

Abstract
Colon cancer incidence is associated with a high-fat diet. Such a diet is linked to elevated levels of bile acids in the gastrointestinal system and the circulation. Secondary bile acids are produced by microorganisms present at high concentrations in the colon. Recent prospective studies and a retrospective study in humans associate high circulating blood levels of secondary bile acids with increased risk of colon cancer. Feeding mice a diet containing a secondary bile acid, so their feces have the bile acid at a level comparable to that in the feces of humans on a high-fat diet, also causes colon cancer in the mice. Studies using human cells grown in culture illuminate some mechanisms by which bile acids cause cancer. In human cells, bile acids cause oxidative stress leading to oxidative DNA damage. Increased DNA damage increases the occurrence of mutations and epimutations, some of which provide a cellular growth advantage such as apoptosis resistance. Cells with such mutations/epimutations increase by natural selection. Apoptosis, or programmed cell death, is a beneficial process that eliminates cells with unrepaired DNA damage, whereas apoptosis-resistant cells are able to survive DNA damage using inaccurate repair processes. This results in apoptosis-resistant cells having more frequent mutations/epimutations, some of which are carcinogenic. The experiments on cultured human cells have provided a basis for understanding at the molecular level the human studies that recently reported an association of bile acids with colon cancer, and the mouse studies showing directly that bile acids cause colon cancer. Similar, but more limited, findings of an association of dietary bile acids with other cancers of the gastrointestinal system suggest that understanding the role of bile acids in colon carcinogenesis may contribute to understanding carcinogenesis in other organs.
Keywords: Carcinogenesis, gastrointestinal cancer, oxidative stress, DNA damage, apoptosis
Impact Statement
Since colon cancer and gastrointestinal system cancers are among the most frequent types of cancer in the world, understanding their underlying cause is important for implementing effective preventive measures. This minireview uniquely integrates recent evidence from several different types of studies. Prospective studies in humans show that colon carcinogenesis is associated with high secondary bile acid exposure. Studies in mice show directly that a secondary bile acid, at a human physiologic level, causes colon cancer. Studies using human cells indicate that bile acids cause oxidative stress, DNA damage, mutations/epimutations and apoptosis resistance, factors leading to cancer. Increasing evidence indicates an etiologic role of bile acids in seven gastrointestinal system cancers. In these cases, increased bile acid exposure from increased reflux or restricted bile flow is likely involved. This integrated perspective should provide a deeper understanding of causation in gastrointestinal system cancers that may assist in prevention.
Introduction
In the United States, colorectal cancer is the third most frequent cancer both in males and in females. 1 The 2022 estimates are 151,030 new colorectal cancer cases and 52,580 deaths. The world-wide estimated number of new colorectal cancer cases and deaths for 2020 for both sexes is 1,880,725 cases and 915,880 deaths. 2 As will be reviewed here, excessive exposure to secondary bile acids is the likely major etiologic factor in colorectal cancer. There is also increasing evidence that bile acids have an etiologic role in other types of gastrointestinal system cancers. These include cancers of the pharynx, esophagus, stomach, pancreas, liver, biliary tract, and small intestine, that combined have a world-wide incidence of over 3 million cases. 2 Considering the societal impact of colorectal cancer and other bile acid–related cancers, it is clearly important to increase our understanding of the carcinogenic action of bile acids so that practical preventive measures can be undertaken.
The following first three sections have often been covered in previous reviews and within the introductory sections of many experimental articles.3–6 They are included to provide a frame of reference to the subsequent sections, some of which are novel in this review.
High-fat diet and colorectal cancer
Colorectal cancer incidence varies by as much as 25-fold among countries. High rates occur in Australia, New Zealand and the United States and low rates occur in African countries and India. 3 People who immigrate from a low to a high-risk country tend to reach the incidence of colorectal cancer of the high-risk country in about one or two generations. 7 The most significant factor influencing colorectal cancer susceptibility in such migrants appears to be the adoption of a Westernized diet. Dietary components can influence colon carcinogenesis by causing epigenetic changes in gene expression and somatic mutation changes in gene sequence that alter the homeostasis of the intestinal mucosa. 7
A number of factors associated with a Western lifestyle may contribute to increased risk of colon cancer, and prominent among these factors is consumption of red and processed meat. 8 Numerous epidemiological studies have reported an association between fat consumption and incidence of colon cancer.9–13 A meta-analysis of 10 prospective studies concluded that high intake of red and processed meat is associated with a significant increased risk of colorectal cancer. 14 In Korean men, risk of colorectal adenoma (a precursor to cancer) increased with higher saturated fatty acid (SFA) intake (SFA are high in red meats). 15 Among Americans of African descent, the rate of colon cancer (65 per 100,000) is much higher than among rural Africans (less than 5 per 100,000). 16 The high rate of colon cancer in African Americans was attributed to a diet high in fat and low in fiber. 16
Bile acids and dietary fat
Bile acids are produced by the liver from cholesterol. The primary bile acids produced, cholic acid and chenodeoxycholic acid, are conjugated to either taurine or glycine and then secreted from the liver to be stored in the gall bladder. Following a high-fat meal, the gall bladder is stimulated to contract and release stored bile acids into the first section of the small intestine (the duodenum) in order to facilitate digestion of saturated fat.17,18 In the small intestinal lumen, bile acids function as lipid solubilizers that facilitate lipid absorption. Most bile acids are actively reabsorbed in the small intestine, especially in the distal ileum, and transported back to the liver. 19 The bile acids transported back to the liver are sent again to the gall bladder, resulting in continuous circulation between the liver and the intestine. Bile acids can be recycled 4–12 times per day, the enterohepatic circulation, between hepatocytes in the liver and enterocytes in the intestine. 20 About 10% of the total bile acid pool reaches the systemic (blood plasma) circulation. 20 About 5% of the bile acids are not reabsorbed in the small intestine and enter the colon. 21 In the colon, the bile acids are transformed by intestinal microorganisms employing processes of deconjugation and dehydroxylation to form the secondary hydrophobic bile acids deoxycholic acid (DOC) and lithocholic acid (LCA).17,18,21 DOC can be reabsorbed from the colon, transported back to the liver, and then re-enter the circulating bile acid pool. LCA is less taken up in the colon and mostly does not re-enter the bile acid pool but is excreted in the stool. The resulting overall enterohepatic circulating bile acid pool then consists of about 30–40% of each of cholic acids and chenodeoxycholic acids, 30% DOC, and about 5% LCA. 21
In humans, a high-fat, high-beef diet leads to a significant increase in the level of secondary bile acids in the colonic contents. 22 In another human-based study, 23 individuals were fed a higher fat diet for six months, where 40% of the energy in their diet now came from soybean oil (compared to 30.6% fat energy in their former diet). In this study, total bile acids and secondary bile acids in the feces of the humans on the higher fat diet increased by 10% and 11%, respectively. In a rat-based study, 24 when lard was added to the diet of rats, this doubled the level of bile acids in the colonic contents, with the secondary bile acids (especially DOC) then being present at roughly twice the level of primary bile acids.
Bile acids and colorectal cancer
Numerous epidemiological studies have reported that fecal bile acid concentrations are elevated in populations with a high incidence of colon cancer.25–28 Patients with colorectal cancer tend to have increased proportions of fecal secondary bile acids. 29 Repeated exposure of the human gastrointestinal tract to high physiologic levels of secondary bile acids, particularly among individuals who have a high dietary fat intake, is an important risk factor for gastrointestinal cancer. 21 The population with the world’s highest recorded incidence of sporadic colorectal cancer are the Native Alaskan people who live in remote communities. 30 The diet of the Native Alaskans was compared to the diet of a rural African population with the lowest colorectal cancer incidence. The Native Alaskan diet was higher in fat and lower in fiber, and fecal samples had a higher level of the secondary bile acid DOC compared to the rural African population. 30
Human prospective and retrospective studies associate bile acids with colon cancer
The study of carcinogenicity of secondary bile acids in humans is inherently more difficult than in animal model systems, partly because of the decades long time frame involved between exposure and tumor development in humans. Yet prospective studies of the association of circulating bile acids with colorectal cancer incidence have recently been reported.31,32 The study by Loftfield et al. 31 was a prospective study in which baseline serum was collected many years prior to any diagnoses of colorectal cancer. Among women, a strong association was found between serum concentrations of bile acids, particularly microbial metabolites of cholic acid, and increased colorectal cancer risk. The bile acids associated with a strong increased risk included the four secondary bile acids DOC, glycodeoxycholic acid, taurodeoxycholic acid, and glycolithocholic acid. 31 A prospective study by Kühn et al. 32 also found that prediagnostic plasma levels of certain conjugated primary and secondary bile acids were positively associated with colon cancer risk.
A 2022 retrospective study of 5589 individuals 33 compared serum total bile acids (sTBAs) of patients without cancer to the sTBAs of patients about to be operated on for colon cancer, esophageal cancer or gastric cancer. There was a high positive association of elevated sTBAs with these gastrointestinal cancers, suggesting that high levels of sTBAs are a risk factor for gastrointestinal cancers.
The principal function of the gallbladder is to store and concentrate bile acids and to provide a buffer for effects of bile acids on the intestinal tract. A meta-analysis of 10 cohort studies showed that cholecystectomy (gallbladder removal), causing a more continuous bathing of the digestive tract with bile acids, is associated with an increased risk of subsequent colon cancer. 34 In a more recent study of the female western population, a strong association was found between colorectal cancer and cholecystectomy. 35 These findings further implicate bile acids in the etiology of colon cancer.
Bile acids cause colon cancer in rodents
As noted above, a high-fat diet can lead to increased production of bile acids to facilitate digestion of the fat. In particular, the secondary bile acids including DOC, taurine conjugated DOC, and the bacterial isomerization product 3 beta-deoxcholic acid (DCA) each increases in the colon of humans on a higher fat diet. 23 To mimic this situation, genetically normal (wild-type) male mice were fed a diet including 0.2% of the bile acid DOC for eight to ten months 36 and wild-type female mice were similarly fed a diet including 0.2% DOC for 10 months. 37 This level of dietary DOC led to fecal concentrations of DOC comparable to fecal levels of DOC in humans consuming a high-fat diet. 37 This DOC supplementation led to colonic tumors in 94% of the male mice including 54% with cancers, and in 91% of the female mice including 45% with cancers. None of the mice fed the same diet, but without DOC, developed tumors. These findings indicated that DOC is a carcinogen in wild-type mice.
In another arm of this study, the antioxidant chlorogenic acid was added to the DOC supplemented diet of the mice. Chlorogenic acid protects DNA against oxidative damage. 38 The addition of chlorogenic acid significantly reduced tumor formation. This finding appears to be analogous to results of a study in humans. Coffee has an abundant level of chlorogenic acid. 39 In a recent human retrospective study, a higher intake of decaffeinated coffee was found to be associated with a reduced risk of colorectal cancers. 40
The types of tumors in the DOC fed mice were histopathologically virtually identical to those in humans. 37 In humans, characteristic aberrant changes in molecular markers are observed both within the colon cancer and in the areas of the colon surrounding a cancer. Such surrounding affected areas are referred to as field defects. In the mice fed a diet including DOC, field defects similar to those in humans were also observed. In these mouse fields, a type of oxidative DNA damage, 8-hydroxydeoxyguanosine (8-OHdG), was increased, and the DNA repair protein excision repair cross-complementing group 1 (ERCC1) was decreased. 37 Other characteristic biomarkers were also changed similarly in both humans and mice. Thus, this diet-related mouse model for colon cancer closely paralleled colon cancer progression in humans both at the level of molecular profile and at the histomorphological level.
In an earlier experiment, rats that spontaneously developed colon cancer were found to have significantly higher levels of DOC in their feces than those rats without colon cancer. 41
Bile acids cause oxidative stress, and carcinogenic oxidative DNA damage and mutations
Bile acids can disrupt cell membranes by their detergent action on membrane lipid components and this disruption promotes the generation of reactive oxygen species. 42 Reactive oxygen species can damage DNA. The oxidative DNA damage 8-OHdG is mutagenic 43 and carcinogenic. 44 When mice were fed a diet containing DOC for eight months, it was found that the DNA in the nuclei of their colonic epithelial cells contained a substantially higher level of 8-OHdG than the nuclei of mice fed the same diet without DOC (Figure 1). 37 This indicates that dietary DOC increases oxidative DNA damage in the nuclei of colonic epithelial cells. Incomplete repair of base alterations such as 8-OHdG can lead to an increase in DNA strand breaks. DOC was found to induce DNA damage including DNA single-strand breaks in human colon epithelial cells at a sufficient level to trigger apoptosis (programmed cell death) of these cells. 45
Figure 1.

Colonic crypts in epithelium from two mice. (a) Photomicrograph of tissue from a mouse fed a standard diet. Scale bar: 50 μm. (b) Photomicrograph of tissue from a mouse fed a diet supplemented with deoxycholate. Scale bar: 50 μm. Cell nuclei are stained dark blue with hematoxylin (for nucleic acid) and immunostained brown for 8-OHdG. The level of 8-OHdG was graded in the nuclei of colonic crypt cells on a scale of 0–4. Mice fed standard diet had crypt 8-OHdG at levels 0 to 2 ((a) level 1) while mice fed diet supplemented with deoxycholate had 8-OHdG in colonic crypts at levels 3 and 4 ((b) level 4). Deoxycholate added to the mouse diet gave a concentration of deoxycholate in the mouse colon similar to the concentration in the colon of humans on a high-fat diet. (A color version of this figure is available in the online journal.)
Source: Modified from Chaya5260 CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=41679524.
Colon cancers occur within field defects
A field defect can be defined as a field of premalignant tissue in which a new cancer is likely to arise. Field defects often appear to be largely histologically normal under the microscope. Such fields are thought to constitute the earliest clones in the carcinogenic process and may include regions with methylation changes 46 or small outgrowths, such as polyps. Thus, colon cancers often arise in a colonic field defect in which a number of polyps can be seen. Surgical resections of colon cancers usually include colon segments that extend more than 10 cm on each side of the cancer (about 13% of an average 150 cm colon) to reduce the possibility of a new cancer arising close to where the previous cancer was removed. Figure 2 shows a colon resection with a number of premalignant polyps.
Figure 2.
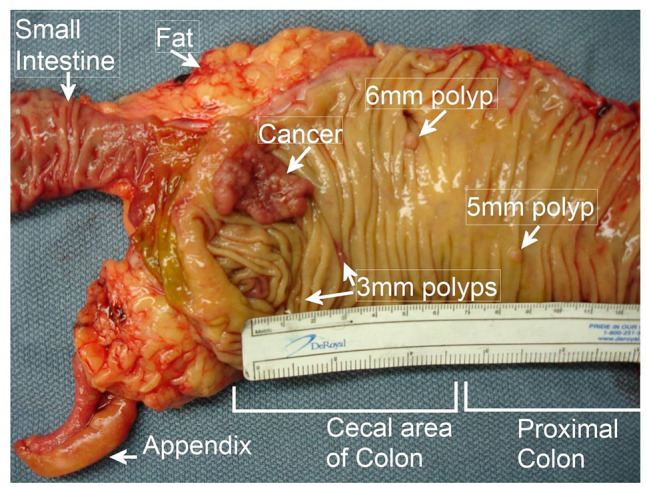
A colon cancer within a field defect in which four precancerous polyps are also arising. This was a patient with sporadic colon cancer. (A color version of this figure is available in the online journal.)
Source: Part of Bernstein0275 CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?>curid=25453056.
Colon resections are usually preceded by a colonoscopy during which polyps are removed, and after surgical resection of the colon cancer, within a year, another colonoscopy is usually performed to remove any further precancerous colonic polyps. During a series of 116 postresection colonoscopies, Paik et al. 47 found that neoplastic (precancerous) polyps were found about 24% of the time, and about 40% of such polyps were found right at the anastomosis (site of the colon resection). This indicates that there were further polyps or incipient polyps in the field defect that extended a bit beyond the cancer plus nearby field defect that had been surgically removed.
Reduced repair of DNA damage in field defects during progression to colon cancer
Oxidative/nitrosative stress is prevalent in the colons of patients with colorectal cancer. 48 Such stress causes DNA damage, and DNA repair processes are necessary to remove this damage. The expression of three DNA repair proteins PMS2, ERCC1, and XPF were found to be substantially reduced in fields surrounding human sporadic colon cancers. 49 Most of the mutations (and epimutations) in tumors arise in the preneoplastic field stage of their development. 50 The finding of field defects for DNA repair proteins surrounding colon cancers 49 suggests that such cancers arose in fields where expression of DNA repair proteins were reduced, and that this reduction facilitated the progression to cancer. PMS2 protein is employed in the process of DNA mismatch repair when DNA damages are moderate, but triggers apoptosis when DNA damages are high. 51 ERCC1 and XPF proteins form a complex that acts as a structure-specific endonuclease, essential for nucleotide excision repair in mammalian cells. 52 The ERCC1–XPF complex functions in the repair of DNA damages induced by reactive oxygen species. 52 Reduced expression of PMS2, ERCC1, and XPF at an early stage would allow the persistence of DNA damages, that upon DNA replication, could generate carcinogenic mutations or epimutations.
In the mouse experiments described above, colon cancer development occurs by about eight to ten months after initiating feeding of DOC. In humans, colon cancer development normally occurs in older individuals, and may be a decade’s long process. Mice have about a fivefold lower level of DNA repair than humans. 53 It was suggested that the earlier occurrence of colon cancer in mice compared to humans could be due, at least in part, to the lower DNA repair activity in mice. 53
Bile acids select for apoptosis resistance – a proliferative advantage
The deficiencies in the DNA repair proteins PMS2, ERCC1 and XPF, described above, appear to arise by epigenetic alteration (epimutation), repressing the expression of the genes encoding these proteins. 49 Epimutations include altered methylation pattern (hypermethylation and hypomethylation) in DNA and post-translational modifications of histones within the nucleosomes located at a gene. One example of an epimutation is the alteration of a promoter methylation pattern that enhances or reduces gene expression. Reduced expression of a protein essential for a particular DNA repair pathway can result in a cell employing an alternative, less accurate pathway of repair. This alternative can lead to further mutations or epimutations in the cells harboring the DNA repair protein deficiency. Such cells are genetically unstable and as a result may acquire mutations/epimutations that provide a growth advantage.
A type of genetic alteration that provides a growth advantage is a mutation/epimutation to apoptosis resistance. Apoptosis is a form of programmed cell death, and acquired resistance to apoptosis is a characteristic of cancers. Apoptosis is an adaptation that removes cells with excessive DNA damage, thus protecting against carcinogenesis. 51 Cells with an apoptosis resistance mutation circumvent this beneficial cell death process and may increase in a growing cell population by a process of natural selection. When colonic epithelial cells were grown in culture and repeatedly exposed to increasing concentrations of DOC, the cells underwent natural selection to develop apoptosis resistance. 54 Analyses of the molecular changes in these apoptosis-resistant cells indicated particular gene expression changes that occurred in response to the persistent bile acid exposure, and several of these could be linked to progression to colon cancer. 54 One such example was increased expression of the antiapoptotic protein B-cell lymphoma 2 (BCL-2). In humans, the colon mucosa adjacent to adenocarcinomas is associated with increased expression of antiapoptotic BCL-2 family proteins, a “field change” that is implicated in tumor carcinogenesis.55,56
Diet, bile acids, gut microbiome and cancer
Diet affects the level of bile acids in the digestive tract. In rats, increased fat consumption stimulates the liver to synthesize more bile acids. 57 In humans, more bile salts are added to the intestinal tract after a high-fat meal than after a low fat meal due to greater contraction of the gall bladder after a high-fat meal. 58 In the case of both rats and humans, a high-fat diet increases the quantity of bile acids that escape enterohepatic recirculation and enter the colon. In humans, those on a high-fat diet have considerably more of both primary and secondary bile acids in their feces than those on a low-fat diet. 59
The increased bile acids in the colon then change the microbiome of the colon. In general, the human gut microbiota are dominated by three primary phyla: Firmicutes (30–50%), Bacteroidetes (20–40%), and Actinobacteria (1–10%). 60
However, two factors, the type of fat and the level of bile acids, can substantially alter the composition of the microbiome in the colon. For instance, wild-type mice were fed one of three diets, each containing 30% fat, but differing in the types of fat. The three different types of fat were lard, safflower oil (mostly polyunsaturated fatty acids), and milk fat. This resulted in the microbiome in the colons of the mice on the lard diet to change to be about 95% Firmicutes, on the safflower diet to be about 60% Bacteroidetes, and on the milk fat diet to be about 50% Bacteroidetes plus a bloom of 10% of a member of the Deltaproteobacteria, Bilophila wadsworthia. 61
Firmicutes are a phylum of bacteria, most of which have Gram-positive cell wall structure. Bacteroidetes are mostly Gram-negative. Gram-positive bacteria are more sensitive to bile acids than Gram-negative bacteria. 62 Bile acids each have a level of bactericidal activity, and DOC, the most active, is about 10-fold more bactericidal than cholic acid. 63 High levels of bile acids in the colon, especially DOC, changes the colonic microbiota, causing dysbiosis.
Both bile acid–induced DNA damage and bacterial dysbiosis can be carcinogenic
Bile acids themselves, at increased levels in the colon, cause DNA damage,45,64 and increased levels of DNA damage increase the likelihood of cancer. 65
Dysbiosis as a cause of cancer
Two interesting reviews on dysbiosis as a cause of colon carcinogenesis are those of Tjalsma et al. 66 and of Sun and Kato. 67 Both reviews point out that bacterial density in the colon is about 1012 cells per mL while bacterial density in the small intestine is about 102 cells per mL. The higher density of bacteria in the colon is thought to account for the more than 10-fold higher incidence of cancer in the colon compared to cancer incidence in the small intestine. Tjalsma et al. proposed that the first step in carcinogenesis is production of DNA damaging agents by some of the bacteria newly more prevalent in the colon after dysbiosis occurs. This will cause DNA damages, followed by mutations in colonic mucosa. One example they give is increased Enterococcus faecalis. E. faecalis produces extracellular superoxide, which has the potential, when converted to hydrogen peroxide, to cause DNA damage in colonic epithelial cells. A second example is an increase in certain Escherichia coli strains that harbor the polyketide synthetase island, which encodes a genotoxin called colibactin, that induces single-strand DNA breaks. The later review by Sun and Kato 67 also proposed that certain newly prevalent bacteria after dysbiosis may be carcinogenic. Their first candidate is Streptococcus bovis, which is a Gram-positive bacterium that can cause systemic infections (endocarditis or bacteremia) in humans. It has the specific ability to grow in 40% bile, so can be selected for in the presence of increased colonic secondary bile acids. About 50–60% of patients diagnosed with S. bovis bacteremia or S. bovis endocarditis were found to have advanced adenoma or cancer in their colons. Another candidate is a strain of aero-anaerobic Gram-negative E. coli carrying the polyketide synthetase island, which encodes a genotoxin called colibactin, that induces single-strand DNA breaks. Epithelial proliferation and E. coli colonization density were significantly correlated in colonic mucosa locations associated with, but distant from, a colorectal cancer. They also showed associations, to some extent, of increased prevalence of Bacteroides fragilis, Fusobacterium nucleatum, and Salmonella enterica with progression to colorectal cancer. In addition, they provided a table of studies on colorectal cancer and indicated the bacterial species that were over- or under-represented in association with those cancers.
Progression to colorectal cancer: DNA damage, mutation, epigenetic alterations, and bile acids as signaling molecules
Progression to colorectal cancer begins with DNA damage. As indicated above, 43 increased levels of DNA damage increase the likelihood of mutation. It has generally been thought that sporadic colorectal cancers develop by the accumulation of a series of mutations in tumor suppressor genes and oncogenes. 68 Mutation frequency, measured by whole genome sequencing in 54 colon cancers, found single nucleotide variants to occur between 2459 and 1,601,093 times in the cancers, somatic insertions or deletions occurred between 360 and 464,252 times in the cancers, and structural variants (inversions and balanced translocations or genomic imbalances) occurred between 6 and 681 times in the cancers. 69
This is much higher than the mutation frequency of 30–70 per diploid genome found in a parent to child generation. Among the thousands of mutations in colon cancers, some alter the protein products of genes. There are about two to eight gene-located driver mutations in colon cancers and about 30 to 70 gene-located passenger mutations. 70 It is likely that numerous further mutations occur in promoters, enhancers, and DNA regions coding for non-coding RNAs such as microRNAs, with such mutations further altering the gene expression landscape.
DNA damage from reactive oxygen species also causes epigenetic alterations such as demethylation of cytosine in a methylated 5’cytosine-3’guanine dinucleotide (a “CpG” site) 71 as indicated in Figure 3. Demethylation of cytosine in a CpG site in the promoter of an oncogene may activate a usually repressed oncogene, promoting progression to cancer. Other DNA damage responses also can result in chromatin modifications that become mitotically stable epigenetic alterations that repress or activate genes. 72 Colon cancers carry about 600 methylations in promoter regions of genes (which can cause repression of those genes) and about 350 demethylations (which can result in activation of the associated genes).73,74
Figure 3.
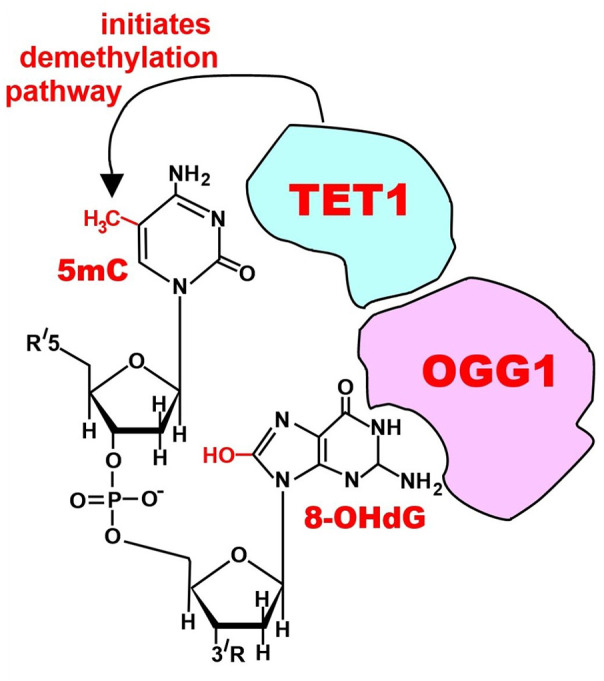
Initiation of DNA demethylation at a CpG site. In adult somatic cells, DNA methylation typically occurs in the context of CpG dinucleotides (CpG sites), forming 5-methylcytosine-pG, or 5mCpG. Reactive oxygen species (ROS) may attack guanine at the dinucleotide site, forming 8-hydroxy-2’-deoxyguanosine (8-OHdG) and resulting in a 5mCp-8-OHdG dinucleotide site. The base excision repair enzyme OGG1 targets 8-OHdG and binds to the lesion without immediate excision. OGG1 present at a 5mCp-8-OHdG site recruits TET1, and TET1 oxidizes the 5mC adjacent to the 8-OHdG. This initiates demethylation of 5mC. (A color version of this figure is available in the online journal.)
Source: Bernstein0275, CC BY-SA 4.0 https://creativecommons.org/licenses/by-sa/4.0.
Bile acids also act as signaling molecules in intestinal epithelium. For instance, taurodeoxycholic acid at 0.05 mmol/L, applied to rat intestinal epithelial cells, increased expression of nuclear factor kappa B (NF-kB) and induced translocation of NF-kB to the nucleus. 75 NF-kB proteins regulate the expression of hundreds of genes, and thereby NF-kB regulates functions including those affecting inflammation, immunity, cell proliferation, and apoptosis. 76
In experiments in our laboratory, 37 we fed mice a control diet or a diet plus deoxycholate. Mice on the diet plus deoxycholate had deoxycholate in their colon at the level in the colons of humans on a high-fat diet. The mice on a control diet, in the stem cell regions of colonic crypts, had beta-catenin expression localized to the membrane regions. The mice on the diet plus deoxycholate, in the stem cell regions of colonic crypts, had high nuclear beta-catenin expression. Beta-catenin nuclear accumulation is found in 40–80% of primary human colon cancers.77,78 Beta-catenin has more than 66 target genes that affect at least 11 pathways. 79
In our experiments with mice fed a diet plus deoxycholate, beclin-1 was strongly expressed in nuclei of the colonic crypts. 37 Beclin-1 activates the autophagic pathway, and this contributes to apoptosis resistance.
Another feeding experiment was carried out by Yde et al. 80 In this experiment, rats were fed for 10 days on a diet with added cholic acid, and this diet generated a total fecal bile acid concentration in the colon similar to the high level found in humans with irritable bowel syndrome with diarrhea. First, they found that the diet plus cholic acid appeared to cause activation of the nuclear bile acid receptor farnesoid X receptor (FXR) as well as activation of the membrane bound bile acid receptor TGR5. Yde et al. then measured the levels of proteins in colonic epithelium cells of the rats fed the added bile acid diet. Of 7717 quantifiable proteins, 183 significantly increased in abundance and 111 decreased in abundance after consuming the added bile acid diet. They carried out a gene ontology analysis of the proteins and found a striking over-representation of proteins involved in chromatid segregation (possibly indicating increased proliferation) in addition to increases in proteins involved in non-coding RNA processing and ribosome generation.
In reviewing work on transplantation of human tumor cells implanted subcutaneously in mice to grow into tumors in mouse models, Guerin et al. 81 pointed out that “it is now clear that several key mutations are necessary but not sufficient for a tumor to develop. Tumor development also requires a favorable, tumor-promoting microenvironment.”
Overall, the mutations and epigenetic alterations accumulating over many years of exposure to increased bile acids, plus the actions of bile acids as signaling molecules (providing a suitable microenvironment), may combine to cause a growing clone derived from a mutated stem cell to progress to form a colon cancer.
Effects of dietary fiber, exercise, and obesity on colon carcinogenesis
Progression to colon cancer is also affected by dietary fiber, exercise, and obesity. The effects of fiber and exercise may counter some of the carcinogenic effects of bile acids, while the effect of obesity may add to the carcinogenic effects.
The short chain fatty acid butyrate is produced in the colonic lumen by bacterial fermentation of undigested dietary carbohydrates, including resistant starch and dietary fiber. High fiber in the diet increases colonic butyrate. 82 Butyrate has many activities in the colon, one of which is to inhibit histone deacetylase in cellular nuclei of the colonic epithelium, leading to increased acetylation of histones. Histone acetylation causes 82 inhibition of cell proliferation, induction of cell differentiation or apoptosis, and induction of expression of particular genes. The effects of butyrate are anticarcinogenic.
Exercise has been shown, in many studies, to have anticarcinogenic effects. 83 For instance, a total of 381 genes in gastrocnemius muscle were upregulated in mice that exercised, including secreted protein acidic and rich in cysteine (SPARC). 84 SPARC protein was secreted into the circulation in response to exercise in both mice and humans. SPARC appears to inhibit colon tumorigenesis by increasing apoptosis.
Frühbeck et al. 85 showed that obesity influences the expression of the inflammasome pathway in visceral adipose tissue, thereby increasing colonic inflammation. This increases an inflammatory cascade that favors a pro-tumorigenic microenvironment.
Bile acids as possible carcinogens throughout the gastrointestinal system
The gastrointestinal tract includes the mouth, pharynx, esophagus, stomach, small intestine, large intestine (cecum, colon, rectum, and anal canal). The gastrointestinal system includes the gastrointestinal tract plus salivary glands, liver, gallbladder, pancreas and biliary tract. There have been numerous reports that bile acids are a likely causal factor in cancers of the gastrointestinal system in addition to colon cancer. High exposure to bile acids has been linked to increased risk of cancer of the laryngopharyngeal tract, esophagus, stomach, pancreas, liver, biliary tract, and small intestine.86–88 Cholecystectomy (gall bladder removal) changes bile flow, and is associated with an increase risk of colon cancer as described above. In a recent large nationwide cohort study in Korea, cholecystectomy was again found to increase the risk of colon cancer, but also was found to increase the risk of cancer at other sites in the gastrointestinal system including the liver, pancreas, biliary tract, pharynx, and oral cavity. 89 The risk of leukemia and thyroid cancer was also increased by cholecystectomy suggesting that changes in circulating bile acids, including bile acids in the blood, may also affect organs outside of the gastrointestinal system.
The evidence for an etiologic role of bile acids in carcinogenesis is most substantial for colon cancer, as reviewed above. However, there is also increasing evidence that bile acids have an etiologic role in other gastrointestinal system cancers. The more well-understood role of bile acids in colon carcinogenesis may help in understanding the etiology of these other bile acid–related cancers.
Hypopharyngeal carcinogenesis
Bile acids are often present in gastrointestinal refluxates and can cause premalignant changes and invasive squamous cell cancer in the hypopharynx.90,91 Bile-induced DNA damage is a likely key step in this process.91,92
Esophageal adenocarcinoma
Gastroesophageal reflux disease can lead to the development of Barrett’s esophagus and subsequent progression to adenocarcinoma of the esophagus. Bile acids present in gastroesophageal reflux appear to contribute to esophageal adenocarcinoma carcinogenesis by induction of oxidative stress, oxidative DNA damage, mutation, and apoptosis.87,88,93 The induction of apoptosis by bile acids can select for cells that are resistant to apoptosis, a characteristic that provides a growth advantage.
Gastric (stomach) cancer
In a human retrospective study, bile reflux was an apparent risk factor for gastric cancer and precancerous lesions. 94 In a mouse model of gastric cancer, a carcinogenic interaction was shown between bile acids and Helicobacter pylori. 95 Iron deficiency linked to altered bile acid metabolism was found to promote H. pylori–induced inflammation-driven gastric carcinogenesis in mice. 96 Among humans, a retrospective study found that use of a bile acid sequestrant was associated with a significant reduction in gastric cancer risk. 96
Pancreatic cancer
About 60% of pancreatic cancers occur in the head of the pancreas that includes the tract of the common bile duct, suggesting that bile acids may be involved in pancreatic carcinogenesis. 97 Bile acids have an association with several of the known risk factors of pancreatic cancer including a diet high in fat and red meat. 97
Hepatocellular cancer
In a prospective study, increased hepatocellular cancer risk was found to be associated with the higher levels of major circulating bile acids that were determined several years prior to tumor diagnosis. 98 Bile acids from the hepatocyte cytoplasm are conveyed by the bile salt export pump into the bile canaliculi. Deficiency in bile salt export due to mutations in the gene ATP-binding cassette, subfamily B member 11 (ABCB11) (encoding the bile salt export pump) can lead to intrahepatic toxic accumulation of bile salts. Among individuals having such mutations, there is an increased incidence of hepatocellular carcinoma or cholangiocarcinoma. 99 FXR plays an essential role in regulating bile acid metabolism. In an FXR−/− mouse model, the absence of this receptor led to an apparent increase in bile acids in the liver, and the spontaneous development of liver tumors. 100 Lowering the bile acid pool in the FXR−/− mice by feeding 2% cholestyramine (a bile acid–sequestering resin) reduced the number and size of the malignant lesions. In another mouse model, liver carcinogenesis induced by a high-fat diet was found to be mediated by altered microbiota that cause sustained retention of hepatic bile acids. 101
Biliary tract
Cholangiosarcomas are malignancies that arise along the biliary tract. Cholestasis (restricted bile flow) can lead to overexposure of cholangiocytes (epithelial cells of the bile duct) to bile acids and subsequently to cholangiocarcinogenesis. 102 The mechanism may involve oxidative DNA damage. 103
Small intestine
The length of the small intestine comprises 75% of the length of the whole gastrointestinal tract. 104 About 50% of small intestinal adenomas occur in the duodenum even though this region comprises just 4% of the length of the small intestine, and these adenomas arise mainly close to the ampulla of Vater, the outlet of the common bile duct where bile acids are released. 105 A human prospective study found a markedly elevated risk for carcinoid tumors of the small intestine with dietary intake of saturated fat. 106 Another human study found that cholecystectomy, which alters bile flow to the small intestine, increases the risk of small intestinal adenocarcinomas, and that the risk declines with increasing distance from the common bile duct. 107
Conclusions
The studies reviewed here critically address the cause of colon cancer. These include (1) recent human prospective and retrospective studies showing an association of bile acids in the circulation with subsequent development of colon cancer and (2) studies demonstrating the induction of colon cancer in mice fed the secondary bile acid, DOC. These more recent findings provide a basis for interpreting earlier population studies that showed an association of a high-fat diet with increased bile acid production and an elevated risk of colon cancer. Additional investigations of human cells grown in culture have provided insight into the biochemical and molecular events underlying colon carcinogenesis. In particular, these investigations showed that bile acids cause oxidative stress resulting in increased DNA damage and then mutations/epimutations, some of which provide a cellular growth advantage such as apoptosis resistance. We have tried to integrate the information from these different approaches including key results that were only recently reported. In addition, we have briefly reviewed emerging evidence indicating that bile acids have a carcinogenic role throughout the gastrointestinal system beyond colon cancer. Excessive cellular exposure to bile acids may arise, aside from a high-fat diet, from increased reflux or restricted bile flow. We consider that the evidence reviewed here indicates that a major cause of colon cancer, and probably other gastrointestinal system cancers, is excessive exposure of cells to bile acids, and that reduction of such exposure is a promising avenue for prevention.
Footnotes
Authors’ Contributions: HB and CB contributed equally to this article.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Carol Bernstein  https://orcid.org/0000-0001-7761-6565
https://orcid.org/0000-0001-7761-6565
References
- 1. American Cancer Society Cancer Facts Figures 2022 . Atlanta, GA: American Cancer Society, 2022 [Google Scholar]
- 2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49 [DOI] [PubMed] [Google Scholar]
- 3. Reddy BS. Role of bile metabolites in colon carcinogenesis. Cancer 1975;36:2401–6 [DOI] [PubMed] [Google Scholar]
- 4. Hill MJ. The role of colon anaerobes in the metabolism of bile acids and steroids, and its relation to colon cancer. Cancer 1975;36:2387–400 [DOI] [PubMed] [Google Scholar]
- 5. Nagengast FM, Grubben MJ, van Munster IP. Role of bile acids in colorectal carcinogenesis. Eur J Cancer 1995;31A:1067–70. [DOI] [PubMed] [Google Scholar]
- 6. Ocvirk S, O’Keefe SJ. Influence of bile acids on colorectal cancer risk: potential mechanisms mediated by diet–gut microbiota interactions. Curr Nutr Rep 2017;6:315–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nystrom M, Mutanen M. Diet and epigenetics in colon cancer. World J Gastroenterol 2009;15:257–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Durko L, Malecka-Panas E. Lifestyle modifications and colorectal cancer. Curr Colorectal Cancer Rep 2014;10:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drasar BS, Irving D. Environmental factors and cancer of the colon and breast. Br J Cancer 1973;27:167–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knox EG. Foods and diseases. Br J Prev Soc Med 1977;31:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller AB, Howe GR, Jain M, Craib KJ, Harrison L. Food items and food groups as risk factors in a case-control study of diet and colo-rectal cancer. Int J Cancer 1983;32:155–61 [DOI] [PubMed] [Google Scholar]
- 12. McKeown-Eyssen GE, Bright-See E. Dietary factors in colon cancer: international relationships. An update. Nutr Cancer 1985;7:251–3 [DOI] [PubMed] [Google Scholar]
- 13. Willett W. The search for the causes of breast and colon cancer. Nature 1989;338:389–94 [DOI] [PubMed] [Google Scholar]
- 14. Chan DS, Lau R, Aune D, Vieira R, Greenwood DC, Kampman E, Norat T. Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies. PLoS ONE 2011;6:e20456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J, Oh SW, Kim YS, Kwon H, Joh HK, Lee JE, Park D, Park JH, Ko AR, Kim YJ. Association between dietary fat intake and colorectal adenoma in Korean adults: a cross-sectional study. Medicine (Baltimore) 2017;96:e5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, Posma JM, Kinross J, Wahl E, Ruder E, Vipperla K, Naidoo V, Mtshali L, Tims S, Puylaert PG, DeLany J, Krasinskas A, Benefiel AC, Kaseb HO, Newton K, Nicholson JK, de Vos WM, Gaskins HR, Zoetendal EG. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun 2015;6:6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ridlon JM, Wolf PG, Gaskins HR. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016;7:201–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016;7:22–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Intestinal absorption of bile acids in health and disease. Compr Physiol 2019;10:21–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mertens KL, Kalsbeek A, Soeters MR, Eggink HM. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front Neurosci 2017;11:617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol 2014;12:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reddy BS, Hanson D, Mangat S, Mathews L, Sbaschnig M, Sharma C, Simi B. Effect of high-fat, high-beef diet and of mode of cooking of beef in the diet on fecal bacterial enzymes and fecal bile acids and neutral sterols. J Nutr 1980;110:1880–7 [DOI] [PubMed] [Google Scholar]
- 23. Wan Y, Wang F, Yuan J, Li J, Jiang D, Zhang J, Li H, Wang R, Tang J, Huang T, Zheng J, Sinclair AJ, Mann J, Li D. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut 2019;68:1417–29 [DOI] [PubMed] [Google Scholar]
- 24. Yoshitsugu R, Kikuchi K, Iwaya H, Fujii N, Hori S, Lee DG, Ishizuka S. Alteration of bile acid metabolism by a high-fat diet is associated with plasma transaminase activities and glucose intolerance in rats. J Nutr Sci Vitaminol (Tokyo) 2019;65:45–51 [DOI] [PubMed] [Google Scholar]
- 25. Hill MJ, Taylor AJ, Thompson MH, Wait R. Fecal steroids and urinary volatile phenols in four Scandinavian populations. Nutr Cancer 1982;4:67–73 [DOI] [PubMed] [Google Scholar]
- 26. Crowther JS, Drasar BS, Hill MJ, Maclennan R, Magnin D, Peach S, Teoh-chan CH. Faecal steroids and bacteria and large bowel cancer in Hong Kong by socio-economic groups. Br J Cancer 1976;34:191–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reddy BS, Wynder EL. Metabolic epidemiology of colon cancer. Fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polyps. Cancer 1977;39:2533–9 [DOI] [PubMed] [Google Scholar]
- 28. Jensen OM, MacLennan R, Wahrendorf J. Diet, bowel function, fecal characteristics, and large bowel cancer in Denmark and Finland. Nutr Cancer 1982;4:5–19 [DOI] [PubMed] [Google Scholar]
- 29. Imray CH, Radley S, Davis A, Barker G, Hendrickse CW, Donovan IA, Lawson AM, Baker PR, Neoptolemos JP. Faecal unconjugated bile acids in patients with colorectal cancer or polyps. Gut 1992;33:1239–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ocvirk S, Wilson AS, Posma JM, Li JV, Koller KR, Day GM, Flanagan CA, Otto JE, Sacco PE, Sacco FD, Sapp FR, Wilson AS, Newton K, Brouard F, DeLany JP, Behnning M, Appolonia CN, Soni D, Bhatti F, Methé B, Fitch A, Morris A, Gaskins HR, Kinross J, Nicholson JK, Thomas TKO’, Keefe SJD. A prospective cohort analysis of gut microbial co-metabolism in Alaska Native and rural African people at high and low risk of colorectal cancer. Am J Clin Nutr 2020;111:406–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loftfield E, Falk RT, Sampson JN, Huang WY, Hullings A, Murphy G, Weinstein SJ, Albanes D, Freedman ND, Sinha R. Prospective associations of circulating bile acids and short-chain fatty acids with incident colorectal cancer. JNCI Cancer Spectr 2022;6:pkac027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kühn T, Stepien M, López-Nogueroles M, Damms-Machado A, Sookthai D, Johnson T, Roca M, Hüsing A, Maldonado SG, Cross AJ, Murphy N, Freisling H, Rinaldi S, Scalbert A, Fedirko V, Severi G, Boutron-Ruault MC, Mancini FR, Sowah SA, Boeing H, Jakszyn P, Sánchez MJ, Merino S, Colorado-Yohar S, Barricarte A, Khaw KT, Schmidt JA, Perez-Cornago A, Trichopoulou A, Karakatsani A, Thriskos P, Palli D, Agnoli C, Tumino R, Sacerdote C, Panico S, Bueno-de-Mesquita B, van Gils CH, Heath AK, Gunter MJ, Riboli E, Lahoz A, Jenab M, Kaaks R. Prediagnostic plasma bile acid levels and colon cancer risk: a prospective study. J Natl Cancer Inst 2020;112:516–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li S, Qu X, Zhang L, Wang N, Chen M, Zhao X, Wang J, Lv H, Qi Y, Zhang L, Liu J, Shi Y. Serum total bile acids in relation to gastrointestinal cancer risk: a retrospective study. Front Oncol 2022;12:859716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Liu H, Li L, Ai M, Gong Z, He Y, Dong Y, Xu S, Wang J, Jin B, Liu J, Teng Z. Cholecystectomy can increase the risk of colorectal cancer: a meta-analysis of 10 cohort studies. PLoS ONE 2017;12:e0181852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aurif F, Kaur H, Chio JPG, Kittaneh M, Malik BH. The association between cholecystectomy and colorectal cancer in the female gender. Cureus 2021;13:e20113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 2011;85:863–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, Bernstein H, Bernstein C. Novel diet-related mouse model of colon cancer parallels human colon cancer. World J Gastrointest Oncol 2014;6:225–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomac I, Šeruga M, Labuda J. Evaluation of antioxidant activity of chlorogenic acids and coffee extracts by an electrochemical DNA-based biosensor. Food Chem 2020;325:126787. [DOI] [PubMed] [Google Scholar]
- 39. Nieber K. The impact of coffee on health. Planta Med 2017;83:1256–63 [DOI] [PubMed] [Google Scholar]
- 40. Um CY, McCullough ML, Guinter MA, Campbell PT, Jacobs EJ, Gapstur SM. Coffee consumption and risk of colorectal cancer in the Cancer Prevention Study-II Nutrition Cohort. Cancer Epidemiol 2020;67:101730. [DOI] [PubMed] [Google Scholar]
- 41. Hayashi E, Amuro Y, Endo T, Yamamoto H, Miyamoto M, Kishimoto S. Fecal bile acids and neutral sterols in rats with spontaneous colon cancer. Int J Cancer 1986;37:629–32 [DOI] [PubMed] [Google Scholar]
- 42. Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol 2009;15:1677–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delaney S, Jarem DA, Volle CB, Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic Res 2012;46:420–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott TL, Rangaswamy S, Wicker CA, Izumi T. Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair. Antioxid Redox Signal 2014;20:708–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol 2001;33:193–203 [DOI] [PubMed] [Google Scholar]
- 46. Teschendorff AE, Gao Y, Jones A, Ruebner M, Beckmann MW, Wachter DL, Fasching PA, Widschwendter M. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun 2016;7:10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paik JH, Jung EJ, Ryu CG, Hwang DY. Detection of Polyps After Resection of Colorectal Cancer. Ann Coloproctol 2015;31:182–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Angelis PM, Dorg L, Pham S, Andersen SN. DNA Repair protein expression and oxidative/nitrosative stress in ulcerative colitis and sporadic colorectal cancer. Anticancer Res 2021;41:3261–70 [DOI] [PubMed] [Google Scholar]
- 49. Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C. Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer. Genome Integr 2012;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rubin H. Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture. Bioessays 2011;33:224–31 [DOI] [PubMed] [Google Scholar]
- 51. Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res 2002;511:145–78 [DOI] [PubMed] [Google Scholar]
- 52. Fisher LA, Samson L, Bessho T. Removal of reactive oxygen species-induced 3’-blocked ends by XPF-ERCC1. Chem Res Toxicol 2011;24: 1876–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cortopassi GA, Wang E. There is substantial agreement among interspecies estimates of DNA repair activity. Mech Ageing Dev 1996;91: 211–8 [DOI] [PubMed] [Google Scholar]
- 54. Crowley-Weber CL, Payne CM, Gleason-Guzman M, Watts GS, Futscher B, Waltmire CN, Crowley C, Dvorakova K, Bernstein C, Craven M, Garewal H, Bernstein H. Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis 2002;23:2063–80 [DOI] [PubMed] [Google Scholar]
- 55. Badvie S, Hanna-Morris A, Andreyev HJ, Cohen P, Saini S, Allen-Mersh TG. A “field change” of inhibited apoptosis occurs in colorectal mucosa adjacent to colorectal adenocarcinoma. J Clin Pathol 2006;59: 942–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ramesh P, Medema JP. BCL-2 family deregulation in colorectal cancer: potential for BH3 mimetics in therapy. Apoptosis 2020;25:305–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Botham KM, Boyd GS. The effect of dietary fat on bile salt synthesis in rat liver. Biochim Biophys Acta 1983;752:307–14 [DOI] [PubMed] [Google Scholar]
- 58. Bisschop PH, Bandsma RH, Stellaard F, ter Harmsel A, Meijer AJ, Sauerwein HP, Kuipers F, Romijn JA. Low-fat, high-carbohydrate and high-fat, low-carbohydrate diets decrease primary bile acid synthesis in humans. Am J Clin Nutr 2004;79:570–6 [DOI] [PubMed] [Google Scholar]
- 59. Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O’Keefe SJ. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 2013;98:111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, Bringer MA, Pezet D, Bonnet M. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 2016;22:501–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Devkota S, Chang EB. Interactions between diet, bile acid metabolism, gut microbiota, and inflammatory bowel diseases. Dig Dis 2015;33: 351–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tian Y, Gui W, Koo I, Smith PB, Allman EL, Nichols RG, Rimal B, Cai J, Liu Q, Patterson AD. The microbiome modulating activity of bile acids. Gut Microbes 2020;11:979–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watanabe M, Fukiya S, Yokota A. Comprehensive evaluation of the bactericidal activities of free bile acids in the large intestine of humans and rodents. J Lipid Res 2017;58:1143–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Glinghammar B, Inoue H, Rafter JJ. Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors Nf-kb and AP-1. Carcinogenesis 2002;23:839–45 [DOI] [PubMed] [Google Scholar]
- 65. Basu AK. DNA damage, mutagenesis and cancer. Int J Mol Sci 2018;19:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 2012;10:575–82 [DOI] [PubMed] [Google Scholar]
- 67. Sun J, Kato I. Gut microbiota, inflammation and colorectal cancer. Genes Dis 2016;3:130–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamagishi H, Kuroda H, Imai Y, Hiraishi H. Molecular pathogenesis of sporadic colorectal cancers. Chin J Cancer 2016;35:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stodolna A, He M, Vasipalli M, Kingsbury Z, Becq J, Stockton JD, Dilworth MP, James J, Sillo T, Blakeway D, Ward ST, Ismail T, Ross MT, Beggs AD. Clinical-grade whole-genome sequencing and 3’ transcriptome analysis of colorectal cancer patients. Genome Med 2021;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z. OGG1 is essential in oxidative stress induced DNA demethylation. Cell Signal 2016;28:1163–71 [DOI] [PubMed] [Google Scholar]
- 72. Ding N, Maiuri AR, O’Hagan HM. The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutat Res Rev Mutat Res 2019;780:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wei J, Li G, Dang S, Zhou Y, Zeng K, Liu M. Discovery and validation of hypermethylated markers for colorectal cancer. Dis Markers 2016; 2016:2192853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Beggs AD, Jones A, El-Bahrawy M, Abulafi M, Hodgson SV, Tomlinson IP. Whole-genome methylation analysis of benign and malignant colorectal tumours. J Pathol 2013;229:697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Strauch ED, Bass BL, Rao JN, Vann JA, Wang JY. NF-kappaB regulates intestinal epithelial cell and bile salt-induced migration after injury. Ann Surg 2003;237:494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zinatizadeh MR, Schock B, Chalbatani GM, Zarandi PK, Jalali SA, Miri SR. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis 2020;8:287–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hugh TJ, Dillon SA, O’Dowd G, Getty B, Pignatelli M, Poston GJ, Kinsella AR. beta-catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer 1999;82:504–11 [DOI] [PubMed] [Google Scholar]
- 78. Kapiteijn E, Liefers GJ, Los LC, Kranenbarg EK, Hermans J, Tollenaar RA, Moriya Y, van de Velde CJ, van Krieken JH. Mechanisms of oncogenesis in colon versus rectal cancer. J Pathol 2001;195:171–8 [DOI] [PubMed] [Google Scholar]
- 79. Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, Kolligs FT. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics 2014;15:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yde J, Wu Q, Borg JF, Fenton RA, Moeller HB. A systems-level analysis of bile acids effects on rat colon epithelial cells. Am J Physiol Gastrointest Liver Physiol 2022;322:G34–48 [DOI] [PubMed] [Google Scholar]
- 81. Guerin MV, Finisguerra V, Van den Eynde BJ, Bercovici N, Trautmann A. Preclinical murine tumor models: a structural and functional perspective. Elife 2020;9:e50740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu H, Wang J, He T, Becker S, Zhang G, Li D, Ma X. Butyrate: a double-edged sword for health? Adv Nutr 2018;9:21–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhong D, Li Y, Huang Y, Hong X, Li J, Jin R. Molecular mechanisms of exercise on cancer: a bibliometrics study and visualization analysis via CiteSpace. Front Mol Biosci 2022;8:797902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aoi W, Naito Y, Takagi T, Tanimura Y, Takanami Y, Kawai Y, Sakuma K, Hang LP, Mizushima K, Hirai Y, Koyama R, Wada S, Higashi A, Kokura S, Ichikawa H, Yoshikawa T. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gut 2013;62:882–9 [DOI] [PubMed] [Google Scholar]
- 85. Frühbeck G, Mentxaka A, Ahechu P, Gómez-Ambrosi J, Ramírez B, Becerril S, Rodríguez A, Unamuno X, Cienfuegos JA, Casado M, Burrell MA, Martín M, Baixauli J, Valentí V, Moncada R, Reina G, Silva C, Catalán V. The differential expression of the inflammasomes in adipose tissue and colon influences the development of colon cancer in a context of obesity by regulating intestinal inflammation. J Inflamm Res 2021;14:6431–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 2005;589:47–65 [DOI] [PubMed] [Google Scholar]
- 87. Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol 2009;15:3329–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Režen T, Rozman D, Kovács T, Kovács P, Sipos A, Bai P, Mikó E. The role of bile acids in carcinogenesis. Cell Mol Life Sci 2022;79:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Choi YJ, Jin EH, Lim JH, Shin CM, Kim N, Han K, Lee DH. Increased risk of cancer after cholecystectomy: a nationwide cohort study in Korea including 123,295 patients. Gut Liver 2022;16:465–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sasaki CT, Doukas SG, Costa J, Vageli DP. Biliary reflux as a causal factor in hypopharyngeal carcinoma: new clinical evidence and implications. Cancer 2019;125:3554–65 [DOI] [PubMed] [Google Scholar]
- 91. Vageli DP, Doukas SG, Doukas PG, Judson BL. Bile reflux and hypopharyngeal cancer (Review). Oncol Rep 2021;46:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sasaki CT, Doukas SG, Doukas PG, Vageli DP. Weakly acidic bile is a risk factor for hypopharyngeal carcinogenesis evidenced by DNA damage, antiapoptotic function, and premalignant dysplastic lesions in vivo. Cancers (Basel) 2021;13:852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jenkins GJ, Cronin J, Alhamdani A, Rawat N, D’Souza F, Thomas T, Eltahir Z, Griffiths AP, Baxter JN. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis 2008;23:399–405 [DOI] [PubMed] [Google Scholar]
- 94. Li D, Zhang J, Yao WZ, Zhang DL, Feng CC, He Q, Lv HH, Cao YP, Wang J, Qi Y, Wu SR, Wang N, Zhao J, Shi YQ. The relationship between gastric cancer, its precancerous lesions and bile reflux: a retrospective study. J Dig Dis 2020;21:222–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Alizadeh M, Raufman JP. Gastrointestinal neoplasia: carcinogenic interaction between bile acids and Helicobacter pylori in the stomach. J Clin Invest 2022;132:e160194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Noto JM, Piazuelo MB, Shah SC, Romero-Gallo J, Hart JL, Di C, Carmichael JD, Delgado AG, Halvorson AE, Greevy RA, Wroblewski LE, Sharma A, Newton AB, Allaman MM, Wilson KT, Washington MK, Calcutt MW, Schey KL, Cummings BP, Flynn CR, Zackular JP, Peek RM., Jr. Iron deficiency linked to altered bile acid metabolism promotes Helicobacter pylori-induced inflammation-driven gastric carcinogenesis. J Clin Invest 2022;132:e147822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Feng HY, Chen YC. Role of bile acids in carcinogenesis of pancreatic cancer: an old topic with new perspective. World J Gastroenterol 2016;22:7463–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stepien M, Lopez-Nogueroles M, Lahoz A, Kühn T, Perlemuter G, Voican C, Ciocan D, Boutron-Ruault MC, Jansen E, Viallon V, Leitzmann M, Tjønneland A, Severi G, Mancini FR, Dong C, Kaaks R, Fortner RT, Bergmann MM, Boeing H, Trichopoulou A, Karakatsani A, Peppa E, Palli D, Krogh V, Tumino R, Sacerdote C, Panico S, Bueno-de-Mesquita HB, Skeie G, Merino S, Ros RZ, Sánchez MJ, Amiano P, Huerta JM, Barricarte A, Sjöberg K, Ohlsson B, Nyström H, Werner M, Perez-Cornago A, Schmidt JA, Freisling H, Scalbert A, Weiderpass E, Christakoudi S, Gunter MJ, Jenab M. Prediagnostic alterations in circulating bile acid profiles in the development of hepatocellular carcinoma. Int J Cancer 2022;150:1255–68 [DOI] [PubMed] [Google Scholar]
- 99. Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, Meier Y, Antoniou A, Stieger B, Arnell H, Ozçay F, Al-Hussaini HF, Bassas AF, Verkade HJ, Fischler B, Németh A, Kotalová R, Shneider BL, Cielecka-Kuszyk J, McClean P, Whitington PF, Sokal E, Jirsa M, Wali SH, Jankowska I, PawÅ‚owska J, Mieli-Vergani G, Knisely AS, Bull LN, Thompson RJ. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008;134:1203–14 [DOI] [PubMed] [Google Scholar]
- 100. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007;67:863–7 [DOI] [PubMed] [Google Scholar]
- 101. Xie G, Wang X, Huang F, Zhao A, Chen W, Yan J, Zhang Y, Lei S, Ge K, Zheng X, Liu J, Su M, Liu P, Jia W. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer 2016; 139:1764–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Labib PL, Goodchild G, Pereira SP. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer 2019;19:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Komichi D, Tazuma S, Nishioka T, Hyogo H, Chayama K. Glycochenodeoxycholate plays a carcinogenic role in immortalized mouse cholangiocytes via oxidative DNA damage. Free Radic Biol Med 2005;39:1418–27 [DOI] [PubMed] [Google Scholar]
- 104. Maguire A, Sheahan K. Primary small bowel adenomas and adenocarcinomas-recent advances. Virchows Arch 2018;473:265–73 [DOI] [PubMed] [Google Scholar]
- 105. Ross RK, Hartnett NM, Bernstein L, Henderson BE. Epidemiology of adenocarcinomas of the small intestine: is bile a small bowel carcinogen. Br J Cancer 1991;63:143–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cross AJ, Leitzmann MF, Subar AF, Thompson FE, Hollenbeck AR, Schatzkin A. A prospective study of meat and fat intake in relation to small intestinal cancer. Cancer Res 2008;68:9274–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lagergren J, Ye W, Ekbom A. Intestinal cancer after cholecystectomy: is bile involved in carcinogenesis. Gastroenterology 2001;121:542–7 [DOI] [PubMed] [Google Scholar]