

Summary
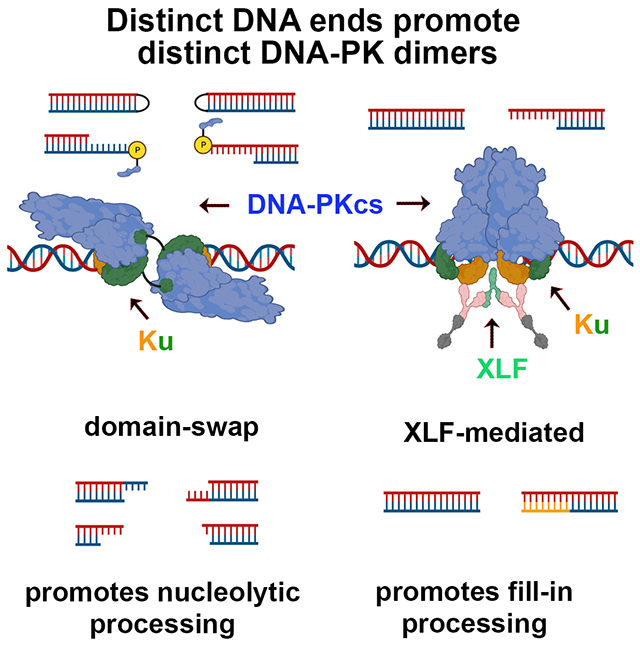
Non-homologous end joining is the major DSBR pathway in mammals. DNA-PK is the hub and organizer of multiple steps in NHEJ. Recent high-resolution structures show how two distinct NHEJ complexes “synapse” two DNA ends. One complex includes a DNA-PK dimer mediated by XLF, whereas a distinct DNA-PK dimer forms via a domain-swap mechanism where the C-terminus of Ku80 from one DNA-PK protomer interacts with another DNA-PK protomer in trans. Remarkably, the distance between the two synapsed DNA ends in both dimers is the same (~115Å), which matches the distance observed in the initial description of an NHEJ long-range synaptic complex. Here, a mutational strategy is used to demonstrate distinct cellular function(s) of the two dimers: one promoting fill-in end-processing, while the other promotes DNA end resection. Thus, the specific DNA-PK dimer formed (which may be impacted by DNA end structure) dictates the mechanism by which ends will be made ligatable.
Graphical Abstract
eTOC
Recent structures show how two distinct DNA-PK dimers synapse DNA ends ~115Å apart before end-alignment in the final ligation complex. To complete NHEJ, DNA end-processing is required before ligation. Buehl et al demonstrate that the XLF-mediated dimer promotes fill-in by DNA synthesis, while the domain-swapped DNA-PK dimer promotes nucleolytic end-processing.
Introduction
The DNA dependent protein complex, DNA-PK, consisting of the Ku70/80 heterodimer and the large catalytic subunit, DNA-PKcs, initiates DNA double strand break repair (DSBR) by the non-homologous end joining pathway (NHEJ). Early models of NHEJ proposed a linear pathway by which 1) Ku70/80 first recognized DNA ends, then 2) recruited DNA-PKcs, which both facilitated DNA end synapsis and regulated DNA end access to end-processing factors, and finally 3) recruitment of the DNA ligase 4 (Lig4) complex, once ligatable ends were available for final ligation (reviewed in1). More recent models favor an iterative mechanism by which synapsis of DNA ends is repeatedly attempted (both before and after end-processing steps) until ligatable ends are detected, allowing the Lig4 complex to resolve the DSB2–5. This iterative strategy would function to minimize alteration of the original sequence. Further advances using single-molecule approaches demonstrate distinct stages of repair that include an initial long-range synaptic complex requiring two DNA-PK protomers6–8. These long-range synaptic complexes proceed to short-range complexes but only with low efficiency. Assembly of short-range complexes requires Ku70/80, the kinase activity of DNA-PKcs, and the three components of the Lig4 complex (XRCC4, XLF, and Lig4), but not the enzymatic activity of Lig46. DNA ends in short-range complexes are positioned close enough to facilitate ligation, and end-processing activities (with the exception of Artemis nuclease activity) are limited to the short-range synaptic complex9. Our recent study documents that catalytically inactive Lig4 promotes NHEJ-like joining in living cells consistent with the Loparo model of a short-range NHEJ complex10. He and colleagues propose that the short-range complex is devoid of DNA-PKcs11, providing an explanation for why Artemis nuclease activity is limited to the long-range complexes. This model is further supported by two reports demonstrating that Ku70/80, XRCC4, Lig4, and XLF all contribute to transient tethering of the two DNA ends7,8 while DNA-PKcs and PAXX further promoted synapsis7. In summary, repetitive progression from long-range to short-range complexes would be consistent with the idea of an iterative mechanism that allows repeated interactions between the two DNA termini, perhaps followed by interaction with processing enzymes, before stable association of compatible ends to facilitate ligation12.
In the past year, several seminal reports have clarified structural characteristics of these NHEJ complexes11,13–16. Two distinct DNA-PK dimers forming long-range complexes have been described that synapse two DNA termini approximately 115Å apart, consistent with previous single molecule experiments6,9. One dimer is formed by the trans-association of Ku80’s C-terminal peptide with its interaction site on DNA-PKcs in a second DNA-PK protomer (termed domain-swap dimer). The second dimer is formed by interaction between two sites in DNA-PKcs (898-900 and 2569-2571) in two DNA-PK protomers and is facilitated by XLF (termed XLF-mediated dimer). Here, using cellular models of NHEJ, we take a mutational approach to show that both DNA-PK dimers are required for efficient NHEJ, and demonstrate that the domain-swap dimer promotes nucleolytic end-processing whereas the XLF-mediated dimer promotes fill-in end-processing. Moreover, although there appears to be an equilibrium between the two dimer forms, the XLF-mediated dimer is essential for final ligation.
Results
The C-terminal helix of Ku80 can interact with DNA-PKcs in both cis and trans; ablating the cis interaction enhances end joining.
We first focused on disrupting the domain-swap dimer. It has been known for decades that the interaction of the DNA-bound Ku70/80 heterodimer with DNA-PKcs is (at least partially) mediated by a short helix at the extreme C-terminus of Ku80 (residues 724-734)17–19. Recent structural studies show that the C-terminal helix of Ku80 may have (at least) two roles15,16. First, in monomeric DNA-PK complexes the C-terminal helix interacts in cis, potentially stabilizing the DNA-PKcs-Ku70/80 interaction, which in turn could stabilize the DNA-PK-DNA interaction. This could be of relevance during the conformational changes that are observed between DNA-end-protection and the DNA-end-processing complex16. The second role of the C-terminal helix of Ku80 is to form DNA-PK dimers via the domain-swap mechanism described by Chaplin et al.
Ideally it would be most informative to disrupt either the cis (or trans) interaction while leaving the trans (or cis) intact. But specific disruption of only the cis or trans interactions presents a challenge as the same interfaces on both polypeptides mediate both, and simple mutation of the interface will likely disrupt both. However, the distance between the C-terminal globular domain and the C-terminal helix in the cis interaction is longer than in the trans interaction (~65Å versus ~40Å, Fig 1A). Unlike the C-terminal helix that is highly conserved (but strikingly only in species that express DNA-PKcs), the linker spanning the Ku80 C-terminal globular domain and the C-terminal helix is not conserved in primary sequence. However, its length is well-conserved in 48 disparate species that also express DNA-PKcs (for example, slime mold, starfish, humans) ranging from 12 to 21 residues (average length 16.6 residues, and the vast majority ranging from 15-17 residues)20. The 17-residue linker in human Ku80 is predicted to be able to span ~60Å in good agreement with the observed distance of ~65Å when interacting in cis; this suggests that the cis interaction may only be possible when the linker is fully extended. We considered that reducing the linker length should exclude or disfavor cis interactions while maintaining the capacity to form intermolecular interactions. Although there is no clear mutational strategy to specifically interfere with the trans interaction, we predicted that lengthening the linker might increase the probability of cis interactions because the natural length of the spacer may limit cis interaction. To this end, eight Ku80 expression plasmids were prepared with different Ku80 C-terminal mutations (Fig 1B and Sup. Table 1). These include two insertional mutants that increase the predicted linker length from 60Å to 80Å (insertion 1) or to 112Å (insertion 2), five mutants that progressively reduce the linker length (resulting in predicted lengths between 7Å and 49Å), and one mutant deleting the entire linker and C-terminal helix.
Figure 1. The C-terminal helix of Ku80 can interact with DNA-PKcs in both cis and trans, but ablating the cis interaction promotes end joining.
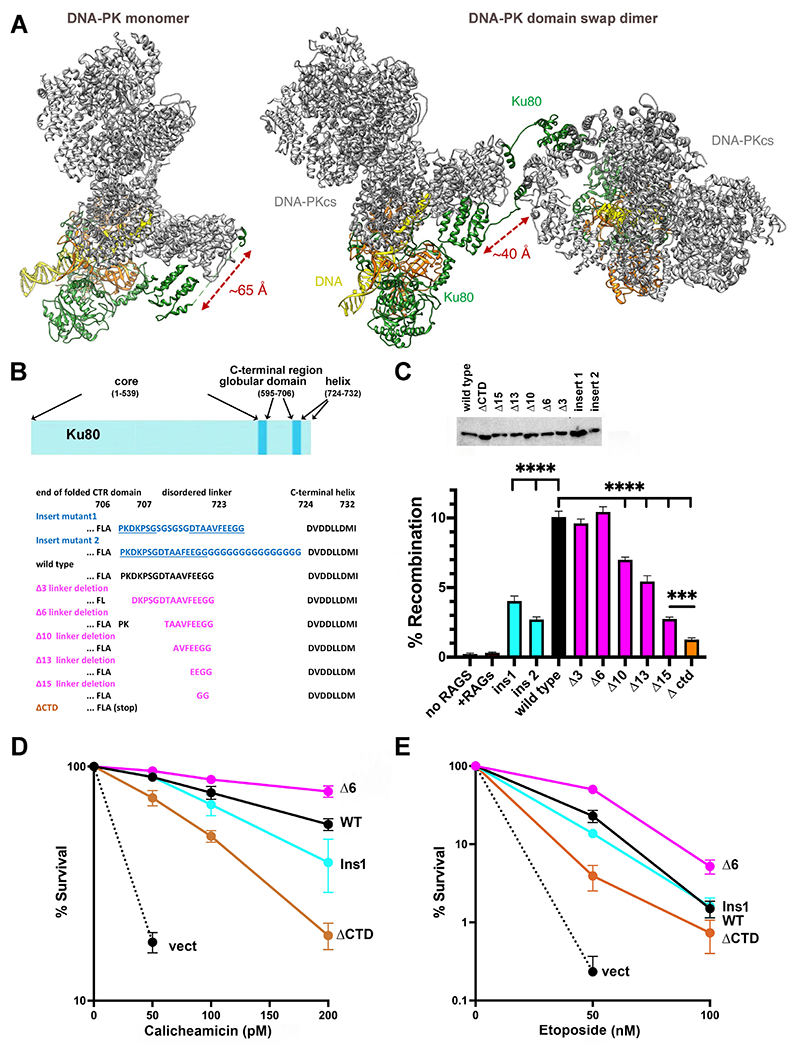
(A) Ribbon diagram of DNA-PK monomer and domain swap dimer. DNA-PKcs is shown in grey, Ku80 in green, Ku70 in orange and DNA in yellow. Diagram representing Ku80. A disordered linker (707-723) separates the C-terminal globular domain (595-706) from the C-terminal helix (724-732). (B) Amino acid sequence of the linker mutants. (C) The fluorescent substrate 290-Crimson/ZS was utilized to detect coding end joining of RAG-induced DSBs. Percent recombination of episomal fluorescent coding-end joining substrate in xrs6 cells transiently transfected with wild-type or mutant Ku80 expression constructs as indicated. Error bars indicate SEM from six independent experiments. ****, P<.0001; ***, P<.001 in two-way A NOVA with Holm-Sidak correction. Relative expression of each Ku80 mutant was assessed by immunoblotting. (D) + (E) xrs6 clonal transfectants expressing wild-type human Ku80 (WT), Δ6 linker shortening mutant (Δ6), insert 1 linker lengthening mutant (Ins1), Ctd linker/C-terminal+helix deletion mutant (ΔCtd), or empty vector (vect) were plated at cloning densities into complete medium with increasing doses of calicheamicin (D) or etoposide (E). Colonies were stained after eight days, and percent survival was calculated. Error bars represent the standard error of the means for six independent experiments.
These mutants were first tested for their capacity to complement the VDJ joining defects of the Ku80 deficient xrs6 CHO cell strain (Fig 1C). Both insertional mutants have modest deficits in coding end joining. Though we recognize that the insertions may have other effects, if increasing the spacer length increases cis interactions (the intent of these mutations), these data suggest that disfavoring trans interactions impedes joining. In contrast, the Δ3 and Δ6 linker shortening mutants (which should span 49Å and 38Å respectively and therefore be unable to form the cis interaction) efficiently complement the VDJ deficits in xrs6 cells, suggesting that the cis interaction is dispensable for joining. Furthermore, thΔA10, Δ13, and Δ15 mutants promote joining levels lower than wild-type Ku80 consistent with the fact that the C-terminal helix would likely be impaired in both cis and trans interactions with DNA-PKcs. The mutant that deletes 15 of 17 residues (Δ15) is nearly as deficient in restoring the VDJ deficits of xrs6 cells as the ΔCtd mutant which completely deletes the C-terminal linker and helix.
To further characterize these mutants, stable xrs6 transfectants were generated and tested for cellular resistance to both calicheamicin and etoposide, two agents known to induce DSBs that require the NHEJ pathway for repair (Fig 1D+E, Sup. Fig 1). [In these assays (and all assays presented herein), at least two independent cell clones for each mutant were tested (Sup. Fig 1), but only a single representative clone is presented for clarity.] Ku80 deficient cells are similarly hyper-sensitive to both calicheamicin and etoposide. Consistent with previous studies17–19, complete deletion of the C-terminal helix and linker (ΔCtd mutant) results in intermediate sensitivity to both drugs (Fig1 D+E). Cell clones expressing the insertional mutant are similarly resistant to both drugs as cells expressing wild-type Ku80 (Fig 1 D+E). Cells expressing the Δ6 mutant are consistently slightly more resistant to both calicheamicin and etoposide than cells expressing wild-type Ku80. The increased resistance to these drugs in cells expressing the Δ6 mutant suggests that increasing the probability of the trans interaction of the Ku80 C-terminus with DNA-PKcs (promoting the formation of the domain-swap dimer) promotes repair. The Δ6 mutant should limit the cis interaction suggesting that the cis interaction of Ku80’s C-terminal helix is dispensable for cellular resistance to both etoposide and calicheamicin.
A single XLF dimer interacts with Ku70, Ku80, and XRCC4 to promote a long-range synaptic complex.
Reports from He and colleagues11 and Blundell and colleagues15 described another DNA-PK dimer complex that was apparent in cryo-EM experiments including the XRCC4/Lig4/XLF complex in addition to DNA-PK (Sup Fig 2A). This DNA-PK dimer includes many protein-protein interactions, with the centrally positioned XLF molecule interacting with both XRCC4 and Ku70, and potentially with Ku80 as well via the Ku70/80 binding motif (KBM) within the disordered C-terminus of XLF21.
We disrupted each of the XLF-mediated interactions either alone or in combination using relatively conservative changes in XLF (mostly alanine substitutions, Sup Table 1). XLF interactions between the head domains of XRCC4 and XLF have been extensively studied22,23; of note, an L115A mutation of XLF completely ablates the interaction with XRCC4 (at physiologic salt concentrations) but has only a modest impact on repair as assessed in both in vitro and cellular assays22,24; in contrast a more disruptive mutation, L115D completely abrogates all XLF function in all cellular assays. Similarly, the highly conserved Ku-binding motif (KBM) in the extreme C-terminus of XLF has also been extensively studied, but ablation of this KBM minimally impacts repair in cellular assays21,25. The interaction observed between the XLF stalk (residues 176-178) and the Ku70 von Willebrand domain (vWA) was first observed in the XLF-mediated dimer11,15; functional studies of this interaction have not been reported. We prepared XLF expression plasmids with mutation of residues 176-178 (RDR) to alanine and used them both as single mutants and in combination with either a KBM mutation (L297W) or the XRCC4 interaction mutant (L115A) to test joining capacity in XLF deficient 293T cells described previously24. XLF with mutations in any of these single mutations is similarly proficient in coding end joining as wild-type XLF (Sup Fig 2B). Similarly, mutating any two of the three interaction sites does not statistically impair VDJ coding end joining. However, in the mutant with all three interaction sites ablated, VDJ coding end joining is impaired. These findings underscore previous conclusions: that multiple protein-protein interactions drive formation and stability of the XLF-mediated dimer11,15. What is new here, is that these interactions function cooperatively to promote end joining. However, even the mutant that disrupts all three XLF-interaction sites retains considerable activity in VDJ joining assays.
We previously generated DNA-PKcs expression constructs that disrupted the 898-900 and 2569-2571 interface (Sup Fig 2A) sites separately and showed that Ala mutants spanning these residues (Sup Table 1) result in partial deficits in VDJ coding and signal end joining15 (Fig 4A). XLF deficient V3 cells were generated to determine whether disrupting the DNA-PKcs-DNA-PKcs interaction concurrently with blocking the XLF-mediated interactions would further impair end joining. Whereas wild-type DNA-PKcs and wild-type XLF substantially complement VDJ deficits, the mutants of either DNA-PKcs or XLF only partially complement and absence or mutation of both more severely reduces levels of VDJ coding end joining (Sup Fig 2C). From this study, it appears that ablating the DNA-PKcs:DNA-PKcs interface has a more substantial impact on end joining than ablating all three XLF-mediated interactions, and we next focused on generating isogenic cell strains with DNA-PKcs mutants that disrupt each dimer to ascertain their function(s) in cellular models of DNA repair.
Figure 4. Disruption of the two long-range synaptic complexes have opposite effects on DNA end-processing.
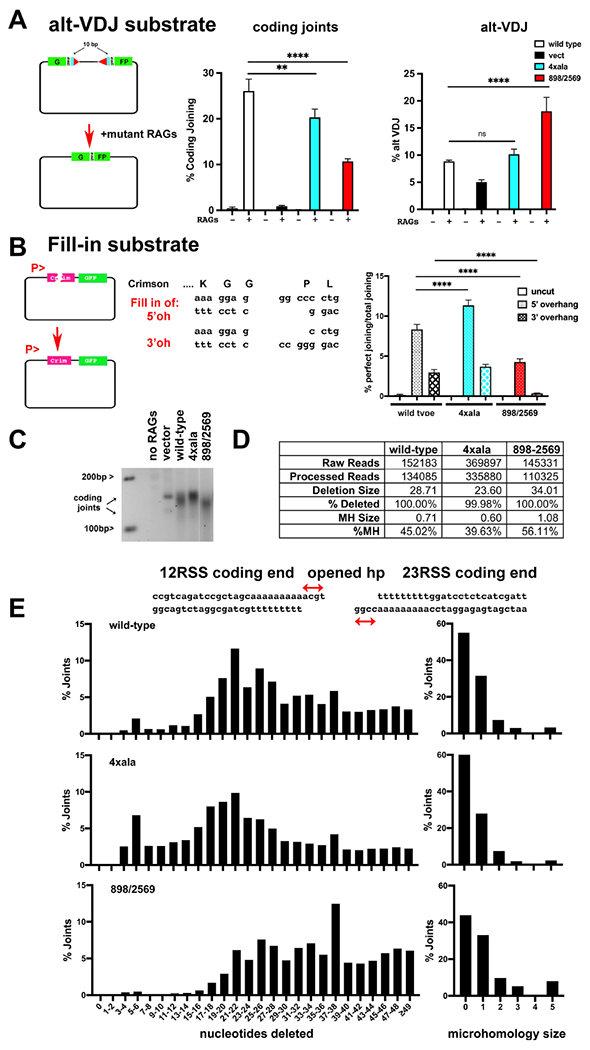
(A) Cartoon depicting the alt-VDJ substrate; restoration of the GFP reading frame requires nucleotide deletions of 10bp from either coding end and utilization of 9bp of short sequence homology. The fluorescent substrate 290-Crimson/ZS (coding joints) or alt-VDJ were utilized to detect coding end joining or alt-VDJ joining of hyper RAG mutant-induced DSBs in V3 cells expressing wild-type, 4Xala mutant, or 898/2659 mutant DNA-PKcs. With the alt-VDJ substrate, dsRED expression was co-transfected to control for transfection efficiency. Percent recombination represents %GFP/%RFP. (B) Cartoon depicting the Fill-in substrate that when cleaved with appropriate restriction enzymes will only restore Crimson expression if over-hanged ends are filled in and re-ligated to blunt end. Uncut or cleaved Fill-in substrate was transfected into cells expressing wild-type DNA-PKcs, the 4Xala, or 898/2569 mutants. Crimson and GFP expression was assessed by Flow cytometry 72 hours later and Crimson/GFP is expressed as %perfect joining. For (A+B) error bars indicate SEM from three independent experiments. **P<0.01; ****P<0.0001; ns=not significant in two-way ANOVA with Holm-Sidak correction. (C) 2.5% agarose electrophoresis of PCR amplification of coding joints from AT coding joint substrate from V3 stable transfectants expressing wild-type or mutant DNA-PKcs and then transfected with substrate and RAG1 and RAG2 expression constructs. Cells were harvested 72 hours after transfection; coding joint substrate was isolated by alkaline lysates, followed by PCR for coding joints. (D+E) Summary of amplicon sequencing of coding joints amplified from V3 transfectants expressing wild type, 4xAla, or 898/2569 DNA-PKcs. Results are averages of two separate experiments.
Ablation of the two DNA-PK mediated dimers has distinct impacts on sensitivity to different radio-mimetic drugs.
In our previous study15, in addition to the mutants disrupting the XLF-mediated dimer discussed above, a DNA-PKcs mutant was constructed in which Ku80’s C-terminal helix interaction site on DNA-PKcs was disrupted by substituting four basic residues with alanine residues (4xala, Fig 2A). Two of the residues in DNA-PKcs that interact with the Ku80 C-terminal helix in cis (K1913, and K1917) are also involved in the trans interaction in the dimeric structure. However, in the trans-mediated domain-swap dimer, R1854 and K1857 also facilitate the interaction, thus all four of these basic residues were replaced with alanine (Sup Table 1). The mutations that disrupt the XLF-mediated dimer are positionally distinct and do not contribute to the domain-swap interaction; similarly, the mutations that disrupt the domain-swap dimer are positionally distinct from the interface observed in the XLF-mediated dimer (Fig 2A+B). To functionally compare cells that are defective in forming either the XLF-mediated or domain-swap dimer, V3 transfectants were generated that express the different DNA-PKcs dimer mutants (Fig 2I).
Figure 2. Ablation of the two DNA-PK mediated dimers has distinct impacts on sensitivity to different radio-mimetic drugs.
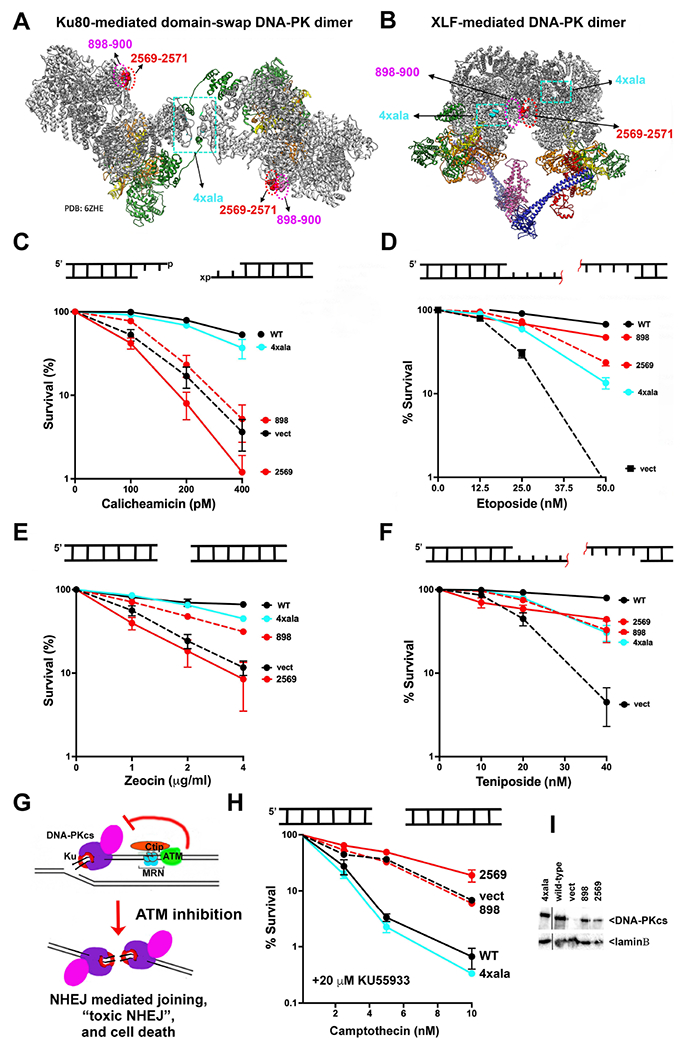
(A + B) Ribbon diagrams of the domain-swap (PDB: 6ZHE) and XLF-mediated (PDB:7NFC) long-range complexes. DNA-PKcs is shown in grey, Ku80 in green, Ku70 in orange and DNA in yellow (left). Insert shows interaction of 4 basic residues with the Ku80 C-terminal helix. (C-H) Cartoon depicting end structures generated by drugs utilized. “x” denotes 3’PG adduct. Red line denotes peptide adduct on 4bp 5’ overhang. V3 clonal transfectants expressing wild-type or mutant DNA-PKcs as indicated were plated at cloning densities into complete medium with increasing doses of zeocin, calicheamicin, teniposide, etoposide, or camptothecin (in the presence of 20 μM KU55933). Colonies were stained after eight days, and percent survival was calculated. Error bars represent the standard error of the mean for at least three independent experiments. (G) Cartoon depicting toxic NHEJ. (I) Immunoblot for DNA-PKcs expression in V3 transfectants.
When we first noted that the two distinct DNA-PK dimers positioned DNA ends ~115Å apart, we contemplated why cells might need two different long-range complexes. We considered that the two dimers might have distinct functions during NHEJ, a process that requires DNA end synapsis, nucleolytic and/or polymerase-dependent end-processing, and ligation. It has been long-appreciated that DNA-PK regulates DNA end-processing through an unknown mechanism26–28, and we considered that the different dimers might promote different types of end-processing events. Thus, we assessed cellular sensitivity of these V3 transfectants to a panel of DNA damaging agents that induce DSBs with distinct terminal modifications that might require different end-processing steps prior to ligation. In most cases, the end-chemistry at DSBs induced by genotoxins is not homogenous; however, the anti-tumor drug zeocin generates mainly blunt-ended DSBs29 whereas calicheamicin generates DSBs with 3’ overhangs, a high percentage of which have 3’ phosphoglycolate (3’PG) adducts30,31 that can be removed by tyrosyl-DNA phosphodiesterase I (TOP1)32. The topoisomerase II (TOP2) inhibitors etoposide and teniposide generate two-ended DSBs primarily in S phase (Zagnoli-Vieira and Caldecott, 2020) that have protein-blocked 5’ ends that are first processed by proteolysis of the trapped TOP2, resulting in peptide-adducted DNA ends. Tyrosyl-DNA phosphodiesterase II (TDP2) restores most of these peptide-adducted DNA ends to 4bp, 5’ phosphorylated overhangs that are repaired by NHEJ (Zagnoli-Vieira and Caldecott, 2020)33,34. It is not known whether TDP2 functions in the NHEJ short-range complex as has been shown for many end-processing factors including TDP19. Finally, when replication forks encounter DNA nicks that result from the TOP1 poison camptothecin, single DNA ends with blunt phosphorylated termini are generated (Fig 2G)35. In normal cells, NHEJ-mediated joining of double-stranded single ends from stalled forks is blocked by an ATM dependent mechanism (Fig 2G). But, if ATM is absent or inhibited, the NHEJ pathway can inappropriately join DNA ends from distinct collapsed forks in a process termed “toxic NHEJ” that results in massive genotoxicity and cell death36. Cells can be rescued from “toxic NHEJ” by ablation of core NHEJ factors (Ku, XLF, XRCC4, and Lig4); we extended the findings of Balmus et al. to show that DNA-PKcs deficiency could also rescue cells from toxic NHEJ37. We included assays with camptothecin and an ATM inhibitor to determine whether either DNA-PK dimer is required for toxic NHEJ.
As expected, cells completely lacking DNA-PKcs are similarly hyper-sensitive to the antitumor drugs and the TOP2 inhibitors (Fig 2C–F). Cells expressing the XLF-mediated dimer mutant are markedly sensitive to both calicheamicin and zeocin, but only modestly sensitive to the TOP2 poisons. In contrast, cells expressing the domain-swap mutant are almost as resistant to zeocin and calicheamicin as cells expressing wild-type DNA-PKcs but are clearly sensitive to both etoposide and teniposide. We tentatively conclude that the XLF-mediated dimer is required for repair of calicheamicin and zeocin-induced damage but is less critical for repair of DNA damage induced by TOP2 poisons. In contrast, the domain-swap dimer is not strictly required for repair of calicheamicin or zeocin-induced damage but is important for repair of DNA damage induced by TOP2 poisons.
To assess toxic NHEJ, cells were treated with increasing doses of camptothecin in the presence of the ATM inhibitor KU55933. Cells expressing wild-type DNA-PKcs are remarkably sensitive to camptothecin in the presence of the ATM inhibitor, whereas cells lacking DNA-PKcs are markedly resistant (Fig 2H). Cells expressing the domain-swap mutants are similarly sensitive to KU55933 + camptothecin as cells expressing wild-type DNA-PKcs, but cells expressing the XLF-mediated dimer mutants rescue cells from KU55933+camptothecin sensitivity to a similar extent as cells lacking DNA-PKcs. We conclude that the XLF-mediated dimer (but not the domain-swap dimer) is required for toxic NHEJ (Fig 2H).
Mutations that disrupt either of the DNA-PK mediated dimers have similar modest deficits in joining VDJ-induced DSBs in episomal assays [15 and Fig 4A). In previous studies, the vast majority of DNA-PKcs mutants that impact joining in episomal assays also impact survival in kill curve assays to a similar extent26,27,38–43. Thus, the observation that cells expressing the XLF-mediated dimer versus the domain-swap dimer have distinct patterns of sensitivity to different DNA damaging agents is totally unexpected, and we next focused on trying to understand the basis for these different drug sensitivities.
The TDP2-independent, Mre11 dependent pathway for processing peptide-blocked 5’overhangs requires the DNA-PK domain-swap dimer.
Elegant work from Caldecott, Pommier, Povirk and their colleagues has provided much clarity as to how TOP2 poison-induced DNA damage is repaired in mammalian cells31,33,34,44–47. Briefly, TOP2 poisons trap the enzyme in the DNA bound state resulting in irreversible protein-blocked 5’ ends that must be processed by protein degradation to generate peptide-adducted DNA ends (Fig 3A); TDP2 can perfectly restore the peptide-adducted ends to 4bp, 5’ phosphorylated overhangs that are efficiently repaired by error-free NHEJ. But not all trapped TOP2 complexes are repaired in this manner; a fraction of TOP2 poison-induced DSBs are repaired in a separate pathway that functions independently of TDP2. In this pathway, a nuclease cleaves the peptide-blocked 5’overhang, leaving non-compatible DNA ends that can be repaired either by error-prone NHEJ, or error-free HR. Recent compelling reports have identified Mre11 as the nuclease that cleaves peptide adducted DNA ends that result from TOP2 poisons48,49. Moreover, Hoa et al demonstrate that Mre11 and Ku have an epistatic relationship in the repair of etoposide induced damage. Finally, Deshpande et al have shown that DNA-PK promotes the nuclease activity of Mre11 in vitro50. Thus, we considered whether one of the DNA-PK dimers might facilitate the Mre11-dependent pathway to repair peptide adducted DNA ends.
Figure 3. Cells expressing the domain-swap mutant are deficient in the TDP2-independent, nuclease dependent pathway for processing peptide-blocked 5’overhangs.
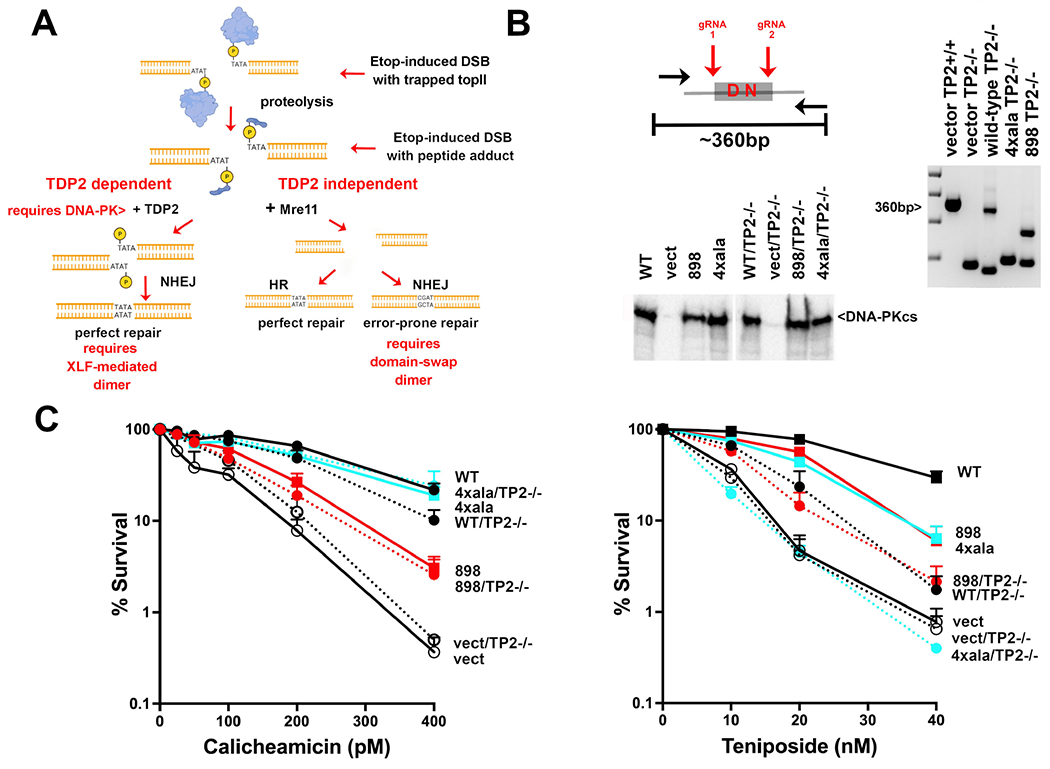
(A) Diagram of repair of TOP2 poison-induced DNA damage45. (B) Diagram of CRISPR strategy to ablate TDP2 from V3 transfectants. (right) Agarose gel electrophoresis of PCR to detect genomic deletions within in V3 cells. (bottom) Immunoblot for DNA-PKcs expression in V3 transfectants before and after TDP2 ablation. (C) V3 clonal transfectants expressing wild-type or mutant DNA-PKcs as indicated were plated at cloning densities into complete medium with increasing doses of calicheamicin or teniposide. Colonies were stained after eight days, and percent survival was calculated. Error bars represent the standard error of the means for six independent experiments.
To help understand how TDP2 and NHEJ cooperate to repair DSBs that result from TOP2 inhibition, TDP2 was ablated from this panel of V3 transfectants (Fig 3B). A CRISPR strategy was utilized to delete the active site in TDP2 using gRNAs to target PAM sites that flank the active site (Fig 3B). [Two independent TDP2 ablated clones were studied for each transfectant, but only one presented.] As expected, ablation of TDP2 has no major impact on cellular calicheamicin resistance in any of the cell types tested (Fig 3C). In contrast, ablation of TDP2 from cells expressing wild-type DNA-PKcs markedly sensitizes cells to teniposide, but these TDP2 ablated cells are still more resistant to teniposide than cells lacking DNA-PKcs (TDP2 proficient or deficient). Ablation of TDP2 from DNA-PKcs deficient cells did not further enhance sensitivity to teniposide, establishing (at least in V3 cells) an epistatic relationship between TDP2 and DNA-PKcs with regards to repair of TOP2 poison-induced damage. Ablation of TDP2 from cells expressing the XLF-mediated dimer mutant results in teniposide sensitivity that is indistinguishable from TDP2 deficient cells expressing wild-type DNA-PKcs; whereas ablation of TDP2 from cells expressing the domain-swap mutant results in cellular teniposide sensitivity that is indistinguishable from cells that completely lack DNA-PKcs (either TDP2 proficient or deficient).
Altogether, these data suggest the following: 1) The XLF-mediated dimer facilitates repair of the perfect 5’ overhang ends generated by TDP2 (potentially by promoting the formation of the short-range complex). In the absence of TDP2 (when all repair must occur by the Mre11-dependent mechanism), cells that lack the XLF-mediated dimer have similar resistance to teniposide as wild-type cells. 2) Since cells expressing either wild-type or the XLF-mediated dimer can both generate the domain-swap dimer, it follows that the domain-swap dimer facilitates repair of peptide adducted ends in the absence of TDP2. In fact, cells expressing the domain-swap mutant that lack TDP2 are similarly hyper-sensitive to teniposide as cells that completely lack DNA-PKcs. Thus, we conclude that the TDP2-independent, Mre11-dependent pathway for processing peptide-blocked 5’overhangs by error-prone NHEJ (initially proposed by Caldecott and colleagues and extended by others) requires the domain-swap dimer. 3) Finally, TDP2 ablation in cells lacking DNA-PKcs does not further sensitize cells to teniposide; these data suggest that both the TDP2 dependent and independent pathways of repair of peptide-adducted ends are completely dependent on DNA-PKcs. These data suggest that whereas Mre11 functions in the context of the domain-swap dimer, TDP2’s capacity to promote repair requires the XLF-mediated long range synaptic complex.
Disruption of either of the long-range synaptic complexes has opposite effects on DNA end-processing as assessed by episomal assays.
The TDP2 ablation experiments suggest that the XLF-mediated dimer facilitates joining of the perfect 5’ overhang ends generated by TDP2 whereas the domain-swap dimer facilitates nucleolytic processing when TDP2 fails. We next assessed whether the mutations that disrupt each DNA-PK dimer impact end-processing using two episomal joining assays that can reveal characteristics of NHEJ-mediated joints. It is well-appreciated that during VDJ recombination, short sequence homologies at opened coding-end termini can facilitate coding end joining51.
To join at sites of internal microhomology, nucleotide loss must occur and (in NHEJ proficient cells) is usually limited to a few nucleotides but can vary depending on the actual gene segments being joined. We have previously utilized a unique VDJ substrate developed by Roth and colleagues to assess highly nucleolytic repair52. This substrate requires nucleotide deletions of 10bp from each coding-end, and then use of a 9 base pair sequence microhomology to restore a GFP open-reading frame (this joining is referred to as alt-VDJ). We reasoned that if loss of one DNA-PK dimer results in increased nucleotide loss during NHEJ, joining at the sites of microhomology within this substrate would be increased. In these assays, a hyper-RAG2 mutant (that destabilizes the RAG post-cleavage complex) was used to increase recombination rates and facilitate detection of alt-VDJ events53. As can be seen, coding end joining (Fig 4A middle panel) using the mutant RAGs is robust, but still strongly dependent on DNA-PKcs. As reported previously15, mutations disrupting either the domain-swap dimer or the XLF-mediated dimer have modest deficits in coding end joining. Using the alt-VDJ substrate, restoration of the GFP reading frame in cells expressing wild-type DNA-PKcs is modest because end-processing is generally limited to less than 10 nucleotides/coding end (Fig 4A, right panel). In cells lacking DNA-PKcs where the only coding end joining is mediated by alternative end joining (a-EJ), detection of alt-VDJ is increased as compared to standard coding end joining because a-EJ is characterized by increased nucleotide loss and strong dependence on the presence of SSH. Thus, in DNA-PKcs deficient cells, joining is enhanced by the presence of the 9bp SSH in the coding ends. Cells expressing the domain-swap mutant (4xala) display a similar level of alt-VDJ as cells expressing wild-type DNA-PKcs. In contrast, cells expressing the mutant that disrupts the XLF-mediated dimer (898/2569) robustly join coding-ends via utilization of the 9bp SSH. These data suggest that whereas nucleotide loss in cells expressing the domain-swap mutant is similar to that of cells expressing wild-type DNA-PKcs, end-processing resulting in nucleotide loss must be enhanced in cells expressing the XLF-mediated dimer mutant.
Although NHEJ is often characterized as an error-prone repair mechanism, numerous studies have also documented its relative fidelity5,9. For example, perfectly cleaved compatible ends are almost always rejoined with complete fidelity; incompatible ends are rejoined to minimize nucleotide loss using fill-in end-processing mechanisms5,9. To assess differences in the capacity to fill-in overhangs during NHEJ in cells expressing the two different dimer mutants, a substrate that measures fidelity of joining was generated (Fig 4B). Briefly, the substrate plasmid is restricted with enzymes generating blunt and over-hang ends as illustrated; if these non-compatible ends are rejoined with no base pair loss via fill-in mechanisms, the crimson (RFP, derivative of red fluorescent protein) open reading frame is restored. In contrast, green fluorescent protein (GFP) is expressed if any joining occurs. In cells expressing wild-type DNA-PKcs, when the substrate with a blunt end and 5’ overhang is transfected, ~8% of the GFP positive cells are also RFP positive. When substrate with a blunt end and 3’ overhang is transfected, ~5% of the GFP positive cells are also RFP positive. With cells expressing the domain-swap mutant (in which the XLF-mediated dimer will predominate) using the 5’ overhang fill-in substrate, ~12% of the GFP positive cells are RFP positive suggesting that cells deficient in generating the domain-swap mutant may be more proficient in fill-in reactions with 5’ overhang ends (Fig 4B). Cells expressing the domain-swap mutant are similarly proficient as cells expressing wild-type DNA-PKcs in perfect rejoining of the 3’ overhang substrate. Cells expressing the XLF-mediated dimer (in which the domain-swap dimer will predominate) are markedly impaired in reconstituting the RFP open-reading frame whether the substrate includes a blunt end paired with either a 5’ or 3’ overhang. These data suggest that the XLF-mediated dimer is required for efficient fill-in end-processing. Cells that disrupt this dimer are inefficient in perfect rejoining of the test substrate; cells that preferentially form more of the XLF-mediated dimer (because the domain-swap dimer is blocked) are more proficient in perfect rejoining of the test substrate (Fig 4B).
Disruptions of the two long-range synaptic complexes have opposite effects on DNA end-processing as assessed by characterization of coding joints from episomal assays.
We next ascertained end-processing characteristics of the DNA-PKcs mutants by sequencing coding joints from episomal assays. As noted above, it has been known for decades that coding end joining can be markedly skewed by the presence of short sequence homology (SSH) at the coding end termini51; thus, if SSH exists near the termini of the opened hairpin coding ends, a single preferentially joined coding joint can sometimes dominate. Thus, for these studies, to limit the impact of SSH, we generated a coding substate that minimizes homology of the opened hairpins for the first 11bp at each end. This substrate contains poly A and T tracks at both coding ends that accentuate nucleotide loss (unpublished data K. Meek and N. Goff). Coding joints from this substrate are therefore diverse without over-representation of a single joint. V3 clones expressing wild-type, mutant, or no DNA-PKcs were transfected with RAG expression vectors and the coding joint substrate; plasmid substrates were isolated 72 hours after transfection and coding joints amplified by PCR (Fig 4C).
Cells lacking DNA-PKcs generate very few coding joints, and only a few uniform joints are amplified by PCR. Joining of coding ends in these episomal assays is robust in cells expressing wild-type DNA-PKcs, and diverse coding joints are readily detected by PCR. Consistent with the more proficient fill-in capacity observed in the fluorescent fill-in assay, cells expressing the domain-swap mutant (4xala) generate diverse coding joints that are consistently slightly longer (fewer nucleotides deleted) than those isolated from wild-type cells. In contrast, (but consistent with the alt-VDJ assays) coding joints isolated from cells expressing the mutant that disrupts the XLF-mediated dimer (898/2569) are clearly shorter than coding joints amplified from cells expressing wild-type DNA-PKcs. Coding joints amplified from cells expressing wild-type DNA-PKcs or the two dimer mutants (4xala or 898/2569) were isolated and subjected to amplicon sequencing (Fig 4D and 4E). Here, 0 nucleotide loss is designated as having maintained the four base-pair hairpin overhangs (P segments) from either coding end. Whereas joints isolated from cells expressing wild-type DNA-PKcs lost on average 28.7 from each coding joint, cells expressing the domain-swap mutant lost on average 23.6 nucleotides/joint, and cells expressing the mutant that disrupts the XLF-mediated dimer lost on average 34.1 nucleotides/joint. Although the substrate was designed to reduce the extent of SSH in the two coding ends, there are still several present. In wild-type cells, joints that utilize SSH comprise 45.02% of joints. In cells expressing the domain-swap or XLF-mediated dimer mutants, joints that utilize SSH comprise 39.63% or 56.11% of joints respectively. In sum, these data further substantiate the conclusion from episomal assays, that the XLF-mediated dimer promotes fill-in end-processing, whereas the domain-swap dimer facilitates nucleotide loss at the site of DNA end joining.
Functional studies of mutants that block both dimer forms suggest that maintaining an equilibrium between the two is required for efficient repair.
We previously generated DNA-PKcs combination mutants that disrupt both long-range synaptic complexes and showed that the combined mutations did not additively impact VDJ coding or signal end joining15. Clonal V3 transfectants expressing each of the double mutants were generated. To our surprise, combining mutations to disrupt both dimer interfaces (either the 898-900 or 2569-2571 with the 4xala mutant, Sup Table 1) substantially reverses cellular sensitivity to calicheamicin, etoposide, and toxic NHEJ as compared to the single mutants alone (Fig 5 A–C). Moreover, the differences in end-processing are also reversed in the combination mutants (Fig 5E). To explain these results, we suggest that the 4xala and 898-900 or 2569-2571 mutants shift the balance of the two long-range synaptic complexes. Although other explanations are possible, we suggest that these alanine mutations that disrupt the stability of both dimers concurrently restore the parity of dimer formation and effectively restore cellular DNA repair.
Figure 5. Cells expressing mutants that disrupt both long-range synaptic complexes are similarly resistant to DNA damaging agents as cells expressing wild-type DNA-PKcs.
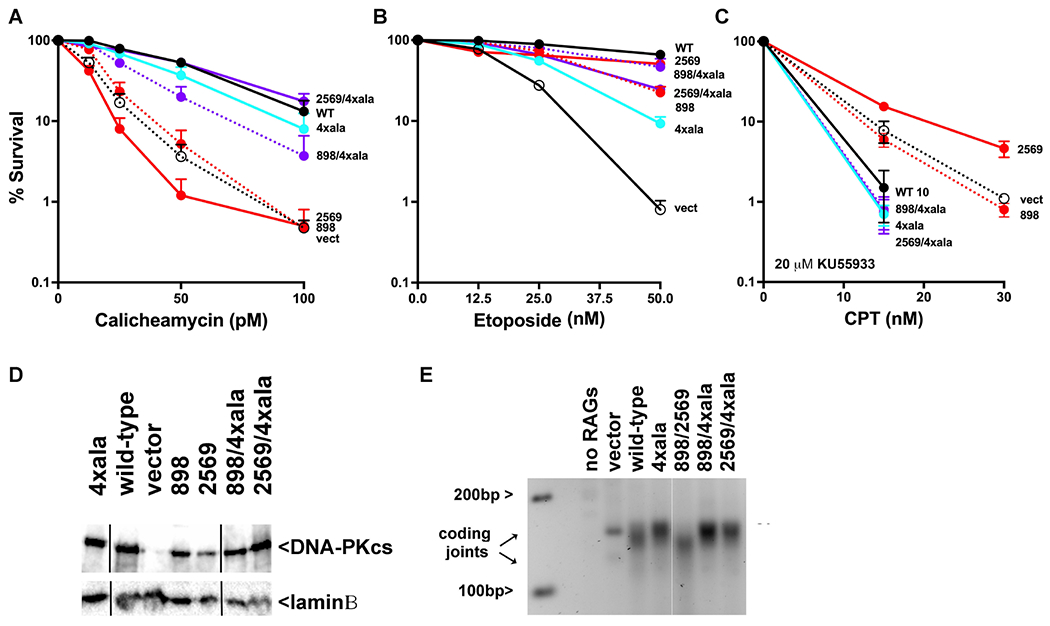
V3 clonal transfectants expressing wild-type human DNA-PKcs, no DNA-PKcs, or mutant DNA-PKcs as indicated were plated at cloning densities into complete medium with increasing doses of calicheamicin (A), etoposide (B), or camptothecin (in the presence of 20 μM KU55933) (C). Colonies were stained after eight days, and percent survival was calculated. Error bars represent the standard error of the means for six independent experiments. (D) Immunoblot for DNA-PKcs expression in V3 transfectants. (E) 2.5% agarose electrophoresis of PCR amplification of coding joints from AT coding joint substrate from V3 stable transfectants expressing or not wild-type or mutant DNA-PKcs and then transfected with substrate and RAG1 and RAG2 expression constructs.
Complete ablation of the XLF-mediated dimer severely impairs NHEJ.
Clearly, the initial alanine mutants only partially impair formation of the two different dimers. This may be analogous to our previous observation that an XLF L115A mutation does not completely disrupt NHEJ but an L115D does24. One can imagine that the negative charge added by L115D prevents positioning XLF between the two XRCC4 dimers (Sup Fig 2). Although the L115A mutant cannot mediate head-to-head interactions with XRCC4 dimers, it can apparently still be positioned within the XLF-mediated dimer, explaining how this mutant retains substantial function. We next constructed DNA-PKcs expression constructs that should more completely ablate each of the DNA-PK mediated dimers, changing the four basic residues that mediate the domain-swap dimer to aspartates (4xasp) and changing two lysine residues (902 and 944) that form a salt bridge in the DNA-PKcs interface in the XLF-mediated dimer to aspartate (898+902asp, Sup Table1). These constructs were first tested in episomal end joining assays (Fig 6A). The 4xasp construct complements joining similarly as the 4xala construct. The 898+902asp construct is severely impaired in promoting coding end joining (as compared to the 898 mutant) and the combined mutant 898+902asp+4xasp is similarly impaired as 898+902asp alone. Stable V3 transfectants were derived and sensitivity to calicheamicin and teniposide assessed. Cells expressing either the 4xasp or 898+902asp mutants are more sensitive to both drugs as compared to the alanine counterparts (Fig 6B+C). Of note, combining the two mutants does not rescue cellular hypersensitivity to either drug type. All together, these data suggest that the more disruptive mutations completely disrupt the two dimer forms and establish a clear functional role for both dimers. Moreover, although other explanations are possible, because cells expressing the combination mutant are not more sensitive to either drug as compared to cells expressing the 898+902asp mutant, this implies that DSBs processed by the domain-swap dimer must progress through the XLF-mediated dimer before ligation in the short-range NHEJ complex.
Figure 6. More disruptive mutants designed to disrupt the domain-swap and XLF-mediated DNA-PK dimers enhance their cellular phenotypes.
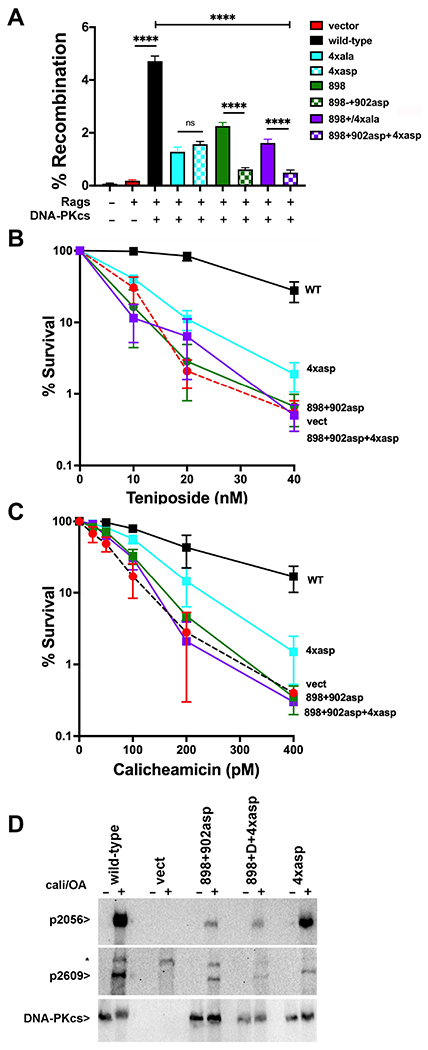
(A) (left) The fluorescent substrate 290-Crimson/ZS (coding joints) was utilized to detect coding end joining of RAG-induced DSBs in V3 cells expressing wild-type or mutant DNA-PKcs as depicted. Percent recombination represents %GFP/%Crimson. **P<0.01; ****P<0.0001; ns=not significant in two-way ANOVA with Holm-Sidak correction, (right) Immunoblot depicting relative DNA-PKcs expression in stable V3 clones as indicated. (B+C) V3 clonal transfectants expressing wild-type DNA-PKcs, vector control or DNA-PKcs mutants as indicated were plated at cloning densities into complete medium with increasing doses of calicheamicin (B) or teniposide (C). Colonies were stained after eight days, and percent survival was calculated. Error bars represent the standard error of the means for six independent experiments. (C) Immunoblot analyses using indicated antibodies of cell extracts from indicated V3 transfectants treated or not with 40nM calicheamicin and 1uM Okadaic acid for thirty minutes. * Indicates a non-specific band.
PQR phosphorylation occurs predominately in the XLF-mediated dimer.
There is a growing consensus in the field that PQR phosphorylation occurs predominately in trans24,54,55; however, our previous study showed no significant deficiency in S2056 phosphorylation (a PQR site) in cells expressing either the domain-swap or XLF-mediated dimer alanine mutants15. The conclusion that the alanine mutants only partially disrupt the dimers provides a potential explanation for the lack of impact on S2056 phosphorylation. We next assessed induction of S2056 phosphorylation in cells expressing the more disruptive mutants. Whereas 40nM and 1uM Okadaic acid promote T2609 phosphorylation (an ABCDE site) in all three mutants (albeit slightly decreased in all three), S2056 phosphorylation is remarkably diminished in cells expressing either the 898+902asp mutant or the combined mutant, but not in the 4xasp mutant (Fig 6D). These data support our interpretation that disruption of the 898-900/2569-2571 dimer interface disrupts formation of the XLF-mediated dimer and blocks trans S2056 phosphorylation. Moreover, the finding that PQR phosphorylation occurs in the XLF-mediated dimer where nucleolytic end-processing is blocked is completely consistent with the biologic outcome of PQR phosphorylation which functions to limit end-processing26.
Discussion
NHEJ is often described as a simple process whereby two DNA double-stranded ends are bound, made compatible, and rejoined56–58. However, each of these steps requires distinct protein complexes and as DNA damaging agents rarely leave perfectly re-ligatable DNA ends, simply rendering ends ligatable often requires iterative biochemical processes. The studies from Loparo and colleagues demonstrating distinct long-range and short-range complexes suggest that a single short-range NHEJ complex might suffice to direct all end-processing events6,9. However, the structural studies11,13,15, support the observation that there are two long-range complexes, both of which are entirely consistent with the long-range complex proposed by Graham et al.6.
The findings that cells limited to one of the two long-range dimers have clear differences in sensitivity to zeocin and calicheamicin, have distinct roles in resolving DNA damage induced by TOP2 poisons, and have pronounced differences in end-processing of VDJ coding joints establish that the two dimer forms have distinct roles in end-processing. The domain-swap dimer facilitates nucleolytic end-processing whereas the XLF-mediated dimer promotes fill-in end-processing. Cells that cannot form the XLF-mediated dimer have severe deficits in autophosphorylation of S2056, a phosphorylation that is believed to occur predominantly in trans24,54,55. S2056 is a site in the “PQR” cluster that limits end-processing when phosphorylated, consistent with the function of the XLF-mediated dimer that promotes fill-in end-processing and prevents nucleolytic end-processing.
In addition to studies documenting distinct NHEJ complexes11,13,15, recent cryo-EM studies have also bolstered our understanding of the structural details of the DNA-PK holoenzyme59–62 providing a growing understanding of the mechanism of kinase activation. Chen and colleagues have shown that after initial DNA end-binding, monomeric DNA-PK complexes proceed through several distinct configurations14; different DNA end structures can promote monomeric DNA-PK structures that protect DNA ends whereas others promote phosphorylation of the ABCDE sites that opens the DNA binding cradle activating Artemis16. These results suggest that Artemis-mediated end-processing may occur in monomeric DNA-PK complexes9,63,64 (Ma et al), and are consistent with an abundance of both cellular and in vivo studies that demonstrate that Artemis end-processing can occur independently of any of the components of the Lig4 complex65–67. However, it is also possible that Artemis could be recruited to and activated within the domain-swap dimer; since it is clear from this study that the domain-swap dimer promotes nucleolytic end processing, we suggest that the ABCDE phosphorylation complex monomers progress to domain swap dimers, that are compatible with Artemis cleavage. Moreover, compelling data from Paull and Sasanuma and colleagues48,49 demonstrate that Mre11 function is strongly enhanced by DNA-PK and that Mre11 functions in an NHEJ dependent manner to facilitate repair of etoposide-induced damage. Our data suggest that Mre11 repair functions in the same pathway as the domain-swap long-range complex, implying that Mre11 functions within this complex. Still, as with Artemis, it is possible that the Mre11 dependent step in processing tyrosyl-adducted DNA termini may occur in monomeric DNA-PK complexes that progress into domain-swap dimers.
The orientation of the two DNA-PK protomers in the domain-swap dimer would likely preclude progression of the domain-swap dimer to the proposed configuration of the short-range complex described by Chen et al.11. Moreover, analysis of the harsher mutant that more completely disables the XLF-mediated dimer suggests that the XLF-mediated dimer is essential for NHEJ. Although other explanations are possible, we suggest that ends processed by the domain-swap dimer must proceed through the XLF-mediated dimer for repair by NHEJ.
Though speculative, we suggest the following embellishments of the model put forth by Chen et al.14: that DNA end structures dictate their path in NHEJ (Fig 7). Ends that require nucleolytic processing (hairpin ends, some peptide adducted ends) promote the ABCDE cis-phosphorylation complex opening the DNA binding cradle in DNA-PK. We suggest that the ABCDE phosphorylation complex monomers progress to domain swap dimers, that promote nucleolytic end-processing by either Mre11 or Artemis (or potentially other nucleases31). In contrast, ends that can be directly ligated, or that can be aligned to promote fill-in end processing, or are appropriate for processing by TDP1, TDP2, or PNKP preferentially form DNA end-protection monomers that progress to XLF-mediated dimers where PQR phosphorylation occurs in trans. In the XLF-mediated dimer, ABCDE phosphorylation [also potentially in trans as suggested by Chen et. al.11] must occur to allow progression to a short-range complex that facilitates fill-in end-processing.
Figure 7.
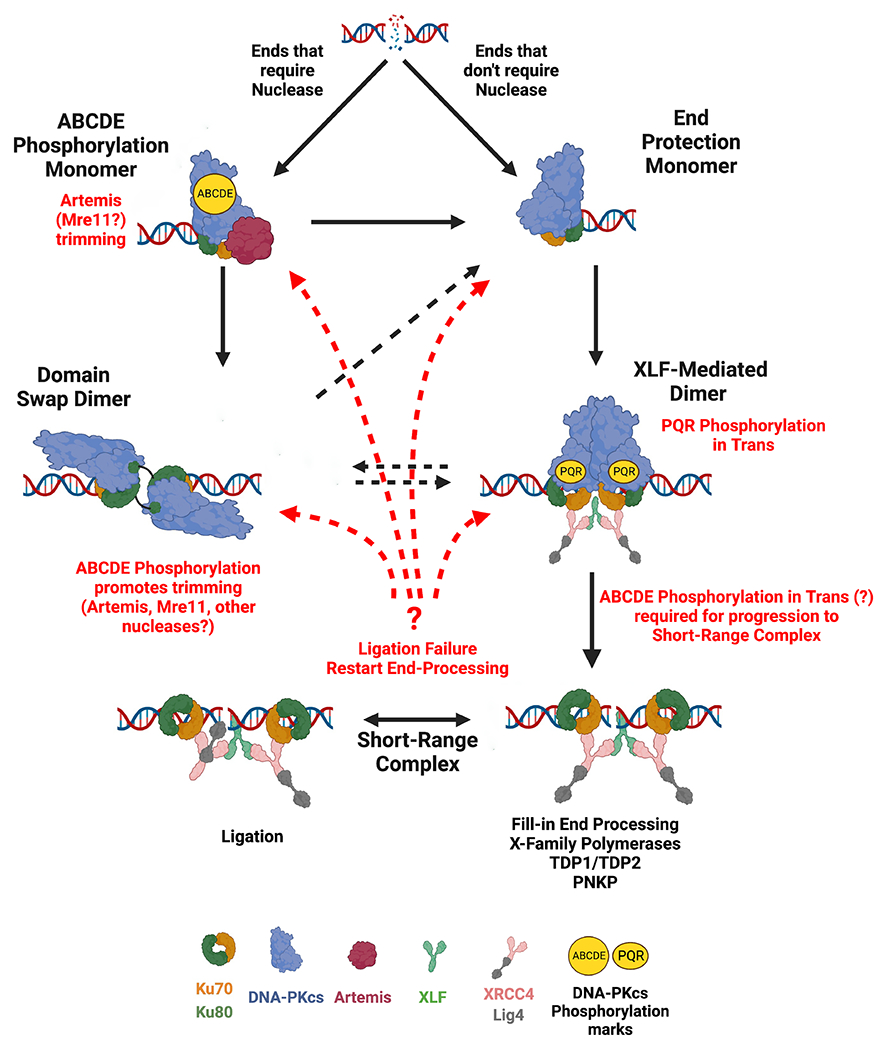
Iterative model of NHEJ.
The synthetic rescue effect of combining the alanine dimer mutations supports the conclusion that there is an equilibrium between the two DNA-PK dimer forms. Moreover, since complete ablation of the XLF-mediated dimer cripples NHEJ, it seems likely that DSBs processed by the domain-swap dimer must eventually be recruited to an XLF-mediated dimer to promote repair in the short-range complex. What is unclear from our research is whether ends bound by either dimer are released and must assemble as monomers that proceed to repair or whether DNA ends can be switched from one long-range complex to another. All in all the data presented are highly supportive of more recent iterative models of NHEJ, that were initially based on observations from Lieber and Ramsden and colleagues showing that the Lig4 complex in the short-range NHEJ complex “samples” DNA termini and maintains alignment until ligatable ends can be positioned for repair2–5. Our data, extend this model to show this iterative characteristic of NHEJ may also apply to the long-range NHEJ complexes.
In summary, data presented here are the first to support the proposal of patently distinct functions for different DNA-PK complexes. The most compelling argument that the two DNA-PK dimers have distinct functions is the observation that each have exactly opposing impacts on end-processing. Thus, DNA-PKcs not only regulates end-processing by regulation of Artemis activity by its own autophosphorylation (of the ABCDE and PQR sites)16,26,27,68, the DNA-PK dimer choice dictates the mechanism by which ends will be made ligatable.
Limitations of the Study
Our understanding of how NHEJ functions to directly repair DSBs has been bolstered by recent studies using cryo-EM and single-molecule methods to define distinct steps of the NHEJ mechanism. Repair appears to progress from two distinct long-range synaptic complexes to short-range synaptic complexes where the final resolution of the DSB occurs by the action of Lig4. The studies presented here provide strong support that both proposed long-range synaptic complexes function in living cells, and that each has a distinct role in promoting DNA end-processing that is required when two DNA ends are not ligatable. However, our studies cannot yet address how DSBs are shuttled between distinct NHEJ complexes, or how either DNA-PK dimer progresses to a short-range, ligation competent complex. Moreover, because biochemical analyses of the DNA-PK dimer mutants are beyond the scope of the current study, the molecular mechanisms by which the domain-swap dimer promotes nucleotide trimming and the XLF-mediated dimer promotes fill-in end-processing are yet to be resolved.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Requests for reagents or information should be directed to and will be fulfilled by the lead contact, Katheryn Meek (kmeek@msu.edu).
Materials Availability
Unique materials from this study will be made available upon request and will be deposited with Addgene upon publication.
Data and Code Availability
All sequencing data (FASTQ files for amplicon sequencing) will be made available upon request.
Analysis of episomal-substrate sequencing for indel and microhomology sizes was performed using a published python script, Junction Analysis (https://github.com/aluthman/Ramsden-Lab/tree/main/Luedeman_et_al_2022)69.
Any additional information required to reanalyze the data reported here will be available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmids.
Expression plasmids for wild-type DNA-PKcs and the dimer mutants have been described previously15,70. An expression plasmid for human Ku80 has been described previously71; Ku80 linker mutants were generated by synthesizing gene blocks (IDT) encoding the mutants described in Fig 1B that included unique XcmI and EcoRI sites for subcloning into the parental plasmid. An expression plasmid for human XLF and the L115A mutant have been described previously24. The XLF KBM mutation (L297W) was synthesized as a double-stranded oligonucleotide spanning unique ClaI and XhoI sites. The RDR>AAA mutation was synthesized as a gene block that spanned unique NheI and NotI sites.
Cell culture, cell strains.
293T cells (female cell strain, CRL-3216) were cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies) and 10 μg/ml ciprofloxacin. Chinese hamster ovary (CHO) cell strains V3 (female cell strain, CVCL_K039) and xrs6 (female cell strain, CVCL_4340) were cultured in alpha-MEM with the same supplements and under the same conditions. Methods to derive V3 and xrs6 transfectants have been previously described70–72. Independent, stable transfectants were selected and maintained in complete medium containing 10 μg/ml puromycin. At least two independently derived clones expressing each mutant were studied.
METHOD DETAILS
Survival assays.
Clonogenic survival assays were performed for V3 and xrs6 transfectants. Briefly, two hundred cells were plated for each transfectant into complete medium containing the indicated dose of the indicated drug in 60 mm diameter tissue culture dishes. After 7 to 10 days, cell colonies were stained with 1% (w/v) crystal violet in ethanol to measure relative survival. A minimum of six independent experiments were performed, and results were averaged.
Cas9-mediated gene disruption.
Cas9-targeted gene disruption was performed using methods similar to those reported by Mali et al.73 to ablate XLF from DNA-PKcs deficient V3 cells. Briefly, a gRNA specific for a PAM in hamster XLF was synthesized as a 455 bp fragment (Integrated DNA Technologies). The sequence of the 22 mer specific for the XLF PAM site is: GGCCTGTTGATGCAGCCATGGG. The synthesized fragment was cloned into pCR2.1 using a TOPO TA cloning kit according to the manufacturers’ instructions (Life Technologies). Cells were transfected with 1 μg gRNA plasmid, 1 μg Cas9 expression plasmid (Addgene), and 0.2 μg of pSuper-Puro (to confer puromycin resistance) in 200ul Opti-MEM (Life Technologies) and 4ul PEI polyethylenimine (PEI, 1 ug/ml, Polysciences). Isolated clones were selected, and DNA isolated for PCR to assess targeting of the XLF alleles in each clone.
To disrupt TDP2, oligos containing PAM sites upstream and downstream of the active site of TDP2 were synthesized (Integrated DNA Technologies) and cloned into the BbsI linearized pSpCas9(BB)-2A-Puro (Addgene). Cells were transfected as above, with 1μg of each Cas9/gRNA expression plasmid. Isolated clones were selected, and DNA isolated for PCR to assess targeting of the TDP2 alleles in each clone.
Episomal end joining assays.
RAG expression plasmids have been described previously53,74. The fluorescent VDJ coding and alt-VDJ substrates have been described52,64,75. Briefly, to assess coding end joining, the coding joint substrate was co-transfected with plasmids encoding RAG1, RAG2, XLF, Ku80, or DNA-PKcs as indicated into V3, xrs6, or 293T cells as indicated. Joining assays were performed on cells plated at 20-40% confluency into 24-well plates in complete medium. Cells were transfected with 0.125 μg substrate, 0.125ug RAG1, RAG2, XLF, Ku80 or .25ug DNA-PKcs as indicated per well using polyethylenimine (PEI, 1 ug/mL, Polysciences) at 2 μL/1 μg DNA. Cells were harvested 72 hours after transfection and analyzed for GFP and RFP expression by flow cytometry. The percentage of recombination was calculated as the percentage of live cells expressing GFP divided by the percentage expressing RFP. Data represents at least three independent experiments, each including triplicate transfections. Alt-VDJ assays were performed similarly, except transfection efficiency was measured by co-transfection of an RFP expression plasmid (dsRED express, Takara). The Fill-in substrate was generated by synthesizing a fragment (IDT) encoding Crimson fluorescent protein including restriction endonuclease sites Eco53KI, PspOMI, and Apa1 that disrupt the Crimson open-reading frame and generate blunt and overhanged ends as described in Fig 4B. The Crimson fragment included NheI and BamHI sites for subcloning this fragment between the promoter and GFP open reading frame in the substrate plasmids. Re-joining of this substrate was measured by transfecting restricted substrate; any plasmid rejoining results in expression of the downstream GFP protein; perfect rejoining will restore crimson expression, and perfect rejoining is noted as %RFP/GFP.
The coding joint substrate was modified so that the coding flanks included the A or T sequences depicted in Fig 4E; in addition, the 23RSS was inverted to provide for inversional joining which facilitates PCR amplification of coding joints. In some assays (Fig 4C–E), transfected plasmids were isolated by alkaline lysates 72 hours after transfection. Coding joints from each transfection were amplified with the tagged primers for amplicon sequencing. PCR products were analyzed by agarose gel electrophoresis and were isolated and subjected to amplicon sequencing (Genewiz). Indel and microhomology analysis was performed using a python script generously provided by Dale Ramsden and Adam Luthman69.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-way ANOVA tests using Holk-Sidek corrections were used to compare recombination rates between cell lines. Statistical analysis was performed using Prism 8 (GraphPad). All details of statistical analysis can be found in the legends of figures 1, 4, and 6. Comparisons were made to wild-type, with the absence of displayed P values indicating no significance.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ku80 mouse monoclonal 111 | Invitrogen | MA5-12933 |
| Anti-XLF Rabbit polyclonal antibody | Invitrogen | PA5-96310 |
| Anti-DNA-PKcs monoclonal antibody 42-27 | Gift from Tim Carter | N/A |
| Anti-laminB Goat polyclonal antibody | Santa Cruz | 6217 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Calicheamicin | Thermo-Fisher | 50-202-9283 |
| Etoposide | Sigma-Aldrich | E1383 |
| Zeocin | Invitrogen | R25001 |
| Teniposide | Millipore Sigma | 28767-20-2 |
| Camptothecin | Sigma-Aldrich | C9911 |
| KU55933 | Tocris | 3544 |
| Okadaic Acid | Thermo-Fisher | J60155 |
| Polyethylenimine | Polysciences | 23966 |
| Critical commercial assays | ||
| TOPO TA cloning kit | Invitrogen | K459540 |
| Deposited data | ||
| Experimental models: Cell lines | ||
| V3 DNA-PKcs deficient CHO cell strain | gift from Martin Gellert | CVCL_K039 |
| xrs6 Ku80 deficient CHO cell strain | gift from David Roth | CVCL_4340 |
| 293T | ATCC | CRL-3216 |
| XLF-deficient 293T cells | Roy et al. 2015 | N/A |
| XLF-deficient V3 cells | This paper | N/A |
| TDP2-deficient V3 cells | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Upstream coding joint primer: CGGTGGGAGGTCTATATAAGCA | IDT | N/A |
| Downstream coding joint primer: CTACACCGTGGTGGAGCAGTA | IDT | N/A |
| TDP2 upstream gRNA top: CACCGCTGTTGTAGGCGAGTGTGGG | IDT | N/A |
| TDP2 upstream gRNA bottom: AAACCCCACACTCGCCTACAACAGC | IDT | N/A |
| TDP2 downstream gRNA top: CACCGCATGAAGTAAGTAACAGAGT | IDT | N/A |
| TDP2 downstream gRNA bottom: AAACACTCTGTTACTTACTTCATGC | IDT | N/A |
| TDP2 5’ PCR primer: GTGCTAACTGTATGGTTTCAGTTG | IDT | N/A |
| TDP2 3’ PCR primer: CCATACCTTTGATAAAAGGAACCC | IDT | N/A |
| XLF 5’ PCR primer: TCTCTCCAGCCTCAGATTCTAA | IDT | N/A |
| XLF 3’ PCR primer: CCAATTCAACCTTCCTTGCCAAC | IDT | N/A |
| Recombinant DNA | ||
| pcDNA6A | Invitrogen | |
| WT Ku80 in pcDNA6 | Douglas et al. 2005 | N/A |
| Ku80 insert 1 mutant in pcDNA6 | This paper | N/A |
| Ku80 Insert 2 mutant in pcDNA6 | This paper | N/A |
| Ku80 Δ3 Ku80 mutant in pcDNA6 | This paper | N/A |
| Ku80 Δ6 Ku80 mutant in pcDNA6 | This paper | N/A |
| Ku80 Δ10 Ku80 mutant in pcDNA6 | This paper | N/A |
| Ku80 Δ13 Ku80 mutant in pcDNA6 | This paper | N/A |
| Ku80 Δ15 Ku80 mutant in pcDNA6 | This paper | N/A |
| Ku80 ΔCTD Ku80 mutant in pcDNA6 | This paper | N/A |
| murine RAG1 Expression Plasmid in pMAL-1 | Landree et al. 1999 | N/A |
| murine RAG2 Expression Plasmid in pMAL-1 | Landree et al. 1999 | N/A |
| murine RAG2 frameshift mutant in pMAL-1 | Corneo et al. 2007 | N/A |
| 290-Crimson/ZS VDJ coding joint substrate plasmid | Meek 2020 | N/A |
| Alt-VDJ substrate | Deriano et al., 2009 | N/A |
| Fill-in substrate | This paper | N/A |
| A/T Coding joint substrate to minimize microhomology | This paper | N/A |
| pDSRED express | Takara | 632539 |
| pEF6V5HisA | Invitrogen | V96120 |
| WT XLF in pEF6 | Roy et al. 2015 | N/A |
| L115A XLF in pEF6 | Roy et al. 2015 | N/A |
| Ku Mut XLF in pEF6 | This paper | N/A |
| L297W XLF in pEF6 | This paper | N/A |
| Ku Mut + L297W XLF in pEF6 | This paper | N/A |
| L115A + Ku Mut XLF in pEF6 | This paper | N/A |
| L115A + L297W XLF in pEF6 | This paper | N/A |
| L115A + Ku Mut + L297W XLF in pEF6 | This paper | N/A |
| pCMV6 expression plasmid | Kienker et al. 2000 | N/A |
| human DNA-PKcs in pCMV6 | Kienker et al. 2000 | N/A |
| human DNA-PKcs in RMCE | Neal et al. 2014 | N/A |
| 898 DNA-PKcs in RMCE | Chaplin et al. 2021 | N/A |
| 2569 DNA-PKcs in RMCE | Chaplin et al. 2021 | N/A |
| 4xala DNA-PKcs in RMCE | Chaplin et al. 2021 | N/A |
| 898 + 4xala DNA-PKcs in RMCE | Chaplin et al. 2021 | N/A |
| 2569 + 4xala DNA-PKcs in RMCE | This paper | N/A |
| 898+2569 DNA-PKcs in RMCE | This paper | N/A |
| 4xasp DNA-PKcs in RMCE | This paper | N/A |
| 898+902asp DNA-PKcs in RMCE | This paper | N/A |
| 898+902asp + 4xasp DNA-PKcs in RMCE | This paper | N/A |
| pSpCas9(BB)-2A-Puro | Addgene | PX459 |
| Software and algorithms | ||
| Junction Analysis | Luedean et al. 2022 | https://github.com/aluthman/Ramsden-Lab/tree/main/Luedeman_et_al_2022 |
| PRISM 9 for MacOS | GraphPad | version 9.4.1 |
| Other | ||
HIGHLIGHTS.
Different DNA ends promote distinct DNA-PK dimers with separate functions
A Ku80 domain-swapped dimer promotes nucleolytic end-processing
An XLF-mediated dimer promotes fill-in end-processing
DNA-PK dimers contribute to the iterative nature of NHEJ
Acknowledgements
We are particularly indebted to both Dale Ramsden and Adam Luthman for their generous and invaluable assistance in analyzing amplicon sequencing projects for differences in junctional diversity and utilization of short sequence homologies. We also appreciate many useful discussions of experimental design and data presentation with both Mauro Modesti and Kefei Yu. Some figures were generated using BioRender.
This work was supported by the USDA National Institute of Food and Agriculture [1019208], and Public Health Service [AI048758, AI147634 to K.M.]. T.L.B. and A. K. C. thank the Wellcome Trust for an Investigator Award (200814/Z/16/Z; 2016–) for support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity Statement
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Meek K, Dang V, and Lees-Miller SP (2008). DNA-PK: the means to justify the ends? Advances in immunology 99, 33–58. 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 2.Pannunzio NR, Watanabe G, and Lieber MR (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. The Journal of biological chemistry 293, 10512–10523. 10.1074/jbc.TM117.000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang HHY, Pannunzio NR, Adachi N, and Lieber MR (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature reviews. Molecular cell biology 18, 495–506. 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y, Lu H, Schwarz K, and Lieber MR (2005). Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: the iterative processing model. Cell cycle 4, 1193–1200. [DOI] [PubMed] [Google Scholar]
- 5.Waters CA, Strande NT, Pryor JM, Strom CN, Mieczkowski P, Burkhalter MD, Oh S, Qaqish BF, Moore DT, Hendrickson EA, and Ramsden DA (2014). The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nature communications 5, 4286. 10.1038/ncomms5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham TG, Walter JC, and Loparo JJ (2016). Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Molecular cell 61, 850–858. 10.1016/j.molcel.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JL, Duboc C, Wu Q, Ochi T, Liang S, Tsutakawa SE, Lees-Miller SP, Nadal M, Tainer JA, Blundell TL, and Strick TR (2018). Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nature structural & molecular biology 25, 482–487. 10.1038/s41594-018-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, and Lieber MR (2019). The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nature communications 10, 3588. 10.1038/s41467-019-11507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stinson BM, Moreno AT, Walter JC, and Loparo JJ (2020). A Mechanism to Minimize Errors during Non-homologous End Joining. Molecular cell 77, 1080–1091 e1088. 10.1016/j.molcel.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goff NJ, Breniere M, Buehl CJ, de Melo AJ, Huskova H, Ochi T, Blundell TL, Mao W, Yu K, Modesti M, Meek K (2022). Catalytically inactive DNA ligase IV promotes DNA repair in living cells. . Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S, Lee L, Naila T, Fishbain S, Wang A, Tomkinson AE, Lees-Miller SP, and He Y (2021). Structural basis of long-range to short-range synaptic transition in NHEJ. Nature 593, 294–298. 10.1038/s41586-021-03458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reid DA, Conlin MP, Yin Y, Chang HH, Watanabe G, Lieber MR, Ramsden DA, and Rothenberg E (2017). Bridging of double-stranded breaks by the nonhomologous end-joining ligation complex is modulated by DNA end chemistry. Nucleic acids research 45, 1872–1878. 10.1093/nar/gkw1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaplin AK, Hardwick SW, Liang S, Kefala Stavridi A, Hnizda A, Cooper LR, De Oliveira TM, Chirgadze DY, and Blundell TL (2021). Dimers of DNA-PK create a stage for DNA double-strand break repair. Nature structural & molecular biology 28, 13–19. 10.1038/s41594-020-00517-x. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Xu X, Chen Y, Cheung JC, Wang H, Jiang J, de Val N, Fox T, Gellert M, and Yang W (2021). Structure of an activated DNA-PK and its implications for NHEJ. Molecular cell 81, 801–810 e803. 10.1016/j.molcel.2020.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaplin AK, Hardwick SW, Stavridi AK, Buehl CJ, Goff NJ, Ropars V, Liang S, De Oliveira TM, Chirgadze DY, Meek K, et al. (2021). Cryo-EM of NHEJ supercomplexes provides insights into DNA repair. Molecular cell 81, 3400–3409 e3403. 10.1016/j.molcel.2021.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Chen X, Li J, Wang H, Buehl CJ, Goff NJ, Meek K, Yang W, and Gellert M (2021). Autophosphorylation transforms DNA-PK from protecting to processing DNA ends. Molecular cell. 10.1016/j.molcel.2021.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gell D, and Jackson SP (1999). Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic acids research 27, 3494–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, Uematsu N, Chen DJ, and van Gent DC (2009). The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Molecular and cellular biology 29, 1134–1142. 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, and Jeggo PA (1999). The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Molecular and cellular biology 19, 3267–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Lees-Miller JP, He Y, and Lees-Miller SP (2021). Structural insights into the role of DNA-PK as a master regulator in NHEJ. Genome Instab Dis 2, 195–210. 10.1007/s42764-021-00047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemoz C, Ropars V, Frit P, Gontier A, Drevet P, Yu J, Guerois R, Pitois A, Comte A, Delteil C, et al. (2018). XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining. Nature structural & molecular biology 25, 971–980. 10.1038/s41594-018-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andres SN, Modesti M, Tsai CJ, Chu G, and Junop MS (2007). Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Molecular cell 28, 1093–1101. 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 23.Malivert L, Ropars V, Nunez M, Drevet P, Miron S, Faure G, Guerois R, Mornon JP, Revy P, Charbonnier JB, et al. (2010). Delineation of the Xrcc4-interacting region in the globular head domain of cernunnos/XLF. The Journal of biological chemistry 285, 26475–26483. 10.1074/jbc.M110.138156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy S, de Melo AJ, Xu Y, Tadi SK, Negrel A, Hendrickson E, Modesti M, and Meek K (2015). XRCC4/XLF Interaction Is Variably Required for DNA Repair and Is Not Required for Ligase IV Stimulation. Molecular and cellular biology 35, 3017–3028. 10.1128/MCB.01503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frit P, Ropars V, Modesti M, Charbonnier JB, and Calsou P (2019). Plugged into the Ku-DNA hub: The NHEJ network. Progress in biophysics and molecular biology 147, 62–76. 10.1016/j.pbiomolbio.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, and Meek K (2005). Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Molecular and cellular biology 25, 10842–10852. 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, and Meek K (2003). Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Molecular and cellular biology 23, 5836–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Pannicke U, Schwarz K, and Lieber MR (2002). Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108, 781–794. [DOI] [PubMed] [Google Scholar]
- 29.Murray V, and Martin RF (1985). Comparison of the sequence specificity of bleomycin cleavage in two slightly different DNA sequences. Nucleic acids research 13, 1467–1481. 10.1093/nar/13.5.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dedon PC, and Goldberg IH (1992). Free-radical mechanisms involved in the formation of sequence-dependent bistranded DNA lesions by the antitumor antibiotics bleomycin, neocarzinostatin, and calicheamicin. Chem Res Toxicol 5, 311–332. 10.1021/tx00027a001. [DOI] [PubMed] [Google Scholar]
- 31.Menon V, and Povirk LF (2016). End-processing nucleases and phosphodiesterases: An elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA repair 43, 57–68. 10.1016/j.dnarep.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Zhou T, Akopiants K, Mohapatra S, Lin PS, Valerie K, Ramsden DA, Lees-Miller SP, and Povirk LF (2009). Tyrosyl-DNA phosphodiesterase and the repair of 3’-phosphoglycolate-terminated DNA double-strand breaks. DNA repair 8, 901–911. 10.1016/j.dnarep.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caldecott KW (2012). Tyrosyl DNA phosphodiesterase 2, an enzyme fit for purpose. Nature structural & molecular biology 19, 1212–1213. 10.1038/nsmb.2455. [DOI] [PubMed] [Google Scholar]
- 34.Gomez-Herreros F, Romero-Granados R, Zeng Z, Alvarez-Quilon A, Quintero C, Ju L, Umans L, Vermeire L, Huylebroeck D, Caldecott KW, and Cortes-Ledesma F (2013). TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS genetics 9, e1003226. 10.1371/journal.pgen.1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, and Pommier Y (2000). Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff. Molecular and cellular biology 20, 3977–3987. 10.1128/MCB.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, Woods M, Fu B, Yang F, Chen E, et al. (2019). ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nature communications 10, 87. 10.1038/s41467-018-07729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neal JA, Dunger K, Geith K, and Meek K (2020). Deciphering the role of distinct DNA-PK phosphorylations at collapsed replication forks. DNA repair 94. ARTN 102925 10.1016/j.dnarep.2020.102925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Convery E, Shin EK, Ding Q, Wang W, Douglas P, Davis LS, Nickoloff JA, Lees-Miller SP, and Meek K (2005). Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Proceedings of the National Academy of Sciences of the United States of America 102, 1345–1350. 10.1073/pnas.0406466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woods T, Wang W, Convery E, Errami A, Zdzienicka MZ, and Meek K (2002). A single amino acid substitution in DNA-PKcs explains the novel phenotype of the CHO mutant, XR-C2. Nucleic acids research 30, 5120–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta S, and Meek K (2005). The leucine rich region of DNA-PKcs contributes to its innate DNA affinity. Nucleic acids research 33, 6972–6981. 10.1093/nar/gki990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, Dobyns WB, Ogi T, Meek K, Davies EG, and Jeggo PA (2013). PRKDC mutations in a SCID patient with profound neurological abnormalities. The Journal of clinical investigation 123, 2969–2980. 10.1172/JCI67349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin YF, Nagasawa H, Little JB, Kato TA, Shih HY, Xie XJ, Wilson PF Jr., Brogan JR, Kurimasa A, Chen DJ, et al. (2014). Differential radiosensitivity phenotypes of DNA-PKcs mutations affecting NHEJ and HRR systems following irradiation with gamma-rays or very low fluences of alpha particles. PloS one 9, e93579. 10.1371/journal.pone.0093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, et al. (2009). A DNA-PKcs mutation in a radiosensitive T-B-SCID patient inhibits Artemis activation and nonhomologous end-joining. The Journal of clinical investigation 119, 91–98. 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caldecott K, Banks G, and Jeggo P (1990). DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer research 50, 5778–5783. [PubMed] [Google Scholar]
- 45.Zagnoli-Vieira G, and Caldecott KW (2020). Untangling trapped topoisomerases with tyrosyl-DNA phosphodiesterases. DNA repair 94. ARTN 102900 10.1016/j.dnarep.2020.102900. [DOI] [PubMed] [Google Scholar]
- 46.Kawale AS, and Povirk LF (2018). Tyrosyl-DNA phosphodiesterases: rescuing the genome from the risks of relaxation. Nucleic acids research 46, 520–537. 10.1093/nar/gkx1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pommier Y, Nussenzweig A, Takeda S, and Austin C (2022). Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol 23, 407–427. 10.1038/s41580-022-00452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, et al. (2016). Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Molecular cell 64, 1010. 10.1016/j.molcel.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 49.Deshpande RA, Lee JH, Arora S, and Paull TT (2016). Nbsl Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Molecular cell 64, 593–606. 10.1016/j.molcel.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Deshpande RA, Myler LR, Soniat MM, Makharashvili N, Lee L, Lees-Miller SP, Finkelstein IJ, and Paull TT (2020). DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci Adv 6, eaay0922. 10.1126/sciadv.aay0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meek K (1990). Analysis of Junctional Diversity during Lymphocyte-B Development. Science 250, 820–823. DOI 10.1126/science.2237433. [DOI] [PubMed] [Google Scholar]
- 52.Deriano L, Stracker TH, Baker A, Petrini JH, and Roth DB (2009). Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Molecular cell 34, 13–25. 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong SY, Arnal S, Holub AJ, Weller GR, Pancake BA, et al. (2007). Rag mutations reveal robust alternative end joining. Nature 449, 483–486. 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- 54.Meek K, Douglas P, Cui X, Ding Q, and Lees-Miller SP (2007). trans Autophosphorylation at DNA-dependent protein kinase’s two major autophosphorylation site clusters facilitates end processing but not end joining. Molecular and cellular biology 27, 3881–3890. 10.1128/MCB.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cottarel J, Frit P, Bombarde O, Salles B, Negrel A, Bernard S, Jeggo PA, Lieber MR, Modesti M, and Calsou P (2013). A noncatalytic function of the ligation complex during nonhomologous end joining. The Journal of cell biology 200, 173–186. 10.1083/jcb.201203128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lees-Miller SP, and Meek K (2003). Repair of DNA double strand breaks by non-homologous end joining. Biochimie 85, 1161–1173. [DOI] [PubMed] [Google Scholar]
- 57.Meek K, Gupta S, Ramsden DA, and Lees-Miller SP (2004). The DNA-dependent protein kinase: the director at the end. Immunological reviews 200, 132–141. 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- 58.Lieber MR (2008). The mechanism of human nonhomologous DNA end joining. The Journal of biological chemistry 283, 1–5. 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 59.Baretic D, Maia de Oliveira T, Niess M, Wan P, Pollard H, Johnson CM, Truman C, McCall E, Fisher D, Williams R, and Phillips C (2019). Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR. Progress in biophysics and molecular biology 147, 4–16. 10.1016/j.pbiomolbio.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Sharif H, Li Y, Dong Y, Dong L, Wang WL, Mao Y, and Wu H (2017). Cryo-EM structure of the DNA-PK holoenzyme. Proceedings of the National Academy of Sciences of the United States of America 114, 7367–7372. 10.1073/pnas.1707386114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sibanda BL, Chirgadze DY, Ascher DB, and Blundell TL (2017). DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355, 520–+. 10.1126/science.aak9654. [DOI] [PubMed] [Google Scholar]
- 62.Yin XT, Liu MJ, Tian Y, Wang JW, and Xu YH (2017). Cryo-EM structure of human DNA-PK holoenzyme. Cell Res 27, 1341–1350. 10.1038/cr.2017.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu H, Shimazaki N, Raval P, Gu J, Watanabe G, Schwarz K, Swanson PC, and Lieber MR (2008). A biochemically defined system for coding joint formation in V(D)J recombination. Mol Cell 31, 485–497. 10.1016/j.molcel.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meek K (2020). Activation of DNA-PK by hairpinned DNA ends reveals a stepwise mechanism of kinase activation. Nucleic acids research 48, 9098–9108. 10.1093/nar/gkaa614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, and Sleckman BP (2011). H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature 469, 245–249. 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, DeVido J, Foy D, Chaudhuri J, Lombard D, and Alt FW (2002). Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Molecular cell 10, 1379–1390. [DOI] [PubMed] [Google Scholar]
- 67.Roth DB, Menetski JP, Nakajima PB, Bosma MJ, and Gellert M (1992). V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell 70, 983–991. [DOI] [PubMed] [Google Scholar]
- 68.Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA, and Lees-Miller SP (2006). DNA-PK autophosphorylation facilitates Artemis endonuclease activity. The EMBO journal 25, 3880–3889. 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luedeman ME, Stroik S, Feng W, Luthman AJ, Gupta GP, and Ramsden DA (2022). Poly(ADP) ribose polymerase promotes DNA polymerase theta-mediated end joining by activation of end resection. Nature communications 13, 4547. 10.1038/s41467-022-32166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kienker LJ, Shin EK, and Meek K (2000). Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic acids research 28, 2752–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Douglas P, Gupta S, Morrice N, Meek K, and Lees-Miller SP (2005). DNA-PK-dependent phosphorylation of Ku70/80 is not required for non-homologous end joining. DNA repair 4, 1006–1018. 10.1016/j.dnarep.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 72.Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, and Meek K (2011). Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Molecular and cellular biology 31, 1719–1733. 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Landree MA, Wibbenmeyer JA, and Roth DB (1999). Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes & development 13, 3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neal JA, Xu Y, Abe M, Hendrickson E, and Meek K (2016). Restoration of ATM Expression in DNA-PKcs-Deficient Cells Inhibits Signal End Joining. Journal of immunology 196, 3032–3042. 10.4049/jimmunol.1501654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data (FASTQ files for amplicon sequencing) will be made available upon request.
Analysis of episomal-substrate sequencing for indel and microhomology sizes was performed using a published python script, Junction Analysis (https://github.com/aluthman/Ramsden-Lab/tree/main/Luedeman_et_al_2022)69.
Any additional information required to reanalyze the data reported here will be available from the lead contact upon request.