

Abstract
Hepatic cholesterol accumulation is an important contributor to hypercholesterolemia, which results in atherosclerosis and cardiovascular disease (CVD). ATP-citrate lyase (ACLY) is a key lipogenic enzyme that converts cytosolic citrate derived from tricarboxylic acid cycle (TCA cycle) to acetyl-CoA in the cytoplasm. Therefore, ACLY represents a link between mitochondria oxidative phosphorylation and cytosolic de novo lipogenesis. In this study, we developed the small molecule 326E with an enedioic acid structural moiety as a novel ACLY inhibitor, and its CoA-conjugated form 326E-CoA inhibited ACLY activity with an IC50 = 5.31 ± 1.2 μmol/L in vitro. 326E treatment reduced de novo lipogenesis, and increased cholesterol efflux in vitro and in vivo. 326E was rapidly absorbed after oral administration, exhibited a higher blood exposure than that of the approved ACLY inhibitor bempedoic acid (BA) used for hypercholesterolemia. Chronic 326E treatment in hamsters and rhesus monkeys resulted in remarkable improvement of hyperlipidemia. Once daily oral administration of 326E for 24 weeks prevented the occurrence of atherosclerosis in ApoE−/− mice to a greater extent than that of BA treatment. Taken together, our data suggest that inhibition of ACLY by 326E represents a promising strategy for the treatment of hypercholesterolemia.
Key words: Hypercholesterolemia, Atherosclerosis, Liver, ATP-Citrate lyase (ACLY), Lipogenesis, Cholesterol efflux, ACLY inhibitor
Graphical abstract
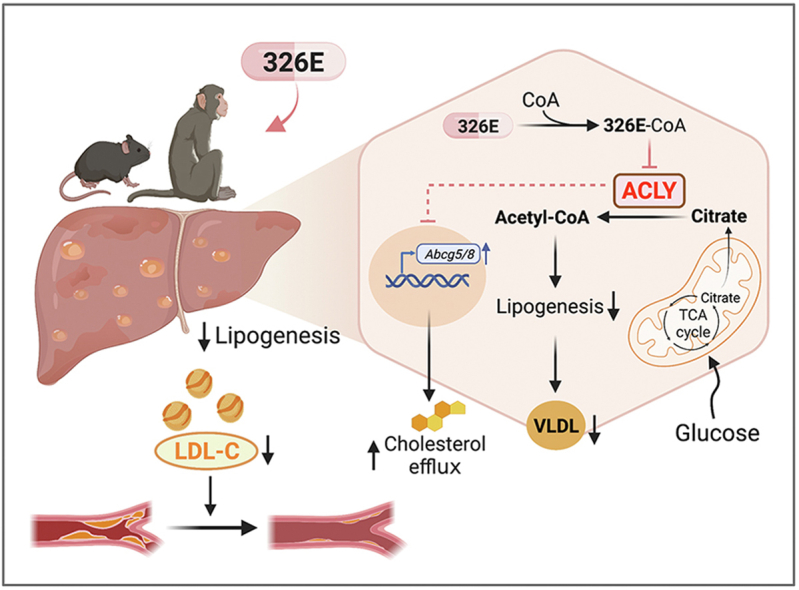
Once daily oral administration of 326E, a novel liver targeted ACLY inhibitor, ameliorates hypercholesterolemia and atherosclerosis by reducing hepatic de novo lipogenesis and increasing hepatic cholesterol efflux into intestine.
1. Introduction
Hypercholesterolemia, defined as excessive plasma cholesterol and low-density lipoprotein cholesterol (LDL-C) levels, is increasing globally prevalent. It has been estimated that over a third of the adult population develops hypercholesterolemia, which has become the lead causes of cardiovascular disease (CVD) worldwide1,2. Thus, effective treatment of hypercholesterolemia has become a public health burden.
The current clinical therapeutic strategy to lower LDL-C is an effective method to control the progression of CVD and atherosclerotic cardiovascular disease (ASCVD)3, 4, 5, 6. Statins inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), the rate-limiting enzyme of hepatic cholesterol synthesis, leading to decrease LDL-C concentrations in hypercholesterolemia patients. The general effectiveness of statins, both in reducing LDL-C levels and decreasing CVD associated complicating disease has led to increasing access to these drugs by patients7, 8, 9. However, many at-risk patients do not achieve sufficient LDL-C lowering effects or are intolerant to statins due to liver-or muscle-related side effects10,11. Furthermore, statin intolerance may lead to a 50% increase in recurrent myocardial infarction or coronary heart disease events compared to those with no statin use12,13. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition lowers LDL-C by reducing the degradation of low-density lipoprotein receptor (LDLR) in hepatocytes14,15. Recently, PCSK9 inhibitors (monoclonal antibodies and small interfering RNA) have emerged as an effective lipid-lowering therapy. However, current PCSK9 inhibitors need to be administered by injection and are costly, which may restrict their clinical use16, 17, 18. For these reasons, there is great interest in identifying novel LDL-C lowering therapeutics associated with a lower percentage of residual side effects that have convenient for administration for hypercholesterolemia treatment19,20.
De novo lipogenesis in the liver is physiologically initiated by dietary carbohydrate intake and promotes energy storage through the insulin receptor substrate (IRS)-AKT signaling axis. Cytosolic acetyl-CoA serves as common substrate for fatty acids and cholesterol synthesis21, 22, 23. Briefly, synthetic citrate from TCA cycle in the mitochondria is transported to the cytoplasm through tricarboxylate transport protein (SLC25a1). Cytoplasmic citrate is then rapidly cleaved by ACLY into acetyl-CoA and oxaloacetate24,25. Acetyl-CoA is subsequently further converted to malonyl-CoA or mevalonate, from which fatty acids or cholesterol are subsequently synthesized21,22. Besides, the elongation of very long chain fatty acids needs additional acetate supplied from gut microbiota or liver production26. As previously study indicated that phosphorylation at Ser455 by AKT increases ACLY activity27. Meanwhile, acetylation of ACLY at Lys540, Lys546, and Lys554 (ACLY-3K) by antagonizes its ubiquitylation and increases ACLY protein stability28,29. Additionally, ACLY inhibition increases ATP binding cassette subfamily G member 5/8 (Abcg5/8) gene expression and is tightly correlated with hepatic cholesterol efflux into the intestine, which may also contribute to its modulation of cholesterol metabolism30,31.
Dysregulation of hepatic ACLY activity or protein expression are connected to hepatic steatosis, type 2 diabetes (T2D) and hyperlipidemia32, 33, 34, 35. Given its key role in hepatic lipid metabolism, ACLY has been considered an attractive target for metabolic syndrome and lipid-lowering treatment36. Despite widespread interest, few ACLY inhibitors have been described to date. Early discovery strategies have primarily focused on synthesizing citrate analogues37. However, most of these candidates did not advance to clinical trials largely due to the insufficient cellular permeability and poor bioavailability. Bempedoic acid (BA, ETC-1002), a prodrug inhibitor of ACLY, has been recently approved by US Food and Drug Administration (FDA) as an additional therapeutic option in high-risk hypercholesterolemia patients who are unable to meet the goals using standard therapy38, 39, 40. Previous studies identified that BA was coupled into the active form BA-CoA responsible for ACLY inhibition by the very long-chain acyl-CoA synthetase-1 (ACSVL1), which is expressed primarily in the liver. The inactive conversion system for the prodrug in muscle explains the lack of myotoxicity41, 42, 43. However, the clinical LDL-C lowering effect induced by BA with maximally tolerated statin therapy treatment was moderated (16.5% placebo-adjusted decrease from baseline at 12 weeks in LDL-C)39.
In this study, we report the identification of a small molecule, 326E, with an enedioic acid structural moiety as a novel ACLY inhibitor and demonstrate its potential therapeutic benefit in hypercholesterolemia. Our results show that ACLY inhibition by 326E effectively improves hyperlipidemia in rodent and primate preclinical models, and intriguingly ameliorates the major hallmarks of atherosclerosis in ApoE−/− mice by reducing hepatic lipogenesis and increasing cholesterol efflux into the intestine.
2. Results
2.1. Synthesis of 326F
Bempedoic acid (BA), a prodrug of the ACLY inhibitor, is approved by FDA for hypercholesterolemia treatment and exerts a modest effect on LDL-C. Among our efforts to explore a more active ACLY inhibitor, we identified a simple dehydrated product of BA in the presence of p-toluenesulfonic acid (TsOH) in toluene and called it 326F as shown in Scheme 1.
Scheme 1.

Synthesis of 326F.
2.2. 326F is more effective than BA on acetyl-CoA derived lipogenesis and gluconeogenesis
ACLY inhibition reduces lipogenesis in hepatocytes. To evaluate the effects of 326F-induced ACLY inhibition on de novo lipogenesis, we treated mouse primary hepatocytes with 326F for 24 h, and the incorporation rates of [1,2-14C]-acetate into fatty acids and cholesterol were determined. Our results showed a promising dose-dependent lipogenesis inhibition in response to 326F treatment, which was more effective than BA treatment (Fig. 1A and B). Oxaloacetate, a carboxylic acid converted from citrate by ACLY, is the key substrate of hepatic gluconeogenesis. We determined that compared to BA treatment, the cumulative glucose production in primary hepatocytes was reduced with a greater tendency after 326F intervention (Fig. 1C). Glutamine-α-ketoglutarate (α-KG) metabolism fuels the tricarboxylic acid cycle and attenuates the oxaloacetate deficiency induced by ACLY inhibition. Our results indicated that additional glutamine supplementation attenuated the inhibition of gluconeogenesis by 326F and BA treatment (Fig. 1D). These data indicate that 326F, a novel potential ACLY inhibitor, reduces de novo lipogenesis and gluconeogenesis to a greater extent than BA mouse primary hepatocytes.
Figure 1.

Identification of 326E inhibits lipogenesis and gluconeogenesis in primary hepatocytes with more effective than BA. (A)–(B) Primary mouse hepatocytes were treated with the indicated concentration of 326F or BA for 24 h. 0.1 μCi/well [1,2-14C]-acetate was added into the culture medium at last 4 h of incubation. The contents of [1,2-14C]-acetate incorporated into fatty acids (A) and cholesterol (B) were measured. The radioactive content of lipids was normalized to DMSO group (n = 4); (C) Primary mouse hepatocytes were treated with the indicated concentration of 326F or BA for 6 h. The rate of gluconeogenesis in the medium induced by pyruvate (2 mmol/L) and lactate (20 mmol/L) was measured. The content of glucose output was normalized to DMSO group (n = 4); (D) Primary mouse hepatocytes were treated with the indicated concentration of 326F or BA for 6 h in the gluconeogenesis medium as (C), with or without suppled with 4.0 mmol/L l-glutamine in the indicated group. The content of glucose output was normalized to DMSO group (n = 4); (E) The structure of 326F, 326Z and 326E; (F)–(G) Primary mouse hepatocytes contain 0.1 μCi/well [1,2-14C]-acetate were treated for 4 h with the indicated concentration of 326F, 326E, 326Z and BA. The contents of [1,2-14C]-acetate incorporated into fatty acids (F) and cholesterol (G) were measured. The radioactive content of lipids was normalized to DMSO group (n = 3–4); (H) The Schematic diagram of the experiment. Male golden hamsters fed an HFHC diet for 2 weeks, then oral administration of 326E (10 mg/kg), 326Z (10 mg/kg) and BA (30 mg/kg) for another 2 weeks (n = 8). HFHC diet, high fat high cholesterol diet (normal chow supplemented with 0.5% cholesterol, 23% fat and 5% Fructose, w/w); (I)–(L) Serum levels of TG (I), TC (J), LDL-C (K) and HDL-C (L) in the groups were determined and plotted at indicated time. The hamsters were fasted overnight before the experiment. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to DMSO or Veh. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to indicated group. ns, not significant.
2.3. Isomer 326E but not 326Z improves hyperlipidemia in a rodent model challenged by a HFHC diet
Considering that 326F is a mixture with the proportion mix of conformations E and Z, we synthesized these two isomers, named 326E and 326Z, respectively, as shown in Scheme 2 (Fig. 1E). Compounds 1 and 2 were protected by 3,4-dihydro-2H-pyran (DHP) respectively, then coupling of alkynyl compound 3 and bromide 4 afforded compound 5, which was then deprotected and bromination using an Appel reaction to give dibromide 6. Alkylation of ethyl isobutyrate with 6 gave ester 7. The subsequent hydrogenation of the alkyne in the presence of nickel acetate and sodium borohydride in ethanol led to the corresponding cis-alkene 8, which was hydrolyzed to obtain 326Z. Compound 5 was reduced with LiAlH4 to obtain trans-alkene 9, then 326E was obtained using similar procedure as for 326Z.
Scheme 2.

Synthesis of 326E and 326Z. Condition: a) PPTS, DHP, DCM. b) n-BuLi, HMPA,THF. c) TsOH, MeOH. d) CBr4, PPh3, DCM. e) ethyl isobutyrate, LDA, THF. f) H2, Ni(OAc)2, NaBH4, ethylene diamine, EtOH. g) KOH, EtOH, H2O. h) LiAlH4, Ethylene glycol diethyl ether.
To probe cellular ACLY inhibition, we further investigated the lipogenesis inhibition effect of 326E and 326Z in vitro. Consistent with the 326F, treatment with the new isomers 326E and 326Z reduced [1,2-14C]-acetate incorporated into fatty acids and cholesterol in a dose dependent manner and was more effective than BA treatment (Fig. 1F and G).
Hepatic ACLY inhibition reduces hepatic lipogenesis and improves dyslipidemia. To investigate the potential lipid-lowering effect of ACLY inhibition by 326E and 326Z, golden hamsters fed a high-fat and high-cholesterol (HFHC) diet for 2 weeks were used as a hyperlipidemia model (Fig. 1H). 326E and 326Z (10 mg/kg) were orally administered for 2 weeks, and BA (30 mg/kg) was used as a positive control. The average serum contents of TG, TC and LDL-C in vehicle-treated animals increased dramatically compared to the normal chow group, and these levels were significantly reduced after daily 326E treatment compared to the vehicle group. The reduction effects after 10 mg/kg 326E treatment were comparable to 30 mg/kg BA treatment (Fig. 1I–K). Although there was objective de novo lipogenesis inhibition in primary hepatocytes, 326Z treatment reflected moderation of the lipid-lowering effects compared to 326E and BA treatment (Fig. 1I–K). The effect on HDL-C after 326E and 326Z treatment was slight (Fig. 1L). Taken together, these results demonstrate that isomer 326E but not 326Z effectively reduces lipogenesis and improves dyslipidemia in a rodent model of HFHC diet-induced hyperlipidemia.
2.4. Identification of the CoA thioester of 326E as a novel ACLY inhibitor
As previously reported, BA rapidly forms a CoA thioester (BA-CoA) through ACSVL1 in the liver, which directly inhibits ACLY with Ki = 2 μmol/L. To gain further insights into ACLY inhibition, we synthesized BA-CoA and 326E-CoA as shown in Scheme 341,44. Thioesters were prepared by activating BA or 326E with 1,1″-carbonyldiimidazole in dry DCM, followed by reaction with the trilithium salt of CoA in an aqueous bicarbonate solution. The resulting product was purified using semi-preparative solid-phase extraction to afford thioesters BA-CoA or 326E-CoA respectively.
Scheme 3.

Synthesis of the CoA thioester of 326E and BA.
To assess the effects of ACLY inhibition in vitro, we evaluated BA-CoA and 326E-CoA for ACLY inhibition in a cell-free system using the ADP-Glo assay. In accordance with previous studies, BA-CoA incubation inhibited recombinant human ACLY activity with an IC50 = 10.56 ± 1.46 μmol/L (Fig. 2A). The 326E-CoA molecule potently inhibited ACLY with an IC50 = 5.31 ± 1.21 μmol/L (Fig. 2B). Furthermore, kinetic analyses of 326E-CoA against ACLY substrates were performed to investigate the mechanism of ACLY inhibition. 326E-CoA was a competitive inhibitor of the CoA substrate with an inhibition constant Ki = 2.51 μmol/L. Meanwhile, 326E-CoA exhibited noncompetitive inhibitor kinetics relative to citrate (Fig. 2C and D, Supporting Information Table S2). In addition, we determined that 326E-CoA affects substantial ACLY stabilization using thermal shift assays (Supporting Information Fig. S1). These results demonstrate that the CoA-conjugated form of 326E is a novel ACLY inhibitor, and that the 326E-CoA-conjugated form competes for CoA binding at ACLY and inhibits catalytic activity.
Figure 2.

326E inhibits ACLY activity and suppresses lipid synthesis in primary hepatocytes isolated from mice. (A)–(B) Dose-dependent inhibition of purified, recombinant ACLY after incubated with BA-CoA (A) and 326E-CoA (B), the activity at control was defined as 100%; (C)–(D) Recombinant human ACLY was incubated with indicated concentrations of 326E-CoA coenzyme A (C) or citrate (D). Ki was calculated by Michaelis-Menten kinetic analysis; (E)–(F) Hepatocytes from mouse were exposed to DMEM with control or multiple doses of 326E for 4 h in the presence of [1,2-14C]-acetate (E) or [14C]-citrate (F) as noted, and IC50 for de novo lipogenesis in the hepatocytes were generated; (G)–(J) Hepatocytes from mouse were exposed to DMEM with control or 326E for 4 h in the presence of [3C]-H2O or [14C]-glucose as noted, fatty acids (G, I) and cholesterol (H, J) biogenesis in the hepatocytes were showed; (K)–(L) ACSLs inhibitor triacsin C attenuates 326E induced lipogenesis in fatty acids (K) and cholesterol synthesis (L). Triacsin C (5 μmol/L) was pre-treated for 30 min before 326E treatment; (M)–(N) Hepatocytes isolated from mice or HepG2 cell lines were exposed to DMEM with control or 326E for 4 h in the presence of [1,2-14C]-acetate as noted, and the radioactive contents of 14C-fatty acids (M) and 14C-cholesterol (N) incorporated by [1,2-14C]-acetate were counted by liquid scintillation counter; (O)–(P) Hepatocytes isolated from Acly f/f mice or Acly f/f:cre mice were exposed to DMEM with control, 326E or BA for 4 h in the presence of 0.1 μCi/well [14C]-citrate as noted, and the radioactive contents of 14C-fatty acids (O) and 14C-cholesterol (P) incorporated by [14C]-citrate were counted by liquid scintillation counter. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared toControl (Con). #P < 0.05, ##P < 0.01, ###P < 0.001 compared to indicated group. ns, not significant.
BA is converted to BA-CoA in the liver by ACSVL1. Firstly, we analyzed the long-chain acyl-CoA synthetases (Acsl) family genes expression. Our results indicated that only Acsvl1 was high expression (100 folds higher than Acsl1) in the liver (Supporting Information Fig. S2). To investigate the potential mechanism of 326E-CoA formation, we performed a series of studies and revealed that 326E was coupled with CoA to form 326E-CoA in vitro and in vivo. Fatty acids (monocarboxylic acids) with C14 to C24 carbon chain lengths, which are potential substrates of ACSLs, were competitive for 326E-CoA formation in the microsomes (Supporting Information Fig. S3). Moreover, to further determine whether 326E-CoA was transformed by 326E treatment in vivo, a single dose of 326E was orally administered to in mice. The content of 326E-CoA was measured using LC–MS/MS. Consistently, 326E-CoA was detected in the liver (2.11 ± 0.84 μg/g liver) after 1 h of 326E treatment, and the coupled rate of 326E to 326E-CoA was 11.89% (Supporting Information Table S3). These results indicate that 326E is converted to 326E-CoA in the liver by ACSLs-related enzymes.
Next, we used primary hepatocytes isolated from mice to further explore the inhibition of de novo lipogenesis. 326E treatment inhibited fatty acids and cholesterol biosynthesis dose dependently when using [1,2-14C]-acetate and [14C]-citrate as precursors. The IC50 values of fatty acids and cholesterol synthesis inhibition were 0.76 ± 0.20 and 1.82 ± 1.89 μmol/L respectively for [1,2-14C]-acetate incorporation (Fig. 2E). Meanwhile, 326E treatment inhibited fatty acids and cholesterol biosynthesis with IC50 values of 13.26 ± 1.89 and 5.53 ± 2.49 μmol/L, respectively after [14C]-citrate incorporation (Fig. 2F). Besides, 326E treatment reduced fatty acids and cholesterol biosynthesis when traced by 3H-H2O and 14C-glucose (Fig. 2G–J). 326E was coupled to 326E-CoA by ACSLs-related enzymes. Furthermore, we utilized triacsin C, an inhibitor of ACSLs family enzymes, including ACSVL1, to confirm the requirement for CoA thioester conjugation. 326E treatment reduced lipogenesis in a dose dependent manner. However, pretreatment with triacsin C for 30 min attenuated de novo lipogenesis inhibition by 326E (Fig. 2K and L). 326E treatment displayed a negligible inhibitory effect on de novo lipogenesis inhibition in the human hepatoma cell line HepG2, due to the slight ACSVL1 expression as reported (Fig. 2M and N, Supporting Information Fig. S4). These results indicate that the reduction of de novo lipogenesis by 326E is dependent on CoA thioester transformation.
Moreover, to specifically investigate the role of ACLY in 326E-attenuated lipogenesis, hepatocytes isolated from ACLY KO mice (Acly f/f:cre) and wild-type mice (Acly f/f) were utilized (Supporting Information Fig. S5). We observed reduced fatty acids and cholesterol biosynthesis as a result of 326E treatment in hepatocytes from Acly f/f but these results were attenuated in Acly f/f:cre hepatocytes, suggesting that ACLY inhibition was required for 326E-facilitated lipogenesis inhibition (Fig. 2O and P). Together, these data suggest that treatment with the small molecule 326E ameliorates lipogenesis in the hepatocytes through ACLY inhibition.
2.5. The in vivo pharmacokinetics of 326E
To increase the solubility and in vivo exposure of 326E based on the weak acid form, we screened the salt types, and sodium salt was selected for further development due to its good solubility and stability. The in vivo pharmacokinetics of 326E were examined in mice and dogs after intravenous (iv) and oral (po) administration (Table 1). Following iv administration, 326E demonstrated low plasma steady-state clearance (CL) in mice (1.31 mL/min/kg) and in dogs (0.13 mL/min/kg). The steady state distribution volumes (Vss) were 0.33 L/kg in mice and 0.29 L/kg in dogs.
Table 1.
Preclinical pharmacokinetics of 326E in mice and dogs.
| Species | Route | Dose (mg/kg) | CL (mL/min/kg) | Vss (L/kg) | t1/2 (h) | Tmax (h) | Cmax (μg/mL) | AUC0–t (μg·h/mL) |
|---|---|---|---|---|---|---|---|---|
| ICR mice | iv | 10 | 1.31 | 0.33 | 4.55 | NA | NA | 138.00 |
| po | 10 | NA | NA | 3.79 | 0.13 | 25.65 | 128.40 | |
| Beagle dog | iv | 5 | 0.13 | 0.29 | 26.20 | NA | NA | 534.00 |
| po | 5 | NA | NA | 27.40 | 0.25 | 39.60 | 419.00 |
Pharmacokinetic parameters were calculated from plasma concentration-time data and were reported as mean value (n = 6, sex in half). Solution formulation for iv pharmacokinetics were conducted using ddH2O. Oral pharmacokinetics were conducted using 0.9% NaCl. NA = not applicable.
Following oral administration at the indicated dose (10 mg/kg in mice and 5 mg/kg in dogs), 326E was rapidly absorbed (Supporting Information Tables S4 and S5). The mean terminal half-life (t1/2) value of 326E was 3.79 h in mice and 27.40 h in dogs (Table 1). These results indicated that 326E is orally bioavailable and has appropriate pharmacokinetic properties for use in mice and dogs.
2.6. Oral administration of 326E reduces hepatic lipogenesis and VLDL-TG secretion
To evaluate its impact on lipogenesis in vivo, we examined the hepatic de novo lipogenesis inhibitory effect of 326E (Fig. 3A). Oral administration of 326E reduced the incorporation of [1,2-14C]-acetate into hepatic fatty acids and cholesterol in a dose dependent manner (Fig. 3B and C). Since 326E was coupled to the active agent 326E-CoA in the liver by ACSLs, we further investigated the effect of lipogenesis after 326E treatment in other tissues. Consistently, our data showed that 326E treatment reduced lipogenesis specifically in the liver but not obviously so in the muscle, heart, ileum, abdominal adipose or kidney tissues (Fig. 3D and E).
Figure 3.

326E treatment reduces hepatic de novo lipogenesis and VLDL-TGs secretion. (A) Schematic diagram of 326E treatment for hepatic de novo lipogenesis. Briefly, male C57BL/6J mice were fasted for 48 h followed by refeeding for another 48h. Animals then received 326E treatment at multi-dose followed by an intraperitoneally injection with 2 μCi [1,2-14C] acetate (in B, C) and 5 μCi [1,2-14C] acetate (in D, E) 1 h later dosing. One hour after [1,2-14C] acetate injection, the mice were sacrificed after anesthetization, and the hepatic lipogenesis was calculated; (B)–(C) The rate of 14C-labeled hepatic de novo lipogenesis after 326E (10, 30, 100 mg/kg) and BA (30 mg/kg) treatment (n = 5–6); (D)–(E) The rate of 14C-labeled de novo lipogenesis after 326E (30 mg/kg) in the liver, leg muscle, ileum, kidney, heart and abdominal fat (a-Fat) (n = 7–8); (F)–(I) Plasma TG (F) and TC (H) concentration under tyloxapol (600 mg/kg) injection were measured after 326E and BA treatment at the indicated dose (n = 10). The area under the curve of TG level (G) and TC (I) in 4 h of VLDL-TGs secretion were shown. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to Veh. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to indicated group. ns, not significant.
The development of hypercholesterolemia is generally due to an imbalance between lipid secretion and elimination. Hepatic VLDL-TG secretion into the circulation is an important mechanism of lipid output from the liver. To determine whether hepatic DNL inhibition by 326E treatment prompted an acute lipid-lowing effect, hepatic VLDL-TG secretion was measured after oral administration of 326E in a fasted-refed mouse model. We discovered that 326E and BA treatment rapidly diminished the increase in plasma TG levels (Fig. 3F and G) and cholesterol levels (Fig. 3H and I) at the indicated times after intraperitoneal injection of the lipoprotein lipase (LPL) inhibitor tyloxapol (Fig. 3F and H). The TG and cholesterol AUC0–4 h were decreased by 75.8% and 58.9% in response to 100 mg/kg 326E treatment respectively (Fig. 3G and I). Consistent with the in vitro effect, these results indicated that oral administration of 326E inhibits hepatic de novo lipogenesis and reduces VLDL-TG secretion in vivo.
2.7. Inhibition of ACLY by 326E and BA increases cholesterol efflux in vitro and in vivo
According to previous studies, hepatic cholesterol efflux is mediated by ACLY30,31. Inhibition of ACLY is relevant to the expression of the cholesterol efflux genes Abcg5/8. To estimate the cholesterol efflux efficacy of ACLY inhibition by 326E, we investigated the effect of the cholesterol efflux rate after 326E treatment in vitro and in vivo. Primary hepatocytes were co-incubated with 15 μg/mL cholesterol (in 420 μg/mL M-β-CD) and BA or 326E for 24 h, and Abcg5/8 mRNA and intracellular cholesterol were measured. Expression of the cholesterol efflux-related genes Abcg5 and Abcg8 was upregulated dose-dependently in response to 326E treatment (Fig. 4A). Consistently, 326E or BA treatment significantly ameliorated intracellular cholesterol accumulation (Fig. 4B). Next, we evaluated the effect of ACLY inhibitors on cholesterol efflux in hepatocytes as assessed by 3H-labelled cholesterol according to the scheme shown in Fig. 4C. 326E or BA treatment markedly promoted cholesterol efflux into the cell culture medium, and the cholesterol efflux rate was much higher than that of the control group (Fig. 4D).
Figure 4.

Treatment of ACLY inhibitors increase cholesterol efflux and reduce hepatic cholesterol in HCD mice. (A)–(B) The mRNA levels of Abcg5 and Abcg8 (A) and intracellular cholesterol (B) were detected in the primary mouse hepatocytes after 326E and BA (50 μmol/L) treatment for 24 h. During the incubation, 15 μg/mL cholesterol (containing 420 μg/mL M-β-CD) was added. The LXR agonist T0901317 (T0, 1 μmol/L) were used as positive control in (B); (C) The schematic diagram of cholesterol efflux in the primary hepatocytes was shown; (D) The cholesterol efflux rate was detected after 326E and BA (50 μmol/L) treatment for 12 h in primary hepatocytes. The LXR agonist T0901317 (1 μmol/L) were used as positive control; (E) Hepatic mRNA of Abcg5 and Abcg8 were measured after a single dose of 326E and BA 30 mg/kg for 24 h in HCD mice (n = 5–6); (F) The content of cholesterol in the gallbladder was detected after a single dose of 326E and BA 30 mg/kg for 24 h in HCD mice (n = 5–6); (G)–(K) 3H-cholesterol and 3H-bile acids content in the feces (H, I) and liver (J, K) were counted with scintillation liquid. Briefly, HCD mice were oral of 326E or BA for 7 days. On the 7th day of treatment, mice were received an intravenous tail vein injection of 2.5 μCi 3H-cholesterol, which was dissolved in 100 uL intralipid (20%). Fecal samples were collected at indicated time after 3H-cholesterol injection. Livers were harvested 24 h later at last dosing (G). The radioactive cholesterol (H, J) and 3H-bile acids (I, K) were detected (n = 5–6); (L)–(M) The HCD mice were oral given with 326E and BA for 7 days. The livers were harvested after 4 h fasting. The representative pictures H&E staining (L) were shown. The contents of hepatic cholesterol (TC) and triglycerides (TG) were measured (M) (n = 5–6). Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared toVeh. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to indicated group. ns, not significant.
Hepatic cholesterol efflux occurs through ABCG5/8 transporters into the gallbladder and is then excreted into the intestine along with bile acids45. To investigate the cholesterol efflux effect of 326E in vivo, high cholesterol diet-induced mice (HCD mice) were orally administered a single dose of 326E or BA for 24 h. Treatment with ACLY inhibitor 326E or BA increased hepatic Abcg5 and Abcg8 mRNA expression (Fig. 4E). Meanwhile, cholesterol content in the gallbladder was much higher than that in the vehicle group, indicated a higher cholesterol efflux rate (Fig. 4F). We next evaluated the cholesterol efflux in HCD mice using 3H-labelled cholesterol after BA and 326E treatment for 7 days (Fig. 4G). The fecal and hepatic radiolabeled cholesterol as well as bile acids were determined. ACLY inhibitor treatment increased radiolabeled cholesterol but not bile acids excretion in feces (Fig. 4H and I). Consistent with this finding, much lower radiant quantities of radiolabeled cholesterol in the liver were observed (Fig. 4J). The radiolabeled 3H-bile acids were reduced in 326E-treated mice compared to vehicle-treated mice, likely due to lower content of hepatic 3H-labeled cholesterol, which was as the result of higher 3H-cholesterol efflux after 326E treatment (Fig. 4K). Similarly, we also observed that 326E and BA treatment for 7 days significantly reduced hepatic cholesterol content in HCD mice (Fig. 4M and L). These findings suggest that the ACLY inhibitor increased cholesterol efflux in HCD mice and reduced hepatic cholesterol content, which at least partly contributed to the lipid-lowering effect in vivo.
2.8. 326E treatment improves hyperlipidemia in rodents and rhesus monkeys
To examine the potential clinical translation of our novel ACLY inhibitor, we administered multiple doses of 326E therapy for 14 days to treat hyperlipidemia in an HFHC diet-induced hamster model (Fig. 5A). Compared to the vehicle group, body weight was significantly decreased during the 2 weeks of 326E treatment at 15 mg/kg and 30 mg/kg, as well as BA treatment (Fig. 5B). In accordance with the reduced body weight, hamsters orally administered 326E exhibited a dose-dependent improvement in the hypolipidemic effect. 326E treatment (30 mg/kg) resulted in a 57.8% reduction in serum TC, 92.9% reduction in serum TG, and 72.8% reduction in serum LDL-C, confirming the hypolipidemic effect of 326E in vivo (Fig. 5C–E). Oral administration of 326E and BA also reduced serum HDL-C levels, likely due to the reduced serum LDL-C concentration (Fig. 5F).
Figure 5.

Effects of 326E on lipid levels in blood of HFHC diet induced hyperlipidemia. (A) Schematic diagram of 326E for hyperlipidemia treatment. Briefly, male golden hamsters were induced by HFHC diet for 2 weeks. Animals then received 326E or BA treatment for another 2 weeks (n = 8). HFHC diet, high fat high cholesterol diet (normal chow supplemented with 0.5% cholesterol, 23% fat and 5% fructose, w/w); (B) Body weight gain were shown after 326E treatment for 2 weeks; (C)–(F) Serum levels of TG (C), TC (D), LDL-C (E), and HDL-C (F) before and during the treatment period were shown. The oral dose of BA was 30 mg/kg. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to Veh.
To predict the efficacy of 326E in treating human hypercholesterolemia, rhesus monkeys with spontaneous hyperlipidemia were used to evaluate the lipid-lowering efficacy of 326E (Fig. 6A). Atorvastatin at the clinically recommended dose (2.4 mg/kg) was used as a positive control (Fig. 6A, n = 3). Body weight was unchanged during treatment with 326E or atorvastatin (Fig. 6B and C). In response to 326E (20 mg/kg) treatment, plasma concentrations of TG and TC were reduced by 20.94% and 31.83%, respectively, compared to those at baseline (Fig. 6D–G). Plasma concentrations of LDL-C and ApoB100 were reduced by 20.11% and 20.88%, respectively, compared to those at baseline (Fig. 6H–K). During treatment, the efficacy of 326E in lowering plasma TC, LDL-C and ApoB100 were comparable with atorvastatin, but TG content reduction after 326E treatment was much effective (Fig. 6D and E). To evaluate the pharmacokinetics, we also examined the in vivo pharmacokinetics of 326E in rhesus monkeys after the first dose administration. 326E was rapidly absorbed (Tmax = 1.33 ± 0.58 h), and the mean terminal half-life (t1/2) value was 49.47 ± 29.34 h in rhesus monkeys. The Cmax of 326E after oral administration was 139.22 ± 48.31 μg/mL (Fig. 6L, Table 2). Accordingly, the AUC0–24 h of individual tendency was similar to the LDL-C lowering effect, suggesting a good relationship between in vivo exposure and lipid lowering efficacy (Fig. 6L and M).
Figure 6.

Lipid lowering efficacy and safety evaluation of 326E in monkeys with hyperlipidemia. (A) Schematic diagram of 326E treatment in rhesus monkey. Briefly, 6–20 years old male rhesus monkey with mild or moderate hyperlipidemia for more than 2 years (LDL-C:1.94–2.45 mmol/L) were selected, and oral treated with Veh, atorvastatin (atorva, 2.4 mg/kg/day) or 326E (20 mg/kg/day). n = 3 monkey per group; (B)–(C) Body weight change after 2 weeks treatment; (D)–(K) Plasma levels of TG (D), TC (F), LDL-C (H), ApoB100 (J) in rhesus monkey before and after 2 weeks treatment; percentage lipid lowering effect were showed compared to baseline TG (E), TC (G), LDL-C (I), ApoB100 (K) were shown; (L)–(M) Pharmacokinetic curve in each monkey (M1, M2, M3) was shown after the first dose of 326E at indicated time (L), and percentage of LDL-C lowering after 326E treated for 2 weeks in each monkey (M); (N)–(O) Plasma levels of ALT (N) and AST (O) before and after 326E or atorvastatin treatment. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, Veh compared to 326E; #P < 0.05, ##P < 0.01, ###P < 0.001, Veh compared to atorva.
Table 2.
Pharmacokinetics of 326E in rhesus monkey with hyperlipidemia.
| Dose (mg/kg) | t1/2 (h) | Tmax (h) | Cmax (μg/mL) | AUC0–t (μg·h/mL) | AUC0–∞ (μg·h/mL) |
|---|---|---|---|---|---|
| 20 | 49.47 ± 29.34 | 1.33 ± 0.58 | 139.22 ± 48.31 | 1777.75 ± 529.58 | 5681.43 ± 1430.25 |
Pharmacokinetic parameters were calculated from plasma concentration–time data related to Fig. 6L and were reported as mean ± SD (n = 3, male).
Encouragingly, during the treatment period of 326E, general organ function, as reflected by plasma creatine kinase (CK) for muscle injury and urea nitrogen for renal function, was similar before and after treatment (Supporting Information Fig. S6). Furthermore, AST and ALT levels in the plasma, which reflect liver function were decreased after 326E treatment (Fig. 6N and O). Therefore, these results indicated that oral administration of 326E might be an effective and safe therapy for hyperlipidemia treatment.
2.9. Inhibition of ACLY by 326E ameliorates atherosclerosis in ApoE−/− mice
Excessive hepatic cholesterol biosynthesis contributes to the development of hypercholesterolemia and atherosclerosis. To assess the anti-atherosclerotic efficacy of chronic ACLY inhibition by 326E in vivo, we investigated the effect of 326E in Western diet (WD)-induced ApoE−/− mice. Once-daily oral administration of 326E significantly decreased body weight gain during treatment with 326E at 15, 30 and 60 mg/kg, but did not affect food intake (Supporting Information Figs. S7A and B). ACLY inhibition had a large impact on hepatic lipid levels. Accordingly, the contents of liver cholesterol and triglycerides were significantly decreased by 326E treatment (Fig. 7A and B). Fasting content of cholesterol and LDL-C levels were lower than those of vehicle mice after 12 weeks of treatment with 30 mg/kg and 60 mg/kg 326E, but not with 15 mg/kg or BA (30 mg/kg) treatment (Figs. S7C and D). To investigate the effect of 326E on atherosclerosis, we stained the entire aortas of WD-induced ApoE−/− mice with Sudan IV. The number and size of aortic plaques were significantly lower in 326E-treated mice than in vehicle-treated mice and BA-treated mice (Fig. 7C and D), and a lower degree of atherosclerotic lesions was observed at the aortic valve in 326E-treated mice as assessed by H&E staining (Fig. 7E and F). BA administration in clinical trials demonstrated a sustained reduction in the content of plasma high sensitivity Creactive protein (hsCRP) levels, so we next investigated the hsCRP lowering effect of 326E treatment. In accordance with the reduced size of aortic plaques, the hsCRP content was reduced by more than 50% in response to multiple doses of 326E (Fig. 7G). Expression of the inflammatory-related genes Cd68, F4/80 and Il-1β was also reduced in a dose dependent manner after 326E treatment (Fig. 7H–J). In addition, gene expression of Abcg5 and Abcg8, which are related to cholesterol efflux was induced after the long-term treatment with 326E, indicating that the antiatherogenic effects of ACLY inhibition are also related to hepatic cholesterol efflux (Fig. 7K and L). Taken together, these results all indicate that treatment with the novel ACLY inhibitor 326E improves atherosclerosis in WD-induced ApoE−/− mice.
Figure 7.

Treatment of 326E decreases atherosclerosis in ApoE−/− mice. (A)–(B) Male 6–8 weeks ApoE−/− mice were induced by Western diet for 23–24 weeks. During the inducement, mice were received 326E or BA treatment once daily (n = 10). Western diet (normal chow supplemented with 0.2% cholesterol, w/w). Cholesterol levels (A) and triglycerides levels (B) in liver after 24 weeks treatment (n = 10); (C)–(D) Representative photographs of aortas from different groups after 24 weeks treatment (C); the quantifications (D) of the lesion areas in C (n = 10); (E)–(F) Representative H&E staining of cross-sections of aortic roots in different groups after 24 weeks treatment (E); the quantifications (F) of the lesion areas in E (n = 8–10); (G) Plasma contents of hsCRP in the indicated groups after 24 weeks treatment (n = 10); (H)–(L) Quantitative mRNA expression of Cd68, F4/80, Il-1β, Abcg5, Abcg8 in the liver after 326E and BA treatment. Data are mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to Veh. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to indicated group. ns, not significant.
3. Discussion
Overnutrition is a driver of hepatic dysfunction and metabolic syndrome46,47. Elevated hepatic de novo lipogenesis, attenuated clearance of circulating atherogenic LDL-C, and inflammatory stimulation has proven difficult to correct CVDs using currently available agents48. Approaches attempting to reduce hepatic lipogenesis and TG-VLDL secretion by targeting ACLY inhibition have been shown to be an attractive approach for combatting hyperlipidemia35,41.
Herein, we identified 326E, a novel ACLY inhibitor, was conjugated to form 326E-CoA in the liver. 326E-CoA inhibited human recombinant ACLY activity in vitro through competing the binding site of CoA. However, the detailed working molecular mechanism needs to be further explored. 326E treatment significantly reduced fatty acids and cholesterol biosynthesis, as well as long chain fatty acids elongation partially dependent on ACLY. Oral administration of 326E improved hyperlipidemia and ameliorated atherosclerosis in animal models. In particularly, oral administration of 326E for 2 weeks in rhesus monkeys resulted in good tolerance and lipid-lowering effects. All these results suggest that the novel ACLY inhibitor 326E is promising for clinical translation.
Cholesterol efflux through cholesterol channels or bile acids into the intestine are the major routes for cholesterol metabolism. Recent in vitro studies have shown that the transcriptional repressor Period 2 (PER2) possesses multiple binding sites within the Abcg5/8 locus. Once ACLY was inhibited by BMS303141, the occupation of PER2 at the Abcg5/8 promoter was reduced, leading to a notable increase in Abcg5/8 expression in primary hepatocytes31. Treatment with the ACLY inhibitor BA significantly increases hepatic Abcg5/8 gene expression in vivo30. Our studies indicated that ACLY inhibition by 326E and BA treatment increases cholesterol efflux in vitro and in vivo, which was associated with improved Abcg5/8 gene expression. Our observation demonstrated that ACLY inhibitor treatment augments hypercholesterolemia not only by inhibiting de novo lipogenesis, but also inducing hepatic cholesterol efflux.
Hepatic de novo lipogenesis reduced by ACLY inhibitor treatment provides a different therapy mechanism from statin therapy. Although having a similar IC50 for ACLY inhibition in vitro, we observed that oral administration of 326E improves WD-induced atherosclerosis in ApoE−/− mice after 24 weeks of treatment, which was much more effective than oral treatment with BA for the same treatment period. 326E treatment reduced aortic plaques by 60%–86.7%, while BA was only reduced plaques by 33.4% after treatment. In addition, compared to vehicle mice, hepatic cholesterol content after 326E treatment was reduced by 50%, which was much stronger than that of BA treatment (37%). The observed dramatic pharmacological therapeutic effects after 326E administration were likely due to the distinctive pharmacokinetic characteristics in vivo.
As in a previous study, the in vitro plasma protein-binding rate of BA was 95%–98% in mouse, rat, monkey and human plasma. Our unpublished data showed that 326E exhibited a much higher plasma protein-binding rate in mice, dogs and humans (>99.8%). Higher blood exposure was detected after oral administration (Cmax>100 μg/mL) of multiple dose's given. Although the free form of 326E was approximately 0.5–1.5 μmol/L, which was close to the IC50 of lipogenesis inhibition, the high plasma protein-binding rate and the tardy plasma steady-state clearance (1.31 mL/min/kg) resulted in continuous ACLY inhibition in the liver, which was closely related to the much greater reduction in hepatic cholesterol and plasma LDL-C. Our studies indicated that the plasma elimination half-life (t1/2) among mice (3.79 h), dogs (27.4 h) and monkey (49.47 h) were extremely different. However, when we are considering the relationship between body size and physiologic function, which is known as allometry ()49, the t1/2 between mice and dogs are comparable (4.55 h in mice, and 21,5 h in dogs). However, in monkey study, the animals are old rhesus monkey with hyperlipidemia (6–20 years old, and with moderate to mild hyperlipidemia for more than 2 years). The age and diseases state may decrease the clearance for drugs, that may be the reason why the t1/2 is longer (49.47 h) in monkey.
Previous studies have determined statins are strong inhibitors of mitochondrial complex III, which is associated with statin-induced myopathies10. In agreement with recent studies for BA41,43, 326E is a substrate of ACSLs (the specific synthetase of ACSLs isoform couples 326E to 326E-CoA needs to be further explored), which is coupled with CoA by ACSLs restricted in the liver. In fact, there was no significant inhibition of muscle de novo lipogenesis after 326E administration in fasted-refed mouse model, and there was good tolerance in rhesus monkeys after 326E treatment, indicating the tissue specificity of the 326E response. Besides, the long term GLP toxicity study in beagle dogs indicated the no observed adverse effect level (NOAEL) was considered to 360 mg/kg (Cmax = 569.50 μg/mL, AUC0–24 h = 2472.50 μg·h/mL). Collectively, the high exposure and good tolerance property further supported that 326E might be an effective and safe therapy for hypercholesterolemia.
4. Conclusions
In summary, our results show that pharmacological ACLY inhibition in the liver by 326E leads to chronically sustainable improvements in lipid homeostasis, including lipid-lowering effects and the amelioration of atherosclerosis by reducing lipogenesis and hepatic cholesterol efflux. These findings demonstrate that 326E has therapeutic potential and may serve as an oral agent for hypercholesterolemia. Recently, we have completed the IND application for this drug and have received approval for a clinical study for hypercholesterolemia (chictr.org.cn, ChiCTR2200057793), the first-in-human (FIH) results will be reported in due course.
5. Experimental
5.1. Chemistry
The experimental procedures and characterization of all compounds are provided in the Supporting Information.
5.2. Animal studies
For hepatocyte isolation and de novo lipogenesis in vivo, 8- to 12-week-old male C57BL/6J mice were purchased from Shanghai Model Organisms (Shanghai, China). Six-to eight-week-old male ApoE−/− mice and six-to eight-week old male golden hamsters were purchased from Charles River (Beijing, China). ApoE−/− mice were fed a high fat, high cholesterol diet (HFHC diet, D12079B, Research Diets, New Jersey, USA) for 23–24 weeks to induce atherosclerosis; hamsters were fed a high fat, high cholesterol, high fructose diet (C11953, Research Diets, New Jersey, USA) for 2–4 weeks to induce hyperlipidemia. C57BL/6J mice were fed a high cholesterol diet (HCD diet, D12109C, Research Diets, New Jersey, USA) for 2–4 weeks followed by analysis of cholesterol efflux. To obtain ACLY KO hepatocytes, ACLY-flox mice (Aclyf/f stock No: 43555, Jackson Laboratory, Bar Harbour, Maine, USA) were crossed with Albumin-Cre mice (stock No: 003574, Jackson Laboratory, Bar Harbor, Maine, USA) to generate liver-specific ACLY KO mice (Aclyf/f:cre). Rodents were housed in caging with 3–4 animals per cage and were maintained at 20–22 °C and at a humidity of 35 ± 5% under a 12 h light-dark cycle (7 AM on, 7 PM off). Food and water were available ad libitum. Age-matched animals were used for all experiments. All animals were sacrificed if needed under anaesthetic conditions, and animal welfare and animal experimental procedures were approved by the Animal Ethics Committee of the Shanghai Institute of Materia Medica, CAS, China.
5.3. In vitro ACLY assays
ACLY activity detection in the cell-free system was conducted as previously reported using an ADP-Glo dependent assay50. Briefly, the reaction was performed in a 384-well plate, containing 2 μL substrate (5 μmol/L ATP, 30 μmol/L citrate, 15 μmol/L CoA) within kinase buffer (40 mmol/L Tris base, 10 mmol/L MgCl2, 5 mmol/L DTT). The reaction was initiated by adding 20 nmol/L recombinant human ACLY protein and multiple doses of compounds into the well, followed by incubation for 30 min at 37 °C. In the reaction, ATPs were converted to ADPs by ACLY. The levels of ADPs in the well, representing the activity of ACLY, were detected using the ADP-Glo kit (V9102, Promega, Madison, USA) according to the manufacturer's instructions. The enzymatic activity curve was analyzed using log (inhibitor) vs. normalized response-variable slope in GraphPad Prism.
5.4. Primary hepatocyte isolation
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL/6J mouse51. Briefly, C57BL/6J mouse was anaesthetized and perfused with 20 mL calcium-free buffer from the inferior vena cava to the liver, and then the buffer containing collagenase I (LS004196, Worthington, USA) was added to breakdown the extracellular matrix and tight junctions at 37 °C. The liver was dissected in a sterile 6-cm cell culture dish containing 10 mL cold DMEM with 10% FBS, followed by filtration through a 70 μm cell strainer and centrifugation at 500 rpm for 3 min. Cells were then centrifuged with Percoll (P1644-100 ML, Sigma–Aldrich, USA) and living cells were resuspended in Hepato ZYME-SFM (17705-021, Thermo Fisher, USA) medium supplemented with 2 mmol/L l-glutamine, 20 units/mL penicillin and 20 μg/mL streptomycin. Then the hepatocytes were plated at 6 × 105 cells/well in 6-well or 1.5 × 105 cells/well in 24-well culture dishes precoated with 0.2% glue.
5.5. In vitro measurement of de novo lipogenesis
Hepatocytes were cultured in 6-well plates (6 × 105 cells/well), and treated with compounds and [1,2-14C]-acetate, [14C]-citrate, [14C]-glucose, [3H]-H2O (0.1 μCi/well) for the indicated times43,52. The cells were washed with cold PBS and dissolved in 0.4–0.6 mL 0.5 mol/L KOH. Cholesterol and fatty acids were separated by saponification at 95 °C for 3 h with 0.4 mL KOH (20% methanol solution). The nonpolar cholesterol was extracted in 0.5 mL petroleum ether 3 times, and the polar fatty acids were extracted in 0.5 mL petroleum ether after adding 0.2 mL H2O and 0.4 mL 5 mol/L H2SO4 for 3 times. The petroleum ether fractions were air-dried overnight at room temperature. Radioactive products were counted using a liquid scintillation counter.
5.6. In vivo measurement of de novo lipogenesis
The lipogenesis effect in vivo was assessed as previously described52. Briefly, male C57BL/6J mice weighing 20–25 g at 8–10 weeks of age were fasted for 48 h followed by refeeding for another 48 h. Next, animals received one dose of vehicle or experimental compounds. One hour after dosing, each mouse was intraperitoneally injected with the indicated dose of [1,2-14C]-acetate (NEC553001MC, PerkinElmer, USA). Mice were sacrificed 1 h after acetate injection and tissues were saponified. Isotope labelled fatty acids and cholesterol were then extracted and counted.
5.7. 326E-CoA conjugated from 326Ein vitro and in vivo
In the in vitro study, mouse liver microsomes were obtained by ultracentrifugation according to a published previously protocol53. The [14C]-326E-CoA conjugation rate was measured according to previously reported experimental methods41. [14C]-326E-CoA was separated from [14C]-326E by diethyl ether with two times of extractions. The contents of [14C]-326E-CoA in the aqueous phase were counted. In the in vivo study, the livers were harvested 1 h after 326E oral treatment, and the contents of 326E and 326E-CoA were measured separately using LC‒MS/MS.
5.8. Plasma and liver lipid determination
Plasma total cholesterol (TC), triglycerides (TG), LDL-C and high-density lipoprotein cholesterol (HDL-C) levels were determined according to the manufacturer's instructions. To measure hepatic contents of triglycerides and cholesterol, liver tissues (30–40 mg) were homogenized in 0.5 mL cold PBS. A 0.4 mL homogenate was added to 1.4 mL of a mixture of CHCl3/CH3OH (2:1, v/v), followed by shaking overnight at room temperature. The suspension was centrifuged at 2500×g for 10 min, and the organic phase was transferred and air-dried. The residual organic substance was resuspended in 800 μL ethanol containing 1% Triton X-100, and the concentrations of triglycerides and cholesterol were determined using commercial kits.
5.9. VLDL-TG secretion experiment
Male C57BL/6J mice weighing 20–25 g at 8–10 weeks of age were fasted for 48 h followed by refeeding for another 48 h. Mice were divided into groups according to body weight during the refeeding period and treated with the indicated compounds for 3 days (one dose daily). Time 0-min blood samples were collected from tails after the last dose of compound treatment 30 min later, followed by intravenous injection of tyloxapol (600 mg/kg, dissolved in saline, T0307-10G, Sigma–Aldrich, USA). Blood samples were collected 1, 2, 3, 4 h after tyloxapol injection and were subjected to centrifugation at 12,000×g for 2 min and stored at –20 °C until TG and cholesterol levels were assayed54.
5.10. In vitro measurement of cholesterol efflux
First, 0.5 μCi/mL 3H-cholesterol (NET139001MC, PerkinElmer, USA) and 15 μg/mL cholesterol (in 420 μg/mL M-β-CD) were added to the primary hepatocytes and cultured for 12 h (37 °C, 5% CO2). The medium containing 3H-cholesterol, was then discarded, and the cells were gently washed 3 times with PBS. Cells were incubated in serum-free medium containing 15 μg/mL cholesterol and 326E (12.5, 25, 50 μmol/L), BA (50 μmol/L) or T0901317 (1 μmol/L) for another 12 h (37 °C, 5% CO2). Cell medium was carefully collected and centrifuged at 14,000×g for 3 min to remove cellular debris, and 200 μL supernatant was transferred into a new microcentrifuge tube. The cells were washed 3 times with 200 μL PBS, and 200 μL 0.25 mol/L NaOH was added to lyse the cells. Transfer 150 μL of cell suspension to a new microcentrifuge tube. The 3H-cholesterol content in the medium supernatant and cell suspension was counted in scintillation liquids (LSC). The cholesterol efflux ratio was determined as Eq. (1):
| Cholesterol efflux ratio (%) = 3H-Cholesterol in the supernatant/(Supernatant 3H-cholesterol + intracellular 3H-cholesterol) × 100 | (1) |
The content of unlabeled intracellular cholesterol was measured using an Amplex™ Red Cholesterol Assay Kit (A12216, Thermo Fisher, MA, USA) according to the manufacturer's protocol.
5.11. In vivo measurement of cholesterol efflux
Male C57BL/6J mice weighing 20‒25 g at 8‒10 weeks of age were induced using a high cholesterol diet (HCD diet, D12109C, Research Diets, USA) for 2 weeks. Then, HCD mice were orally administered ETC-1002 (HY-12357, MedChemExpress, USA) or 326E at 30 mg/kg for 7 days. On the 7th day of ACLY inhibitor treatment, mice were received an intravenous tail vein injection of 2.5 μCi 3H-cholesterol, which was dissolved in 100 μL intralipid (20%). Fecal samples were collected 1, 2, 3, 4 days after 3H-cholesterol injection. The feces samples were soaked and homogenized in ddH2O (w/v = 1:10) overnight at 4 °C. Then, 100 μL homogenates were mixed with 400 μL KOH (20% in methanol), and saponified at 95 °C for 3 h. The neutral cholesterol fraction was extracted 2 times using 800 μL petroleum ether. The extracts were dried overnight and counted in scintillation liquids. Feces were prepared by mixing with ddH2O (w/v = 1:10) for 12 h at 4 °C. An equal volume of tert-butanol was added, and samples were extracted for 45 min at 37 °C. The supernatant, which contained bile acids, was transferred after centrifugation at 2000×g for 15 min. The 3H-bile acid content in the supernatants was measured in scintillation liquids55.
5.12. Analysis of atherosclerotic lesions
To quantify the atherosclerotic area along the entire aorta, the aortic tree isolated from ApoE−/− mice was dissected out and fixed in 70% ethanol for 10 min. Then, the lesions were stained with Sudan IV for 10 min, and rinsed twice with 70% ethanol (1.5 min/wash). The aortic tree was rinsed with PBS for 5 min to remove the residual ethanol, and the sudanophilic lesion area was assessed using Image J Pro Plus56.
5.13. Histology
Tissue specimens were fixed in 4% paraformaldehyde (PFA), and paraffin-embedded sections were subjected to standard haematoxylin-eosin (H&E) staining.
5.14. Pharmacokinetic evaluation
ICR mice and beagle dogs were orally administered or intravenously injected with the indicated compounds, and serial blood samples were collected at the indicated times. Blood samples were centrifuged at 12,000 rpm for 2 min at 4 °C to generate plasma. All plasma samples were kept frozen until analysis. The concentration of compounds in the plasma was detected using LC–MS/MS.
5.15. Rhesus monkey studies
Six to twenty-year old male rhesus monkeys with spontaneous mild or moderate hyperlipidemia for more than 2 years (LDL-C: 1.94–2.45 mmol/L) were selected, and then divided into three groups (n = 3). 326E (20 mg/kg) or atorvastatin (2.4 mg/kg) was administered by oral gavage once daily for 2 weeks at Sichuan Primed Shines Bio-tech Co., Ltd., China. A comprehensive evaluation of clinical laboratory measurements was conducted, including TC, TG, LDL-C, ApoB100, ALT, AST, CK and BUN. After the first oral administration of 326E, the PK study was performed.
5.16. Statistical analysis
All results are reported as the means ± SEM. Differences between two groups were analyzed using a two-tailed unpaired Student's t-test. Differences in multiple groups were analyzed using one-way ANOVA (GraphPad Prism version 6.0 for Windows). Differences were considered significant when P < 0.05.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (92057116 and 82170872); the Medical Guidance Project of Shanghai Science and Technology Commission (20S11903400, China); the “Personalized Medicines-Molecular Signature-based Drug Discovery and Development” Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12040328, China); the “State Key Laboratory of Drug Research” Shanghai Institute of Materia Medica, Chinese Academy of Sciences (SIMM2105KF-02, China); Natural Science Foundation of Shanghai's 2021 “Science and Technology Innovation Action Plan” (21ZR1475300, China); the Lingang Laboratory (LG-QS-202205-01, China).
Author contributions
Fajun Nan, Jingya Li, Zhifu Xie and Mei Zhang participated in the research design. Qian Song, Long Cheng, Xinwen Zhang, Gaolei Song, Yangming Zhang, Xinyu Sun, Min Gu, Chendong Zhou, Jianpeng Yin, Kexin Zhu and Xiaoyan Chen performed the experiments and data analysis. Zhifu Xie, Mei Zhang and Qian Song contributed to the preparation of this paper. Fajun Nan and Jingya Li contributed to discussion, review and editing the manuscript.
Conflicts of interest
Yangming Zhang received consultant allowance from Burgeon Therapeutics Co., Ltd. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.06.011.
Contributor Information
Jingya Li, Email: jyli@simm.ac.cn.
Fajun Nan, Email: fjnan@simm.ac.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Ference B.A., Ginsberg H.N., Graham I., Ray K.K., Packard C.J., Bruckert E., et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the european atherosclerosis society consensus panel. Eur Heart J. 2017;38:2459–2472. doi: 10.1093/eurheartj/ehx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman M.G., Ference B.A., Im K., Wiviott S.D., Giugliano R.P., Grundy S.M., et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316:1289–1297. doi: 10.1001/jama.2016.13985. [DOI] [PubMed] [Google Scholar]
- 3.Stone N.J., Robinson J.G., Lichtenstein A.H., Bairey Merz C.N., Blum C.B., Eckel R.H., et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the american college of cardiology/american heart association task force on practice guidelines. Circulation. 2014;129:S1–S45. doi: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 4.Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23:S139–S148. [PubMed] [Google Scholar]
- 5.Xiao C., Dash S., Morgantini C., Hegele R.A., Lewis G.F. Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes. 2016;65:1767–1778. doi: 10.2337/db16-0046. [DOI] [PubMed] [Google Scholar]
- 6.Brautbar A., Ballantyne C.M. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol. 2011;8:253–265. doi: 10.1038/nrcardio.2011.2. [DOI] [PubMed] [Google Scholar]
- 7.Uspst Force, Bibbins-Domingo K., Grossman D.C., Curry S.J., Davidson K.W., Epling J.W., Jr., et al. Statin use for the primary prevention of cardiovascular disease in adults: us preventive services task force recommendation statement. JAMA. 2016;316:1997–2007. doi: 10.1001/jama.2016.15450. [DOI] [PubMed] [Google Scholar]
- 8.Ford I., Murray H., McCowan C., Packard C.J. Long-term safety and efficacy of lowering low-density lipoprotein cholesterol with statin therapy: 20-year follow-up of west of scotland coronary prevention study. Circulation. 2016;133:1073–1080. doi: 10.1161/CIRCULATIONAHA.115.019014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Istvan E.S., Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 10.Schirris T.J., Renkema G.H., Ritschel T., Voermans N.C., Bilos A., van Engelen B.G., et al. Statin-induced myopathy is associated with mitochondrial complex iii inhibition. Cell Metab. 2015;22:399–407. doi: 10.1016/j.cmet.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Golomb B.A., Evans M.A. Statin adverse effects: a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs. 2008;8:373–418. doi: 10.2165/0129784-200808060-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins R., Reith C., Emberson J., Armitage J., Baigent C., Blackwell L., et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388:2532–2561. doi: 10.1016/S0140-6736(16)31357-5. [DOI] [PubMed] [Google Scholar]
- 13.Ashen M.D., Foody J.M. Evidence-based guidelines for cardiovascular risk reduction: the safety and efficacy of high-dose statin therapy. J Cardiovasc Nurs. 2009;24:429–438. doi: 10.1097/JCN.0b013e3181b4bab4. [DOI] [PubMed] [Google Scholar]
- 14.Lagace T.A. Pcsk9 and ldlr degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol. 2014;25:387–393. doi: 10.1097/MOL.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of african descent resulting from frequent nonsense mutations in pcsk9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 16.Stein E.A., Mellis S., Yancopoulos G.D., Stahl N., Logan D., Smith W.B., et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 17.Ray K.K., Landmesser U., Leiter L.A., Kallend D., Dufour R., Karakas M., et al. Inclisiran in patients at high cardiovascular risk with elevated ldl cholesterol. N Engl J Med. 2017;376:1430–1440. doi: 10.1056/NEJMoa1615758. [DOI] [PubMed] [Google Scholar]
- 18.Navarese E.P., Kolodziejczak M., Schulze V., Gurbel P.A., Tantry U., Lin Y., et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med. 2015;163:40–51. doi: 10.7326/M14-2957. [DOI] [PubMed] [Google Scholar]
- 19.Krahenbuhl S., Pavik-Mezzour I., von Eckardstein A. Unmet needs in LDL-C lowering: when statins won't do. Drugs. 2016;76:1175–1190. doi: 10.1007/s40265-016-0613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gotto A.M., Jr., Moon J.E. Pharmacotherapies for lipid modification: beyond the statins. Nat Rev Cardiol. 2013;10:560–570. doi: 10.1038/nrcardio.2013.117. [DOI] [PubMed] [Google Scholar]
- 21.Sanders F.W., Griffin J.L. De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc. 2016;91:452–468. doi: 10.1111/brv.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu S., Mulligan K., Schwarz J.M. Dietary carbohydrates and fatty liver disease: de novo lipogenesis. Curr Opin Clin Nutr Metab Care. 2018;21:277–282. doi: 10.1097/MCO.0000000000000469. [DOI] [PubMed] [Google Scholar]
- 23.Hellerstein M.K., Schwarz J.M., Neese R.A. Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr. 1996;16:523–557. doi: 10.1146/annurev.nu.16.070196.002515. [DOI] [PubMed] [Google Scholar]
- 24.Srere P.A. The citrate cleavage enzyme. I. Distribution and purification. J Biol Chem. 1959;234:2544–2547. [PubMed] [Google Scholar]
- 25.Tan M., Mosaoa R., Graham G.T., Kasprzyk-Pawelec A., Gadre S., Parasido E., et al. Inhibition of the mitochondrial citrate carrier, SLC25A1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020;27:2143–2157. doi: 10.1038/s41418-020-0491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kindt A., Liebisch G., Clavel T., Haller D., Hörmannsperger G., Yoon H., et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat Commun. 2018;9:3760. doi: 10.1038/s41467-018-05767-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez Calejman C., Trefely S., Entwisle S.W., Luciano A., Jung S.M., Hsiao W., et al. Mtorc2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Commun. 2020;11:575. doi: 10.1038/s41467-020-14430-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo L., Guo Y.Y., Li B.Y., Peng W.Q., Chang X.X., Gao X., et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J Biol Chem. 2019;294:11805–11816. doi: 10.1074/jbc.RA119.008708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin R., Tao R., Gao X., Li T., Zhou X., Guan K.L., et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samsoondar J.P., Burke A.C., Sutherland B.G., Telford D.E., Sawyez C.G., Edwards J.Y., et al. Prevention of diet-induced metabolic dysregulation, inflammation, and atherosclerosis in LDLR‒/‒ mice by treatment with the ATP-citrate lyase inhibitor bempedoic acid. Arterioscler Thromb Vasc Biol. 2017;37:647–656. doi: 10.1161/ATVBAHA.116.308963. [DOI] [PubMed] [Google Scholar]
- 31.Molusky M.M., Hsieh J., Lee S.X., Ramakrishnan R., Tascau L., Haeusler R.A., et al. Metformin and amp kinase activation increase expression of the sterol transporters ABCG5/8 (ATP-binding cassette transporter g5/g8) with potential antiatherogenic consequences. Arterioscler Thromb Vasc Biol. 2018;38:1493–1503. doi: 10.1161/ATVBAHA.118.311212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beigneux A.P., Kosinski C., Gavino B., Horton J.D., Skarnes W.C., Young S.G. ATP-citrate lyase deficiency in the mouse. J Biol Chem. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q., Jiang L., Wang J., Li S., Yu Y., You J., et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology. 2009;49:1166–1175. doi: 10.1002/hep.22774. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q., Li S., Jiang L., Zhou Y., Li Z., Shao M., et al. Deficiency in hepatic ATP-citrate lyase affects VLDL-triglyceride mobilization and liver fatty acid composition in mice. J Lipid Res. 2010;51:2516–2526. doi: 10.1194/jlr.M003335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinkosky S.L., Groot P.H.E., Lalwani N.D., Steinberg G.R. Targeting ATP-citrate lyase in hyperlipidemia and metabolic disorders. Trends Mol Med. 2017;23:1047–1063. doi: 10.1016/j.molmed.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Ference B.A., Ray K.K., Catapano A.L., Ference T.B., Burgess S., Neff D.R., et al. Mendelian randomization study of acly and cardiovascular disease. N Engl J Med. 2019;380:1033–1042. doi: 10.1056/NEJMoa1806747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Granchi C. Atp citrate lyase (acly) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018;157:1276–1291. doi: 10.1016/j.ejmech.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Feng X., Zhang L., Xu S., Shen A.Z. ATP-citrate lyase (acly) in lipid metabolism and atherosclerosis: an updated review. Prog Lipid Res. 2020;77 doi: 10.1016/j.plipres.2019.101006. [DOI] [PubMed] [Google Scholar]
- 39.Ray K.K., Bays H.E., Catapano A.L., Lalwani N.D., Bloedon L.T., Sterling L.R., et al. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med. 2019;380:1022–1032. doi: 10.1056/NEJMoa1803917. [DOI] [PubMed] [Google Scholar]
- 40.Goldberg A.C., Leiter L.A., Stroes E.S.G., Baum S.J., Hanselman J.C., Bloedon L.T., et al. Effect of bempedoic acid vs placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease: the clear wisdom randomized clinical trial. JAMA. 2019;322:1780–1788. doi: 10.1001/jama.2019.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinkosky S.L., Newton R.S., Day E.A., Ford R.J., Lhotak S., Austin R.C., et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun. 2016;7 doi: 10.1038/ncomms13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burke A.C., Telford D.E., Sutherland B.G., Edwards J.Y., Sawyez C.G., Barrett P.H.R., et al. Bempedoic acid lowers low-density lipoprotein cholesterol and attenuates atherosclerosis in low-density lipoprotein receptor-deficient (LDLR+/‒ and LDLR‒/‒) yucatan miniature pigs. Arterioscler Thromb Vasc Biol. 2018;38:1178–1190. doi: 10.1161/ATVBAHA.117.310676. [DOI] [PubMed] [Google Scholar]
- 43.Pinkosky S.L., Filippov S., Srivastava R.A., Hanselman J.C., Bradshaw C.D., Hurley T.R., et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. 2013;54:134–151. doi: 10.1194/jlr.M030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pal M., Khanal M., Marko R., Thirumalairajan S., Bearne S.L. Rational design and synthesis of substrate-product analogue inhibitors of α-methylacyl-coenzyme a racemase from mycobacterium tuberculosis. Chem Commun (Camb) 2016;52:2740–2743. doi: 10.1039/c5cc08096g. [DOI] [PubMed] [Google Scholar]
- 45.Graf G.A., Yu L., Li W.P., Gerard R., Tuma P.L., Cohen J.C., et al. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. [DOI] [PubMed] [Google Scholar]
- 46.Whaley-Connell A., Sowers J.R. Indices of obesity and cardiometabolic risk. Hypertension. 2011;58:991–993. doi: 10.1161/HYPERTENSIONAHA.111.180406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farzadfar F., Finucane M.M., Danaei G., Pelizzari P.M., Cowan M.J., Paciorek C.J., et al. National, regional, and global trends in serum total cholesterol since 1980: systematic analysis of health examination surveys and epidemiological studies with 321 country-years and 3.0 million participants. Lancet. 2011;377:578–586. doi: 10.1016/S0140-6736(10)62038-7. [DOI] [PubMed] [Google Scholar]
- 48.Michos E.D., McEvoy J.W., Blumenthal R.S. Lipid management for the prevention of atherosclerotic cardiovascular disease. N Engl J Med. 2019;381:1557–1567. doi: 10.1056/NEJMra1806939. [DOI] [PubMed] [Google Scholar]
- 49.Grover A., Benet L.Z. 4th ed. CRC Press; 2013. Clinical pharmacokinetics and pharmacodynamics. Encyclopedia of pharmaceutical science and technology; pp. 473–487. [Google Scholar]
- 50.Wei J., Leit S., Kuai J., Therrien E., Rafi S., Harwood H.J., Jr., et al. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature. 2019;568:566–570. doi: 10.1038/s41586-019-1094-6. [DOI] [PubMed] [Google Scholar]
- 51.Li W.C., Ralphs K.L., Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- 52.Sun S.M., Xie Z.F., Zhang Y.M., Zhang X.W., Zhou C.D., Yin J.P., et al. Ampk activator c24 inhibits hepatic lipogenesis and ameliorates dyslipidemia in HFHC diet-induced animal models. Acta Pharmacol Sin. 2021;42:585–592. doi: 10.1038/s41401-020-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell R.L., Suppnick J.D., Hettrick J.M., Nigro N.D. Rat liver microsome-mediated N-demethylation and mutagenicity of azoxymethane. Cancer Res. 1978;38:4585–4590. [PubMed] [Google Scholar]
- 54.Lam T.K., Gutierrez-Juarez R., Pocai A., Bhanot S., Tso P., Schwartz G.J., et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med. 2007;13:171–180. doi: 10.1038/nm1540. [DOI] [PubMed] [Google Scholar]
- 55.Xie Y., Kennedy S., Sidhu R., Luo J., Ory D.S., Davidson N.O. Liver x receptor agonist modulation of cholesterol efflux in mice with intestine-specific deletion of microsomal triglyceride transfer protein. Arterioscler Thromb Vasc Biol. 2012;32:1624–1631. doi: 10.1161/ATVBAHA.112.246066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang J.J., Li J.G., Qi W., Qiu W.W., Li P.S., Li B.L., et al. Inhibition of srebp by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011;13:44–56. doi: 10.1016/j.cmet.2010.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.