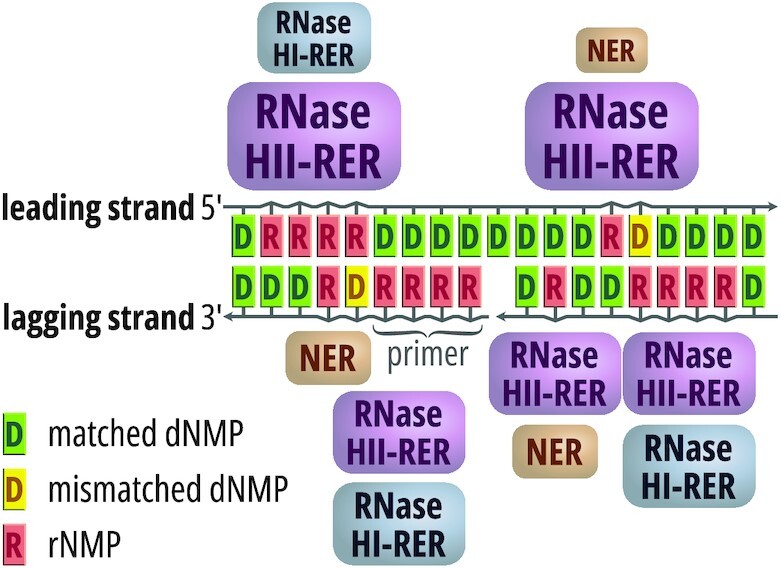
Model of strand-specific ribonucleotide excision repair in Escherichia coli cells. RNase HII is the major enzyme involved in RER with a particularly important role on the leading strand. In the absence of RNase HII, backup RER pathways are triggered: RER dependent on RNase HI, and under SOS activation conditions, NER. These pathways operate with higher efficiency on the lagging DNA strand. RNase HI can also remove primers synthesized by the primase, and its activity on the lagging strand can indirectly stimulate removal of single ribonucleotides by pol I on this strand.
Biological significance of increased RER of the lagging DNA strand
In bacteria, essential genes are frequently oriented on the chromosome such that their transcription is carried out in the same direction as replisome progression, so as to avoid head-on collisions between replication and transcription machineries (84). During co-directional transcription, the template strand for transcription is also used for synthesis of the leading strand. The same template strand has been replicated as a lagging strand during the previous replication cycle. The impact of ribonucleotide incorporation on transcription has not been thoroughly studied, however it has been suggested that ribonucleotides persisting in DNA may affect the fidelity of RNA polymerase (85). Therefore, it is possible that tighter control of the number of ribonucleotides incorporated during lagging strand synthesis might be an additional gatekeeper ensuring cellular fitness and homeostasis (86).
DATA AVAILABILITY
The HydEn-seq sequencing data have been deposited in the Gene Expression Omnibus database under accession number: GSE141315.
Supplementary Material
ACKNOWLEDGEMENTS
We dedicate this manuscript to our colleagues, Alexandra Vaisman and Piotr Jonczyk, both of whom sadly passed away during the course of these studies which could not have been conducted, nor completed, without their intellectual and experimental input. We thank Anna Bebenek (IBB) and John McDonald (NICHD) for careful reading of the manuscript and helpful comments. We also thank Martin Reijns (MRC) for stimulating discussions and helpful comments/suggestions about our study and Scott Lujan (NIEHS) for help with mutational spectra analysis.
Notes
Present address: Mahmood Faraz, Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala 75185, Sweden.
Contributor Information
Krystian Łazowski, Laboratory of DNA Replication and Genome Stability, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw 02-106, Poland.
Mahmood Faraz, Department of Medical Biochemistry and Cell Biology, University of Gothenburg, Gothenburg 40530, Sweden.
Alexandra Vaisman, Laboratory of Genomic Integrity, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892-3371, USA.
Nicholas W Ashton, Laboratory of Genomic Integrity, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892-3371, USA.
Piotr Jonczyk, Laboratory of DNA Replication and Genome Stability, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw 02-106, Poland.
Iwona J Fijalkowska, Laboratory of DNA Replication and Genome Stability, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw 02-106, Poland.
Anders R Clausen, Department of Medical Biochemistry and Cell Biology, University of Gothenburg, Gothenburg 40530, Sweden.
Roger Woodgate, Laboratory of Genomic Integrity, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892-3371, USA.
Karolina Makiela-Dzbenska, Laboratory of DNA Replication and Genome Stability, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw 02-106, Poland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health, National Institute of Child Health and Human Development Intramural Research Program (to R.W.); National Science Centre, Poland [HARMONIA Grant 2015/18/M/NZ3/00402 to K.M.D., PRELUDIUM Grant 2019/35/N/NZ1/03402 to K.Ł.]; Swedish Research Council [2018-05121_VR to A.R.C.]. Funding for open access charge: NIH/NICHD Intramural Research Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. McHenry C.S. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 2003; 49:1157–1165. [DOI] [PubMed] [Google Scholar]
- 2. Yao N., O’Donnell M. Bacterial and eukaryotic replisome machines. JSM Biochem. Mol. Biol. 2016; 3:1013. [PMC free article] [PubMed] [Google Scholar]
- 3. Zechner E.L., Wu C.A., Marians K.J.. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. III. A polymerase-primase interaction governs primer size. J. Biol. Chem. 1992; 267:4054–4063. [PubMed] [Google Scholar]
- 4. Ogawa T., Okazaki T.. Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Mol. Gen. Genet. 1984; 193:231–237. [DOI] [PubMed] [Google Scholar]
- 5. Randall J.R., Nye T.M., Wozniak K.J., Simmons L.A.. RNase HIII is important for Okazaki fragment processing in Bacillus subtilis. J. Bacteriol. 2019; 201:e00686-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fijalkowska I.J., Schaaper R.M., Jonczyk P.. DNA replication fidelity in Escherichia coli: a multi-DNA polymerase affair. FEMS Microbiol. Rev. 2012; 36:1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fijalkowska I.J., Jonczyk P., Maliszewska-Tkaczyk M., Bialoskorska M., Schaaper R.M.. Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:10020–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maslowska K.H., Makiela-Dzbenska K., Mo J.-Y., Fijalkowska I.J., Schaaper R.M.. High-accuracy lagging-strand DNA replication mediated by DNA polymerase dissociation. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:4212–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makiela-Dzbenska K., Maslowska K.H., Kuban W., Gawel D., Jonczyk P., Schaaper R.M., Fijalkowska I.J.. Replication fidelity in E. coli: differential leading and lagging strand effects for dnaE antimutator alleles. DNA Repair (Amst.). 2019; 83:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maliszewska-Tkaczyk M., Jonczyk P., Bialoskorska M., Schaaper R.M., Fijalkowska I.J.. SOS mutator activity: unequal mutagenesis on leading and lagging strands. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:12678–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vandewiele D., Fernández de Henestrosa A.R., Timms A.R., Bridges B.A., Woodgate R.. Sequence analysis and phenotypes of five temperature sensitive mutator alleles of dnaE, encoding modified α-catalytic subunits of Escherichia coli DNA polymerase III holoenzyme. Mutat. Res. 2002; 499:85–95. [DOI] [PubMed] [Google Scholar]
- 12. Banach-Orlowska M., Fijalkowska I.J., Schaaper R.M., Jonczyk P.. DNA polymerase II as a fidelity factor in chromosomal DNA synthesis in Escherichia coli. Mol. Microbiol. 2005; 58:61–70. [DOI] [PubMed] [Google Scholar]
- 13. Kuban W., Banach-Orlowska M., Schaaper R.M., Jonczyk P., Fijalkowska I.J.. Role of DNA polymerase IV in Escherichia coli SOS mutator activity. J. Bacteriol. 2006; 188:7977–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makiela-Dzbenska K., Jaszczur M.M., Banach-Orlowska M., Jonczyk P., Schaaper R.M., Fijalkowska I.J.. Role of Escherichia coli DNA polymerase I in chromosomal DNA replication fidelity. Mol. Microbiol. 2009; 74:1114–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joyce C.M. Choosing the right sugar: how polymerases select a nucleotide substrate. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams J.S., Kunkel T.A.. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst.). 2014; 19:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaisman A., Woodgate R.. Redundancy in ribonucleotide excision repair: competition, compensation, and cooperation. DNA Repair (Amst.). 2015; 29:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clausen A.R., Lujan S.A., Burkholder A.B., Orebaugh C.D., Williams J.S., Clausen M.F., Malc E.P., Mieczkowski P.A., Fargo D.C., Smith D.J.et al.. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol. 2015; 22:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown J.A., Suo Z.. Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry. 2011; 50:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parasuram R., Coulther T.A., Hollander J.M., Keston-Smith E., Ondrechen M.J., Beuning P.J.. Prediction of active site and distal residues in E.coli DNA polymerase III alpha polymerase activity. Biochemistry. 2018; 57:1063–1072. [DOI] [PubMed] [Google Scholar]
- 21. Vaisman A., Łazowski K., Reijns M.A.M., Walsh E., McDonald J.P., Moreno K.C., Quiros D.R., Schmidt M., Kranz H., Yang W.et al.. Novel Escherichia coli active site dnaE alleles with altered base and sugar selectivity. Mol. Microbiol. 2021; 116:909–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vaisman A., Kuban W., McDonald J.P., Karata K., Yang W., Goodman M.F., Woodgate R.. Critical amino acids in Escherichia coli UmuC responsible for sugar discrimination and base-substitution fidelity. Nucleic Acids Res. 2012; 40:6144–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vaisman A., McDonald J.P., Noll S., Huston D., Loeb G., Goodman M.F., Woodgate R.. Investigating the mechanisms of ribonucleotide excision repair in Escherichia coli. Mutat. Res. 2014; 761:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cerritelli S.M., Crouch R.J.. The balancing act of ribonucleotides in DNA. Trends Biochem. Sci. 2016; 41:434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Itaya M. Isolation and characterization of a second RNase H (RNase HII) of Escherichia coli K-12 encoded by the rnhB gene. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:8587–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tadokoro T., Kanaya S.. Ribonuclease H: molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009; 276:1482–1493. [DOI] [PubMed] [Google Scholar]
- 27. Hyjek M., Figiel M., Nowotny M.. RNases H: structure and mechanism. DNA Repair (Amst.). 2019; 84:102672. [DOI] [PubMed] [Google Scholar]
- 28. Reijns M.A.M., Rabe B., Rigby R.E., Mill P., Astell K.R., Lettice L.A., Boyle S., Leitch A., Keighren M., Kilanowski F.et al.. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012; 149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tannous E., Kanaya E., Kanaya S.. Role of RNase H1 in DNA repair: removal of single ribonucleotide misincorporated into DNA in collaboration with RNase H2. Sci. Rep. 2015; 5:9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee H., Cho H., Kim J., Lee S., Yoo J., Park D., Lee G.. RNase H is an exo- and endoribonuclease with asymmetric directionality, depending on the binding mode to the structural variants of RNA:DNA hybrids. Nucleic Acids Res. 2022; 50:1801–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gowrishankar J., Krishna Leela J., Anupama K.. R-loops in bacterial transcription: their causes and consequences. Transcription. 2013; 4:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drolet M., Brochu J.. R-loop-dependent replication and genomic instability in bacteria. DNA Repair (Amst.). 2019; 84:102693. [DOI] [PubMed] [Google Scholar]
- 33. Naito S., Uchida H.. RNase H and replication of ColE1 DNA in Escherichia coli. J. Bacteriol. 1986; 166:143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wendel B.M., Hernandez A.J., Courcelle C.T., Courcelle J.. Ligase A and RNase HI participate in completing replication on the chromosome in Escherichia coli. DNA. 2021; 1:13–25. [Google Scholar]
- 35. Cerritelli S.M., Frolova E.G., Feng C., Grinberg A., Love P.E., Crouch R.J.. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell. 2003; 11:807–815. [DOI] [PubMed] [Google Scholar]
- 36. Arora R., Lee Y., Wischnewski H., Brun C.M., Schwarz T., Azzalin C.M.. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014; 5:5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maduike N.Z., Tehranchi A.K., Wang J.D., Kreuzer K.N.. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol. Microbiol. 2014; 91:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kouzminova E.A., Kadyrov F.F., Kuzminov A.. RNase HII saves rnhA mutant Escherichia coli from R-loop-associated chromosomal fragmentation. J. Mol. Biol. 2017; 429:2873–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kouzminova E.A., Kuzminov A.. Ultraviolet-induced RNA:DNA hybrids interfere with chromosomal DNA synthesis. Nucleic Acids Res. 2021; 49:3888–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walsh E., Henrikus S.S., Vaisman A., Makiela-Dzbenska K., Armstrong T.J., Łazowski K., McDonald J.P., Goodman M.F., van Oijen A.M., Jonczyk P.et al.. Role of RNase H enzymes in maintaining genome stability in Escherichia coli expressing a steric-gate mutant of pol VICE391. DNA Repair (Amst.). 2019; 84:102685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vaisman A., McDonald J.P., Huston D., Kuban W., Liu L., Van Houten B., Woodgate R.. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet. 2013; 9:e1003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Faraz M., Woodgate R., Clausen A.R.. Tracking Escherichia coli DNA polymerase V to the entire genome during the SOS response. DNA Repair (Amst.). 2021; 101:103075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fijalkowska I.J., Dunn R.L., Schaaper R.M.. Genetic requirements and mutational specificity of the Escherichia coli SOS mutator activity. J. Bacteriol. 1997; 179:7435–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Curti E., McDonald J.P., Mead S., Woodgate R.. DNA polymerase switching: effects on spontaneous mutagenesis in Escherichia coli. Mol. Microbiol. 2009; 71:315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niccum B.A., Coplen C.P., Lee H., Mohammed Ismail W., Tang H., Foster P.L.. New complexities of SOS-induced “untargeted” mutagenesis in Escherichia coli as revealed by mutation accumulation and whole-genome sequencing. DNA Repair (Amst.). 2020; 90:102852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watanabe-Akanuma M., Woodgate R., Ohta T.. Enhanced generation of A:T → T:A transversions in a recA730 lexA51(Def) mutant of Escherichia coli. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 1997; 373:61–66. [DOI] [PubMed] [Google Scholar]
- 47. Gawel D., Maliszewska-Tkaczyk M., Jonczyk P., Schaaper R.M., Fijalkowska I.J.. Lack of strand bias in UV-induced mutagenesis in Escherichia coli. J. Bacteriol. 2002; 184:4449–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sweasy J.B., Witkin E.M., Sinha N., Roegner-Maniscalco V.. RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J. Bacteriol. 1990; 172:3030–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wanner B.L. Novel regulatory mutants of the phosphate regulon in Escherichia coli K-12. J. Mol. Biol. 1986; 191:39–58. [DOI] [PubMed] [Google Scholar]
- 50. Cupples C.G., Miller J.H.. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fijalkowska I.J., Schaaper R.M.. Effects of Escherichia coli dnaE antimutator alleles in a proofreading- deficient mutD5 strain. J. Bacteriol. 1995; 177:5979–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swerdlow S.J., Schaaper R.M.. Mutagenesis in the lacI gene target of E. Coli: improved analysis for lacId and lacO mutants. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 2014; 770:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng Q. New algorithms for Luria-Delbrück fluctuation analysis. Math. Biosci. 2005; 196:198–214. [DOI] [PubMed] [Google Scholar]
- 54. Zheng Q. rSalvador: an R package for the fluctuation experiment. G3: Genes Genomes Genet. 2017; 7:3849–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zheng Q. New approaches to mutation rate fold change in Luria–Delbrück fluctuation experiments. Math. Biosci. 2021; 335:108572. [DOI] [PubMed] [Google Scholar]
- 56. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10. [Google Scholar]
- 57. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goodman M.F., McDonald J.P., Jaszczur M.M., Woodgate R.. Insights into the complex levels of regulation imposed on Escherichia coli DNA polymerase V. DNA Repair (Amst.). 2016; 44:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Henrikus S.S., van Oijen A.M., Robinson A.. Specialised DNA polymerases in Escherichia coli: roles within multiple pathways. Curr. Genet. 2018; 64:1189–1196. [DOI] [PubMed] [Google Scholar]
- 60. Creighton S., Huang M.M., Cai H., Arnheim N., Goodman M.F.. Base mispair extension kinetics. Binding of avian myeloblastosis reverse transcriptase to matched and mismatched base pair termini. J. Biol. Chem. 1992; 267:2633–2639. [PubMed] [Google Scholar]
- 61. Rossetti G., Dans P.D., Gomez-Pinto I., Ivani I., Gonzalez C., Orozco M.. The structural impact of DNA mismatches. Nucleic Acids Res. 2015; 43:4309–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schaaper R.M. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 1993; 268:23762–23765. [PubMed] [Google Scholar]
- 63. Niccum B.A., Lee H., MohammedIsmail W., Tang H., Foster P.L.. The spectrum of replication errors in the absence of error correction assayed across the whole genome of Escherichia coli. Genetics. 2018; 209:1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reijns M.A.M., Kemp H., Ding J., De Procé S.M., Jackson A.P., Taylor M.S.. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015; 518:502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lujan S.A., Williams J.S., Kunkel T.A.. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 2016; 26:640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oka A., Sugimoto K., Takanami M., Hirota Y.. Replication origin of the Escherichia coli K-12 chromosome: the size and structure of the minimum DNA segment carrying the information for autonomous replication. Mol. Gen. Genet. 1980; 178:9–20. [DOI] [PubMed] [Google Scholar]
- 67. Duggin I.G., Bell S.D.. Termination structures in the Escherichia coli chromosome replication fork trap. J. Mol. Biol. 2009; 387:532–539. [DOI] [PubMed] [Google Scholar]
- 68. Kornberg A., Baker T.A.. DNA Replication. 2nd edn. 1992; California, NY: W.H. Freeman & Co. [Google Scholar]
- 69. O’Donnell M. Replisome architecture and dynamics in Escherichia coli. J. Biol. Chem. 2006; 281:10653–10656. [DOI] [PubMed] [Google Scholar]
- 70. Pomerantz R.T., O’Donnell M. Replisome mechanics: insights into a twin DNA polymerase machine. Trends Microbiol. 2007; 15:156–164. [DOI] [PubMed] [Google Scholar]
- 71. Yao N.Y., O’Donnell M. Replisome dynamics and use of DNA trombone loops to bypass replication blocks. Mol. Biosyst. 2008; 4:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Langston L.D., Indiani C., O’Donnell M. Whither the replisome: emerging perspectives on the dynamic nature of the DNA replication machinery. Cell Cycle. 2009; 8:2686–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McHenry C.S. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011; 15:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Williams J.S., Clausen A.R., Nick McElhinny S.A., Watts B.E., Johansson E., Kunkel T.A.. Proofreading of ribonucleotides inserted into DNA by yeast DNA polymerase ϵ. DNA Repair (Amst.). 2012; 11:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Clausen A.R., Zhang S., Burgers P.M., Lee M.Y., Kunkel T.A.. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair (Amst.). 2013; 12:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cronan G.E., Kouzminova E.A., Kuzminov A.. Near-continuously synthesized leading strands in Escherichia coli are broken by ribonucleotide excision. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Petzold C., Marceau A.H., Miller K.H., Marqusee S., Keck J.L.. Interaction with single-stranded DNA-binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. J. Biol. Chem. 2015; 290:14626–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wolak C., Ma H.J., Soubry N., Sandler S.J., Reyes-Lamothe R., Keck J.L.. Interaction with single-stranded DNA-binding protein localizes ribonuclease HI to DNA replication forks and facilitates R-loop removal. Mol. Microbiol. 2020; 114:495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kuban W., Banach-Orlowska M., Bialoskorska M., Lipowska A., Schaaper R.M., Jonczyk P., Fijalkowska I.J.. Mutator phenotype resulting from DNA polymerase IV overproduction in Escherichia coli: preferential mutagenesis on the lagging strand. J. Bacteriol. 2005; 187:6862–6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997; 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pomerantz R.T., O’Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008; 456:762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dimude J.U., Stockum A., Midgley-Smith S.L., Upton A.L., Foster H.A., Khan A., Saunders N.J., Retkute R., Rudolph C.J.. The consequences of replicating in the wrong orientation: bacterial chromosome duplication without an active replication origin. mBio. 2015; 6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Scholz S.A., Diao R., Wolfe M.B., Fivenson E.M., Lin X.N., Freddolino P.L.. High-resolution mapping of the Escherichia coli chromosome reveals positions of high and low transcription. Cell Syst. 2019; 8:212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rocha E.P.C. Order and disorder in bacterial genomes. Curr. Opin. Microbiol. 2004; 7:519–527. [DOI] [PubMed] [Google Scholar]
- 85. Mehta A.P., Wang Y., Reed S.A., Supekova L., Javahishvili T., Chaput J.C., Schultz P.G.. Bacterial genome containing chimeric DNA–RNA sequences. J. Am. Chem. Soc. 2018; 140:11464–11473. [DOI] [PubMed] [Google Scholar]
- 86. Xu L., Wang W., Zhang L., Chong J., Huang X., Wang D. Impact of template backbone heterogeneity on RNA polymerase II transcription. Nucleic Acids Res. 2015; 43:2232–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The HydEn-seq sequencing data have been deposited in the Gene Expression Omnibus database under accession number: GSE141315.