

Abstract
Diabetic foot ulceration is a devastating diabetic complication with unmet needs. We explored the efficacy of calcium-crosslinked alginate dressings in topically delivering primary macrophages and their secretome to diabetic wounds. The alginate bandages had a microporous structure that enabled even cell loading with pro-longed cell survival and egress following wound placement. In vitro experiments showed that we could successfully differentiate and polarize primary murine bone marrow derived monocytes into M0, M1, M2a and M2c defined states with distinct gene expression, surface protein and secretome profiles. The primary macrophages were delivered in the bandages, migrated within the wounds and were still present for as long as 16 days post-injury. In wounds of db/db mice, treatment with all macrophage subtypes and their secretome, when compared to control, accelerated wound healing. Bulk RNA sequencing analysis and multiplex protein quantification of wound lysates revealed that M2c macrophages conditioned media had the most impact in wound healing affecting processes like neurogenesis, while M1 conditioned media promoted keratinization and epidermal differentiation. Collectively, our results indicate that alginate dressings can serve as a delivery platform for topical treatment of diabetic wounds and that conditioned media from distinctly polarized macrophages is equally or more effective than their parental cells in advancing wound healing and could therefore be a promising and technically advantageous alternative to cell therapy.
Keywords: Alginate, Hydrogel, Macrophage, Secretome, Polarization, Diabetic wound healing
1. Introduction
The development of chronic diabetic foot ulcers (DFUs) is a devastating complication of diabetes mellitus with a long-term impact on patients’ morbidity, mortality and quality of life. Non-healing DFUs lead to 70,000 lower extremity amputations per year in the USA [1–3]. The lifetime incidence of DFUs is approximately 19% among persons with diabetes and the 5-year risk of death among people with DFUs is 2.5 times higher than in those with diabetes alone [2–5]. The steep rise in the diabetic patient population urgently necessitates investigations aiming to develop new strategies to accelerate healing of diabetic wounds.
Wound healing is a well-orchestrated process that involves epidermal, dermal, endothelial, and immune cells, and can be divided into distinct overlapping phases [6,7]. Immediately after injury, a blood clot forms and immune cells infiltrate the wound site (inflammatory phase). The cells from the epidermis and dermis begin proliferating and migrate into the wound bed to close the defect (proliferative phase). Then, dermal cells deposit and restructure the extracellular matrix in the wound bed (remodeling phase). Unlike acute wounds, DFUs and other chronic wounds lack the linear progression from one phase of wound healing to the next and are mainly characterized by the presence of chronic inflammation [8–11]. Macrophages are essential for effective wound repair and they play an important role in the normal progression from one phase of wound healing to the next [12–16]. More specifically, macrophage phenotype can be broadly characterized as classically activated pro-inflammatory (M1) or alternatively activated immuno-modulatory (M2), despite the existence of numerous phenotypes that cover the full spectrum between these two phenotypes [17–19]. M1 activation is required during the acute inflammatory phase but is also prevalent in chronic wounds [20] while M2 activation during the proliferation phase promotes angiogenesis and collagen production [21]. Studies in our group have demonstrated the implication of macrophages in the persistent pro-inflammatory state in the skin of diabetic patients and various animal models of diabetes, with an elevated pro-inflammatory cytokine and gene expression profile contributing to dysregulated wound healing, but have also highlighted the importance of mounting an acute-like inflammatory response to stimulate the stagnating chronic state [22–26].
Previous reports have explored the application of monocytes or distinctly polarized macrophages to cutaneous wounds with mixed results showing either impairment [27] or acceleration [28–30] of wound closure. Here, we sought to determine how using fabricated hydrogel dressings to apply in vitro unpolarized “M0”, or polarized “M1”, “M2a” and “M2c” macrophages and their secretome on diabetic mouse wounds influenced healing. We systematically characterized primary monocyte derived macrophages and we showed that calcium-crosslinked alginate bandages could successfully deliver cells or their secretome in vivo. In a model of diabetic wound healing lasting for 16 days where bandages were left unperturbed for 13 days all macrophage subtypes improved wound healing, while in a shorter 10 day long model where wound size was measured every two days M2c macrophages and all conditioned media types accelerated wound closure. Detailed analysis of tissues with histology, immunofluorescence staining, transcriptomics and proteomics revealed different modes of action for the treatments. Our results suggest that an alginate dressing based delivery of macrophage secretome could be an alternative to cell therapy for treating non-healing chronic DFUs.
2. Results
2.1. Alginate bandage fabrication and optimization of cell seeding
Alginate bandages with micron-sized pores were fabricated by a process of lyophilization, calcium crosslinking, and a second lyophilization step to yield dry, off-the-shelf available materials (Fig. 1A). Fabricated bandages demonstrated consistent porosity across their cross-section (Fig. 1B), with continuous pores running throughout the volume (Fig. 1C). The calcium crosslinking and second lyophilization did not markedly alter the structure nor porosity resulting from the initial bandage lyophilization (Figs. S1A–D). The swelling ratio of the bandages was measured at 44 ± 3 (Fig. S1E). These results indicate minimal changes in the bandage physical properties from initial casting through completion of processing.
Fig. 1. Alginate dressing fabrication and characterization.
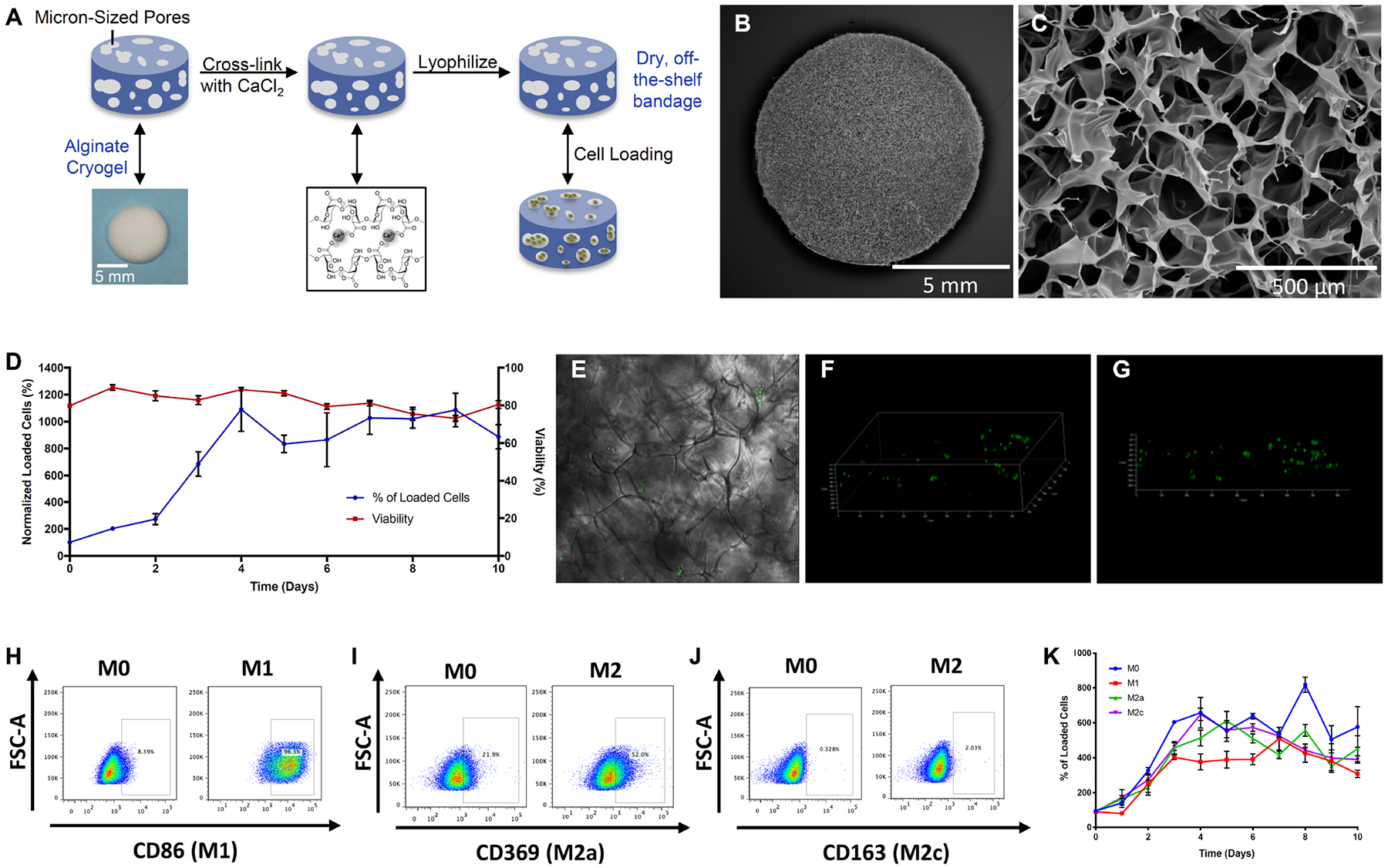
(A) Descriptive schematic of the dressing fabrication showing cross-linking, lyophilization and cell loading. (B,C) Representative SEM images revealing the porous structure of the dressing. (D) Line graph demonstrating viability and number of loaded RAW 264.7 macrophages over time. (E–G) Representative confocal image (E) and 3D reconstruction from z-stacks (F,G) of cell loaded dressing showing cell distribution within the gel. (H–J) Representative flow cytometric dot plots of non-polarized M0 and polarized M1 stained for marker CD86 (H), M2a stained for marker CD369 (I) and M2c stained for marker CD163. (J) RAW macrophages within the dressings. (K) Line graph demonstrating number of RAW macrophages polarized subtypes over time.
To determine appropriate cell loading methodology, the RAW macrophage line was used in initial studies. Unpolarized RAW cells were seeded into bandages at varying loading volumes and cell densities, with a range of resulting cell seeding efficiencies and initial cell viabilities (Fig. S2). Based on these results, a loading volume of 50 μl was used in all further studies. Analysis of cell-seeded bandages over time in culture revealed the cells proliferated to ~10-fold the initial seeding number by day 4, and maintained high viability for the 10 day duration of the study (Fig. 1C). Bright field microscopy observation revealed groups of cells distributed throughout the pores of the bandages at this time (Fig. 1E), an observation confirmed by confocal microscopy visualization of flu-orescently labeled cells (Fig. 1F–G). Subsequently, macrophages were polarized to the M1, M2a and M2c states (Fig. 1H–J; Fig. S3), and seeded onto bandages. Cells polarized to all of the states similarly first expanded over the first four days of culture, and then maintained an approximately constant number (Fig. 1K).
2.2. Generation and characterization of primary polarized macrophages
Primary bone marrow cells were collected from femurs and tibias of C57BL/6 or mRFP1 mice and passed through a MACS column to yield monocytes with high purity. Monocytes attached and grew in culture, were differentiated into macrophages and subsequently polarized into distinct populations with the addition of macrophage colony stimulating factor (M-CSF) for M0 state; M-CSF, lipopolysaccharides (LPS) and interferon gamma (IFNγ) for M1; M-CSF, and interleukins 4 and 13 (IL-4, IL-13) for M2a; M-CSF and interleukin 10 (IL-10) for M2c. The experimental workflow is outlined in Fig. S4. To confirm the appropriate polarization of macrophages into the desired subtypes we profiled the cells’ secretome using a multiplex panel on their concentrated conditioned media, which allows simultaneous measurement of 32 cytokines and chemokines. As depicted in the heat map and hierarchical clustering analysis, as well as in Table S1, the CM profiles clustered according to polarization and M1 showed high expression of all featured proteins, including IFNγ and IL-1β. M2a CM was enriched in IL-4, IL-13 and Eotaxin (CCL11), while M2c CM contained more IL-10, IL-17 and MIP-1β (CCL4) (Fig. 2A and B). We also assessed the transcriptome levels of established macrophage markers with qRT-PCR and found M1 cells to express Tnf and Nos2, M2a cells Fizz1 and Arg1 and M2c Il10 and Cd163 (Fig. 2C). To corroborate the findings at the protein level we performed flow cytometry staining for known cell surface markers CD86 for M1, CD369 and MGL2 (CD301b) for M2a and CD163 for M2c. All macrophages were positive for pan macrophage markers CD11b and F4/80 and were uniquely enriched for their respective polarization marker (Fig. 2D), except for M2c which was not enriched for CD163 as expected. Notably, M1 polarized cells displayed overexpression in markers typically associated with the M2c state. Collectively, these results demonstrate that we were able to successfully produce distinctly polarized primary macrophages in vitro: M1 high in Nos2, CD86 and others, M2a high in IL-4, Fizz1, CD369 and MGL2, and M2c high in IL-10 and CCL4.
Fig. 2. Primary bone marrow derived macrophages polarization and characterization.
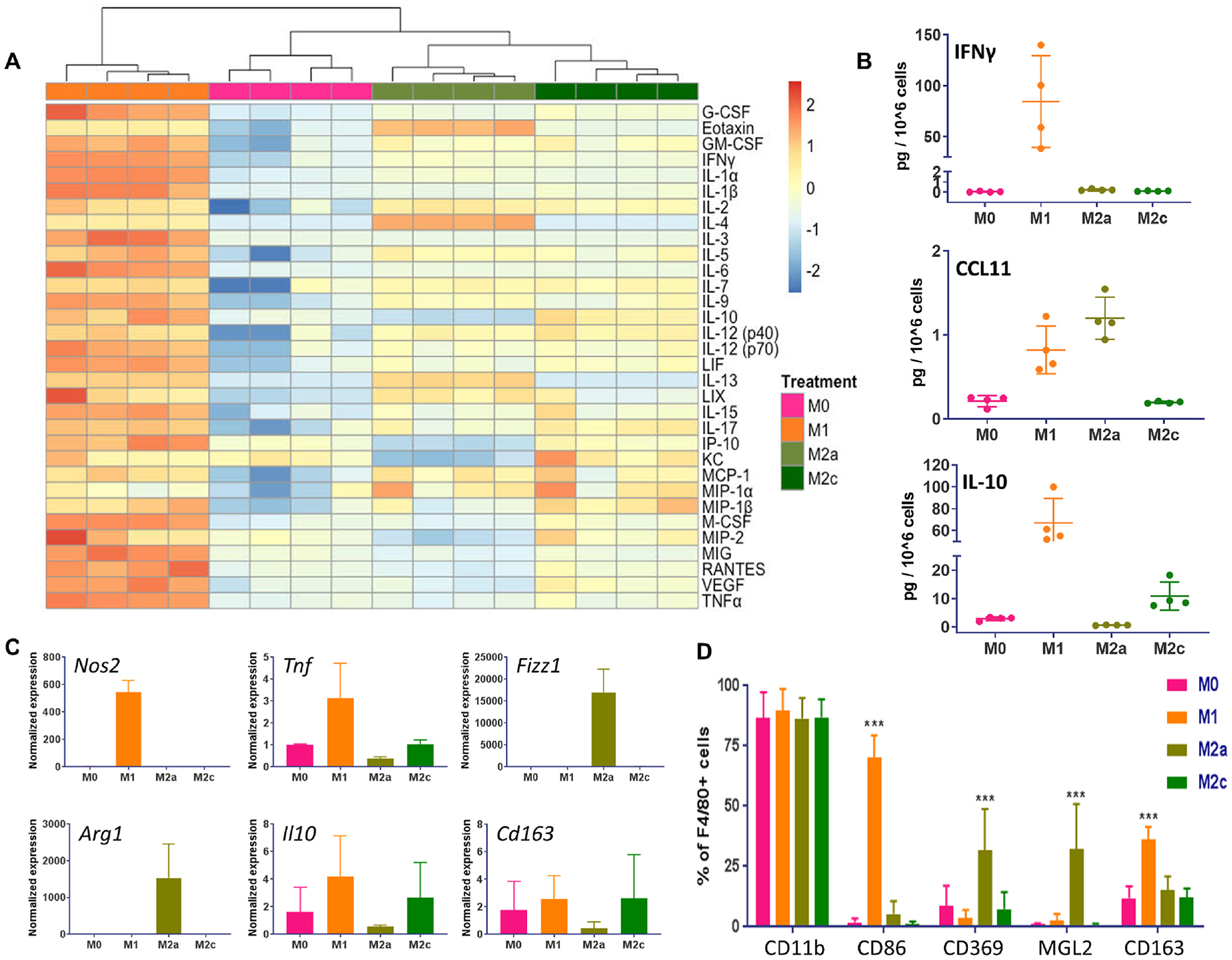
(A) Heatmap of 32 cytokines and chemokines measured with a Luminex assay in the concentrated conditioned media of polarized macrophages, n = 4, values were log2 transformed, scaled per row and clustered using Euclidian distance hierarchical clustering. Orange denotes high and blue low expression with light/dark shades representing smaller/larger levels of expression. (B) Selected set of cytokines and chemokines measured in polarized macrophages’ secretome, n = 4. (C) Quantitative RT-PCR analyses of macrophage marker genes Nos2, Tnf, Fizz1, Arg1, Il10 and Cd163 with Actb used as housekeeping gene, n = 2. (D) Quantification of multicolor flow cytometry staining of polarized macrophages (F4/80+ cells) for surface markers CD11b, CD86, CD369, MGL2 (CD301b) and CD163, n = 5, two-way ANOVA with Tukey’s post-hoc test, ***P < 0.001 compared to the other polarization states. Data are represented as mean ± s.d.
Application of macrophage-loaded bandages to diabetic mouse wounds accelerated healing after 16 days.
The primary cells could be loaded into the bandages with an efficiency of 50% after 24 h and retained their polarization within the bandages (Fig. S5). For in vivo wound delivery we loaded one million cells on each bandage expecting half of the cells to be maintained within the bandage. The stability of the bandages was evaluated both in vitro, where minimal differences in the mass of bandages soaked in RPMI over 16 days were detected (Fig. S6A), and in vivo after being placed on the wounds for 10 days. The bandage weight gradually decreased as the wound healed (Fig. S6B) and consequently the material dried out. The release profile of CM loaded onto the bandages was measured focusing on a similarly abundant protein in all conditions: MCP-1. There was a burst release in the first 24 h with almost all the protein having been liberated by day 3 (Fig. S6C). To assess the effect of applying polarized macrophages on diabetic wounds, two 6-mm biopsy punch excisional injuries were inflicted on the back of 16-week old male db/db mice and control (Ctrl), or M0, M1, M2a and M2c macrophage-loaded bandages were placed onto the wounds. We used RFP expressing cells to enable tracing in the wounds [31]. The bandages were changed for freshly loaded ones on day 3 post-wounding and then left undisturbed until wounds were collected for analyses on day 16 (Fig. 3A and B). Treatment with any of the cells significantly enhanced wound closure (Fig. 3C and D). Immunofluorescent staining for RFP revealed that the cells had migrated from the bandages and were populating the wounds (Fig. 3E and F) after 16 days. In addition, the total number of macrophages was elevated in the wounds of mice treated with cell-loaded bandages as evidenced by F4/80 staining, especially for condition M2c (Fig. 3G and H). To evaluate the polarization of macrophages within the wounds we stained for M1 markers TNFa and iNOS (Figs. S7F and I) and M2 markers Ym1 (Fig. 3I and J) and CD206 (Fig. S7J) with pan macrophage markers CD11b and CD68 as co-stains. Multiple sets of markers were used for increased accuracy. In the M2a and M2c treated wounds there were significantly more M2 positive cells, while there was no difference in the number of M1 positive cells throughout the treatment groups. There were no differences in the quantity of CD31+ blood vessels, neutrophils, mast cells, aSMA + cells and Ki67+ proliferating cells and a slight reduction in CD3+ cells for M2a treated wounds (Fig. S7). To further dissect the wounds’ cellular composition we compared the numbers of all counted macrophages and those of exogenous cells: all macrophages across different treatments were found to be between 300 and 400 cells/mm2 (Fig. 3H), while RFP + cells were between 150 and 250 cells/mm2 (after subtracting the control stained samples which represent autofluorescent cells and are ~150 cells/mm2) (Fig. 3F). These measurements suggest that at least 50% of the observed macrophages within the wounds were of an exogenous source even after 16 days. As a whole, these findings indicate that macrophages delivered with alginate bandages ameliorate healing regardless of polarized state, penetrate into the wounds and persist after 16 days and in the case of M2 cells either retain or promote similar polarization within the tissue.
Fig. 3. Application of macrophage-loaded bandages to diabetic mouse wounds enhanced healing after 16 days.
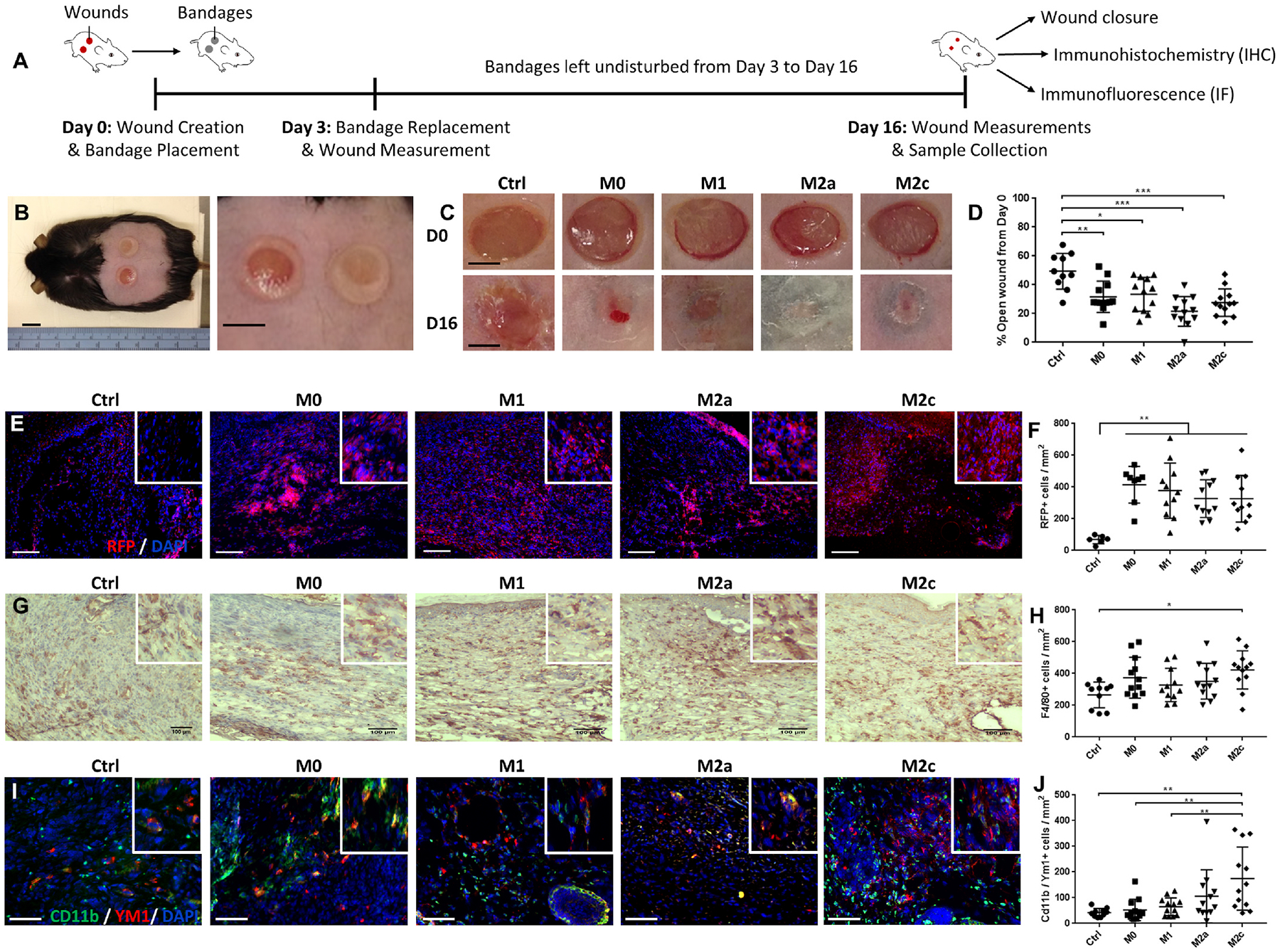
(A) Experimental design for wound healing experiment: two 6-mm biopsy punch wounds were inflicted on the back of 16-week old male db/db mice and control (Ctrl) or macrophage-loaded bandages were placed onto the wounds. Treatment groups included M0, M1, M2a and M2c polarized macrophages. The bandages were changed on day 3 post-wounding and wounds were collected for analyses on day 16. (B) Representative image of db/db mouse with bandage covered dorsal wounds (right, zoomed in). (C) Representative macroscopic views of wounds on day 0 (D0) and day 16 (D16) for the different treatment groups. (D) Quantification of wound closure on D16 expressed as % of open wound compared to D0, n = 10–12 wounds. (E) Representative immunofluorescence staining for RFP + cells (red) in D16 wounds. Nuclei are stained with DAPI (blue). (F) Quantification of RFP + cells, n = 6–11 wounds. (G) Representative images from D16 wounds of immunohistochemical staining for macrophage marker F4/80. (H) Quantification of F4/80+ cells, n = 10–12 wounds. (I) Representative immunofluorescence staining of D16 wound sections for pan-macrophage marker CD11b (green) and M2-associated marker YM1 (red). Nuclei are stained with DAPI (blue). (J) Quantification of CD11b and YM1 double positive cells, n = 10–12 wounds. Data are represented as mean ± s.d., *P < 0.05; **P < 0.01; ***P < 0.001, one-way ANOVA with Tukey’s post-hoc test. Scale bars represent 10 mmx2009; (B), 2 mm (C), 100 μm (E, G, I). Insets on (E, G, I) are 4X zoom.
Application of macrophage conditioned media-loaded bandages to diabetic mouse wounds enhanced healing after 10 days.
In an effort to more closely follow the wound healing process, we repeated the previous in vivo experiment but measured the wound size every two days until day 10. Furthermore, we included the concentrated conditioned media of the polarized cells as additional treatment groups (Fig. 4A). Estimating half a million cells to be loaded into each bandage we calculated the equivalent concentrated CM to be approximately 50 μl, which we deposited onto the bandages. The wound healing was improved for M2c cells treated wounds and for all CM groups (Fig. 4B, C, D). Application of the CMC loaded bandages had the most striking effect with fastest closure rates on days 3, 5 and 7 (Figs. S8C and D). The total number of macrophages in the wounds was increased only for the M2a condition (Figs. S9A and B). A similar comparative analysis as the Day 16 study showed that 210–300 cells/mm2 were RFP+ (Fig. S10G), while there were 215–325 F4/80+ cells/mm2 (Fig. S9B) averaged across all conditions suggesting that almost all macrophages within the cell-treated wounds were exogenous at that stage of healing. Immunofluorescent staining for the assessment of polarization revealed that there were fewer M1 macrophages in the M2a and M2c groups and all CM groups (Fig. 4E, H) compared to the control, while we found no differences in M2 macrophage numbers (Figs. S9D, E, F). Moreover, we observed more aSMA + cells for M0, M2a, M2c, CM0 and CM1 conditions (Fig. 4F, I), while wound revascularization was amplified for the CMA group as measured by blood vessel density with CD31 staining (Fig. 4G, J). For the cell treated wounds, we found that the M0 group had the most mast cells and M2c the least, while M1 had the lowest percentage of mast cell degranulation and the highest number of CD3+ cells. No change was observed in neutrophil or Ki67+ cell abundance (Fig. S10). On the other hand, the CM0 group had the fewest mast cells and CM1 and CMA showed increase in neutrophils. There were no differences in mast cell degranulation, CD3+ or Ki67+ cells, or in additional indicators of healing progression like granulation tissue area or thickness of the newly formed epidermis (Figs. S11B, D, H, I). Overall, these results suggest that, unlike their parental cells, CM treatment from any polarized macrophage subtype similarly promotes healing in a model of 10 days but the wounds contain distinct cellular infiltrates pointing to different mechanisms affected. Considering that the release of most of the protein (MCP-1) from the bandages was shown to be largely completed by day 3, we replenished the bandages with freshly loaded ones after three days. The beneficial effects likely relate to the continuous presence of the released agent with this approach.
Fig. 4. Application of macrophage and their conditioned media loaded bandages to diabetic mouse wounds and assessment of healing over 10 days.

(A) Experimental design for wound healing experiment: two 6-mm biopsy punch wounds were inflicted on the back of 16-week old male db/db mice and control (Ctrl), macrophage-loaded or conditioned media (CM)-loaded bandages were placed onto the wounds. Treatment groups included M0, M1, M2a and M2c polarized macrophages and their respective CM: CM0, CM1, CMA and CMC. The bandages were changed on day 3 post-wounding, healing was measured on Days 5 and 7 and wounds were collected for analyses on day 10. (B) Representative macroscopic views of wounds on day 0 (D0), day 5 (D5) and day 10 (D10) for the different treatment groups. (C) Quantification of wound closure for the cell treated mice on D5 and D10 expressed as % of open wound compared to D0, n = 10–14 wounds. (D) Quantification of wound closure for the CM treated mice on D5 and D10 expressed as % of open wound compared to D0, n = 9–14 wounds. (E) Quantification of immunofluorescently stained pan-macrophage marker CD68 and M1-associated marker TNFa double positive cells in the wounds of cell treated mice, n = 6–13 wounds. (F) Quantification of immufluorescencent staining for α-smooth muscle actin (aSMA) positive cells in the wounds of cell treated mice, n = 9–13 wounds. (G) Quantification of number of blood vessels from immunohistochemical staining for endothelial cell marker CD31 in the wounds of cell treated mice, n = 9–14 wounds. (H) Quantification of CD68 and TNFa double positive cells in the wounds of CM treated mice, n = 6–13 wounds. (I) Quantification of aSMA positive cells in the wounds of CM treated mice, n = 9–13 wounds. (J) Quantification of number of blood vessels in the wounds of CM treated mice, n = 9–14 wounds. Data are represented as mean ± s.d., *P < 0.05; **P < 0.01; ***P < 0.001, one-way ANOVA with Tukey’s post-hoc test. Scale bars represent 2 mm.
2.3. Transcriptomic analysis identifies differences in the mode of action of various CM
In order to better understand the mechanisms for wound healing acceleration with the CM treatments, we performed standard bulk RNA-seq analysis on D10 wound samples. We found no detectable differences in CM0 and CMA treated wounds’ transcriptomes, potentially due to biological variability, insufficient number of replicates and the advanced stage of the healing process, but principal component analysis (PCA) revealed segregation of Ctrl, CM1 and CMC treated wounds, indicating distinct gene expression profiles (Fig. 5A). This was also evident from the dendrogram of the heat map hierarchical clustering analysis where samples were grouped according to treatment (Fig. 5D). Differential gene expression analysis with log(fold change)<1 or >1 and FDR <0.05 on CM1 treated samples versus control identified 59 significantly modified genes (33 upregulated) (Figs. 5B) and 619 genes (177 upregulated) in CMC versus control (Fig. 5C). The volcano plots and heat map illustrate the most highly expressed genes, while the complete lists are included in Tables S2 and 3. Furthermore, overrepresentation analysis with the significantly differentially expressed genes highlighted the activation of distinct wound healing processes in CM1 vs Ctrl, including keratinization, anchoring junction and keratinocyte and epidermal cell differentiation (Fig. 5E, top). On the other hand, glutamatergic synapse, axonogenesis, synapse and extracellular structure organization were enriched processes in CMC vs Ctrl comparison (Fig. 5F, top). Congruent with our histological observation of increased numbers of M1 macrophages in the Ctrl group, multiple inflammatory processes were activated: regulation of inflammatory response, leukocyte and granulocyte migration, regulation of leukocyte activation and myeloid cell differentiation (Fig. 5E and F, bottom). These findings imply that Ctrl wounds remain at an unresolved inflammatory state, while CM1 treatment amplifies epidermal differentiation and CMC stimulates neurogenesis.
Fig. 5. Application of CM1-and CMC-loaded bandages to db/db mouse wounds significantly altered the global transcriptome of Day 10 post-wounding tissues.
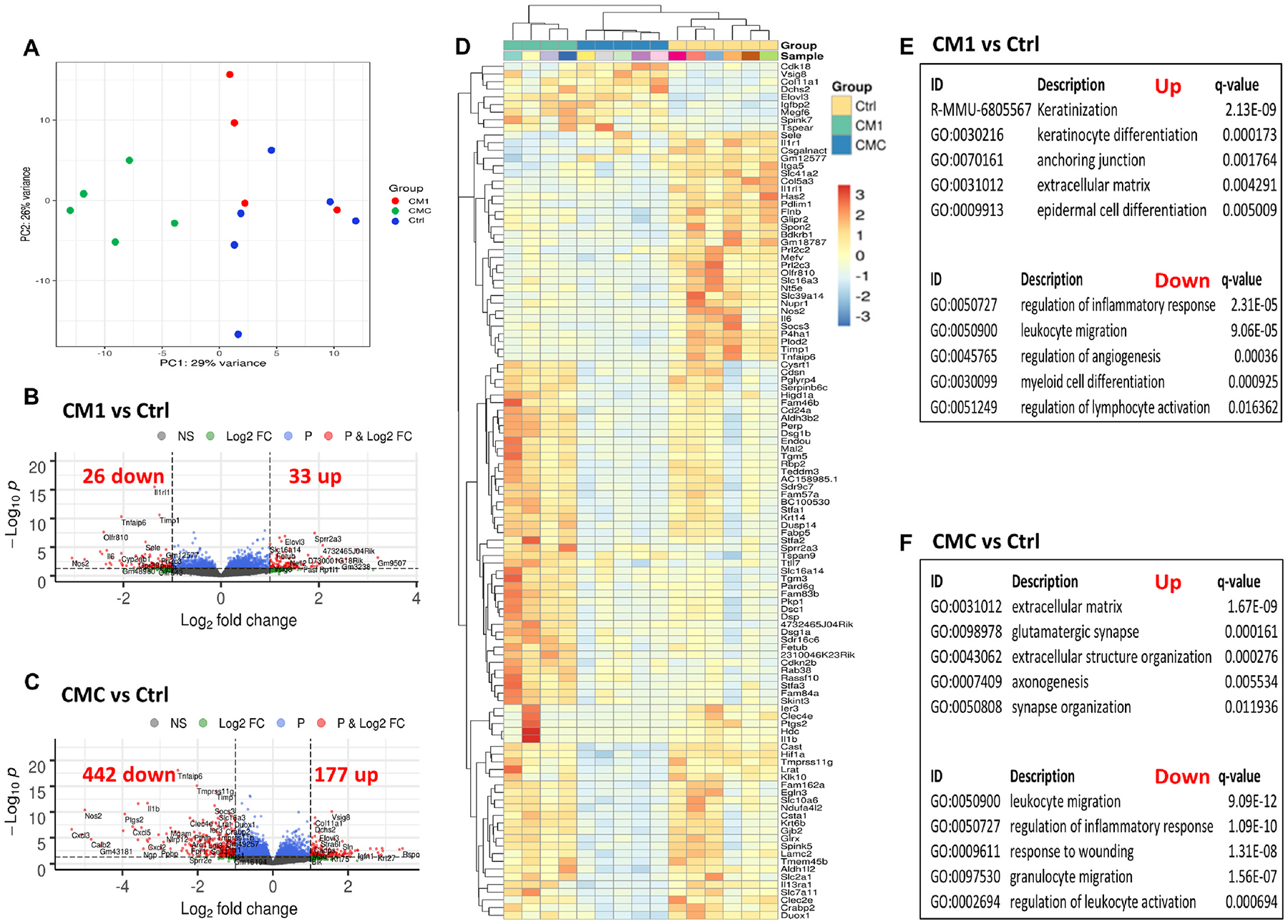
(A) Principal component analysis (PCA) plot illustrating the variances of the Ctrl (blue dots, n = 6), CM1 (red dots, n = 4) and CMC (green dots, n = 5) datasets. (B–C) Volcano plots displaying gene expression profiles when comparing CM1 against Ctrl (B) and CMC against Ctrl (C) treatments. Red colored data points represent genes that meet the thresholds of fold change (FC) above 1 or under −1, False Discovery Rate (FDR) < 0.05. (D) Heatmap depicting the expression level z-scores of the top 40 differentially expressed genes per pairwise comparison: CM1 vs Ctrl, CMC vs Ctrl, and CMC vs CM1. Higher expression levels correspond to warmer colors. (E–F) Overrepresentation analysis results for CM1 vs Ctrl (E) and CMC vs Ctrl (F) up- (top) and down-regulated (bottom) features sorted by q-value in ascending order
Application of macrophage CM-loaded bandages alters diabetic mouse wounds’ proteomic landscape.
Next, to gain further insight into how CM treatment improves wound healing, we utilized multiplex protein arrays with the Luminex platform on wound lysates from day 10 to quantitate cytokines, chemokines and growth factors with known involvement in wound repair. We thus used panels for detection of TGF-beta signaling molecules, angiogenesis factors and MMPs (Table S4). CMC treated wounds were the most impacted and contained smaller amounts of GCSF, MMP3 and soluble PECAM-1. As CMC-loaded bandages showed the most consistently significant beneficial effect in wound closure, the lower levels of these molecules could be a reflection of the more advanced healing status of the wounds. Notably, Ctrl treated wounds displayed the highest quantities of MMP3 vs most other conditions. In addition, CM0 treated tissues had more prolactin compared to CM1 and CM1 had less TGFb1 compared to control. Taken as a whole, these results confirm that CMC is the most impactful treatment and demonstrate that multiplex assays can be employed to appreciate changes in wound tissue proteome.
3. Discussion
In the present study, we first showed that alginate dressings with a microporous structure enable cell loading, and are an appropriate delivery platform for macrophages and their conditioned media. A microporous structure was desirable, in order to enable cell loading throughout the dressing volume, and subsequent cellular egress into wounds after placement. These dressings further enabled cell proliferation and maintenance of viability. In the lyophilized state, these dressings can be readily stored, and will rapidly absorb aqueous solutions, allowing for simple loading of cell suspensions and conditioned media. Alginate is widely utilized to fabricate wound dressing materials (e.g., Algicell™, AlgiSite™), potentially simplifying the clinical translation of the dressings developed here. Their uncomplicated fabrication process, cost, biocompatibility and extended shelf-life make them ideal for topical treatment of DFUs [32–34].
We subsequently described the isolation of primary purified bone marrow derived monocytes and differentiation into distinct macrophage phenotypes in vitro, which displayed unique secretome and gene and protein expression profiles. M1 macrophages exhibited a striking upregulation of most markers examined, even ones considered as typical M2c markers such as IL-10 and CD163, while M2c macrophages subtly differed from unpolarized cells and also had increased levels of pro-inflammatory cytokines IL-17 and CCL4, underscoring the plasticity and dual nature of macrophages even in defined tissue culture and activation conditions. Additional profiling of the cells’ phenotypes with next generation sequencing will further elucidate these differences. Bone marrow derived cells which are typically used in macrophage research might still end up as 100% macrophages after culture but the initial inclusion of the entire bone marrow on a culture plate with many different immune cell types present, such as neutrophils and mesenchymal stem cells, results in a panoply of activation stimuli that could potentially affect macrophage phenotype. Our approach was to define as many parameters as possible in the culture and polarization conditions, as well as work with purified monocytes that would more accurately emulate the initial cell infiltration in the wound during early inflammation [11].
Our experimental design differs from that of similar studies in several aspects. Jetten et al. [27] used 15% L-929 cell conditioned medium as a source of M-CSF, without defining its other constituents. The polarization cytokines’ concentrations were also lower than ours, while they only added IL-4 and not IL-13 for the M2a condition and they injected the cells in the wounds on days 1 and 3 as opposed to delivery with alginate dressings on days 0 and 3. This could explain the dissimilarities in our results, where we found M2 macrophages displaying the best effect in wound closure, in contrast with their report of exacerbated healing with M2 cells treatment. Our studies more closely resemble that of Hu et al. [29], albeit still with several differences. They employed a different mouse strain (FVB vs B6 backgrounds) and a different wound healing model, the cells were not derived from purified monocytes, were not pre-polarized and they were delivered with pullulan-collagen hydrogels that was embedded in the wound and has been previously shown to promote healing [35]. Consistent with our observations, the macrophages in that study displayed plasticity even before wound application, with simultaneous expression of both pro- and anti-inflammatory markers, and the delivered cells were present within diabetic wounds for 19 days as measured with luciferase bioluminescence. By mapping the gene expression of the delivered cells they demonstrated the phenotype shifted as the wounds healed, but did not quantify the amount of exogenous compared to native macrophages. Based on our analyses, the macrophages we administered into the wounds constituted a large component of all macrophages present (50–100%), effectively substituting the native macrophages but their post-implantation phenotypes appear similar except for the M2c condition on Day 16 and the M2a and M2c conditions on Day 10. This would suggest that the wound microenvironment directs available cells towards specific phenotypes and M2 pre-polarized states are more resistant to these cues. Importantly, additional studies are necessary to specifically track the fate, expression profiles and cellular cross-talk of exogenous cells, together with resident macrophages and recruited monocytes at the wound site.
Employing a shorter duration model of wound healing, we detected an improvement in wound healing only with M2c macrophages treatment, but also generally enhanced closure rates for all conditions including the control. Measuring the wound size every two days and changing the Tegaderm dressing could have stimulated healing. Nevertheless, the use of the respective conditioned media loaded dressings led to enhanced wound healing for all conditions. The divergence in wound healing outcomes between live cells and conditioned media treatments could be attributed to the macrophages not maintaining their pre-polarized states as they enter the wounds and not secreting the same factors they did when cultured in vitro. Further analyses with immunofluorescent staining, transcriptomic and proteomic profiling demonstrated that the healed wounds were composed of different cellular infiltrates and exhibited distinct gene expression programs and proteome profiles. For instance, the CM1 treated group showed processes associated with keratinization and keratinocyte differentiation as significantly enriched, reflecting the proven crosstalk of keratinocytes and inflammatory cells including macrophages [36,37]. On the other hand, the CMC treated group was enriched for several neurogenesis related processes, which is in line with an established M2 macrophage role in promotion of neuronal regeneration [38,39]. Secretome from various cell sources including endothelial progenitor cells [40], peripheral blood mononuclear cells [41], mesenchymal stem cells [42,43] and congruent with our findings M2 macrophages [44] has been shown to significantly promote wound healing. Our findings suggest that conditioned media from macrophages is equally or more effective than their parental cells in advancing wound healing. The fact that conditioned media from distinct polarized states had disparate effects could be leveraged in combination treatments, where for example CMA could facilitate angiogenesis and CM1 keratinocyte differentiation. However, given the highly complex composition of conditioned media, extensive studies are required to ensure minimized side-effects, standardization in production and treatment regimens before it could be harnessed therapeutically in the clinic.
Future investigations will focus on systematically characterizing the secretome contents with high-throughput proteomics [45–47] to unveil growth factors, cytokines, chemokines or other molecules that could serve as therapeutic targets. Additionally, extracellular vesicles which have also been shown as key components of conditioned media and important intercellular communication facilitators in wound healing [48–52], will be analyzed. Furthermore, the techniques of single cell RNA-seq [25,53,54] and spatial transcriptomics [55] employed at integral time points following injury will shed light into the specific cell types and signaling pathways affected by different treatments and offer novel insights into the progression of wound healing.
4. Materials and methods
4.1. Animals
All animals were housed and treated in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experimental procedures were approved by the IACUC at Beth Israel Deaconess Medical Center. Male C57BL/6 J (stock # 000664), CAG:mRFP1 [B6. Cg-Tg(CAG-mRFP1)1F1Hadj/J, stock # 005884)] and db/db [B6·BKS(D)-Leprdb/J, stock # 000697] mice were obtained from Jackson Laboratories and were acclimated to the animal facility for at least 1 week before being processed. Diabetic mice were routinely weighed and their blood glucose was assessed with a glucometer (Contour, Bayer).
4.2. Alginate bandage fabrication, characterization and loading with cells/conditioned medium
MVG alginate (NovaMatrix, Sandvika, Norway) was reconstituted in deionized water to 2% (w/v). Then, 100 μl of this solution was transferred into acrylic molds and frozen at 20 °C overnight. Frozen suspensions were lyophilized to create microporous materials. Dried polymer meshes were placed in 100 mM CaCl2 solution for 15 min to crosslink the alginate, and subsequently lyophilized again for storage before use. To load bandages with cells, lyophilized, calcium cross-linked bandages were combined with concentrated cell suspensions added dropwise onto each bandage. The RAW 264.7 (ATCC. TIB-71TM) cell line was used to optimize conditions for loading macrophages onto bandages. Cell-loaded bandages were placed in cell culture incubators, and culture medium added around the cell-loaded bandages after 15 min. To determine cell seeding efficiency and viability, bandages were dissolved by exposure to EDTA, and cell number analyzed by direct counting; cell viabilities were determined via Trypan blue exclusion. To load bandages with cell-conditioned medium, a similar process was followed, with 50 μl of the conditioned medium added to each bandage and allowed to soak into the material, and bandages were subsequently implanted into animals or analyzed for release of proteins. Pore size was determined from SEM images with ImageJ. Swelling ratio (Q) was measured by weighing (W) the drying bandages, then submerging them in 3 mL of RPMI media for 48 h at 37° and then re-weighing and using formula Q = (Wswollen – Wdry)/Wdry.
4.3. Alginate bandage stability assessment
For the in vitro stability evaluation, the gels were submerged in 3 mL of serum free RPMI 1640 medium and placed at 37 °C in 5% CO2 and 95% humidity. The swollen gel weight was measured at designated time points. For in vivo stability, 50 μl of RPMI serum free media was loaded onto the bandages to replicate the wound healing studies and then the gels were placed onto db/db mouse wounds and their weight measured every two days post-injury.
4.4. Conditioned media release study
Bandages were loaded with 50 μl of conditioned media from each polarization condition and left undisturbed for 20 min. They were then immersed in 0.5 mL of release buffer (HBSS with 2 mM CaCl2 and 2% BSA) in a 24-well plate and placed at 37 °C on a shaker. On designated time points the bandages were transferred into fresh release buffer filled wells and the release buffer was collected and immediately frozen at 80 °C. After completion of the experiment, all collected buffer samples were thawed and analyzed with a Luminex assay for MCP-1.
4.5. Generation of mouse bone marrow derived primary macrophage cultures
Male C57BL/6 or mRFP1 mice were sacrificed and bone marrow was rapidly isolated from tibiae and femora via centrifugation as described [56]. Cell suspensions were manually counted on a hemocytometer or with a K2 Cellometer automated cell counter (Nexcelom Bioscience) and immediately processed or aliquoted with appropriate freezing media (90% FBS, 10% DMSO) and stored in liquid N2. To establish primary macrophage cultures, the Monocyte Isolation Kit (Miltenyi Biotec, 130-100-629) was used per the manufacturer’s instructions. Briefly, monocytes were isolated by depletion of non-target cells; non-target cells were indirectly magnetically labeled with a cocktail of biotin-conjugated monoclonal antibodies, as primary labeling reagent, and anti-biotin monoclonal antibodies conjugated to MicroBeads as secondary labeling reagent. The unwanted cells were depleted by retaining them within a MACS Column in the magnetic field of a Mid-iMACS Separator, while the unlabeled monocytes passed through the column. The monocytes were seeded on non-tissue culture treated 6-well plates (ThermoFisher, 1,256,680) at a density of 0.25 × 106 cells per well and grown at 37 °C in 5% CO2 and 95% humidity with RPMI 1640 medium (ThermoFisher, 22,400,105) supplemented with 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, 10% heat inactivated FBS (MilliporeSigma, F4135, Lot # 17H165), 1% Pen/Strep (ThermoFisher, 15,140,122), 1% MEM non-essential amino acids (ThermoFisher, 11,140,050) at a pH of 7.2 and 50 ng/mL of freshly thawed M-CSF (Peprotech, 315–02). Medium, referred to as complete RPMI, was changed every three days until the cells were confluent after 9–10 days in culture. FBS from the same lot was utilized for all the studies and it was confirmed that it didn’t induce polarization of the cells with flow cytometry staining. Primary cells for characterization or in vivo studies were only used at passage 0.
4.6. Macrophage polarization and flow cytometry characterization
To polarize the cells to different phenotypes the following cytokines were added into complete RPMI for 24 h. For M1: 100 ng/mL LPS (Sigma, L4391) and 200 ng/mL IFNγ (Peprotech, 315-05); for M2a: 80 ng/mL IL-4 13 (Peprotech, 214-14) and 40 ng/mL IL-13 (Peprotech, 210-13) and for M2c: 80 ng/mL IL-10 (Peprotech, 210-10). For M0 complete RPMI with no addition of other cytokines was used. To detach the cells, the plates were incubated with Accutase (ThermoFisher, A1110501) for 15 min at 37 °C and then washed 2x with sterile DPBS (ThermoFisher, 14,190,250). Cells suspensions were centrifuged 350 rcf, 5 min, 4 °C, then pellets were resuspended in FACS buffer (auto-MACS rinsing solution with 0.05% BSA, Miltenyi Biotec, 130-091-222 and 130-091-376) and processed immediately for flow cytometry or in Trizol and stored in −80 °C for subsequent RNA isolation. To determine the expression of established polarization markers, we performed flow cytometry analysis. For all cell surface staining experiments, cells were used at a concentration of 106 cells/mL. A LIVE/DEAD fixable dead cell stain kit was used to exclude dead cells from the analysis (ThermoFisher, L-34959). AbC total antibody and amine reactive ArC compensation beads kits (ThermoFisher, A10497 and A10346) were included for single stain controls. After completing the viability stain per the manufacturer’s instructions, cells were washed 1x and blocked (FACS buffer with 0.05% anti-mouse CD16/32 Biolegend, 101,320 and 0.05% Tru-stain monocyte blocker Biolegend, 426,103) for 10 min at room temperature (RT). Then the following antibodies cocktail was added for 30 min at RT in the dark: CD11 b PE/Dazzle 594 (Biolegend 101,255, 1:100 dilution), F4/80 APC/Cy7 (Biolegend 123,117, 1:100), CD86 FITC (Biolegend 105,109, 1:100), CD369 APC (Biolegend 144,305, 1:200), CD301b(MGL2) PerCP/Cy5.5 (Biolegend 146,809, 1:100) and CD163 PE (Bioss BS-2527R-PE, 1:50). Controls included unstained cells, single stains and corresponding isotype controls. Cells were washed 2x and then resuspended in FACS buffer with 0.4% PFA and stored at 4 °C in the dark until analysis the next day. The samples were run on a three laser - 10 color Gallios flow cytometry analyzer (Beckman Coulter) and data was processed with Kaluza software (Beckman Coulter) at the BIDMC Flow Cytometry Core.
4.7. Quantitative reverse transcription PCR (qRT-PCR) on polarized macrophages
To assess the expression of specific macrophage marker genes, RNA purification and cDNA synthesis was performed as previously described [57]. Briefly, total RNA was isolated from approximately 0.5 × 106 cells using the RNeasy Mini Kit (QIAGEN), quantified on a NanoDrop 2000c (ThermoFisher) and 1 μg of RNA was reverse-transcribed using the iScript cDNA Synthesis Kit (BioRad) according to the manufacturer’s instructions. cDNA was then used for qRT-PCR on a Stratagene Mx3005P (Agilent Technologies) using KAPA SYBR FAST Master Mix (Kapa Biosystems). The forward (fw) and reverse (rv) primer sequences used for each gene were designed with primer-BLAST (NCBI, NIH) or obtained from Harvard primer bank (https://pga.mgh.harvard.edu/primerbank/) and were the following: Nos2 GTTCTCAGCCCAACAATACAAGA (fw) and GTGGACGGGTCGATGTCAC (rv), Tnf CCCTCACACTCAGATCATCTTCT (fw) and GCTACGACGTGGGCTACAG (rv), Fizz1 CCAATCCAGCTAACTATCCCTCC (fw) and ACCCAGTAGCAGTCATCCCA (rv), Arg1 CTCCAAGCCAAAGTCCTTAGAG (fw) and AGGAGCTGTCATTAGGGACATC (rv), Il10 GCTCTTACTGACTGGCATGAG (fw) and CGCAGCTCTAGGAGCATGTG (rv), Cd163 ATGGGTGGACACAGAATGGTT (fw) and CAGGAGCGTTAGTGACAGCAG (rv) and Actb GGCACCACACCTTCTACAATG (fw) and GGGGTGTTGAAGGTCTCAAAC (rv). Expression data were normalized to the expression of housekeeping gene beta-actin (Actb) and were analyzed using the 2 −ΔΔCT method as previously described [58].
4.8. Cytokine profiling analysis on polarized macrophages’ conditioned media using Luminex multiplex assays
We examined the impact of different polarization treatments on the inflammatory profiles by performing cytokine profiling on conditioned media from primary macrophage cultures. Conditioned media from M0, M1, M2a and M2c macrophages was collected after seeding 350,00–400,000 monocytes on 60 mm petri dishes (ThermoFisher, 12-565-95), keeping them in culture with complete RPMI for 7-8 days until confluent, then adding polarization cytokines for 24 h. Plates were next washed 3x with DPBS and serum free complete RPMI was added for 24 more hours and the media containing cells’ secreted factors was collected. This was centrifuged for 10 min at 3000 rcf at 4 °C to remove dead cells and debris and the supernatant was immediately concentrated (~13x) using Amicon Ultra centrifugal filters with a 3 kDa cut off (Millipore Sigma, UFC900308) for 30 min at 10,000 rcf at 4 °C, aliquoted, and stored at −80 °C. For all of these conditions, final collected concentrated conditioned media quantity as well as cell number was determined for each dish by measuring the media volume and counting the cells for normalization purposes. Cell- and serum-free RPMI was used to assess background. The cytokine profiling was performed using a Milliplex mouse cytokine/chemokine 32-plex magnetic bead panel (Millipore Sigma, MCYTMAG-70K-PMX) that detects 32 secreted proteins per the manufacturer’s instructions and analyzed on a Luminex MAGPIX instrument with xponent software version 4.2. All samples were measured in technical duplicates and four biological replicates. Mean fluorescent intensity (MFI) values were converted to pg/mL with a five parameter logistic curve fitting model. Values were normalized per million cells and volume concentrated. Normalized values were imported to R, log2 transformed for optimal visualization and processed with package pheatmap, with scaling per cytokine and including hierarchical clustering dendrogram [59].
4.9. Wounding experiments
Animals were anesthetized with isoflurane, hairs were clipped and depilated, skin site was disinfected with betadine and ethanol and two full thickness excisional wounds were created on the sides of their dorsum using a sterile disposable 6 mm punch biopsy (Integra Miltex). Male db/db mice at 16 weeks of age were used with 5–7 animals per group. Each individual wound was considered as a biological replicate [60]. Alginate gels containing cells or conditioned media were placed on the wounds immediately after wounding and the wounds were covered with Tegaderm dressing (3 M) to secure the gels into place. Wounds were let to heal by secondary intention. Following wounding, all animals were housed individually until completion of the study. The gels and dressings were changed after 3 days and the wounds were photographed using a standard iphone 5 camera secured on a stand and also measured with digital calipers (ThermoFisher, 14-648-17) right after wounding and at designated time points (days 3 and 16 or days 3, 5, 7 and 10). A ruler was placed beside the wounds as a scale bar for area calculation and size. Wound closure was quantified in a blinded fashion using both Image J and caliper measurements and expressed as percentage healed compared to day 0.
4.10. Histology, immunohistochemistry and immunofluorescence
Unwounded skin removed for wounding on day 0 and wound tissues following sacrifice on day 10 or 16 were bisected at the center of the wound. One half, to be used for protein and RNA isolation was snap frozen in liquid N2 and stored at −80 °C, while the other was frozen in OCT (Andwin Scientific, NC9806257) for histology and immunohistochemistry/fluorescence. Frozen tissues were sectioned at 5–7 μm thickness. Tissue sections were blocked in either 1% BSA, 5% Donkey serum, or 3% mouse IgG from Mouse on Mouse (MOM) kit (Vector Laboratories). Mast cell degranulation was assessed with toluidine blue assay as previously described [57]. The primary antibodies used were rabbit anti-CD3 (1:200; Abcam, ab16669), rat anti-Neutrophil Marker NIMP-R14 (1:100; Santa Cruz Biotechnology, sc-59338), rat anti-F4/80 (1:100; Abcam, ab6640 and Santa Cruz Biotechnology, sc-52664), rabbit anti-CD11b (1:200; Abcam, ab133357), mouse anti-iNOS (1:150; ThermoFisher, ma3–030), goat anti-YM1 (1:100; R&D Systems, AF2446); rabbit anti-CD68 (1:100; Abcam, ab125212), rat anti-CD206 (1:100; Santa Cruz Biotechnology, sc-58987), goat anti-TNFa (1:200; R&D Systems, AF410NA), rabbit anti-Alpha Smooth Muscle Actin (1:100; Abcam, ab5694), rabbit anti-Ki67 (1:300; Abcam, ab15580), rat anti-RFP (1:50; Chromotek, ABIN334653) and rabbit anti-CD31 (1:100; Abcam, ab28364). After staining with primary antibodies of interest overnight at 4 °C, NovaRED (Vector Laboratories) or Alexa Fluor 488-, 555- and 568-conjugated secondary antibodies (Abcam) were added for 1 h at RT. DAPI (1:400; 5 mg/mL in double distilled H2O, ThermoFisher, D1306) was used for nuclear staining. TrueVIEW Autofluorescence Quenching Kit (Vector Labs, SP-8400) treatment was included to enhance the quality of immunofluorescence staining. Tissue sections were mounted in Permount (ThermoFisher, SP15–100) or in ProLong Gold Antifade (ThermoFisher, P36930) mountant, were visualized with Nikon Eclipse E200 brightfield (Nikon) and Zeiss Axio Imager. A2m fluorescence (Carl Zeiss) upright microscopes and images processed with Motic Images Plus 3.0 (Motic), ZEN 2011 (Carl Zeiss) and ImageJ/FIJI software packages. All images were taken at the center of the wounds, guided by the absence of hair follicles and the characteristic density of the granulation tissue. Images were quantified in a blinded fashion by two independent observers and presented as positive (single or double stained) cells per mm2.
4.11. Protein isolation and Luminex quantitation
Day 10 snap frozen wound samples were transferred into T-PER™ Tissue Protein Extraction Reagent buffer (ThermoFisher, 98,510) containing HALT™ Phosphatase and Protease Inhibitor Cocktail (ThermoFisher, 78,440) and tissues were homogenized on a TissueLyser LT (Qiagen) per the manufacturer’s instructions. Total protein was quantitated with a Pierce™ BCA Protein Assay Kit (ThermoFisher, 23,227). Different Luminex panels were used to measure concentrations of TGFB (Millipore Sigma, 3-plex TGFBMAG-64K-03) or MMP family members (Millipore Sigma, 5-plex MMMP3MAG-79K) and angiogenesis and growth factor associated molecules (Millipore Sigma, 27-plex MAGPMAG-24K) following the manufacturer’s instructions. Values per sample were normalized per total protein contained and expressed as pg/total mg of protein.
4.12. Protein statistical analysis
We have included in our analyses all the proteins that were detectable in at least 90% of the samples, which was the case for all proteins, but MCP1 and TNF. The latter two were not included in the further analyses. For each of the 30 remaining proteins, half of the lowest detectable concentration was imputed for non-detectable values. The analyses of variance were performed on values transformed to their base 10 logarithms under α = 0.05. Associations that remained significant after Bonferroni correction for multiple testing were listed in the legend of Supplementary Table 3 (α = 0.0016). Statistical analyses were performed with SAS 9.4 (SAS, Cary, NC).
4.13. Transcriptome sequencing and analysis
Total RNA was isolated from day 10 snap frozen wound samples treated with Ctrl or CM loaded bandages using the Qiagen miRNEasy kit (Qiagen, 217,004) on a TissueLyser LT (Qiagen) per the manufacturer’s instructions. RNA Sample QC, library preparations and sequencing reactions were conducted at GENEWIZ, LLC. (South Plainfield, NJ, USA).
RNA samples were quantified using Nanodrop 2000 spectrophotometer (ThermoFisher) and Qubit 2.0 Fluorometer (Life Technologies) and RNA integrity was checked with 4200 TapeStation (Agilent Technologies). Because RNA quality was suboptimal (average RIN valuê5) we proceeded with ribosomal RNA (rRNA) negative selection before library preparation [61]. rRNA depletion was performed using Ribozero rRNA Removal Kit (Illumina). RNA sequencing library preparation used NEBNext Ultra RNA Library Prep Kit for Illumina by following the manufacturer’s recommendations (NEB). Briefly, enriched RNAs were fragmented for 15 min at 94 °C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapter was ligated to cDNA fragments, followed by index addition and library enrichment with limited cycle PCR. Sequencing libraries were validated using the Agilent Tapestation 4200 and quantified by using Qubit 2.0 Fluorometer as well as by quantitative PCR (Applied Biosystems). The sequencing libraries were multiplexed and clustered on two lanes of a flowcell and loaded on the Illumina HiSeq 2000 instrument according to the manufacturer’s instructions. The samples were sequenced using a 2 × 150 Paired End configuration. The HiSeq Control Software (HCS) conducted image analysis and base calling. Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software.
Read quality was evaluated using FastQC [62] and data were pre-processed with Cutadapt [63] for adapter removal following best practices [64]. Gene expression against the GRCm38 transcriptome (Ensembl 93 version) [65] was quantified using STAR [66] and featureCounts [67]. Differential gene expression analysis was performed using DESeq2 [68], while ClusterProfiler [69] was utilized for downstream functional investigations. Genes with log2 |Fold Change| ≥1 and False Discovery Rate (FDR) ≤ 0.05 were considered statistically significant.
4.14. Statistics
Data are presented as mean ± standard deviation (s.d.). Statistical power analysis was based on preliminary data or data from other studies in our unit. Student’s t-test and one or two-way ANOVA with Tukey’s post hoc test were employed using Minitab and GraphPad Prism 7.04.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health Grant DP3DK108224 (D.M. and A.V.). A.V. received funding from the National Rongxiang Xu Foundation. G.T. received a George and Marie Vergottis Foundation Postdoctoral Fellowship.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2022.121692.
CRediT author statement
Conceptualization: G.T, D.M., A.V.; Formal analysis: G.T, S.R, X.K, S. M., M.N.; Funding acquisition: D.M., A.V.; Investigation: G.T, S.R, S.L., Z.L., A.L, K.K, P.W.; Methodology: G.T., S.R., S.L.; Supervision: I.V., M. N., D.M., A.V.; Validation: G.T., S.R.; Visualization: G.T., S.R.; Writing -original draft: G.T.; Writing - review & editing: G.T., M.N., D. M., and A. V.
Data availability
The RNA-seq data used in the present study were deposited in the Gene Expression Omnibus (GEO) with accession number ‘GSE149419’.
References
- [1].Gregg EW, Li Y, Wang J, et al. , Changes in diabetes-related complications in the United States, 1990–2010, N. Engl. J. Med 370 (2014) 1514–1523. [DOI] [PubMed] [Google Scholar]
- [2].Armstrong DG, Boulton AJM, Bus SA, Diabetic foot ulcers and their recurrence, N. Engl. J. Med 376 (2017) 2367–2375. [DOI] [PubMed] [Google Scholar]
- [3].Jeffcoate WJ, Vileikyte L, Boyko EJ, Armstrong DG, Boulton AJM, Current challenges and opportunities in the prevention and management of diabetic foot ulcers, Diabetes Care 41 (2018) 645–652. [DOI] [PubMed] [Google Scholar]
- [4].Singh N, Armstrong DG, Lipsky BA, Preventing foot ulcers in patients with diabetes, JAMA 293 (2005) 217–228. [DOI] [PubMed] [Google Scholar]
- [5].Veves A, Manes C, Murray HJ, Young MJ, Boulton AJ, Painful neuropathy and foot ulceration in diabetic patients, Diabetes Care 16 (1993) 1187–1189. [DOI] [PubMed] [Google Scholar]
- [6].Eming SA, Martin P, Tomic-Canic M, Wound repair and regeneration: mechanisms, signaling, and translation, Sci. Transl. Med 6 (2014) 265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shaw TJ, Martin P, Wound repair: a showcase for cell plasticity and migration, Curr. Opin. Cell Biol 42 (2016) 29–37. [DOI] [PubMed] [Google Scholar]
- [8].Falanga V, Wound healing and its impairment in the diabetic foot, Lancet 366 (2005) 1736–1743. [DOI] [PubMed] [Google Scholar]
- [9].Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E, Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds, J. Invest. Dermatol 111 (1998) 850–857. [DOI] [PubMed] [Google Scholar]
- [10].Brem H, Tomic-Canic M, Cellular and molecular basis of wound healing in diabetes, J. Clin. Investig 117 (2007) 1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kimball A, Schaller M, Joshi A, et al. , Ly6C(Hi) blood monocyte/macrophage drive chronic inflammation and impair wound healing in diabetes mellitus, Arterioscler. Thromb. Vasc. Biol 38 (2018) 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goren I, Kampfer H, Podda M, Pfeilschifter J, Frank S, Leptin and wound inflammation in diabetic ob/ob mice: differential regulation of neutrophil and macrophage influx and a potential role for the scab as a sink for inflammatory cells and mediators, Diabetes 52 (2003) 2821–2832. [DOI] [PubMed] [Google Scholar]
- [13].Lucas T, Waisman A, Ranjan R, et al. , Differential roles of macrophages in diverse phases of skin repair, J. Immunol 184 (2010) 3964–3977. [DOI] [PubMed] [Google Scholar]
- [14].Mirza R, DiPietro LA, Koh TJ, Selective and specific macrophage ablation is detrimental to wound healing in mice, Am. J. Pathol 175 (2009) 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Leibovich SJ, Ross R, The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum, Am. J. Pathol 78 (1975) 71–100. [PMC free article] [PubMed] [Google Scholar]
- [16].Maruyama K, Asai J, Ii M, Thorne T, Losordo DW, D’Amore PA, Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing, Am. J. Pathol 170 (2007) 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mosser DM, Edwards JP, Exploring the full spectrum of macrophage activation, Nat. Rev. Immunol 8 (2008) 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, Gallagher KA, Macrophage-mediated inflammation in normal and diabetic wound healing, J. Immunol 199 (2017) 17–24. [DOI] [PubMed] [Google Scholar]
- [19].Louiselle AE, Niemiec SM, Zgheib C, Liechty KW, Macrophage polarization and diabetic wound healing, Transl. Res 236 (2021) 109–116. [DOI] [PubMed] [Google Scholar]
- [20].Sindrilaru A, Peters T, Wieschalka S, et al. , An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice, J. Clin. Investig 121 (2011) 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Novak ML, Koh TJ, Phenotypic transitions of macrophages orchestrate tissue repair, Am. J. Pathol 183 (2013) 1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pradhan Nabzdyk L, Kuchibhotla S, Chun M, et al. , Expression of neuropeptides and cytokines in a rabbit model of diabetic neuroischemic wound-healing, J. Vasc. Surg 58 (2013) 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tellechea A, Kafanas A, Leal EC, et al. , Increased skin inflammation and blood vessel density in human and experimental diabetes, Int. J. Low. Extrem. Wounds 12 (2013) 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Theocharidis G, Baltzis D, Roustit M, et al. , Integrated skin transcriptomics and serum multiplex assays reveal novel mechanisms of wound healing in diabetic foot ulcers, Diabetes 69 (2020) 2157–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Theocharidis G, Thomas BE, Sarkar D, et al. , Single cell transcriptomic landscape of diabetic foot ulcers, Nat. Commun 13 (2022) 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smith A, Watkins T, Theocharidis G, et al. , A novel three-dimensional skin disease model to assess macrophage function in diabetes, Tissue Eng. C Methods 27 (2021) 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jetten N, Roumans N, Gijbels MJ, et al. , Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses, PLoS One 9 (2014), e102994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gu XY, Shen SE, Huang CF, et al. , Effect of activated autologous monocytes/macrophages on wound healing in a rodent model of experimental diabetes, Diabetes Res. Clin. Pract 102 (2013) 53–59. [DOI] [PubMed] [Google Scholar]
- [29].Hu MS, Walmsley GG, Barnes LA, et al. , Delivery of monocyte lineage cells in a biomimetic scaffold enhances tissue repair, JCI Insight 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zuloff-Shani A, Adunsky A, Even-Zahav A, et al. , Hard to heal pressure ulcers (stage III-IV): efficacy of injected activated macrophage suspension (AMS) as compared with standard of care (SOC) treatment controlled trial, Arch. Gerontol. Geriatr 51 (2010) 268–272. [DOI] [PubMed] [Google Scholar]
- [31].Long JZ, Lackan CS, Hadjantonakis AK, Genetic and spectrally distinct in vivo imaging: embryonic stem cells and mice with widespread expression of a monomeric red fluorescent protein, BMC Biotechnol 5 (2005) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kong HJ, Smith MK, Mooney DJ, Designing alginate hydrogels to maintain viability of immobilized cells, Biomaterials 24 (2003) 4023–4029. [DOI] [PubMed] [Google Scholar]
- [33].Matoori S, Veves A, Mooney DJ, Advanced bandages for diabetic wound healing, Sci. Transl. Med (2021) 13. [DOI] [PubMed] [Google Scholar]
- [34].Rowley JA, Madlambayan G, Mooney DJ, Alginate hydrogels as synthetic extracellular matrix materials, Biomaterials 20 (1999) 45–53. [DOI] [PubMed] [Google Scholar]
- [35].Chen K, Sivaraj D, Davitt MF, et al. , Pullulan-Collagen hydrogel wound dressing promotes dermal remodelling and wound healing compared to commercially available collagen dressings, Wound Repair Regen 30 (2022) 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Piipponen M, Li D, Landen NX, The immune functions of keratinocytes in skin wound healing, Int. J. Mol. Sci 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tohyama M, Shirakara Y, Yamasaki K, Sayama K, Hashimoto K, Differentiated keratinocytes are responsible for TNF-alpha regulated production of macrophage inflammatory protein 3 alpha/CCL20, a potent chemokine for Langerhans cells, J. Dermatol. Sci 27 (2001) 130–139. [DOI] [PubMed] [Google Scholar]
- [38].Yuan J, Ge H, Liu W, et al. , M2 microglia promotes neurogenesis and oligodendrogenesis from neural stem/progenitor cells via the PPARgamma signaling pathway, Oncotarget 8 (2017) 19855–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang K, Zheng J, Bian G, et al. , Polarized macrophages have distinct roles in the differentiation and migration of embryonic spinal-cord-derived neural stem cells after grafting to injured sites of spinal cord, Mol. Ther 23 (2015) 1077–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim JY, Song SH, Kim KL, et al. , Human cord blood-derived endothelial progenitor cells and their conditioned media exhibit therapeutic equivalence for diabetic wound healing, Cell Transplant 19 (2010) 1635–1644. [DOI] [PubMed] [Google Scholar]
- [41].Mildner M, Hacker S, Haider T, et al. , Secretome of peripheral blood mononuclear cells enhances wound healing, PLoS One 8 (2013), e60103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ormazabal V, Nova-Lampeti E, Rojas D, et al. , Secretome from human mesenchymal stem cells-derived endothelial cells promotes wound healing in a type-2 diabetes mouse model, Int. J. Mol. Sci 23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park SR, Kim JW, Jun HS, Roh JY, Lee HY, Hong IS, Stem cell secretome and its effect on cellular mechanisms relevant to wound healing, Mol. Ther 26 (2018) 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Savitri C, Kwon JW, Drobyshava V, Ha SS, Park K, M2 macrophage-derived concentrated conditioned media significantly improves skin wound healing, Tissue Eng. Regen. Med 19 (2022) 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tegel H, Dannemeyer M, Kanje S, et al. , High throughput generation of a resource of the human secretome in mammalian cells, Nat. Biotechnol 58 (2020) 45–54. [DOI] [PubMed] [Google Scholar]
- [46].Weng Y, Sui Z, Shan Y, et al. , In-depth proteomic quantification of cell secretome in serum-containing conditioned medium, Anal. Chem 88 (2016) 4971–4978. [DOI] [PubMed] [Google Scholar]
- [47].Kehl D, Generali M, Mallone A, et al. , Proteomic analysis of human mesenchymal stromal cell secretomes: a systematic comparison of the angiogenic potential, NPJ Regen. Med 4 (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Born LJ, Chang KH, Shoureshi P, et al. , HOTAIR-loaded mesenchymal stem/stromal cell extracellular vesicles enhance angiogenesis and wound healing, Adv. Healthc. Mater 11 (2022), e2002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shen Y, Xu G, Huang H, et al. , Sequential release of small extracellular vesicles from bilayered thiolated alginate/polyethylene glycol diacrylate hydrogels for scarless wound healing, ACS Nano 15 (2021) 6352–6368. [DOI] [PubMed] [Google Scholar]
- [50].Oh EJ, Gangadaran P, Rajendran RL, et al. , Extracellular vesicles derived from fibroblasts promote wound healing by optimizing fibroblast and endothelial cellular functions, Stem Cell 39 (2021) 266–279. [DOI] [PubMed] [Google Scholar]
- [51].Liu W, Yuan Y, Liu D, Extracellular vesicles from adipose-derived stem cells promote diabetic wound healing via the PI3K-AKT-mTOR-HIF-1 alpha signaling pathway, Tissue Eng. Regen. Med 18 (2021) 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang P, Theocharidis G, Vlachos IS, et al. , Exosomes derived from epidermal stem cells improve diabetic wound healing, J. Invest. Dermatol (2022). [DOI] [PubMed] [Google Scholar]
- [53].Theocharidis G, Tekkela S, Veves A, McGrath JA, Onoufriadis A, Single-cell transcriptomics in human skin research: available technologies, technical considerations and disease applications, Exp. Dermatol 31 (2022) 655–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tellechea A, Bai S, Dangwal S, et al. , Topical application of a mast cell stabilizer improves impaired diabetic wound healing, J. Invest. Dermatol 140 (2020) 901–911 e11. [DOI] [PubMed] [Google Scholar]
- [55].Vickovic S, Eraslan G, Salmen F, et al. , High-definition spatial transcriptomics for in situ tissue profiling, Nat. Methods 16 (2019) 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Amend SR, Valkenburg KC, Pienta KJ, Murine hind limb long bone dissection and bone marrow isolation, JoVE 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tellechea A, Leal EC, Kafanas A, et al. , Mast cells regulate wound healing in diabetes, Diabetes 65 (2016) 2006–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [59].Kolde R, Pheatmap: pretty heatmaps, R Pack. Vers 61 (2012) 915. [Google Scholar]
- [60].Ansell DM, Campbell L, Thomason HA, Brass A, Hardman MJ, A statistical analysis of murine incisional and excisional acute wound models, Wound Repair Regen 22 (2014) 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schuierer S, Carbone W, Knehr J, et al. , A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples, BMC Genom 18 (2017) 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Andrews S FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010.
- [63].Martin M, Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads, vol. 17, 2011, p. 3, 2011. [Google Scholar]
- [64].Conesa A, Madrigal P, Tarazona S, et al. , A survey of best practices for RNA-seq data analysis, Genome Biol 17 (2016) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cunningham F, Achuthan P, Akanni W, et al. , Ensembl 2019. Nucleic Acids Research, vol. 47, 2019, pp. D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dobin A, Davis CA, Schlesinger F, et al. , STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29 (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features, Bioinformatics 30 (2014) 923–930. [DOI] [PubMed] [Google Scholar]
- [68].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yu G, Wang LG, Han Y, He QY, clusterProfiler: an R package for comparing biological themes among gene clusters, OMICS A J. Integr. Biol 16 (2012) 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data used in the present study were deposited in the Gene Expression Omnibus (GEO) with accession number ‘GSE149419’.