

Abstract
Although an essential step, cell functional annotation often proves particularly challenging from single-cell transcriptional data. Several methods have been developed to accomplish this task. However, in most cases, these rely on techniques initially developed for bulk RNA sequencing or simply make use of marker genes identified from cell clustering followed by supervised annotation. To overcome these limitations and automatize the process, we have developed two novel methods, the single-cell gene set enrichment analysis (scGSEA) and the single-cell mapper (scMAP). scGSEA combines latent data representations and gene set enrichment scores to detect coordinated gene activity at single-cell resolution. scMAP uses transfer learning techniques to re-purpose and contextualize new cells into a reference cell atlas. Using both simulated and real datasets, we show that scGSEA effectively recapitulates recurrent patterns of pathways’ activity shared by cells from different experimental conditions. At the same time, we show that scMAP can reliably map and contextualize new single-cell profiles on a breast cancer atlas we recently released. Both tools are provided in an effective and straightforward workflow providing a framework to determine cell function and significantly improve annotation and interpretation of scRNA-seq data.
INTRODUCTION
Single-cell RNA sequencing (scRNA-seq) delivers unprecedented opportunities for measuring gene expression at genome-wide scale and single-cell resolution. It provides a cost-effective way to study cellular tissue composition (1–5), dynamic processes during cell developmental stages (6) or the role of transcriptional heterogeneity in pathological conditions and in response to treatments (7). Indeed, scRNA-seq is becoming the leading technique for transcriptome profiling with large atlases of cells now routinely released from labs worldwide (1–8). Although scRNA-seq confers the advantage of measuring gene expression at the granular resolution of individual cells, the data produced with this sequencing technique are extremely noisy and zero-inflated (9). This property arises from both the low amount of mRNA available in a single cell and the limited capture efficiency of the technology. Thus, computational tools initially built to analyse bulk RNA-seq are often inadequate for scRNA-seq, and ad-hoc tools need to be developed to address the peculiarities of single-cell data. The significant increase in scRNA-seq studies raised several computational challenges, such as the development of automatic methods for functional annotation of scRNA-seq helpful for cell type assignment or disease diagnosis.
In the last decade, a plethora of tools have been developed to summarize regulated gene expression profiles into simplified functional categories useful for the annotation and interpretation of bulk RNA-seq data (10–16). Among these, the gene set enrichment analysis (GSEA) (11) is probably the most used. GSEA is a statistical tool that aims to measure coordinated activity of a priori defined gene set (i.e. pathway) starting from a ranked list of genes usually obtained from differential expression (DE) analysis. By aggregating individual DE genes into pathways, GSEA projects DE results in a robust and easily interpretable biological space where additional analytics tools can be later applied. Currently, the application of GSEA to single-cell data remains challenging (17) and only a few methods are available but still not efficient (18,19). Therefore, there is an urgent need to develop novel computational methods capable of scoring the activity of a pathway within a reasonable time scale. On the other hand, the availability of large single-cell reference atlases comprising up to millions of cells across different species, conditions, tissues, or organs provides an unprecedented opportunity to use ‘re-purposing’ techniques for the functional annotation of cells. Indeed, transfer learning (TL) techniques (20) are becoming increasing popular in the analysis of high-throughput -omics datasets including single-cell datasets (21–24). TL is a machine learning technique that use a joint embedding to integrate a query set of objects into a given reference and to contextualize them with the metadata associated with the reference elements.
Here, we present two novel ready-to-use pipelines tailored to scRNA-seq data that can be used to automatize cell functional annotation: scGSEA (single-cell gene set enrichment analysis) and scMAP (single-cell Mapper). scGSEA is a statistical framework for scoring coordinated gene activity in individual cells to automatically determine the pathways that are active in a cell. scMAP is a TL algorithm to map a query set of cell transcriptional profiles on top of an existing reference atlas and contextualize the new data with the reference metadata. Both methods are based on non-negative matrix factorization (NMF) (25), a popular matrix decomposition method, that can be solved in a very computationally efficient manner (26). We validated scGSEA to identify pathway activity in both a simulated dataset and a real dataset comprising cells during various reprogramming stages (6) or drug treatment (27). We also tested the ability of scMAP in mapping novel sequenced cells from several conditions, including cells sequenced in different batches or another lab with a different sequencing technique. Both tools were developed in the framework of the gficf package (9,28), an R package we recently developed for the normalization, visualization and clustering of single-cell data that takes advantage of text-mining approaches and available at https://github.com/gambalab/gficf.
MATERIALS AND METHODS
Cell culture
MDAMB468 and CAL51 cell lines used in this study were obtained from commercial providers and cultured in ATCC recommended complete media at 37°C and 5% CO2.
scRNA library preparation, sequencing and alignment
Single-cell transcriptomics of the MDAMB468 and CAL51 cell lines were performed with DROP-seq technology (29) and library preparation as described in Gambardella et al. (7). scRNA libraries were sequenced with NovaSeq 6000 machine using an SP 100 cycles flow cell. Raw reads pre-processing was performed using Drop-seq tools v2.3.0 and following the Dropseq Core Computational Protocol reported at http://mccarrolllab.org/dropseq. Briefly, raw reads were first filtered to remove all read pairs with at least one base in their cell barcode or UMI with a quality score <10. Then, read 2 was trimmed at the 5’ end to remove any TSO adapter sequence, and at the 3’ end to remove polyA tails. Filtered reads were then fed to STARsolo tool v2.7.10a (https://github.com/cellgeni/STARsolo) to perform alignment, UMI deduplication and gene expression quantification. Hg38 human genome (primary assembly v40) downloaded from GENCODE (30) was used as a reference genome for read alignment. Only high depth cells with at least 2500 UMI were retained and used to test our cell mapping tool. Alignment pipeline can be found at https://github.com/gambalab/dropseq.
Single-cell gene set enrichment analysis (scGSEA)
To perform scGSEA, raw count matrix was first normalized with gficf package (28). Then, NMF was used to decompose gficf scores (or normalized log(CPM + 1) expression) into two positive related matrices containing gene weights (W) and cell weights (H) where each column of W or row of H defines a latent actor 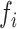 . Non-negative matrix factorization was performed by using the fast parallel implementation that can be found in RcppML R package (26) available at https://github.com/zdebruine/RcppML. NMF implementation in the RcppML package is based on Alternating Least Square (ALS) approach and diagonalized NMF to enable symmetric factorization and to reduce bias related random initialization (26). Gene set enrichment analysis was performed against each column of the W matrix using as input a pre-defined list of gene sets
. Non-negative matrix factorization was performed by using the fast parallel implementation that can be found in RcppML R package (26) available at https://github.com/zdebruine/RcppML. NMF implementation in the RcppML package is based on Alternating Least Square (ALS) approach and diagonalized NMF to enable symmetric factorization and to reduce bias related random initialization (26). Gene set enrichment analysis was performed against each column of the W matrix using as input a pre-defined list of gene sets 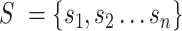 . GSEA was performed using the R package fgsea (31) available at https://github.com/ctlab/fgsea. At the end of this process, a novel matrix P with the same number of columns (i.e. factors) of W and with the number of rows equal to the number of inputs used gene set is obtained. Each element of Pi,j contains the normalized enrichment scores of the pathway
. GSEA was performed using the R package fgsea (31) available at https://github.com/ctlab/fgsea. At the end of this process, a novel matrix P with the same number of columns (i.e. factors) of W and with the number of rows equal to the number of inputs used gene set is obtained. Each element of Pi,j contains the normalized enrichment scores of the pathway 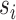 related to the factor
related to the factor 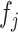 . Next, only positives and significant normalized enrichment scores with an FDR < 0.05 were retained while all the other elements of P are put to zero. Finally, since elements of P describe the pathways’ contribution in each latent factor, the pathway's activity level in each profiled cell is computed as the weighted sum of the normalized pathway's enrichment scores across the factors shared by the cell. This corresponds simply to the dot product of the P and H matrices. The described pipeline is implemented in the function scGSEA of the gficf package.
. Next, only positives and significant normalized enrichment scores with an FDR < 0.05 were retained while all the other elements of P are put to zero. Finally, since elements of P describe the pathways’ contribution in each latent factor, the pathway's activity level in each profiled cell is computed as the weighted sum of the normalized pathway's enrichment scores across the factors shared by the cell. This corresponds simply to the dot product of the P and H matrices. The described pipeline is implemented in the function scGSEA of the gficf package.
Simulated scRNA-seq profiles and gene sets
Splatter R package (32) with default parameters was used to generate a zero-inflated count dataset composed of 5000 cells and 1000 genes with cells grouped in four distinct populations. Next, six gene sets were simulated to be exclusively expressed in each group of simulated cells (i.e. 6 gene sets × 4 groups of cells = 24 simulated gene sets). The 24 gene sets comprised 2432 unique genes that we added to the simulated dataset generated with splatter. A zero-inflate Poisson distribution with success probability equal to 50% and lambda value of 10 was used to simulate the expression of a gene set in a specific set of cells. This to simulate a moderate dropout and a relatively low gene count expression for each gene set.
Estimation of scGSEA computational time
The computation time of the scGSEA tool was estimated using three collections of different pathways (i.e. 50 Hallmarks gene sets, 186 KEGG pathways and 7763 GOBP gene sets) and 10 000 cells randomly selected from the single-cell breast cancer atlas we recently published (7). Next, scGSEA pipeline was performed using a number of CPU cores ranging from 1 to 16. This process was repeated five times to obtain an average computational time representative of each execution for each specific number of CPUs used. Simulations were performed using a PC equipped with a 16-cores AMD Ryzen 9 3950X and 128GB of RAM.
Pseudo-time analysis
scGSEA method was run using as input the 1044 PC9 single-cell transcriptional profiles and the 50 hallmarks gene sets (v2022.1) downloaded from MSigDB (www.gsea-msigdb.org/gsea/msigdb). The obtained pathways’ activity matrix was used to run psupertime function of psupertime R package and infer the pseudo-time order of cells. Psupertime function was run with default parameters and using as labels their sequencing day (i.e. 0, 1, 2, 4, 9 or 11)
Single-cell mapper (scMAP)
UMAP embedding space of reference single-cell breast cancer cell-line atlas or the reference single-cell breast cancer patient atlas were built from scratch with gficf package (28). Then, new sequenced cells are mapped on the reference atlases following the strategy depicted in Supplementary Figure S2. Briefly, new scRNA-seq profiles are first normalized with gficf strategy but using the ICF weight learned on the reference atlas and then projected to the existing NMF (or PC) sub-space using gene loadings learned from the reference atlas. These values are then used as input of the umap_transform function of uwot package, which uses the UMAP estimated model to map the new cells into the reference UMAP space. Finally, the cell line of origin associated with each mapped cell, or its cancer subtype was predicted by using k nearest-neighbor algorithm from KernelKnn package (https://mlampros.github.io/KernelKnn). For all assignments, k parameter was set to 101. Single-cell mapping pipeline is implemented into the function scMAP of the gficf package.
Public single-cell transcriptional dataset
The raw counts of the 35 276 single-cell transcriptional profiles of the 32 breast cancer cell (7) used in this study were downloaded from figshare (https://doi.org/10.6084/m9.figshare.15022698). The 25 1203 single-cell transcriptional profile from pluripotent stem cells (6) were obtained from GEO database with accession number GSE122662. The single-cell transcriptional profiles of MCF7 cells and derived clones (33) were obtained from GEO database with accession number GSE114462. The 2311 single cell the transcriptional profile across the eleven breast cancer cell lines (8) were obtained from GEO database with accession number GSE157220. PC9 lung cells were obtained from GEO database with accession number GSE149383 but only 1044 cells with a total number of UMI >2500 were used in this work. Patients derived single-cell transcriptional profiles of 34 treatment-naïve breast primary tumors (34) were obtained from GEO database with accession number GSE161529. A subset of this dataset was used, including 47 692 epithelial cells (35) with a minimum of 5000 UMIs, from 24 breast cancer patients. These single-cell transcriptional profiles comprised 7 Triple Negative (TN) breast cancer patients, 5 HER2-amplified (HER2+) breast cancer patients and 12 ER + breast cancer patients.
RESULTS
Gifcf package overview
We recently introduced an R package named gficf useful for normalization of 3’ single-cell transcriptional data and the identification of biomarker genes across multiple experimental conditions or cell types (7,9,28). Our tool builds on a data transformation model named Gene Frequency – Inverse Cell Frequency (i.e. gf-icf) derived from the Term Frequency - Inverse Document Frequency (i.e. tf-idf) approach. TF-IDF is a statistical measure extensively used in the fields of text analysis and machine learning applied to Natural Language Processing (NLP) for quantifying the relevance of a word (i.e. gene) in a document (i.e. cell) amongst a comprehensive collection of documents (i.e. scRNA-seq dataset) (36,37). When applying this model to scRNAseq data, the relevance of a gene increases proportionally to its expression in the cell but is offset by the frequency of the gene in the population of sequenced cells (28), so that only genes highly expressed in a small fraction of the cells are selected as the most relevant.
Briefly, the gf-icf pipeline (9,28) starts from a set of single-cell transcriptional profiles and consists of the following steps: (i) cell quality control (QC) and filtering, (ii) rescaling of gene expression profiles of each cell to sum one (GF step) after raw count normalization (38), (iii) cross-cell normalization, to assign higher scores to rarely expressed genes than commonly expressed genes within each cell (ICF step), (iv) an L2 rescaling step to normalize gf-icf values; (v) linear dimensionality reduction of the data (i.e. PCA(39) or NMF(26)) to condense the complexity of the dataset into a lower-dimensional space, (vi) non-linear dimensionality reduction (i.e. t-SNE (40) or UMAP (41)) of the data for its visualization and finally (vii) several downstream analyses including cell clustering and differential expression (9) (Figure 1A). GF-ICF method is implemented as an open-source R package, freely available at https://github.com/gambalab/gficf.
Figure 1.

Single-cell gene set enrichment analysis overview and performances. (A) GFICF package overview. (B) Single-cell gene set enrichment analysis pipeline. (C) UMAP plot of 5000 simulated cells grouped in four distinct groups. (D) Reconstructed activity of 24 simulated pathways across the 5000 cells in (C). In the heatmap pathways are along rows while simulated cells along columns. Cells are ordered according to their group of origin. (E) Comparison between scGSEA pathway scores and signature scores originally computed by Schiebinger et al. on 25 1203 single-cell profiles collected during differentiation stages. First row shows original gene set scores computed by Schiebinger et al. using wot phyton package. Second row shows gene set scores computed with scGSEA tool in the gficf R package. Each column represents a different gene set. Scores were plotted on the original FLE (force-directed layout) coordinates published by Schiebinger et al. (F) Spearman Correlation Coefficient (SCC) between scGSEA scores and wot package signature scores across the 25 1203 single-cell transcriptional profiles in (E). (G) UMAP representation of 1044 cells subject to eleven days of consecutive erlotinib treatment. Cells are color-coded according to sequenced day (i.e. 0, 1, 2, 4, 9 and 11 days). Single-cell transcriptional profiles were normalized with gficf package. (H) EMT activity scores against inferred cell pseudo-time using the activity scores of 50 hallmark gene sets downloaded from MSigDB. Cells are color-coded as in (G). (I–K) Same as (H) but for wnt, cholesterol and fatty acid pathways respectively.
Here we updated the gf-icf package and implemented two novel functionalities: scGSEA and scMap.
scGSEA for the reconstruction of pathway activity at single-cell resolution
We aimed to construct a bioinformatics method that could measure the activity of an a priori defined collection of gene sets (i.e. pathways) at the single-cell resolution. To this end, we developed a robust and fast single-cell Gene Set Enrichment Analysis (scGSEA) algorithm that takes advantage of the informative biological signals spreading across the latent factors of gene expression values obtained from non-negative matrix factorization (see Materials and methods) (25,26). The scGSEA method starts from a set of single-cell expression profiles and a collection of gene sets and scores their cumulative expression (i.e. pathway activity) in each of the profiled cells and it is divided into three main steps (Figure 1B). In the first step, we use NMF to decompose GF-ICF normalized data into two positive related matrices containing gene weights (W) and cell weights (H). Each column of W or row of H defines a factor representing complex biological processes that recur throughout the set of sequenced cells (42,43). While, at the same time, all features define the latent space amongst genes and cells. In the second step, we infer which biological processes each latent factor is associated with by performing GSEA (31) against each column of the W matrix. Since the values in each column of the W matrix are continuous weights describing the relative contribution of a gene in each inferred factor, a positive and significant value of the enrichment score (ES) for a specific pathway implies the factor is related to it. At the end of this step, the W matrix is transformed into a novel positive matrix P describing the pathways’ contribution in the form of normalized enrichment scores (NES) for each latent factor. Finally, in the third step, we infer the pathway’s activity level in each profiled cell by multiplying the P matrix reconstructed in step two with the H matrix obtained using the NMF method in step one. The rationale behind this last step is that each column of the matrix H describes the relative contribution of a cell across the factors. Thus, a cell with a high weight for a specific factor is assumed to share the phenotype or biological process related to that factor (43). Consequently, the activity level of a pathway in a cell can be computed as the weighted sum of the normalized pathway’s enrichment scores across the factors shared by the cell.
We performed simulations to assess the effectiveness of scGSEA in recapitulating the activity level of a pathway at single-cell level (see methods for details). By using the splatter package (32) we generated a zero-inflated count dataset composed of 5000 cells grouped in four distinct populations (Figure 1C) through a gamma-Poisson distribution using parameters inferred from a real dataset. Then, we assigned 24 overlapping gene sets of different sizes (Supplementary Table S1) to be exclusively expressed in each cluster (i.e. six specific gene sets per cluster). As shown in Figure 1D, we found that scGSEA can identify in each group of cells the activity of the six specific simulated pathways with some of these correctly predicted active also in other groups of cells due to their partial overlap with other genes sets (Supplementary Figure S1). We found similar results also when applied NFM step was performed on log-normalized CPM counts (Supplementary Figure S2A). Next, to demonstrate the reliability of the scGSEA method on a real dataset, we applied it to the cell reprogramming scRNA-seq data in Schiebinger et al. (6). This dataset comprises 25 1203 single-cell transcriptional profiles collected at half-day intervals across 18 days of reprogramming by ectopic expression of OKSM (a.k.a. Oct4, Klf4, Sox2 and Myc) transcription factors. In this work, the authors constructed seven curated gene signatures to score cells as pluripotent-, epithelial-, trophoblast-, neural-, MEF-like and proliferative. Hence, we applied our scGSEA method to this dataset with these seven gene sets as the input. As shown in Figure 1E,F, we found a high degree of correlation (avg. 0.8) between the predicted scGSEA pathway's activity level and the pathway expression computed by Schiebinger et al. (6). We obtained similar results also when NFM step was performed starting from log-normalized CPM counts (Supplementary Figure S2B,C). Finally, we estimated the average computational time required by the scGSEA tool with 10 000 cells and different pathway collections from MSigDB, including the 50 Hallmarks gene sets, the 186 KEGG pathways and the 7763 GOBP gene sets (see Materials and methods). As shown in Supplementary Figure S3, scGSEA analysis always requires a few minutes to be completed when the number of pathways is small, like in the case of KEGG or the Hallmarks pathway collections. While about 5 h are needed when using a larger pathway collection like the GOBP and only one CPU core. However, as Supplementary Figure S3 shows, in this scenario scGSEA computational time quickly decrease to about one hour using four CPUs.
Next, we investigated whether we could use scGSEA scores to infer cell trajectories and reconstruct dynamics of the key pathways driving resistance to EGFR inhibitors in non-small-cell lung carcinoma (NSCLC). Several studies have suggested that acquired resistance mechanisms to EGFR inhibitors involve the compensatory activation of redundant signalling pathways that share effectors or downstream modulators of the EGFR signalling cascade, thus bypassing EGFR inhibition. Different pathways can serve as alternative routes for reactivation of signalling downstream of inhibited EGFR, including MET, IGF-1R, PI3K-AKT-mTOR, BRAF/RAS and Wnt signalling pathways (44–47), all able to sustain cell survival, proliferation, migration and epithelial–mesenchymal transition (EMT). Therefore, we downloaded the scRNA-seq dataset published by Aissa et al. (27) comprising 1044 PC9 lung cells (Figure 1G) that were subject to eleven days of consecutive erlotinib treatment and sequenced at six different time points (i.e. 0, 1, 2, 4, 9 and 11 days). We then applied scGSEA to score the activity of 50 MSigDB hallmark pathways (48) across the cells. The resulting scores were used as input to reconstruct the dynamic activity of these pathway by applying a pseudotime algorithm (49) (Supplementary Figure S4). As shown in Figure 1H,I we found strong upregulation of EMT and Wntb-catenin signalling pathways in erlotinib tolerant cells, while erlotinib tolerant cells showed a potent inhibition of genes related to cholesterol and fatty acid metabolism, as also reported by Aissa et al. (Figure 1J,K).
These results show how scGSEA could recapitulate recurrent patterns of pathways’ activity shared by hundreds of thousands of cells from multiple conditions and during dynamic processes like cell reprogramming or drug treatments.
Mapping a single-cell transcriptional profile on a reference atlas
We developed scMAP (single-cell Mapper), a transfer learning algorithm that combines text mining data transformation and a k-nearest neighbours’ (KNN) classifier to map a query set of single-cell transcriptional profiles on top of a reference atlas (see Materials and methods). Our strategy consists of three main steps, as schematised in Supplementary Figure S5: (i) we first normalize the query cell profiles with the GF-ICF method by using the ICF weights learned by the reference atlas; (ii) we then project normalized cell profiles to the NMF (or PC) sub-space of the reference atlas before mapping them onto its UMAP embedding space; and (iii) finally, we use the KNN algorithm to contextualize mapped cells using available metadata.
To test scMAP, we took advantage of the single-cell atlas of breast cancer we have recently released (7). This atlas comprises 35 276 individual cells from 32 breast cancer cell lines covering all four major breast tumour subtypes (i.e. LuminalA, LuminalB, Her2-positive and Basal Like). Hence, we first applied GF-ICF tool (28) on these cells to generate a reference UMAP embedding space from either the top 100 NMF factors (Figure 2A) or the top 50 PC (Figure 2B). Next, we used three approaches to test the accuracy of our method in correctly mapping the very cells in the breast cancer atlas. First, we used a cross-validation approach where we randomly divided the 35 276 single-cell transcriptional profiles in different proportions of training and test cells (i.e. from 10 to 90% of the cells in each cell line). With this approach, we use training cells to reconstruct the reference BC cell-line atlas and the remaining cells to measure the performances of the mapping algorithm. Second, we re-sequenced with the DROP-seq platform 1683 cells of two cell lines included in the atlas and mapped the transcriptome of these cells onto it. Third, we tested our mapping strategy on 16 683 single-cell transcriptional profiles from 11 cell lines included in the atlas but sequenced by other laboratories with 10X genomics platform (8,33). In all cases the performances of the mapping algorithm were tested by using either NMF or PCA as cell sub-space before remapping them into the UMAP embedding space. After remapping, the label of a cell was assigned using the closest 101 cells (7) (see Materials and methods). As shown in Figure 2C, the median cross-validation approach’s accuracy of the mapping algorithm was 97% when using NFM as cell sub-space and 99% when using PC as sub-space. However, with the cross-validation approach we always use cells sequenced in the same batch. Thus, to avoid this possible bias, we re-sequenced 306 and 1377 cells of MDAMB468 and CAL51 cell lines, respectively (see Materials and methods). As shown in Figure 2D, 96% of MDAMB468 and 97.3% for CAL51 cells were recognized and labelled correctly when using NFM as cell sub-space. While when using PC as subspace mapping accuracy was of 79.7% and 98.1% for MDAM468 and CAL51 respectively (Figure 2E).
Figure 2.

Single-cell mapping accuracy evaluation. (A) UMAP representation of 35 276 cells from 32 breast cancer cell-lines using as cell subspace NMF. Cells are color-coded according to their cell-line of origin. Single-cell transcriptional profiles were normalized with gficf package. (B) Same as (A) but using PC as cell sub-space (C) Accuracy evaluation of mapping method using cross-validation approach using as cell sub-space either NMF (orange) or PCA (green). Each boxplot display accuracy distribution in classifying the 32 cell lines but using as a training set the percentage of cells indicated on the x-axis. Accuracy is defined as the number of correctly classified cells over the total number of mapped cells. (D) Left plot; mapping of the MDAMB468 and CAL51 cells after they were re-sequenced with drop-SEQ technology. Right plot; accuracy of the mapping method in classifying re-sequenced MDAMB468 and CAL51 cells. NMF cell subspace is used. (E) Same as (D) but using PC sub-space. (F) Accuracy evaluation of the mapping method on 14 372 single-cell transcriptomes sequenced with 10x Chromium method for MCF7 parental (P) cell line and three derived subclones (C1,C2,C3) using as cell sub-space either NMF (left) or PCA (right). (G) Performance of the mapping method in classifying 2311 single-cell transcriptomes sequenced using the 10x Chromium method from eleven distinct breast cell lines cell. Performances are reported in terms ROC curve and AUC is also displayed. (H) Same as (G) but using PC cell sub-space.
Next, we investigated whether the sequencing technology could affect the efficacy of the mapping algorithm. Thus, we started mapping into the reference breast cancer cell-line atlas 14 372 single-cell transcriptomes sequenced using the 10x Chromium method from the MCF7 cell line and three subclones derived from it (33). Considering that KPL1 has been documented as an MCF7 derivative cell line (50,51), we found that 13 699 of the 14 372 cells (95.3%) were recognized correctly (Figure 2F) when using NFM as subspace. Using PC instead of NFM as a cell sub-space, the total accuracy was of 94.2% (Figure 2F). Finally, we mapped 2311 single-cell transcriptomes sequenced using the 10x Chromium method from 11 additional breast cell lines included in our atlas and published by Kinker et al. (8). Figure 2G shows classification performance on these cells in terms of ROC (receiver operating characteristic) curve with an AUC of 0.975. The AUC of the ROC curve was 0.876 when the PC subspace was used instead of NFM (Figure 2H).
Next, to demonstrate the reliability of the scMAP tool in correctly contextualize novel sequenced cells on a reference atlas we used it to perform automatic breast cancer subtype classification from single-cell data of patient’s tissue biopsy. To this end, we used 47 692 single-cell transcriptional profiles from 24 treatment-naive primary tumours comprising 7 Triple Negative (TN), 5 HER2-amplified (HER2+) and 12 ER + breast cancer patients (34) (see Materials and methods). Next, we used leave-one-out cross-validation approach to test the accuracy of scMAP in correctly mapping novel patients’ cells and predicting their cancer subtype. With this approach, at each iteration, cells from 23 patients are used to reconstruct the reference BC patient atlas and the remaining cells of the left-out patient mapped and contextualized onto it. As Supplementary Figure S6 shows we obtained an average accuracy of tumour classification of 81% when using NMF as cell sub-space and of 76% when the PC subspace was used instead of NFM.
Overall, these analyses shows that the mapping approach and transfer learning strategy we developed provide reliable results with good accuracy for both mapping and contextualize new cells in a reference atlas.
DISCUSSION
Single-cell RNA-seq provides a cost-effective way to study cell composition of tissues (1–5), cell developmental stages (6), and to elucidate the role of transcriptional heterogeneity in pathological conditions or in response to drug treatments (7). Indeed, large atlases of cells with their relative associated metadata are now routinely released. However, computational methods for the reconstruction of pathway activity at single-cell level useful for their annotation or to contextualize novel sequenced cells in an already available and annotated atlas of cells remain challenging.
Finding an effective way to capture coordinated gene activity at the single-cell level is crucial for single-cell data transformation and subsequent analyses. Such transformation allows representation of cellular state in terms of activity levels of biological processes (i.e. set of genes) rather than through the expression levels of individual genes. Thus, allowing the projection of single-cell data to a quickly biologically interpretable space in which analytic approaches can later be applied. For example, such representations could be used to detect within the same cell type shifts in the portion of cells exhibiting an altered biological process between two phenotypes of interest. Thus improving the identification of dysregulated signalling pathways across pathological conditions, otherwise identified from differential expression (38,52–56) or co-expression (19,57,58–69) analyses that are still challenging for single-cell datasets due to their zero-inflated nature (70–76).
Here, we introduced scGSEA, a novel efficient ready-to-use method that provides an effective and simple workflow for the measurement of pathway activity at single-cell level. Our scGSEA takes advantage of the informative biological signals spreading across the colinearly optimized and additive collection of factors of gene expression values obtained from an NMF model. Indeed, when applied on bulk transcriptional datasets, NMF factors have already been shown to better capture patterns of coordinated gene activities compared to other matrix decomposition methods like SVD ranks or PCA components (43) Recently, NMF has been shown to greatly improve scRNA-seq data clustering and visualization thanks to its inbuilt ability to impute missing values and decompose data into additive parts (26). In applications, we demonstrated that our scGSEA has high accuracy in capturing coordinated gene activity at the single-cell level in both simulated and real dataset comprising hundreds of thousands of cells.
The ‘double dipping’ problem is referred as the problem to use the same dataset for selection and selective analysis that can lead to invalid statistical inferences (77). However, scGSEA tool directly leverages NMF expression latent factors to infer pathway activity at a single-cell level thus avoiding the double dipping problem. On the other hand, since based on NMF, scGSEA also inherits limitations of the NMF model. For example, a limit of this model, like other matrix decomposition techniques, is the choice of the exact number of factors to use. This choice can be made by either using a subjective approach like finding an inflection point in a curve of ranks against an objective (i.e. elbow plot) or with higher precision, but computationally demanding approaches, like jackstraw or k-fold cross-validation. However, when choosing the number of factors to use, to avoid information loss, it is advisable to adopt a cautious approach by selecting a higher number. Thus, we generally recommend using at least 100 NMF factors on a large dataset comprising >10 000 cells, otherwise 50 should suffice.
In this study we also presented scMAP, a transfer learning algorithm that combines text mining data transformation and the k-nearest neighbours’ algorithm to map a query set of cell transcriptional profiles on top of an existing cell atlas. Finding an effective way to ‘re-purpose’ sequenced cells in an already annotated dataset of cells could be helpful in the study of different diseases. For example, we have already shown that we can use breast cancer cell lines for automatic cancer subtype classification starting from the single-cell transcriptomic dataset of patient biopsies (7). Recently, it has also been shown that with transfer learning, we can use the knowledge of sensitive drugs for each cell line to predict the patient's treatment once the patient’s cells were confidently mapped on the reference atlas (78). Here, we demonstrated our method has high accuracy in cell mapping even when we profile cells’ transcriptomes after several culture passages in a different batch or with a different sequencing technique. The proposed mapping method may be applicable in several scenarios; however, it is best suited when the query cells consist of cell types and experimental protocols close to the reference data. Finally, the number of shared genes between the query and reference cells can also impact mapping accuracy. We recommend using all available genes in the reference-building step to guarantee more extensive feature overlap between reference and query cells, which naturally increases the mapping quality.
In summary, we have updated our gficf R package with two novel functionalities, both useful for cell functional annotation. One allows to capture coordinated gene activity at the single-cell level, and another that can re-purpose newly sequenced cells into an already annotated reference dataset. Both tools are provided in an effective and straightforward workflow and are implemented in the framework of our open-source R package gficf available at https://github.com/gambalab/gficf.
CODE AVAILABILITY
R package of gf-icf pipeline and examples of use are available at the following address https://github.com/gambalab/gficf. While scripts to reproduce main figures and analyses are available at the following address https://github.com/gambalab/scGSA_scMAP_manuscript.
DATA AVAILABILITY
All datasets used in the manuscript have been deposited on fighshare at the following DOI https://doi.org/10.6084/m9.figshare.22109414, while the gficf version to reproduce manuscript results can be found on zenodo at the following DOI https://doi.org/10.5281/zenodo.7646881. Single-cell RNA-seq of MDAMB468 and CAL51 cell lines can be found on the GEO database with accession number GSE214827.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: G.G. wrote the manuscript, conceived, and developed the tools. S.P. and G.V. performed NGS experiments while M.F. performed bioinformatics analysis.
Contributor Information
Melania Franchini, Telethon Institute of Genetics and Medicine, Pozzuoli 80078 Naples, Italy; Department of Electrical Engineering and Information Technologies, University of Naples Federico II, 80125 Naples, Italy.
Simona Pellecchia, Telethon Institute of Genetics and Medicine, Pozzuoli 80078 Naples, Italy.
Gaetano Viscido, Telethon Institute of Genetics and Medicine, Pozzuoli 80078 Naples, Italy.
Gennaro Gambardella, Telethon Institute of Genetics and Medicine, Pozzuoli 80078 Naples, Italy; Department of Chemical Materials and Industrial Engineering, University of Naples Federico II, 80125 Naples, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
AIRC [23162]; iPC project [H2020 826121]; Fondazione Telethon.
Conflict of interest statement. None declared.
REFERENCES
- 1. Schaum N., Karkanias J., Neff N.F., May A.P., Quake S.R., Wyss-Coray T., Darmanis S., Batson J., Botvinnik O., Chen M.B.et al.. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018; 562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F.et al.. Mapping the mouse cell atlas by Microwell-Seq. Cell. 2018; 172:1091–1107. [DOI] [PubMed] [Google Scholar]
- 3. Almanzar N., Antony J., Baghel A.S., Bakerman I., Bansal I., Barres B.A., Beachy P.A., Berdnik D., Bilen B., Brownfield D.et al.. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020; 583:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W.et al.. Construction of a human cell landscape at single-cell level. Nature. 2020; 581:303–309. [DOI] [PubMed] [Google Scholar]
- 5. Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M.et al.. The Human cell atlas. Elife. 2017; 6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schiebinger G., Shu J., Tabaka M., Cleary B., Subramanian V., Solomon A., Gould J., Liu S., Lin S., Berube P.et al.. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell. 2019; 176:928–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gambardella G., Viscido G., Tumaini B., Isacchi A., Bosotti R., di Bernardo D.. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 2022; 13:1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kinker G.S., Greenwald A.C., Tal R., Orlova Z., Cuoco M.S., McFarland J.M., Warren A., Rodman C., Roth J.A., Bender S.A.et al.. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020; 52:1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Slovin S., Carissimo A., Panariello F., Grimaldi A., Bouché V., Gambardella G., Cacchiarelli D.. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol. Biol. 2021; 2284:343–365. [DOI] [PubMed] [Google Scholar]
- 10. Shi Jing L., Fathiah Muzaffar Shah F., Saberi Mohamad M., Moorthy K., Deris S., Zakaria Z., Napis S.. A review on bioinformatics enrichment analysis tools towards functional analysis of high throughput gene set data. Curr. Proteom. 2015; 12:14–27. [Google Scholar]
- 11. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oron A.P., Jiang Z., Gentleman R.. Gene set enrichment analysis using linear models and diagnostics. Bioinformatics. 2008; 24:2586–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Camp J.G., Badsha F., Florio M., Kanton S., Gerber T., Wilsch-Bräuninger M., Lewitus E., Sykes A., Hevers W., Lancaster M.et al.. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. 2015; 112:15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goeman J.J., van de Geer S.A., de Kort F., van Houwelingen H.C.. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004; 20:93–99. [DOI] [PubMed] [Google Scholar]
- 15. Kim S.-Y., Volsky D.J.. PAGE: parametric analysis of gene set enrichment. BMC Bioinf. 2005; 6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu D., Smyth G.K.. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012; 40:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noureen N., Ye Z., Chen Y., Wang X., Zheng S.. Signature-scoring methods developed for bulk samples are not adequate for cancer single-cell RNA sequencing data. Elife. 2022; 11:e71994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pont F., Tosolini M., Fournié J.J.. Single-cell signature Explorer for comprehensive visualization of single cell signatures across scRNA-seq datasets. Nucleic Acids Res. 2019; 47:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J.et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan S.J., Yang Q.. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2010; 22:1345–1359. [Google Scholar]
- 21. Lin Y., Wu T.-Y., Wan S., Yang J.Y.H., Wong W.H., Wang Y.X.R.. scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning. Nat. Biotechnol. 2022; 40:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng M., Li Y., Wamsley B., Wei Y., Roeder K.. Integration and transfer learning of single-cell transcriptomes via cFIT. Proc. Natl. Acad. Sci. 2021; 118:e2024383118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lotfollahi M., Naghipourfar M., Luecken M.D., Khajavi M., Büttner M., Wagenstetter M., Avsec Ž., Gayoso A., Yosef N., Interlandi M.et al.. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2022; 40:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stein-O’Brien G.L., Clark B.S., Sherman T., Zibetti C., Hu Q., Sealfon R., Liu S., Qian J., Colantuoni C., Blackshaw S.et al.. Decomposing cell identity for transfer learning across cellular measurements, platforms, tissues, and species. Cell Syst. 2019; 8:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee D.D., Seung H.S.. Learning the parts of objects by non-negative matrix factorization. Nature. 1999; 401:788–791. [DOI] [PubMed] [Google Scholar]
- 26. DeBruine Z.J., Melcher K., Triche T.J.. Fast and robust non-negative matrix factorization for single-cell experiments. 2021; bioRxiv doi:01 September 2021, preprint: not peer reviewed 10.1101/2021.09.01.458620. [DOI]
- 27. Aissa A.F., Islam A.B.M.M.K., Ariss M.M., Go C.C., Rader A.E., Conrardy R.D., Gajda A.M., Rubio-Perez C., Valyi-Nagy K., Pasquinelli M.et al.. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 2021; 12:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gambardella G., di Bernardo D.. A tool for visualization and analysis of single-cell RNA-seq data based on text mining. Front. Genet. 2019; 10:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M.et al.. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015; 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S.et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A.. Fast gene set enrichment analysis. 2021; bioRxiv doi:01 February 2021, preprint: not peer reviewed 10.1101/060012. [DOI]
- 32. Zappia L., Phipson B., Oshlack A.. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 2017; 18:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C.A., Dempster J., Lyons N.J., Burns R.et al.. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018; 560:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pal B., Chen Y., Vaillant F., Capaldo B.D., Joyce R., Song X., Bryant V.L., Penington J.S., Di Stefano L., Tubau Ribera N.et al.. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021; 40:e107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y., Pal B., Lindeman G.J., Visvader J.E., Smyth G.K.. R code and downstream analysis objects for the scRNA-seq atlas of normal and tumorigenic human breast tissue. Sci. Data. 2022; 9:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leskovec J., Rajaraman A., Ullman J.D.. Mining of Massive Datasets Cambridge. 2014; Cambridge: University Press. [Google Scholar]
- 37. Robertson S.E., Jones K.S.. Relevance weighting of search terms. J. Am. Soc. Inf. Sci. 1976; 27:129–146. [Google Scholar]
- 38. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pearson K. LIII. On lines and planes of closest fit to systems of points in space. London, Edinburgh, Dublin Philos. Mag. J. Sci. 1901; 2:559–572. [Google Scholar]
- 40. Laurens van der Maaten, Hinton G.. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 41. McInnes L., Healy J., Melville J.. UMAP: uniform manifold approximation and projection for dimension reduction. 2018; arxiv doi:18 September 2020, preprint: not peer reviewedhttps://arxiv.org/abs/1802.03426.
- 42. Clark B.S., Stein-O’Brien G.L., Shiau F., Cannon G.H., Davis-Marcisak E., Sherman T., Santiago C.P., Hoang T.V., Rajaii F., James-Esposito R.E.et al.. Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron. 2019; 102:1111–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stein-O’Brien G.L., Arora R., Culhane A.C., Favorov A.V., Garmire L.X., Greene C.S., Goff L.A., Li Y., Ngom A., Ochs M.F.et al.. Enter the matrix: factorization uncovers knowledge from omics. Trends Genet. 2018; 34:790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J., Jia J., Zhu F., Ma X., Han B., Wei X., Tan C., Jiang Y., Chen Y.. Analysis of bypass signaling in EGFR pathway and profiling of bypass genes for predicting response to anticancer EGFR tyrosine kinase inhibitors. Mol. Biosyst. 2012; 8:2645–2656. [DOI] [PubMed] [Google Scholar]
- 45. Shi K., Wang G., Pei J., Zhang J., Wang J., Ouyang L., Wang Y., Li W.. Emerging strategies to overcome resistance to third-generation EGFR inhibitors. J. Hematol. Oncol. 2022; 15:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He J., Huang Z., Han L., Gong Y., Xie C.. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 2021; 59:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L., Zhu H., Liao Y., Wu W., Liu L., Liu L., Wu Y., Sun F., Lin H.. Inhibition of wnt/β-catenin pathway reverses multi-drug resistance and EMT in Oct4+/Nanog+ NSCLC cells. Biomed. Pharmacother. 2020; 127:110225. [DOI] [PubMed] [Google Scholar]
- 48. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Macnair W., Gupta R., Claassen M.. psupertime: supervised pseudotime analysis for time-series single-cell RNA-seq data. Bioinformatics. 2022; 38:i290–i298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Demichelis F., Greulich H., Macoska J.A., Beroukhim R., Sellers W.R., Garraway L., Rubin M.A.. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008; 36:2446–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Capes-Davis A., Theodosopoulos G., Atkin I., Drexler H.G., Kohara A., MacLeod R.A.F., Masters J.R., Nakamura Y., Reid Y.A., Reddel R.R.et al.. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int. J. Cancer. 2010; 127:1–8. [DOI] [PubMed] [Google Scholar]
- 52. Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M.et al.. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015; 16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kharchenko P.V., Silberstein L., Scadden D.T.. Bayesian approach to single-cell differential expression analysis. Nat Meth. 2014; 11:740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Van den Berge K., Perraudeau F., Soneson C., Love M.I., Risso D., Vert J.-P., Robinson M.D., Dudoit S., Clement L.. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018; 19:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deshpande T.M., Pandey A.K., Shyama S.K.. Review: breast cancer and etiology. Trends Med. 2017; 17:1–7. [Google Scholar]
- 58. Specht A.T., Li J.. LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics. 2017; 33:764–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matsumoto H., Kiryu H., Furusawa C., Ko M.S.H., Ko S.B.H., Gouda N., Hayashi T., Nikaido I.. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-seq during differentiation. Bioinformatics. 2017; 33:2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chan T.E., Stumpf M.P.H., Babtie A.C.. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017; 5:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Papili Gao N., Ud-Dean S.M.M., Gandrillon O., Gunawan R.. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics. 2018; 34:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanchez-Castillo M., Blanco D., Tienda-Luna I.M., Carrion M.C., Huang Y.. A Bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data. Bioinformatics. 2018; 34:964–970. [DOI] [PubMed] [Google Scholar]
- 63. Woodhouse S., Piterman N., Wintersteiger C.M., Göttgens B., Fisher J.. SCNS: a graphical tool for reconstructing executable regulatory networks from single-cell genomic data. BMC Syst. Biol. 2018; 12:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qiu X., Rahimzamani A., Wang L., Ren B., Mao Q., Durham T., McFaline-Figueroa J.L., Saunders L., Trapnell C., Kannan S.. Inferring causal gene regulatory networks from coupled single-cell expression dynamics using scribe. Cell Syst. 2020; 10:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gambardella G., Moretti M.N.M.N., De Cegli R., Cardone L., Peron A., Di Bernardo D.. Differential network analysis for the identification of condition-specific pathway activity and regulation. Bioinformatics. 2013; 29:1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gambardella G., Peluso I., Montefusco S., Bansal M., Medina D.L., Lawrence N., di Bernardo D.. A reverse-engineering approach to dissect post-translational modulators of transcription factor's activity from transcriptional data. BMC Bioinf. 2015; 16:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huynh-Thu V.A., Sanguinetti G.. Combining tree-based and dynamical systems for the inference of gene regulatory networks. Bioinformatics. 2015; 31:1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim S. ppcor: an R package for a fast calculation to semi-partial correlation coefficients. Commun. Stat. Appl. Methods. 2015; 22:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moerman T., Aibar Santos S., Bravo González-Blas C., Simm J., Moreau Y., Aerts J., Aerts S.. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019; 35:2159–2161. [DOI] [PubMed] [Google Scholar]
- 70. Pratapa A., Jalihal A.P., Law J.N., Bharadwaj A., Murali T.M.. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods. 2020; 17:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kang Y., Thieffry D., Cantini L.. Evaluating the reproducibility of single-cell gene regulatory network inference algorithms. Front. Genet. 2021; 12:617282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen S., Mar J.C.. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinf. 2018; 19:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aubin-Frankowski P.-C., Vert J.-P.. Gene regulation inference from single-cell RNA-seq data with linear differential equations and velocity inference. Bioinformatics. 2020; 36:4774–4780. [DOI] [PubMed] [Google Scholar]
- 74. Squair J.W., Gautier M., Kathe C., Anderson M.A., James N.D., Hutson T.H., Hudelle R., Qaiser T., Matson K.J.E., Barraud Q.et al.. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021; 12:5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang T., Li B., Nelson C.E., Nabavi S.. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinf. 2019; 20:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Soneson C., Robinson M.D.. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods. 2018; 15:255–261. [DOI] [PubMed] [Google Scholar]
- 77. Neufeld A., Gao L.L., Popp J., Battle A., Witten D. Inference after latent variable estimation for single-cell RNA sequencing data. Biostatistics. 2022; 10.1093/biostatistics/kxac047. [DOI] [PubMed] [Google Scholar]
- 78. Osorio D., McGrail D.J., Sahni N., Yi S.S.. Drug combination prioritization for cancer treatment using single-cell RNA-seq based transfer learning. 2022; bioRxiv doi:09 April 2022, preprint: not peer reviewed 10.1101/2022.04.06.487357. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used in the manuscript have been deposited on fighshare at the following DOI https://doi.org/10.6084/m9.figshare.22109414, while the gficf version to reproduce manuscript results can be found on zenodo at the following DOI https://doi.org/10.5281/zenodo.7646881. Single-cell RNA-seq of MDAMB468 and CAL51 cell lines can be found on the GEO database with accession number GSE214827.