

Abstract
Conventional gene expression quantification approaches, such as microarrays or quantitative PCR, have similar variations of estimates for all genes. However, next-generation short-read or long-read sequencing use read counts to estimate expression levels with much wider dynamic ranges. In addition to the accuracy of estimated isoform expression, efficiency, which measures the degree of estimation uncertainty, is also an important factor for downstream analysis. Instead of read count, we present DELongSeq, which employs information matrix of EM algorithm to quantify uncertainty of isoform expression estimates to improve estimation efficiency. DELongSeq uses random-effect regression model for the analysis of DE isoform, in that within-study variation represents variable precision in isoform expression estimation and between-study variation represents variation in isoform expression levels across samples. More importantly, DELongSeq allows 1 case versus 1 control comparison of differential expression, which has specific application scenarios in precision medicine (such as before versus after treatment, or tumor versus stromal tissues). Through extensive simulations and analysis of several RNA-Seq datasets, we show that the uncertainty quantification approach is computationally reliable, and can improve the power of differential expression (DE) analysis of isoforms or genes. In summary, DELongSeq allows for efficient detection of differential isoform/gene expression from long-read RNA-Seq data.
INTRODUCTION
The application of long-read RNA sequencing technologies has gained popularity due to its potential to allow isoform profiling in an unbiased manner, including novel isoform detection and quantification (1–3). It has become a promising platform for transcriptomics studies because it enables more accurate and a wider range of measurement of expression at isoform level. One of major applications of long-read RNA-Seq is to detect differential isoform expression across experimental conditions, which is of great biological interest due to its direct relevance to protein function and disease pathogenesis (4–6). Previous evidence suggests that almost all human multi-exon genes have more than one isoform (7), and different isoforms are often differentially expressed (DE) across tissues, developmental stages, disease conditions, and even across cells from the same tissue (8,9). Therefore, detection of DE isoforms is important for understanding complicated biological mechanisms and for mapping disease susceptibility genes (10–22).
In recent years, long-read methods designed for isoform expression quantification has emerged (23), such as Mandalorion (24), TALON (25), LIQA (26), FLAIR (27), NanoCount (28), ScanExitronLR (29) and Mini-IsoQLR (30). Among them, TALON compares reads to existing gene and transcript models to create novel models, LIQA corrects sequencing bias in expression estimation and FLAIR clusters alignments into groups and collapses them into isoforms. However, detection of differentially expressed (DE) isoforms using long-read RNA-Seq is still challenging because of the uncertainty in isoform expression estimation owing to ambiguous reads alignment and the variability in precision of the estimates across samples. Popular short-read analysis methods such as DESeq (31), DESeq2 (32) and EdgeR (33) expect read counts as input. These approaches treat the number of fragments originating from each isoform using other software as fixed observed value when detecting differential expressed isoforms. However, ignorance of the variability of expression estimation can result in increased false positive detection because the isoform expression estimation may be systematically biased. Existing short-read approaches such as Cufflinks (34), BitSeq (35) and EBSeq (36) can appropriately account for uncertainty in isoform expression estimates. However, these programs cannot be used to perform 1 case versus 1 control comparison of differential expression which has specific application scenarios in precision medicine (such as before versus after treatment, or tumor versus stromal tissues). Recognizing these limitations of existing methods, we assume that a powerful approach for detecting DE isoforms ideally should be able to account for isoform expression estimation uncertainty and variation in the precision of isoform expression estimates across biological replicates, and compatible to variety of application scenarios such as 1 case versus 1 control comparison.
In this article, we developed DELongSeq, which employs information matrix of expectation–maximization (EM) algorithm to quantify the uncertainty of isoform expression estimates. It can be naturally paired with LIQA which uses EM algorithm to quantify gene or isoform expression levels. Rather than read count, DELongSeq accurately detect DE isoform based on expression and uncertainty estimates using random-effect regression model. This model is to synthesize results from multiple studies while accounting for varying standard errors of the effect estimates by explicitly allowing for different sources of variability: within- and between-study variation. Its mathematical model matches perfectly with the analysis of DE isoform in that within-study variation represents variable precision in isoform expression estimation and between-study variation represents variation in isoform expression levels across samples.
MATERIALS AND METHODS
Weighted complete likelihood function of DELongSeq
Given a gene of interest, let 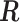 denote the set of reads that are mapped to the gene of interest, and
denote the set of reads that are mapped to the gene of interest, and 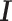 denote the set of known isoforms. For a specific isoform
denote the set of known isoforms. For a specific isoform 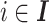 , let
, let 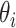 denote its relative abundance, with
denote its relative abundance, with 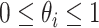 and
and 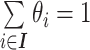 and
and 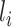 denote its length. For each single-molecule long-read
denote its length. For each single-molecule long-read 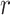 , let
, let 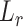 denote its length. The probability that a read originates from isoform
denote its length. The probability that a read originates from isoform 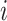 is
is 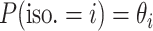 . We define
. We define 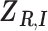 as a
as a  matrix with
matrix with 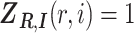 if long-read
if long-read  is generated from a molecule that is originated from isoform
is generated from a molecule that is originated from isoform 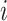 , and
, and 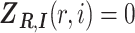 otherwise. For isoform quantification, our goal is to estimate
otherwise. For isoform quantification, our goal is to estimate 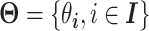 based on RNA-seq long-reads mapped to the gene.
based on RNA-seq long-reads mapped to the gene.
With the notation above, the complete data likelihood of the RNA-seq data can be written as
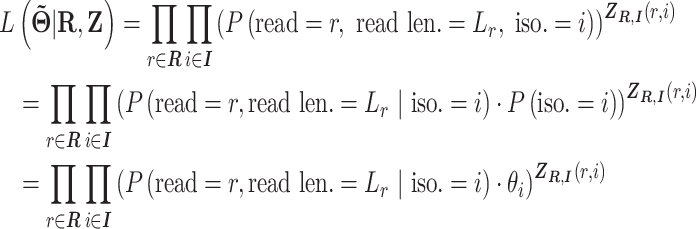 |
This formula is based the fact that given the isoform origin, the probability of observing read alignment can be inferred. The conditional probability of read  derived from isoform
derived from isoform 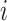 with length
with length  can be directly computed from LIQA or similar software tools. For LIQA, the complete data likelihood is
can be directly computed from LIQA or similar software tools. For LIQA, the complete data likelihood is
 |
and the update procedure of the EM algorithm is as follows:
E-step: We calculate function
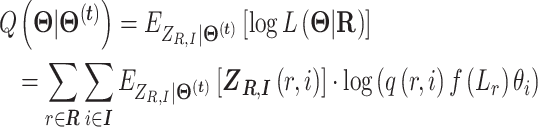 |
where 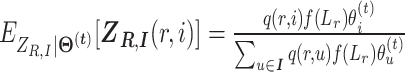 .
.
M-step: We maximize function 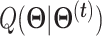 and have
and have
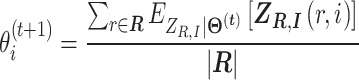 |
The EM algorithm consists of alternating between the E- and M-steps until convergence. We start the algorithm with 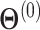 assuming all isoforms are equally expressed and stop when the log likelihood is no longer increasing significantly.
assuming all isoforms are equally expressed and stop when the log likelihood is no longer increasing significantly.
Quantifying isoform expression uncertainty of EM estimator
Variance of parameter 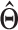 will be estimated by inferring observed information
will be estimated by inferring observed information 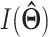 . The complete data likelihood is
. The complete data likelihood is
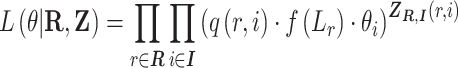 |
and the procedure of computing observed information in EM algorithm (37) is as follows:
Let 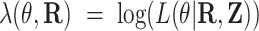 ,
, 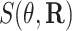 be the gradient of
be the gradient of 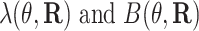 be the negative of the associated second derivative.
be the negative of the associated second derivative.
By differentiation:
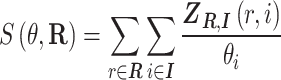 |
 |
Then, we define 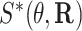 and
and 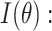
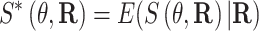 |
 |
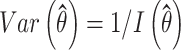 |
Therefore, the 95% confidence interval of 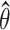 is
is 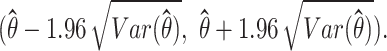
Random-effects regression for testing differential expression
Let 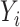 denote the estimated expression level of an isoform of a gene, as represented by transcript per million (TPM) value for subject i, and
denote the estimated expression level of an isoform of a gene, as represented by transcript per million (TPM) value for subject i, and 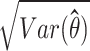 denote the standard error of the estimated TPM.
denote the standard error of the estimated TPM. 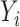 can be obtained from software tools such as LIQA that estimate isoform-specific gene expression (
can be obtained from software tools such as LIQA that estimate isoform-specific gene expression (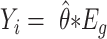 , where
, where 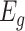 represents the TPM of gene g). The variable of interest (e.g. disease status, treatment group, etc.) is denoted by
represents the TPM of gene g). The variable of interest (e.g. disease status, treatment group, etc.) is denoted by  . In differential expression analysis, the TPM values are usually log-transformed so that their distribution is approximately normal. A random-effects regression model is used to account for variable uncertainty in isoform expression estimation when testing the relationship between isoform expression and covariate
. In differential expression analysis, the TPM values are usually log-transformed so that their distribution is approximately normal. A random-effects regression model is used to account for variable uncertainty in isoform expression estimation when testing the relationship between isoform expression and covariate 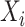 :
:
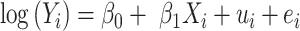 |
where 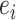 represents random error and
represents random error and 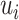 is the estimation uncertainty for
is the estimation uncertainty for 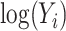 . For this random-effects model, we have: 1)
. For this random-effects model, we have: 1) 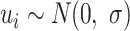 , 2)
, 2)  , 3)
, 3) 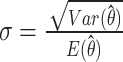 based on delta method and 4) the n observations are independent.
based on delta method and 4) the n observations are independent.
With random-effects model, we can detect differential expression both at the gene level and the isoform level. Specifically, we test the null hypothesis of no differential expression between cases and controls, i.e. 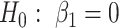 versus
versus 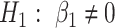 . Rejection of this null hypothesis indicates that isoform/gene is differentially utilized between cases and controls. This hypothesis can be tested using likelihood ratio test with test statistic
. Rejection of this null hypothesis indicates that isoform/gene is differentially utilized between cases and controls. This hypothesis can be tested using likelihood ratio test with test statistic 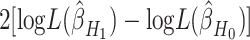 . Under the null hypothesis, this test statistic approximately follows a
. Under the null hypothesis, this test statistic approximately follows a 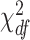 distribution in which
distribution in which 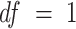 . Statistical inference was implemented using metatest (38) package in R.
. Statistical inference was implemented using metatest (38) package in R.
Datasets and evaluations
We first conducted simulations to evaluate the performance of DELongSeq and compared it with other methods for DE isoforms analysis based on GENCODE v24 annotation. To simulate a realistic dataset with known ground truth, we used NanoSim (39) to generate RNA-seq data. The NanoSim program assigns expression value for each isoform following a mixed power/exponential law. Additionally, it simulates common sources of systematic bias in the abundance and distribution of produced reads by in silico library preparation and sequencing. The use of NanoSim facilitates the comparison of different methods under a more realistic setting than evaluations based on simulating count data directly without the full RNA-seq protocol. To simulate RNA-seq reads using NanoSim, the human genome sequence (hg38) was downloaded from UCSC Genome Browser (https://genome.ucsc.edu/). We simulated Nanopore reads for 10 cases and 10 controls (∼1 million reads per subject). To make our simulated data close to those seen in real studies, the isoform relative abundances of each gene were sampled from a Dirichlet distribution in which the mean and variance parameters were determined from a real human acute myeloid leukemia (AML) dataset (case versus control). The simulated RNA-seq reads were mapped to the hg38 reference human genome using minimap2 (40) with default options. In all results presented in this paper, we only considered genes with at least two isoforms. Additionally, we required a gene to have at least 3 mapped reads on average across all RNA-seq samples. We also evaluated the impact of sample size by analyzing a subset of n cases and n controls (n = 5) randomly chosen from the full simulated dataset. There were 6321 genes (18 478 isoforms) for the GENCODE annotation (Supplementary Figure S1).
Next, we evaluated the performance of DELongSeq in DE isoform detection using an RNA-seq dataset generated from esophageal squamous epithelial cell (ESCC) (41). This dataset includes PacBio SMRT reads generated from normal immortalized and cancerous esophageal squamous epithelial cell lines. The RNA-seq data were downloaded from Gene Expression Omnibus (PRJNA515570). We applied DELongSeq, FLAIR and TALON to detect differential isoform usage between normal-like and cancer cells. Specifically, given read counts of 8365 endogenous GENCODE genes with at least two annotated isoforms, we applied DELongSeq to quantify isoform expression and variations, yielding accurate estimation uncertainty. Then, differential expression isoform comparison was conducted. Known splicing differences in existing studies were treated as ground truth to evaluate DELongSeq's performance in characterizing isoform usage across samples.
Another real data used for evaluation was acquired from Huang et al. (42), which includes PacBio sequencing data of 10 Gastric Cancer (GC) cell lines. These cell lines were predicted to cover four major GC molecular subtypes (chromosomal unstable (CIN), Epstein–Barr positive (EBV), genome stable (GS) and microsatellite unstable (MSI)), where same GC molecular subtypes are considered to be relatively homogenous. In this study, raw data in FASTQ format were downloaded from SRA [PRJNA635275] and GEO [GSE157750] and aligned to hg38 reference genome with GENCODE gene annotation using minimap2 (40). For each sample, we processed the BAM file and quantify isoform expression and uncertainty using DELongSeq for 3367 genes with 12 923 isoforms. Given expression estimates and variance, DE analysis between each pair of 10 GC cell lines was conducted. To validate the performance of DELongSeq, we compared alternative promoter expression difference between cell lines at gene MET and FGFR4, since these two genes were previously reported to have differential promoter usage between conditions.
RESULTS
We evaluated the performance of DELongSeq on both simulated and real RNA-Seq data and compared it with two existing algorithms, including FLAIR and TALON. To make a fair comparison, all programs used alignment file (BAM format) as input. However, these two methods do not explicitly model estimation uncertainty for each isoform. In our analysis, we used the default option in these two methods. A transcript was declared to be DE if its FDR-adjusted p-value was less than the nominal FDR level or if its posterior probability of DE was greater than one – nominal FDR level. While TALON is not specifically designed for differential expression analysis of isoforms, given the popularity of this method, it might be tempting for an end user to use it identify DE isoforms using read counts per transcript. Therefore, we included TALON in our comparisons by using Mann–Whitney U test (thereafter referred to as TALON + MW) because TALON only estimates isoform expression without estimation uncertainty. This test takes isoform expression estimates from TALON as input, and uses Mann–Whitney U test to identify DE isoform and genes.
Expression and uncertainty quantification on simulation data
Reliable estimation of isoform expression level is critical for DE isoform detection. In DELongSeq, a key step is the uncertainty quantification. In simulation study, we computed a set of measurements to evaluate the estimation accuracy of each method. First, we measured the similarity between the estimated isoform relative abundance and the ground truth by calculating Spearman's correlation. Second, we measured the length and true value coverage probability of 95% confidence interval (95% CI). Figure 1A shows the scatter plot between estimated and true values of isoform expression (global isoform expression and isoform relative abundances). Spearman's correlations were calculated. Figure 1B gives 95% CIs length distribution and true value coverage probability of relative abundance estimates for the DELongSeq. For the uncertainty quantification, the median length of 95% CI ranges from 0.57 to 2.6 TPM at different sequencing depths. The coverage probability ranges from 93% to 99%. Lower read coverage yields longer 95% CI length (measured in TPM) due to less informative reads for statistical inference. To evaluate the robustness of DELongSeq to a low read coverage, we analyzed major isoforms (the isoform with highest fraction among all isoforms for a gene) from genes with read coverage <10. DELongSeq yields similar Spearman's correlation (0.73 and 0.68) of TPM and relative abundance estimates compared to higher read coverage genes. For major isoform, we find that the expression and uncertainty quantification accuracy of DELongSeq is nearly unchanged (5% less). For other isoforms, the accuracy is 20% lower when 10 less reads were mapped to the gene in the analysis.
Figure 1.

Performance of DELongSeq on isoform expression and uncertainty quantification using simulation data. (A) Scatter plots of true isoform expression and estimated isoform expression, global isoform expression (left) and isoform relative abundances (right). Spearman's correlation between true value and estimation were calculated. (B) Violin plots of 95% CI length (measured in TPM) in log-scale for genes with at least 5, 10 and 20 coverage reads. True value coverage probabilities of DELongSeq were calculated.
Performance of isoform-based tests on simulation data
Next, we evaluated the performance of DELongSeq in DE isoform detection, and compared it with other methods including FLAIR, TALON + MW. All methods were run with the same input dataset. We set a threshold 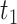 on mean isoform level difference between cases and controls, to define the ground truth of DE isoform. An isoform was considered to be a DE event if the mean expression level difference, denoted by
on mean isoform level difference between cases and controls, to define the ground truth of DE isoform. An isoform was considered to be a DE event if the mean expression level difference, denoted by 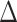 , was greater than
, was greater than 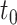 . To evaluate power with different effect sizes, the value of
. To evaluate power with different effect sizes, the value of 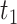 was set at 0.5 and 1 in TPM, respectively. To evaluate type I error rate, a true non-DE event was defined as an isoform with Δexon = 0. In practice, we encountered many instances of exons that showed statistically significant DE events but exhibited exon-inclusion level difference that is too small to warrant biological significance. Therefore, we required an isoform to show expression difference >0.5 in order to be declared as a DE event.
was set at 0.5 and 1 in TPM, respectively. To evaluate type I error rate, a true non-DE event was defined as an isoform with Δexon = 0. In practice, we encountered many instances of exons that showed statistically significant DE events but exhibited exon-inclusion level difference that is too small to warrant biological significance. Therefore, we required an isoform to show expression difference >0.5 in order to be declared as a DE event.
Since different methods use different criteria to filter out isoforms with invalid results (failure of numerical algorithms, or small number of alignment reads, etc.), the numbers of tests returned by each method are quite different. To make a fair comparison, we calculated type I error rates and power in two ways. In the first approach, the calculations were based on the true number of DE and non-DE events in the input data, which include those failed to be analyzed by each method. Including all events in the input data allows us to better assess each method's sensitivity and specificity. In real studies, it is desirable to have a method that yields valid results for all events. In the second approach, the type I error rates and power of each method were calculated based on its own returned test results. The denominators in these calculations can be substantially different among methods.
Figure 2A and B show the comparison results based on isoform when DELongSeq and FLAIR were evaluated using the approach when 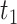 was 0.5. The number of DE events detected by DELongSeq ranged from 2623 to 2860, and only 1.58 to 2.52% of these events were false positives. FLAIR detected 2170–2419 events with type I error rates 2.61 to 2.83%. To control for multiple testing, we also evaluated the false discovery rate of each method. Both DELongSeq and FLAIR had false discovery rate controlled at the 5% level. In power comparison, all methods had increased power as the threshold value
was 0.5. The number of DE events detected by DELongSeq ranged from 2623 to 2860, and only 1.58 to 2.52% of these events were false positives. FLAIR detected 2170–2419 events with type I error rates 2.61 to 2.83%. To control for multiple testing, we also evaluated the false discovery rate of each method. Both DELongSeq and FLAIR had false discovery rate controlled at the 5% level. In power comparison, all methods had increased power as the threshold value 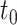 increased because differentially expressed isoforms with larger relative abundance level difference were easier to detect. The power of DELongSeq was higher than FLAIR under different sample sizes because model based on isoform relative abundance allows DELongSeq to select out most confidence isoforms, whereas count-based method only use reads mapped and ignore estimation ambiguity elsewhere even if there are less informative reads mapped to the gene. We observed similar patterns at other threshold values of
increased because differentially expressed isoforms with larger relative abundance level difference were easier to detect. The power of DELongSeq was higher than FLAIR under different sample sizes because model based on isoform relative abundance allows DELongSeq to select out most confidence isoforms, whereas count-based method only use reads mapped and ignore estimation ambiguity elsewhere even if there are less informative reads mapped to the gene. We observed similar patterns at other threshold values of 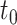 = 1. Figure 2(C) (D) show the comparison results of DELongSeq and FLAIR based on the second approach. Next, we compared with TALON + MW. To make a fair comparison with TALON, we focused on those differential expressed isoforms that were analyzed by TALON. The type I error rates of TALON + MW (5.12–8.67%) exceeded the nominal level, especially when sample size was small. Additionally, DELongSeq had greater power than TALON + MW for all scenarios we considered (Supplementary Figure S2). Moreover, we evaluated the performance difference between DELongSeq using random effect model and fixed effect model. Figure S7A shows the comparison of power in detecting differential expression isoform for all scenarios. Random effect model has 1%-8% power improvement compared to fixed effect model. Random effect model is more robust to lower sample size and small expression difference (Supplementary Tables S1 and S2). Also, we assessed the impact of isoform length on DELongSeq. We divided the isoforms into two categories based on median of the length (isoform length > median, isoform length < median) and summarized the power for each group of isoforms. Supplementary Figure S7B shows that DELongSeq has 0.18 higher power on short isoform in average. This is not surprising because isoforms with longer length are more challenging to estimate expression, which is due to the fact that they are less likely to be fully covered by sequencing reads compared short isoform.
= 1. Figure 2(C) (D) show the comparison results of DELongSeq and FLAIR based on the second approach. Next, we compared with TALON + MW. To make a fair comparison with TALON, we focused on those differential expressed isoforms that were analyzed by TALON. The type I error rates of TALON + MW (5.12–8.67%) exceeded the nominal level, especially when sample size was small. Additionally, DELongSeq had greater power than TALON + MW for all scenarios we considered (Supplementary Figure S2). Moreover, we evaluated the performance difference between DELongSeq using random effect model and fixed effect model. Figure S7A shows the comparison of power in detecting differential expression isoform for all scenarios. Random effect model has 1%-8% power improvement compared to fixed effect model. Random effect model is more robust to lower sample size and small expression difference (Supplementary Tables S1 and S2). Also, we assessed the impact of isoform length on DELongSeq. We divided the isoforms into two categories based on median of the length (isoform length > median, isoform length < median) and summarized the power for each group of isoforms. Supplementary Figure S7B shows that DELongSeq has 0.18 higher power on short isoform in average. This is not surprising because isoforms with longer length are more challenging to estimate expression, which is due to the fact that they are less likely to be fully covered by sequencing reads compared short isoform.
Figure 2.

Type I error and power of DELongSeq, FLAIR and TALON in DE isoform detection with different sample sizes and thresholds. Calculations were based on all DE and non-DE isoforms in the input data. Significance was evaluated at the 5% significance level. An isoform with true TPM difference > 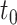 was defined as a true DE gene. (A) 10 versus 10 with
was defined as a true DE gene. (A) 10 versus 10 with 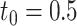 . (B) 5 versus 5 with
. (B) 5 versus 5 with 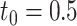 . (C) 10 versus 10 with
. (C) 10 versus 10 with 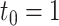 . (D) 5 versus 5 with
. (D) 5 versus 5 with 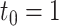 .
.
Performance of gene-based tests on simulation data
Next, we evaluated the performance of DELongSeq in gene-based analysis and compared it with FLAIR and TALON + MW. A gene was considered DE gene when the log fold change between cases and controls, denoted by Δgene, was greater than t0. Log fold change, which represents the mean gene expression difference in log scale, is determined based on a real human acute myeloid leukemia (AML) dataset. The threshold values of t0 varied at 0.5 and 1, respectively. We compared the type I error rates and power among different methods using two ways, similar to those employed in isoform-based comparisons. Figure 3 shows that empirical type I error rates and power of DELongSeq, FLAIR and TALON + MW based on true DE and non-DE genes in the input data under Ensembl annotations. All methods had type I error rates controlled at the 5% significance level when n was 10; however, when n was 5, the type I error rate of TALON + MW was above 5%. Furthermore, DELongSeq had higher power than the other methods for most situations, and method-specific results indicate a similar pattern. The power of TALON is lower than the other methods.
Figure 3.

Type I error and power of DELongSeq, FLAIR and TALON in DE gene detection with different sample sizes and thresholds. Calculations were based on all DE and non-DE genes in the input data. Significance was evaluated at the 5% significance level. A gene with true TPM difference > 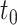 was defined as a true DE gene. (A) 10 versus 10 with
was defined as a true DE gene. (A) 10 versus 10 with 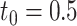 . (B) 5 versus 5 with
. (B) 5 versus 5 with 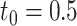 . (C) 10 versus 10 with
. (C) 10 versus 10 with  . (D) 5 versus 5 with
. (D) 5 versus 5 with 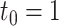 .
.
Evaluation of DELongSeq on 1 versus 1 comparisons
An important feature of DELongSeq is 1 case versus 1 control comparison because it can give estimate of variation even when there is only one sample. Since there are no biological replicates, the null hypothesis would be different than typical gene expression studies, but 1 versus 1 comparisons have specific application scenarios in practice. Since linear regression model requires multiple samples to perform the test, we performed 5 versus 5 (linear regression) and compare with 1 versus 1 pairwise comparison between case and control groups using DELongSeq. Figure 4A and B show that DELongSeq performed better than simple linear regression model in 5 versus 5 comparison. The power of DELongSeq was relatively stable as the sample size decreased, especially when sample size is 1. For DELongSeq, the average power of DELongSeq is higher in 10 versus 10 compared to 5 versus 5 for both isoform- and gene-level but not significantly (P-value = 0.47 and 0.19). These results demonstrate the advantage of DELongSeq in DE gene and isoform analysis. The poor performance of simple linear regression-based method is possibly due to the fact that it requires relatively high sample size, which might limit the performance in real study. Indeed, this challenge motivated DELongSeq to quantify expression estimate uncertainty. Compared to count-based regression model, variance estimates model is more robust to low sample size and allows 1 case versus 1 control comparison.
Figure 4.

Evaluation of 1 case versus 1 control comparison and impact of expression uncertainty on DE isoform and gene detection using DELongSeq (red), FLAIR (green), TALON (blue) and simple linear regression(grey). (A, B) Simulation results power of DE isoform(A) and gene (B) detection using DELongSeq 1 versus 1 pairwise comparison and simple linear regression model based on different sample sizes. The average and standard deviation of power for DELongSeq were calculated. (C, D) Simulation results power of DE isoform (C) and gene (D) detection using DELongSeq, FLAIR and TALON. Isoforms and genes are stratified by the expression estimate uncertainty (variance < 33% quantile, 33% ≤ variance < 66% quantile, variance ≥ 66%).
Moreover, we evaluated the impact of uncertainty level on DE isoform detection. Specifically, we divided isoforms in the simulated data into three groups based on the quantile of expression estimate variance (low: [0, 33%); medium: [33, 66%); high: [66, 100%]). We compared with FLAIR and TALON + MW. Figure 4C and D show that all methods tend to have reduced power for isoforms with higher level of uncertainty. However, TALON + MW dropped significantly as the uncertainty level increased. In contrast, DELongSeq and FLAIR were relatively stable as uncertainty level increased. These results show that DELongSeq and FLAIR are more robust to the impact of estimation uncertainty (Supplementary Figures S3 and S4).
Application to PacBio data on esophageal squamous epithelial cells (ESCC)
Next, we evaluated the performance of DELongSeq in DE isoform detection using an RNA-seq dataset generated to study esophageal squamous cell carcinoma (ESCC). This dataset includes PacBio SMRT reads generated from normal immortalized and cancerous esophageal squamous epithelial cell lines. The RNA-seq data were downloaded from Gene Expression Omnibus (PRJNA515570). We applied DELongSeq, FLAIR and TALON + MW to detect differential isoform usage between normal-like and cancer cells. Known splicing differences in existing studies were treated as ground truth to evaluate DELongSeq's performance in characterizing isoform usage across samples.
Figure 5A and B show the number of DE isoforms detected by each method. 89% of the genes detected by DELongSeq were detected by at least another method (FLAIR and TALON + MW), and the corresponding numbers were 90% for FLAIR, 74% for TALON + MW. Given the high percentage of unique isoforms detected by FLAIR and TALON + MW, we next examined if the Sashimi plots of corresponding genes detected by FLAIR and TALON + MW show empirical evidence supporting the conclusion. Our results indicate that the average number of read coverage at these genes are 8.2, whereas this number of unique detected genes by DELongSeq was 21.6. This indicates the lack of concordance for FLAIR and TALON + MW with other methods is likely due to its inflated false positive rates (43).
Figure 5.

Performance of DELongSeq using ESCC data. (A, B) DE genes and isoforms detected by different methods for ESCC versus normal cells. (C) Examination of DE isoform usage detected by DELongSeq but missed by FLAIR and TALON. Sashimi plots of gene INTS12. Informative exonic regions were red squares. (D) Examination of non-DE isoform usage for DELongSeq. Sashimi plots of gene ACSL3. Informative exonic regions were red squares.
For isoforms that were detected by DELongSeq but missed by FLAIR and TALON + MW, we searched for empirical evidence in RNA-seq coverage plot. Among the 356 genes detected by DELongSeq only, 35.1% have more than three isoforms, which is significantly higher than the corresponding percentage among the remaining 4302 qualified genes (21.5%). We randomly picked five genes among the 356 genes, and generated coverage plots using the Sashimi plot feature in IGV to verify our results (Supplementary Figures S5 and S6). For example, gene INTS12 has more than two annotated isoforms and multiple alternatively spliced exons. The boxed areas in Figure 5C shows visual evidence of DE between normal-like and cancer cells in this gene. The P-value from DELongSeq was 0.012, whereas this gene was missed by FLAIR and TALON + MW. For gene ACSL3, we generated coverage plot for alternatively spliced exon. DELongSeq detected no DE events with p-value = 0.323, estimated isoform expression level difference = 0.025 and 95% CI = [0.07, 0.31]). The read coverage difference of Sashimi plot between normal-like and cancerous ESCC in Sashimi plots indicates the equal usage of isoforms (NM_001134422, NM_0011134423, NM_001282765) which include this exon in both ESCC and normal cells. Both results were consistent, suggesting better performance of DELongSeq.
Application to PacBio data on gastric cancer (GC)
Although designed primarily to compare gene/isoform expression between two groups of samples, DELongSeq can also perform 1 versus 1 DE comparison as shown in simulation studies above, which typically treats uncertainty estimates as statistical test variance. There are many real-world scenarios where 1 versus 1 DE comparison would be important, but is typically overlooked by traditional studies: for example, the comparison of stromal tissues versus cancerous tissues in an individual patient, or the comparison of splicing differences in two isogenic cell lines with the knockout/knockdown of one specific gene. To the best of our knowledge, the only other approach to perform this analysis explicitly is Cuffdiff (34). Due to the sparsity of splicing informative reads, few methods are able to quantify estimation uncertainty. To evaluate the performance of DELongSeq in 1 versus 1 comparison in real data, we analyzed a gastric cancer cell data set, which are Iso-seq data generated from 10 different cell lines. These cell lines are selected to represent four TCGA GC cells subtypes (CIN—chromosomal unstable, EBV—Epstein-Barr virus positive, GS—genome stable and MSI—microsatellite unstable). We performed 1 versus 1 isoform DE analysis for these four GC subtypes across the 10 cell lines in this data set. 1 versus 1 sample comparison requires high estimation accuracy with less estimation uncertainty, making it challenging to detect DE isoform. However, by selecting out high coverage genes and appropriate statistical modeling, DELongSeq is able to detect DE isoform in 1 versus 1 comparison. Figure 6A shows the heatmap of proportion of the detected DE isoforms for each pairwise comparison across all 10 cell lines by DELongSeq. Four GC subtypes are clearly differentiated from each other from this heatmap. To better understand these results, we treated the proportions of significant DE isoforms for each pairwise comparison as similarity metric and performed hierarchical clustering analysis. The separation of clusters agrees well with label GC subtypes. These clustering results demonstrate the reliability of DE isoform detection across cell types using DELongSeq in 1 versus 1 comparison.
Figure 6.

Performance of DELongSeq using GC data (A, B) and SH-SY5Y data (C, D). (A) Pairwise DE isoform comparison across 10 cell lines from 4 GC subtypes using DELongSeq. Colors on the top and left strip indicate four different GC subtypes. Colors on the heatmaps show the proportion of detected DE isoform for each pairwise comparison between cell lines. (B) Isoform relative abundance quantification from gene FGFR4 and MET using DELongSeq. Barplots show the expression level estimates of isoform NM_022963 from gene FGFR4 and XM_006715990 from gene MET across different GC subtypes. Colors indicate different GC subtypes. (C) Scatter plot of P-values between multi-sample and 1 versus 1 comparison. (D) Realtive abundance estimates of major isoforms (ENST00000344425, ENST00000626839) from gene KCNQ2 in differentiated and undifferentiated IMR-32 cells.
Furthermore, we examined GC subtype-specific isoform expression, which offers higher resolution of expression heterogeneity than total gene expression. As shown in Figure 6(B), isoform expression showed more distinct pattern across GC subtypes for genes MET and FGFR4, which is consistent with observations made in the original publication. For example, expression of isoforms NM_022963 in gene FGFR4 are up-regulated in the CIN and MSI subtypes, but down-regulated in EBV compared to other GC subtypes. Similarly, isoforms XM_006715990 are highly expressed in the CIN subtype. These isoforms initiated from the new promoter sites are predicted to disrupt signal peptide sequences required for localization to the cell membrane. Similar mechanisms had been reported whereby alternative promoter usage leads to protein localization at different cellular compartment.
Application to nanopore data on human SH-SY5Y (5Y) neuroblastoma cells
Moreover, we applied DELongSeq to Nanopore RNA-seq data generated from human brain (28). This dataset includes two undifferentiated and five differentiated SH-SY5Y cell samples. We performed DE analysis both at isoform and gene level after filtering out lowly expressed genes using DELongSeq. For multi-sample comparison, we have detected 185 genes and 356 isoforms that shows differential expression between differentiated and undifferentiated 5Y cells. In addition, we performed pairwise 1 versus 1 DE comparison between samples. Among detected DE genes and isoforms in multi-sample comparison, most (153 out of 185 genes and 302 out of 356 isoforms) had P-values <0.05, suggesting the consistence of 1 versus 1 DE comparison. Figure 6C shows that the Spearman's correlation of P-values between multi-sample and 1 versus 1 comparison is 0.68 for DELongSeq. This demonstrates the robustness of DELongSeq's performance to relatively small sample size.
We further examined the 302 DE isoforms that were detected in both multi-sample and 1 versus 1 comparison by DELongSeq. Several of these isoforms are from genes that have been implicated in human SH-SY5Y cell differentiation. For example, Smith et al. (44) reported that gene KCNQ2 is required during neuronal development. Their findings showed that isoform ENST00000344425 was preferentially utilized in undifferentiated IMR-32 neuroblastoma cells, whereas isoform ENST00000626839 is up-regulated in differentiated IMR-32 cells. As shown in Figure 6D, the 95% CI of isoform ENST00000344425 relative abundance is [0.82, 0.88] in undifferentiated IMR-32, whereas the 95% CI is [0.34, 0.41] in differentiated IMR-32 cells. These results are consistent with PCR measurement in Smith et al. It is worth noting that among the genes for which the 302 DE transcripts originate from, only about half were detected to be DE at gene level analysis by DELongSeq. This suggests that if one were to perform gene-only analysis, signals at the isoform level would have been missed.
DISCUSSION
Detection of genes and isoforms with mis-regulated expression is a critical step in transcriptomics studies. In expression quantification and differential detection, it is important to account for the uncertainty of estimation and various precision across samples, which may play a role to influence the accuracy of statistical inference. Existing methods either model read count directly or ignore the variance of expression estimates. In this article, we have proposed a flexible statistical model, utilizing well-established variance estimation approach of EM-based model to quantify uncertainty. Through simulation study and the real study of cancer patient samples, we demonstrated that this proposed method allows 1 case versus 1 control comparison and can improve the power of gene and isoform differential analysis while controlling for false positive rate. We have provided a software package (https://github.com/WGLab/DELongSeq), with detailed instructions, example data sets, and reproducible workflows on how to analyze raw long-read RNA-seq data directly.
Compared to existing methods, DELongSeq has several advantages. First, uncertainty of isoform estimate leads to more accurate expression profiling. Existing methods ignore estimate variance in differential expression analysis, which may lead to poor detection or false positive results in the context of long-read sequencing when every sample can vary greatly from another. Given gene and isoform uncertainty score, we filtered out the transcript with large variance estimate in regression model. Since the isoforms that were covered by less reads tended to have higher expression variance than those with more reads, the genes and isoform filtered out would higher read coverage. This is confirmed in our simulation study. The power of our approach would have been higher without the filter, but at the expense of inflated false positive results. We are still searching for the threshold which could maximize the power of DE gene and isoform detection while minimizing false positive rate.
Second, regression model based on expression estimates is more flexible to adjust covariates and account for the variation across samples. FLAIR, TALON and DESeq2 are count based models to detect DE isoforms. FLAIR and TALON calculate fold change directly without accounting for variation. DESeq2 applies negative binomial regression to allow the dispersion parameter to vary from gene to gene. It shrinks estimation to estimate the dispersion parameters and fold change. In our simulation study, although FLAIR could control false positive rate, it was conservative in all comparisons. It also had lower power to detect DE genes compared to other methods. In contrast, TALON + MW showed inflated false positive rate for non-DE isoforms, especially when sample size was small. DELongSeq is more robust to the impact of sample size and covariates. We note these three methods cannot take into account the uncertainty in isoform expression uncertainty, which may lead to biased testing results.
Third, by quantifying isoform expression uncertainty, DELongSeq avoids sample related confounding effect in DE isoform detection. It allows 1 case versus 1 control comparison in the same sample instead of pooling within- and cross-sample variation together in regression model. Since DELongSeq estimates sample-specific variance 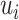 beforehand and treat it as input in regression model, more information can be utilized to estimate cross-sample variance
beforehand and treat it as input in regression model, more information can be utilized to estimate cross-sample variance 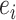 , hence lead to more accurate DE isoform detection. In our simulations, we found that DELongSeq was more powerful than other methods.
, hence lead to more accurate DE isoform detection. In our simulations, we found that DELongSeq was more powerful than other methods.
The current model of DELongSeq is based on read length correction of LIQA. Since LIQA is robust to read length bias, it is of our interest to evaluate the contribution of LIQA to the good performance of DELongSeq. Therefore, we divided genes in our simulated data into two groups based on the degree of non-uniformity of read coverage. Figure S3 shows that DELongSeq consistently outperforms other methods for both isoform-based and gene-based tests regardless of the degree of non-uniformity, suggesting that when read coverage is not uniform, DELongSeq would benefit from the use of an isoform expression estimation method that is robust to non-uniformity. Moreover, we compared the performance with the approach that uses simple linear regression model based on LIQA’s output. Figure 5 and 6 also show the robust performance of DELongSeq in real dataset, suggesting that expression uncertainty level is important in DE isoform detection.
In summary, we have developed a flexible regression framework to detect DELongSeq at both isoform and gene levels. Through extensive simulations and the analysis of several real RNA-seq dataset generated on the PacBio and Nanopore platform, we showed that DELongSeq outperformed competing methods, particularly when sample size is small or the difference between groups under comparison are small. We believe that DELongSeq can contribute to future transcriptomics studies using the long-read RNA-Seq techniques.
DATA AVAILABILITY
The ESCC dataset underlying this acritical are available in GEO at https://www.ncbi.nlm.nih.gov/geo, and can be accessed with PRJNA515570. The Gastric Cancer dataset underlying this article are available in SRA at https://www.ncbi.nlm.nih.gov/sra, and can be accessed with PRJNA635275. The Human Neuroblastoma dataset underlying this article are available in ENA at https://www.ebi.ac.uk/ena, and can be accessed with PRJEB39347.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Wang lab members for insightful comments and for testing the software tools. We also thank the developers of the NanoSim software tool, and the generators of the long-read data on the ESCC, GC and human SH-SY5Y (5Y) neuroblastoma cells datasets for making the data publicly available for benchmarking studies.
Author contribution: Y.H. and K.W. initiated and designed the project, formulated the model. Y.H. developed and implemented the algorithm. Y.H. and A.G. conducted the analysis. Y.H. and K.W. wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yu Hu, Raymond G. Perelman Center for Cellular and Molecular Therapeutics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Anagha Gouru, Raymond G. Perelman Center for Cellular and Molecular Therapeutics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Kai Wang, Raymond G. Perelman Center for Cellular and Molecular Therapeutics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA; Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
NIH/NIGMS [GM132713]; NIH/NICHD [HD105354]; CHOP Research Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sharon D., Tilgner H., Grubert F., Snyder M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013; 31:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weirather J.L., de Cesare M., Wang Y., Piazza P., Sebastiano V., Wang X.J., Buck D., Au K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res. 2017; 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Glinos D.A., Garborcauskas G., Hoffman P., Ehsan N., Jiang L., Gokden A., Dai X., Aguet F., Brown K.L., Garimella K. Transcriptome variation in human tissues revealed by long-read sequencing. Nature. 2022; 608:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chung M., Bruno V.M., Rasko D.A., Cuomo C.A., Munoz J.F., Livny J., Shetty A.C., Mahurkar A., Dunning Hotopp J.C. Best practices on the differential expression analysis of multi-species RNA-seq. Genome Biol. 2021; 22:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dong X., Tian L., Gouil Q., Kariyawasam H., Su S., De Paoli-Iseppi R., Prawer Y.D.J., Clark M.B., Breslin K., Iminitoff M. et al. The long and the short of it: unlocking nanopore long-read RNA sequencing data with short-read differential expression analysis tools. NAR Genom Bioinform. 2021; 3:lqab028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reixachs-Sole M., Eyras E. Uncovering the impacts of alternative splicing on the proteome with current omics techniques. Wiley Interdiscip. Rev. RNA. 2022; 13:e1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013; 498:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng Q., Ramskold D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014; 343:193–196. [DOI] [PubMed] [Google Scholar]
- 10. Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J., Carlson S., Helgason A., Walters G.B., Gunnarsdottir S. et al. Genetics of gene expression and its effect on disease. Nature. 2008; 452:423–428. [DOI] [PubMed] [Google Scholar]
- 11. Lee T.I., Young R.A. Transcriptional regulation and its misregulation in disease. Cell. 2013; 152:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sui X., Kong N., Ye L., Han W., Zhou J., Zhang Q., He C., Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014; 344:174–179. [DOI] [PubMed] [Google Scholar]
- 13. Han J., Xiong J., Wang D., Fu X.D. Pre-mRNA splicing: where and when in the nucleus. Trends Cell Biol. 2011; 21:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H.K., Pham M.H.C., Ko K.S., Rhee B.D., Han J. Alternative splicing isoforms in health and disease. Pflugers Arch. 2018; 470:995–1016. [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Morley M., Brandimarto J., Hannenhalli S., Hu Y., Ashley E.A., Tang W.H., Moravec C.S., Margulies K.B., Cappola T.P. et al. RNA-seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015; 105:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montes M., Sanford B.L., Comiskey D.F., Chandler D.S. RNA splicing and disease: animal models to therapies. Trends Genet. 2019; 35:68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scotti M.M., Swanson M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016; 17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., Szczesniak M.W., Gaffney D.J., Elo L.L., Zhang X. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corchete L.A., Rojas E.A., Alonso-Lopez D., De Las Rivas J., Gutierrez N.C., Burguillo F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020; 10:19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soneson C., Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinf. 2013; 14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng R., Xu Z., Luo M., Wang P., Cao H., Jin X., Zhou W., Xiao L., Jiang Q. Identification of alternative splicing-derived cancer neoantigens for mRNA vaccine development. Brief Bioinform. 2022; 23:bbab553. [DOI] [PubMed] [Google Scholar]
- 22. Jia C., Guan W., Yang A., Xiao R., Tang W.H., Moravec C.S., Margulies K.B., Cappola T.P., Li M., Li C. MetaDiff: differential isoform expression analysis using random-effects meta-regression. BMC Bioinf. 2015; 16:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Au K.F. The blooming of long-read sequencing reforms biomedical research. Genome Biol. 2022; 23:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Byrne A., Beaudin A.E., Olsen H.E., Jain M., Cole C., Palmer T., DuBois R.M., Forsberg E.C., Akeson M., Vollmers C. Nanopore long-read rnaseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017; 8:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wyman D., Balderrama-Gutierrez G., Reese F., Jiang S., Rahmanian S., Forner S., Matheos D., Zeng W., Williams B.A., Trout D A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. 2020; bioRxiv doi:18 June 2019, preprint: not peer reviewedhttps://doi.org/10.1101/672931.
- 26. Hu Y., Fang L., Chen X., Zhong J.F., Li M., Wang K. LIQA: long-read isoform quantification and analysis. Genome Biol. 2021; 22:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang A.D., Soulette C.M., van Baren M.J., Hart K., Hrabeta-Robinson E., Wu C.J., Brooks A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020; 11:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gleeson J., Leger A., Prawer Y.D.J., Lane T.A., Harrison P.J., Haerty W., Clark M.B. Accurate expression quantification from nanopore direct RNA sequencing with NanoCount. Nucleic Acids Res. 2022; 50:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fry J.L., Li Y., Yang R. ScanExitronLR: characterization and quantification of exitron splicing events in long-read RNA-seq data. Bioinformatics. 2022; 38:4966–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Núñez-Moreno G., Tamayo A., Cortón M., Mínguez P. Mini-IsoQLR: a pipeline for isoform quantification using long-reads sequencing data for single locus analysis. 2022; bioRxiv doi:04 March 2022, preprint: not peer reviewed 10.1101/2022.03.01.482488. [DOI]
- 31. Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Glaus P., Honkela A., Rattray M. Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics. 2012; 28:1721–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M., Haag J.D., Gould M.N., Stewart R.M., Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013; 29:1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Louis A.T. Finding the observed information matrix when using the EM algorithm. J. R. Stat. Soc. B. 1982; 44:226–233. [Google Scholar]
- 38. Huizenga M.H., Visser I., Dolan V.C. Testing overall and moderator effects in random effects meta-regression. Br. J. Math. Stat. Psychol. 2011; 64:1–19. [DOI] [PubMed] [Google Scholar]
- 39. Yang C., Chu J., Warren R.L., Birol I. NanoSim: nanopore sequence read simulator based on statistical characterization. Gigascience. 2017; 6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng Y.W., Chen Y.M., Zhao Q.Q., Zhao X., Wu Y.R., Chen D.Z., Liao L.D., Chen Y., Yang Q., Xu L.Y. et al. Long read single-molecule real-time sequencing elucidates transcriptome-wide heterogeneity and complexity in esophageal squamous cells. Front. Genet. 2019; 10:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang K.K., Huang J., Wu J.K.L., Lee M., Tay S.T., Kumar V., Ramnarayanan K., Padmanabhan N., Xu C., Tan A.L.K. et al. Long-read transcriptome sequencing reveals abundant promoter diversity in distinct molecular subtypes of gastric cancer. Genome Biol. 2021; 22:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y., Ge X., Peng F., Li W., Li J.J. Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biol. 2022; 23:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith J.S., Iannotti C.A., Dargis P., Christian E.P., Aiyar J. Differential expression of kcnq2 splice variants: implications to m current function during neuronal development. J. Neurosci. 2001; 21:1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ESCC dataset underlying this acritical are available in GEO at https://www.ncbi.nlm.nih.gov/geo, and can be accessed with PRJNA515570. The Gastric Cancer dataset underlying this article are available in SRA at https://www.ncbi.nlm.nih.gov/sra, and can be accessed with PRJNA635275. The Human Neuroblastoma dataset underlying this article are available in ENA at https://www.ebi.ac.uk/ena, and can be accessed with PRJEB39347.