

Abstract
Since the discovery of recurrent mutations in histone H3 variants in pediatric brain tumours, so-called ‘oncohistones’ have been identified in various cancers. While their mechanism of action remains under active investigation, several studies have shed light on how they promote genome-wide epigenetic perturbations. These findings converge on altered post-translational modifications on two key lysine (K) residues of the H3 tail, K27 and K36, which regulate several cellular processes, including those linked to cell differentiation during development. We will review how these oncohistones affect the methylation of cognate residues, but also disrupt the distribution of opposing chromatin marks, creating genome-wide epigenetic changes which participate in the oncogenic process. Ultimately, tumorigenesis is promoted through the maintenance of a progenitor state at the expense of differentiation in defined cellular and developmental contexts. As these epigenetic disruptions are reversible, improved understanding of oncohistone pathogenicity can result in needed alternative therapies.
Keywords: H3, oncohistones, epigenome, development, differentiation
Graphical Abstract
A number of cancers carry recurrent, somatic, gain-of-function, heterozygous mutations in different histone 3 (H3)-encoding genes, which lead to amino acid substitutions on key residues of the H3 tail. These hotspot H3 mutations, oncohistones as we label them, were first identified in a deadly brain cancer, pediatric high-grade gliomas (pHGGs), where they account for a large proportion of these tumours. Notably, they show remarkable spatio-temporal specificity, indicating that their pathogenesis may be closely linked to aberrant development [1, 2]. Indeed, HGGs of the central nervous system midline (which includes the pons, thalamus and spine) target younger children, and show a high frequency (~80%) of H3 lysine to methionine, or rarely to isoleucine, substitutions (K27M/I). These K27M/I mutations occur in either canonical H3.1/H3.2 variants mainly in the pons, or in the non-canonical H3.3 variant across all brain midline structures (Figure 1) [2–6]. By contrast, in HGGs of the brain hemispheres, glycine 34 to arginine or valine (G34R/V) substitutions are specific to H3F3A, which encodes the H3.3 variant, and target primarily the temporo-parietal cortex in adolescents and young adults, where they account for ~30% of these tumours [2–6]. Other hemispheric H3 wild-type gliomas of adolescents and young adults (mean age of 33 years) occur primarily in fronto-parietal lobes and carry non-overlapping truncating mutations in SETD2 [7], the only H3K36 tri-methyltransferase in humans, and/or hotspot somatic mutations in isocitrate dehydrogenases 1/2 (IDH1/2) [3, 6, 8–10]. IDH mutations generate a neomorphic enzyme and excess production of the oncometabolite 2-hydroxyglutarate, which competitively inhibits histone and DNA demethylases to affect the methylation of K residues on the H3 tail and promote a CpG island methylator phenotype (CIMP). Together, these findings reinforce that epigenetic perturbations are a major initiating factor in the development of pediatric and young adult HGGs. Furthermore, the regional and temporal specificity of the mutations, also reflected molecularly in distinct DNA methylation and gene expression profiles [5, 11], suggests different cellular origins during their development.
Figure 1. Histone mutations in cancers.
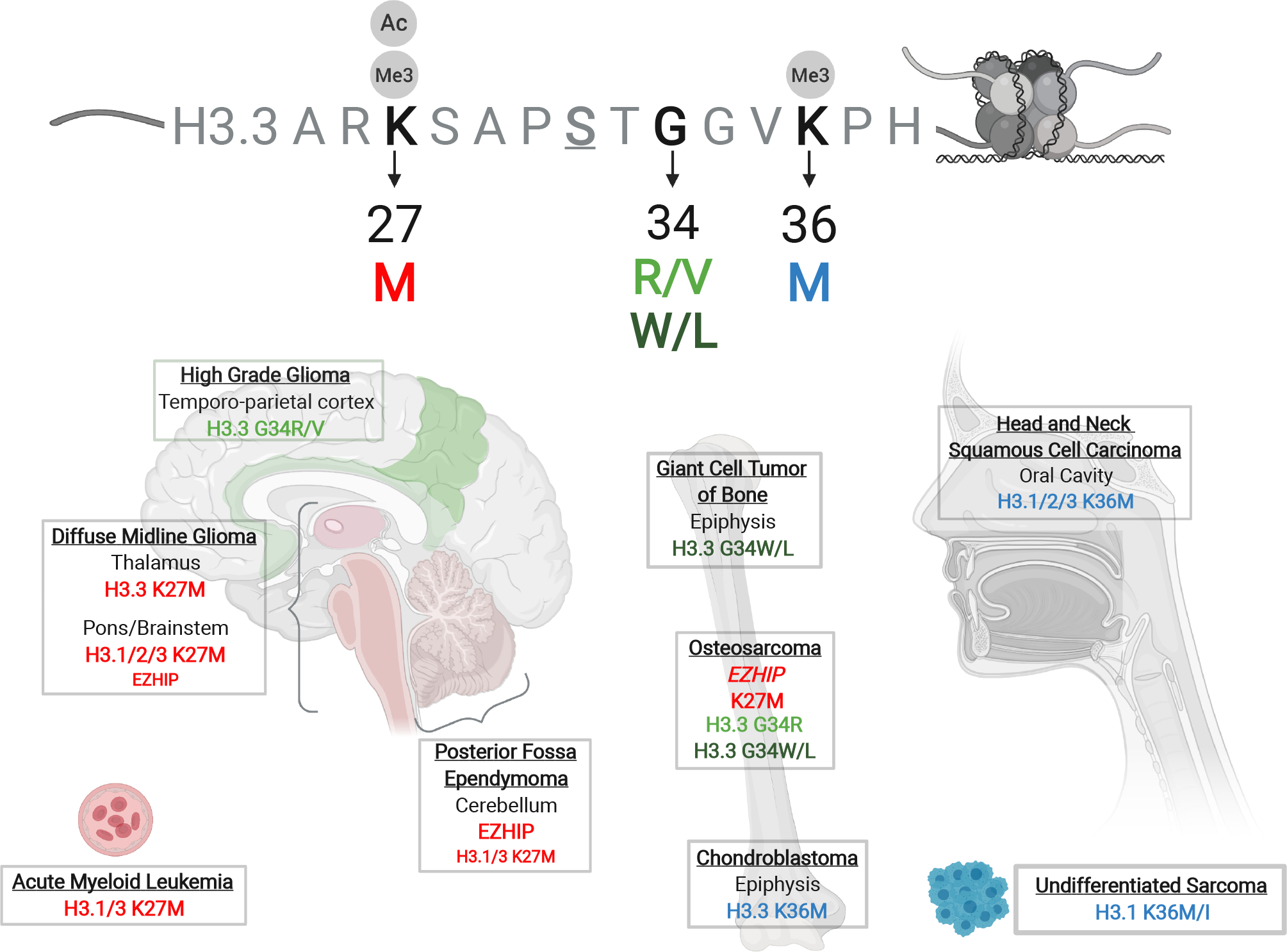
Schematic of the histone H3.3 tail above, highlighting key residues (K27, G34, K36) recurrently mutated in cancers and their associated post-translational modifications. Depicted below is the regional tissue specificity of oncohistone mutations and their occurrence in specific cancer types.
Outside of HGGs, oncohistones are the initiating driver event in other cancers. H3K27M/I mutations occur in rare cases of acute myeloid leukemia [12, 13] and a subgroup of ependymoma, Group A posterior fossa ependymomas (PFA-EPN) [14, 15]. Strikingly, recent studies identified that high expression of the uncharacterized gene CXorf67 (now designated EZH2 inhibitory protein - EZHIP) predominates in PFA-EPN and are mutually exclusive with the rare H3K27M mutations identified in these tumours [16–18]. Furthermore, rare H3 wild-type diffuse midline gliomas also exhibit high expression of EZHIP, with data reviewed below suggesting the encoded protein has similar effects as H3K27M on the epigenome [16–19].
In giant cell tumour of bone (GCTB), H3.3G34 mutations constitute the singular driver mutation, with ~ 92% characterized by H3F3A or H3F3B (the other gene encoding for H3.3) glycine to tryptophan (G34W) substitutions, and rarely by leucine, arginine, valine or methionine (G34L/R/V/M) substitutions [20]. Moreover, K36 to methionine (K36M) substitutions in H3F3B occur in 95% of chondroblastomas [20], while a subset of undifferentiated soft tissue sarcomas [21], and of human papillomavirus (HPV)-negative head and neck squamous cell carcinomas (HNSCCs) [22] carry H3K36M/I mutations. Additionally, ~6% of osteosarcomas carry either K27M but mostly G34R or G34W H3 mutations [23]. Last, various somatic histone mutations were recently identified at a low frequency across many different cancers, on residues other than H3K27, H3G34 or H3K36, as well as in non-H3 core histones [24]; however, for some, their role as oncogenic drivers remains to be experimentally validated.
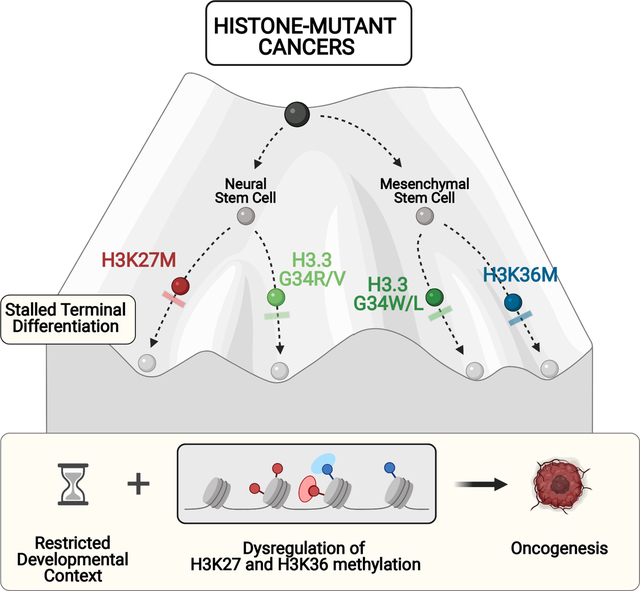
The exquisite temporo-spatial distribution and the specificity of distinct histone mutations to certain cancer types suggests the presence of permissive windows during development where the cell/lineage-of-origin is vulnerable to epigenomic perturbation. We will review herein how post-translational modifications (PTM) on K27 and K36 are established and the crosstalk between these marks and Polycomb repressive complexes (PRC1/2) deposition and function. We will discuss recent findings on how these oncohistones and the oncohistone-mimic EZHIP disrupt this crosstalk to stall differentiation, promoting tumour formation through a novel mechanism that involves deregulation of PRC functions during normal development.
Polycomb repressive complexes and H3K27 and H3K36 PTM
The epigenetic effects of H3K27, H3G34 and H3K36 mutations are mediated through H3K27 and H3K36 PTM, which regulate transcriptional activity by recruiting distinct reader proteins to chromatin (Figure 2). H3K27 can be acetylated by CBP and p300 acetyltransferases, or methylated by PRC2 which comprises the catalytic EZH2 or EZH1 component, as well as EED, SUZ12 and RBBP4/7 core subunits. H3K27ac is an active histone mark associated with cis regulatory elements such as promoters and enhancers, promoting the expression of genes implicated in cell differentiation [25]. The highest methylation state of H3K27, H3K27me3, is a repressive mark commonly occurring in facultative heterochromatin, and plays an important role in cell fate determination and differentiation during development [26, 27]. The functions of the lower methylation states of H3K27 (H3K27me1/2) are less understood, although the broad genomic distribution of H3K27me2 may serve as a protective mechanism against inappropriate activation of distal cis regulatory elements [28]. One of the mechanisms by which H3K27me3 is known to repress transcription is by recruiting canonical PRC1, which deposits H2AK119 monoubiquitylation and contains a reader chromobox (CBX) component that recognizes H3K27me3 and compacts adjacent chromatin. Similarly, PRC2 is allosterically activated through the recognition of its own H3K27me3 product by the EED subunit, enabling propagation of repressive H3K27me3 [29]. H3K27me3 genomic distribution varies considerably by cell type and developmental stage and is impacted by the presence of other opposing epigenetic marks including H3K9me3 (marking constitutive heterochromatin), DNA methylation, H3K27ac, and H3K36me2/3.
Figure 2. Relationships between H3K27, H3K36 and DNA methylation.
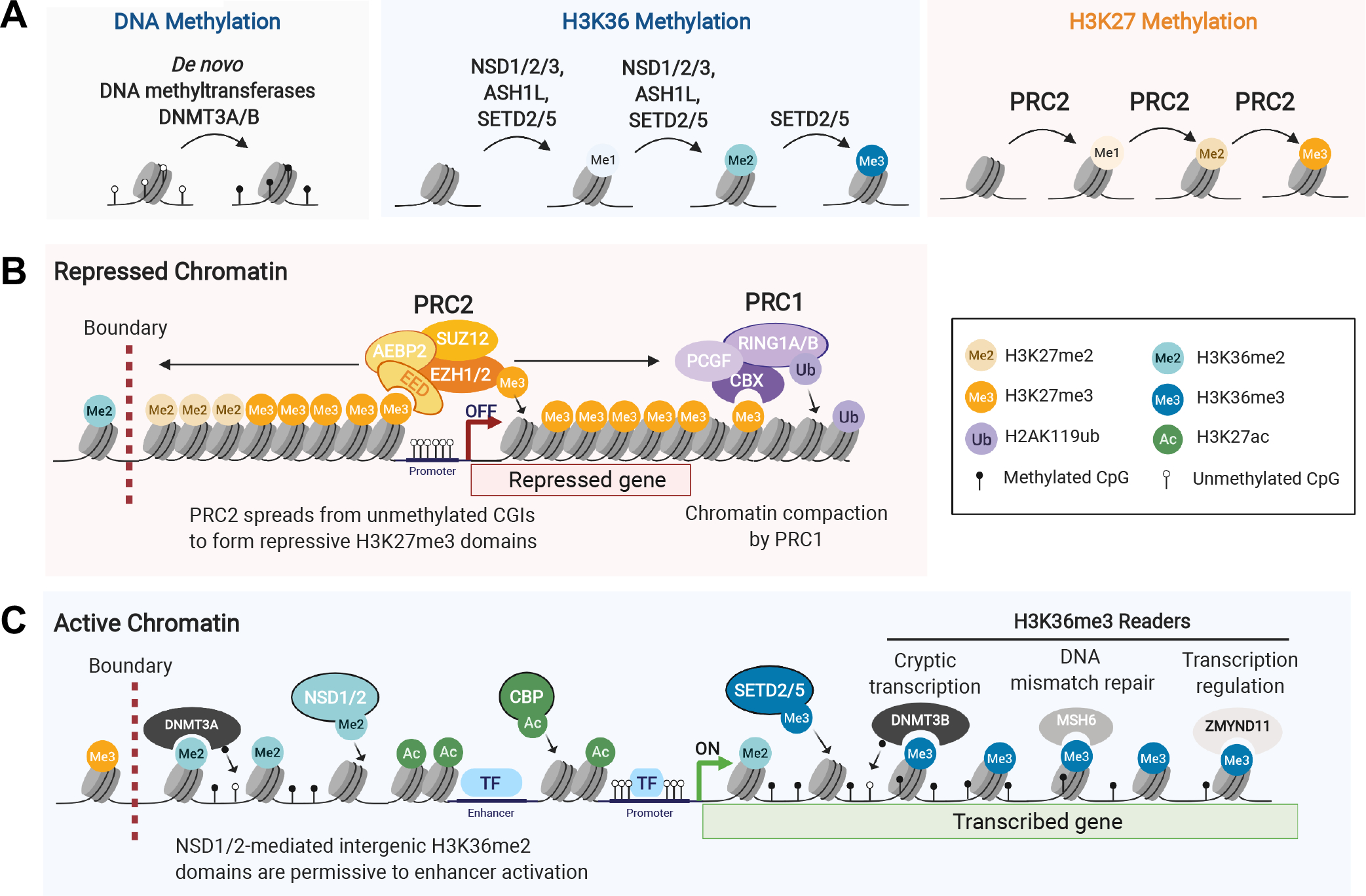
A. Methyltransferases performing the steps of de novo DNA methylation, H3K36 and H3K27 methylation.
B. An example of repressed chromatin mediated by the PRC2 and PRC1 complexes, as initiated by PRC2 recruitment to unmethylated CpG islands (CGIs) and consequent spread of H3K27 methylation, and followed by chromatin compaction by canonical PRC1 which recognizes H3K27me3 through its CBX subunit.
C. An example of active chromatin, illustrated by co-regulation of intergenic domains by H3K36me2- and H3K27ac-depositing enzymes, whereas genic deposition of H3K36me3 recruits various readers with distinct functions.
Several methyltransferases perform H3K36 methylation in mammalian cells, including NSD1, NSD2, NSD3, ASH1L, and SETD2. Although H3K36me3 is considered an active histone mark, its deposition is likely coupled to transcription: the H3K36 tri-methyltransferase Set2 in yeast is recruited by RNA polymerase II during transcription elongation along gene bodies. H3K36me3 is absent on intronless genes, more abundant on transcribed exons, and represses specific splicing events, suggesting it may regulate alternative splicing and spurious intragenic transcription. H3K36 methylation is recognized by various reader proteins containing PWWP domains. For example, the PWWP domain of the de novo DNA methyltransferase DNMT3B preferentially mediates binding to H3K36me3 at gene bodies [30], while the PWWP domain of DNMT3A preferentially recognizes H3K36me2 [31], a mark widely distributed across active intergenic regions [32]. H3K36me3-mediated recruitment of DNMT3B and the resulting genic DNA methylation may reduce aberrant, or cryptic, transcription initiation at sites other than at the canonical promoter [33]. Other proteins with H3K36-recognizing PWWP domains include the DNA mismatch repair protein MSH6 [34], and the transcriptional repressor ZMYND11 [35], suggesting varied roles for H3K36 methylation.
Importantly, there is considerable crosstalk between H3K27 and H3K36 methylation states. Patients with germline mutations in H3K27/K36 methyltransferases present with overlapping developmental syndromes characterized by varying degrees of intellectual disability, and aberrant skeletal development. Notably, neuroectoderm-derived brain and mesenchymal-derived bone tissues are the ones commonly implicated in H3-mutant cancers, suggesting their specific susceptibility to perturbations of H3K27/K36 methylation. Molecularly, H3K27/K36 crosstalk is exemplified by mutual exclusivity between higher methylation states, such that H3K27me3 and H3K36me3 and, even if less stringently, H3K36me2 do not coexist on the same H3 tail. NSD1 loss in mouse embryonic stem cells (ESCs) leads to H3K36me2 reduction genome-wide and concurrent expansion of H3K27me3 domains [36]. By contrast, NSD2 overexpression in multiple myeloma promotes expansion of intergenic H3K36me2 domains and contraction of H3K27me3 domains [32, 37]. This H3K27/K36 crosstalk may be mediated through a sensing pocket in EZH2 adjacent to its catalytic site, promoting the enzyme’s catalytic activity in the presence of unmodified H3K36 but hindering it when H3K36 is methylated [38]. A deeper understanding of the chromatin dynamics of H3K27 and H3K36 PTMs and their impact on transcription has been provided as shown below by studies on oncohistone pathogenesis (Figure 3).
Figure 3. Epigenetic mechanisms of oncohistone mutants.
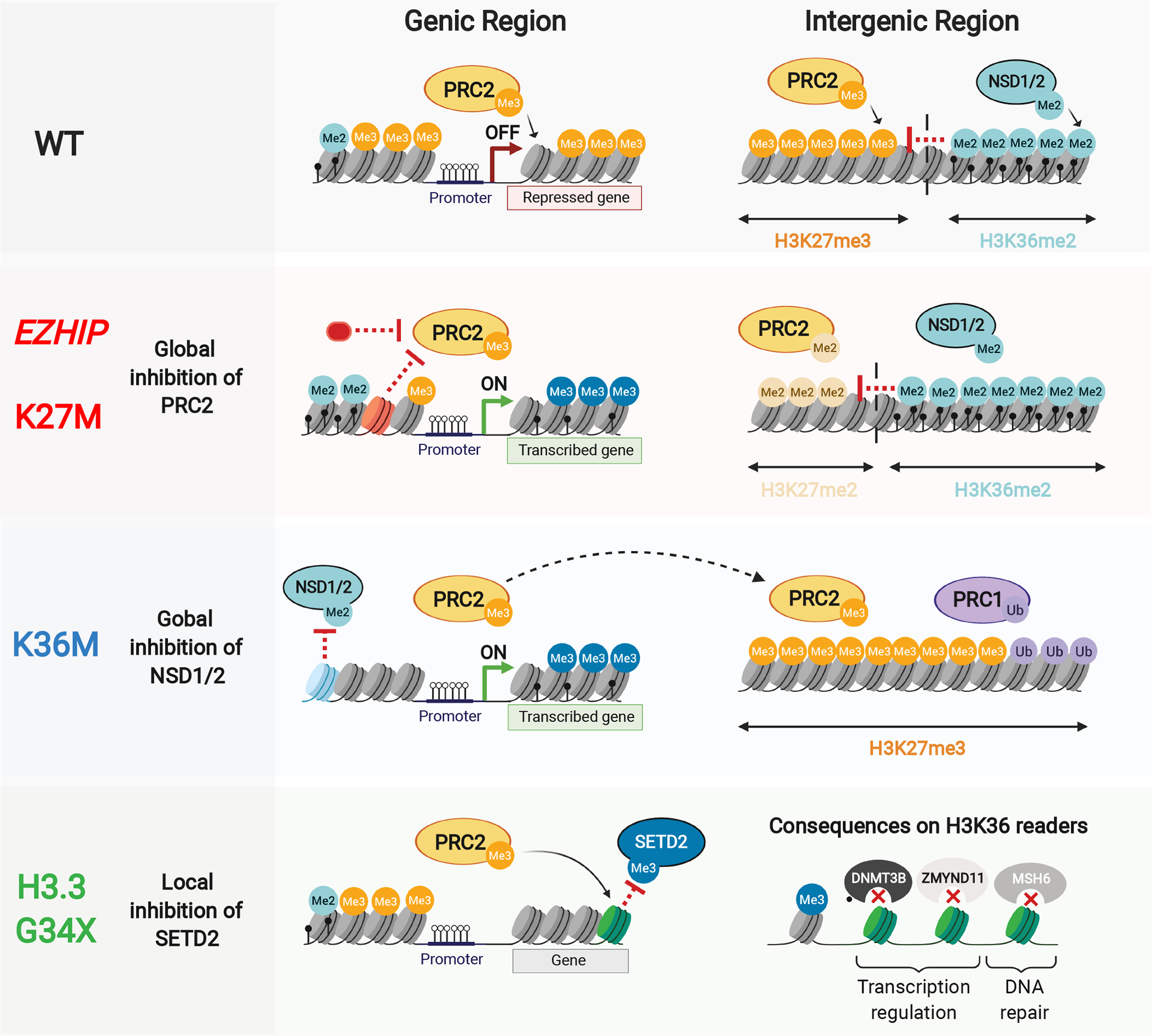
Schematic illustrating immediate consequences of oncohistone mutations on methyltransferase function (left), followed by downstream effects (right) resulting from disrupted boundaries and genomic redistribution of methyltransferases, or disruption of local reader recruitment.
K-to-M oncohistones
K-to-M oncohistones have been the most extensively studied, as they have a dominant negative effect on levels of their cognate lysine PTMs even if they only contribute 3–18% of the total H3 pool [39]. Indeed, global levels of H3K27 and H3K36 methylation are consistently reduced in K27M and K36M-mutant cells respectively, regardless of whether the mutation occurs in canonical H3.1/2 or variant H3.3 genes [21, 39–43]. Canonical H3.1/2 histones are deposited ubiquitously in the genome in a DNA replication-coupled manner, while variant H3.3 histones are deposited in transcriptionally active genic and regulatory regions in a DNA replication-independent manner by distinct chaperones. These profiles reflect the deposition patterns of H3.1K27M versus H3.3K27M oncohistones [43, 44]. However, the differing locations of H3.1K27M and H3.3K27M pHGGs in the brain, their distinct partner mutations, enhancer, DNA methylation and gene expression landscapes [44, 45] suggest a distinct cell-of-origin that has made unraveling the effects of H3.1K27M and H3.3K27M on chromatin challenging.
The mechanisms underlying K-to-M inhibition of cognate methyltransferases remain incompletely understood. Structural modelling of H3K36M/I with SETD2’s catalytic domain (well conserved in NSD1/2 and ASH1L) predicted enhanced association with the mutant H3 compared to wild-type [46]. While some structural modelling and in vitro studies suggest that the H3K27M histone binds with greater avidity to EZH2 than wild-type H3 [39, 47], other studies indicate that this effect of H3K27M on PRC2 binding is minor [48]. Indeed, multiple studies mapping H3K27M and PRC2 on chromatin indicate that they do not co-localize [43, 49–52]. These data argue against sequestration of PRC2 onto H3K27M nucleosomes. However, transient association between PRC2 and K27M on chromatin has been shown to impair EZH2 catalytic activity even after PRC2 dissociates from H3K27M [53], possibly through persistent reduction of EZH2 automethylation in K27M cells [54]. While this persistent inhibition of PRC2 activity by K27M could explain its dominant negative effect on H3K27 methylation, the unique chromatin profile of H3K27M pHGGs continues to be an area of active investigation.
Mutations in PRC2 components in malignant peripheral nerve sheath tumours result in near-complete loss of H3K27me3 genome-wide and frequently overlap with CDKN2A deletions [55, 56]. In contrast, genome-wide profiling of H3K27M pHGG cells revealed that despite a profound global loss of H3K27me3, residual deposition of the mark persists, and is restricted to narrow PRC2 recruitment sites in mutant cells [41, 51, 57]. These are mainly at dense, unmethylated CpG islands (CGIs), which are known preferred PRC2 nucleation sites from where the complex normally proceeds along adjacent nucleosomes, propelled by allosteric EED activation, to form broad H3K27me3 domains [58]. The long-range spread of the repressive H3K27 mark from PRC2 nucleation sites is thus impaired in K27M cells, but focal deposition of H3K27me3 at specific genomic loci in response to various stimuli remains possible [51, 52, 57, 59]. Notably, H3K27me2 can still be deposited widely in H3K27M cells in regions comprising H3K27me3 domains in H3 wild-type cells [50, 51]. This observation further argues against chromatin sequestration of PRC2 on mutant nucleosomes [47], and suggests that H3K27M most severely impairs the complex’s ability to perform the time-consuming conversion of H3K27me2 to H3K27me3 [58, 60], possibly precluding mutant cells from achieving levels needed to trigger allosteric EED activation over the course of a cell division cycle [50, 51, 61]. The residual H3K27me3 peaks that maintain repression of genes such as CDKN2A are likely essential for cell survival since H3K27M-mutant cells are more sensitive to their further depletion using EZH2 inhibitors [52, 57]. Last, H3K27M is needed for initiation but also for tumour maintenance and its effects are reversible as experimental knock-out of the mutation in pHGG cell lines restores H3K27 methylation levels and deposition and strongly reduces tumour formation in murine orthotopic xenograft models [49, 51, 62].
H3K36M-mutant cells present inverse effects than H3K27M on the epigenetic landscape. They are characterized by global loss of H3K36me2/3 methylation through inhibition of NSD1/2 and SETD2 methyltransferase activity, and a corresponding increase in intergenic H3K27me3 deposition [21, 42]. Replacement of intergenic H3K36me2 with H3K27me3 in H3K36M cells prompts redistribution of canonical PRC1 away from its genic targets to new intergenic H3K27me3 domains, resulting in complex downstream effects on transcription, including de-repression of PRC1/2 targets and a block in mesenchymal differentiation [21]. In contrast H3K27me3 loss correlates with gene activation in H3K27M pHGGs and residual H3K27me3 peaks maintain undue gene silencing, together producing a block in neural differentiation [51, 63, 64]. The transcriptional consequences of K-to-M mutations are however still incompletely understood. The few transcriptional changes observed in H3K27M cells, for instance, might be explained by complex compensatory epigenetic mechanisms [51], such as spread of antagonistic H3K36me2 in intergenic regions [53], or an increase in H3K27ac levels and its distribution genome-wide leading to pervasive acetylation and baseline increased expression of the silent genome, including transposable elements [65]. Undue de-repression of repetitive elements can be further enhanced using DNA methylation inhibitors, which removes another layer controlling the silent genome to further increase expression of these repeat elements. This in turn was shown to selectively stimulate an endogenous anti-viral response in H3K27M cells, a finding which may provide novel therapeutic strategies for H3K27M pHGGs [65].
EZHIP, an endogenous H3K27M mimic
EZH1/2 inhibitory protein’s (EZHIP) involvement in cancer has provided surprising insights into H3K27M oncogenicity (Figure 3). While aberrant EZHIP expression occurs primarily in PFA-EPN, the presence of rare, mutually exclusive H3K27M-mutant PFA-EPN supports a convergence of their effects on chromatin. The EZHIP protein is present in placental mammals and is largely unstructured. It contains however 12 conserved amino acids at the C-terminus that show similarity to the histone H3 N-terminal tail surrounding K27 [18]. Curiously, the residue in EZHIP corresponding to H3K27 is a methionine (M406), which inhibits PRC2 in a similar manner as H3K27M [17, 18]. Indeed, EZHIP interacts with, and has a high affinity for, allosterically activated PRC2 [61]. Like K27M, EZHIP promotes global loss of H3K27me3 and retention of the mark selectively at unmethylated CGIs [18, 66]. Unlike H3K27M, EZHIP is likely not incorporated into chromatin, favoring a model where transient association of EZHIP or H3K27M with PRC2 can persistently impair catalytic function. During normal development, EZHIP is selectively expressed in germ cells, where it acts as an endogenous mechanism aimed at restricting PRC2 activity; thus, inactivation of EZHIP in oocytes leads to reduced fertility [67]. The mechanisms enabling aberrant EZHIP expression in PFA-EPN and rare diffuse midline gliomas remain unknown, even if EZHIP’s effects on the epigenome mimic to a large extent what is observed in H3K27M.
H3.3G34 oncohistones
The H3.3G34R/V/W/L mutations occur on a residue which does not undergo PTM. They are further distinguished from K-to-M mutations by their occurrence solely in noncanonical H3.3, which implies a specific role for this H3 variant in their pathogenicity. H3.3 differs from canonical H3.1 and H3.2 by a mere four and five amino acids respectively and has several distinct properties. Unlike H3.1/H3.2, it is synthesized in a replication-independent manner and needs chaperones for deposition on chromatin, in euchromatin at actively transcribed genes, the HIRA complex, and in telomeric/pericentromeric heterochromatin and endogenous retroviral elements, a complex containing the Alpha Thalassemia Mental Retardation Syndrome X-linked (ATRX) chromatin remodeler. The epigenetic effects of H3.3G34 mutations are largely unknown but are presumed to arise from their impact on H3K36 methylation (Figure 3). Structural studies suggest that mutation of the small glycine residue to bulky arginine, valine or tryptophan residues creates steric interference within a narrow channel of the catalytic domain of H3K36 methyltransferases like SETD2 or H3K36 demethylases like KDM2A. Indeed, H3.3G34R/V/W/L histones display local loss of H3K36me3 and resultant gain of H3K27me3 specifically in cis on the mutant histone tail, contrasting with the dominant-negative effects of K-to-M oncohistones [39, 68]. The mutual exclusivity between H3.3G34R/V-mutant HGGs and the subset of hemispheric HGGs carrying SETD2 loss-of-function mutations further supports a convergence of effect on H3K36me3.
The diversity of H3K36me3’s downstream effects has prompted several lines of investigation for H3.3G34 oncohistones. Splicing defects were identified in H3.3G34W isogenic lines, likely through increased interaction between H3.3G34W and components of the spliceosome [69]. H3.3G34R/V/D mutations were also shown to prevent H3K36me3 recognition by DNA mismatch repair machinery, resulting in an elevated mutation rate [70]. Moreover, binding of the H3.3K36me3-specific reader and transcriptional repressor ZMYND11 was impaired by H3.3G34R/V mutations [35], whereas H3.3G34R promoted aberrant interaction with the enhancer-associated ZMYND8/RACK7 repressor [71]. H3.3G34R was additionally suggested to inhibit activity of the KDM4 family of H3K9/K36 demethylases, promoting increased H3K36me3 and H3K9me3 at select loci [72]. Recent data indicate that, H3.3G34R/V in neuronal progenitors [73], or H3.3G34W in mesenchymal progenitors may promote altered splicing [74]. A unifying mechanism reconciling these disparate models of H3.3G34-mutation induced epigenetic perturbation is still lacking. In a recent study, loss of H3.3K36me3 induced by H3.3G34W promoted H3.3 and H3K27me3 redistribution, resulting in dilution of PRC2 from the intergenome and enrichment at gene bodies, thereby blocking differentiation programs in the mesenchymal progenitors carrying this mutation [75]. In H3.3G34R/V HGGs, similar redistribution of H3K27me3 resulted in blocking terminal differentiation of the mutant interneuron progenitor cells [76], suggesting that aberrant PRC2 recruitment and titration of this complex from its original targets promotes stalled development.
In all, oncohistones and the oncohistone mimic EZHIP seem to converge on stalling differentiation and impairing normal development. This is through undue retention of PRC2 at its nucleation sites at CGIs for H3K27M/I and EZHIP, or redistribution and titration of this complex following H3.3K36me3 loss in H3.3G34R/V/W and H3K36M/I mutants. The crosstalk of these changes with the redistribution of other opposing chromatin marks also plays a major role in the oncogenic process and is being actively investigated.
Intersection of oncohistone mutations with developmental lineages and oncogenic partners
The remarkable tissue, regional and temporal specificity of histone-mutant cancers has led to an appreciation of the developmental and cellular contexts permissive to oncohistone-mediated tumorigenesis (Figure 4). Characterization of H3K27M gliomas using single-cell transcriptomics revealed a proliferative population of malignant cells resembling pontine oligodendrocyte precursor cells (OPCs), with the potential to differentiate to oligodendrocyte-like or astrocyte-like cells [64, 77]. Experimental data confirm that H3K27M impairs differentiation to maintain the progenitor state [49, 63]. Similarly, bulk and single-cell transcriptomes of EZHIP-expressing PFA-EPN share features of prenatal gliogenic progenitors [78]. Interestingly, the age of diagnosis of pediatric H3K27M gliomas coincides with a period of pontine expansion during normal development linked primarily to increased myelination from proliferating and differentiating OPCs [79]. This normal proliferative activity may be key to acquiring additional oncogenic partner mutations since rare incidental reports of H3K27M-mutant diffuse midline gliomas in asymptomatic individuals [80], or in initial low-grade gliomas that transform on recurrence [81], suggest that there may be a significant latency between acquisition of the early clonal H3K27M mutation [82] and the rapid tumour growth associated with high-grade gliomas.
Figure 4. Intersection of oncohistone mutations with developmental lineages and oncogenic partner mutations.
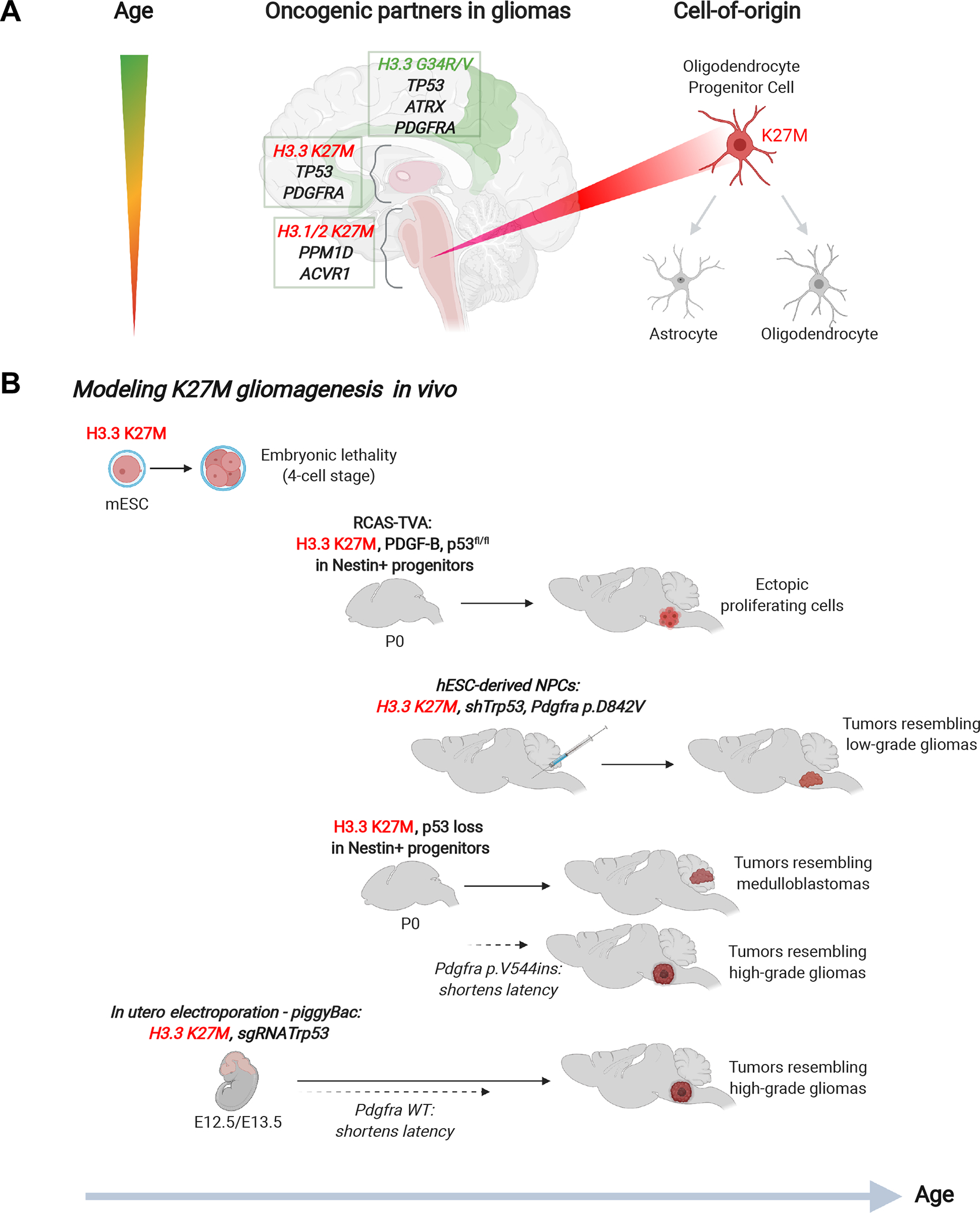
A. Oncohistone mutations occurring in high-grade gliomas follow a specific temporal and regional pattern with specific oncogenic partners, consistent with a distinct cell-of-origin.
B. Murine models of H3.3 K27M using different techniques and in combination with oncogenic partner mutations, to achieve similarity with H3.3 K27M high-grade gliomas.
The unique association of oncohistone mutation with specific tissue/cell contexts is further supported by the consistent co-occurrence of frequent or obligate oncogenic partners (Figure 4A). These partnerships are exemplified by histone-mutant HGGs which are commonly associated with specific loss-of-function mutations in the TP53 cell-cycle pathway (e.g. TP53, PPM1D, CHECK2) and activating mutations in genes encoding growth factors (e.g. ACVR1, PDGFRA, PIK3CA) [3, 6, 83]. TP53 mutations, PDGFRA and EGFR activation more commonly occur in H3.3K27M gliomas, whereas PPM1D and ACVR1 mutations preferentially associate with canonical H3.1/2 K27M gliomas [10, 19]. This is consistent with distinct enhancer landscapes identified in H3.3 and H3.1/2 K27M gliomas supporting a distinct cell-of-origin [44]. Furthermore, some cooperating mutations are clonal events that are present throughout disease progression, underscoring their importance for both tumour development and maintenance [82].
Similarly, H3.3G34R/V HGGs invariably co-occur with mutations of TP53 and the chromatin remodeler ATRX [1]. Inactivating ATRX mutations enable neoplastic cells to achieve immortality by promoting an alternative lengthening of telomeres phenotype [84], likely through destabilization of telomeric nucleosomal organization upon H3.3 loss. These mutations frequently occur in thalamic and hemispheric HGGs of all subtypes; however, their obligate partnership in H3.3G34R/V HGGs is unique. ATRX deficiency may serve to potentiate the in cis epigenetic effects of H3.3G34 mutations by promoting HIRA-mediated deposition of H3.3G34R/V oncohistones into active transcriptional or regulatory regions of the genome. By contrast, H3.3G34W GCTBs do not require TP53 or ATRX partners for tumorigenesis, but are also less aggressive tumours occurring in cells of mesenchymal origin [75]. Last, recent data indicate that G34R/V HGGs are in fact neuronal in origin, as they arise in interneuron progenitors [73, 76] in the ventral forebrain during early development or in the sub-ventricular zone shortly after birth [76]. The chromatin conformation in these progenitors, in combination with H3.3G34R/V mediated effects on specific chromatin marks, co-opt PDGFRA by allowing aberrant overexpression and mutations of this growth factor receptor, such that half of all H3.3G34R/V HGGs carry mutations of PDGFRA. Ultimately, mutant-PDGFRA provides the astrocytic features that classify these tumours as glial and is a potent oncogene, as the H3.3G34R/V mutations are poorly tumorigenic on their own [76].
Thus, the dependence of histone-mutant cancers on specific oncogenic partners reinforces the importance of the cell-of-origin/tissue context, which is best illustrated by attempts to model H3K27M gliomagenesis in vivo (Figure 4B). Endogenous knock-in of H3.3K27M in mouse ESC resulted in embryonic lethality at the four-cell stage [59]. Neonatal expression of H3.3K27M together with p53 loss in murine nestin+ neural progenitor cells (NPCs) resulted in ectopic clusters of proliferating cells in the brainstem [39, 59]. Notably, in utero electroporation of H3.3K27M and p53 loss in murine hindbrain and cortex at E12.5 or E13.5 promoted fully penetrant tumours in mice resembling phenotypically and molecularly H3K27M HGGs; additional overexpression of wild-type Pdgfra in this context significantly reduced tumour latency [59]. Transplantation of human ESC-derived NPCs carrying H3.3K27M, shRNA against Trp53, and mutant PDGFRA (p.D842V) into the pons of immunocompromised mice promoted the formation of tumours resembling low-grade gliomas, with some transcriptional similarities to H3K27M diffuse midline gliomas [63]. In another model, neonatal induction of H3.3K27M expression alone at the endogenous locus in nestin+ NPCs produced tumours predominantly resembling medulloblastomas when combined with p53 loss [85]. Addition of mutant Pdgfra (p.V544ins) in this case pushed gliogenesis, resembling HGGs. In summary, these different in vivo models highlight the complexity of H3K27M gliomagenesis and reinforce the importance of selecting the appropriate developmental period, cell-of-origin, and necessary oncogenic partners to accurately recapitulate the oncogenic process in patient tumours.
Concluding remarks
Many unanswered questions related to oncohistone pathogenicity remain and range from their predicted molecular effects on the epigenome to their exquisite partnership of genetic alterations and cell/tissue specificities. Further studies will undoubtedly clarify the manner of epigenetic dysregulation mediated by H3.3G34 onco-mutations, while providing insights into the exquisite preference for H3.3G34R/V mutations in HGGs and H3.3G34W/L in GCTBs. Similarly, deeper characterization of neural cell types during normal development and comparison with neoplastic cell signatures may shed light on the origin of high frequency of EZHIP overexpression in PFA-EPN and why these tumours do not seem to need additional genetic alterations compared to H3K27M in diffuse midline gliomas, despite both having seemingly similar effects on the epigenome. Analysis of comprehensive single-cell transcriptomic atlases of the developing brain and experimentally induced loss in specific cell types may also reveal the cell types, developmental periods, and brain regions that are most susceptible to epigenetic perturbation causing brain tumours. The rare cancers driven by H3 mutations are likely intimately linked with restricted developmental contexts and cell type lineages that are permissive to their chromatin remodeling effects, as also suggested by a recent study [86]. These altered chromatin states may be promoting indefinite progenitor cell renewal, cell proliferation and acquisition of subsequent genetic alterations, ultimately leading to tumour formation.
Mutations in epigenetic modifiers have also provided insights into convergent mechanisms in oncohistone-mutant cancers. For instance, the presence of SETD2 mutations in non-G34R/V hemispheric HGGs suggests convergence through H3K36me3 loss in both HGG subtypes. NSD1 mutations in non-K36M HNSCCs similarly implies convergence through H3K36me2 loss for K36M HNSCCs. The paucity of mutations in components of the PRC2 complex in HGGs or PFA-EPN suggests that H3K27M and EZHIP have distinct and more complex effects on the epigenome than a complete loss of H3K27me3 levels. Therefore, further studies of relevant epigenetic modifiers and the dynamics of H3K27/K36 PTMs will undoubtedly expand our understanding of oncohistone-mutant cancers.
Finally, insights into oncohistone-mediated epigenetic perturbations and obligate oncogenic partnerships in specific developmental contexts will be essential to developing targeted therapies for these cancers. Indeed, several clinical trials are ongoing: using an H3.3K27M peptide vaccine (NCT02960230), histone deacetylase inhibitors like panobinostat and vorinostat targeting increased acetylation (e.g. H3K27ac) observed in H3K27M gliomas (NCT02717455, NCT02420613, NCT01189266, NCT03566199), or inhibitors of the oncogenic partner PDGFRA (NCT03352427). EZH2 inhibitors like tazemetostat are currently being tested in lymphomas carrying gain-of-function EZH2 mutations; potential future applications of EZH2 inhibitors could include the suppression of residual H3K27me3 peaks in H3K27M gliomas.
Last, oncohistones are a seminal discovery as they can also serve as tools to reveal physiological patterns for multiple chromatin marks and provide new insight into PRC regulation and function that would have been otherwise very difficult to assess. Indeed, their discovery and data generated on their pathogenesis go beyond the cancers they drive and will benefit treatment of other disorders where the epigenome is misregulated. These range from genetic overgrowth syndromes, other neurodevelopmental diseases where in rare cases germline H3.3 mutations have been recently identified [87], to aging and neurodegenerative diseases where PRC functions seem deregulated. As more becomes known about oncohistones, it is likely that targeted therapies will translate to the bedside and help improve clinical outcomes for patients with these disorders.
Acknowledgements
This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium. It was supported by funding from: A Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l’Économie, de la Science et de l’Innovation du Québec, US National Institutes of Health (NIH grant P01-CA196539); the Canadian Institutes for Health Research (CIHR grant MOP-286756 and FDN-154307) and support from the Fondation Charles Bruneau and We Love You Connie Foundation. NJ is a member of the Penny Cole Laboratory.
Abbreviations:
- ATRX
alpha thalassemia mental retardation syndrome X-linked
- CBX
chromobox
- CGI
CpG islands
- CIMP
CpG island methylator phenotype
- ESC
embryonic stem cell
- EZHIP
EZH2 inhibitory protein
- GCTB
giant cell tumour of bone
- H3
histone H3
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papillomavirus
- IDH
isocitrate dehydrogenase
- NPC
neural progenitor cell
- OPC
oligodendrocyte progenitor cell
- pHGG
pediatric high-grade glioma
- PFA-EPN
Group A posterior fossa ependymoma
- PRC
Polycomb repressive complex
- PTM
post-translational modification
Footnotes
Conflicts of Interests: There are no conflicting interests to declare.
REFERENCES
- 1.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM & Jabado N (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma, Nature. 482, 226–31. [DOI] [PubMed] [Google Scholar]
- 2.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ & St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome, P. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas, Nat Genet. 44, 251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, Johann P, Tomita T, Goldman S, Nagib M, Bendel A, Goumnerova L, Bowers DC, Leonard JR, Rubin JB, Alden T, Browd S, Geyer JR, Leary S, Jallo G, Cohen K, Gupta N, Prados MD, Carret AS, Ellezam B, Crevier L, Klekner A, Bognar L, Hauser P, Garami M, Myseros J, Dong Z, Siegel PM, Malkin H, Ligon AH, Albrecht S, Pfister SM, Ligon KL, Majewski J, Jabado N & Kieran MW (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma, Nat Genet. 46, 462–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N & Hawkins C (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas, Acta Neuropathol. 124, 439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Fruhwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Durken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N & Pfister SM (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma, Cancer Cell. 22, 425–37. [DOI] [PubMed] [Google Scholar]
- 6.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, Lu C, Nagahawatte P, Hedlund E, Rusch M, Pounds S, Lin T, Onar-Thomas A, Huether R, Kriwacki R, Parker M, Gupta P, Becksfort J, Wei L, Mulder HL, Boggs K, Vadodaria B, Yergeau D, Russell JC, Ochoa K, Fulton RS, Fulton LL, Jones C, Boop FA, Broniscer A, Wetmore C, Gajjar A, Ding L, Mardis ER, Wilson RK, Taylor MR, Downing JR, Ellison DW, Zhang J & Baker SJ (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma, Nat Genet. 46, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S, Fleming A, Hadjadj D, Busche S, Lepage P, Montpetit A, Staffa A, Gerges N, Zakrzewska M, Zakrzewski K, Liberski PP, Hauser P, Garami M, Klekner A, Bognar L, Zadeh G, Faury D, Pfister SM, Jabado N & Majewski J (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas, Acta Neuropathol. 125, 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE & Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme, Science. 321, 1807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, Forrest WF, Pujara K, Carrillo JA, Pandita A, Ellingson BM, Bowers CW, Soriano RH, Schmidt NO, Mohan S, Yong WH, Seshagiri S, Modrusan Z, Jiang Z, Aldape KD, Mischel PS, Liau LM, Escovedo CJ, Chen W, Nghiemphu PL, James CD, Prados MD, Westphal M, Lamszus K, Cloughesy T & Phillips HS (2011) Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin, J Clin Oncol. 29, 4482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, Temelso S, Popov S, Molinari V, Raman P, Waanders AJ, Han HJ, Gupta S, Marshall L, Zacharoulis S, Vaidya S, Mandeville HC, Bridges LR, Martin AJ, Al-Sarraj S, Chandler C, Ng HK, Li X, Mu K, Trabelsi S, Brahim DH, Kisljakov AN, Konovalov DM, Moore AS, Carcaboso AM, Sunol M, de Torres C, Cruz O, Mora J, Shats LI, Stavale JN, Bidinotto LT, Reis RM, Entz-Werle N, Farrell M, Cryan J, Crimmins D, Caird J, Pears J, Monje M, Debily MA, Castel D, Grill J, Hawkins C, Nikbakht H, Jabado N, Baker SJ, Pfister SM, Jones DTW, Fouladi M, von Bueren AO, Baudis M, Resnick A & Jones C (2017) Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma, Cancer Cell. 32, 520–537 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capper D Jones DT. Sill W, Hovestadt M, Schrimpf V, Sturm D, Koelsche D, Sahm C, Chavez F, Reuss L, Kratz DE, Wefers A, Huang AK, Pajtler K, Schweizer KW, Stichel L, Olar D, Engel A, Lindenberg NW, Harter K, Braczynski PN, Plate AK, Dohmen KH, Garvalov H, Coras BK, Holsken R, Hewer A, Bewerunge-Hudler E, Schick M, Fischer M, Beschorner R, Schittenhelm R, Staszewski J, Wani O, Varlet K, Pages P, Temming M, Lohmann P, Selt D, Witt F, Milde H, Witt T, Aronica O, Giangaspero E, Rushing F, Scheurlen E, Geisenberger W, Rodriguez C, Becker FJ, Preusser A, Haberler M, Bjerkvig C, Cryan R, Farrell J, Deckert M, Hench M, Frank J, Serrano S, Kannan J, Tsirigos K, Bruck A, Hofer W, Brehmer S, Seiz-Rosenhagen S, Hanggi M, Hans D, Rozsnoki V, Hansford S, Kohlhof JR, Kristensen P, Lechner BW, Lopes M, Mawrin B, Ketter C, Kulozik R, Khatib A, Heppner Z, Koch F, Jouvet A, Keohane A, Muhleisen C, Mueller H, Pohl W, Prinz U, Benner M, Zapatka A, Gottardo M, Driever NG, Kramm PH, Muller CM, Rutkowski HL, von Hoff S, Fruhwald K, Gnekow MC, Fleischhack A, Tippelt G, Calaminus S, Monoranu G, Perry CM, Jones A, C., et al. (2018) DNA methylation-based classification of central nervous system tumours, Nature. 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehnertz B, Zhang YW, Boivin I, Mayotte N, Tomellini E, Chagraoui J, Lavallee VP, Hebert J & Sauvageau G (2017) H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations, Blood. 130, 2204–2214. [DOI] [PubMed] [Google Scholar]
- 13.Boileau M, Shirinian M, Gayden T, Harutyunyan AS, Chen CCL, Mikael LG, Duncan HM, Neumann AL, Arreba-Tutusaus P, De Jay N, Zeinieh M, Rossokhata K, Zhang Y, Nikbakht H, Mouawad C, Massoud R, Frey F, Nasr R, El Cheikh J, El Sabban M, Kleinman CL, Mahfouz R, Minden MD, Jabado N, Bazarbachi A & Eppert K (2019) Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness, Nat Commun. 10, 2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gessi M, Capper D, Sahm F, Huang K, von Deimling A, Tippelt S, Fleischhack G, Scherbaum D, Alfer J, Juhnke BO, von Hoff K, Rutkowski S, Warmuth-Metz M, Chavez L, Pfister SM, Pietsch T, Jones DT & Sturm D (2016) Evidence of H3 K27M mutations in posterior fossa ependymomas, Acta Neuropathol. 132, 635–7. [DOI] [PubMed] [Google Scholar]
- 15.Ryall S, Guzman M, Elbabaa SK, Luu B, Mack SC, Zapotocky M, Taylor MD, Hawkins C & Ramaswamy V (2017) H3 K27M mutations are extremely rare in posterior fossa group A ependymoma, Childs Nerv Syst. 33, 1047–1051. [DOI] [PubMed] [Google Scholar]
- 16.Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hubner JM, Ramaswamy V, Jia S, Dalton JD, Haupfear K, Rogers HA, Punchihewa C, Lee R, Easton J, Wu G, Ritzmann TA, Chapman R, Chavez L, Boop FA, Klimo P, Sabin ND, Ogg R, Mack SC, Freibaum BD, Kim HJ, Witt H, Jones DTW, Vo B, Gajjar A, Pounds S, Onar-Thomas A, Roussel MF, Zhang J, Taylor JP, Merchant TE, Grundy R, Tatevossian RG, Taylor MD, Pfister SM, Korshunov A, Kool M & Ellison DW (2018) Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas, Acta Neuropathol. 136, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hubner JM, Muller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, Ellison DW, Pfister SM, Pajtler KW & Kool M (2019) EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma, Neuro Oncol. 21, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, Bajic A, Juretic N, Deshmukh S, Venneti S, Muir TW, Garcia BA, Jabado N & Lewis PW (2019) PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism, Nat Commun. 10, 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, Hench J, Frank S, Krskova L, Vicha A, Zapotocky M, Bison B, Castel D, Grill J, Debily MA, Harter PN, Snuderl M, Kramm CM, Reifenberger G, Korshunov A, Jabado N, Wesseling P, Wick W, Solomon DA, Perry A, Jacques TS, Jones C, Witt O, Pfister SM, von Deimling A, Jones DTW & Sahm F (2021) A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR, Neuro Oncol. 23, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goodie V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ & Flanagan AM (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone, Nat Genet. 45, 1479–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, Wunder JS, Dickson BC, Lundgren SM, Jani KS, De Jay N, Papillon-Cavanagh S, Andrulis IL, Sawyer SL, Grynspan D, Turcotte RE, Nadaf J, Fahiminiyah S, Muir TW, Majewski J, Thompson CB, Chi P, Garcia BA, Allis CD, Jabado N & Lewis PW (2016) Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape, Science. 352, 844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA, Weinreb I, Goldstein D, Lewis PW, Dancu OM, Dhaliwal S, Stecho W, Howlett CJ, Mymryk JS, Barrett JW, Nichols AC, Allis CD, Majewski J & Jabado N (2017) Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas, Nat Genet. 49, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koelsche C, Schrimpf D, Tharun L, Roth E, Sturm D, Jones DTW, Renker EK, Sill M, Baude A, Sahm F, Capper D, Bewerunge-Hudler M, Hartmann W, Kulozik AE, Petersen I, Flucke U, Schreuder HWB, Buttner R, Weber MA, Schirmacher P, Plass C, Pfister SM, von Deimling A & Mechtersheimer G (2017) Histone 3.3 hotspot mutations in conventional osteosarcomas: a comprehensive clinical and molecular characterization of six H3F3A mutated cases, Clin Sarcoma Res. 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, Soshnev AA, Kundra R, Schultz N, Muir TW & Allis CD (2019) The expanding landscape of ‘oncohistone’ mutations in human cancers, Nature. 567, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA & Wysocka J (2011) A unique chromatin signature uncovers early developmental enhancers in humans, Nature. 470, 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pengelly AR, Copur O, Jackle H, Herzig A & Muller J (2013) A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb, Science. 339, 698–9. [DOI] [PubMed] [Google Scholar]
- 27.Margueron R & Reinberg D (2011) The Polycomb complex PRC2 and its mark in life, Nature. 469, 343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Fischle W, Bonaldi T & Pasini D (2014) Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity, Mol Cell. 53, 49–62. [DOI] [PubMed] [Google Scholar]
- 29.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D & Gamblin SJ (2009) Role of the polycomb protein EED in the propagation of repressive histone marks, Nature. 461, 762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A & Schubeler D (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation, Nature. 520, 243–7. [DOI] [PubMed] [Google Scholar]
- 31.Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, Lemiesz AE, Marchione DM, Marunde MR, Meiners MJ, Cheek MA, Keogh MC, Bareke E, Djedid A, Harutyunyan AS, Jabado N, Garcia BA, Li H, Allis CD, Majewski J & Lu C (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape, Nature. 573, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lhoumaud P, Badri S, Rodriguez-Hernaez J, Sakellaropoulos T, Sethia G, Kloetgen A, Cornwell M, Bhattacharyya S, Ay F, Bonneau R, Tsirigos A & Skok JA (2019) NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains, Nat Commun. 10, 4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F & Oliviero S (2017) Intragenic DNA methylation prevents spurious transcription initiation, Nature. 543, 72–77. [DOI] [PubMed] [Google Scholar]
- 34.Li F, Mao G, Tong D, Huang J, Gu L, Yang W & Li GM (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha, Cell. 153, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, Li B, Barton MC, Li W, Li H & Shi X (2014) ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression, Nature. 508, 263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, Dillon E, Wynne K, Pasini D, Krogan NJ, Bracken AP & Cagney G (2018) The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells, Mol Cell. 70, 371–379 e5. [DOI] [PubMed] [Google Scholar]
- 37.Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, Hua Y, Bulic M, Jiang Y, Carrara M, Calogero RA, Kath WL, Kelleher NL, Wang JP, Elemento O & Licht JD (2014) Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation, PLoS Genet. 10, e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jani KS, Jain SU, Ge EJ, Diehl KL, Lundgren SM, Muller MM, Lewis PW & Muir TW (2019) Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase, Proc Natl Acad Sci U S A. 116, 8295–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ & Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma, Science. 340, 857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Hojfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Seker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, von Deimling A, Jabado N, Puget S, Grill J, Helin K, Korshunov A, Lichter P, Monje M, Plass C, Cho YJ & Pfister SM (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas, Cancer Cell. 24, 660–72. [DOI] [PubMed] [Google Scholar]
- 41.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J & Zhang Z (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression, Genes Dev. 27, 985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang D, Gan H, Lee JH, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, Zhang H, Yang N, Bradley EW, Ho TH, Rubin BP, Bridge JA, Thibodeau SN, Ordog T, Chen Y, van Wijnen AJ, Oliveira AM, Xu RM, Westendorf JJ & Zhang Z (2016) The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas, Science. 352, 1344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarthy JF, Meers MP, Janssens DH, Henikoff JG, Feldman H, Paddison PJ, Lockwood CM, Vitanza NA, Olson JM, Ahmad K & Henikoff S (2020) Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones, Elife. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagaraja S, Quezada MA, Gillespie SM, Arzt M, Lennon JJ, Woo PJ, Hovestadt V, Kambhampati M, Filbin MG, Suva ML, Nazarian J & Monje M (2019) Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State, Mol Cell. 76, 965–980 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castel D, Philippe C, Kergrohen T, Sill M, Merlevede J, Barret E, Puget S, Sainte-Rose C, Kramm CM, Jones C, Varlet P, Pfister SM, Grill J, Jones DTW & Debily MA (2018) Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location, Acta Neuropathol Commun. 6, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang S, Zheng X, Lu C, Li GM, Allis CD & Li H (2016) Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase, Genes Dev. 30, 1611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, Wilson JR & Gamblin SJ (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2, Nat Commun. 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW & Cech TR (2017) Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA, Nat Struct Mol Biol. 24, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, Shaw TI, Zhu X, Larson JD, Easton J, Shao Y, Yergeau DA, Rosencrance C, Boggs K, Rusch MC, Ding L, Zhang J, Finkelstein D, Noyes RM, Russell BL, Xu B, Broniscer A, Wetmore C, Pounds SB, Ellison DW, Zhang J & Baker SJ (2019) H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo, Acta Neuropathol. 137, 637–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, Krug B, Russo C, Bareke E, Marchione DM, Coradin M, Garcia BA, Jabado N & Majewski J (2020) H3K27M in Gliomas Causes a One-Step Decrease in H3K27 Methylation and Reduced Spreading within the Constraints of H3K36 Methylation, Cell Rep. 33, 108390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, Marchione DM, Li R, Nikbakht H, Hu B, Cagnone G, Cheung WA, Mohammadnia A, Bechet D, Faury D, McConechy MK, Pathania M, Jain SU, Ellezam B, Weil AG, Montpetit A, Salomoni P, Pastinen T, Lu C, Lewis PW, Garcia BA, Kleinman CL, Jabado N & Majewski J (2019) H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis, Nat Commun. 10, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, Misharin AV, Abshiru NA, Lulla RR, Saratsis AM, Kelleher NL, James CD & Shilatifard A (2017) Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas, Nat Med. 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, Modrek AS, Bayin NS, Placantonakis DG, Karajannis MA, Snuderl M, Ueberheide B & Reinberg D (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma, Sci Adv. 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee CH, Yu JR, Granat J, Saldana-Meyer R, Andrade J, LeRoy G, Jin Y, Lund P, Stafford JM, Garcia BA, Ueberheide B & Reinberg D (2019) Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma, Genes Dev. 33, 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wojcik JB, Marchione DM, Sidoli S, Djedid A, Lisby A, Majewski J & Garcia BA (2019) Epigenomic Reordering Induced by Polycomb Loss Drives Oncogenesis but Leads to Therapeutic Vulnerabilities in Malignant Peripheral Nerve Sheath Tumors, Cancer Res. 79, 3205–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR & Chi P (2014) PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors, Nat Genet. 46, 1227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, Tvardovskiy A, Jensen ON, Olaciregui NG, Lavarino C, Sunol M, de Torres C, Mora J, Carcaboso AM & Helin K (2017) EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas, Nat Med. 23, 483–492. [DOI] [PubMed] [Google Scholar]
- 58.Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, Skok JA & Reinberg D (2018) Capturing the Onset of PRC2-Mediated Repressive Domain Formation, Mol Cell. 70, 1149–1162 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, Costa JR, Hebert S, Khazaei S, Ibrahim NS, Herrero J, Riccio A, Albrecht S, Ketteler R, Brandner S, Kleinman CL, Jabado N & Salomoni P (2017) H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas, Cancer Cell. 32, 684–700 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hojfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON & Helin K (2018) Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2, Nat Struct Mol Biol. 25, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jain SU, Rashoff AQ, Krabbenhoft SD, Hoelper D, Do TJ, Gibson TJ, Lundgren SM, Bondra ER, Deshmukh S, Harutyunyan AS, Juretic N, Jabado N, Harrison MM & Lewis PW (2020) H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2, Mol Cell. 80, 726–735 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen KY, Bush K, Klein RH, Cervantes V, Lewis N, Naqvi A, Carcaboso AM, Lechpammer M & Knoepfler PS (2020) Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling, Commun Biol. 3, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Funato K, Major T, Lewis PW, Allis CD & Tabar V (2014) Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation, Science. 346, 1529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, Poreau B, De Jay N, Hebert S, Monlong J, Farmer WT, Donovan LK, Hu Y, McConechy MK, Cavalli FMG, Mikael LG, Ellezam B, Richer M, Allaire A, Weil AG, Atkinson J, Farmer JP, Dudley RWR, Larouche V, Crevier L, Albrecht S, Filbin MG, Sartelet H, Lutz PE, Nagy C, Turecki G, Costantino S, Dirks PB, Murai KK, Bourque G, Ragoussis J, Garzia L, Taylor MD, Jabado N & Kleinman CL (2019) Stalled developmental programs at the root of pediatric brain tumors, Nat Genet. 51, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, Bertrand KC, Mikael LG, McConechy MK, Chen CCL, Khazaei S, Koncar RF, Agnihotri S, Faury D, Ellezam B, Weil AG, Ursini-Siegel J, De Carvalho DD, Dirks PB, Lewis PW, Salomoni P, Lupien M, Arrowsmith C, Lasko PF, Garcia BA, Kleinman CL, Jabado N & Mack SC (2019) Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas, Cancer Cell. 35, 782–797 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, Helmin KA, Ryan CA, Murray DC, Rickels RA, Yilmaz BD, Rendleman EJ, Savas JN, Singer BD, Bulun SE & Shilatifard A (2019) CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism, Sci Adv. 5, eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ragazzini R, Perez-Palacios R, Baymaz IH, Diop S, Ancelin K, Zielinski D, Michaud A, Givelet M, Borsos M, Aflaki S, Legoix P, Jansen P, Servant N, Torres-Padilla ME, Bourc’his D, Fouchet P, Vermeulen M & Margueron R (2019) EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells, Nat Commun. 10, 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi L, Shi J, Shi X, Li W & Wen H (2018) Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis, J Mol Biol. 430, 1562–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lim J, Park JH, Baude A, Yoo Y, Lee YK, Schmidt CR, Park JB, Fellenberg J, Zustin J, Haller F, Krucken I, Kang HG, Park YJ, Plass C & Lindroth AM (2017) The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing, Sci Rep. 7, 13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang J, Huang Y, Mao G, Yang S, Rennert G, Gu L, Li H & Li GM (2018) Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction, Proc Natl Acad Sci U S A. 115, 9598–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiao F, Li Z, He C, Xu W, Yang G, Liu T, Shen H, Cai J, Anastas JN, Mao Y, Yu Y, Lan F, Shi YG, Jones C, Xu Y, Baker SJ, Shi Y & Guo R (2020) RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma, Sci Adv. 6, eaba2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, Das PP, Mann JR & Wong LH (2018) Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma, Nat Commun. 9, 3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Funato K, Smith RC, Saito Y & Tabar V (2021) Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma, Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lutsik P, Baude A, Mancarella D, Oz S, Kuhn A, Toth R, Hey J, Toprak UH, Lim J, Nguyen VH, Jiang C, Mayakonda A, Hartmann M, Rosemann F, Breuer K, Vonficht D, Grunschlager F, Lee S, Schuhmacher MK, Kusevic D, Jauch A, Weichenhan D, Zustin J, Schlesner M, Haas S, Park JH, Park YJ, Oppermann U, Jeltsch A, Haller F, Fellenberg J, Lindroth AM & Plass C (2020) Globally altered epigenetic landscape and delayed osteogenic differentiation in H3.3-G34W-mutant giant cell tumor of bone, Nat Commun. 11, 5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khazaei S, De Jay N, Deshmukh S, Hendrikse LD, Jawhar W, Chen CCL, Mikael LG, Faury D, Marchione DM, Lanoix J, Bonneil E, Ishii T, Jain SU, Rossokhata K, Sihota TS, Eveleigh R, Lisi V, Harutyunyan AS, Jung S, Karamchandani J, Dickson BC, Turcotte R, Wunder JS, Thibault P, Lewis PW, Garcia BA, Mack SC, Taylor MD, Garzia L, Kleinman CL & Jabado N (2020) H3.3 G34W Promotes Growth and Impedes Differentiation of Osteoblast-Like Mesenchymal Progenitors in Giant Cell Tumor of Bone, Cancer Discov. 10, 1968–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, Faury D, Jawhar W, Dali R, Suzuki H, Pathania M, A D, Dubois, Woodward, Hebert, Coutelier, Karamchandani, Albrecht, Brandner, De Jay N, Gayden T, Bajic A, Harutyunyan AS, Marchione DM, Mikael LG, Juretic N, Zeinieh M, Russo C, Maestro N, Bassenden AV, Hauser P, Virga J, Bognar L, Klekner A, Zapotocky M, Vicha A, Krskova L, Vanova K, Zamecnik J, Sumerauer D, Ekert PG, Ziegler DS, Ellezam B, Filbin MG, Blanchette M, Hansford JR, Khuong-Quang DA, Berghuis AM, Weil AG, Garcia BA, Garzia L, Mack SC, Beroukhim R, Ligon KL, Taylor MD, Bandopadhayay P, Kramm C, Pfister SM, Korshunov A, Sturm D, Jones DTW, Salomoni P, Kleinman CL & Jabado N (2020) Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis, Cell. 183, 1617–1633 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, Haberler C, Yizhak K, Gojo J, Egervari K, Mount C, van Galen P, Bonal DM, Nguyen QD, Beck A, Sinai C, Czech T, Dorfer C, Goumnerova L, Lavarino C, Carcaboso AM, Mora J, Mylvaganam R, Luo CC, Peyrl A, Popovic M, Azizi A, Batchelor TT, Frosch MP, Martinez-Lage M, Kieran MW, Bandopadhayay P, Beroukhim R, Fritsch G, Getz G, Rozenblatt-Rosen O, Wucherpfennig KW, Louis DN, Monje M, Slavc I, Ligon KL, Golub TR, Regev A, Bernstein BE & Suva ML (2018) Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq, Science. 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, Ramaswamy V, Hendrikse LD, Kumar S, Mack SC, Lee JJY, Fong V, Juraschka K, Przelicki D, Michealraj A, Skowron P, Luu B, Suzuki H, Morrissy AS, Cavalli FMG, Garzia L, Daniels C, Wu X, Qazi MA, Singh SK, Chan JA, Marra MA, Malkin D, Dirks P, Heisler L, Pugh T, Ng K, Notta F, Thompson EM, Kleinman CL, Joyner AL, Jabado N, Stein L & Taylor MD (2019) Childhood cerebellar tumours mirror conserved fetal transcriptional programs, Nature. 572, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tate MC, Lindquist RA, Nguyen T, Sanai N, Barkovich AJ, Huang EJ, Rowitch DH & Alvarez-Buylla A (2015) Postnatal growth of the human pons: a morphometric and immunohistochemical analysis, J Comp Neurol. 523, 449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walsh M, Hamilton W, Casler V & Yachnis A (2018) An Incidental Diffuse Midline Glioma Found at Autopsy, J Forensic Sci. 63, 316–317. [DOI] [PubMed] [Google Scholar]
- 81.Ishibashi K, Inoue T, Fukushima H, Watanabe Y, Iwai Y, Sakamoto H, Yamasaki K, Hara J, Shofuda T, Kanematsu D, Yoshioka E & Kanemura Y (2016) Pediatric thalamic glioma with H3F3A K27M mutation, which was detected before and after malignant transformation: a case report, Childs Nerv Syst. 32, 2433–2438. [DOI] [PubMed] [Google Scholar]
- 82.Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho CY, Kambhampati M, Hwang EI, Faury D, Siu A, Papillon-Cavanagh S, Bechet D, Ligon KL, Ellezam B, Ingram WJ, Stinson C, Moore AS, Warren KE, Karamchandani J, Packer RJ, Jabado N, Majewski J & Nazarian J (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma, Nat Commun. 7, 11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, Zagzag D, Foreman NK, Donson A, Hegert JV, Smith A, Chan J, Lafay-Cousin L, Dunn S, Hukin J, Dunham C, Scheinemann K, Michaud J, Zelcer S, Ramsay D, Cain J, Brennan C, Souweidane MM, Jones C, Allis CD, Brudno M, Becher O & Hawkins C (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations, Nat Genet. 46, 451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N & Meeker AK (2011) Altered telomeres in tumors with ATRX and DAXX mutations, Science. 333, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, Fan Y, Shaw TI, Silveira AB, Qu C, Xu R, Zhu X, Zhang J, Russell HR, Peters JL, Finkelstein D, Xu B, Lin T, Tinkle CL, Patay Z, Onar-Thomas A, Pounds SB, McKinnon PJ, Ellison DW, Zhang J & Baker SJ (2019) Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression, Cancer Cell. 35, 140–155 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bressan RB, Southgate B, Ferguson KM, Blin C, Grant V, Alfazema N, Wills JC, Marques-Torrejon MA, Morrison GM, Ashmore J, Robertson F, Williams CAC, Bradley L, von Kriegsheim A, Anderson RA, Tomlinson SR & Pollard SM (2021) Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants, Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bryant L Li, Cox D, Marchione SG, Joiner D, Wilson EF, Janssen K, Lee K, March P, Nair ME, Sherr D, Fregeau E, Wierenga B, Wadley KJ, Mancini A, Powell-Hamilton GMS, van de Kamp N, Grebe J, Dean T, Ross J, Crawford A, Powis HP, Cho Z, Willing MT, Manwaring MC, Schot L, Nava R, Afenjar C, Lessel A, Wagner D, Klopstock M, Winkelmann T, Catarino J, Retterer CB, Schuette K, Innis JL, Pizzino JW, Luttgen A, Denecke S, Strom J, Monaghan TM, Study KG, Yuan DDD, Dubbs ZF, Bend H, Lee R, Lyons JA, Hoefele MJ, Gunthner J, Reutter R, Keren H, Radtke B, Sherbini K, Mrokse O, Helbig C, Odent KL, Cogne S, Mercier B, Bezieau S, Besnard S, Kury T, Redon S, Reinson R, Wojcik K, Ounap MH, Ilves K, Innes P, Kernohan AM, K. D.Care4Rare Canada, Costain C, Meyn G, Chitayat MS, Zackai D, Lehman E, Kitson A, Study H, Martin C, Martinez-Agosto MG, J. A.Undiagnosed Diseases, Nelson N, Palmer SF, Papp CGS, Parker JC, Sinsheimer NH, Vilain JS, Wan E, Yoon J, Zheng AJ, Brimble A, Ferrero E, Radio GB, Carli FC, Barresi D, Brusco S, Tartaglia A, Thomas M, Umana JM, Weiss L, Gotway MM, Stuurman G, E. K, et al. (2020) Histone H3.3 beyond cancer: Germline mutations in Histone 3 Family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients, Sci Adv. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]