

Abstract
Fibrosis affects millions of people with cardiac disease. We developed a therapeutic approach to generate transient anti-fibrotic chimeric antigen receptor (CAR) T cells in vivo by delivering modified mRNA in T cell targeted lipid nanoparticles. The efficacy of these in vivo reprogrammed CAR T cells was evaluated by injecting CD5-targeted lipid nanoparticles into a mouse model of heart failure. Efficient delivery of modified mRNA encoding the CAR to T lymphocytes was observed, which produced transient, effective CAR T cells in vivo. Anti-fibrotic CAR T cells exhibited trogocytosis and retained the target antigen as they accumulated in the spleen. Treatment with modified mRNA targeted lipid nanoparticles reduced fibrosis and restored cardiac function after injury. In vivo generation of CAR T cells holds promise as a therapeutic platform to treat various diseases.
One Sentence Summary:
CD5-targeted lipid nanoparticles deliver therapeutic mRNA to lymphocytes in vivo where they form transient anti-fibrotic chimeric antigen receptor T cells which significantly improve cardiac function in a mouse model of heart failure.
Cardiac fibroblasts become activated in response to various myocardial injuries through well-studied mechanisms including TGFβ-SMAD2/3, interleukin-11 and other interactions with the immune system (1–6). In many chronic heart diseases, these fibroblasts fail to quiesce and secrete excessive extracellular matrix resulting in fibrosis (7). Fibrosis both stiffens the myocardium and negatively impacts cardiomyocyte health and function (8). Despite in-depth understanding of activated cardiac fibroblasts, clinical trials of anti-fibrotic therapeutics have only demonstrated a modest effect (5, 7) at best. Furthermore, these interventions aim to limit fibrotic progression and are not designed to remodel fibrosis once it is established. To address this significant clinical problem, we recently demonstrated the use of chimeric antigen receptor (CAR) T cells to specifically eliminate activated fibroblasts as a therapy for heart failure (9). Elimination of activated fibroblasts in a mouse model of heart disease resulted in a significant reduction of cardiac fibrosis and improved cardiac function (9). One caveat of that work is the indefinite persistence of engineered T cells similar to CAR T cell therapy currently used in the oncology clinic (10). Fibroblast activation is part of a normal wound-healing process in many tissues and persistent anti-fibrosis CAR T cells could pose a risk in the setting of future injuries. Therefore, we leveraged the power of nucleoside-modified mRNA technology to develop a transient anti-fibrotic CAR T therapeutic.
Therapeutic messenger RNAs can be stabilized through incorporation of modified nucleosides, synthetic capping, the addition of lengthy poly-A tails and enhanced with codon optimization (11–13). 1-Methylpseudouridine integration also boosts translation (13, 14). Direct introduction of mRNA into T cells ex vivo by electroporation has been used successfully by our group and others to make CAR T cells (15); however, this process carries significant cost and risk and requires extensive infrastructure. Thus, we developed an approach that could be used to avoid removing T cells from the patient and by packaging modified mRNAs in lipid nanoparticles (LNP) capable of producing CAR T cells in vivo after injection. LNP-mRNA technology underlies recent successes in COVID-19 vaccine development and holds exceptional promise for additional therapeutic strategies (16–20). Once in the body, mRNA-loaded LNP, absent of any specific targeting strategies, are endocytosed by various cell types (especially hepatocytes if injected intravenously) (21, 22). Shortly after cellular uptake, the mRNA escapes the endosome, releasing the mRNA into the cytoplasm where it is transiently transcribed before degrading (11). Targeting antibodies can be decorated on the surface of the LNP in order to direct uptake (and mRNA expression) to specific cell types (23, 24). We hypothesized that an LNP directed to T lymphocytes could deliver sufficient mRNAs to produce functional CAR T cells in vivo (Fig. 1A). Since mRNA is restricted to the cytoplasm, incapable of genomic integration, intrinsically unstable, and diluted during cell division, these CAR T cells will be, by design, transient.
Fig. 1. CD5-targeted lipid nanoparticles produce functional, mRNA-based FAPCAR T cells in vitro.
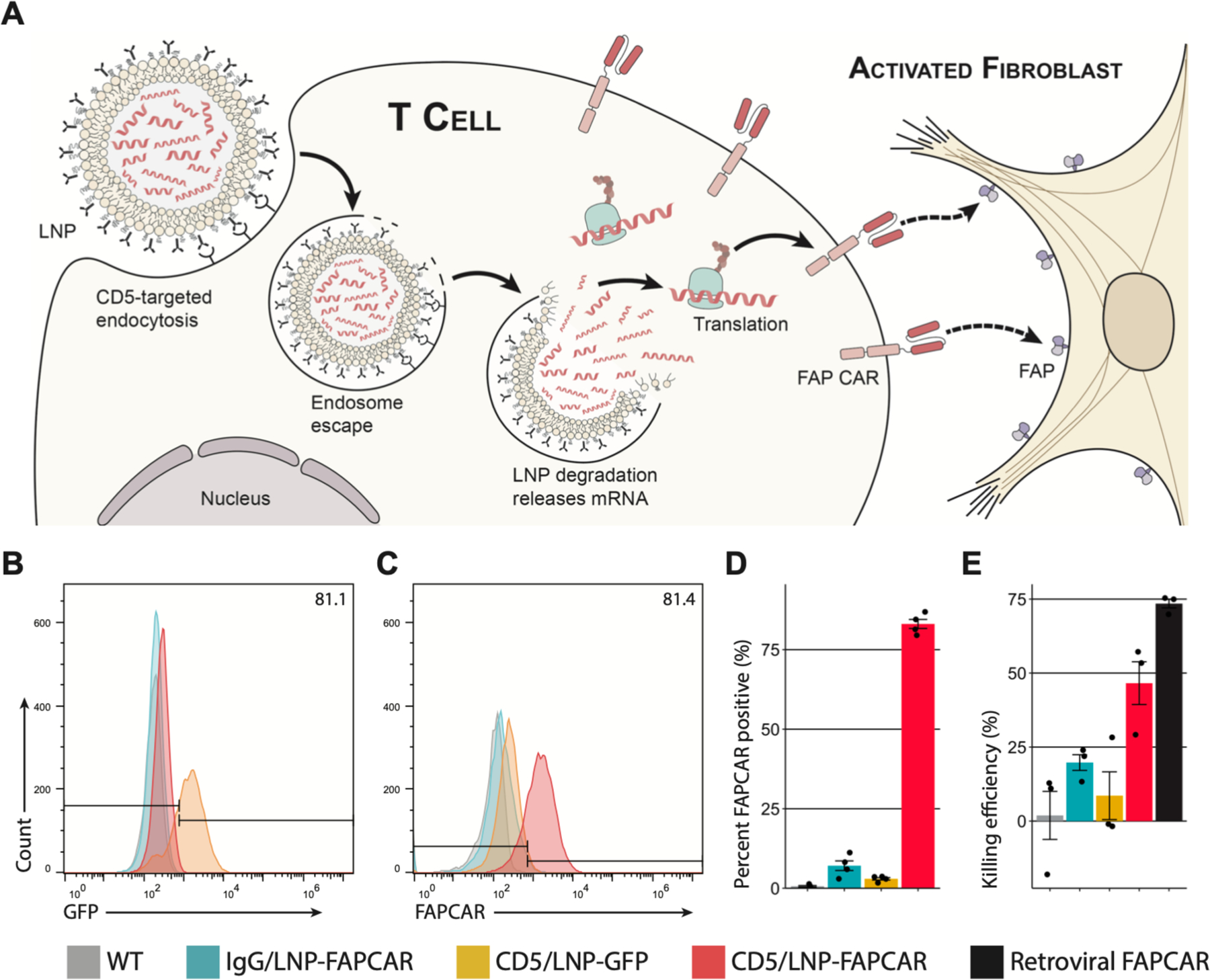
(A) Schematic outlining the molecular process to create transient FAPCAR T cells using CD5-targeted LNP. Representative flow cytometry analysis of (B) GFP and (C) FAPCAR expression in murine T cells 48 hours after incubation with either IgG/LNP-FAPCAR, CD5/LNP-GFP, or CD5/LNP-FAPCAR. (D) Quantification of murine T cells (percent) staining positive for FAPCAR from biologically independent replicates (n = 4). (E) FAPCAR T cells were mixed with FAP-expressing target HEK293T cells overnight and assayed for killing efficiency in biologically independent replicates (n = 3). Data are mean +/− s.e.m.
We generated modified nucleoside-containing mRNA encoding a CAR designed against fibroblast activation protein (FAP) (a marker of activated fibroblasts) and packaged it in CD5-targeted LNP (referred to as “targeting antibody/LNP-mRNA cargo” or CD5/LNP-FAPCAR) (Fig. 1A) (9, 25). CD5 is naturally expressed by T cells and a small subset of B cells, and is not required for T cell effector function (26, 27). As a first proof-of-concept experiment, we incubated CD5/LNP containing modified mRNA encoding either FAPCAR or GFP with freshly isolated, activated murine T cells in vitro for 48 hours. CD5-targeted LNP delivered their mRNA cargo to the vast majority of T cells in culture, where 81% expressed GFP after exposure to CD5/LNP-GFP (Fig. 1B) and 83% of T cells expressed FAPCAR after exposure to CD5/LNP-FAPCAR (Fig. 1C and D) as measured by flow cytometry (fig. S1A). In vitro, CAR expression peaks at 24 hours and rapidly abates over ensuing days (fig. S1B). LNP decorated with isotype control (IgG) antibodies and thus not explicitly directed to lymphocytes, were only able to deliver mRNA to a small fraction (7%) of T cells in vitro (Fig. 1C and D). These LNP-generated CAR T cells were able to effectively kill FAP-expressing target cells in vitro (Fig. 1E) in a dose-dependent manner (fig. S1C) similar to virally engineered FAPCAR T cells. Gene transfer via targeted LNP in vitro is also possible and efficient (89–93%) in human T cells as demonstrated by targeting ACH2 cells with CD5/LNP-GFP (fig. S1D).
We next assessed whether CD5-targeted lipid nanoparticle mRNA could also efficiently reprogram T cells in vivo. Mice that were intravenously injected with CD5/LNP containing luciferase mRNA (CD5/LNP-Luc) were found to express abundant luciferase activity in their splenic T cells, while mice injected with isotype-control (non-targeting) IgG/LNP-Luc did not (Fig. 2A). Bioluminescence imaging demonstrated spleen targeting only in CD5/LNP-Luc treated animals (fig. S2A). Liver expression of LNP-delivered mRNA was observed in both CD5/LNP-Luc and IgG/LNP-Luc treated animals as expected mainly due to normal hepatic clearance of LNP, as reported previously (22, 24). In another experiment, CD5/LNP were loaded with mRNA encoding Cre recombinase (CD5/LNP-Cre) and injected into Ai6 Cre-reporter mice (Rosa26CAG-LSL-ZsGreen). We found evidence of genetic recombination (ZsGreen expression) specifically in CD3+ T cells (both CD4+ and CD8+ subsets) from CD5/LNP-Cre-injected animals but little evidence of Cre recombinase activity in CD3–(non-T) cells (mainly representing B cells, dendritic cells and macrophages) or in IgG/LNP-Cre-injected mice (Fig. 2B). We next asked whether targeted LNP could deliver FAPCAR mRNA (CD5/LNP-FAPCAR) to T cells in an established murine hypertensive model of cardiac injury and fibrosis produced by constant infusion of angiotensin II and phenylephrine (AngII/PE) via implanted 28-day osmotic mini-pumps (9, 28). Mice were injured for one week to allow fibrosis to be established before injecting CD5/LNP-FAPCAR (9). Forty-eight hours after LNP injection, we found a consistent population of FAPCAR+ T cells (17.5–24.7%) exclusively in mice that received CD5/LNP-FAPCAR (Fig. 2C, D and fig S2B). In contrast, non-targeted (IgG/LNP-FAPCAR) and targeted LNP containing GFP (CD5/LNP-GFP) did not produce FAPCAR T cells (Fig. 2C, D and fig. S2B). We observed FAPCAR expression in each major T cell subset with a slight enrichment in CD4+ T cells above their prevalence in the spleen (of all FAPCAR T positive cells, 87% were CD4+ and 9–10% CD8+, with the majority of both classes portraying a naïve phenotype; 25–37% of Tregs are FAPCAR+ (fig. S2C and table S1). A mix of CAR+ T cell subtypes has been shown to benefit CAR effectiveness (29). We did not observe significant FAPCAR expression in splenic B cells or NK cells (fig. S2C). No FAPCAR expression was found in splenic T cells one week after injection, demonstrating the transient nature of FAPCAR expression in this model (table S1).
Fig. 2. CD5-targeted lipid nanoparticles produce mRNA-based FAPCAR T cells in vivo.
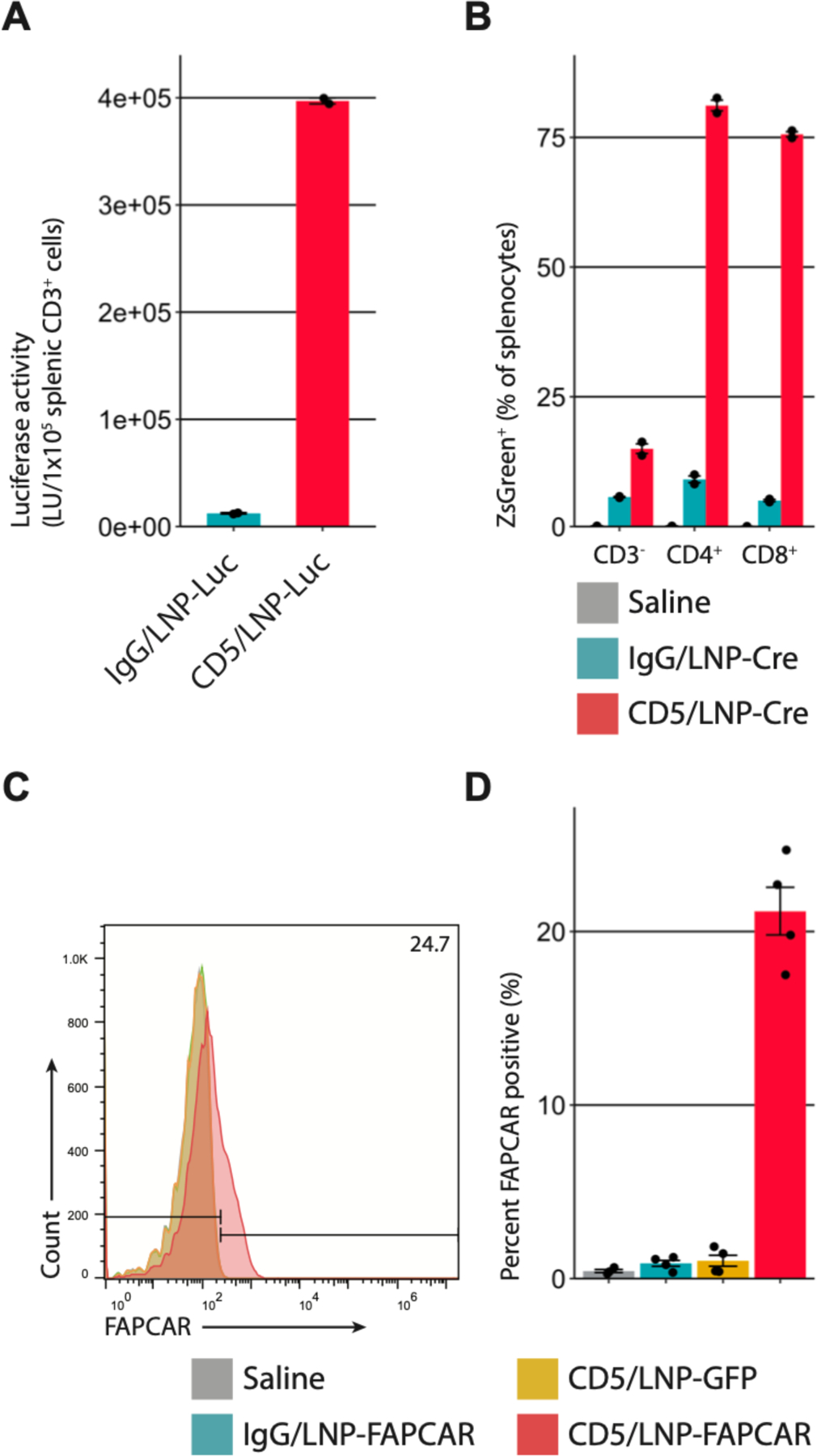
(A) Luciferase activity in CD3+ splenocytes 24 hours after intravenous injection of 8µg of control IgG/LNP-Luc or CD5/LNP-Luc. Bar graphs represent two biologically independent replicates. (B) Ai6 mice (Rosa26CAG-LSL-ZsGreen) were injected with 30µg of non-targeted/LNP-Cre (NT), IgG/LNP-Cre, or CD5/LNP-Cre. After 24 hours ZsGreen expression was observed in (81.1%) CD4+ and (75.6%) CD8+, but not in many (15.0%) CD3–splenocytes. Bar graphs represent two biologically independent replicates. (C) T cells were isolated from the spleens of AngII/PE injured mice, 48 hours after injection of 10µg of LNP. Representative flow cytometry analysis shows FAPCAR expression in animals injected with CD5/LNP-FAPCAR, but not in control saline, IgG/LNP-FAPCAR, or CD5/LNP-GFP animals. (D) Quantification of murine T cells staining positive for FAPCAR in C. n = 4 biologically independent mice in two separate cohorts. Data are mean +/− s.e.m.
CAR T cell therapy has previously been associated with a process called trogocytosis in which lymphocytes extract surface molecules through the immunological synapse from antigen-presenting cells (30–32) (Fig. 3A). We sought to determine if FAPCAR T cells, produced either in vivo with CD5/LNP-FAPCAR mRNA or adoptively transferred ex vivo virally engineered CAR T cells exhibit evidence of trogocytosis as further support that functional FAPCAR T cells are produced in situ. First, we mixed retrovirus-engineered FAPCAR T cells with HEK293T cells overexpressing RFP-tagged FAP in vitro and observed trogocytosis with live-imaging confocal microscopy (Fig. 3B and movie S1). Immunofluorescence analysis of spleens from AngII/PE injured animals treated with adoptively transferred, virally transduced GFP-tagged FAPCAR T cells revealed extensive FAP staining in the white pulp regions of the spleen, that was not seen in injured animals treated with control T cells or uninjured animals (Fig. 3C and fig. S3). The FAP+ cells in the spleens of injured and treated animals co-stained for GFP which indicates that they were transduced cells (Fig. 3D). Furthermore, the FAP staining appeared as cytoplasmic punctae consistent with trogocytosis (Fig. 3D). We observed some rare FAP+/GFP-negative cells in the spleens of injured, treated animals that was not observed in controls (Fig. 3D arrow). CD3+ lymphocytes containing FAP+ punctae were also seen in the spleens of injured animals treated with CD5/LNP-FAPCAR therapy but not in those treated with IgG/LNP-FAPCAR control (Fig. 3E). We are not aware of prior reports of CAR T cells exhibiting trogocytosis in the spleen after therapy, perhaps because prior studies have focused on CAR T cells directed against lymphocytic markers that would be difficult to distinguish from endogenous expression in the spleen. These findings are consistent with functional anti-FAP CAR T cells being produced in vivo following CD5/LNP-FAPCAR treatment.
Fig. 3. FAPCAR T cells trogocytose FAP from activated cardiac fibroblasts and return FAP to the spleen only in AngII/PE injured, FAPCAR T-treated animals.
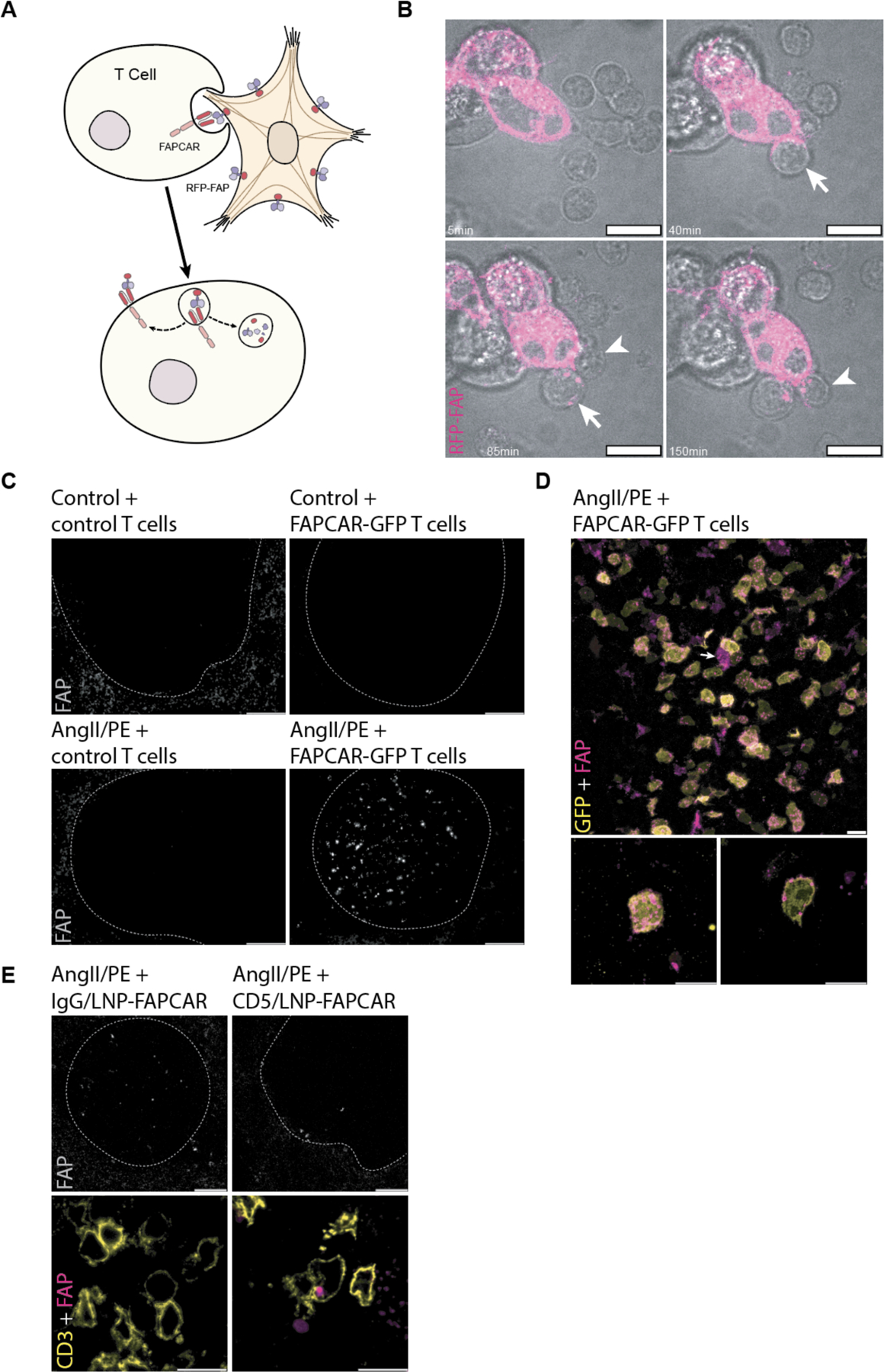
(A) Schematic representation of FAPCAR-expressing T cells trogocytosing FAP from activated fibroblasts. (B) Confocal time-lapse micrographs of two FAPCAR T cells first forming an immunological synapse at 40min (arrow), and 85min (arrowhead) then trogocytosing RFP-FAP (magenta) from HEK293T cells (punctae seen at 85min, arrow, and 150min, arrowhead within FAPCAR T cells). Scale bars: 10μm. (C) Widefield images of FAP-stained spleens (white pulp regions highlighted by the dashed line) of an uninjured animal 24 hours after adoptive transfer of 107 MigR1-control T cells, an uninjured animal 24 hours after adoptive transfer of 107 FAPCAR-GFP T cells, an AngII/PE-injured (7 days) animal 48 hours after adoptive transfer of 107 MigR1-control T cells, and an AngII/PE-injured (7 days) animal 48 hours after adoptive transfer of 107 FAPCAR-GFP T cells. Scale bars: 100μm. (D) Confocal micrograph of FAP (magenta) and FAPCAR-GFP (yellow) in a white pulp region of the spleen of an AngII/PE-injured (7 days) animal 48 hours after adoptive transfer of 107 FAPCAR-GFP T cells. Max-Z projection (lower left subpanel) and a single Z slice (lower right subpanel) of a representative FAP+/FAPCAR+ T cell. Scale bars: 10μm. (E) Confocal micrographs of a white pulp region (dashed outline) of FAP-stained spleens from AngII/PE-injured (7 days) animals injected with 10μg of IgG/LNP-FAPCAR or CD5/LNP-FAPCAR for 48 hours. FAP (grey and magenta) and CD3 (yellow) overlap specifically in CD5/LNP-FAPCAR-treated condition. Scale bars: 100μm (top row; greyscale) or 10μm (bottom row; merged pseudo-colored).
We next assessed whether CD5/LNP-FAPCAR treatment was able to improve cardiac function in injured mice as observed previously (9). To test this, we induced cardiac injury in mice with AngII/PE delivered via 28-day osmotic mini-pumps. After one week, when fibrosis is apparent (9), 10μg of LNP were injected intravenously. Two weeks after injection, cardiac function was analyzed by echocardiography (Fig. 4A, fig. S4A and B). We observed marked functional improvements in injured mice treated with in vivo-produced, transient FAPCAR T cells, consistent with our previous studies using adoptively transferred viral FAPCAR T cells (movie S2 to 5). AngII/PE-injured mice treated with CD5/LNP-FAPCAR exhibited normalized left ventricular (LV) end diastolic and end systolic volumes (Fig. 4B and C). Also, consistent with our previous study (9), body weight-normalized LV mass (estimated in M-mode) did not show statistically significant differences following CD5/LNP-FAPCAR injection although a trend in improvement compared to control injured mice was noted (Fig. 4D). Importantly, LV diastolic function (E/e’) returned to uninjured levels (Fig. 4E). LV systolic function was also noticeably improved as measured by ejection fraction (Fig. 4F) and global longitudinal strain (Fig. 4G and H). Injection of non-targeting IgG/LNP-FAPCAR did not alter LV function (fig. S4C). In CD5/LNP-FAPCAR injected animals, but not in controls, we observed an accumulation of CD3+ T cells within regions occupied by FAP+ fibroblasts (fig. S4D) (9). Furthermore, many of these CD3+ T cells are FAPCAR+ (80 of 137 or 58% of CD3+ T cells observed in 25 highly magnified fields of view in five histologic sections) indicating that they had been transduced with FAPCAR mRNA, while CD3+ T cells from control animals did not co-stain for the FAPCAR (fig. S4E). Consistent with our prior results (9), we observed a statistically significant improvement of the heart weight to body weight ratio (a measure of cardiac hypertrophy) in treated animals (fig. S5A).
Fig. 4. In vivo generation of transient FAPCAR T cells improves cardiac function after injury.
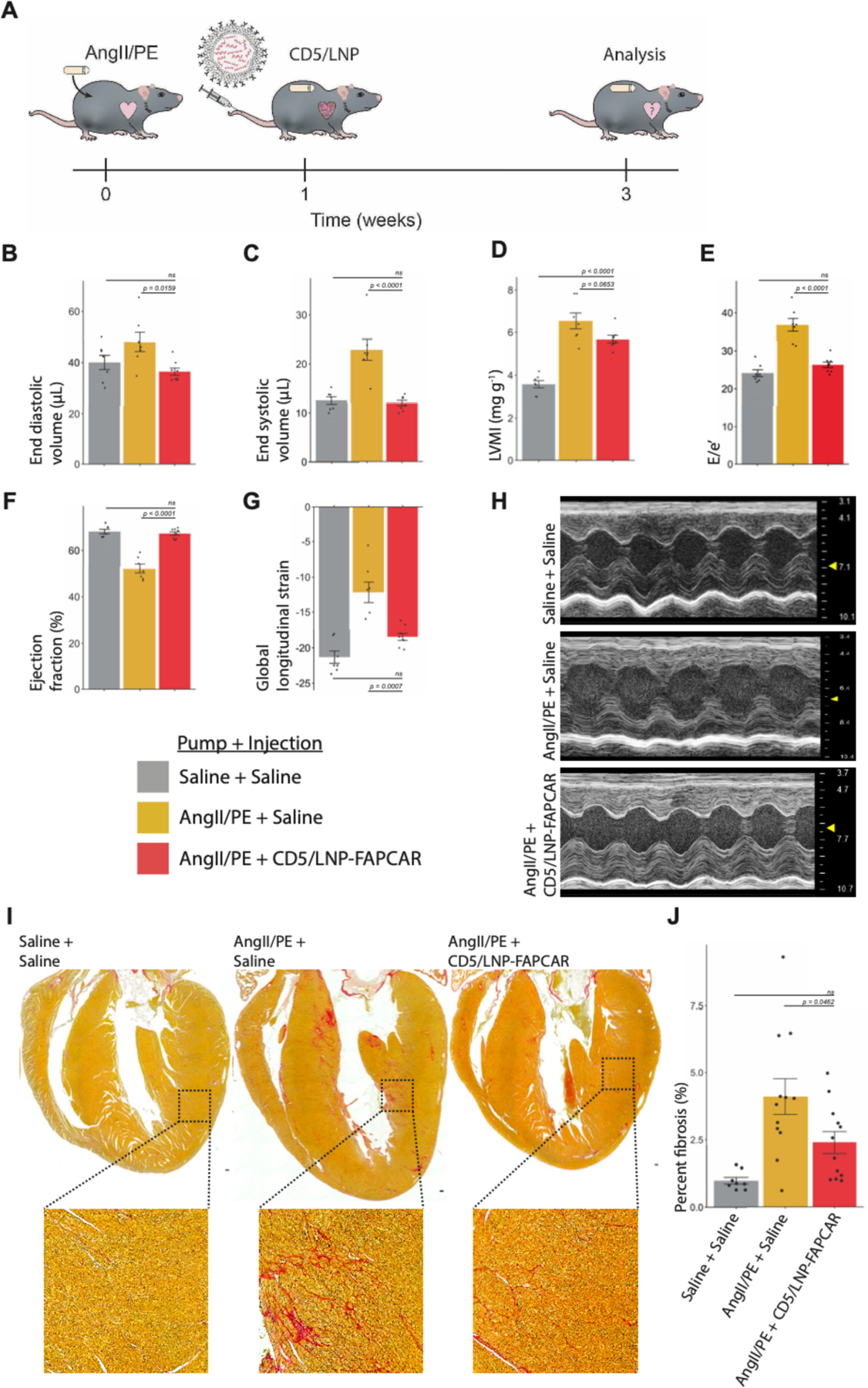
Wild-type adult C57BL/6 mice were continually dosed with saline or AngII/PE via implanted 28-day osmotic minipump. After one week of cardiac pressure-overload injury, CD5-targeted LNP were injected. Mice were analyzed after an additional two weeks. (A) Schematic representation of experimental timeline. Echocardiograph measurements show improvements in left ventricle (LV) volumes, diastolic and systolic function following a single injection of 10μg of CD5/LNP-FAPCAR. Measurements of (B) end diastolic and (C) end systolic volumes (μL). (D) M-mode estimate of weight-normalized LV mass (mg/g). (E) Diastolic function (E/e’, an estimate of LV filling pressure) (F) ejection fraction (%) and (G) global longitudinal strain. (H) Representative m-mode echocardiography images. Echocardiograph data represent n = 7, 7, 8 biologically independent mice per condition, spread over three cohorts. (I) Picrosirius red (PSR) staining highlights collagen (pink) in coronal cardiac sections of mock uninjured animals (3 weeks after saline pump implant + saline injection at week 1), injured control animals (AngII/PE + saline), isotype non-targeted LNP control (AngII/PE + IgG/LNP-FAPCAR) and treated animals (AngII/PE + CD5/LNP-FAPCAR). Inset shows magnification of left ventricular myocardium. Scale bar: 100μm. (J) Quantification of percent fibrosis of the ventricles seen in (I). Histology data represent n = 8, 11, 12, biologically independent mice per condition, spread over five cohorts. Data are mean +/− s.e.m. Displayed p-values are from Tukey’s post-hoc test following one-way ANOVA p<0.05.
Histologic analysis, as assessed by staining with picrosirius red, highlighted a significant improvement in the overall burden of extracellular matrix between injured mice treated with CD5/LNP-FAPCAR and those treated with saline or IgG/LNP-FAPCAR controls (Fig. 4I and J and fig. S5B and C). Furthermore, a subset of treated animals (5 of 12) was indistinguishable from uninjured controls, apart from persistent perivascular fibrosis which results from activated fibroblasts that do not express FAP (9) (fig. S5D arrows). Prior studies, in which FAP-expressing activated fibroblasts were eliminated by genetic ablation or treatment with virally-transduced CAR T cells, have also shown persistence of perivascular fibrosis (9, 28). Thus, CD5/LNP-FAPCAR treatment resulted in improved function and decreased interstitial fibrosis. Importantly, we did not observe any gross histological changes in non-cardiac organs or weight loss following CD5/LNP-FAPCAR injection (fig. S6A and B).
These experimental results provide a proof of concept that modified mRNA encapsulated in targeted LNP can be delivered intravenously to produce functional engineered T cells in vivo. The remarkable success and safety of modified mRNA/LNP SARS-CoV-2 vaccines has stimulated broad efforts to extend this therapeutic platform to address numerous pathologies. By targeting LNP to specific cell types, as we demonstrate here for lymphocytes, modified mRNA therapeutics are likely to have far-reaching applications. Generation of engineered T cells in vivo using mRNA is attractive for certain disorders because the transient nature of the produced CAR T cells is likely to limit toxicities – including risks incurred by lymphodepletion prior to injection – and allow for precise dosing. Unlike patients with cancer, those suffering from fibrotic disorders may not require a complete elimination of pathologic cells (activated fibroblasts) but may symptomatically benefit from an overall reduction in burden of disease. Furthermore, targeted LNP/mRNA technology affords the advantageous ability to titrate dosing and to re-dose as needed. Future studies will be needed to optimize the dosing strategy, LNP composition, and targeting approaches to further enhance therapeutic effects and limit potential toxicities. Nevertheless, the possibility of an “off the shelf” universal therapeutic capable of engineering specific immune functions provides promise for a scalable and affordable avenue to address the enormous medical burden of heart failure and other fibrotic disorders.
Supplementary Material
Acknowledgements
We thank A. Kiseleva for technical support and essential pandemic comradery, C. Smith for manuscript comments, N. Olimpo for troubleshooting advice, and the Pathology Core Laboratory at the Children’s Hospital of Philadelphia Research Institute for providing picrosirius red staining services.
Funding:
This research was supported by NIH AI142596, HL134839, and AI124429 to D.W., the Penn Center for AIDS Research (CFAR) an NIH-funded program (P30 AI 045008) to H.P., and NIH R35 HL140018, the Cotswold Foundation, and the WW Smith endowed chair to J.A.E.
National Institutes of Health grant AI142596 (D.W.)
National Institutes of Health grant HL134839 (D.W.)
National Institutes of Health grant AI124429 (D.W.)
The Penn Center for AIDS Research (CFAR) an NIH-funded program grant P30 AI 045008 (H.P.)
National Institutes of Health grant R35 HL140018 (J.A.E.)
The Cotswold Foundation (J.A.E.)
The WW Smith endowed chair (J.A.E.)
Footnotes
Competing Interests: S.M.A., E.P., C.H.J., H.A., D.W., H.P., and J.A.E. are scientific founders and hold equity in Capstan Therapeutics. Y.K.T. and B.L.M. are employees and hold equity in Acuitas Therapeutics. S.M.A. is on the scientific advisory boards of Verismo and Bioardis. C.H.J. is a scientific founder and has equity in Tmunity Therapeutics and DeCART Therapeutics and reports grants from Tmunity Therapeutics and is on the scientific advisory boards of BluesphereBio, Cabaletta, Carisma, Cellares, Celldex, ImmuneSensor, Poseida, Verismo, Viracta Therapeutics, WIRB Copernicus Group and Ziopharm Oncology. D.W. receives research support from BioNTech. S.M.A., E.P., and C.H.J. are inventors (University of Pennsylvania, Wistar Institute) on a patent for a FAP CAR (US Utility Patent 9,365,641 issued 14 June 2016, WIPO Patent Application PCT/US2013/062717). S.M.A., E.P., H.A., and J.A.E. are inventors (University of Pennsylvania) on a patent for the use of CAR T therapy in heart disease (US Provisional Patent Application 62/563,323 filed 26 September 2017, WIPO Patent Application PCT/US2018/052605). J.G.R., I.T., H.A., D.W., H.P., and J.A.E. are inventors (University of Pennsylvania) on a patent for the use of CD5/LNP-FAPCAR as an anti-fibrotic therapy (US Provisional Patent Application 63/090,998 filed 13 September 2020, WIPO Patent Application PCT/ US21/54764 filed 13 October 2021). I.T., D.W., and H.P. are inventors (University of Pennsylvania) on a patent for the in vivo targeting of T cells for mRNA therapeutics (US Provisional Patent Application 63/090,985 filed 13 October 2020, WIPO Patent Application PCT/US21/54769 filed 13 October 2021). I.T., D.W., and H.P. are inventors (University of Pennsylvania) on a patent for the in vivo targeting of CD4+ T cells for mRNA therapeutics (US Provisional Patent Application 63/091,010 filed 13 October 2020, WIPO Patent Application PCT/US21/54775). In accordance with the University of Pennsylvania policies and procedures and our ethical obligations as researchers, D.W. is named on additional patents that describe the use of nucleoside-modified mRNA and targeted lipid nanoparticles as platforms to deliver therapeutic proteins and vaccines. C.H.J. is named on additional patents that describe the creation and therapeutic use of chimeric antigen receptors. These interests have been fully disclosed to the University of Pennsylvania, and approved plans are in place for managing any potential conflicts arising from licensing these patents.
Data and materials availability: All data is available in the main manuscript or the supplementary materials. Correspondence and requests for materials can be addressed to H.A., D.W., H.P., or J.A.E.
Supplementary Materials:
References 9, 25, 33–36
References and Notes
- 1.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD, Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127, 3770–3783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schafer S, Viswanathan S, Widjaja AA, Lim W-W, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimarães-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong B, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA, IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552, 110–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J Clin Invest 124, 2921–2934 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, Kurmangaliyev YZ, Nasiri R, Ahadian S, Nguyen T, Tan XHM, Zhou Y, Wu R, Rodriguez A, Cohn W, Wang Y, Whitelegge J, Ryazantsev S, Khademhosseini A, Teitell MA, Chiou PY, Birk DE, Rowat AC, Crosbie RH, Pellegrini M, Seldin M, Lusis AJ, Deb A, Type V Collagen in Scar Tissue Regulates the Size of Scar after Heart Injury. Cell 182, 545–562.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rurik JG, Aghajanian H, Epstein JA, Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ Res 128, 1766–1779 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Widjaja AA, Viswanathan S, Jinrui D, Singh BK, Tan J, Wei Ting JG, Lamb D, Shekeran SG, George BL, Schafer S, Carling D, Adami E, Cook SA, Molecular Dissection of Pro-Fibrotic IL11 Signaling in Cardiac and Pulmonary Fibroblasts. Front Mol Biosci 8, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henderson NC, Rieder F, Wynn TA, Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.González A, Schelbert EB, Díez J, Butler J, Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J Am Coll Cardiol 71, 1696–1706 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher C, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Puré E, Albelda SM, Epstein JA, Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH, T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci Transl Med 3, 95ra73-95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weissman D, mRNA transcript therapy. Expert Rev Vaccines 14, 265–281 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Karikó K, Buckstein M, Ni H, Weissman D, Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D, Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16, 1833–1840 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T, N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release 217, 337–344 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH, Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70, 9053–9061 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardi N, Hogan MJ, Porter FW, Weissman D, mRNA vaccines-a new era in vaccinology. Nat Rev Drug Discov 17, 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizvi F, Everton E, Smith AR, Liu H, Osota E, Beattie M, Tam Y, Pardi N, Weissman D, Gouon-Evans V, Murine liver repair via transient activation of regenerative pathways in hepatocytes using lipid nanoparticle-complexed nucleoside-modified mRNA. Nat Commun 12 (2021), doi: 10.1038/s41467-021-20903-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, Akilli-Öztürk Ö, Kranz LM, Berger H, Petschenka J, Diken M, Kreiter S, Yogev N, Waisman A, Karikó K, Türeci Ö, Sahin U, A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science (80-). 371, 145–153 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Szőke D, Kovács G, Kemecsei É, Bálint L, Szoták-Ajtay K, Aradi P, Styevkóné Dinnyés A, Mui BL, Tam YK, Madden TD, Karikó K, Kataru RP, Hope MJ, Weissman D, Mehrara BJ, Pardi N, Jakus Z, Nucleoside-modified VEGFC mRNA induces organ-specific lymphatic growth and reverses experimental lymphedema. Nat Commun 12, 3460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, Phillips J, Xu Y, Amaral A, Boyd AP, Cehelsky JE, McKee MD, Schiermeier A, Harari O, Murphy A, Kyratsous CA, Zambrowicz B, Soltys R, Gutstein DE, Leonard J, Sepp-Lorenzino L, Lebwohl D, CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med, 1–10 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D, Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 217, 345–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev KG, Cantley WL, Dorkin JR, Butler JS, Qin L, Racie T, Sprague A, Fava E, Zeigerer A, Hope MJ, Zerial M, Sah DW, Fitzgerald K, Tracy MA, Manoharan M, Koteliansky V, De Fougerolles A, Maier MA, Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther 18, 1357–1364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parhiz H, Shuvaev VV, Pardi N, Khoshnejad M, Kiseleva RY, Brenner JS, Uhler T, Tuyishime S, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D, Muzykantov VR, PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J Control Release 291, 106–115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tombácz I, Laczkó D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A, Papp TE, Alameh M-G, Shuvaev V, Mui BL, Tam YK, Muzykantov V, Pardi N, Weissman D, Parhiz H, Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNP. Mol Ther (2021), doi: 10.1016/j.ymthe.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Puré E, Albelda SM, Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res 2, 154–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boumsell L, Coppin H, Pham D, Raynal B, Lemerle J, Dausset J, Bernard A, An antigen shared by a human T cell subset and B cell chronic lymphocytic leukemic cells. Distribution on normal and malignant lymphoid cells. J Exp Med 152, 229–234 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soldevila G, Raman C, Lozano F, The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr Opin Immunol 23, 310–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H, Looso M, Troidl C, Offermanns S, Wettschureck N, Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res 118, 1906–1917 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, Li Z, Cunanan KM, Huse M, Hendrickson RC, Wang X, Rivière I, Sadelain M, CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joly E, Hudrisier D, What is trogocytosis and what is its purpose? Nat Immunol 4, 815–815 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Martínez-Martín N, Fernández-Arenas E, Cemerski S, Delgado P, Turner M, Heuser J, Irvine DJ, Huang B, Bustelo XR, Shaw A, Alarcón B, T Cell Receptor Internalization from the Immunological Synapse Is Mediated by TC21 and RhoG GTPase-Dependent Phagocytosis. Immunity 35, 208–222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newick K, O’brien S, Sun J, Kapoor V, Maceyko S, Lo A, Puré E, Moon E, Albelda SM, Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase a localization. Cancer Immunol Res 4, 541–551 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baiersdörfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U, Karikó K, A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol Ther - Nucleic Acids 15, 26–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, Tam YK, Ansell SM, Kumar V, Qin J, Zhang X, Wang Q, Panesar S, Hutabarat R, Carioto M, Hettinger J, Kandasamy P, Butler D, Rajeev KG, Pang B, Charisse K, Fitzgerald K, Mui BL, Du X, Cullis P, Madden TD, Hope MJ, Manoharan M, Akinc A, Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther 21, 1570–1578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER, Estrogen inhibits cardiac hypertrophy: Role of estrogen receptor-β to inhibit calcineurin. Endocrinology 149, 3361–3369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.