

SUMMARY
Cachexia is a systemic wasting syndrome that increases cancer-associated mortality. How cachexia progressively and differentially impacts distinct tissues is largely unknown. Here, we find that the heart and skeletal muscle undergo wasting at early stages and are the tissues transcriptionally most impacted by cachexia. We also identify general and organ-specific transcriptional changes that indicate functional derangement by cachexia even in tissues that do not undergo wasting, such as the brain. Secreted factors constitute a top category of cancer-regulated genes in host tissues, and these changes include upregulation of the angiotensin-converting enzyme (ACE). ACE inhibition with the drug lisinopril improves muscle force and partially impedes cachexia-induced transcriptional changes, although wasting is not prevented, suggesting that cancer-induced host-secreted factors can regulate tissue function during cachexia. Altogether, by defining prevalent and temporal and tissue-specific responses to cachexia, this resource highlights biomarkers and possible targets for general and tissue-tailored anti-cachexia therapies.
In brief
Graca et al. examine the transcriptional and cellular changes that occur during the progression of cancer cachexia in mice and identify general and organ-specific transcriptional changes that underlie the differential propensity and temporal dynamics of cancer-induced tissue wasting and dysfunction. This resource highlights biomarkers and possible targets for anti-cachexia therapies.
Graphical Abstract
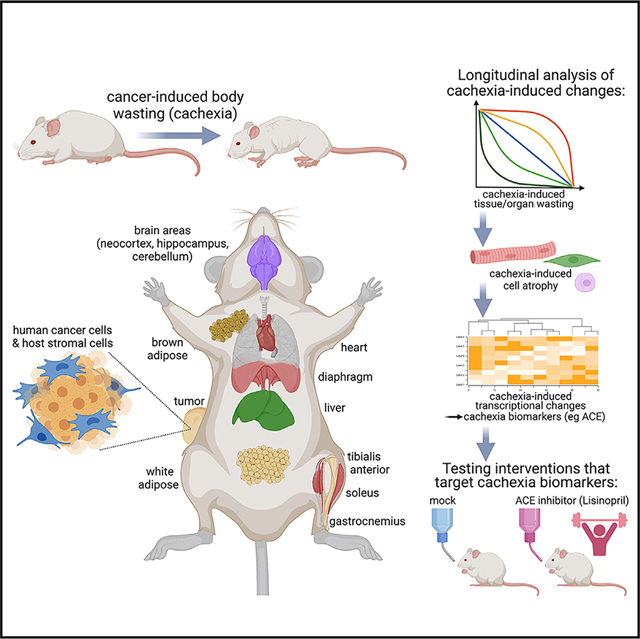
INTRODUCTION
In cancer patients, an unintentional loss of >5% body weight is an important disease complication known as cachexia,1–5 a multifactorial condition that negatively affects survival.6–9 Consistently, several studies have demonstrated that cachexia worsens disease outcome. For example, preventing muscle mass loss in tumor-bearing mice improves prognosis and prolongs their survival even if cancer progression per se is not halted.6–9 Moreover, cachexia frequently occurs in conjunction with metastasis,8,10 and thus it is a component of malignant and typically less treatable cancers. Although anti-cachexia treatments have the potential to improve the prognosis of some of the most aggressive tumors, no therapy is currently available. Overall, the National Cancer Institute (NCI) estimates that cancer cachexia occurs in ~50% of all tumors, and that it is responsible for one-third of all cancer deaths.11
Cachexia arises from behavioral and systemic changes induced by circulating cytokines secreted by cancer cells and associated stromal cells,1,2 as well as by immune cells, including those that infiltrate the tumor and host tissues.12,13 The local and systemic impact of such inflammatory signaling can be modified by host-dependent variables such as the responsiveness of target tissues to inflammatory signaling and the concurrent activation of compensatory mechanisms to mitigate cachexia-induced changes.1,2,14
Although it is well known that cachexia induces wasting and functional changes in skeletal muscle, heart, and white adipose tissue (WAT),4,5,15,16 relatively less is known about the impact of cachexia on other tissues and organs. It is also largely uncharted how cachexia develops over time and whether the temporal dynamics and magnitude of cachexia-induced changes differ across target organs. Moreover, it is likely that many more tissues than currently appreciated are affected by cachexia. Specifically, although body and tissue wasting is a defining and well-documented result of cachexia,4,5,15,16 it remains relatively unexplored whether cachexia impacts the function of target tissues, including those that are typically considered not to be affected by cachexia because they do not undergo wasting. For example, despite the finding that cachexia affects brain functions such as feeding behavior,17,18 relatively little is known about the molecular adaptations induced by cachexia in the brain.19 Similarly, beyond molecular changes that have been mechanistically linked to wasting of target tissues such as the skeletal muscle and WAT,4,5,15,16 little is known about cachexia-induced changes that impact tissue function independently from wasting.
Recently, we have identified a pediatric melanoma xenograft from the Childhood Solid Tumor Network20,21 that induces profound body wasting (~25%) compared with controls.22 Here, we have utilized this model to examine the systemic impact of cachexia on diverse tissues and organs. By following the progressive development of cachexia, we find that distinct magnitudes and temporal patterns of tissue wasting and transcriptional changes define the response to cachectic signaling of the heart, liver, brown and white adipose, skeletal muscles (soleus, tibialis anterior [TA], and gastrocnemius), and brain regions (cerebellum, hippocampus, neocortex). Interestingly, secreted factors are highly modulated by cachexia across tissues. These include the angiotensin-converting enzyme (ACE): ACE expression increases during cachexia, and this occurs most prominently in skeletal muscles. ACE inhibition with the anti-hypertension drug lisinopril does not impede wasting but preserves muscle strength, and this occurs by impeding cachexia-induced transcriptional changes. Altogether, this study provides a resource that outlines how cachexia differentially impacts host tissues, which may help identify leads for anti-cachexia therapies.
RESULTS
Comparison of pediatric melanoma xenografts with distinct capacity to induce cachexia reveals defining transcriptional changes in cancer cells and the associated microenvironment
To examine the mechanistic basis underlying the body wasting capacity of cachectic versus non-cachectic melanoma xenografts, we injected ~106 pediatric melanoma xenograft cells subcutaneously in the flank of NCI athymic NCr-nu/nu female mice and allowed them to grow for 2, 4, 6, and 8 weeks. Tumors were retrieved at these time points and examined via RNA sequencing (RNA-seq) to determine (1) the differences in gene expression that characterize the cachectic versus the non-cachectic melanoma, and (2) whether tumor growth is associated with major gene expression changes over time that could contribute to the progressive development of body wasting. Although the same number of cells was injected for the cachexia-inducing (“cachectic”) melanoma (MAST360B/SJMEL030083_X2) and for the cachexia non-inducing (“non-cachectic”) melanoma (MAST552A/SJMEL031086_X3) at day 0, there was a significantly lower growth of the non-cachectic melanomas, which could therefore be analyzed only at 8 weeks after cancer cell injection.
Principal-component analysis (PCA) of RNA-seq from the cachectic tumors at 2, 4, 6, and 8 weeks of age indicates that there are limited changes in gene expression over time in cancer cells. However, the non-cachectic melanoma radically differs from the cachectic melanoma (Figure 1A), as indicated by a cluster of 7,071 genes (Figure 1B) and by upregulated (Figure 1C) and downregulated (Figure 1D) gene categories.
Figure 1. The secretome of cachectic melanomas includes signaling factors with known roles in body wasting.
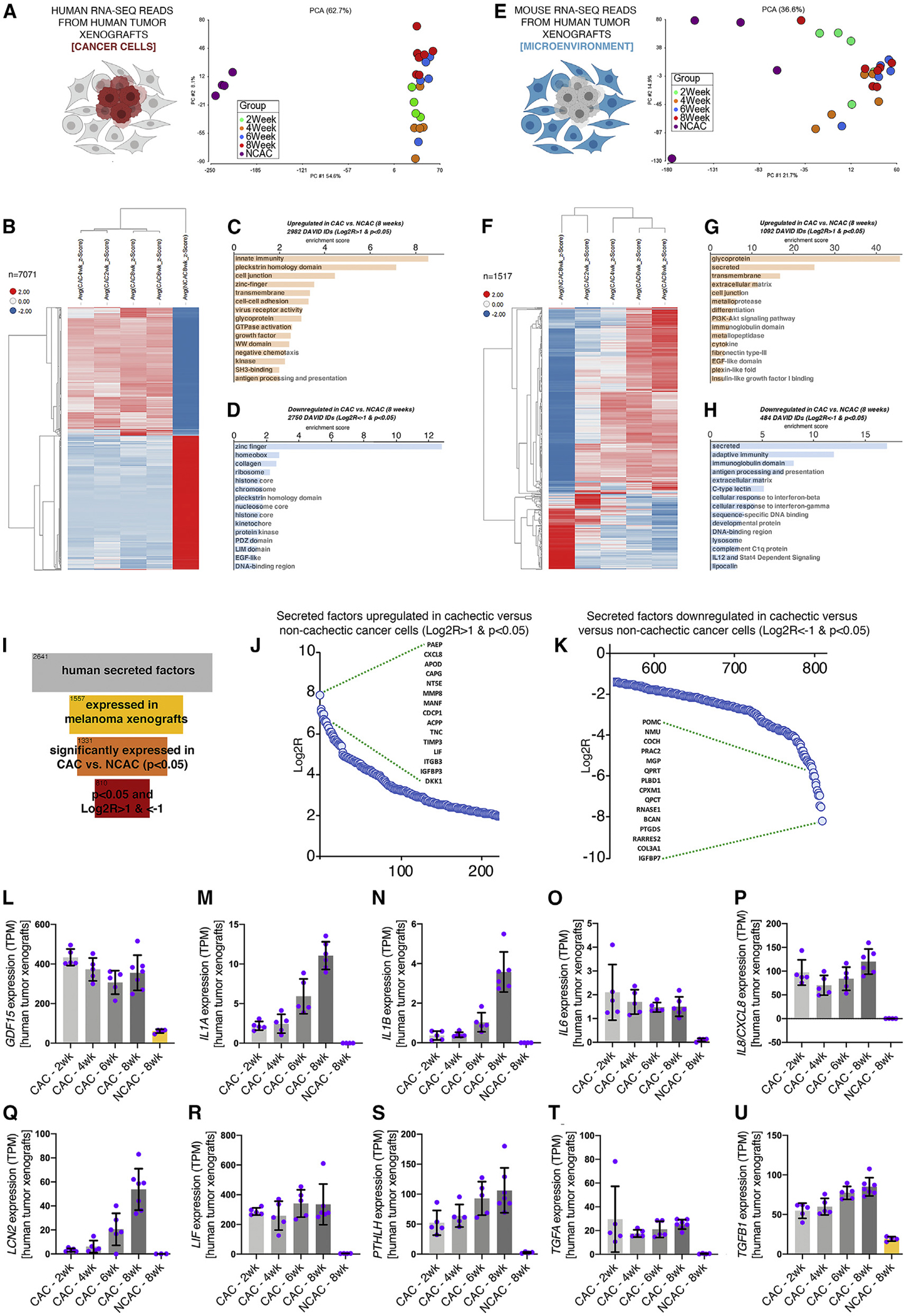
(A) PCA of human RNA-seq reads from tumor xenografts reveals substantial differences between cachectic versus non-cachectic melanomas. There are limited changes that occur in cachectic melanomas over time.
(B) Heatmap of 7,071 genes that are highly modulated in cachectic versus non-cachectic melanomas.
(C and D) Genes modulated with p < 0.05 and Log2R > 1 (C) and Log2R < −1 (D) in cachectic versus non-cachectic melanomas at 8 weeks after tumor implantation are shown.
(E and F) PCA of mouse RNA-seq reads from melanoma xenografts, indicative of host mouse cells of the stroma (microenvironment) associated with human cancer cells (E). There are progressive changes in the stroma from 2 to 8 weeks after melanoma injection, including a core set of 1,517 genes (F).
(G and H) Several categories are represented among upregulated (G) and downregulated (H) genes.
(I) Out of 2,641 secreted proteins, 1,557 are expressed by melanoma cells, 1,331 are significantly modulated (p < 0.05) in cachectic versus non-cachectic melanomas, and 810 are differentially expressed with p < 0.05 and Log2R > 1 or Log2R < −1.
(J and K) Secreted factors that are most highly upregulated (J) and downregulated (K) in cachectic versus non-cachectic melanomas. Log2R values are shown on the y axis (p < 0.05).
(L–U) Several signaling factors known to drive body wasting are significantly upregulated in cachectic versus non-cachectic melanomas, including GDF15 (L), IL1A (M), IL1B (N), IL6 (O), IL8/CXCL8 (P), LCN2 (Q), LIF (R), PTHLH (S), TGFA (T), and TGFB1 (U).
Bars represent SD; n is indicated. All comparisons are significant (p < 0.05) in cachectic versus control at 8 weeks after tumor cell injection. TPM, transcripts per million reads. See also Figure S1.
In addition to human cancer cells, the xenografts retrieved from mice include mouse host cells of the tumor microenvironment, which contribute to cancer progression.23 Because human transcripts can be easily differentiated from mouse RNA-seq reads, we examined gene expression changes in these cancer-associated stromal cells. PCA revealed that the microenvironment of cachectic melanomas differs from that of non-cachectic melanomas (Figure 1E), and that a cluster of 1,517 genes most stringently defines the transcriptional changes that occur in the microenvironment of cachectic versus non-cachectic melanomas (Figure 1F), and that several gene categories are upregulated (Figure 1G) and downregulated (Figure 1H), including secreted and extracellular matrix proteins, which suggests that secreted factors produced by host stromal cells contribute to cachexia.
Secreted factors are differentially expressed in cachectic versus non-cachectic tumor cells and the host stroma
Cachexia is induced by inflammatory cytokines secreted by cancer cells, as well as by stromal and immune cells.4,5,24 Out of the 2,641 secreted factors encoded by the human genome,25 1,557 were detected by RNA-seq in the melanoma xenografts: 1,331 were significantly (p < 0.05) regulated in cachectic versus non-cachectic melanoma cells, and 810 of these had at least a 2-fold regulation (Log2R > 1 or Log2R < −1), indicating that cachectic cells radically differ from non-cachectic cancer cells for the expression of secreted factors (Figure 1I).
Closer analysis of the most highly upregulated (Figure 1J) and downregulated (Figure 1K) secreted factors in cachectic versus non-cachectic melanomas (8 weeks post-injection) indicates that some of them had been previously implicated in cancer-induced body wasting. Upregulated factors include CXCL8/IL826–28 and LIF29–31 (Figure 1J), as well as the lipocalin PAEP (Figure 1J), which may contribute to cancer-induced anorexia, as inferred from the function of lipocalin-2 (LCN2),17 and to melanoma growth and immune evasion,32,33 similar to NT5E34 (Figure 1J). Conversely, downregulated factors included extracellular matrix proteins (collagen [COL3A1] and brevican [BCAN]) and the insulin-like growth factor binding protein 7 (IGFBP7), which impairs insulin receptor signaling35 (Figure 1K).
We next examined the expression timeline of a few cytokines upregulated in cachectic versus non-cachectic melanomas and previously implicated in cachexia4,5 (Figures 1L–1U). IL1A, IL1B, LCN2, PTHLH, and TGFB1 expression increases progressively from 2 to 8 weeks after cancer cell injection (Figures 1M, 1N, 1Q, 1S, and 1U). However, the expression of other wasting factors (GDF15, IL6, IL8/CXCL8, LIF, and TGFA) does not substantially change over time (Figures 1L, 1O, 1P, 1R, and 1T). Altogether, these analyses highlight the different temporal modulation of cachectic factors produced by cancer cells during melanoma progression.
In addition to cancer cells, tumor-associated host stromal cells also produce cachectic cytokines.27,36 Secreted factors upregulated by stromal cells of cachectic versus non-cachectic melanomas include matrix metalloproteases (Mmp3, Mmp9, Mmp10, Mmp13, and Mmp16) and chemokines (Cxcl2 and Cxcl5) (Figure S1A). Other chemokines (Ccl5, Ccl17, Cxcl9, and Xcl1), collagens (Col8a1, Col8a2, and Col10a1), and extracellular matrix proteins (Nov/Ccn3, Fmod, and Ogn) are downregulated (Figure S1B). Cross-comparison of secreted factors expressed by cancer and stromal cells reveals minimal overlap (R2 = 0.1384; Figure S1C), indicating that cancer cells and the microenvironment contribute different sets of secreted factors, some of which may be causally involved in systemic wasting.
Wasting induced by cachectic melanoma xenografts distinctly impacts different organ systems
Despite the well-known impact of cancer cachexia,4,5,18 whether and how tumor-initiated inflammatory signaling differentially affects tissue and organ wasting remains incompletely understood. To address this question, we have examined the weight of tissues and organs at 2, 4, 6, and 8 weeks after subcutaneous (orthotopic) injection of cachexia non-inducing (“non-cachectic”) melanomas, cachexia-inducing (“cachectic”) melanomas, and mock (PBS) into 2-month-old NCI athymic NCr-nu/nu female mice (Figure 2A).
Figure 2. Melanoma-induced cachexia differentially impacts tissue and organ wasting in mice.
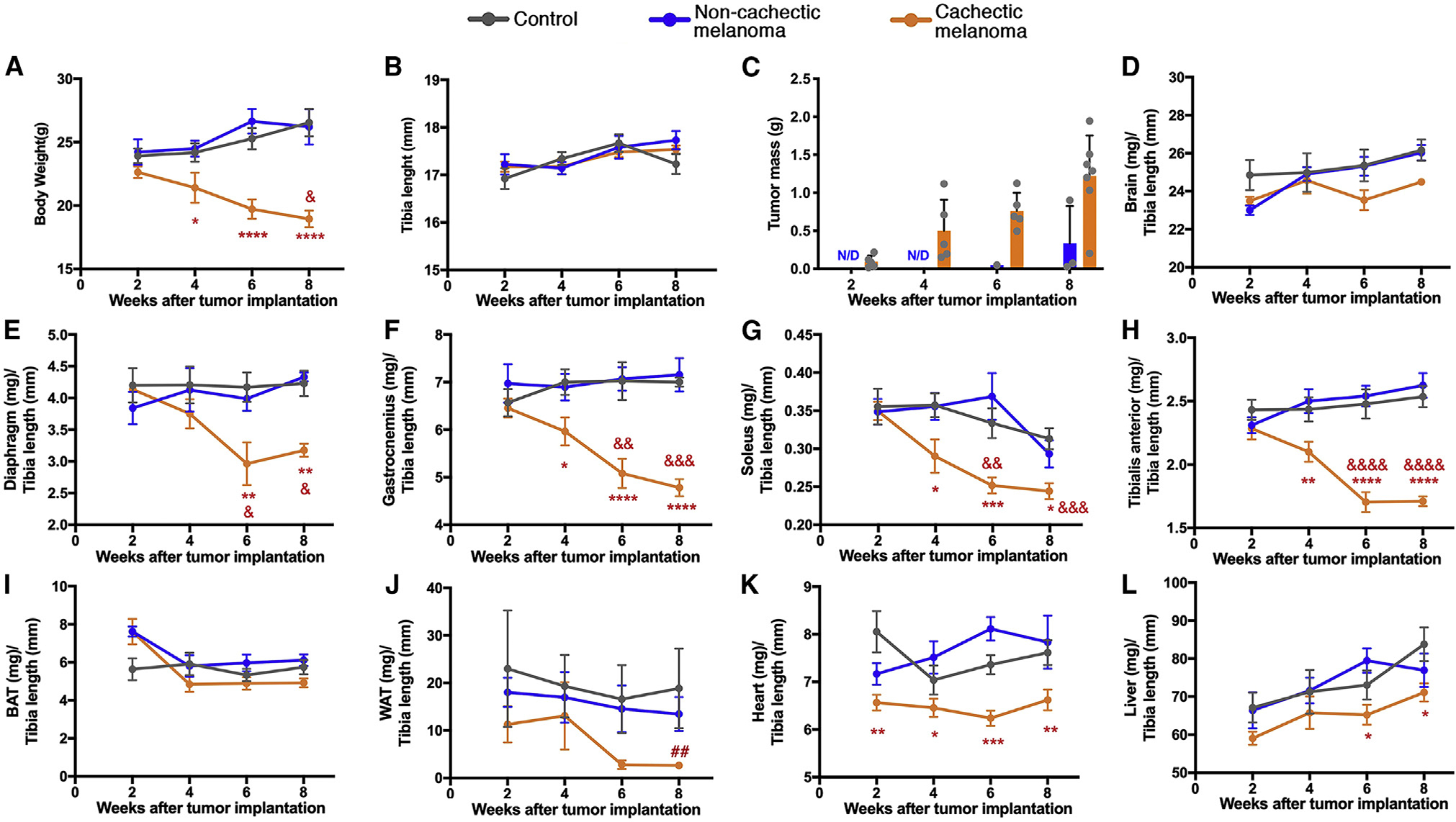
Orthotopic melanoma xenografts were established via the subcutaneous injection of cachectic cancer cells and compared with mock (PBS) and non-cachectic melanomas. Organs and tissues were harvested at 2, 4, 6, and 8 weeks after implantation and processed for histology and RNA-seq.
(A) Cachectic melanomas progressively induce body wasting, which is significant from week 4. No decline in body weight is seen in mice injected with non-cachectic melanomas.
(B) The length of the tibia bone does not change between groups, and it is used for normalization.
(C) Cachectic melanomas grow more than non-cachectic xenografts.
(D) There is no change in brain weight.
(E–H) Cachexia leads to profound wasting of the diaphragm (E), gastrocnemius (F), soleus (G), and tibialis anterior (TA) (H) skeletal muscles.
(I and J) The weight of the interscapular brown adipose tissue (BAT) marginally declines with cachexia (I), whereas the gonadal white adipose tissue (WAT) undergoes wasting (J).
(K) Heart mass declines already at 2 weeks after cancer cell injection.
(L) Whereas liver weight increases in controls, such post-natal growth is stunted by cachexia.
SEM with *p < 0.05, **p < 0.01, and ***p < 0.001 compared with controls. Ampersands (&) indicate the statistical significance within each group compared with the 2-week time point. The hashtag symbol (#) indicates significance when comparing cachectic melanomas versus controls within the same time point: ##p < 0.01. ND, not detected. See also Figure S2.
As expected based on our previous study with these models,22 progressive body weight loss occurs in mice injected with cachectic melanomas (Figure 2A). This wasting occurs without overall changes in the length of the tibia bone (Figure 2B). The cachectic melanoma xenografts displayed a higher tumor mass compared with the non-cachectic melanoma xenografts at 8 weeks of age (Figure 2C), indicating that cachectic melanoma cells have a growth advantage. Although theoretically such higher tumor mass could explain why cachectic melanoma xenografts induce body weight loss compared with slow-growing non-cachectic melanoma cells, this does not appear to be the case here because body weight loss did not occur in mice injected with non-cachectic melanoma cells even after 13 weeks from tumor implantation, a time point at which the mass of non-cachectic melanomas surpassed that of cachectic melanomas (~4 g versus ~1.5 g; Figures S2 and 2C) at 8 weeks (the humane endpoint for mice implanted with cachectic melanomas). Therefore, the capacity to induce body wasting does not merely correlate with the tumor burden but rather likely depends on pro-wasting factors secreted by cachectic versus non-cachectic melanomas (Figures 1 and S1), although other variables (such as the rate of tumor growth) may also contribute.
Although cachexia arises from systemic adaptations that lead to body weight loss,4,5,18 it remains largely unknown how different organs progressively respond to cachexia and distinctly contribute to body wasting. To address this question, we have examined the weight of a panel of tissues and organs over the course of cachexia development. Because the length of the tibia bone does not significantly change over the 8-week time course of tumor growth (Figure 2B), it was used as a normalization factor.
There was no significant difference in brain weight across mice injected with cachectic and non-cachectic cancer cells and controls (Figure 2D). However, there was a significant decline in the weight of several skeletal muscles that differ for function and myofiber-type composition, i.e., the diaphragm (Figure 2E), the gastrocnemius (Figure 2F), the soleus (Figure 2G), and the TA (Figure 2H). There was no major effect of cachectic melanomas on the mass of the interscapular brown adipose tissue (BAT; Figure 2I), whereas the mass of the gonadal WAT declined (Figure 2J), although with high variability.
The weight of the heart was reduced at all time points in response to cachectic melanomas compared with controls (Figure 2K). Although decline in heart weight appears to be an early event in cachexia (i.e., 2 weeks after cancer cell injection), it did not substantially further decline over time, possibly because of compensatory heart hypertrophy37,38 that could be induced to counteract cachexia-associated anemia.39,40
Lastly, although liver weight increased in all groups over time (as a result of post-natal development41), it was reduced in mice injected with cachectic melanomas at 6–8 weeks post-injection, compared with time-matched controls (Figure 2L). Altogether, distinct temporal dynamics characterize the impact of cachexia on organs and tissues.
Differential impact of cachexia on cell atrophy across tissues
To investigate whether cancer-induced tissue wasting arises from a reduction in cell size (atrophy), we utilized hematoxylin and eosin (H&E) staining and image analysis to examine cell size in the liver, BAT and WAT, heart, and skeletal muscles (gastrocnemius, soleus, and TA). These analyses identified varying degrees of cancer-induced cell atrophy (Figure 3).
Figure 3. Different degrees of cancer-induced cell atrophy in distinct tissues.
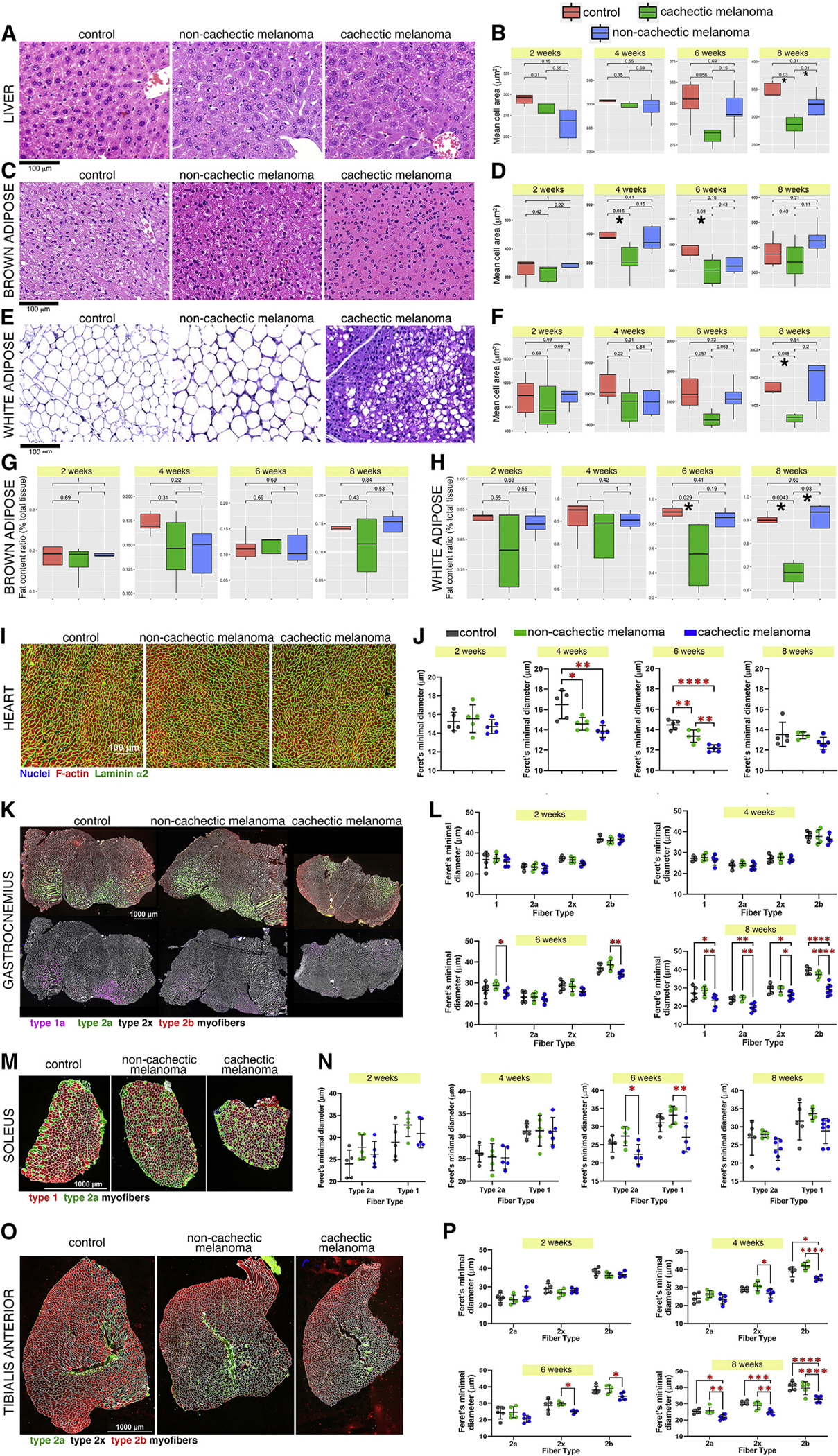
(A) H&E staining of liver sections after 8 weeks from melanoma injection. Scale bar, 100 μm.
(B) Cachexia reduces hepatocyte size at 6 and 8 weeks. Control values are shown in red, whereas cachectic and non-cachectic values are shown in green and blue, respectively. *p < 0.05.
(C–F) Cachexia leads to cell atrophy at 4 and 6 weeks (brown adipocytes; C and D) and at 8 weeks (white adipocytes; E and F).
(G and H) There is also a trend toward decreased fat content of brown adipocytes (G), and such delipidation is significant for white adipocytes (H).
(I) Analysis of cardiomyocyte size at 8 weeks after melanoma injection; nuclei are shown in blue, F-actin in red, and laminin α2 in green. Scale bar, 100 μm.
(J) For each time point, the Feret’s minimal diameter provides an estimate of the average cell size. There are no substantial changes at 2 weeks, whereas cardiomyocyte size is significantly reduced at 4–6 weeks.
(K) Myofiber size in the gastrocnemius at 8 weeks after tumor implantation. Scale bar, 1000 μm.
(L) Atrophy of type 1 and 2b myofibers occurs significantly at 6 weeks, whereas general decline in myofiber size is found at week 8.
(M and N) The soleus muscle is relatively resistant to wasting, and a significant or trending decline in type 1 and 2a myofiber size is found only at 6–8 weeks.
(O and P) On the contrary, atrophy of type 2x and 2b myofibers occurs at 4 and 6 weeks and is further exacerbated by 8 weeks in the tibialis anterior muscle, which is prone to wasting.
In (I)–(P), SD is shown with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
In agreement with other models that have found cachexia-induced liver dysfunction,18,24 hepatocyte cell size was significantly reduced 8 weeks after implantation of cachectic melanomas, compared with controls (Figures 3A and 3B). It was previously found that cancer increases BAT function,42 and that it induces the loss of WAT mass.2,4,5,16,18,24 We find that cachectic melanomas have an overall minor and variable impact on brown adipocyte cell size (Figures 3C and 3D), whereas that of white adipocytes is significantly reduced at 8 weeks (Figures 3E and 3F). Moreover, fat stores of white adipocytes are lower in mice that carry cachectic melanomas versus controls (Figures 3G and 3H).
In addition to liver and adipose tissue changes, patients with cancer cachexia display signs of chronic heart failure, including fatigue and decreased exercise capacity, and this is considered a major cause of cachexia-induced mortality.30,43–48 Although we found that cardiac cell size (as indicated by the Feret’s minimal diameter) is not affected at the 2- and 8-week time points, it significantly declines at 4–6 weeks (Figures 3I and 3J). Although cachectic melanomas strongly induce cardiac atrophy, non-cachectic melanomas also induce some level of cardiac cell atrophy compared with mock-treated controls (Figures 3I and 3J). This suggests that cancers, including those that do not induce overt cachexia, may impact the heart. However, apart from the 6-week time point, there was no substantial difference in cardiac cell size, suggesting that cachexia-induced heart wasting (Figure 2K) may result from apoptosis and/or necroptosis.44–48
Beyond the heart, previous studies have shown that different skeletal muscles have distinct propensity to atrophy induced by cachexia.49–51 To determine the impact of melanoma-induced cachexia on skeletal muscles, we have investigated wasting of the gastrocnemius (Figures 3K and 3L), soleus (Figures 3M and 3N), and TA skeletal muscles (Figures 3O and 3P), which differ for their myofiber-type composition.52–54 Immunostaining with antibodies specific for myosin heavy chain (MHC) isoforms (specific for different myofiber types) revealed that there are minimal changes in myofiber size at 2–4 weeks after xenograft implantation; there was, however, a decline in myofiber sizes at 6–8 weeks (Figures 3L, 3N, and 3P) after tumor implantation, with no changes in the % of myofiber types and number of myofibers, as expected based on previous studies.49–51 Contrary to the relative resistance to cancer-induced wasting of the soleus (Figure 3N), the TA displayed significant myofiber atrophy at early stages of cancer progression (Figure 3P). Altogether, these studies identify varying degrees of cell atrophy in distinct tissues in response to cancer-induced wasting.
Transcriptional profiling identifies molecular determinants of organ and tissue wasting induced by cachectic melanoma xenografts
To determine the mechanistic basis of differential responses of organs and tissues to cachexia, we have examined the transcriptional changes induced by cachectic melanomas compared with controls.
PCA revealed that although all samples clustered together at 2 weeks of age, the samples obtained from cachectic animals progressively diverged from the respective controls for the same tissue at later time points, i.e., 4, 6, and 8 weeks post-injection (Figure S3). Cluster analysis of the transcriptional changes associated with cachexia further indicated that body wasting was linked with upregulation, and to a lower extent downregulation, of five gene clusters (for a total of 1,094 genes) across multiple tissues and organs (Figures 4A–4C). Cluster 1 consisted primarily of genes downregulated during cachexia, and these included genes encoding for secreted proteins, proteins involved in antigen processing and presentation, and mitochondrial matrix proteins (Figure 4C). Clusters 2–4 consisted of genes moderately upregulated during cachexia: these included genes that regulate lipid metabolism, peroxisomes, endoplasmic reticulum (ER), and glutathione metabolism (cluster 2); ribosomal components, mitochondrial proteins, and splicing factors (cluster 3); and secreted factors and extracellular matrix proteins (cluster 4). Lastly, cluster 5 (the largest cluster, n = 440) consisted of genes highly upregulated during cachexia, which included inflammatory factors such as regulators of neutrophil chemotaxis, innate immunity, mast cell activation, and complement factors (Figure 4C).
Figure 4. Gradual transcriptional changes define cachexia progression in tissues and organs of mice implanted with cachectic melanoma xenografts.
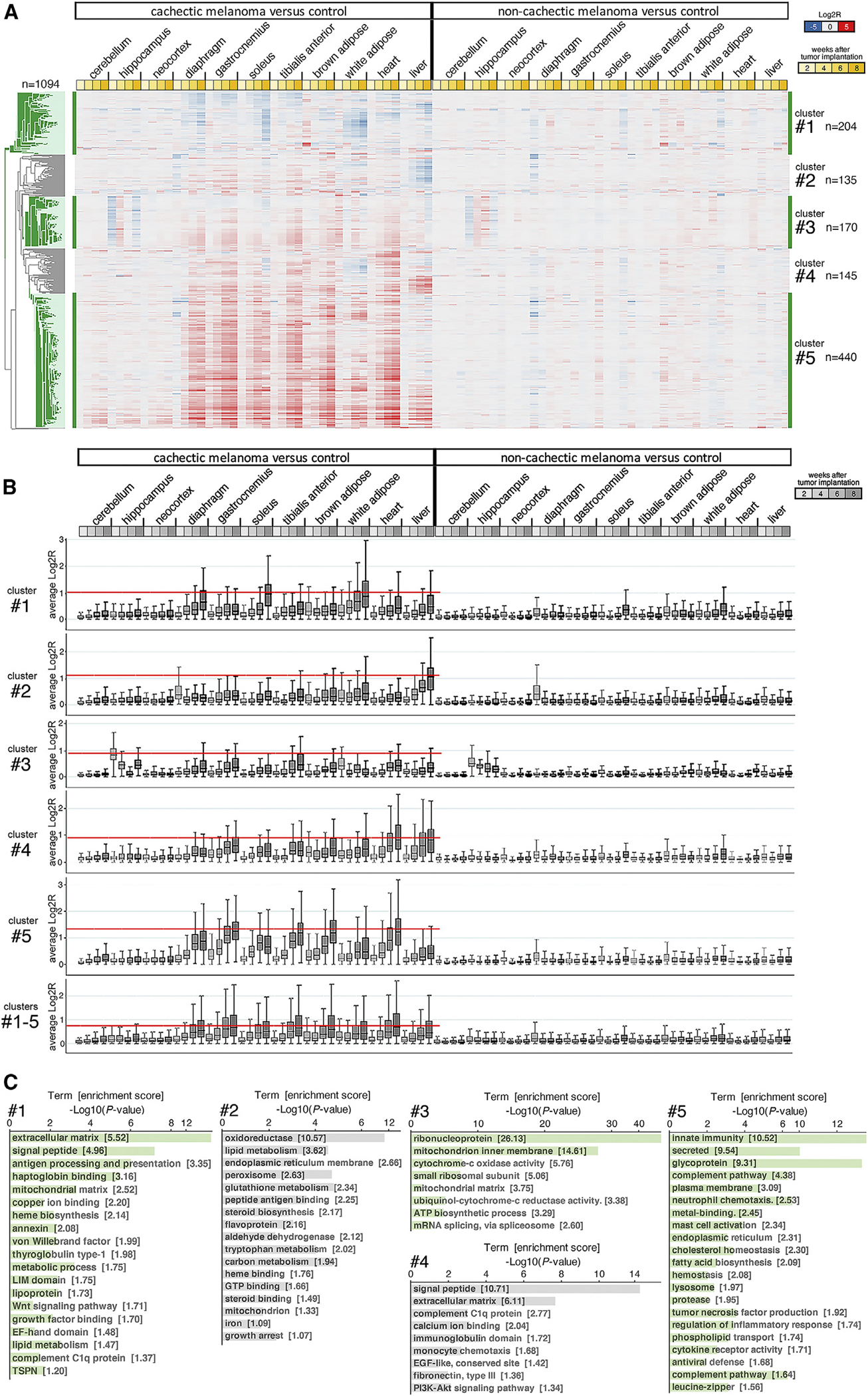
(A) Heatmap of 1,094 genes (five clusters) that are most highly associated with cachexia development. Most of these genes are upregulated by cachexia, particularly in skeletal muscles and the heart. Downregulation of gene expression occurs primarily in the WAT and liver. Few of these genes are modulated in brain areas (cerebellum, hippocampus, and neocortex). No expression changes occur in the tissues/organs of mice implanted with non-cachectic melanomas, compared with controls. The mean Log2R values are color coded (downregulated genes are shown in blue, upregulated genes in red). Different shades of yellow indicate 2, 4, 6, and 8 weeks after tumor cell injection.
(B) Cumulative gene expression changes induced by cachexia in distinct tissues for each gene cluster, analyzed separately (clusters 1, 2, 3, 4, and 5) and cumulatively (clusters 1–5). The absolute Log2R values are shown. The red line identifies the tissues where cachexia induces the most prominent gene expression changes. The boxes indicate the 25th and 75th percentiles, whereas the whiskers indicate the range.
(C) GO term analyses for gene clusters 1–5 identify different gene categories that are modulated by cachexia.
See also Figures S3–S6.
Although genes in clusters 1–5 were typically regulated in a consistent manner across tissues during cachexia, there were some noticeable exceptions. Specifically, cluster 2 genes were moderately upregulated in all tissues during cachexia but were downregulated in the liver (Figures 4A and 4B). Likewise, genes in cluster 4 were upregulated by cachexia across tissues but downregulated in the white adipose, and partly downregulated and upregulated in the liver (Figures 4A and 4B). Moreover, a fraction of cluster 5 genes was upregulated in the liver during cachexia (Figures 4A and 4B). Altogether, gene expression profiling identifies a cachexia signature that generally occurs across tissues but that also has tissue-specific features.
The analysis of transcriptional changes associated with cachexia identifies different magnitudes of gene modulation that occur in tissue-specific fashions. Specifically, analysis of the average absolute Log2R values for clusters 1–5 indicates that the heart and the gastrocnemius are the tissues most impacted by cachexia, followed by the TA, the BAT, and the diaphragm (Figure 4B). Gene expression in the soleus, WAT, and liver appears to be moderately affected, whereas there is relatively little impact of cachexia on gene expression in the hippocampus, neocortex, and cerebellum (Figure 4B, clusters 1–5). Analysis of the average absolute Log2R values indicates that the most affected tissues are the soleus and white adipose in cluster 1, the liver in cluster 2, the hippocampus in cluster 3, the heart and liver in cluster 4, and the heart and gastrocnemius in cluster 5 (Figure 4B).
Interestingly, whereas the progressive changes in gene expression observed in skeletal muscle (gastrocnemius, diaphragm, TA, and soleus; Figure 4A) mirror the progressive wasting that occurs during cachexia in this tissue (Figures 2E–2H), the correlation between transcriptional changes and weight loss is less noticeable or even absent for other tissues. Specifically, the interscapular BAT undergoes profound gene expression changes during cachexia (Figure 4A) but little weight loss (Figure 2I). Moreover, heart mass is reduced early during cachexia (i.e., 2 weeks), but not substantially further in the following weeks (Figure 2K), whereas cachexia-induced transcriptional changes occur progressively in the heart from 2 to 8 weeks (Figure 4A).
A possible explanation for the disconnect in the dynamics of tissue weight loss and transcriptional changes is that cachexia impacts not only size but also organ/tissue metabolism and function. Therefore, many of the cachexia-associated transcriptional changes are likely involved in modulating tissue function independently from size. For example, cancer-downregulated genes in the liver included those necessary for lipid metabolism, fatty acid and tryptophan metabolism, and peroxisomes (Figures S4A–S4D), consistent with previous findings that cancer reduces hepatic functions.55,56 Conversely, melanoma-induced cachexia may increase brown adipocyte function (i.e., fatty acid β-oxidation and thermogenesis), as previously reported in other models42 (Figures S4E–S4H). In agreement with this scenario, cachexia upregulates expression in the brown adipose of the Ctsb and Ctsc cathepsins, perhaps indicative of increased lysosomal functions, and of Scd1, which is necessary for mitochondrial fatty acid oxidation (Figure S4H).
Beyond the liver, brown adipose, and white adipose (Figures S4I–S4L), the heart provides a remarkable example of cancer-induced transcriptional changes that are likely involved in tissue dysfunction rather than in wasting. Specifically, there are several voltage-gated ion channels that are downregulated and that may contribute to reduce heart contractile function during cachexia (Figure S5A). These include Scn4a, a sodium voltage-gated channel subunit that is mutant in Brugada syndrome and arrhythmias57; Cacna1s, a calcium voltage-gated channel mutant in malignant hyperthermia and hypokalemic periodic paralysis58,59; Hcn2, a pacemaker channel that is mutant in arrhythmias60,61; and Clcn1, which is mutant in myotonia congenita62,63 (Figure S5A). Although autophagy and ubiquitin ligases are highly modulated by melanoma-induced cachexia (Figures S5B–S5D and S6A–S6C) and are considered key drivers of muscle mass loss,49,50,64–66 they may also alter muscle metabolic and functional properties by tagging for degradation regulators of these processes (Figures S6A–S6C). Altogether, these examples highlight cachexia-induced transcriptional changes that are likely involved in altering tissue function, rather than size, in cancer-bearing animals.
Cancer cachexia impacts the expression of secreted proteins and of genes with metabolic functions in the cerebellum, hippocampus, and neocortex
Systemic inflammation is a key component of cachexia.2,4,5 Although inflammation promotes neurodegeneration and modulates brain function,67–70 little is known about the impact of cancer cachexia on the brain.19 Previously, it was found that pancreatic cancer cachexia alters cholinergic and glutamatergic brain metabolites,71 and that hepatoma-induced cachexia decreases the expression of brain voltage-gated potassium channels.72 However, beyond these few genes, it remains unknown how the brain is globally affected by cancer cachexia.
To address this question, we have examined the transcriptional changes induced in three different brain areas (cerebellum, hippocampus, and neocortex) at 2–8 weeks after tumor implantation. Overall, relatively few genes were modulated by cachexia in the cerebellum (87 genes), hippocampus (165 genes), and neocortex (88 genes) (Figures 5A, 5D, and 5G). These gene sets are largely upregulated by cachexia in the cerebellum and neocortex (Figures 5A and 5G). Conversely, downregulation of gene expression occurs early (2 weeks) in the hippocampus (Figure 5D) and includes genes encoding for ribosomal components, mitochondrial proteins, histones, and secreted proteins.
Figure 5. Cachexia induces transcriptional changes in the brain.
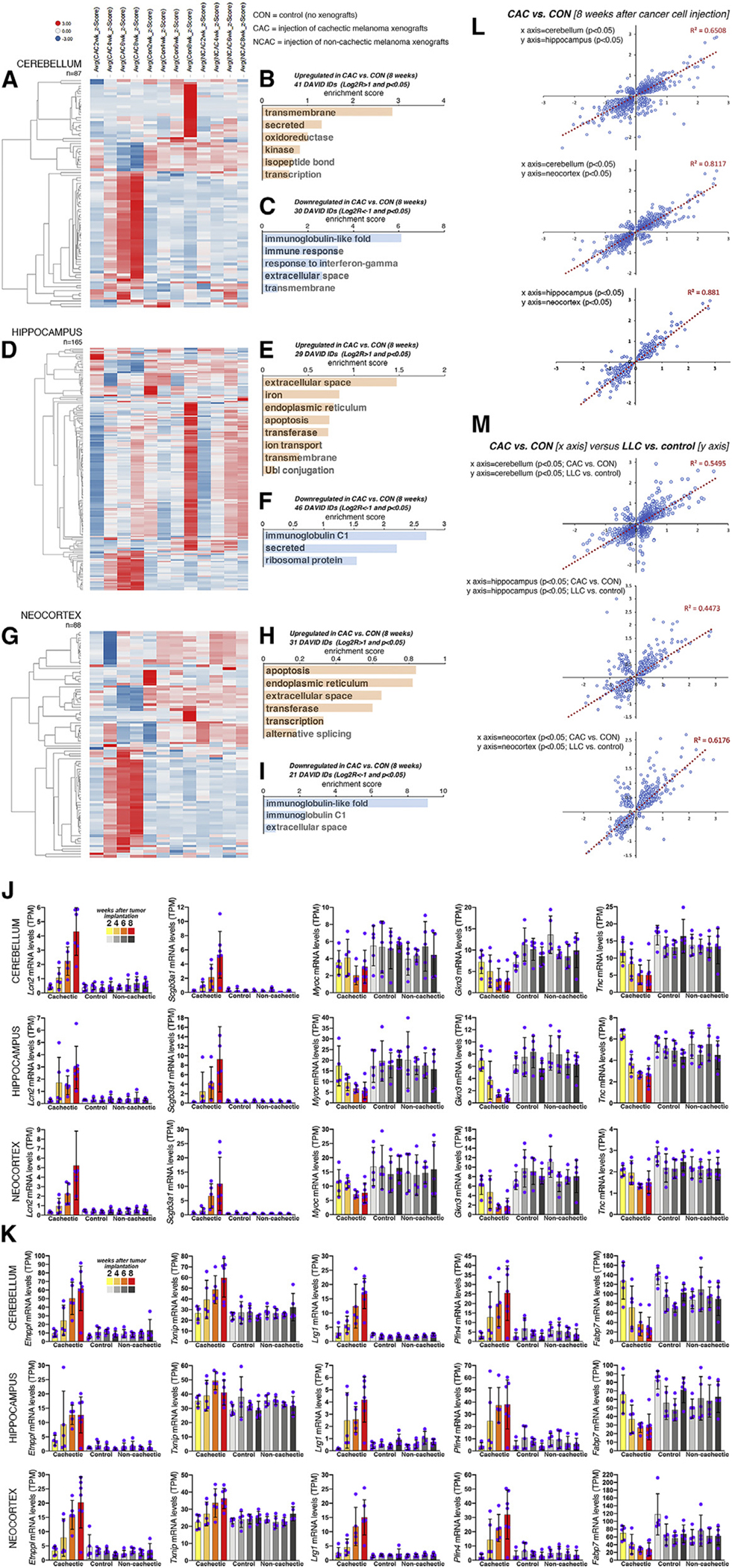
(A–I) Heatmap of 87, 165, and 88 genes that define the response of the cerebellum (A), hippocampus (D), and neocortex (G) to cachexia induced by melanoma xenografts. The average Z scores are color coded (downregulated genes are shown in blue, upregulated genes in red). Gene categories that are upregulated (p < 0.05 and Log2R > 1; B, E, and H) and downregulated (p < 0.05 and Log2R < −1; C, F, and I) are shown.
(J and K) Extracellular matrix and secreted proteins (J) are modulated by cachexia in the cerebellum, hippocampus, and neocortex, as well as genes with metabolic functions (K). Bars represent SD. All comparisons are significant (p < 0.05) in cachectic versus control at 8 weeks after tumor cell injection.
(L) There is high similarity in the responses induced by cachectic melanomas across brain regions.
(M) There is also a moderate degree of similarity (R2 = 0.44–0.61) in the changes induced by cachectic melanomas versus LLC cells.
See also Figure S6.
Although the impact of cachexia on the brain appears minimal compared with other organs (Figure 4), many genes important for brain function are modulated by melanoma-induced cachexia in the cerebellum (Figures 5B and 5C), hippocampus (Figures 5E and 5F), and neocortex (Figures 5H and 5I). Some of these transcriptional changes (such as those for secreted/extracellular proteins) likely reflect cachexia-induced neuroinflammation, which has been previously found to depend on neutrophil infiltration in the brain via CCL2-CCR2 signaling.73
Beyond secreted factors involved in immunity, there are other cachexia-modulated secreted factors that could have signaling roles (Figure 5J). For example, the Lcn2 lipocalin and the Scgb3a1 secretoglobin increase during cachexia in the cerebellum, hippocampus, and neocortex (Figure 5J), whereas Myocilin (Myoc), Gastrokine 3 (Gkn3), and Tenascin C (Tnc) decline (Figure 5J). Although the function of many of these secreted factors is unknown, cancer cell-secreted Lcn2 (Figure 1Q) induces anorexia,17 which is a component of cachexia.18,19,74,75 Therefore, in addition to cancer cell-secreted Lcn2, also cachexia-induced Lcn2 expression in the brain could regulate feeding behavior.
Several genes with metabolic functions were also upregulated in the brain of mice implanted with cachectic tumors, including Etnppl (which catalyzes phosphoethanolamine breakdown), Txnip (which inhibits glucose uptake76–78), Lrg1 (a glucose-responsive glycoprotein that promotes apoptosis and reduces brain-derived neurotrophic factor [BDNF] signaling79–81), and Plin4 (a perilipin involved in intracellular lipid storage), whereas the fatty acid-binding protein Fabp7 was downregulated (Figure 5K).
Altogether, these findings indicate that cachexia impacts the expression of key metabolic genes in the brain and suggest a possible overlap between cachexia-induced changes in the brain and pathways that drive neurodegeneration. For example, decreased glucose uptake is a key feature of Alzheimer’s disease and other neurodegenerative diseases.82,83 Our finding that Txnip is upregulated by cachexia in the brain (Figure 5K), together with previous knowledge that Txnip inhibits intracellular glucose import,76–78 suggests that impairment of glucose uptake is a common feature and a possible point of synergy between cancer-induced cachexia and neurodegeneration.
Derangement of gene expression in the brain is a common feature of cachexia induced by pediatric melanoma xenografts and by Lewis lung carcinoma (LLC) cells
Subcutaneous injection of LLC cells is an established model to investigate cachexia, which develops ~3 weeks after cancer cell injection.84–87 Although the impact on the adipose and skeletal muscle is well known,84–86 it remains unexplored whether LLC tumors regulate gene expression in the brain, as we have found for melanoma-induced cachexia (Figures 5A–5K).
To address this point, we have examined the cerebellum, hippocampus, and neocortex from mice after 3 weeks from LLC cancer cell injection. RNA-seq indicates that 586 genes define the impact of LLC-induced cachexia on the brain (Figure S6D). Glycoproteins, extracellular matrix, and secreted proteins are among the genes that are upregulated in control versus LLC cerebellar samples (cluster 1), whereas other proteins of the same gene categories are conversely regulated (cluster 2; Figure S6E). Peroxidases and proteins involved in immunity and chemotaxis are upregulated in LLC samples from all brain regions (cerebellum, hippocampus, and neocortex) compared with controls (cluster 3). Other clusters (4–5) consist of genes that differ across brain regions (Figure S6E). Altogether, these findings indicate that alteration of brain gene expression is a common feature of cachexia induced by different tumor types.
Next, we examined the overall similarity in the gene expression changes induced in different brain areas by melanoma xenografts. There was overall similarity in the gene expression changes (p < 0.05) induced by melanomas in the cerebellum versus the hippocampus (R2 = 0.65) and even more so in the cerebellum versus the neocortex (R2 = 0.81) and the hippocampus versus the neocortex (R2 = 0.88; Figure 5L). Moreover, cross-comparison of the transcriptional changes (p < 0.05) induced by melanomas versus LLC revealed a good correlation in the cerebellum (R2 = 0.56), hippocampus (R2 = 0.44), and neocortex (R2 = 0.61) (Figure 5M). Altogether, these analyses indicate that the brain is a target of cachectic signaling induced by different cancer types.
The expression of secreted factors is profoundly remodeled by cachexia across tissues
By analyzing gene sets regulated across tissues, “secreted factors” and related categories emerge as prominently modulated by cancer cachexia (Figures 4, S4, and S5). Further analysis of the 2,641 genes encoding for secreted factors25 indicates that cachexia primarily upregulates the expression of secreted factors in most tissues, although it overall reduces their expression in the soleus, white adipose, and stromal cells (Figure 6A). Comparison of the similarity in the cancer-induced gene expression changes for secreted factors reveals that, as expected, tissues that are related (such as different skeletal muscles) display a high degree of similarity (Figure 6B). Modulated secreted factors include cytokines, growth factors, growth factor-binding proteins, and secreted enzymes (Figure 6C). Interestingly, the expression of ACE (also known as ACE1) progressively increases during melanoma-induced cancer cachexia across tissues (Figure 6C), including the TA muscle (Figure 6D), which undergoes profound cancer-induced wasting (Figures 3O and 3P). ACE protein levels similarly increase with cachexia in TA muscles (Figure 6E). Further analysis of a published gene expression dataset (GEO: GSE41726) consisting of rectus abdominis muscle biopsies from cancer patients88,89 further indicates that ACE mRNA levels significantly correlate with the expression of the known atrophy markers FBX O 32 (atrogin-1) and TRIM63 (MuRF1) in female cancer patients but minimally in males (Figure 6F). Altogether, these findings indicate that ACE transcriptional upregulation can occur (albeit with varying degrees) in wasting skeletal muscles.
Figure 6. Cachexia remodels the expression of secreted factors across tissues.
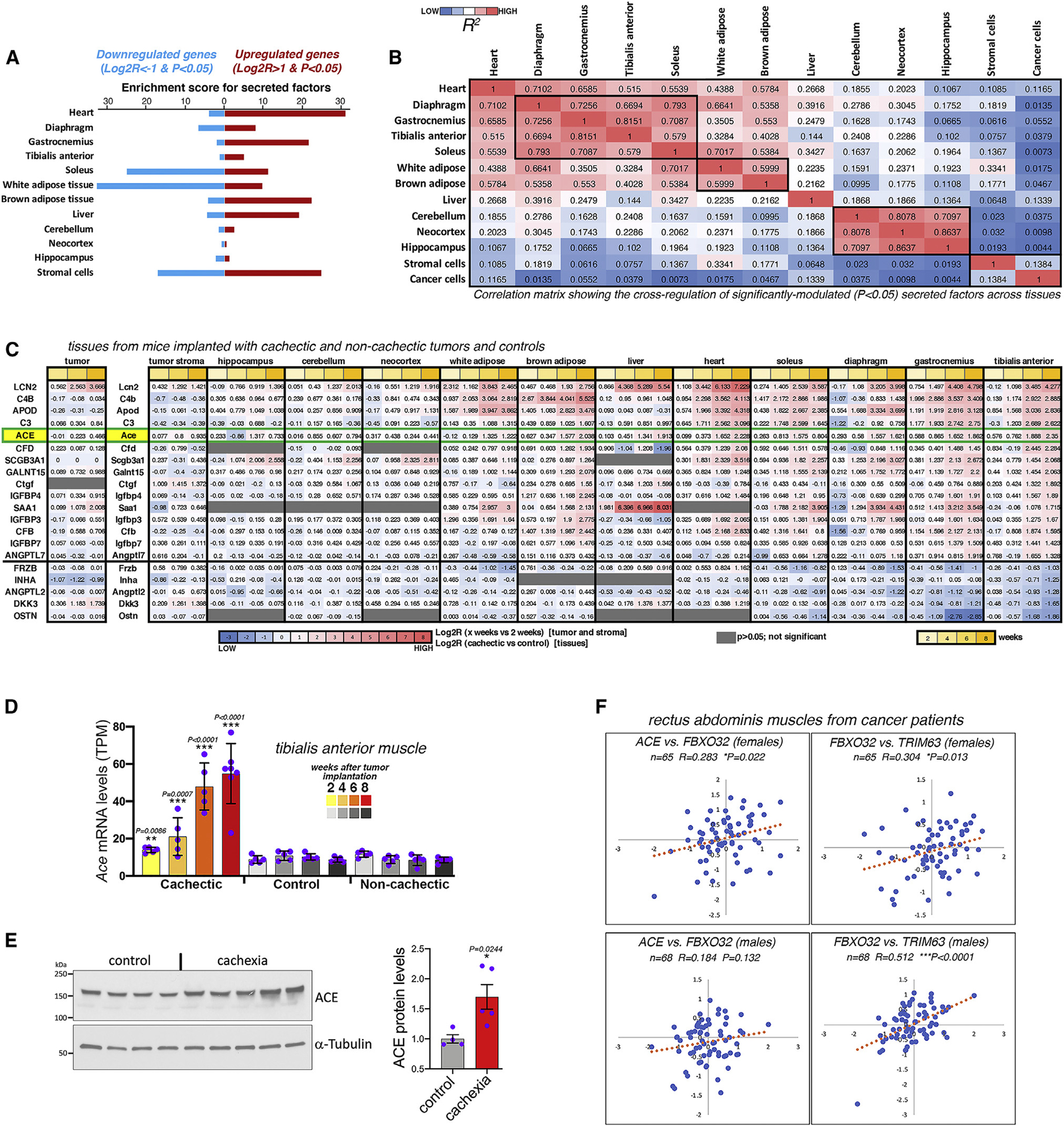
(A) Expression of secreted factors is regulated across tissues by cancer cachexia.
(B) Comparison of the similarity in the cancer-induced transcriptional changes for secreted factors.
(C) Cachexia-induced secreted factors include cytokines, growth factors, growth factor-binding proteins, and enzymes, such as ACE.
(D) ACE expression progressively increases in the TA muscle during cancer-induced wasting. SD with **p < 0.01 and ***p < 0.001.
(E) Western blots indicate that cachexia increases ACE protein levels in TA muscles. SEM with *p < 0.05 and n = 4–5.
(F) ACE expression correlates with that of FBX O 32 (atrogin-1) and TRIM63 (MuRF1) in muscle biopsies from female cancer patients.
Pharmacological inhibition of the cachexia-induced secreted factor ACE does not halt body and muscle wasting but partially preserves skeletal muscle function
ACE is a secreted enzyme that converts the hormone angiotensin I to the vasoconstrictor hormone angiotensin II, and that therefore is key for regulating blood pressure as part of the renin-angiotensin system.90 On this basis, ACE inhibitors are widely used to treat hypertension.91
We have found that ACE is progressively upregulated during cancer cachexia across tissues (Figure 6). An ACE gene variant encoding for a protein with lower enzymatic activity was previously found to enhance skeletal muscle performance in humans.92,93 Moreover, it was suggested that ACE inhibition might counteract cachexia.94 On this basis, we next examined whether pharmacological ACE inhibition during cancer progression impedes cachexia. To this purpose, the US Food and Drug Administration (FDA)-approved ACE inhibitor lisinopril was administered from day 0 (i.e., from the day of cancer cell injection) in the drinking water (at 40 mg/L), as previously done.95 As expected, there was a progressive decline in body weight (Figure 7A) and in the tumor-free body mass (Figure 7B) of mice bearing cachectic melanomas. However, this occurred to a similar extent also in mice treated with lisinopril (Figures 7A and 7B), indicating that ACE inhibition does not block cancer-induced body wasting. The tumor size and mass were also similar (Figures 7C and 7D), indicating that lisinopril does not impact tumor growth in this system. As previously found (Figure 2), the length of the tibia bone does not change during cachexia development (Figure 7E) and was therefore used as a normalization factor for the analysis of tissue weight. Cancer-induced atrophy occurred in the heart, liver, pancreas, and WAT, and this equally occurred with or without lisinopril treatment (Figures 7F–7I). Interestingly, although there was a reduction in the weight of the brown adipose in response to cancer, this decline was in part prevented by lisinopril (Figure 7J). The weight of distinct skeletal muscles (gastrocnemius, soleus, diaphragm, and TA) declined in response to the injection of cachectic melanomas (Figures 7K–7N) independently from lisinopril treatment. There was no significant effect of either cachexia or lisinopril on the normalized tetanic force (Figure 7O), i.e., the maximal muscle contraction normalized by the TA muscle mass.96,97 However, ACE inhibition improved the normalized twitch force produced by TA muscles (Figure 7P), which is indicative of its physiological contraction on stimulation.96,97 Specifically, pairwise comparisons indicate that cachexia significantly reduces the normalized twitch force of TA muscles, and that this is mitigated by lisinopril (Figure 7P). Altogether, these findings suggest that lisinopril helps maintain skeletal muscle function during cancer cachexia.
Figure 7. ACE inhibition with lisinopril does not prevent cancer-induced wasting but preserves muscle force.
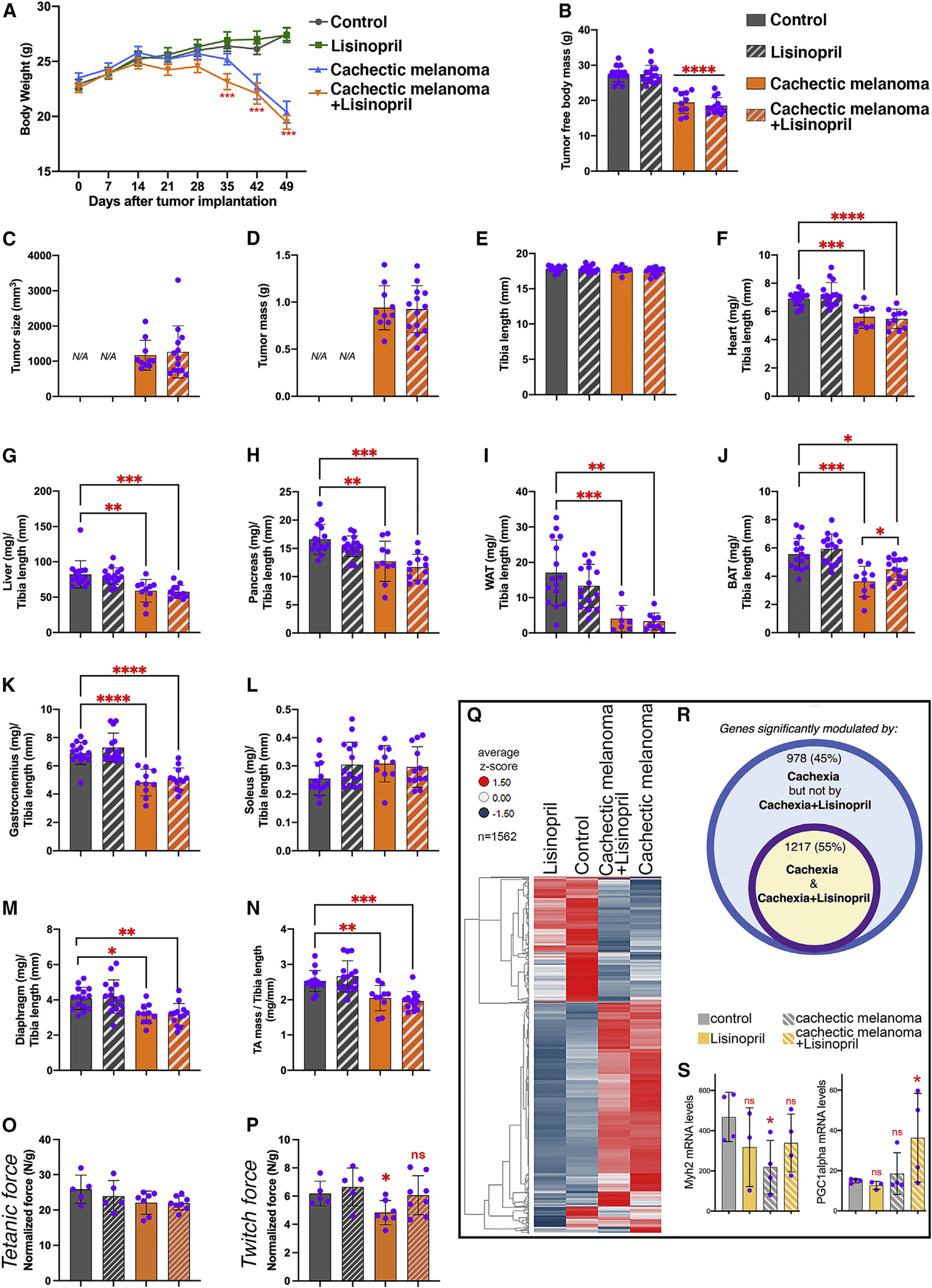
(A and B) Cachectic melanomas induce progressive decline in body weight (A) and tumor-free body mass (B), and this is not prevented by the ACE inhibitor lisinopril.
(C and D) The tumor size and mass are similar in lisinopril- versus mock-treated mice.
(E and F) Tibia length (E) is unchanged, whereas heart mass (F) declines with cachexia irrespective of lisinopril treatment.
(G–I) Cancer-induced wasting occurs in the liver, pancreas, and WAT and is not modulated by lisinopril.
(J) Reduction in the weight of the BAT is partly prevented by lisinopril.
(K–N) The weight of several skeletal muscles (including the TA) declines with cachexia, and this is not rescued.
(O) There is no substantial effect of cachexia or lisinopril on the tetanic force normalized by the TA mass.
(P) ACE inhibition improves muscle force production, as indicated by the normalized twitch force of TA muscles. Specifically, pairwise comparisons indicate that cachexia significantly reduces the normalized twitch force of the TA, and this is mitigated by lisinopril.
(Q) Heatmap of 1,562 genes that define the response of TA muscles to cachexia and lisinopril. The average Z scores are color coded.
(R) 2,195 genes are differentially modulated by cachexia, but lisinopril impedes the significant modulation of 978 (~45%) of them.
(S) Examples of differentially modulated genes include Myh2, i.e., myosin heavy chain 2a, which is significantly downregulated by cachexia but less so with concomitant lisinopril treatment. PGC-1α expression is not modulated by cachexia, but it is significantly higher on concomitant lisinopril treatment.
In (A)–(P), SD with *p < 0.05, **p < 0.01, and ***p < 0.001. In (S), SD with *p < 0.05 and **p < 0.01; n(TA) = 3–4. See also Figure S7.
Lisinopril partially prevents cachexia-induced transcriptional changes and promotes the expression of genes that support muscle contraction
To determine its mechanisms of action, we utilized RNA-seq to determine the transcriptional changes induced by cachexia with and without concomitant treatment with lisinopril (Figure 7Q). Globally, melanoma-induced cachexia resulted in the significant modulation of 2,195 genes, but lisinopril prevented the significant regulation of 978 (~45%) of them (Figure 7R). Differentially regulated genes possibly connected with the lisinopril-mediated improvement of muscle function include Myh2 and PGC-1α (Figure 7S). Myh2 encodes for the contractile protein MHC 2a, and it is significantly downregulated by cachexia, but not if mice are concomitantly treated with lisinopril (Figure 7S). Previous studies have reported a cancer-induced general decline in MHC protein abundance and sarcomere disintegration,98,99 which impairs muscle contraction. In our model, Myh2 is the only MHC protein that is transcriptionally downregulated by cachexia, and this is prevented by lisinopril. Interestingly, cachexia tends to reduce the relative proportion of type 2a myofibers (Figure S7D), and this might explain why Myh2 (i.e., MHC 2a) levels decline on cachexia. However, this occurs also in cachectic mice treated with lisinopril (Figure S7D), and therefore preservation of Myh2 expression by lisinopril does not arise from changes in the number of type 2a myofibers. The capacity of lisinopril to promote Myh2 expression may therefore, at least in part, explain how lisinopril preserves muscle function in cachectic mice. Because human muscles contain type 2a myofibers,52–54 the protective effects of lisinopril on MHC 2a expression may have therapeutic relevance for humans.
Another gene that was differentially modulated by cachexia and lisinopril is PGC-1α (Figure 7S), which is a transcription factor responsible for exercise-induced muscle adaptations and that is transcriptionally upregulated by muscle contraction.100–102 Although cachexia does not modulate PGC-1α mRNA levels, its expression is significantly induced by lisinopril during cachexia (Figure 7S), and this may contribute to the improvement in muscle function caused by lisinopril and/or be a readout of increased contractile capacity (Figure 7P).
Altogether, these studies indicate that lisinopril opposes several of the transcriptional changes induced by cachexia, some of which may be responsible for decreased muscle force production during cachexia.
DISCUSSION
Cachexia is an important medical condition, but the mechanisms underlying its progression are incompletely understood. Here, we have examined the cellular and transcriptional changes that progressively occur in skeletal muscles, heart, white and brown adipose, liver, and brain during cachexia. By analyzing tissue weight over time, we find that some tissues undergo atrophy (such as skeletal muscles), whereas others like the liver and brain are less affected or not affected by cachexia (Figure 2). There are also differences in the dynamics of wasting. For example, there is heart atrophy already after 2 weeks from tumor implantation, a time point at which there are minimal (if any) signs of wasting in other tissues. However, heart weight remains constant in the following weeks, whereas the weight of other tissues and their cell size progressively declines (Figures 2 and 3). These studies therefore indicate a differential response of organs and tissues to cachectic signaling and that this is characterized by different degrees and timelines of wasting.
As expected based on previous studies, we find that cachexia induces the expression of genes that are likely drivers of tissue wasting, such as proteases, ubiquitin ligases, and components of the autophagy/lysosome system.49,50,64–66 However, many transcriptional changes are temporally disconnected from wasting. For example, heart wasting occurs at early time points (2–4 weeks), but its weight remains stable in the following weeks of cachexia progression (6–8 weeks). In contrast, heart transcriptional changes gradually increase over time from 2 to 8 weeks and consist primarily of genes that regulate heart function, such as downregulation of voltage-gated ion channels that are necessary for myocardial contraction and that cause arrythmias when mutant (Figure S5). Consistent with this disconnect between tissue wasting and transcriptional changes, we find that cachexia prominently modulates gene expression also in tissues and organs that do not undergo wasting, such as the brain, and that these include several genes with metabolic functions, such as Txnip (Figure 5), an inhibitor of glucose import,76,78 suggesting that, similar to neurodegeneration,70,103 cachexia deranges brain glucose metabolism. Although cachexia induces transcriptional changes associated with decreased function in many tissues (heart, muscle, brain, liver), the converse occurs for the brown and white adipose, where cachexia increases the expression of genes necessary for fatty acid β-oxidation, as previously observed.104 Altogether, our study pinpoints cachexia-induced gene expression changes that are involved in tissue wasting, as well as others that are likely drivers of tissue functional changes.
Although many gene sets are regulated by cancer-initiated signaling, secreted factors constitute a top category of regulated genes in host tissues. Because inter-tissue signaling is a known driver of cachexia,2,104 it is possible that the induction of such secreted factors in host tissues in turn contributes to the cancer-initiated systemic signaling that drives tissue dysfunction and wasting. For example, the heart is one of the tissues that displays early wasting and that is transcriptionally most affected by cachexia (Figures 2 and 4). Interestingly, chronic heart failure has been found to induce systemic wasting in non-cancerous systems,105–107 suggesting that some of the transcriptional changes and tissue wasting observed in other tissues during cachexia may derive from cancer-induced heart dysfunction.
To probe the relevance of secreted factors that are induced in host tissues by cancer cells, we have utilized an FDA-approved pharmacological inhibitor of the ACE, a secreted factor that is induced by cachectic melanomas across tissues, including skeletal muscles (Figure 6). ACE inhibition via lisinopril provides some benefits: although it does not impede tissue wasting (Figures 7A–7N), lisinopril partially preserves muscle force production (Figure 7P), and this occurs, at least in part, by hindering cachexia-induced transcriptional changes (Figures 7Q–7S). A series of previously published studies indicates that cancer cachexia reduces the integrity and function of pre- and post-synaptic portions of the neuromuscular junction (NMJ), which can result in a denervation-like phenotype.108–111 Therefore, it is possible that lisinopril-mediated improvements in muscle force arise also from NMJ preservation. Interestingly, ACE inhibitors attenuate tumor-induced myocardial dysfunction112 and even tumor growth in other systems.113,114 Although we have not observed any effects on tumor growth, these studies indicate that ACE inhibition may be beneficial in diverse ways depending on the cancer context. Altogether, we propose that lisinopril may help maintain motor function in cachectic patients.
In summary, our longitudinal analysis of transcriptional changes induced by cachexia suggests that paracrine and endocrine signaling via cachexia-induced secreted factors released by non-cancerous host tissues contributes to systemic cachexia.
Limitations of the study
This study was done in athymic nude mice that lack T lymphocytes to avoid the immune rejection of the human melanoma xenografts used here. Previous studies have found that different populations of T cells can induce or rather protect from cachexia.13,115,116 Therefore, a limitation of this study is that the progressive development of cachexia has been observed in the absence of T lymphocytes. Nonetheless, in combination with studies in immunocompetent mice, this study’s limitation could be exploited as a vantage point to define T cell-independent versus -dependent mechanisms of cachexia.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Fabio Demontis (Fabio.Demontis@stjude.org).
Materials availability
There are no restrictions to the availability of tools generated in this study.
Data and code availability
All data supporting the findings of this study are available within the paper, the supplemental information, and the Data S1, which reports the source data. The 747 RNA-seq samples reported in this publication have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GEO: GSE183613 and GEO: GSE214981.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mouse husbandry
Mice were handled following animal ethics guidelines and the protocol was approved by the IACUC at St. Jude Children’s Research Hospital. All mice were housed in a ventilated rodent-housing system with a controlled temperature (22–23°C) and given free access to food and water.
Mice that carry LLC (Lewis Lung Carcinoma) tumors
LLC (Lewis Lung Carcinoma) cells were obtained from the American Type Culture Collection (ATCC #CRL-1642) and regularly screened to exclude the presence of mycoplasma before experimental use. LLC cells were cultured at 37°C with 5% CO2 in DMEM (high glucose DMEM, with Glutamax, GIBCO, #10566016) containing 10% fetal bovine serum (GIBCO, #10437–028), and penicillin/streptomycin (10,000 U/ML, GIBCO, #15140122). Male C57BL/6J (The Jackson Laboratory, JAX#000664) mice were utilized at 4 months of age. 106 LLC cells were injected into the right and left flank16,85,86 and tumors were allowed to grow up to ~3 weeks, at which time tumor-bearing mice were euthanized.
Mice bearing orthotopic pediatric melanoma xenografts
MAST360B/SJMEL030083_X2 and MAST552A/SJMEL031086_X3 are patient-derived melanoma xenografts22 from the Childhood Solid Tumor Network collection at St. Jude Children’s Research Hospital.20,21,117–119 In preparation for injection, melanoma cells were thawed, gently washed in RPMI, resuspended in Matrigel (Corning #354234) at 0.1 mL Matrigel/106 cells, and kept on ice before the injection. Subsequently, 106 melanoma cells were inoculated subcutaneously into the flank of female 2-month-old NCI Ath/nude mice (Charles River Laboratory #553NCIATH/NU). Tumors were monitored and allowed to grow typically for 2, 4, 6, and 8 weeks.
METHOD DETAILS
Tissue collection
At each time point of cachexia progression, mice were euthanized and the body weight recorded. All tissues and organs were dissected and weighed. The tumors were removed, weighed, and immediately snap-frozen for RNA-seq. A portion of the liver, gonadal white adipose tissue (WAT), and interscapular brown adipose tissue (iBAT) was fixed in 10% formalin for histology (H&E) and another portion was snap-frozen for RNA-seq. The brain was dissected, weighed, and the hippocampus, neocortex and cerebellum were removed and snap-frozen. Skeletal muscles were bisected at the mid-belly whereas the heart was transversally cut. One part was mounted onto tragacanth gum and frozen in liquid nitrogen-cooled isopentane (Sigma-Aldrich, #277258) for subsequent histological analyses; a second part was snap-frozen for RNA-seq. Schemes were drawn with BioRender.
RNA preparation
Snap-frozen tissues and organs were homogenized with a NextAdvance bullet blender in TRIzol at 4°C.120 RNA was extracted by isopropanol precipitation from the aqueous phase.121 The integrity and purity of RNA was assessed with a bioanalyzer.
RNA sequencing
RNA sequencing libraries for each sample were prepared from total RNA by using the Illumina TruSeq RNA Sample Prep v2 Kit per the manufacturer’s instructions, and sequencing was completed on the Illumina NovaSeq 6000. The 100-bp paired-end reads were trimmed, filtered against quality (Phred-like Q20 or greater) and length (50-bp or longer), and aligned to the mouse reference genome GRCm38/mm10 (for reads obtained from the mouse host tissues and from the mouse stromal cells of the tumor xenografts) and to the human reference genome GRCh38/hg38 (for reads obtained from the human melanoma cells of the tumor xenografts) by using CLC Genomics Workbench v12.0.1 (Qiagen). For gene expression comparisons, we obtained the TPM (transcript per million) counts from the CLC RNA-Seq Analysis tool. The differential gene expression analysis was performed by applying the non-parametric ANOVA using the Kruskal-Wallis and Dunn’s tests on log-transformed TPM between three to five replicates of experimental groups, implemented in Partek Genomics Suite v7.0 software (Partek Inc.). The gene sets were analyzed by DAVID (v6.8, https://david.ncifcrf.gov).
The 747 RNA-seq samples discussed in this publication have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GEO: GSE183613 and GEO: GSE214981.
Hierarchical clustering of RNA-seq data
Protein-coding genes with significantly different expression between sample groups (p value <0.01 and log2R > 1 in more than one tissue type, compared to its corresponding control samples) and protein coding genes with significantly different expression through the time course of cachexia progression (p value <0.01 and log2R > 1 between any two different timepoints from any tissue type) were hierarchically clustered into heatmaps by using the UPGMA clustering method (unweighted pair group method with arithmetic mean) with similarity measure of correlation, implemented in Spotfire v7.5.0 software (TIBCO).
Hematoxylin and eosin (H&E) staining
Formalin-fixed tissues and organs were processed and embedded in paraffin by standard techniques, sectioned at 4-μm, mounted on positively charged glass slides (Superfrost Plus; Thermo Fisher Scientific, Waltham, MA), and dried at 60°C for 20 min. Tissue sections were then stained with H&E according to standard procedures.122
Image analysis of H&E slides
The mean cell size was estimated from H&E slides that were scanned with a Zeiss Axioscan and a 20x objective. For these image analyses, StarDist123,124 was used to define for each image the mean cell size (in μm2 units), defined as the tissue area divided by the number of nuclei. StarDist is a deep learning segmentation method with pre-trained weights for bright-field images and can be used to segment the cell nuclei in H&E slides. In addition, QuPath 0.2.3125 was used to draw ROIs and batch segment the images.
For the quantitation of cachexia-induced delipidation, we defined areas occupied by lipid droplets in white adipocytes as those that have intensities greater than 185. We defined all regions within WAT with intensities less than 185 to be the rest of the adipose tissue (non-fat regions). We defined the fat and non-fat regions in the brown adipose tissue (BAT) by using a machine-learning based pixel classifier trained with the QuPath software.125 Because the ROIs used for the quantification include white background regions consisting of glass, we used a machine-learning-based pixel classifier to detect glass regions; subsequently, glass areas were subtracted from the total ROI area to accurately estimate the total area of tissue within the ROIs. We then estimated the relative fat content of WAT and BAT by calculating the ratio of the area occupied by fat normalized to the total tissue area.
All analyses were done in a consistent, automated manner for each image, tissue type, and time point. Statistical significance was estimated by performing pairwise Mann-Whitney U tests between cohorts for each tissue type and time point. In the boxplots reporting this data, the top and bottom of the box represent the 25th and 75th percentile, and the centerline is the 50th percentile; the lower whisker is defined by the smallest observation greater than or equal to the lower hinge −1.5*IQR; the upper whisker is defined by the largest observation less than or equal to the upper hinge +1.5*IQR (IQR = the inter-quartile range between the first and third quartiles).
Analysis of myofiber type, size, and number in the soleus, tibialis anterior, and gastrocnemius skeletal muscles
Unfixed slides with sections of skeletal muscles were incubated for 1h with blocking buffer (PBS with 2% BSA and 0.1% Triton X-100) prior to overnight immunostaining with primary antibodies at 4°C. The primary antibodies used were mouse IgG2b anti-myosin heavy chain type 1 (DSHB, cat. no. BA-F8), mouse IgG1 anti-myosin heavy chain type 2a (DSHB, cat. no. SC-71), and rat anti-laminin α2 (4H8–2; Santa Cruz, cat. no. sc-59854). The slides were washed and incubated with the secondary antibodies including anti-mouse AlexaFluor555-conjugated IgG2b (Invitrogen, cat. no. A21147), anti-mouse AlexaFluor488-conjugated IgG1 (Invitrogen, cat. no. A21121), and anti-rat AlexaFluor647-conjugated IgG (Invitrogen, cat. no. A21247). DAPI (Sigma Aldrich, cat. no. 10-236-276-001) was used to identify nuclei. The soleus was imaged on the Nikon C2 confocal microscope with a 10X objective. Myosin heavy chain immunostaining was used to classify type 1 myofibers (red), type 2a myofibers (green), and presumed type 2x/2b myofibers (black) for which no MHC type 1 or type 2a immunoreactivity was detected. Myofiber type and size were analyzed with the Nikon Elements software by using the inverse threshold of laminin α2 staining to determine myofiber boundaries. To categorize myofiber types, the intersections of the inverse of laminin and the myosin heavy chain-specific staining was used. The myofiber size was measured by using the inverse laminin immunostaining for boundaries, excluding myofibers with diameters of <10 and >150 μm and areas of <100 and >10,000 μm2. The Feret’s minimal diameter was used as the myofiber measurement size as this is a geometrical parameter for the analysis of unevenly shaped or cut objects.54 The analysis was performed by using the Nikon Elements software and the “Object Count” function.
Similar procedures were used for the analysis of tibialis anterior muscles. However, the following primary antibodies were used to detect the myofiber types that compose tibialis anterior muscles: antibodies against type 2a (SC-71) and 2b myosin heavy chain (BF-F3). The sections were then washed and incubated with secondary antibodies to detect primary antibodies for type 2a (anti-mouse AlexaFluor488-conjugated IgG1) and type 2b myosin heavy chain (anti-mouse AlexaFluor555-conjugated IgM). Myosin heavy chain immunostaining was used to classify type 2b myofibers (red), type 2a myofibers (green), and presumed type 2x myofibers (black) that were not stained for type 2a or 2b MHC isoforms.
For the analysis of the gastrocnemius, the primary antibodies used were mouse IgG2b anti-myosin heavy chain type 1 (DSHB, cat. no. BA-F8), mouse IgG1 anti-myosin heavy chain type 2a (DSHB, cat. no. SC-71), mouse IgM anti-myosin heavy chain type 2b (DSHB, cat. no. BF-F3), and rat anti-laminin α2 (4H8–2; Santa Cruz, cat. no. sc-59854). Staining for MHC type 1 was done separately (i.e. on different slides) from staining for MHC type 2a and 2b isoforms. Myosin heavy chain immunostaining was used to classify type 1 myofibers (pink), type 2a myofibers (green), type 2b myofibers (red), and presumed type 2x myofibers (black) that were not stained for type 1, 2a, or 2b.
For the quantification of the number of myofibers composing soleus, tibialis anterior, and gastrocnemius skeletal muscles, all myofibers in muscle cross-sections were counted based on the myofiber borders identified by laminin α2 immunostaining.
Western blot
The Western blot analysis of ACE1 levels was done by utilizing the KO-validated rabbit anti-ACE1 antibody (Abcam, cat. no. ab254222) and normalized by α-Tubulin levels (Cell Signaling Technologies, cat. no. 2125S), which were detected according to standard procedures.87,126
Lisinopril treatment
Lisinopril was administered from day 0 (i.e. from the day of cancer cell injection) in drinking water at 40 mg/L.95 The water was changed regularly as for the mock-treated mice.
Muscle force measurements
The measurement of the twitch and tetanic force of the tibialis anterior (TA) muscle85,126,127 was normalized by the TA mass. Mice were deeply anesthetized via isofluorane and monitored throughout the experiment. The distal tendon of the tibialis anterior was carefully dissected and individually tied with 4.0 braided surgical silk. The sciatic nerve was exposed and all branches were cut except for the common peroneal nerve. The foot was secured to a platform and the knee immobilized using a stainless-steel pin. The body temperature was monitored and maintained at 37°C. The suture from the tendon was individually attached to the lever arm of a 305B dual-mode servomotor transducer (Aurora Scientific, ON, Canada). Muscle contractions were then elicited by stimulating the distal part of the sciatic via bipolar electrodes, using supramaximal square-wave pulses of 0.2 msec (701A stimulator; Aurora Scientific). Data acquisition and control of the servomotor were conducted by using a Lab-View-based DMC program (version 5.202; Aurora Scientific). Optimal muscle length (Lo) was determined by incrementally stretching the muscle until the maximum isometric twitch force was achieved. The maximum isometric tetanic force (Po) was acquired by using a train of 150Hz supramaximal electrical pulses for 500 ms at the optimal length in the muscles and the highest Po was recorded. A 2-min resting period was allowed between each tetanic contraction. Lo was measured using digital calipers. The relative force (N/g) was normalized by dividing the force by the muscle weight.
Analysis of cardiomyocyte size
Unfixed slides with the transversally cross-sectioned heart were incubated for 1h with blocking buffer (PBS with 2% BSA and 0.1% Triton X-100) prior to overnight immunostaining at 4°C with rat anti-laminin α2 (4H8–2; Santa Cruz, cat. no. sc-59854) and AlexaFluor635-conjugated phalloidin (Invitrogen, cat. no. A22284). The slides were washed and incubated with anti-rat AlexaFluor488-conjugated IgG (Invitrogen, cat. no. A48262). DAPI (Sigma Aldrich, cat. no. 10-236-276-001) was used to identify nuclei.
For the analysis of cardiomyocyte size, the left ventricle of the heart was identified and imaged on the Nikon C2 confocal microscope with a 20X objective. Phalloidin was used to classify the individual cardiomyocytes (red), while the inverse threshold of laminin α2 was used to determine cardiomyocyte boundaries. Cardiomyocyte size was determined with the Nikon Elements software by using the intersection of inverse laminin and the phalloidin staining. Cardiomyocytes with an area of <100 and >1000 μm2 were excluded. The Feret’s minimal diameter was used to measure cardiomyocyte size and this analysis was performed by using the Nikon Elements software and the “Object Count” function.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data organization, scientific graphing, and statistical analyses were performed with Microsoft Excel (version 14.7.3) and GraphPad Prism (version 6). The unpaired two-tailed Student’s t-test was used to compare the means of two independent groups to each other. One-way ANOVA with post hoc testing was used for multiple comparisons of more than two groups of normally distributed data. Two-way ANOVA with post hoc testing was used for multiple comparisons of more than two groups of normally distributed data in presence of two independent variables. Data in Figure 6F was statistically analyzed with a Pearson correlation test. The n for each experiment can be found in the figures and represents independently generated samples from individual mice. Tissue weight graphs in Figure 2 display the mean ± SEM. In all other figures, bar graphs represent the mean ± SD. A significant result was defined as p < 0.05. Throughout the figures, asterisks and ampersand symbols indicate the significance of p values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical information of RNA-seq data is described in the RNA-seq method section.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse IgG2b anti-myosin heavy chain type 1 | DSHB | BA-F8; RRID: AB_10572253 |
| Mouse IgG1 anti-Myosin heavy chain type 2a | DSHB | SC-71; RRID:AB_2147165 |
| Mouse IgM anti-Myosin heavy chain type 2b | DSHB | BF-F3; RRID:AB_2266724 |
| Rat anti-laminin α-2 (4H8-2) | Santa Cruz | Sc-59854; RRID:AB_784266 |
| Rabbit anti-ACE1 (KO-validated) | Abcam | ab254222 |
| Rabbit anti-α-Tubulin | Cell Signaling Technologies | 2125S; RRID:AB_2619646 |
| Anti-rabbit IgG, HRP-linked | Cell Signaling Technologies | 7074S; RRID:AB_2099233 |
| Alexa Fluor 488 anti-mouse IgG1 | Life Technologies | A21121; RRID:AB_2535764 |
| Alexa Fluor 555 anti-mouse IgM | Life Technologies | A21426; RRID:AB_2535847 |
| Alexa Fluor 647 anti-rat | Life Technologies | A21247; RRID:AB_141778 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Alexa Fluor 635 Phalloidin | Life Technologies | A22284 |
| DAPI | Roche | 10236276001 |
| TRIzol | Ambion | 15596018 |
| 6-well plates | Corning | REF3516 |
| Transparent 96-well plates | Corning | REF3599 |
| High glucose DMEM, with glutamax | Gibco | 10566016 |
| Fetal bovine serum | Gibco | 10437-028 |
| Penicillin streptomycin | Gibco | 15140122 |
| PBS | Gibco | 10010023 |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| Isopentane | Sigma-Aldrich | 277258 |
| Matrigel | Corning | 354234 |
| Qbrelis (Lisinopril), oral solution, 1 mg/mL | Azurity pharmaceuticals | NDC52652-3001-1 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data of tissues and organs from mice implanted with cachexia-inducing and non-inducing melanoma xenografts and controls, at different timepoints of cachexia progression | This study | Gene Expression Omnibus, GEO: GSE183613 |
| RNA-seq data of brain regions from mice implanted with LLC cancer cells and controls | This study | Gene Expression Omnibus, GEO: GSE183613 |
| RNA-seq data of muscles from mice implanted with cachexia-inducing melanoma xenografts and controls, and either treated with Lisinopril or mock | This study | Gene Expression Omnibus, GEO: GSE183613 and GEO: GSE214981 |
| Microarray data of muscle biopsies from cancer patients with cachexia | (Martinelli et al.; Johns et al.)88,89 | Gene Expression Omnibus, GEO: GSE41726 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: LLC cells | ATCC | CRL-1642 |
| Human: melanoma xenograft MAST360B/SJMEL030083_X2 | Childhood Solid Tumor Network collection at St. Jude Children’s Research Hospital |
N/A |
| Human: melanoma xenograft MAST552A/SJMEL031086_X3 | Childhood Solid Tumor Network collection at St. Jude Children’s Research Hospital |
N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX000664 |
| Mouse: NCI Ath/nude | Charles River Laboratory | 553NCIATH/NU |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | Graphpad | https://www.graphpad.com/ |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software |
| Photoshop CSX | Adobe | https://www.adobe.com/products/photoshop.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Dynamic Muscle Control LabBook 610A | Aurora Scientific | https://aurorascientific.com/ |
Highlights.
Cancer cachexia induces prevalent, as well as temporal and tissue-specific, changes
Cachexia causes dysfunction also in tissues that do not atrophy, such as the brain
Secreted factors are a top category of cancer-regulated genes in host tissues
Inhibition of the secreted factor ACE impedes cachexia-induced muscle weakness
ACKNOWLEDGMENTS
We thank the DSHB for antibodies and the Light Microscopy facility, the Pathology Core, and the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital. We also thank Yong Ha Youn and Young-Goo Han for help in the dissection of brain regions, and the Childhood Solid Tumor Network, Åsa Karlström, Brittney Gordon, and the CIVIT for assistance with xenograft experiments. This work was supported primarily by a research grant to F.D. from The Hartwell Foundation and by the NCI (P30CA021765, developmental funds from St. Jude Comprehensive Cancer Center). F.D. is also supported by the National Institute on Aging (R01AG055532 and R21AG079267) and the Alzheimer’s Association (AARG-NTF-22-973220). M.L. is supported by the NCI (R01CA245301). Research at St. Jude Children’s Research Hospital is supported by the ALSAC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111934.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Argilés JM, Busquets S, Stemmler B, and López-Soriano FJ (2014). Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–762. 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 2.Tsoli M, and Robertson G (2013). Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 24, 174–183. 10.1016/j.tem.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 4.Baracos VE, Martin L, Korc M, Guttridge DC, and Fearon KCH (2018). Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 17105. 10.1038/nrdp.2017.105. [DOI] [PubMed] [Google Scholar]
- 5.Fearon KCH, Glass DJ, and Guttridge DC (2012). Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166. 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo G, et al. (2015). Targeting of Fn14 prevents cancer-induced cachexia and prolongs survival. Cell 162, 1365–1378. 10.1016/j.cell.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543. 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, Hollingsworth MA, Jain R, Tanji K, López-Pintado S, et al. (2018). Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 24, 770–781. 10.1038/s41591-018-0054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts BM, Ahn B, Smuder AJ, Al-Rajhi M, Gill LC, Beharry AW, Powers SK, Fuller DD, Ferreira LF, and Judge AR (2013). Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J 27, 2600–2610. 10.1096/fj.12-222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biswas AK, and Acharyya S (2020). Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 20, 274–284. 10.1038/s41568-020-0251-4. [DOI] [PubMed] [Google Scholar]
- 11.NCI. Cancer Cachexia: After Years of No Advances, Progress Looks Possible. https://www.cancer.gov/about-cancer/treatment/research/cachexia.
- 12.Pillon NJ, Bilan PJ, Fink LN, and Klip A (2013). Cross-talk between skeletal muscle and immune cells: muscle-derived mediators and metabolic implications. Am. J. Physiol. Endocrinol. Metab. 304, E453–E465. 10.1152/ajpendo.00553.2012. [DOI] [PubMed] [Google Scholar]
- 13.VanderVeen BN, Murphy EA, and Carson JA (2020). The impact of immune cells on the skeletal muscle microenvironment during cancer cachexia. Front. Physiol. 11, 1037. 10.3389/fphys.2020.01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Lerma Barbaro A (2015). The complex liaison between cachexia and tumor burden (Review). Oncol. Rep. 34, 1635–1649. 10.3892/or.2015.4164. [DOI] [PubMed] [Google Scholar]
- 15.Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, Gerber MH, George TJ Jr., Behrns KE, Hughes SJ, et al. (2017). Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 8, 1177–1189. 10.18632/oncotarget.13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talbert EE, Cuitiño MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, Leone GW, Ostrowski MC, Paleo B, Weisleder N, et al. (2019). Modeling human cancer-induced cachexia. Cell Rep. 28, 1612–1622.e4. 10.1016/j.celrep.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson B, Zhu X, Norgard MA, Levasseur PR, Butler JT, Buenafe A, Burfeind KG, Michaelis KA, Pelz KR, Mendez H, et al. (2021). Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat. Commun. 12, 2057. 10.1038/s41467-021-22361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohm M, Zeigerer A, Machado J, and Herzig S (2019). Energy metabolism in cachexia. EMBO Rep. 20, e47258. 10.15252/embr.201847258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molfino A, Gioia G, Rossi Fanelli F, and Laviano A (2015). Contribution of neuroinflammation to the pathogenesis of cancer cachexia. Mediators Inflamm. 2015, 801685. 10.1155/2015/801685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart E, Federico S, Karlstrom A, Shelat A, Sablauer A, Pappo A, and Dyer MA (2016). The Childhood Solid Tumor Network: a new resource for the developmental biology and oncology research communities. Dev. Biol. 411, 287–293. 10.1016/j.ydbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, Karlstrom A, Twarog NR, Clay MR, Bahrami A, et al. (2017). Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 549, 96–100. 10.1038/nature23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graca FA, Rai M, Hunt LC, Stephan A, Wang Y-D, Gordon B, Wang R, Quarato G, Xu B, Fan Y, et al. (2022). The myokine Fibcd1 is an endogenous determinant of myofiber size and mitigates cancer-induced myofiber atrophy. Nat. Commun. 13, 2370. 10.1038/s41467-022-30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petruzzelli M, and Wagner EF (2016). Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 30, 489–501. 10.1101/gad.276733.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uhlén M, Karlsson MJ, Hober A, Svensson AS, Scheffel J, Kotol D, Zhong W, Tebani A, Strandberg L, Edfors F, et al. (2019). The human secretome. Sci. Signal. 12, eaaz0274. 10.1126/scisignal.aaz0274. [DOI] [PubMed] [Google Scholar]
- 26.Hou YC, Wang CJ, Chao YJ, Chen HY, Wang HC, Tung HL, Lin JT, and Shan YS (2018). Elevated serum interleukin-8 level correlates with cancer-related cachexia and sarcopenia: an indicator for pancreatic cancer outcomes. J. Clin. Med. 7, 502. 10.3390/jcm7120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Callaway CS, Delitto AE, Patel R, Nosacka RL, D’Lugos AC, Delitto D, Deyhle MR, Trevino JG, Judge SM, and Judge AR (2019). IL-8 released from human pancreatic cancer and tumor-associated stromal cells signals through a CXCR2-ERK1/2 Axis to induce muscle atrophy. Cancers 11, 1863. 10.3390/cancers11121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cury SS, de Moraes D, Freire PP, de Oliveira G, Marques DVP, Fernandez GJ, Dal-Pai-Silva M, Hasimoto ÉN, Dos Reis PP, Rogatto SR, and Carvalho RF (2019). Tumor transcriptome reveals high expression of IL-8 in non-small cell lung cancer patients with low pectoralis muscle area and reduced survival. Cancers 11, 1251. 10.3390/cancers11091251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora GK, Gupta A, Narayanan S, Guo T, Iyengar P, and Infante RE (2018). Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. JCI Insight 3, e121221. 10.1172/jci.insight.121221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kandarian SC, Nosacka RL, Delitto AE, Judge AR, Judge SM, Ganey JD, Moreira JD, and Jackman RW (2018). Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 1109–1120. 10.1002/jcsm.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seto DN, Kandarian SC, and Jackman RW (2015). A key role for leukemia inhibitory factor in C26 cancer cachexia. J. Biol. Chem. 290, 19976–19986. 10.1074/jbc.M115.638411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren S, Chai L, Wang C, Li C, Ren Q, Yang L, Wang F, Qiao Z, Li W, He M, et al. (2015). Human malignant melanoma-derived progestagen-associated endometrial protein immunosuppresses T lymphocytes in vitro. PLoS One 10, e0119038. 10.1371/journal.pone.0119038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren S, Howell PM Jr., Han Y, Wang J, Liu M, Wang Y, Quan G, Du W, Fang L, and Riker AI (2011). Overexpression of the progestagen-associated endometrial protein gene is associated with microphthalmia-associated transcription factor in human melanoma. Ochsner J. 11, 212–219. [PMC free article] [PubMed] [Google Scholar]
- 34.Kordaß T, Osen W, and Eichmüller SB (2018). Controlling the immune suppressor: transcription factors and MicroRNAs regulating CD73/NT5E. Front. Immunol. 9, 813. 10.3389/fimmu.2018.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruan W, Kang Z, Li Y, Sun T, Wang L, Liang L, Lai M, and Wu T (2016). Interaction between IGFBP7 and insulin: a theoretical and experimental study. Sci. Rep. 6, 19586. 10.1038/srep19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kasprzak A (2021). The role of tumor microenvironment cells in colorectal cancer (CRC) cachexia. Int. J. Mol. Sci. 22, 1565. 10.3390/ijms22041565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metivier F, Marchais SJ, Guerin AP, Pannier B, and London GM (2000). Pathophysiology of anaemia: focus on the heart and blood vessels. Nephrol. Dial. Transplant. 15, 14–18. 10.1093/oxfordjournals.ndt.a027970. [DOI] [PubMed] [Google Scholar]
- 38.Naito Y, Tsujino T, Matsumoto M, Sakoda T, Ohyanagi M, and Masuyama T (2009). Adaptive response of the heart to long-term anemia induced by iron deficiency. Am. J. Physiol. Heart Circ. Physiol. 296, H585–H593. 10.1152/ajpheart.00463.2008. [DOI] [PubMed] [Google Scholar]
- 39.Wyart E, Hsu MY, Sartori R, Mina E, Rausch V, Pierobon ES, Mezzanotte M, Pezzini C, Bindels LB, Lauria A, et al. (2022). Iron supplementation is sufficient to rescue skeletal muscle mass and function in cancer cachexia. EMBO Rep. 23, e53746. 10.15252/embr.202153746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarz S, Prokopchuk O, Esefeld K, Gröschel S, Bachmann J, Lorenzen S, Friess H, Halle M, and Martignoni ME (2017). The clinical picture of cachexia: a mosaic of different parameters (experience of 503 patients). BMC Cancer 17, 130. 10.1186/s12885-017-3116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ober EA, and Lemaigre FP (2018). Development of the liver: insights into organ and tissue morphogenesis. J. Hepatol. 68, 1049–1062. 10.1016/j.jhep.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Tsoli M, Moore M, Burg D, Painter A, Taylor R, Lockie SH, Turner N, Warren A, Cooney G, Oldfield B, et al. (2012). Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. 72, 4372–4382. 10.1158/0008-5472.CAN-11-3536. [DOI] [PubMed] [Google Scholar]
- 43.Anker MS, Sanz AP, Zamorano JL, Mehra MR, Butler J, Riess H, Coats AJS, and Anker SD (2021). Advanced cancer is also a heart failure syndrome: a hypothesis. J. Cachexia Sarcopenia Muscle 12, 533–537. 10.1002/jcsm.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barkhudaryan A, Scherbakov N, Springer J, and Doehner W (2017). Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail. 4, 458–467. 10.1002/ehf2.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belloum Y, Rannou-Bekono F, and Favier FB (2017). Cancer-induced cardiac cachexia: pathogenesis and impact of physical activity (Review). Oncol. Rep. 37, 2543–2552. 10.3892/or.2017.5542. [DOI] [PubMed] [Google Scholar]
- 46.Murphy KT (2016). The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 310, H466–H477. 10.1152/ajpheart.00720.2015. [DOI] [PubMed] [Google Scholar]
- 47.Rausch V, Sala V, Penna F, Porporato PE, and Ghigo A (2021). Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 10, 1. 10.1038/s41389-020-00288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, Kaschina E, Palus S, Pötsch M, von Websky K, et al. (2014). Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur. Heart J. 35, 932–941. 10.1093/eurheartj/eht302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonaldo P, and Sandri M (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6, 25–39. 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piccirillo R, Demontis F, Perrimon N, and Goldberg AL (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 243, 201–215. 10.1002/dvdy.24036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demontis F, Piccirillo R, Goldberg AL, and Perrimon N (2013). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis. Model. Mech. 6, 1339–1352. 10.1242/dmm.012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciciliot S, Rossi AC, Dyar KA, Blaauw B, and Schiaffino S (2013). Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 45, 2191–2199. 10.1016/j.biocel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Schiaffino S, and Reggiani C (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531. 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 54.Bloemberg D, and Quadrilatero J (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7, e35273. 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narsale AA, Enos RT, Puppa MJ, Chatterjee S, Murphy EA, Fayad R, Pena MO, Durstine JL, and Carson JA (2015). Liver inflammation and metabolic signaling in ApcMin/+ mice: the role of cachexia progression. PLoS One 10, e0119888. 10.1371/journal.pone.0119888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones A, Friedrich K, Rohm M, Schäfer M, Algire C, Kulozik P, Seibert O, Müller-Decker K, Sijmonsma T, Strzoda D, et al. (2013). TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol. Med. 5, 294–308. 10.1002/emmm.201201869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bissay V, Van Malderen SCH, Keymolen K, Lissens W, Peeters U, Daneels D, Jansen AC, Pappaert G, Brugada P, De Keyser J, and Van Dooren S (2016). SCN4A variants and Brugada syndrome: phenotypic and genotypic overlap between cardiac and skeletal muscle sodium channelopathies. Eur. J. Hum. Genet. 24, 400–407. 10.1038/ejhg.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider T, Dibue-Adjei M, Neumaier F, Akhtar I, Hescheler J, Kamp MA, and Tevoufouet EE (2015). R-type voltage-gated Ca(2)(+) channels in cardiac and neuronal rhythmogenesis. Curr. Mol. Pharmacol. 8, 102–108. 10.2174/1874467208666150507093845. [DOI] [PubMed] [Google Scholar]
- 59.Isackson PJ, Wang J, Zia M, Spurgeon P, Levesque A, Bard J, James S, Nowak N, Lee TK, and Vladutiu GD (2018). RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 19, 1235–1249. 10.2217/pgs-2018-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rivolta I, Binda A, Masi A, and DiFrancesco JC (2020). Cardiac and neuronal HCN channelopathies. Pflugers Arch. 472, 931–951. 10.1007/s00424-020-02384-3. [DOI] [PubMed] [Google Scholar]
- 61.Whitaker GM, Angoli D, Nazzari H, Shigemoto R, and Accili EA (2007). HCN2 and HCN4 isoforms self-assemble and co-assemble with equal preference to form functional pacemaker channels. J. Biol. Chem. 282, 22900–22909. 10.1074/jbc.M610978200. [DOI] [PubMed] [Google Scholar]
- 62.Damar IH, and Eroz R (2019). Evaluation of cases with myotonia congenita for cardiovascular risk. Medeni. Med. J. 34, 374–379. 10.5222/MMJ.2019.93357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Imbrici P, Altamura C, Pessia M, Mantegazza R, Desaphy JF, and Camerino DC (2015). ClC-1 chloride channels: state-of-the-art research and future challenges. Front. Cell. Neurosci. 9, 156. 10.3389/fncel.2015.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, and Goldberg AL (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483. [DOI] [PubMed] [Google Scholar]
- 65.Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, Tesfahun AN, Moestue S, Kim J, Nonstad U, et al. (2017). Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 7, 2046. 10.1038/s41598-017-02088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bodine SC, and Baehr LM (2014). Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 307, E469–E484. 10.1152/ajpendo.00204.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dantzer R, O’Connor JC, Freund GG, Johnson RW, and Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, and Medeiros R (2018). Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 15, 276. 10.1186/s12974-018-1313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schrepf A, Kaplan CM, Ichesco E, Larkin T, Harte SE, Harris RE, Murray AD, Waiter GD, Clauw DJ, and Basu N (2018). A multi-modal MRI study of the central response to inflammation in rheumatoid arthritis. Nat. Commun. 9, 2243. 10.1038/s41467-018-04648-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chitnis T, and Weiner HL (2017). CNS inflammation and neurodegeneration. J. Clin. Invest. 127, 3577–3587. 10.1172/JCI90609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winnard PT Jr., Bharti SK, Sharma RK, Krishnamachary B, Mironchik Y, Penet MF, Goggins MG, Maitra A, Kamel I, Horton KM, et al. (2020). Brain metabolites in cholinergic and glutamatergic pathways are altered by pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 11, 1487–1500. 10.1002/jcsm.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vicente R, Escalada A, Coma M, Fuster G, Sánchez-Tilló E, López-Iglesias C, Soler C, Solsona C, Celada A, and Felipe A (2003). Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J. Biol. Chem. 278, 46307–46320. 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- 73.Burfeind KG, Zhu X, Norgard MA, Levasseur PR, Huisman C, Buenafe AC, Olson B, Michaelis KA, Torres ER, Jeng S, et al. (2020). Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. Elife 9, e54095. 10.7554/eLife.54095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inui A (1999). Cancer anorexia-cachexia syndrome: are neuropeptides the key? Cancer Res. 59, 4493–4501. [PubMed] [Google Scholar]
- 75.Inui A (2002). Cancer anorexia-cachexia syndrome: current issues in research and management. CA. Cancer J. Clin. 52, 72–91. 10.3322/canjclin.52.2.72. [DOI] [PubMed] [Google Scholar]
- 76.Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, et al. (2007). TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 4, e158. 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hunt LC, Xu B, Finkelstein D, Fan Y, Carroll PA, Cheng PF, Eisenman RN, and Demontis F (2015). The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 29, 2475–2489. 10.1101/gad.267419.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, et al. (2013). AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 49, 1167–1175. 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alsina FC, Hita FJ, Fontanet PA, Irala D, Hedman H, Ledda F, and Paratcha G (2016). Lrig1 is a cell-intrinsic modulator of hippocampal dendrite complexity and BDNF signaling. EMBO Rep. 17, 601–616. 10.15252/embr.201541218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin J, Sun H, Liu D, Wang H, Liu Q, Chen H, Zhong D, and Li G (2019). LRG1 promotes apoptosis and autophagy through the TGFbetasmad1/5 signaling pathway to exacerbate ischemia/reperfusion injury. Neuroscience 413, 123–134. 10.1016/j.neuroscience.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 81.Cartier J, Piyasena C, Sparrow SA, Boardman JP, and Drake AJ (2018). Alterations in glucose concentrations affect DNA methylation at Lrg1 in an ex vivo rat cortical slice model of preterm brain injury. Eur. J. Neurosci. 47, 380–387. 10.1111/ejn.13825. [DOI] [PubMed] [Google Scholar]
- 82.Shah K, Desilva S, and Abbruscato T (2012). The role of glucose transporters in brain disease: diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 13, 12629–12655. 10.3390/ijms131012629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niccoli T, Cabecinha M, Tillmann A, Kerr F, Wong CT, Cardenes D, Vincent AJ, Bettedi L, Li L, Grönke S, et al. (2016). Increased glucose transport into neurons rescues abeta toxicity in Drosophila. Curr. Biol. 26, 2550. 10.1016/j.cub.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, and Spiegelman BM (2014). Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513, 100–104. 10.1038/nature13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunt LC, Stover J, Haugen B, Shaw TI, Li Y, Pagala VR, Finkelstein D, Barton ER, Fan Y, Labelle M, et al. (2019). A key role for the ubiquitin ligase UBR4 in myofiber hypertrophy in Drosophila and mice. Cell Rep. 28, 1268–1281.e6. 10.1016/j.celrep.2019.06.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Puppa MJ, Gao S, Narsale AA, and Carson JA (2014). Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009. 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hunt LC, Graca FA, Pagala V, Wang YD, Li Y, Yuan ZF, Fan Y, Labelle M, Peng J, and Demontis F (2021). Integrated genomic and proteomic analyses identify stimulus-dependent molecular changes associated with distinct modes of skeletal muscle atrophy. Cell Rep. 37, 109971. 10.1016/j.celrep.2021.109971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martinelli GB, Olivari D, Re Cecconi AD, Talamini L, Ottoboni L, Lecker SH, Stretch C, Baracos VE, Bathe OF, Resovi A, et al. (2016). Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 35, 6212–6222. 10.1038/onc.2016.153. [DOI] [PubMed] [Google Scholar]
- 89.Johns N, Stretch C, Tan BHL, Solheim TS, Sørhaug S, Stephens NA, Gioulbasanis I, Skipworth RJE, Deans DAC, Vigano A, et al. (2017). New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J. Cachexia Sarcopenia Muscle 8, 122–130. 10.1002/jcsm.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, and Bader M (2019). The renin-angiotensin system: going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 316, H958–H970. 10.1152/ajpheart.00723.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi L, Mao C, Xu Z, and Zhang L (2010). Angiotensin-converting enzymes and drug discovery in cardiovascular diseases. Drug Discov. Today 15, 332–341. 10.1016/j.drudis.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Williams AG, Rayson MP, Jubb M, World M, Woods DR, Hayward M, Martin J, Humphries SE, and Montgomery HE (2000). The ACE gene and muscle performance. Nature 403, 614. 10.1038/35001141. [DOI] [PubMed] [Google Scholar]
- 93.Woods DR, Humphries SE, and Montgomery HE (2000). The ACE I/D polymorphism and human physical performance. Trends Endocrinol. Metab. 11, 416–420. 10.1016/s1043-2760(00)00310-6. [DOI] [PubMed] [Google Scholar]
- 94.Schanze N, and Springer J (2012). Evidence for an effect of ACE inhibitors on cancer cachexia. J. Cachexia Sarcopenia Muscle 3, 139. 10.1007/s13539-012-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao DY, Giani JF, Veiras LC, Bernstein EA, Okwan-Duodu D, Ahmed F, Bresee C, Tourtellotte WG, Karumanchi SA, Bernstein KE, and Khan Z (2021). An ACE inhibitor reduces bactericidal activity of human neutrophils in vitro and impairs mouse neutrophil activity in vivo. Sci. Transl. Med. 13, eabj2138. 10.1126/scitranslmed.abj2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Enoka RM, and Duchateau J (2017). Rate coding and the control of muscle force. Cold Spring Harb. Perspect. Med. 7, a029702. 10.1101/cshperspect.a029702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sweeney HL, and Hammers DW (2018). Muscle contraction. Cold Spring Harb. Perspect. Biol. 10, a023200. 10.1101/cshperspect.a023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamada T, Ashida Y, Tatebayashi D, Abe M, and Himori K (2020). Cancer cachexia induces preferential skeletal muscle myosin loss when combined with denervation. Front. Physiol. 11, 445. 10.3389/fphys.2020.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shum AMY, Poljak A, Bentley NL, Turner N, Tan TC, and Polly P (2018). Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: alterations in sarcomeric and mitochondrial protein expression. Oncotarget 9, 22001–22022. 10.18632/oncotarget.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, and Spiegelman BM (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 103, 16260–16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, and Moraes CT (2009). Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 106, 20405–20410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, and Spiegelman BM (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 103.Niccoli T, Cabecinha M, Tillmann A, Kerr F, Wong CT, Cardenes D, Vincent AJ, Bettedi L, Li L, Grönke S, et al. (2016). Increased glucose transport into neurons rescues abeta toxicity in Drosophila. Curr. Biol. 26, 2291–2300. 10.1016/j.cub.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, and Spiegelman BM (2016). PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 23, 315–323. 10.1016/j.cmet.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anker SD, and Sharma R (2002). The syndrome of cardiac cachexia. Int. J. Cardiol. 85, 51–66. 10.1016/s0167-5273(02)00233-4. [DOI] [PubMed] [Google Scholar]
- 106.Loncar G, Springer J, Anker M, Doehner W, and Lainscak M (2016). Cardiac cachexia: hic et nunc. J. Cachexia Sarcopenia Muscle 7, 246–260. 10.1002/jcsm.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lena A, Ebner N, and Anker MS (2019). Cardiac cachexia. Eur. Heart J. Suppl. 21, L24–L27. 10.1093/eurheartj/suz241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brown JL, Lawrence MM, Ahn B, Kneis P, Piekarz KM, Qaisar R, Ranjit R, Bian J, Pharaoh G, Brown C, et al. (2020). Cancer cachexia in a mouse model of oxidative stress. J. Cachexia Sarcopenia Muscle 11, 1688–1704. 10.1002/jcsm.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sartori R, Hagg A, Zampieri S, Armani A, Winbanks CE, Viana LR, Haidar M, Watt KI, Qian H, Pezzini C, et al. (2021). Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci. Transl. Med. 13, eaay9592. 10.1126/scitranslmed.aay9592. [DOI] [PubMed] [Google Scholar]
- 110.Daou N, Hassani M, Matos E, De Castro GS, Costa RGF, Seelaender M, Moresi V, Rocchi M, Adamo S, Li Z, et al. (2020). Displaced myonuclei in cancer cachexia suggest altered innervation. Int. J. Mol. Sci. 21, 1092. 10.3390/ijms21031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huot JR, Pin F, and Bonetto A (2021). Muscle weakness caused by cancer and chemotherapy is associated with loss of motor unit connectivity. Am. J. Cancer Res. 11, 2990–3001. [PMC free article] [PubMed] [Google Scholar]
- 112.Stevens SCW, Velten M, Youtz DJ, Clark Y, Jing R, Reiser PJ, Bicer S, Devine RD, McCarthy DO, and Wold LE (2015). Losartan treatment attenuates tumor-induced myocardial dysfunction. J. Mol. Cell. Cardiol. 85, 37–47. 10.1016/j.yjmcc.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sanders PM, Russell ST, and Tisdale MJ (2005). Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 93, 425–434. 10.1038/sj.bjc.6602725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murphy KT, Chee A, Trieu J, Naim T, and Lynch GS (2013). Inhibition of the renin-angiotensin system improves physiological outcomes in mice with mild or severe cancer cachexia. Int. J. Cancer 133, 1234–1246. 10.1002/ijc.28128. [DOI] [PubMed] [Google Scholar]
- 115.Baazim H, Schweiger M, Moschinger M, Xu H, Scherer T, Popa A, Gallage S, Ali A, Khamina K, Kosack L, et al. (2019). CD8(+) T cells induce cachexia during chronic viral infection. Nat. Immunol. 20, 701–710. 10.1038/s41590-019-0397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Z, Zhao C, Moya R, and Davies JD (2008). A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 181, 4676–4684. 10.4049/jimmunol.181.7.4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bahrami A, and Barnhill RL (2018). Pathology and genomics of pediatric melanoma: a critical reexamination and new insights. Pediatr. Blood Cancer 65, e26792. 10.1002/pbc.26792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Newman S, Fan L, Pribnow A, Silkov A, Rice SV, Lee S, Shao Y, Shaner B, Mulder H, Nakitandwe J, et al. (2019). Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat. Med. 25, 597–602. 10.1038/s41591-019-0373-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Newman S, Pappo A, Raimondi S, Zhang J, Barnhill R, and Bahrami A (2019). Pathologic characteristics of spitz melanoma with MAP3K8 fusion or truncation in a pediatric cohort. Am. J. Surg. Pathol. 43, 1631–1637. 10.1097/PAS.0000000000001362. [DOI] [PubMed] [Google Scholar]
- 120.Rai M, Curley M, Coleman Z, Nityanandam A, Jiao J, Graca FA, Hunt LC, and Demontis F (2021). Analysis of proteostasis during aging with western blot of detergent-soluble and insoluble protein fractions. STAR Protoc. 2, 100628. 10.1016/j.xpro.2021.100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rai M, Coleman Z, Curley M, Nityanandam A, Platt A, Robles-Murguia M, Jiao J, Finkelstein D, Wang YD, Xu B, et al. (2021). Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging. Cell Metab. 33, 1137–1154.e9. 10.1016/j.cmet.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, Zhou X, Jeon J, Griffiths L, Nguyen R, et al. (2020). MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 11, 913. 10.1038/s41467-020-14682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fazeli E, Roy NH, Follain G, Laine RF, von Chamier L, Hänninen PE, Eriksson JE, Tinevez JY, and Jacquemet G (2020). Automated cell tracking using StarDist and TrackMate. F1000Res. 9, 1279. 10.12688/f1000research.27019.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schmidt U, Weigert M, Broaddus C, and Myers G (2018). Cell detection with star-convex polygons. In International Conference on Medical Image Computing and Computer-Assisted Intervention (Springer), pp. 265–273. [Google Scholar]
- 125.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hunt LC, Schadeberg B, Stover J, Haugen B, Pagala V, Wang YD, Puglise J, Barton ER, Peng J, and Demontis F (2021). Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat. Commun. 12, 1418. 10.1038/s41467-021-21738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu M, Hammers DW, Barton ER, and Sweeney HL (2016). Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS One 11, e0166803. 10.1371/journal.pone.0166803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper, the supplemental information, and the Data S1, which reports the source data. The 747 RNA-seq samples reported in this publication have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GEO: GSE183613 and GEO: GSE214981.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.