

Abstract
Our previous studies on nascent transcription across the human β-globin gene cluster revealed the presence of intergenic transcripts in addition to the expected genic transcripts. We now show that transcription into the β-globin locus control region (LCR) begins within an ERV9 endogenous retroviral long terminal repeat upstream of DNase I hypersensitive site 5. However, in a transgenic mouse, which has the human β-globin LCR but lacks the ERV9 LTR, transcription begins upstream of the transgenic locus. We postulate that in this transgenic mouse nearby endogenous mouse promoters are activated by the LCR. Intergenic transcription is also detected across the whole transgenic globin gene locus independently of the stage of erythroid development. Intergenic transcription in the β-globin cluster is erythroid specific; however, it can be induced in nonerythroid cells by several means: by transinduction with a plasmid transcribing part of the cluster, by exogenous addition of transcription factors, and by treatment with the histone deacetylase inhibitor trichostatin A.
The human β-globin cluster, which extends over 70 kb of chromosome 11, contains the five β-globin genes (ε, Gγ, Aγ, δ, and β) arranged in the same orientation and in the order of their expression during development. These genes are under the influence of a number of erythroid-specific transcription factors (TFs) including GATA-1 and its cofactor FOG-1 (30), NF-E2, and EKLF (reviewed in reference 21).
Several kilobases upstream of the ɛ-globin gene are at least five DNase I-hypersensitive sites (HS1 to HS5) which constitute the locus control region (LCR). These contain binding sites for GATA-1 and NF-E2 as well as CACC sites that bind a variety of general TFs including Sp1 (21). In transgenic mice the LCR is necessary for position-independent, copy-number-dependent regulated expression of the globin genes (13). Although HS2 has classical enhancer activity (27), recent experiments have shown that the LCR as a whole is orientation dependent and therefore does not fulfil the classical definition of an enhancer (28). It has been suggested that the LCR is responsible for the open chromatin structure of the β-globin locus. Thus, in the naturally occurring Hispanic chromosome, from which 35 kb including most of the LCR is deleted, the entire cluster is DNase I resistant and the globin genes are transcriptionally inactive (11). However, recent discrete targeted deletions of both human and mouse LCRs in their natural contexts suggest that sequences outside the region covered by the five HSs may also play a role in the generation of a permissive chromatin structure throughout the cluster (10, 22).
Our previous studies of transcription in the β-globin cluster showed that in addition to transcription of the five genic regions, there is also transcription in the regions between the globin genes and across the LCR (3). Transcription of the intergenic regions of the β-globin cluster occurs on the same strand as that of the globin genes and is specific to erythroid cells. Further work on the phenomenon of intergenic transcription by Gribnau et al. (12) has suggested that the level of intergenic transcription can be correlated with the DNase I sensitivity in the β-globin cluster. We have now extended our studies of intergenic transcription in the human β-globin cluster. We have mapped the most 5′ extent of LCR transcription into the endogenous locus to a retroviral element (ERV9) 5′ to HS5. In contrast, intergenic transcripts downstream of two of the globin genes (ɛ and Aγ) do not correspond to any definable promoter and no precise transcription start site could be identified. We further show that intergenic transcription occurs across the entire human globin cluster in the adult erythroid cells of a transgenic mouse. Although intergenic transcription of the β-globin cluster is specific to erythroid cells, it can be induced in nonerythroid cells by the expression of transiently transfected globin genes, a process known as transinduction. Even though the mechanism of transinduction is unknown, the transfected plasmids are seen to colocalize with the genomic copies of the cluster, and it is thought that this in some way releases the intergenic regions for transcription (3). We now describe two other means of inducing intergenic transcription in a nonerythroid environment, by the introduction of exogenous TFs and by the histone deacetylase inhibitor trichostatin A.
MATERIALS AND METHODS
Plasmids and probes.
Probes were generated from GenBank sequence accession no. AF064190 as follows: A (positions 1 to 512), B (512 to 1122), D (1941 to 2645), E (2645 to 3222), F (3222 to 3971), G (3971 to 4819), L17 (4818 to 5286), L18 (5286 to 5819), L19 (5819 to 6192), and L20 (6192 to 6509). Other globin probes were as previously described (3). Control probes for M13 (background), VA, and HIS and 5S (adenovirus VAI gene, mouse histone H4, and Xenopus laevis 5S DNA, respectively) have also been described (see references listed in reference 3).
Plasmids VA, which expresses the adenovirus VA1 gene, and Tat (1) have been described. HIVβback contains a promoterless β-globin fragment (positions 62185 to 64301 of GenBank sequence accession no. U01317) fused to the human immunodeficiency virus (HIV) promoter in inverse orientation, so the antisense strand of the gene is transcribed. In pβF+ a fragment corresponding to probe F replaces the β-globin promoter at position 62211. pβEF+ also contains the simian virus 40 (SV40) enhancer in reverse orientation with respect to β-globin. Aγ and ɛ 3′ flanking regions tested for promoter activity were fragments from positions 21362 to 23325, 42152 to 43185, and 42152 to 43785. Stuart Orkin kindly donated erythroid TF expression plasmids pXM GATA-1 (29), pMT2 FOG (30), and pEF1α-neo NF-E2 (14). Shona Murphy provided a plasmid which expresses the GAL4 binding domain fused to the OCT-1 activation domain (P+NC in reference 20).
Tissue culture.
Subconfluent HeLa cells were transfected using Effectene (Qiagen) with 1 μg of VA plasmid, 1.5 μg of Tat plasmid and 5 μg of the globin and TF plasmids 24 h prior to nuclear run-on analysis (NRO). Hemin (40 μM) and trichostatin A (TSA) (3 μM) inductions were for 24 and 18 h prior to NRO, respectively.
NRO.
Nuclear isolation and run-on analyses of cultured cells were performed as previously described (3). The line 72 transgenic mouse containing the human β-globin cluster has been described (26). Mice were made anemic by treatment with 0.04 mg of acetylphenylhydrazine per g of body weight. An 80 to 95% pure population of erythroblasts was generated on the sixth day of treatment by gently scraping the surface of the excised spleen. These cells were washed in Tris-buffered saline (pH 7), and nuclei were prepared for NRO as for tissue culture cells.
RNA extraction and analysis.
Cytoplasmic RNA was isolated 48 h after transfection as previously described (9). A BanII-AccI fragment encompassing the putative ERV9 promoter and β-globin exon 1 in pβF+ was cloned into pGEM4 and transcribed into an antisense riboprobe. Five hundred counts per second of riboprobe was hybridized to 20 μg of cytoplasmic RNA in 80% formamide–40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.4)–1 mM EDTA–400 mM NaCl at 56°C overnight. RNase protection was as described previously (24).
An oligonucleotide complementary to β-globin exon 1 (GTAACGGCAGACTTCTCCTCAGG) was end labeled (24) and purified using a G25 spin column. One thousand counts per second of primer was hybridized to 20 μg of RNA at 55°C for 5 h and reverse transcribed with avian myeloblastosis virus reverse transcriptase as described previously (32). A sequencing ladder was generated with labeled primer using Sequenase.
RT-PCR analysis.
One microgram of cytoplasmic RNA was reverse transcribed using primer GRT (ATCTCCACATTATTTTAGTC) with Superscript II (Gibco BRL) according to the manufacturer's instructions. cDNA was amplified using Pfu Turbo (Stratagene) and the following primers spanning the ERV9 LTR: G3′ (TTAATTGCAAGGGCTGTTGGAAA), FDOWN (TCGGGTCCCCTTCCACACTGT), FDS (CTCTCTACCAATCAGCAGGATGT), FU3 (ACCTTTGTGTGGACACTCTG), and F600 (TTGCTGGGGATGCGAAGAAC).
5′ rapid amplification of cDNA ends (RACE) was carried out using the SMART RACE cDNA amplification kit (Clontech) according to the manufacturer's instructions. GRT was used for reverse transcription (RT), and amplification was performed with G3′. Genuine RACE products were identified by blotting and hybridization to probe G and were reamplified using nested primers prior to cloning and sequencing.
RESULTS
The 5′ extent of LCR transcription.
Previous studies of the human β-globin cluster showed that all regions of the LCR up to and including HS5 are transcribed (3). Using a variety of techniques, we now map the 5′ extent of transcription beyond the LCR in the human erythroleukemic cell line K562, which endogenously expresses both ɛ- and γ-globin genes.
We have generated six additional single-stranded DNA probes upstream of and contiguous with the previous panel of probes. NRO in hemin-induced K562 cells shows transcription across the more downstream probes (E, F, and G) but not over probes A, B, and D (Fig. 1A). All signals were abolished by pretreatment of the nuclei with low concentrations of α-amanitin, indicating that they are due to transcription by RNA polymerase II.
FIG. 1.
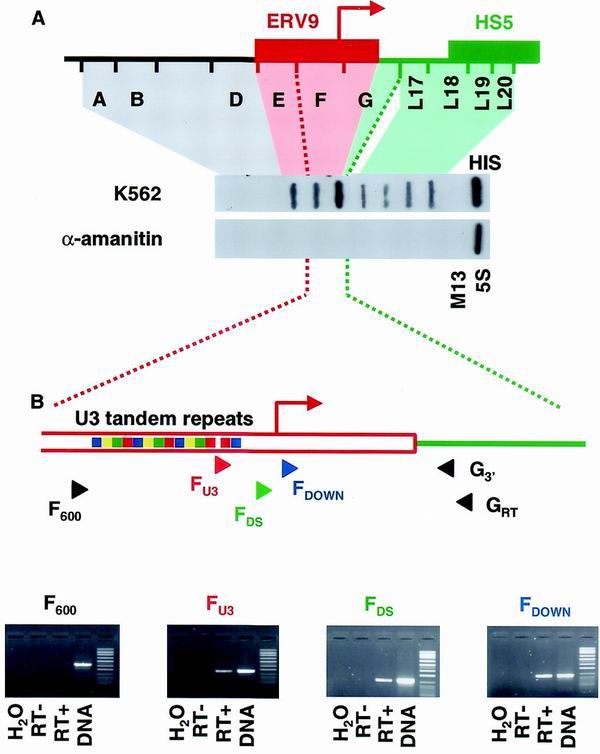
Transcription of the 5′ region of the β-globin LCR. (A) NRO analysis of hemin-induced K562 cells, in the absence and presence of α-amanitin. The positions of single-stranded M13 probes relative to the ERV9 retroviral LTR and HS5 are shown. Probes for M13 background, histone H4 (HIS), and 5S RNA are also shown. (B) Analysis of endogenous LCR transcripts by RT-PCR. Primer positions are shown relative to the features of the ERV9 LTR. Red arrow, putative transcription start site; colored boxes, U3 tandem repeats. RT-PCR was carried out on cytoplasmic RNA extracted from K562 cells, and in each case water, RT-negative, and DNA controls were included. Green, LCR; red, ERV9 LTR.
The region encompassed by probes E through G is a member of the ERV9 family of endogenous retroviral elements (18). A 1.6-kb region contained within probes E through G shows 80 to 90% homology to the long terminal repeat (LTR) of ERV9 and includes a putative promoter with a transcription start site 35 nucleotides before the 3′ end of probe F. The ERV9 family has ≈50 members as well as ≈4,000 single LTRs (15). It therefore seems likely that at least some of the signals we observe over probes E through G in K562 cells are derived from other ERV9 elements.
To determine whether any part of the signal we observe over probes E through G is due to transcription of the ERV9 element upstream of the β-globin cluster, we carried out RT-PCR analysis of this region. RNA was reverse transcribed using a primer located in the unique region downstream of the ERV9 LTR (GRT). The downstream (nested) PCR primer (G3′) was also located within the unique region. Of four 5′ PCR primers that hybridize to sites across the ERV9 LTR, three gave RT-PCR products (Fig. 1B), indicating that the β-globin ERV9 element is transcribed in erythroid cells. However, as two of these primers lie upstream of the LTR promoter, it is unclear whether this promoter itself contributes to LCR transcription. Our most upstream primer failed to give an RT-PCR product, suggesting that the signal we detected over probe E in our run-on experiments is derived from transcribed ERV9 elements elsewhere in the genome.
The ERV9 family of endogenous retroviruses has previously been studied by La Mantia et al. (16), who defined a minimal promoter, mapped transcription start sites, and studied the expression pattern of the ERV9 elements. To confirm that the ERV9 LTR promoter upstream of the β-globin cluster is active, we used a transient reporter gene assay. A DNA fragment corresponding to probe F, which contains the putative promoter and transcription start site, was fused to a promoterless β-globin gene both in the presence and in the absence of the SV40 enhancer (pβEF+ and pβF+, respectively). The β-globin gene provides a stable 3′ end for the ERV9 LTR transcript, and following transfection into HeLa cells, steady-state RNA was analyzed quantitatively by RNase protection (Fig. 2A) and by primer extension to accurately map the 5′ ends (Fig. 2B). A cluster of transcription start sites similar to those mapped by La Mantia et al. was found in the β-globin ERV9 LTR promoter. When normalized to the VA transfection control, the SV40 enhancer provided an approximately threefold stimulation of transcription, although the β-cluster ERV9 LTR promoter alone was able to efficiently initiate transcription in HeLa cells. The ERV9 promoter activity was also confirmed in both K562 and HeLa cells using a luciferase reporter gene system (data not shown).
FIG. 2.
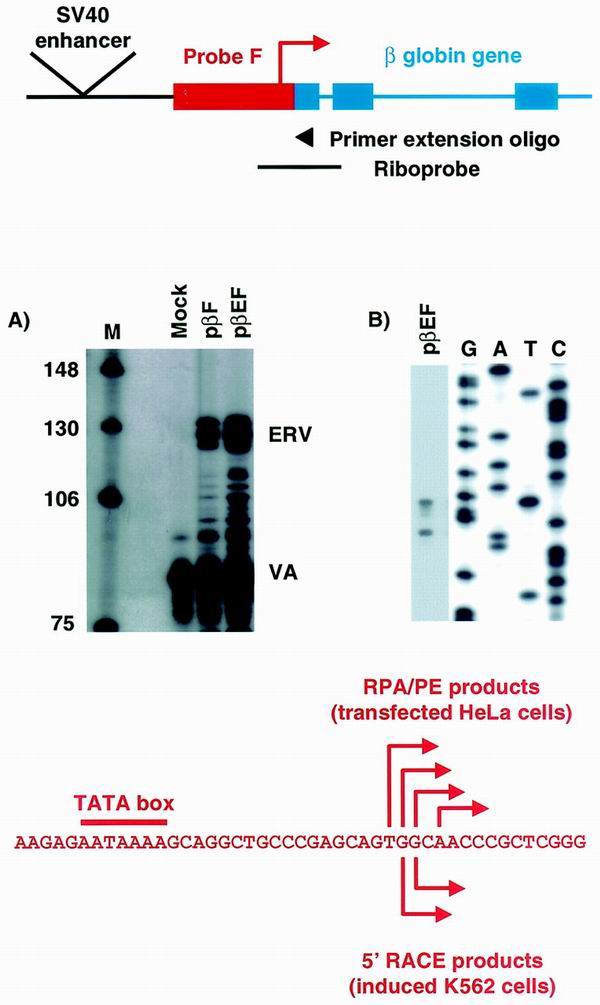
Reporter constructs, whose structure is shown in the upper panel, were transiently transfected into HeLa cells and analyzed by RNase protection (A) and primer extension (B). The transcription start sites identified by primer extension are shown below panels A and B along with the positions of the 5′ RACE products identified from endogenous LCR expression in hemin-induced K562 cells.
Finally we carried out 5′ RACE analysis of endogenously expressed LCR RNA from induced K562 cells using the SMART cDNA Amplification Kit (Clontech). During this procedure the 5′ adapter is specifically added to cDNAs which have reached the 5′ end of the corresponding RNA, enriching for full-length capped cDNAs (19). Using the primers GRT and G3′ as specific RT-PCR and 3′ PCR primers, respectively, we carried out several independent RACE experiments and identified, reamplified, cloned, and sequenced the genuine products. 5′ RACE products corresponding to two of the ERV9 LTR promoter transcription start sites seen in our transient assays were generated from K562 RNA (Fig. 2). These data demonstrate that this promoter is endogenously active in erythroid cells and contributes to LCR transcription. However, a number of RACE products which extend upstream of this promoter into the U3 region of the LTR were also generated. These findings confirm our RT-PCR analysis by indicating that at least some LCR transcripts initiate within the ERV9 LTR upstream of its promoter.
Definition of other intergenic transcripts.
We have also attempted to assign start sites and promoter activity to the intergenic transcripts downstream of the human ɛ and Aγ genes. NRO suggests that intergenic transcription begins approximately 3.5 kb downstream of both ɛ and Aγ (3) (Fig. 4). Using the luciferase reporter gene system we found no evidence for promoter activity in either of these regions, despite testing a variety of fragments in the presence of an enhancer and in both erythroid and nonerythroid cells. We also attempted to detect 5′ ends from endogenous intergenic transcripts in K562 cells using 5′ RACE; however, we were able to detect only a low-level population of random ends (data not shown). Since some of these apparent 5′ ends lie upstream of the region suggested by NRO, it is not clear whether they represented random initiation events. It is possible that in embryonic and fetal erythroid cells such as K562 a small amount of read-through from the expressed ɛ and Aγ genes occurs. Random premature termination of RT of such transcripts could then produce the population of transcripts we detected.
FIG. 4.
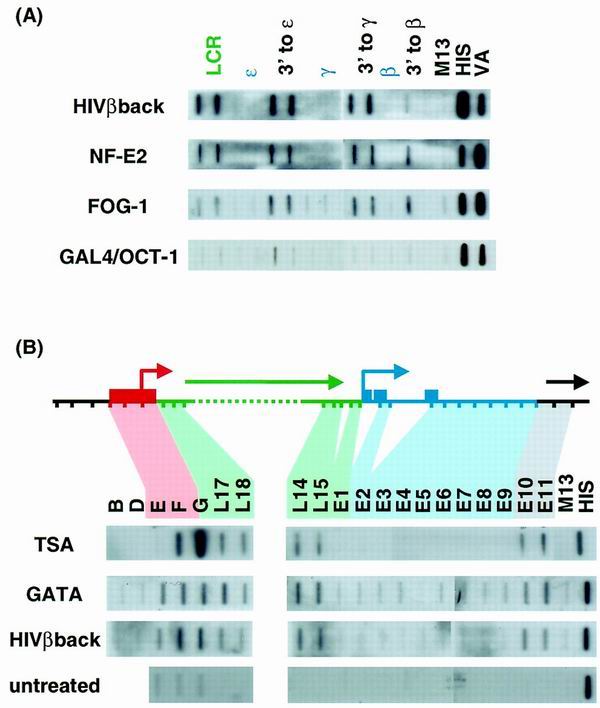
Induction of transcription across the β-globin cluster in nonerythroid cells. HeLa cells were transfected with a plasmid expressing the antisense β-globin gene or with clones expressing the tethered NF-E2 heterodimer, GATA-1, FOG-1, or a GAL4/OCT-1 fusion protein plus the VA1 gene as a transfection control. (A) Cells were analyzed by NRO, and the resulting RNA was hybridized to a panel of probes taken from the LCR (green), intergenic (black), and genic (blue) regions of the entire cluster. (B) A more extended series of probes showing the beginning and end of sense strand transcription across the LCR is shown for HeLa cells expressing GATA-1, transinduced or induced with TSA, as well as for uninduced HeLa cells. Note that the three repetitive probes (E, F, and G) also give signals in the uninduced cells, presumably from copies elsewhere in the genome.
Intergenic transcription in a transgenic β-globin cluster.
We have also detected intergenic transcription in mice carrying the human β-globin cluster as a transgene. These data therefore provide the first description of intergenic transcripts detected by NRO reading across the human β-globin gene cluster in an authentic erythroid tissue. Previously, intergenic transcripts were analyzed in erythroid tissue only by in situ hybridization.
Line 72 is a transgenic mouse line that has previously been analyzed in detail (26) and shows normal expression and DNase I sensitivity pattern in the human β-globin cluster. The single copy of the human β-globin cluster stretches from just upstream of HS5 (probe L17) to several kilobases downstream of the β-globin gene and notably lacks the ERV9 LTR. Erythroblasts isolated from the spleen of anemic adult mice were analyzed by NRO. Erythroblasts from nontransgenic mice were analyzed alongside, so that any contributions to NRO signals made by the mouse β-globin cluster could be determined. Transcription was seen across the entire LCR (Fig. 3), including the most 5′ region of the transgene (i.e., probe 17). We postulate that in the absence of the ERV9 LTR, transcription in the line 72 transgenic mouse can initiate from endogenous mouse promoter(s) activated by the transgenic LCR, since even the most upstream part of the transgene is transcribed. Transcription of the human β-globin gene was also apparent (probes B5 and B6), while ɛ and the γ genes were silent (probes E6 and AG3), as would be expected in adult tissue. Note that the signal detected by probe E2 is due to cross-hybridization to both the human β and mouse βMAJ genes as this probe is within the conserved exon 2 of the gene. These data demonstrate that there are relatively even levels of intergenic transcription across the human cluster. Consequently they are at variance with the results of Gribnau et al. (10), who reported that in adult erythroid cells intergenic transcription was greatly reduced in the regions surrounding the embryonic and fetal ɛ and γ genes, as shown by RNA fluorescent in situ hybridization (FISH) analysis. As a further control we also compared the ɛ and β intergenic transcription levels in K562 cells (Fig. 3, lower panels) and again found them to be similar in this embryonic erythroid cell line.
FIG. 3.

NRO analysis of intergenic transcription in a human β-globin cluster transgene in the mouse. Probe positions are indicated (LCR probes are in green, genic probes are in blue, and intergenic probes are in black). The NRO profile of the transgenic mouse line 72 was compared to that of a nontransgenic mouse and with that of the human erythroleukemic cell line K562. Signals were quantitated by phosphorimager and corrected for U content and with respect to HIS and are shown in the lower panels. We have not looked for antisense LCR transcription in this mouse.
Induction of LCR transcription in nonerythroid cells.
We have previously described one way of inducing intergenic transcription in the β-globin cluster in nonerythroid cell lines, namely by transinduction. Transiently transfected plasmids expressing sequences within the β-globin cluster colocalize with the chromosomal copy of the cluster and activate intergenic transcription in an as yet undetermined way (3). All regions of the β-globin cluster tested (genic and intergenic) are able to transinduce intergenic transcription, and this effect is independent of the orientation of the expression plasmid (Fig. 4). This is entirely consistent with our previous suggestions that transinduction is mediated at the DNA level by the colocalization of the actively transcribed plasmid and the endogenous β-globin locus (3).
We have now looked at the effects of erythroid TFs on transcription of the β-globin cluster in HeLa cells. Expression plasmids were obtained for the NF-E2 heterodimer, GATA-1, and FOG-1, and the GAL4 DNA binding domain fused to the OCT-1 activation domain provided a nonerythroid control. Following transfection of these plasmids into HeLa cells we used NRO to look for induction of transcription at the nascent level. All three erythroid TFs were able to stimulate transcription of all the intergenic regions and the LCR (Fig. 4), although no combination of transfected plasmids was able to activate the globin genes themselves (combinations not shown). The GAL4/OCT-1 fusion protein did not induce globin intergenic transcription, although it has been previously shown to activate transcription linked to GAL4 binding sites (20).
A third way in which intergenic transcription can be induced in a nonerythroid environment is by treatment with TSA. TSA is a histone deacetylase inhibitor and as such causes a global increase in the level of acetylated histones (35). Although the exact method by which histone acetylation leads to an opening of chromatin is not clear (25), it has recently been shown that treatment with TSA can activate retroviral promoters, presumably by releasing them from a repressive chromatin environment (6). Treatment of HeLa cells with TSA induces all of the globin intergenic transcripts but fails to stimulate transcription of the globin genes (Fig. 4B and data not shown). The signals observed for the LTR probes (F and especially G) are disproportionately high following TSA induction, presumably because ERV9 elements elsewhere in the genome are also activated.
We have noticed that when transcription of the β-cluster is induced by any of the means described above, patchy antisense transcription can be detected in the LCR (data not shown). This is at variance with the previously reported strand-specific LCR transcription of K562 cells (3) and may suggest that during induction intergenic transcription occurs as a consequence of a general opening of the chromatin domain. However, it is also clear that on the sense strand the initiation and termination sites of induced transcription coincide with the intergenic transcripts of erythroid cells (Fig. 4B) and the ɛ gene transcript (probes E2 to E9, as shown by hybrid selection NRO [8]) is completely absent.
DISCUSSION
These studies extend our knowledge of the nature and significance of nongenic transcripts derived from the human β-globin gene cluster. We have used several approaches to determine where transcription of the β-globin LCR begins in the human erythroleukemic cell line K562. These experiments reveal that there is no sense strand transcription upstream of an ERV9 endogenous retroviral element 1.1 kb upstream of HS5. This isolated LTR includes the retroviral promoter, which in transient assays was active in both erythroid and nonerythroid cells. However, although our analysis of endogenous RNA confirms that the ERV9 promoter is active and contributes to transcription of the LCR, transcripts that may initiate within the U3 repeats of the LTR were also detected. Our results extend the preliminary transcription analysis which accompanied a detailed sequence analysis of the region by Long et al. (18). However, these authors failed to detect transcripts initiating within the U3 region, and in this respect our analysis has been more refined. In view of the more extended region of DNase I sensitivity surrounding the β-globin gene cluster, we have also looked for transcription on the antisense strand, towards the odorant receptor genes embedded within the same chromatin domain (5). A small amount of antisense transcription was seen over the repetitive probes (E through G) but did not extend into unique probes A through D or 17 through 20 (data not shown) and was likely to derive from ERV9 elements elsewhere in the genome. However, two additional probes which lie close to HOR5′β 1 (GenBank accession no. AF137396, positions 29195 to 29737 and 30600 to 31024) did show erythroid-specific antisense transcription, which will be the subject of further studies.
Long et al. also suggest that the ERV9 LTR promoter has inherently erythroid properties. Although it is difficult to compare transfection experiments in different cell lines, our transient reporter gene analysis shows that ERV9 can be active in a nonerythroid environment at a level approaching its activity in erythroid cells. Furthermore, transinduction of HeLa cells shows that transcription from the endogenous β-globin ERV9 LTR can be activated to quite high levels (comparable to those seen in K562 cells) in the absence of erythroid TFs, again suggesting that the promoters driving intergenic transcription are not inherently erythroid specific. By increasing the global level of histone acetylation using a histone deacetylase inhibitor (TSA), we were again able to induce intergenic transcription, which suggests that in nonerythroid cells the block to transcription is at the level of chromatin structure. A number of erythroid-specific TFs (NF-E2, GATA-1, and FOG-1) can also independently activate intergenic transcription. There are potential binding sites for GATA-1 within the U3 repeats of the ERV9 LTR (18) which may contribute to this activation. However, it is also possible that exogenously supplied TFs act through the LCR to open the chromatin which normally represses intergenic transcription in nonerythroid cells. We have not been able to activate transcription of the globin genes themselves, despite using a combination of the above induction methods. This may be because we have not accounted for all of the erythroid factors necessary for authentic globin gene expression, although it is also possible that the globin promoters are repressed in nonerythroid cells.
The finding that transcription of the LCR in part starts at the ERV9 LTR promoter is interesting because this region is not present in many transgenic mice which express the β-globin genes normally. It is also absent from the endogenous mouse locus, the LCR of which is transcribed as shown by FISH analysis (3). It is therefore apparent that the ERV9 LTR is not required per se for LCR transcription. We have looked for transcription of the LCR in the transgenic mouse line 72, which has previously been described (13, 26) and which lacks the ERV9 LTR. Transcription of the human LCR was detected up to the most 5′ probe present in the transgene (L17). We suggest that in this mouse the human LCR activates nearby endogenous mouse promoter(s) to serve as a transcription start site. However, it should be noted that our results do not rule out the possibility of additional transcription initiation events within the LCR, for example from HS2 (4, 31) and HS3 (17), as has been reported previously. It may be that initiation as far upstream as we observe is not essential for transcription of the LCR and that in some transgenic mice initiation at sites within the LCR provides the sole means by which it is transcribed. However, it is also possible that the absence of these upstream transcripts accounts for some of the minor position effects still seen in LCR-containing mice (2).
Our model of the ways in which transcription across the human β-globin locus can be activated is shown in Fig. 5. In nonerythroid cells (Fig. 5a) the globin chromatin domain is normally closed so that no transcription occurs. In erythroid cells nearby promoters, which are not inherently erythroid specific, are activated, possibly through enhancer-type interactions with the LCR or else due to the partial opening of the β-globin chromatin domain by the binding of erythroid TFs at the HSs (Fig. 5b). In the case of the endogenous human β-globin gene cluster the most upstream promoter is in the ERV9 LTR, but in transgenic mice other promoters may be recruited. The globin genes are also activated by the combination of erythroid TFs binding to sites within their promoters as well as enhancer interactions with the LCR and the overall opening of the chromatin structure. Intergenic transcription can be induced in nonerythroid cells by at least three means: during transinduction by an unknown mechanism involving colocalization of the transfected plasmid and endogenous locus; by inhibition of histone deacetylases, resulting in a global opening of chromatin allowing access to these intergenic promoters (Fig. 5c); and by exogenously supplied erythroid TFs again either through enhancer-type interactions with the LCR or because of improved access due to the opening of the β-globin chromatin domain (Fig. 5d). In each case the globin genes themselves are not transcribed, as the full complement of erythroid TFs are not present.
FIG. 5.
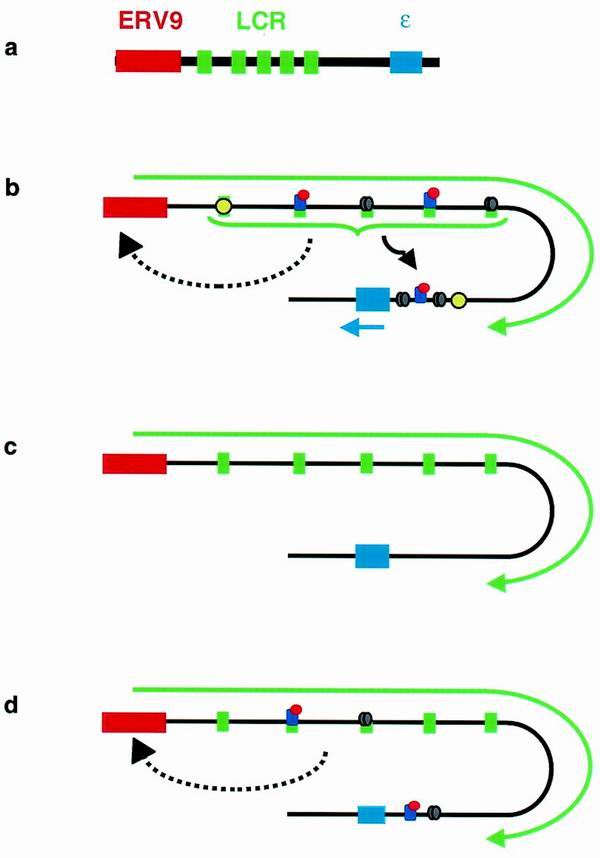
Transcription and chromatin structure in the globin locus in nonerythroid cells (a), in erythroid cells (b), following inhibition of HDACs with TSA (c), and following exogenous addition of erythroid TFs (d). The thickness of the solid black line represents the chromatin status: the thicker line indicates closed chromatin, while thinner lines indicate a more open structure. Green and blue arrows show intergenic and genic transcripts. Erythroid TFs (variously colored ovals) are shown bound to the 5 HSs of the LCR (green boxes) and to the promoter of the ɛ gene (blue box). The solid and dashed black arrows indicate likely and possible, respectively, enhancer-type activations by the LCR.
While it has long been appreciated that the degree of chromatin condensation affects the ability of a region of DNA to be transcribed, it has recently become apparent that the opposite is also true and that transcription can affect the nature of chromatin. Thus, it has been demonstrated that the Saccharomyces cerevisiae elongator complex, which is integral to the transcribing polymerase II complex, has histone acetylase activity which may disrupt the chromatin structure facilitating rounds of transcription (34). Chromatin-remodeling factors have also been isolated as part of the polymerase II complex in both mammalian and yeast cells (7, 33). Such data suggest that intergenic transcription could maintain or extend the DNase I sensitivity initiated by the binding of TFs. Indeed, a recent report (17) shows that when erythroid TFs bind to the β-globin HS2 and HS3 hypersensitivity is established over the core of the HSs in the absence of chromatin. Transcripts initiating within the core of both hypersensitive regions were also generated. A role for intergenic transcription in propagating DNase sensitivity in the β-globin locus is clearly an attractive conclusion.
Support for this idea and for the importance of transcripts initiating upstream of HS5 comes from recent work by Reik et al. (22). A targeted deletion of HS2 to HS5 (Δ2-5) in the human β-globin LCR was generated so that the role of the LCR in globin gene expression could be assessed in its natural chromosomal location. Although globin gene expression was disrupted by this deletion, the cluster retained its erythroid-specific DNase I sensitivity. In contrast, the Hispanic thalassemia deletion causes the locus to become DNase I resistant in addition to affecting globin expression (11). Significantly, the Hispanic deletion includes the ERV9 LTR, whereas the 5′ limit of the targeted deletion lies between HS5 and ERV9. We suggest that the DNase I sensitivity of the globin cluster is retained in the Δ2-5 mutation because transcription initiating within the ERV9 LTR can be activated by elements outside the deleted region; however, it has not yet been shown whether what remains of the LCR is transcribed following either of these deletions.
In a recent study of the human β-globin cluster in transgenic mice, Gribnau et al. (12) suggest that the cluster can be divided into three differentially active subdomains: the embryonic and fetal εγ subdomain, the fetal and adult δβ subdomain, and an LCR subdomain active at all developmental stages. The production of intergenic transcripts in each case coincided with enhanced DNase I sensitivity and in the case of the ɛγ and δβ subdomains coincided with the activities of the genes themselves. Although the causal link between transcription and open chromatin suggested by these observations is attractive, our data are somewhat contradictory. We see equivalent levels of intergenic transcription in all three subdomains in adult (line 72 spleen) and embryonic (K562) cells and also during any of the induction methods that we have described. The discrepancy between our data and those of Gribnau et al. is likely to reflect the different sensitivities of the two techniques used, with NRO using several different probes providing a more quantitative measure of nascent transcription than does FISH.
Ultimate proof of a role for intergenic and LCR transcription in maintaining an open chromatin structure and through this mechanism affecting β-globin gene expression must come from disrupting these transcripts in a whole-locus setting (either in transgenic mice or by targeted disruption of the endogenous locus in cell lines). While those experiments are currently in progress, the data presented here and recent work by others lead us to predict that nongenic transcription will have an important role in these processes. Furthermore, the demonstration that the intergenic regions of other gene clusters (e.g., IL-4/IL-13 [23]) are transcribed leads us to anticipate that intergenic transcription will be a feature of at least some mammalian gene loci.
ACKNOWLEDGMENTS
This work was supported by Wellcome Project grant 051855 and a Wellcome Prize Studentship (052044) to S.J.E.R.
We gratefully acknowledge Joan Monks for technical assistance. Stuart Orkin and Shona Murphy generously provided TF clones. Thanks to Frances Mortimer for work on the Aγ region, to Simon Brackenridge, Mick Dye, and Allie Binnie for helpful discussions, and to Bill Wood for help with the transgenic mice.
K.E.P. and S.J.E.R. contributed equally to this work.
REFERENCES
- 1.Adams S E, Johnson I D, Braddock M, Kingsman A J, Kingsman S M, Edwards R M. Synthesis of a gene for the HIV transactivator protein Tat by a novel single stranded approach involving invivo gap repair. Nucleic Acids Res. 1988;16:4287–4298. doi: 10.1093/nar/16.10.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alami R, Greally J M, Tanimoto K, Hwang S, Feng Y Q, Engel J D, Fiering S, Bouhassira E E. Beta-globin YAC transgenes exhibit uniform expression levels but position effect variegation in mice. Hum Mol Genet. 2000;9:631–636. doi: 10.1093/hmg/9.4.631. [DOI] [PubMed] [Google Scholar]
- 3.Ashe H L, Monks J, Wijgerde M, Fraser P, Proudfoot N J. Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev. 1997;11:2494–2509. doi: 10.1101/gad.11.19.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohl D, Kong S, Li C, Wickrema A, Krantz S, Tuan D. The locus control region far upstream of the human P-like globin genes is transcribed in erythroid cells in a direction toward the globin genes. Blood. 1995;86:2501. [Google Scholar]
- 5.Bulger M, vonDoorninck J H, Saitoh N, Telling A, Farrell C, Bender M A, Felsenfeld G, Axel R, Groudine M. Conservation of sequence and structure flanking the mouse and human beta-globin loci: the beta-globin genes are embedded within an array of odorant receptor genes. Proc Natl Acad Sci USA. 1999;96:5129–5134. doi: 10.1073/pnas.96.9.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W Y, Townes T M. Molecular mechanism for silencing virally transduced genes involves histone deacetylation and chromatin condensation. Proc Natl Acad Sci USA. 1999;97:377–382. doi: 10.1073/pnas.97.1.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Orphanides G, Sun X, Yang X J, Ogryzko V, Lees E, Nakatani Y, Reinberg D. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol Cell Biol. 1998;18:5355–5363. doi: 10.1128/mcb.18.9.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dye M J, Proudfoot N J. Multiple transcript cleavage precedes polymerase release in termination by RNA polymerase II. Cell. 2001;105:669–681. doi: 10.1016/s0092-8674(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 9.Eggermont J, Proudfoot N J. Poly(A) signals and transcriptional pause sites combine to prevent interference between RNA polymerase-II promoters. EMBO J. 1993;12:2539–2548. doi: 10.1002/j.1460-2075.1993.tb05909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epner E, Reik A, Cimbora D, Telling A, Bender M A, Fiering S, Enver T, Martin D I K, Kennedy M, Keller G, Groudine M. The beta-globin LCR is not necessary for an open chromatin structure or developmentally regulated transcription of the native mouse beta-globin locus. Mol Cell. 1998;2:447–455. doi: 10.1016/s1097-2765(00)80144-6. [DOI] [PubMed] [Google Scholar]
- 11.Forrester W C, Epner E, Driscoll M C, Enver T, Brice M, Papayannopoulou T, Groudine M. A deletion of the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev. 1990;4:1637–1649. doi: 10.1101/gad.4.10.1637. [DOI] [PubMed] [Google Scholar]
- 12.Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta globin locus. Mol Cell. 2000;5:377–386. doi: 10.1016/s1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- 13.Grosveld F, Vanassendelft G B, Greaves D R, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 14.Kotkow K J, Orkin S H. Dependence of globin gene-expression in mouse erythroleukemia-cells on the NF-E2 heterodimer. Mol Cell Biol. 1995;15:4640–4647. doi: 10.1128/mcb.15.8.4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Mantia G, Maglione D, Pengue G, Dicristofano A, Simeone A, Lanfrancone L, Lania L. Identification and characterization of novel human endogenous retroviral sequences preferentially expressed in undifferentiated embryonal carcinoma-cells. Nucleic Acids Res. 1991;19:1513–1520. doi: 10.1093/nar/19.7.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.La Mantia G, Majello B, DiCristofano A, Strazzullo M, Minchiotti G, Lania L. Identification of regulatory elements within the minimal promoter region of the human endogenous ERV9 proviruses: accurate transcription is controlled by an Inr-like element. Nucleic Acids Res. 1992;20:4129–4136. doi: 10.1093/nar/20.16.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leach K M, Nightingale K, Igrashi K, Levings P P, Engel J D, Becker P B, Bungert J. Reconstitution of human beta-globin locus control region hypersensitive sites in the absence of chromatin assembly. Mol Cell Biol. 2001;21:2629–2640. doi: 10.1128/MCB.21.8.2629-2640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long Q M, Bengra C, Li C H, Kutlar F, Tuan D. A long terminal repeat of the human endogenous retrovirus ERV-9 is located in the 5′ boundary area of the human beta-globin locus control region. Genomics. 1998;54:542–555. doi: 10.1006/geno.1998.5608. [DOI] [PubMed] [Google Scholar]
- 19.Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L, Chenchik A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–1560. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy S. Differential in vivo activation of the class II and class III snRNA genes by the POU-specific domain of Oct-1. Nucleic Acids Res. 1997;25:2068–2076. doi: 10.1093/nar/25.11.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orkin S H. Transcription factors and hematopoietic development. J Biol Chem. 1995;270:4955–4958. doi: 10.1074/jbc.270.10.4955. [DOI] [PubMed] [Google Scholar]
- 22.Reik A, Telling A, Zitnik G, Cimbora D, Epner E, Groudine M. The locus control region is necessary for gene expression in the human beta-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol Cell Biol. 1998;18:5992–6000. doi: 10.1128/mcb.18.10.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogan D F, Cousins D J, Staynov D Z. Intergenic transcription occurs throughout the human IL-4/IL-13 gene cluster. Biochem Biophys Res Commun. 1999;255:556–561. doi: 10.1006/bbrc.1999.0241. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Press; 1989. [Google Scholar]
- 25.Strahl B D, Allis C D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 26.Strouboulis J, Dillon N, Grosveld F. Developmental regulation of a complete 70-Kb human beta-globin locus in transgenic mice. Genes Dev. 1992;6:1857–1864. doi: 10.1101/gad.6.10.1857. [DOI] [PubMed] [Google Scholar]
- 27.Talbot D, Grosveld F. The 5′HS2 of the globin locus-control region enhances transcription through the interaction of a multimeric complex binding at 2 functionally distinct NF-E2 binding-sites. EMBO J. 1991;10:1391–1398. doi: 10.1002/j.1460-2075.1991.tb07659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanimoto K, Liu Q H, Bungert J, Engel J D. Effects of altered gene order or orientation of the locus control region on human beta-globin gene expression in mice. Nature. 1999;398:344–348. doi: 10.1038/18698. [DOI] [PubMed] [Google Scholar]
- 29.Tsai S F, Martin D I K, Zon L I, Dandrea A D, Wong G G, Orkin S H. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian-cells. Nature. 1989;339:446–451. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- 30.Tsang A P, Visvader J E, Turner C A, Fujiwara Y, Yu C N, Weiss M J, Crossley M, Orkin S H. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109–119. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 31.Tuan D, Kong S M, Hu K. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci USA. 1992;89:11219–11223. doi: 10.1073/pnas.89.23.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams J G, Mason P J. Hybridisation in the analysis of RNA. In: Hames B D, Higgins S J, editors. Nucleic acid hybridisation: a practical approach. New York, N.Y: Academic Press, Inc.; 1985. pp. 139–160. [Google Scholar]
- 33.Wilson C J, Chao D M, Imbalzano A N, Schnitzler G R, Kingston R E, Young R A. RNA polymerase II holoenzyme contains SWI/SNF regulators involved in chromatin remodeling. Cell. 1996;84:235–244. doi: 10.1016/s0092-8674(00)80978-2. [DOI] [PubMed] [Google Scholar]
- 34.Wittschieben B O, Otero G, deBizemont T, Fellows J, ErdjumentBromage H, Ohba R, Li Y, Allis C D, Tempst P, Svejstrup J Q. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell. 1999;4:123–128. doi: 10.1016/s1097-2765(00)80194-x. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]