

Abstract
Implantation of biomaterials or devices into soft tissue often leads to the development of the foreign body response (FBR), an inflammatory condition that can cause implant failure, tissue injury, and death of the patient. Macrophages accumulate and fuse to generate destructive foreign body giant cells (FBGCs) at the tissue-implant interface, leading to the development of fibrous scar tissue around the implant that is generated by myofibroblasts. We previously showed that the FBR in vivo and FBGC formation in vitro require transient receptor potential vanilloid 4 (TRPV4), a mechanosensitive ion channel. Here, we report that TRPV4 was required specifically for the FBR induced by implant stiffness independently of biochemical cues and for intracellular stiffening that promotes FBGC formation in vitro. TRPV4 deficiency reduced collagen deposition and the accumulation of macrophages, FBGCs, and myofibroblasts at stiff, but not soft, implants in vivo and inhibited macrophage-induced differentiation of wild-type fibroblasts into myofibroblasts in vitro. Atomic force microscopy demonstrated that TRPV4 was required for implant-adjacent tissue stiffening in vivo and for cytoskeletal remodeling and intracellular stiffening induced by fusogenic cytokines in vitro. Together, these data suggest a mechanism whereby a reciprocal functional interaction between TRPV4 and substrate stiffness leads to cytoskeletal remodeling and cellular force generation to promote FBGC formation during the FBR.
INTRODUCTION
Implantable biomaterials and medical devices are used in millions of procedures each year worldwide (1–4). It is estimated that about $170 billion per year is spent in the United States on various biomedical implants, including prostheses in orthopedic, dental, cardiovascular, and reconstructive surgery, in ophthalmological procedures, in angioplasty and hemodialysis, and as controlled drug release devices and sensors (1–6). However, the implantation of biomedical devices into soft host tissue is linked to the development of the foreign body response (FBR), a chronic inflammatory response (1–6). FBR results in the development of an outer layer of collagenous fibrous capsule around the implant and an inner layer of implant-adherent macrophages and foreign body giant cells (FBGCs), leading to implant failure and rejection, and is associated with substantial morbidity and mortality (1–6). FBR poses an unmet challenge for both scientist and patient because there are no therapeutic strategies that can mitigate the unwanted side effects of implantation (1–6). FBR-dependent implant failure costs the U.S. health care system more than $10 billion per year (7–10). Despite decades of research in bioengineering and cell biology, the molecular mechanisms underlying the FBR remain elusive.
Macrophages are recognized as the key orchestrators of the FBR due to their ability to secrete inflammatory proteins, undergo fusion to form FBGCs to degrade the implants, synthesize matrix components, promote the differentiation and recruitment of the myofibroblasts that remodel the extracellular matrix (ECM), and form the fibrous capsule around the implant (4, 11–14). A key objective in the design of next-generation implantable biomaterials to enhance biocompatibility and functionality is to determine how to modulate macrophage activity without inducing FBR and FBGC formation. Thus, it is essential to identify the inter- and intracellular signaling pathways that mediate macrophage-biomaterial interactions and macrophage responses to biomaterials.
Emerging data support a role for substrate stiffness (rigidity) of the extracellular and intracellular matrices in numerous cellular processes including gene expression, cell migration, and differentiation (15–28). Conversely, it is also recognized that soluble modulators such as cytokines increase both tissue stiffness and cell elasticity, although the mechanisms by which mechanical and soluble signals are integrated to drive biological functions are unclear (29–32). Cells detect changes in substrate stiffness and adjust their functions in response, a capacity that has increasingly become a subject of research because of its importance in diverse biological processes including development, inflammation, wound healing, fibrogenesis, and oncogenesis (15–37). Previous reports by our group and others have documented that various macrophage functions including differentiation, adhesion, motility, phagocytosis, spreading, proliferation, and cytoskeletal remodeling are sensitive to substrate stiffness, suggesting that stiffness may play a role in the FBR (14, 19, 23, 25, 38–43). Emerging data suggest that the severity of the FBR is determined, in part, by inherent mechanical cues from the biomaterial such as stiffness and surface topography and by consequent microenvironmental cues after implantation (23, 44, 45).
Cell fusion is an essential process in numerous normal and pathophysiological conditions such as fertilization, muscle development, generation of bone-resorbing osteoclasts, and response to pathogens and implants (46–48). Homotypic fusion of macrophages in response to an implant generates multinucleated FBGCs, which subsequently become inflammatory and destructive, causing harm to the implant and surrounding tissues (4, 49). Several plasma membrane receptors have been shown to participate in macrophage fusion, including CD36, CD44, CD47, CD200, signal regulatory protein 1-a, interleukin-4 (IL-4) receptor, E-cadherin, and mannose receptor (4, 50–52). In addition, blockade of molecules including DNAX-activating protein of 12-kDa dendritic cell–specific transmembrane protein, the small guanosine triphosphatase Rac1, and the transcription factor signal transducer and activator of transcription 6 have been shown to abrogate macrophage fusion (51, 53–56).
Despite the wealth of knowledge relating to macrophage activation and FBR, the elicited intracellular pathway in macrophage-implant interactions and the identification of potential mechanosensitive receptors that integrate the mechanical cues and their associated downstream pathways have not been identified. We previously published data showing that transient receptor potential vanilloid 4 (TRPV4), a mechanosensitive plasma membrane channel, is required for FBR and FBGC formation (14). TRPV4, a member of the TRPV superfamily, is widely present in numerous cell types including macrophages (14, 19, 41, 42, 57–65). Previous reports by our group and others have shown that TRPV4 is activated by a range of chemical and mechanical stimuli including changes in matrix and substrate stiffness, osmolarity, mechanical stresses, growth factors, and metabolites of arachidonic acid (14, 20, 24, 27, 59–65). TRPV4 is associated with numerous physiological functions including osmolarity sensing in kidneys, shear stress detection in arteries, neurological function, and control of osteogenesis (57, 58, 61, 65–67). In mice, TRPV4 deficiency is linked to altered vasodilatory responses and osmosensing, sensory and motor neuropathies, and lung and dermal fibrosis (20, 57, 58, 65–68). Here, we report that both the FBR induced by the inherent stiffness of the implant and the consequent augmentation of stiffness in implant-adjacent tissues depend on TRPV4. Residues 100 to 130 in the N-terminal domain of TRPV4 are essential for biomechanical stimulus–induced TRPV4 activation and FBGC formation. We also show that TRPV4-dependent F-actin generation enhances fusogenic cytokine–induced stiffness generation in macrophages and FBGC formation.
RESULTS
Implant stiffness–dependent FBR depends on TRPV4
To assess the impact of TRPV4 on stiffness-dependent FBR in vivo, we used a widely accepted subcutaneous biomaterial implantation model in which collagen-coated polyacrylamide (PA) gel discs of soft (1 kPa) and rigid (50 kPa) stiffness were implanted into wild-type (WT) and Trpv4−/− mice (14, 23, 69). We selected this stiffness range to mimic the stiffness of normal skin and breast tissue (~1 kPa) and fibrotic skin (~8 to 50 kPa). We found that 4 weeks after implantation, the amount of collagen deposition in the area surrounding the implant was 4.5-fold higher in WT mice receiving 50-kPa matrix compared to 1 kPa, suggesting that implant-elicited fibrosis, an indication of FBR, was augmented by increased implant stiffness (Fig. 1A). In contrast, similar amounts of collagen deposition were detected in Trpv4−/− mice receiving either 1- or 50-kPa implants, which was 4.5-fold reduced compared to 50-kPa implants in WT mice, suggesting that stiffness-induced fibrosis development in vivo was significantly diminished in the absence of TRPV4 (Fig. 1A). Implantation of biomaterials in soft tissues sparks the recruitment and accumulation of inflammatory cells, specifically macrophages, on the implant surface and surrounding areas. These accumulating cells ultimately fuse and develop into implant-damaging FBGCs. Macrophage accumulation on the surface of the implant, another important hallmark of FBR, was increased ninefold in WT mice receiving 50-kPa implants compared to 1-kPa implants; however, in Trpv4−/− mice, no stiffness-induced differences in macrophage accumulation was found (Fig. 1B). These results suggest that stiffness-induced FBR in vivo is severely impaired in the absence of TRPV4. Thick accumulation of macrophages at the tissue-implant interface made it difficult to precisely determine the number of FBGCs at that area. However, increased numbers of clusters of giant cells, which are multinucleate and positive for the macrophage marker CD68, were detected in 50-kPa implant–containing WT mice compared to Trpv4−/− mice, suggesting a critical role for TRPV4 in stiffness-induced FBGC formation in vivo. Together, our findings of reduced collagen deposition, reduced macrophage accumulation, and diminished FBGCs in soft implant–carrying WT mice and in Trpv4−/− mice suggested that biomaterial stiffness–induced FBR depends on TRPV4.
Fig. 1. TRPV4 is required for the FBR induced by stiff implants.
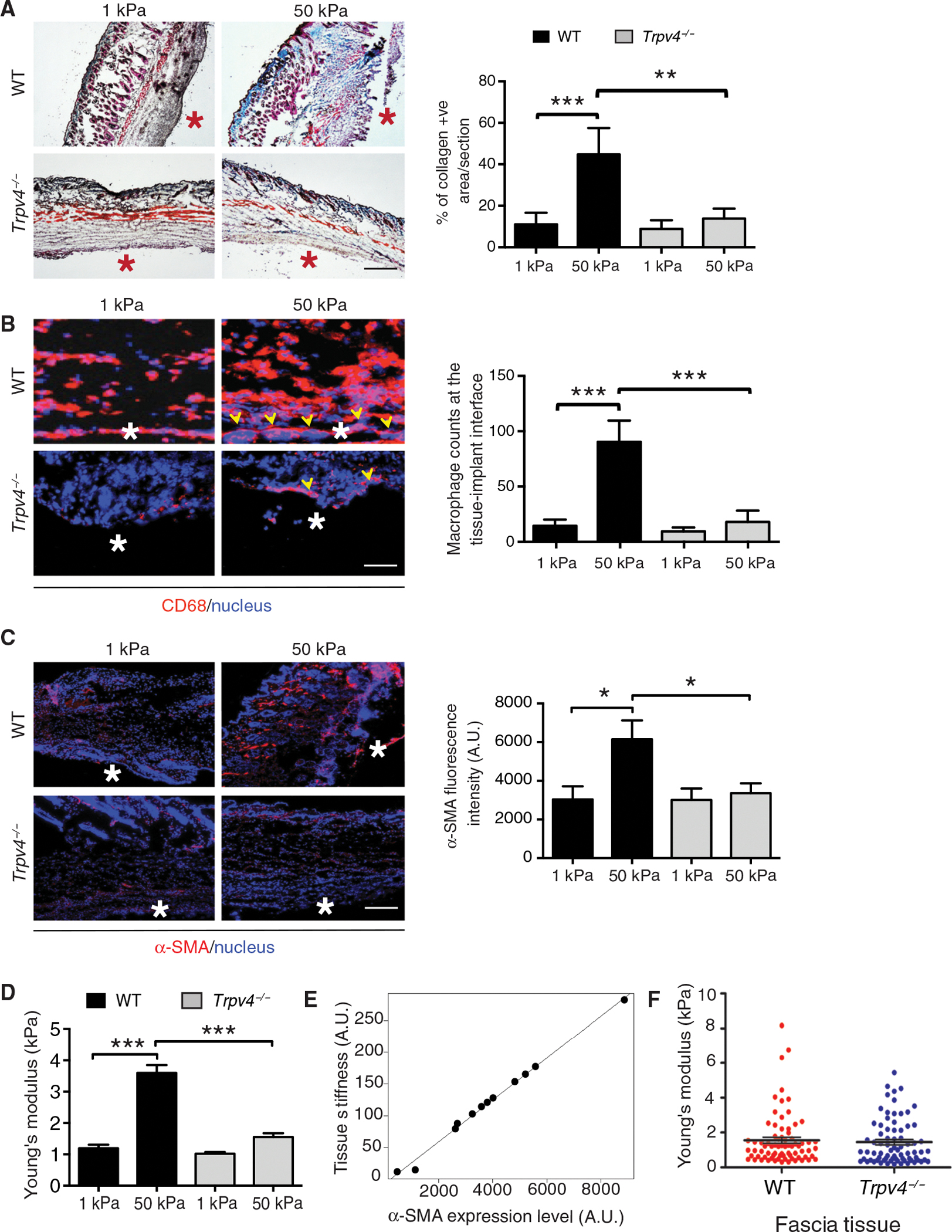
(A to C) Imaging and quantification of collagen (A), the macrophage marker CD68 (B), and the myofibroblast marker α-SMA (C) in representative skin sections from wild-type (WT) and Trpv4−/− mice implanted subcutaneously with 1- and 50-kPa collagen-coated polyacrylamide (PA) gels for 28 days. Asterisks indicate tissue-implant interface. Collagen (blue) and keratin (red) in (A) were visualized with Masson’s trichrome. Arrowheads in (B) indicate FBGCs near the tissue-implant interface. CD68 and α-SMA were visualized by immunofluorescence. Scale bars, 50 μm. n = 5 mice per group and 2 implants per mouse. *P < 0.001, Student’s t test. (D) Quantification of Young’s modulus in tissue adjacent to implants by AFM analysis. n = 5 tissue samples per group and 70 force curves per sample. ***P < 0.001, one-way ANOVA followed by Bonferroni test. (E) Scatterplot with linear regression analysis showing the average tissue stiffness relative to α-SMA abundance in five samples from each condition plus two data points from sham-operated mice. r = 0.997, regression P = 1.03 × 10−12. (F) Quantification of Young’s modulus of the fascia tissue from sham-operated WT and Trpv4−/− mice (no implants). n = 5 tissue samples per group and 50 force curves per sample. One-way ANOVA followed by Bonferroni test. A.U., arbitrary units.
To assess the role of fibroblasts and myofibroblasts in tissue stiffness generation, we performed studies in a subcutaneous implant model in WT and Trpv4−/− mice using collagen-coated PA hydrogel discs of soft (1 kPa) and rigid (50 kPa) stiffness. We found that 4 weeks after implantation, the number of myofibroblasts, as determined by the abundance of α–smooth muscle actin (α-SMA), in the area surrounding the implant was 2.5-fold higher in WT mice receiving the 50-kPa matrix compared to the 1-kPa matrix, suggesting that increased implant stiffness augmented implant-elicited myofibroblast recruitment and differentiation (Fig. 1C). In contrast, similar accumulations of myofibroblasts were detected in Trpv4−/− mice receiving either 1- or 50-kPa implants, and these were reduced 2.5-fold compared to 50-kPa implants in WT mice, suggesting that the stiffness-induced myofibroblast differentiation in vivo was significantly diminished in the absence of TRPV4 (Fig. 1C). To test whether implant-adjacent tissue stiffening is associated with myofibroblast differentiation in vivo in mice, we directly measured fibrous capsule tissue stiffness using atomic force microscopy (AFM) in PA hydrogel–implanted skin tissues. At 28 days after implantation, skin tissue sections surrounding the implant were subjected to AFM analysis to obtain force curves of the tissues. We found that 4 weeks after implantation, the fibrous capsule tissue stiffness in the area surrounding the implant was 3.5-fold higher in WT mice receiving 50-kPa matrix compared to mice receiving 1-kPa matrix, suggesting that implant-elicited tissue stiffness was augmented by stiff matrix (Fig. 1D). In contrast, similar increases in stiffness were detected in Trpv4−/− mice receiving either 1- or 50-kPa implants, and these increases were reduced threefold compared to the increases in WT mice receiving 50-kPa implants, suggesting that implant stiffness–dependent tissue stiffening in vivo was significantly diminished in the absence of TRPV4 (Fig. 1D). Analysis of fibrous capsule tissue stiffness induction and myofibroblast differentiation showed a nearly perfect correlation between the two variables (r = 0.997, regression P = 1.03 × 10−12) (Fig. 1E), suggesting a role for myofibroblasts in tissue stiffening under FBR. As a control, we determined stiffness of the fascia underlying the dorsal skin tissue in unimplanted (sham-operated) WT and Trpv4−/− mice. We found fascia from WT and Trpv4−/− mice to have similar stiffness, suggesting that deficiency of TRPV4 did not affect the basal stiffness of the fascia (Fig. 1F).
We characterized the biophysical properties of PA gel implants by three distinct methods (fig. S1, A to D): cryo-fractured scanning electron microscopy to examine the integrity of PA gels and distribution of collagen on the gel surface (fig. S1A), AFM analysis to confirm the stiffness of the PA gels (fig. S1B), and immunofluorescence staining to show amounts of collagen I adsorption to both stiff and soft substrates (fig. S1C). Fibrosis is a critical pathological feature in FBR to biomaterials. Fibrosis, regardless of its cause or location, is associated with excess deposition of collagens, primarily collagen I, and other ECM proteins in the injured or inflamed tissue, which can lead to matrix remodeling and permanent scarring (70). For these reasons, we chose collagen to coat plates and coverslips. Our immunofluorescence data showed the presence of collagens at the interface of implant and skin tissue and other tissue areas that may result from both newly synthesized collagens and collagens present on the implanted gels (fig. S1D). We found increased collagen in WT tissue implanted with stiff matrix compared to Trpv4−/− tissue, which is consistent with results from Masson trichrome staining (Fig. 1A).
TRPV4 augments implant-adjacent tissue stiffness during FBR
Accumulating data suggests that besides the biomaterial’s inherent stiffness, consequent changes in the adjacent tissue stiffness after implantation can also promote FBR in a feed-forward manner (11–13, 23, 44, 71). To determine whether TRPV4 promotes tissue stiffening after implantation of cellular ester (a commonly used biomaterial) and to assess the clinical relevance of our data generated using PA gels (Fig. 1, A to H), we directly measured tissue stiffness (Young’s modulus) surrounding the implant using AFM in skin tissue from WT and Trpv4−/− mice (72, 73). Discs of mixed cellulose ester membrane were implanted subcutaneously into WT and Trpv4−/− mice (14). At 28 days after implantation, 30-μm skin tissue sections surrounding the implant were subjected to AFM analysis to obtain force maps of the tissues (Fig. 2, A to D). The scanned areas were chosen to be representative of the heterogenous nature of the tissue while being sufficiently localized to identify changes to the ECM due to implantation. A detailed inspection of the distribution of tissue stiffness revealed increased rigidity in the tissues in WT mice in response to nearby cellulose implants (Fig. 2B). However, loss of TRPV4 resulted in skewed distribution to softer regions (≤2 kPa) with a loss of tissue stiffness heterogeneity (Fig. 2B). In WT mice receiving cellulose implants, the mean tissue stiffness was 3.8 kPa as compared to 1.43 kPa in Trpv4−/− mice, possibly due to reduced ECM deposition. In WT sham-operated mice, the mean tissue stiffness was 1.58 kPa as compared to a mean value of 0.83 kPa in Trpv4−/− sham-operated mice, suggesting that TRPV4 positively regulates basal skin tissue stiffness (Fig. 2, C and D). Together, these results provide compelling evidence that TRPV4 is a key mediator of tissue stiffness during FBR and contributes to basal skin stiffness. These results contrast with the observation that there was no difference in the fascia stiffness between WT and Trpv4−/− mice (Fig. 1F). These different stiffness values may be the result of dissimilar tissue composition for fascia versus skin. However, in agreement with the results from collagen-coated PA implants, we found that the abundance of myofibroblasts and the induction of tissue stiffness in the area surrounding cellulose ester membrane implants 4 weeks after implantation was 3.5- and 3-fold higher, respectively, in WT mice than in Trpv4−/− mice, suggesting a link between myofibroblast generation and tissue stiffness in FBR (Fig. 2, C to E). These results are consistent with the well-established role of myofibroblasts in driving tissue stiffening in fibrosis not caused by implants (21, 22).
Fig. 2. TRPV4 promotes tissue stiffness during the FBR.
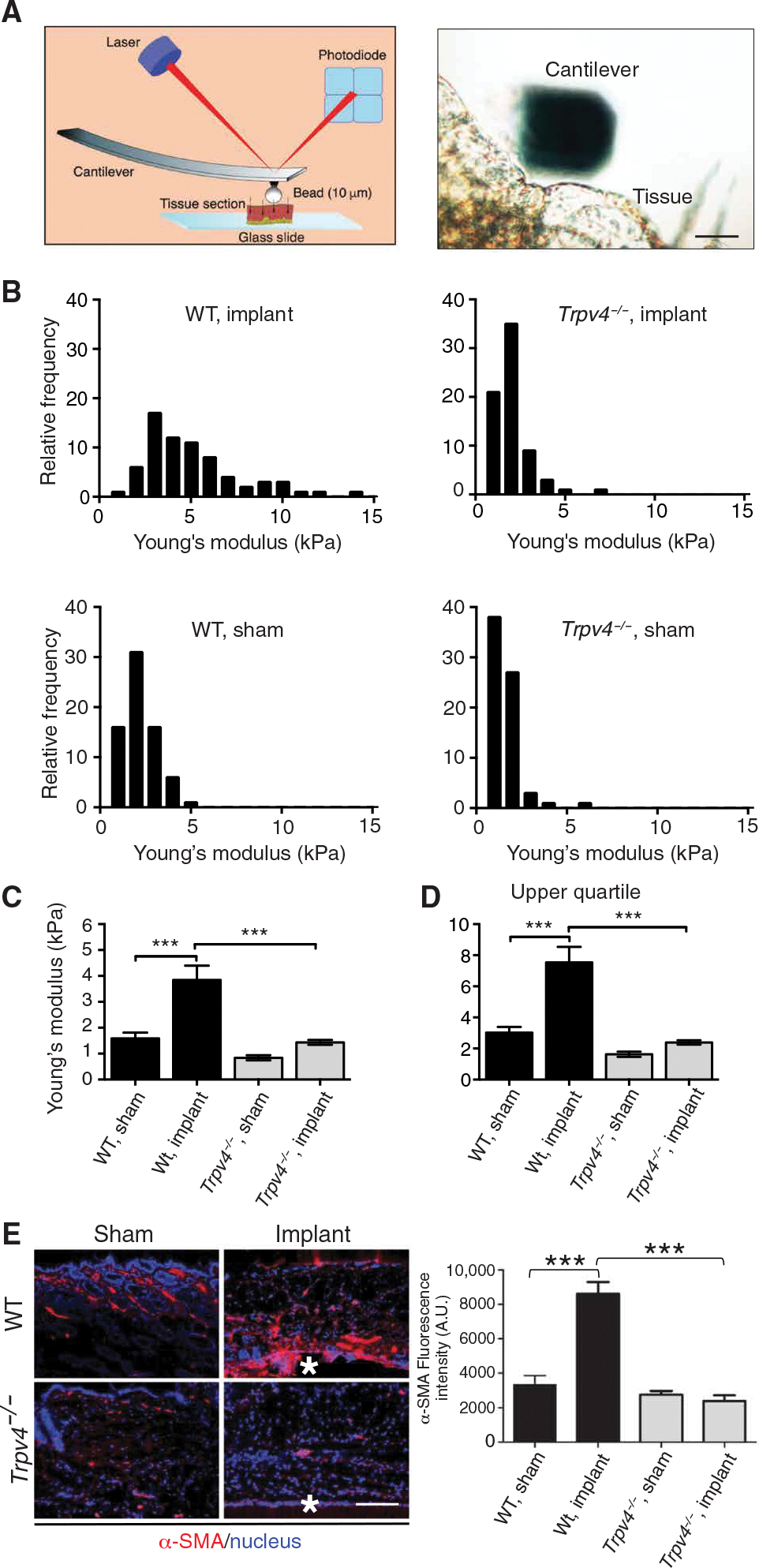
(A) Schematic diagram of the AFM system for measuring skin tissue stiffness (Young’s modulus) and a micrograph showing a representative example of the cantilever with an attached bead in contact with a tissue sample. Scale bar, 20 μm. A photodiode detector recorded the deflection of a laser beam by deformation of the cantilever attached to a 10-μm borosilicated glass bead. Force curves generated by this system were fitted to the Hertz model to calculate Young’s modulus. (B) Representative frequency distributions of force curves measured in individual skin tissue sections adjacent to cellulose ester implants from WT and Trpv4−/− mice and sham-operated mice. (C and D) Quantification of Young’s modulus (C) and upper quartile (D) data points from all samples. n = 5 tissue samples per group and 70 force curves per sample. ***P < 0.001, one-way ANOVA followed by Bonferroni test. (E) Imaging and quantification of the myofibroblast marker μ-SMA near the tissue-implant interface (asterisks) in skin tissue sections from WT and Trpv4−/− mice with or without cellulose implants. Scale bar, 50 μm. n = 5 mice per group and 2 implants per mouse. ***P < 0.001, Student’s t test. A.U., arbitrary units.
TRPV4 promotes FBGC formation and macrophage-dependent myofibroblast differentiation
The recruitment of macrophages to the implant and their fusion to generate FBGCs precedes myofibroblast differentiation, tissue stiffening, and encapsulation in the FBR. Because Trpv4 knockout reduced macrophage recruitment and FBGC formation at implants in vivo, we hypothesized that TRPV4 may play a role in macrophage recruitment or FBGC formation in a stiffness-dependent manner. To assess the importance of TRPV4 in implant-induced FBGC formation, we used an established in vitro assay for FBGC formation in response to the cytokines IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) (14). Bone marrow–derived macrophages (BMDMs) from WT and Trpv4−/− mice were plated on physiologically relevant soft (1 kPa) and stiff (50 kPa) collagen-coated PA hydrogels in the absence or presence of the cytokines. We found a significant increase in FBGC formation on high-stiffness PA gels compared to low-stiffness gels in WT macrophages in the absence of IL-4 and GM-CSF (Fig. 3, A to C). Cytokine priming further increased the numbers of FBGCs and the percentage of macrophages that underwent fusion (Fig. 3, A to C). These changes in FBGC formation mediated by substrate stiffness and soluble factors were absent in Trpv4−/− macrophages (Fig. 3, A to C), suggesting a critical role for TRPV4 in physiologically relevant stiffness-induced FBGC formation.
Fig. 3. TRPV4 promotes the functional interaction of macrophages with fibroblasts in response to fusogenic cytokines on physiologically relevant stiff substrates.

(A) Merged bright-field and fluorescence imaging of multinucleated FBGCs in WT or Trpv4−/− BMDMs plated on PA gels of different stiffness and treated with the fusogenic cytokines IL-4 and GM-CSF. Cell nuclei were stained with DAPI. (B and C) Quantification of the number of FBGCs/high-power field (hpf) (B) and the percentage of BMDMs involved in fusion events (C). Scale bar, 20 μm. n = 3 biological replicates and 5 images per group. **P < 0.01 and ***P < 0.001, Student’s t test. (D) Representative immunofluorescence images showing α-SMA, phalloidin, and nuclei in WT mouse dermal fibroblasts after 48 hours of stimulation with CM collected from WT or Trpv4−/− BMDMs plated on collagen-coated PA gels of 1- or 50-kPa stiffness. Scale bar, 20 μm. (E) Quantification of myofibroblast differentiation from the experiment shown in (D). n = 3 independent experiments and 10 cells per condition. **P < 0.01 and ***P < 0.001, Student’s t test. Ut, untreated. A.U., arbitrary units.
To determine whether TRPV4 was required for macrophage-induced differentiation of fibroblasts into myofibroblasts, we assessed whether conditioned medium (CM) collected from WT or Trpv4−/− BMDMs grown on defined stiffness PA gels with or without IL-4 and GM-CSF modulated myofibroblast differentiation. We treated primary mouse dermal fibroblasts with CM collected from WT or Trpv4−/− BMDMs grown on varied stiffness gels with or without IL-4 and GM-CSF, using fresh medium as a control. We found a significant increase in myofibroblast differentiation with CM from macrophages cultured on high-stiffness substrate compared to those cultured on low-stiffness substrate (Fig. 3, D and E). Priming the macrophages with IL-4 plus GM-CSF further increased the number of myofibroblasts (Fig. 3, D and E). These stiffness- and soluble factor–mediated changes in myofibroblast differentiation were absent in response to CM collected from Trpv4−/− macrophages (Fig. 3, D and E), suggesting a critical role for TRPV4 in physiologically relevant stiffness-induced functional interactions between myofibroblasts and macrophages.
TRPV4 residues 100 to 130 are required for FBGC formation in response to mechanical stimulation
We mapped TRPV4 for functional domains that could support mechanical stimulus–induced TRPV4 activation in macrophages during FBR. Three N-terminal deletion mutants of different lengths (TRPV4-Δ1-30, TRPV4-Δ1-130, and TRPV4-Δ100-130) were created and cloned into an adenovirus (Ad) expression system (Fig. 4A). These three mutants were specifically chosen because earlier studies in a different cell type indicated that these regions were functionally relevant for activation of TRPV4 in response to biomechanical stimuli (74). We confirmed successful expression of WT TRPV4 (Ad-TRPV4-WT) and the mutant constructs in Trpv4−/− BMDMs by Western blot analysis analyzing both cytosolic and plasma membrane fractions (Fig. 4B). To determine which domain(s) of TRPV4 supported its activation by a mechanical stimulus in BMDMs, we assayed Ca2+ influx induced by 70% hypotonic (90 mosM) and isotonic (300 mosM) solutions in Trpv4−/− BMDMs transfected with the WT or mutant TRPV4 constructs or with empty vector (Ad-Vec). In a hypotonic environment, Trpv4−/− BMDMs transfected with Ad-TRPV4-WT exhibited high Ca2+ influx as expected, whereas the Ad-TRPV4-Δ1-30 mutant showed a twofold increase in Ca2+ influx compared to the Ad-TRPV4-WT despite the fact that TRPV4-Δ1-30 was poorly expressed on the plasma membrane (Fig. 4, B to D). Ca2+ influx was reduced to about 60% of WT in Ad-TRPV4-Δ1-130 mutant transfectants (Fig. 4, C and D). Whereas Ca2+ influx was similarly reduced in the Ad-TRPV4-Δ100-130 deletion construct, no Ca2+ influx was recorded for any of the constructs in an isotonic environment (Fig. 4, C and D). Together, the requirement for residues 100 to 130 to support hypotonicity-induced Ca2+ influx indicates that this region is indispensable for TRPV4 mechanosensing in BMDMs.
Fig. 4. TRPV4 residues 100 to 130 mediate the response to biomechanical stimulus.

(A) Schematic diagram of adenovirus (Ad) expression constructs encoding WT TRPV4 and three mutant forms with deletions of N-terminal residues 1 to 130, 1 to 30, or 100 to 130. PRD, proline-rich domain; ANK, ankyrin repeats. (B) Immunoblotting for TRPV4, flotillin, and tubulin in membrane and cytosolic fractions of Trpv4−/− BMDMs transfected with the indicated Ad expression constructs. Ad-Vec is an empty vector control. Image is representative of n = 3 biological replicates. (C) Representative FlexStation 3 recording of Ca2+ influx in Trpv4−/− BMDMs transfected with the indicated Ad expression constructs and treated with isotonic (300 mosM) or 70% hypotonic (90 mosM) solution. (D) Quantification of Ca2+ influx from the experiment shown in (C). Experiments were repeated three times in quadruplicate. A23, Ca2+ ionophore A23187. RFU, relative fluorescence unit. ***P < 0.001, Student’s t test.
To assess whether attenuation of the TRPV4 mechanosensing response seen in Ad-TRPV4-Δ1-130 and Ad-TRPV4-Δ100-130 transfectants associated with a reduction in FBGC formation, we used a gain-of-function strategy as used previously (14, 24). We overexpressed TRPV4-WT, TRPV4-Δ1-30, TRPV4-Δ1-130, and TRPV4-Δ100-130 in Trpv4−/− BMDMs by transfection of each Ad expression construct. In these experiments, Ad-Vec was used as a negative control, and untransfected WT BMDMs were used as a positive control. We assessed the transfectants and WT controls in the IL-4 plus GM-CSF–induced FBGC formation assay (Fig. 5, A to D). The WT BMDM control and both Ad-TRPV4-WT and Ad-TRPV4-Δ1-30 transfectants showed a mean number of five FBGCs per field; this number was significantly reduced in Trpv4−/− BMDMs transfected with Ad-Vec, Ad-TRPV4-Δ1-130, or Ad-TRPV4-Δ100-130 (Fig. 5, A and B). The percentage of cells involved in fusion events was reduced by 28 and 37%, respectively, in Ad-TRPV4-Δ1-130 and Ad-TRPV4-Δ100-130 transfectants compared to Ad-TRPV4-WT transfectants (Fig. 5, A and C). These results were also congruent with the average size of the FBGCs, which were significantly smaller in the Ad-TRPV4-Δ1-130 and Ad-TRPV4-Δ100-130 transfectants (by 35 and 45%, respectively) compared to Ad-TRPV4-WT transfectants (Fig. 5, A and D). Consistent with the biomechanical stimuli–induced Ca2+ influx data, these results suggested that residues 100 to 130 were crucial for FBGC formation in response to mechanical stimulation, whereas residues 1 to 30 had no noticeable effect on FBGC formation.
Fig. 5. TRPV4 residues 100 to 130 are required for FBGC formation.

(A) Representative images of multinucleated FBGCs in Giemsa-stained untreated WT or Trpv4−/− BMDMs transfected with Ad-TRPV4-WT, Ad-TRPV4-Δ1-30, AdTRPV4-Δ1-130, Ad-TRPV4-Δ100-130, or Ad-Vec (empty vector). (B to D) Quantification of the number of FBGCs/hpf (B), the percentage of BMDMs undergoing fusion (C), and the average size of FBGCs (D). Scale bar, 100 μm. n = 3 biological replicates and 6 images per group. **P < 0.01 and ***P < 0.001, Student’s t test.
TRPV4 deficiency does not affect macrophage receptors known to be involved in the FBR or FBGC formation
Macrophage scavenger receptor CD36 has been linked to the generation of multinucleated giant cells through macrophage cell fusion in vitro (50). To investigate whether TRPV4-mediated FBGC formation required CD36, we treated WT and Trpv4−/− BMDMs with IL-4 and GM-CSF for 48 hours. Immunofluorescence analysis showed that cytokine stimulation of WT cells caused increased colocalization of CD36 and TRPV4 on the plasma membrane in multinucleated cells compared to untreated WT BMDMs (Fig. 6A). We saw no change in the abundance or plasma membrane localization of CD36 in Trpv4−/− cells with or without cytokine stimulation (Fig. 6A). Additional analysis by fluorescence-activated cell sorting (FACS) detected no difference in CD36 abundance on the cell surface of either WT or Trpv4−/− BMDMs with or without IL-4 and GM-CSF stimulation, although a small increase in surface TRPV4 was seen in cytokine-stimulated WT cells (Fig. 6, B and C). We also assessed CD36 and other macrophage membrane receptors [scavenger receptor A-1 (SRA-1), cadherin-1, CD47, and TLR4 (Toll-like receptor 4)] associated with FBR, FBGC formation, macrophage fusion, and phagocytosis by Western blot analysis. Our results showed similar amounts of each receptor in both WT and Trpv4−/− BMDMs in the presence or absence of IL-4 and GM-CSF (Fig. 6D). Together, these results suggest that TRPV4 deficiency in BMDMs does not alter the abundance or localization of membrane receptors currently known to be involved in the FBR or FBGC formation under fusogenic cytokine stimulation.
Fig. 6. Absence of TRPV4 does not perturb BMDM receptors known to be involved in the FBR or FBGC formation.

(A) Representative images showing CD36, TRPV4, and nuclei (DAPI) in WT and Trpv4−/− BMDMs after treatment with IL-4 + GM-CSF or untreated. Cells were also stained with isotype control IgGs. Scale bars, 20 μm. n = 3 independent experiments and 10 cells per condition. (B) Representative log plot of flow cytometric data showing cell surface abundance of CD36 in WT and Trpv4−/− BMDMs and TRPV4 in WT BMDMs after incubation with IL-4 + GM-CSF for 24 hours. (C) Quantification of data from (B). n = 3 independent experiments and 20,000 cells per condition; Student’s t test. (D) Immunoblotting and quantification of the BMDM receptors CD36, ECAD1, SRA-1, TLR4, and CD47 at 24 and 48 hours after treatment of WT and Trpv4−/− BMDMs with IL-4 + GM-CSF. GAPDH is a loading control. n = 3 biological replicates. Two-way ANOVA followed by Bonferroni test. A.U., arbitrary units.
TRPV4 promotes intracellular stiffening in macrophages under fusogenic conditions
To assess the role of TRPV4 in the regulation of cell stiffness under fusogenic conditions, we measured the intracellular stiffness of WT BMDMs by AFM after growing cells in the absence or presence of IL-4 plus GM-CSF for 48 hours (Fig. 7, A to H). The calculated stiffness correlated well with previously reported values for different indenters (75–80), where a slight overestimation of Young’s modulus could be expected because of the use of nanometer-sized tips. A detailed inspection of the distribution of cell stiffness showed a significantly increased Young’s modulus heterogeneously distributed near lamellipodial areas in WT BMDMs treated with IL-4 plus GM-CSF compared to untreated cells (Fig. 7, B to D and G). To assess whether TRPV4 was required for the increase in cell stiffness elicited by IL-4 plus GM-CSF treatment, we also measured intracellular stiffness in Trpv4−/− BMDMs expressing Ad-Vec or Ad-TRPV4-WT and treated with IL-4 plus GM-CSF. AFM analysis revealed that the relative frequency of the Young’s modulus of Trpv4−/− cells transfected with Ad-Vec showed reduced heterogeneity, with most values skewed toward the softer region (<20 kPa). Transfection of Ad-TRPV4-WT restored the diminished stiffness and lack of heterogeneity in Trpv4−/− BMDMs (Fig. 7, B and E to G). Upper quartile values followed the same trend, suggesting that the difference of Young’s modulus remained the same at the stiffer region in all four experimental conditions (Fig. 7H). Together, these results suggest that TRPV4 is directly involved in intracellular stiffness induction in BMDMs under fusogenic conditions.
Fig. 7. TRPV4 is directly involved in intracellular stiffness induction in BMDMs stimulated with fusogenic cytokines.

(A) Schematic diagram of the AFM system for determining the Young’s modulus of BMDMs and a micrograph showing a BMDM in contact with a cantilever. Scale bar, 20 μm. A laser beam deflected by the deformation of a cantilever attached to a 30-nm circular symmetric quartz probe was captured by a photodetector. Young’s modulus values (500 per condition) were acquired by fitting individual force curves to the Hertz model. (B) AFM micrographs using heat mapping to represent variations in stiffness in the cell body and near lamellipodia-filopodial areas of WT and Trpv4−/− BMDMs treated as indicated. Scale bar, 5 μm. (C to F) Frequency distribution of Young’s modulus values of WT and Trpv4−/− BMDMs from the experiment shown in (B). n = 3 independent experiments, 10 cells per group, and 3 to 4 areas per cell scanned. (G) Quantification of Young’s modulus for each group and (H) upper quartile data points from the histograms of the experiment in (B). ***P < 0.001, one-way ANOVA followed by Bonferroni test.
Throughout this study, we used several strategies to reduce confounding effects from the stiffness of the underlying substrate. First, we used a quartz AFM probe with a tip diameter of 60 nm and applied a force of 0.5 nN to ensure lesser indentation of the cell. In this configuration, the 60-nm tip is blunt enough to ensure accurate contact point determination during the measurement while allowing us to obtain structural information from the cell surface. Using this probe, AFM was operated in advanced force spectroscopy–based mode, referred to as “quantitative imaging,” which allows nanomechanical characterization and simultaneous imaging of the cell under various treatment conditions (Fig. 7B). Second, to limit the contribution from the underlying substrate, which could be substantial with very thin samples such as the spread area of a cell, we calculated the Young’s modulus by fitting data from only the first 25% of the indentation depth (~250 nm) into the Hertz model. Moreover, published stiffness values for macrophages and similar cell types vary significantly (75–80). This can be attributed, in part, to substrate stiffness because macrophages have been shown to adjust their mechanosensing activity in response to changes in surrounding physical cues (79). However, several of the reported stiffness studies vary in terms of indenter shapes and sizes, ranging from nanometer-scale sharp tips to micrometer-scale spheres, which have been shown to influence measured stiffness values (80). Typically, with larger micrometer spherical indenters, cell stiffness heterogeneity, originating from the different stiffnesses of the individual components, is averaged down. In contrast, with nanometer-sized sharper indenters, as we have used here (Fig. 7A), it is possible to distinguish between the contributions of individual structures, such as cytoskeletal filaments or podosomes, which lead to higher force pressure exerted over a much smaller area of the measured structures and which result in stiffness values in the range of 20 to 50 kPa.
TRPV4-dependent actin polymerization and intracellular stiffening is critical for FBGC formation
Cytoskeletal remodeling governs numerous physiological processes including cell fusion (69, 81). Cell stiffness is predominantly mediated by cytoskeletal remodeling that generates F-actin (82). To test whether TRPV4-dependent FBGC formation was associated with cytoskeletal remodeling, we determined the amount of F-actin generation in FBGCs induced by IL-4 plus GM-CSF. We found enrichment of F-actin rings in multinucleated FBGCs in WT BMDMs but no F-actin enrichment and no FBGCs in Trpv4−/− BMDMs (Fig. 8A). We also found increased colocalization of TRPV4 and F-actin in multinucleated WT BMDMs after treatment with IL-4 plus GM-CSF compared to untreated cells (Fig. 8B). Examination of the lamellipodia and filopodia, key phenotypic features of FBGCs (54), also revealed a dense colocalization of TRPV4 and F-actin in multinucleated WT BMDMs after treatment with IL-4 plus GM-CSF (Fig. 8B). Using high-resolution AFM imaging, we showed that TRPV4 deficiency impaired lamellipodia and filopodia formation in the presence or absence of IL-4 plus GM-CSF in BMDMs (Fig. 8, C and D). Reconstitution of Trpv4−/− BMDMs with Ad-TRPV4-WT followed by IL-4 plus GM-CSF treatment, in part, reversed impaired lamellipodia and filopodia formation compared to Ad-Vec–transfected cells, suggesting a direct role of TRPV4 in this process (Fig. 8, C and D). Additional results showed that Ad-TRPV4-WT–dependent reversion of lamellipodia and filopodia formation in Trpv4−/− BMDMs was blocked by treatment with the F-actin inhibitor cytochalasin D (5 μM), suggesting that F-actin–supported lamellipodia and filopodia formation was, in part, mediated by TRPV4 (Fig. 8, C and D).
Fig. 8. TRPV4-dependent FBGC formation depends on an increase in intracellular stiffness driven by F-actin–dependent cytoskeletal remodeling.

(A) Phalloidin staining to show F-actin (red) in WT and Trpv4−/− BMDMs stimulated with IL-4 + GM-CSF. Cells were counterstained with DAPI (blue). Dashed boxes in the left images indicate the areas shown in higher magnification on the right. Arrowheads indicate the accumulation of F-actin. Scale bars, 50 μm (left) and 10 μm (right). (B) Immunofluorescence images showing WT BMDMs stained for TRPV4 and F-actin 7 days after IL-4 + GM-CSF treatment or no stimulation. Scale bars, 10 μm. In (A) and (B), n = 50 cells per group from two independent experiments. (C) High-resolution AFM micrographs showing the distribution of lamellipodia-filopodial areas of the indicated cell groups. Scale bar, 2 μm. n = 3 independent experiments, 10 cells per group, and 2 scanned areas per cell. (D) Quantification of percent area of filopodia in BMDMs from the experiment shown in (C). **P < 0.01 and ***P < 0.001, Student’s t test. (E) Quantification of Young’s modulus and (F) upper quartile data points acquired from the experiment shown in (C). n = 3 independent experiments and 500 data points per group. ***P < 0.001, one-way ANOVA followed by Bonferroni test. (G) Giemsa staining to show FBGC formation by Trpv4−/− BMDMs transfected with Ad-Vec (empty vector) or Ad-TRPV4-WT with or without cytochalasin D (Cyt D) treatment after 8 days of stimulation with fusogenic cytokines (IL-4 + GM-CSF). Scale bar, 50 μm. (H to J) Quantification of the number of FBGC/hpf (H), percent fusion (I), and the average size of FBGCs (J) from the experiment shown in (G). n = 3 independent experiments and 5 images per group. **P < 0.01 and ***P < 0.001, Student’s t test.
We also measured the generation of intracellular stiffness in BMDMs using AFM under similar conditions as above and found that cellular stiffness decreased by fourfold in the presence of cytochalasin D in Ad-TRPV4-WT–overexpressing Trpv4−/− BMDMs compared to vehicle-treated Ad-TRPV4-WT–overexpressing Trpv4−/− or WT BMDMs (Fig. 8, E and F). Because the generation of both lamellipodia and filopodia and matrix stiffness were associated with cell fusion and both processes were shown to depend on TRPV4, we examined whether TRPV4-induced FBGC formation was reliant on the generation of F-actin. Trpv4−/− BMDMs transfected with Ad-TRPV4-WT showed an increase in the number of FBGCs, in the percentage of macrophages that underwent fusion, and in the average size of FBGCs compared to the Ad-Vec–transfected counterparts; these factors were all significantly diminished by the presence of cytochalasin D (Fig. 8, G to J). Together, these results suggest that F-actin–dependent remodeling of the cytoskeleton and consequent induction of intracellular stiffness plays an essential role in TRPV4-dependent FBGC formation.
DISCUSSION
Macrophages are important in the FBR, and emerging data support a role for the stiffness of natural and engineered substrates, the ECM, and intracellular matrices in macrophage differentiation and inflammation (15–32, 81–84). The FBR and FBGC formation and development depend on the integration of biomechanical and biochemical signals (4). We previously showed that TRPV4, a mechanosensitive ion channel, is required for FBR and FBGC formation (14). The data presented here demonstrate that TRPV4 was required for the enhanced differentiation of FBGCs and myofibroblasts on stiff compared to soft implants and was essential for augmenting implant-adjacent tissue stiffness under FBR. Although TRPV4 did not affect the abundance or localization of receptors known to be required for the FBR or FBGC formation in vivo, it was critical for fusogenic cytokines to stimulate BMDM fusion and for paracrine factors from stimulated BMDMs to promote myofibroblast activation in vitro. Furthermore, we provide evidence that TRPV4 activity increased the intracellular stiffness of macrophages by promoting F-actin–dependent cytoskeletal remodeling.
Tissue stiffness correlates directly with ECM deposition (72). Increased matrix stiffness triggers FBR (23), which is consistent with our previous report of reductions in the FBR and in collagen deposition in implant-adjacent tissue in Trpv4 knockout mice (14). Here, we extended those findings by demonstrating that stiff implants (50 kPa) induced a heightened FBR compared to soft implants (1 kPa), as evidenced by increased collagen deposition, fibrosis, and accumulation of macrophages and FBGCs at the tissue-implant interface in WT mice (Fig. 1, A to C). These enhanced responses to stiffer implants depended on TRPV4. The late stage of FBR involves prolonged ECM remodeling that leads to exuberant fibrosis and hardening of the tissue, encompassing the implant through the deposition of collagen and other ECM proteins (1–4, 11, 84). Data presented here suggest a direct role for TRPV4 in promoting tissue stiffness in FBR (Fig. 2, B to D). Because TRPV4 is sensitive to both intracellular tension and surrounding matrix stiffness (18, 19, 24, 27, 85), our finding that stiffer implants lead to increased FBR and consequent tissue stiffening in a TRPV4-dependent manner suggests a feed-forward mechanism in which a reciprocal functional interaction between TRPV4 and matrix stiffness propels FBR.
During FBR, macrophages are likely subjected to a complex array of biophysical cues in vivo because of their interactions with other cell types, such as fibroblasts, rigidity of the tissue matrix, and the inherent stiffness of the implant, all of which can modify their responses (44, 86, 87). For example, macrophages preferentially engulf stiffer particles over softer particles of the same chemical composition (88), and stiffer three-dimensional polyethylene glycol–RGD (Arg-Gly-Asp) hydrogels elicit stronger FBR and larger numbers of activated macrophages at the tissue-implant interface (23). Macrophages, similar to other cells, use adhesion molecules such as integrins and cadherins to sense changes in substrate stiffness, and these molecules are associated with processes such as stress fiber development and cytoskeletal remodeling (89). In our effort to identity a mechanosensing plasma membrane receptor and associated mechanisms by which mechanical signals are transduced into cells to drive FBR processes, we reported that TRPV4 is involved in FBR and FBGC formation (14). Using deletion strategies, we have extended these findings to show that N-terminal residues 100 to 130 of TRPV4 are indispensable for its mechanosensing activity in macrophages, as revealed by the requirement for these residues in executing hypotonicity-induced Ca2+ influx (Fig. 4, C and D). Consistent with this Ca2+ influx data, we further found that these residues were critical for FBGC formation (Fig. 5, A to D). It is not clear why the deletion of residues 1 to 30 of TRPV4 increased Ca2+ influx and FBGC formation in our assays; therefore, this question merits further investigation. Activation of the TRPV4 channel requires phosphatidylinositol 4,5-bisphosphate (PIP2) binding in the N-terminal region (residues 121 to 125) of the protein (74). Deletion of residues 100 to 130 was thought to interfere with PIP2-TRPV4 interaction, subsequently causing suppression of activation of the channel (90). Whether similar PIP2-TRPV4 binding is also required for FBGC formation remains to be determined.
Cell fusion is an integral part of several normal and pathophysiological processes including fertilization, muscle development, bone homeostasis, and the response to implants (46–48). To date, a small number of plasma membrane receptors have been shown to take part in macrophage fusion, including CD36, CD47, and E-cadherin (4, 50–52). However, we found that TRPV4 deficiency did not alter CD36 expression or its translocation to the plasma membrane in BMDMs stimulated with fusogenic cytokines (Fig. 6, A to D). The expression of another surface molecule associated with macrophage fusion, CD47 (52), was also unaffected by the absence of TRPV4 (Fig. 6D). We also found no change in the abundances of cadherin-1, SRA-1, or TLR4 in Trpv4−/− cells compared to those in WT BMDMs (Fig. 6D), raising the possibility that impaired FBGC formation seen in the absence of TRPV4 function may be independent of these receptors.
We found that 4 weeks after implant, the abundance of myofibroblasts in the area surrounding the implant was sensitive to the stiffness of the implants in a TRPV4-dependent manner (Fig. 1C), suggesting that stiffness-induced myofibroblast differentiation in vivo under FBR was modulated by TRPV4. Moreover, our AFM analysis revealed that the tissue stiffness in the area surrounding the implant was 3.5-fold higher in WT mice receiving stiff implants compared to soft implants, whereas tissue stiffening in response to stiff implants in vivo was diminished in the absence of TRPV4 (Fig. 1, D and F). Similarly, the abundance of myofibroblasts and the induction of tissue stiffness in the area surrounding the cellulose ester filter implant was 3.5- and 3-fold higher, respectively, in WT mice compared to Trpv4−/− (Fig. 2, B to E), suggesting that the association between myofibroblast generation and tissue stiffness is also evident in a clinically relevant biomaterial implantation model. In addition, we found a nearly perfect correlation between tissue stiffening and myofibroblast differentiation. Together, these results are consistent with TRPV4-dependent myofibroblast generation and consequent remodeling of the tissue matrix contributing to tissue stiffening and FBGC formation under FBR. Furthermore, we found a critical role for TRPV4 in physiologically relevant substrate stiffness–induced FBGC formation, which was further augmented in the presence of fusogenic soluble factors (Fig. 3, A to C). Macrophages and fibroblasts are both sensitive to changes in matrix stiffness, and these cells coexist in inflamed fibrotic tissue, which creates the opportunity for functional interaction between these two cell types. We found a significant increase of myofibroblast differentiation by CM from macrophages grown on physiologically relevant high-stiffness (50 kPa) substrate compared to macrophages grown on CM from low-stiffness (1 kPa) substrate, and priming the macrophages with fusogenic cytokines further increased the numbers of myofibroblasts (Fig. 3, D and E). These stiffness- and biochemical factor–mediated changes in myofibroblast differentiation were absent in response to CM collected from Trpv4−/− macrophages, suggesting a critical role of TRPV4 in stiffness-induced functional interactions between myofibroblasts and macrophages. Thus, we speculate that under FBR, a concerted action of TRPV4-dependent biochemical and soluble factors as well as implant and ECM stiffness determine the macrophage and fibroblast responses.
Because TRPV4 activation in fibroblasts promotes their differentiation into myofibroblasts (24), some of the reduction in myofibroblast recruitment to implants that we observed in Trpv4 knockout mice may be due to a role for TRPV4-mediated fibroblast activation during the FBR. Furthermore, TRPV4 is proinflammatory in various cell types, including macrophages (83, 91), making it likely that factors other than substrate stiffness sensing play a role in the regulation of FBR by TRPV4. Examining the FBR in the context of macrophage- or fibroblast-specific Trpv4 knockout would shed more light on the relative contribution of TRPV4-mediated signaling in these cell types to the FBR. This work is currently underway in our laboratory. We also cannot exclude the possibility that the reduced FBR and FBGC formation in macrophages in global Trpv4-null mice is associated with developmental compensation or unknown secondary compensation. However, previous studies from our group showed that deficiency of TRPV4 did not affect basal macrophage maturation in vivo (14). Future studies using macrophage- or fibroblast-specific Trpv4-null mice will shed more light on this area.
Macrophages are central to the development and progression of the FBR (4, 11–14). Previous reports by our laboratory and others have shown that various FBR-related macrophage functions including differentiation, adhesion, motility, phagocytosis, spreading, and cytoskeletal remodeling are affected by matrix stiffness, suggesting that stiffness may be a critical biomechanical cue in the development of the FBR (14, 19, 23, 25, 38–43). Macrophage fusion leading to the formation of FBGCs exhibits multiple features of phagocytosis such as actin polymerization and the involvement of mannose receptor activity (4, 11, 12). It is also recognized that failed phagocytosis of macroscopic implants by macrophages leads to FBGC generation, chronic inflammation, and consequent development of the FBR (4, 11, 12). In this respect, a previous report showed that inhibition of Rac activation or Rac1 knockdown can prevent lamellipodia formation and macrophage fusion without interfering with the ability of macrophages to take up microspheres, implying that fusion and phagocytosis may be partially decoupled (54). TRPV4 mechanosensing has been linked to macrophage phagocytic function (41), implying that impairment of TRPV4-dependent phagocytosis may be involved in the FBR.
Lack of TRPV4 function abrogated the fusogenic cytokine–induced increase in intracellular stiffness in BMDMs (Fig. 7, C to D), demonstrating that TRPV4 is absolutely required for intracellular stiffness induction in BMDMs under fusogenic conditions. Cell stiffness is mostly regulated by F-actin generation (82), and we showed that TRPV4 deficiency decreased F-actin generation and lamellipodia and filopodia formation in response to fusogenic cytokines (Fig. 8, A to F). Cytochalasin D inhibited the reversion of both intracellular stiffness and lamellipodia and filopodia formation in TRPV4-reconstituted knockout BMDMs (Fig. 8, D to F), demonstrating that both responses depended on TRPV4-mediated increases in F-actin. Lamellipodia formation through the Rac1 pathway has been previously implicated in cell fusion and FBGC formation (54), and we previously reported that TRPV4-dependent fusion of BMDMs in vitro also depends on the Rac1 pathway (92). The findings reported here are consistent with TRPV4-mediated generation of F-actin being mediated by Rac1.
Because TRPV4 proteins have numerous physiological functions, directly and nonspecifically targeting TRPV4 is likely not a desirable strategy for treating the FBR to biomaterials. TRPV4 has been linked to sheer stress detection in blood vessels, osmolarity sensing in kidneys, sensing of bladder filling, maintenance of epithelial barrier function in lungs, and pain sensation (59–61, 65). TRPV4 also plays an essential role in osteoclastogenesis by modulating the terminal differentiation and activity of osteoclasts, which are also a type of multinucleated giant cell (67). Regarding the function of TRPV4 in cell types involved in the FBR, it is required for phagocytosis in macrophages (41), and it promotes the differentiation of fibroblasts into myofibroblasts (24), which contribute not only to fibrosis but also to normal repair and regenerative processes. A molecular or pharmacological strategy targeting the downstream signaling events specific to the FBR- and FBGC-promoting functions of TRPV4 without disrupting its functions in other contexts would have great therapeutic potential.
In summary, the results of our study support the notion that a feed-forward functional interaction between TRPV4 and matrix stiffness leads to cytoskeletal remodeling and cellular force generation in macrophages that promote FBGC formation in vitro and the FBR in mice. This process is likely to be involved in the FBR to stiff implants in humans. These findings provide insights into macrophage-fibroblast and macrophage-implant interactions, which might be exploited in the development of immune-competent bioimplants and therapeutics.
MATERIALS AND METHODS
Reagents and antibodies
The following primary antibodies were purchased: TRPV4 (Alomone Labs), CD36, SRA-1 (R&D Systems), collagen I, CD68 (Bio-Rad), CD47, TLR4, epithelial cadherin 1 (ECAD1), flotillin-1, tubulin (Cell Signaling Technology), α-SMA (Thermo Fisher Scientific), phycoerythrin (PE)–conjugated CD36, and glyceraldehyde phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology). Secondary goat, mouse, and rabbit antibodies were obtained from Jackson ImmunoResearch. Alexa Fluor 488– and Alexa Fluor 594–conjugated immunoglobulin G (IgG), ProLong Diamond 4′,6-diamidino-2-phenylindole (DAPI), Alexa Fluor 488 and Alexa Fluor 594 Phalloidin, Western blot reagents, and the Mem-PER Plus Membrane Protein Extraction Kit were acquired from Thermo Fisher Scientific. PE-conjugated goat and rabbit IgG, recombinant murine IL-4, M-CSF, and GM-CSF proteins were purchased from R&D Systems. A23187 (calcium ionophore), Giemsa solution, hematoxylin solution, and Masson’s trichrome staining kit were purchased from Millipore Sigma. Dulbecco’s modified Eagle’s medium (DMEM), antibiotic-antimycotic, fetal bovine serum (FBS), and other cell culture reagents were purchased from Gibco. FLIPR calcium 6 assay kits for Ca2+ influx analysis were obtained from Molecular Devices. Cytochalasin D was obtained from Tocris Bioscience. The following Ad constructs were generated by Vector Biolabs: Ad (RGD)–mouseTRPV4-WT (Ad-TRPV4-WT), three deletion constructs of different lengths of mouse TRPV4 gene (Ad-TRPV4-Δ1-30, Ad-TRPV4-Δ1-130, and Ad-TRPV4-Δ100-130), and Ad-Vec control.
Mice and stiffness-induced FBR model
Trpv4 knockout mice (Trpv4−/−) on a C57BL/6 background were originally created by M. Suzuki (Jichi Medical University, Tochigi, Japan). Congenic WT mice were purchased from Charles River Laboratories. Institutional Animal Care and Use Committee guidelines were followed for each experiment, and animal protocols were approved by the University of Maryland review committee. All mice used for experiments were 8 to 12 weeks old. For generating FBR, mixed cellulose ester (0.45-μm pore size filters, 12 mm2; Millipore, St. Louis, MO) or collagen-coated PA gel of varied stiffness (1 and 50 kPa; Matrigen) were purchased, and 6-mm-diameter implant discs were embedded subcutaneously in each flank of the mice, with each mouse receiving two implants of the same stiffness. The implants and the surrounding skin tissue were surgically removed from the experimental mice 28 days after the initial implantation (14). Masson’s trichrome analysis, DAPI staining, and CD68 immunostaining were used to determine the development of the fibrotic area, FBGC, and macrophage abundance, respectively, near the implanted area in the skin tissue section (14, 68).
In vitro FBGC formation from BMDMs and Ad transfection
Bone marrow was harvested from mouse femur and incubated with M-CSF (25 ng/ml) supplemented DMEM for 7 to 8 days to obtain mature BMDMs (14, 19, 42). BMDMs were seeded on Permanox plastic slides (Lab-Tek chamber slides; Nunc) with DMEM, 10% FBS, and infected with Ad TRPV4 constructs or empty vector control (working concentration: 108 plaque-forming units/ml) for 48 hours followed by a medium replacement and another 24 hours of incubation with 10% serum containing DMEM. Fresh DMEM containing murine IL-4 plus GM-CSF (25 ng/ml) was added to the cells and maintained for 6 to 7 days until fusions reached maximum levels (14, 91). Slides were stained with Giemsa solution, and the numbers and average size of FBGCs per field were counted, and the total area occupied by FBGCs per field was measured. The percentage of BMDMs taking part in cell fusion was calculated from the number of giant cell nuclei (>5 nuclei) divided by the number of total nuclei (14).
Myofibroblast differentiation induced by CM
Primary normal mouse dermal fibroblasts were obtained from skin biopsies from armpit of 8- to 10-week-old WT and Trpv4−/− mice as described previously (20). For giant cell formation, fully differentiated BMDMs were incubated on collagen-coated 1- or 50-kPa petri plates (Matrigen) in DMEM containing 10% serum for 24 hours. Cells were then treated with murine IL-4 plus GM-CSF (25 ng/ml) or phosphate-buffered saline (PBS) in DMEM with 10% serum and maintained for 6 to 7 days until 40% fusion was reached. Cells were fixed and stained with DAPI, and fluorescence and bright-field images were captured. In a parallel experiment, once the giant cells reached 40% fusion, the cells were incubated in serum-free DMEM for 48 hours, and the medium was collected as CM. The mouse dermal fibroblasts were incubated in CM for 48 hours and then stained with Alexa Fluor–conjugated phalloidin for F-actin and α-SMA antibody along with DAPI to confirm myofibroblast differentiation.
Intracellular Ca2+ influx measurement
FLIPR Calcium 6 Assay Kit was used to detect changes in the intercellular Ca2+ influx. After seeding on 96-well plates (20,000 cells per well in DMEM containing 1% FBS), BMDMs were transduced with Ad TRPV4 expression constructs or vector controls for 48 hours. Cells were washed and incubated for 45 min with a FLIPR kit reagent (calcium 6 dye in 1× Hanks’ balanced salt solution containing 20 mM Hepes and 2.5 mM probenecid) at 37°C and then transferred to the FlexStation 3 System for measuring the Ca2+ influx. During the experiment, 300 mosM (isotonic) or 90 mosM (70% hypotonic) solution was used as a stimulus to induce Ca2+ influx, which was recorded by measuring ΔF/F (max-min). A23187 (calcium ionophore) was used as a positive control. Data are shown as relative fluorescence units (19, 24, 42, 57, 74).
Western blotting and FACS analysis
To isolate cytosolic and membrane proteins, cells were seeded on Permanox plastic plates and transduced with Ad TRPV4 constructs or vector control for 48 hours. Protein extracts were isolated using the Mem-PER Plus Membrane Protein Extraction Kit following the manufacturer’s instructions. In a separate experiment, to directly determine the expression levels of various receptors, BMDMs were seeded on Permanox plastic plates and treated with murine IL-4 plus GM-CSF (25 ng/ml) for 24 and 48 hours. Whole-cell extracts were made using radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor cocktail. Extracts were separated on 10% SDS–PA gel electrophoresis, transferred to a nitrocellulose membrane, and probed with antibodies against TRPV4 (1:1000), CD36 (1:1000), SRA-1 (1:1000), ECAD1 (1:1000), CD47 (1:1000), TLR4 (1:1000), flotillin-1 (1:1000), tubulin (1:1000), or GAPDH (1:3000), respectively. The blots were visualized using anti-rabbit (1:2000) or anti-mouse (1:2000) horseradish peroxidase–conjugated secondary IgG and developed in UVP BioSpectrum. Images were quantified using UVP data processing software (20, 24). For FACS analysis of CD36 and TRPV4, WT and TRPV4−/− BMDMs were grown with or without murine IL-4 plus GM-CSF (25 ng/ml) for 24 hours on Permanox plastic plates. Cells were blocked with 2% bovine serum albumin (BSA) in PBS and incubated at room temperature with PE-conjugated CD36 antibody or TRPV4 antibody followed by PE-conjugated mouse secondary antibody. The specificity of antibody binding was confirmed by the use of isotype-matched control antibodies. BD FACSCanto II flow cytometer was used to analyze all samples (93).
Histology and immunofluorescence
Skin tissue sections (7 μm) were fixed with 10% formalin and stained with Masson’s trichrome stain as per the instructions of the manufacturer. To measure the level of collagen-1 in collagen-coated stiffness coverslips or skin tissue sections, the coverslips/tissue sections were fixed and stained with collagen-1 primary antibody (1:200) followed by Alexa Fluor–labeled anti-rabbit secondary antibody. Some sections were stained with anti-CD68 IgG (1:100) at 4°C overnight to identify macrophage presence and location in the tissue. Anti-rat IgG conjugated with Alexa Fluor 594 (1:200) was used as secondary antibody for immunofluorescence. For staining, BMDMs were grown on Permanox slides and treated with murine IL-4 plus GM-CSF (25 ng/ml) for 7 to 8 days. After fixing the cells with 3% paraformaldehyde, permeabilizing with 0.1% Triton X-100, and blocking with 1% BSA for 1 hour, cells were incubated overnight at 4°C with antibodies to TRPV4 (1:200) or CD36 (1:200). Anti-rabbit or anti-mouse IgG conjugated with Alexa Fluor 488/594 (1:500) was used as the secondary antibody. Alexa Fluor–labeled phalloidin was used to stain F-actin. DAPI was used to stain nuclei.
Atomic force microscopy
We used a JPK NanoWizard 4 AFM (Bruker Nano GmbH, Berlin, Germany) to measure/image the stiffness and topography of BMDMs and stiffness of implant-adjacent skin tissues and PA gels. For stiffness measurements on living cells, the AFM was operated in advanced force spectroscopy–based mode, referred to as quantitative imaging (QI), which allows nanomechanical characterization and simultaneous imaging of the sample. For this, BMDMs were transduced with Ad TRPV4 constructs or vector controls for 48 hours. Cells were then seeded on glass-bottom, poly-d-lysine–coated 35-mm petri dish (FD35PDL, World Precision Instruments) and treated with or without IL-4 and GM-CSF (25 ng/ml) for 24 hours. For stiffness measurements, cells were kept in PBS buffer at 37°C and 5% CO2 and imaged by acquiring QI maps of 128 × 128 pixels. For high-resolution imaging, the cells were fixed with 3% paraformaldehyde after 24 hours of treatment with or without IL-4/GM-CSF and imaged by QI at a resolution of 512 × 512 pixels. For all QI measurements, qp-BioAC-CI-CB2 cantilevers (NANOSENSORS) with a nominal resonance frequency of 50 kHz in air, the spring constant of 0.1 N/m, partial gold coating on the detector side, and a circular symmetric quartz probe with a 30-nm radius were used. We imaged a total of 10 cells per condition, scanning three to four areas per cell by applying an imaging set point of 0.5 nN. To verify the stiffness of the 1- and 50-kPa PA gels used for implantation, we used qp-BioAC-CI-CB2 cantilever to acquire QI maps. For skin and fascia tissue stiffness measurements, 30-μm-thick microtome sections were immobilized on glass microscope slides in PBS, whereas their stiffness was measured by recording of individual contact mode force spectroscopy curves. For tissues, CP-qp-CONT-BSG colloidal probes (sQube) with a nominal resonance frequency of 30 kHz in air, spring constant of 0.1 N/m, gold coating on the detector side, and attached borosilicate glass sphere with a diameter of 10 μm were used. We generated 70 force curves across five tissue areas adjacent to the implant with an approach speed of 2 μm/s and a maximum set point of 0.7 nN per condition. The sensitivity and spring constant of each cantilever was individually calibrated before measurements. The Young’s modulus was calculated using JPK Data Processing software by fitting a Hertz contact mechanics model with a spherical indenter shape to the acquired force curves. To limit the contribution of the underlying matrix that could arise from very thin samples, such as the spread area of a cell, we restricted the Hertz fit to the initial 25% of the indentation depth in our calculation of the Young’s modulus.
Scanning electron microscopy
Collagen-1–coated PA gels of varied stiffness (1 and 50 kPa) were fixed in 2.5% glutaraldehyde for 1 hour, washed in PBS, and incubated in 1% osmium tetroxide for 1 hour at room temperature. After washing with double-distilled water, samples were gradually dehydrated by sequential immersion in 70, 90, and 100% ethanol and cryo-fractured by an ethanol-cryofracture technique to expose the vertical layers of the collagen-coated PA gel (92). Fractured gel pieces were thawed in 100% ethanol, subsequently treated with 1:1 and 1:2 ethanol:hexamethyldisilazane mixtures, and dried overnight in 100% hexamethyldisilazane in a vacuum desiccator. The dried gel pieces were mounted on specimen stubs, and samples were sputter-coated with ~10-nm layer of gold-palladium alloy before imaging on a Hitachi S-4700 field-emission scanning electron microscope.
Statistical analysis
All data are displayed as means ± SEM. Student’s t test or one-way analysis of variance (ANOVA) followed by Bonferroni test was used to compare different groups; “ns” denotes not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. All experiments with animals both in vivo and in vitro were performed in a randomized blinded manner.
Supplementary Material
Funding:
This work was supported by an NIH (R01EB024556) grant to S.O.R.
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scisignal.abd4077
View/request a protocol for this paper from Bio-protocol.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Reagents used in this article will be made available following University of Maryland guidelines.
REFERENCES AND NOTES
- 1.Ratner BD, A pore way to heal and regenerate: 21st century thinking on biocompatibility. Regen. Biomater. 3, 107–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Major MR, Wong VW, Nelson ER, Longaker MT, Gurtner GC, The foreign body response: At the interface of surgery and bioengineering. Plast. Reconstr. Surg. 135, 1489–1498 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Velnar T, Bunc G, Klobucar R, Gradisnik L, Biomaterials and host versus graft response: A short review. Bosn. J. Basic Med. Sci. 16, 82–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson JM, Rodriguez A, Chang DT, Foreign body reaction to biomaterials. Semin. Immunol. 20, 86–100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang G, Zhou DD, Technology advances and challenges in hermetic packaging for implantable medical devices, in Biological and Medical Physics, Biomedical Engineering (Springer, 2010), pp. 27–61. [Google Scholar]
- 6.MedPAC, “Report to the Congress: Medicare and the Health Care Delivery System” (2017).
- 7.Padmanabhan J, Kyriakides TR, Nanomaterials, inflammation, and tissue engineering. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 7, 355–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacík I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG, Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater. 14, 643–651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolan EB, Varela CE, Mendez K, Whyte W, Levey RE, Robinson ST, Maye E, O’Dwyer J, Beatty R, Rothman A, Fan Y, Hochstein J, Rothenbucher SE, Wylie R, Starr JR, Monaghan M, Dockery P, Duffy GP, Roche ET, An actuatable soft reservoir modulates host foreign body response. Sci. Robot. 4, eaax7043 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Sperli A, Bersou A Jr., de Freitas JOG, Michalany N, Complications in breast augmentation. Rev. Bras. Cir. Plást. 15, 33–46 (2000). [Google Scholar]
- 11.Moore LB, Kyriakides TR, Molecular characterization of macrophage-biomaterial interactions. Adv. Exp. Med. Biol. 865, 109–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Browne S, Pandit A, Biomaterial-mediated modification of the local inflammatory environment. Front. Bioeng. Biotechnol. 3, 67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF, Macrophage polarization: An opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials 33, 3792–3802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goswami R, Arya RK, Biswas D, Zhu X, Rahaman SO, Transient receptor potential vanilloid 4 is required for foreign body response and giant cell formation. Am. J. Pathol. 189, 1505–1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S, Cellular mechanotransduction: Stiffness does matter. Nat. Mater. 13, 918–920 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS, Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 23, 781–791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma S, Goswami R, Zhang DX, Rahaman SO, TRPV4 regulates matrix stiffness and TGFβ1-induced epithelial-mesenchymal transition. J. Cell. Mol. Med. 23, 761–774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S, Goswami R, Rahaman SO, The TRPV4-TAZ mechanotransduction signaling axis in matrix stiffness- and TGFβ1-induced epithelial-mesenchymal transition. Cell. Mol. Bioeng. 12, 139–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goswami R, Merth M, Sharma S, Alharbi MO, Aranda-Espinoza H, Zhu X, Rahaman SO, TRPV4 calcium-permeable channel is a novel regulator of oxidized LDL-induced macrophage foam cell formation. Free Radic. Biol. Med. 110, 142–150 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Sharma S, Goswami R, Merth M, Cohen J, Lei KY, Zhang DX, Rahaman SO, TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 312, C562–C572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tschumperlin DJ, Fibroblasts and the ground they walk on. Physiol. Ther. 28, 380–390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irianto J, Pfeifer CR, Xia Y, Discher DE, SnapShot: Mechanosensing matrix. Cell 165, 1820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blakney AK, Swartzlander MD, Bryant SJ, The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J. Biomed. Mater. Res. A 100, 1375–1386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahaman SO, Tschumperlin DJ, Mitchell A, Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, Scheraga RG, Ghosh S, Thodeti CK, Zhang DX, Moran MM, Schilling WP, Tschumperlin DJ, Olman MA, TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Invest. 124, 5225–5238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adlerz KM, Aranda-Espinoza H, Hayenga HN, Substrate elasticity regulates the behavior of human monocyte-derived macrophages. Eur. Biophys. J. 45, 301–309 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Hind LE, Dembo M, Hammer DA, Macrophage motility is driven by frontal-towing with a force magnitude dependent on substrate stiffness. Integr. Biol. 7, 447–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE, Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface β1 integrins. Integr. Biol. 2, 435–442 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stroka KM, Aranda-Espinoza H, Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood 118, 1632–1640 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wheeler AP, Jesmok G, Brigham KL, Tumor necrosis factor’s effects on lung mechanics, gas exchange, and airway reactivity in sheep. J. Appl. Physiol. 68, 2542–2549 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Erzurum SC, Downey GP, Doherty DE, Schwab B, Elson EL, Worthen GS, Mechanisms of lipopolysaccharide-induced neutrophil retention: Relative contributions of adhesive and cellular mechanical properties. J. Immunol. 149, 154–162 (1992). [PubMed] [Google Scholar]
- 31.Pi J, Li T, Liu J, Su X, Wang R, Yang F, Bai H, Jin H, Cai J, Detection of lipopolysaccharide induced inflammatory responses in RAW264.7 macrophages using atomic force microscope. Micron 65, 1–9 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, Gardel ML, Allen MJ, Birukov KG, Birukova AA, Attenuation of lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am. J. Respir. Cell Mol. Biol. 52, 152–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, Ahad A, Luu M, Moon S, Caesar J, Cardoso WV, Grant MB, Chaqour B, CCN1–Yes-associated protein feedback loop regulates physiological and pathological angiogenesis. Mol. Cell. Biol. 39, 107–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos-Lewis W, Page-McCaw A, Basement membrane mechanics shape development: Lessons from the fly. Matrix Biol. 75–76, 72–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacolley P, Regnault V, Segers P, Laurent S, Vascular smooth muscle cells and arterial stiffening: Relevance in development, aging, and disease. Physiol. Rev. 97, 1555–1617 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Bonnans C, Chou J, Werb Z, Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mammoto T, Jiang E, Jiang A, Mammoto A, Extracellular matrix structure and tissue stiffness control postnatal lung development through the lipoprotein receptor-related protein 5/Tie2 signaling system. Am. J. Respir. Cell Mol. Biol. 49, 1009–1018 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Féréol S, Fodil R, Labat B, Galiacy S, Laurent VM, Louis B, Isabey D, Planus E, Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil. Cytoskeleton 63, 321–340 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Previtera ML, Sengupta A, Substrate stiffness regulates proinflammatory mediator production through TLR4 activity in macrophages. PLOS ONE 10, e0145813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hind LE, Lurier EB, Dembo M, Spiller KL, Hammer DA, Effect of M1–M2 polarization on the motility and traction stresses of primary human macrophages. Cell. Mol. Bioeng. 9, 455–465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, McDonald C, Hamilton TA, Olman MA, TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J. Immunol. 196, 428–436 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta N, Goswami R, Alharbi MO, Biswas D, Rahaman SO, TRPV4 is a regulator in P. gingivalis lipopolysaccharide-induced exacerbation of macrophage foam cell formation. Physiol. Rep. 7, e14069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul NE, Skazik C, Harwardt M, Bartneck M, Denecke B, Klee D, Salber J, Zwadlo-Klarwasser G, Topographical control of human macrophages by a regularly microstructured polyvinylidene fluoride surface. Biomaterials 29, 4056–4064 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Zaveri TD, Lewis JS, Dolgova NV, Clare-Salzler MJ, Keselowsky BG, Integrin-directed modulation of macrophage responses to biomaterials. Biomaterials 35, 3504–3515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nemir S, West JL, Synthetic materials in the study of cell response to substrate rigidity. Ann. Biomed. Eng. 38, 2–20 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Sapir A, Avinoam O, Podbilewicz B, Chernomordik LV, Viral and developmental cell fusion mechanisms: Conservation and divergence. Dev. Cell 14, 11–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oren-Suissa M, Podbilewicz B, Cell fusion during development. Trends Cell Biol. 17, 537–546 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Chen EH, Olson EN, Unveiling the mechanisms of cell-cell fusion. Science 308, 369–373 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Vignery A, Macrophage fusion: The making of osteoclasts and giant cells. J. Exp. Med. 202, 337–340 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Helming L, Winter J, Gordon S, The scavenger receptor CD36 plays a role in cytokine-induced macrophage fusion. J. Cell Sci. 122, 453–459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moreno JL, Mikhailenko I, Tondravi MM, Keegan AD, IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: Contribution of E-cadherin. J. Leukoc. Biol. 82, 1542–1553 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Han X, Sterling H, Chen Y, Saginario C, Brown EJ, Frazier WA, Lindberg FP, Vignery A, CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 275, 37984–37992 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T, DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202, 345–351 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jay SM, Skokos E, Laiwalla F, Krady MM, Kyriakides TR, Foreign body giant cell formation is preceded by lamellipodia formation and can be attenuated by inhibition of Rac1 activation. Am. J. Pathol. 171, 632–640 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helming L, Tomasello E, Kyriakides TR, Martinez FO, Takai T, Gordon S, Vivier E, Essential role of DAP12 signaling in macrophage programming into a fusion-competent state. Sci. Signal. 1, ra11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Helming L, Gordon S, The molecular basis of macrophage fusion. Immunobiology 212, 785–793 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN, An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med. 4, 159ra148 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC, TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, 923–932 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Elias A, Mrkonjić S, Jung C, Pardo-Pastor C, Vicente R, Valverde MA, The trpv4 channel. Handb. Exp. Pharmacol. 222, 293–319 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Everaerts W, Nilius B, Owsianik G, The vanilloid transient receptor potential channel TRPV4: From structure to disease. Prog. Biophys. Mol. Biol. 103, 2–17 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Liedtke W, Molecular mechanisms of TRPV4-mediated neural signaling. Ann. N. Y. Acad. Sci. 1144, 42–52 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Liedtke W, Tobin DM, Bargmann CI, Friedman JM, Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 100, 14531–14536 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK, TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 54, 45–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F, TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. U.S.A. 111, 1316–1321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I, TRPV4: Molecular conductor of a diverse orchestra. Physiol. Rev. 96, 911–973 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T, Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc. Natl. Acad. Sci. U.S.A. 107, 19084–19089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K, Vanden Bosch A, Bouillon R, Nilius B, Carmeliet G, TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 8, 257–265 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Goswami R, Cohen J, Sharma S, Zhang DX, Lafyatis R, Bhawan J, Rahaman SO, TRPV4 ion channel is associated with scleroderma. J. Invest. Dermatol. 137, 962–965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kyriakides TR, Foster MJ, Keeney GE, Tsai A, Giachelli CM, Clark-Lewis I, Rollins BJ, Bornstein P, The CC chemokine ligand, CCL2/MCP1, participates in macrophage fusion and foreign body giant cell formation. Am. J. Pathol. 165, 2157–2166 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wynn TA, Ramalingam TR, Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Damanik FFR, Rothuizen TC, van Blitterswijk C, Rotmans JI, Moroni L, Towards an in vitro model mimicking the foreign body response: Tailoring the surface properties of biomaterials to modulate extracellular matrix. Sci. Rep. 4, 6325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butcher DT, Alliston T, Weaver VM, A tense situation: Forcing tumour progression. Nat. Rev. Cancer 9, 108–122 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Southern BD, Grove LM, Rahaman SO, Abraham S, Scheraga RG, Niese KA, Sun H, Herzog EL, Liu F, Tschumperlin DJ, Egelhoff TT, Rosenfeld SS, Olman MA, Matrix-driven myosin II mediates the pro-fibrotic fibroblast phenotype. J. Biol. Chem. 291, 6083–6095 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia-Elias A, Mrkonjić S, Pardo-Pastor C, Inada H, Hellmich UA, Rubio-Moscardó F, Plata C, Gaudet R, Vicente R, Valverde MA, Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc. Natl. Acad. Sci. U.S.A. 110, 9553–9558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasas S, Radotic K, Longo G, Saha B, Alonso-Sarduy L, Dietler G, Roduit C, A universal fluid cell for the imaging of biological specimens in the atomic force microscope. Microsc. Res. Tech. 76, 357–363 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Labernadie A, Thibault C, Vieu C, Maridonneau-Parini I, Charrière GM, Dynamics of podosome stiffness revealed by atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 21016–21021 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charrière GM, Maridonneau-Parini I, Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat. Commun. 5, 5343 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Schaefer A, te Riet J, Ritz K, Hoogenboezem M, Anthony EC, Mul FPJ, de Vries CJ, Daemen MJ, Figdor CG, van Buul JD, Hordijk PL, Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J. Cell Sci. 127, 4985 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ, Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 89, 47–59 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Sicard D, Fredenburgh LE, Tschumperlin DJ, Measured pulmonary arterial tissue stiffness is highly sensitive to AFM indenter dimensions. J. Mech. Behav. Biomed. Mater. 74, 118–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Defife KM, Jenney CR, Colton E, Anderson JM, Disruption of filamentous actin inhibits human macrophage fusion. FASEB J. 13, 823–832 (1999). [DOI] [PubMed] [Google Scholar]
- 82.Gavara N, Chadwick RS, Relationship between cell stiffness and stress fiber amount, assessed by simultaneous atomic force microscopy and live-cell fluorescence imaging. Biomech. Model. Mechanobiol. 15, 511–523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dutta B, Arya RK, Goswami R, Alharbi MO, Sharma S, Rahaman SO, Role of macrophage TRPV4 in inflammation. Lab. Investig. 100, 178–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sheikh Z, Brooks PJ, Barzilay O, Fine N, Glogauer M, Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 8, 5671–5701 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, Ingber DE, TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 104, 1123–1130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen L, Tredget EE, Wu PYG, Wu Y, Wu Y, Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLOS ONE 3, e1886 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Engler AJ, Sen S, Sweeney HL, Discher DE, Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Beningo KA, Wang YL, Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J. Cell Sci. 115, 849–856 (2002). [DOI] [PubMed] [Google Scholar]
- 89.Schwartz MA, DeSimone DW, Cell adhesion receptors in mechanotransduction. Curr. Opin. Cell Biol. 20, 551–556 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hinz B, Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 (2007). [DOI] [PubMed] [Google Scholar]
- 91.Dutta B, Goswami R, Rahaman SO, TRPV4 plays a role in matrix stiffness-induced macrophage polarization. Front. Immunol. 11, 570195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arya RK, Goswami R, Rahaman SO, Mechanotransduction via a TRPV4-Rac1 signaling axis plays a role in multinucleated giant cell formation. J. Biol. Chem. 296, 100129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alharbi MO, Dutta B, Goswami R, Sharma S, Lei KY, Rahaman SO, Identification and functional analysis of a biflavone as a novel inhibitor of transient receptor potential vanilloid 4-dependent atherogenic processes. Sci. Rep. 11, 8173 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.