

Key Points
Question
Does olamkicept, a selective inhibitor of the soluble interleukin 6 (sIL-6R)/IL-6 complex, increase the likelihood of clinical response in patients with active ulcerative colitis?
Findings
In this randomized clinical trial that included 91 patients with active ulcerative colitis and an inadequate response to conventional therapy, biweekly intravenous infusion with olamkicept 600 mg, olamkicept 300 mg, and placebo resulted in clinical response rates of 58.6%, 43.3%, and 34.5%, respectively, at 12 weeks. Only the difference between 600 mg and placebo was statistically significant.
Meaning
Intravenous olamkicept 600 mg biweekly, compared with placebo, increased the likelihood of clinical response at 12 weeks in patients with active ulcerative colitis, but further research is needed for replication and to assess longer-term efficacy and safety.
Abstract
Importance
Olamkicept, a soluble gp130-Fc-fusion-protein, selectively inhibits interleukin 6 (IL-6) trans-signaling by binding the soluble IL-6 receptor/IL-6 complex. It has anti-inflammatory activities in inflammatory murine models without immune suppression.
Objective
To assess the effect of olamkicept as induction therapy in patients with active ulcerative colitis.
Design, Setting, and Participants
Randomized, double-blind, placebo-controlled phase 2 trial of olamkicept in 91 adults with active ulcerative colitis (full Mayo score ≥5, rectal bleeding score ≥1, endoscopy score ≥2) and an inadequate response to conventional therapy. The study was conducted at 22 clinical study sites in East Asia. Patients were recruited beginning in February 2018. Final follow-up occurred in December 2020.
Interventions
Eligible patients were randomized 1:1:1 to receive a biweekly intravenous infusion of olamkicept 600 mg (n = 30) or 300 mg (n = 31) or placebo (n = 30) for 12 weeks.
Main Outcomes and Measures
The primary end point was clinical response at week 12 (defined as ≥3 and ≥30% decrease from baseline total Mayo score; range, 0-12 [worst] with ≥1 decrease and ≤1 in rectal bleeding [range, 0-3 {worst}]). There were 25 secondary efficacy outcomes, including clinical remission and mucosal healing at week 12.
Results
Ninety-one patients (mean age, 41 years; 25 women [27.5%]) were randomized; 79 (86.8%) completed the trial. At week 12, more patients receiving olamkicept 600 mg (17/29 [58.6%]) or 300 mg (13/30 [43.3%]) achieved clinical response than placebo (10/29 [34.5%]), with adjusted difference vs placebo of 26.6% (90% CI, 6.2% to 47.1%; P = .03) for 600 mg and 8.3% (90% CI, −12.6% to 29.1%; P = .52) for 300 mg. Among patients randomized to receive 600 mg olamkicept, 16 of 25 secondary outcomes were statistically significant compared with placebo. Among patients randomized to receive 300 mg, 6 of 25 secondary outcomes were statistically significant compared with placebo. Treatment-related adverse events occurred in 53.3% (16/30) of patients receiving 600 mg olamkicept, 58.1% (18/31) receiving 300 mg olamkicept, and 50% (15/30) receiving placebo. The most common drug-related adverse events were bilirubin presence in the urine, hyperuricemia, and increased aspartate aminotransferase levels, and all were more common in the olamkicept groups compared with placebo.
Conclusions and Relevance
Among patients with active ulcerative colitis, biweekly infusion of olamkicept 600 mg, but not 300 mg, resulted in a greater likelihood of clinical response at 12 weeks compared with placebo. Further research is needed for replication and to assess longer-term efficacy and safety.
Trial Registration
ClinicalTrials.gov Identifier: NCT03235752
This randomized clinical trial compares clinical response at 12 weeks with olamkicept vs placebo in treating adult patients with active ulcerative colitis.
Introduction
Ulcerative colitis, one of the 2 major forms of inflammatory bowel disease, had an annual incidence of 8.8 to 23.1 per 100 000 person-years in North America and 0.97 to 57.9 per 100 000 person-years in Europe between 1990 and 2016.1 The incidence of ulcerative colitis is increasing in countries such as China and Korea.2,3 In China, it is anticipated that by 2035, there will be more than 2 million patients with ulcerative colitis, but few highly-effective therapies are available.4,5,6 Ulcerative colitis is characterized by inflammatory pathophysiology and mucosal immune dysregulation, involving a dynamic, chronic activation of immune cells associated with increases in proinflammatory cytokines and intestinal mucosal injury. Many anticytokine therapies like anti-TNF therapy have significant adverse effects.
Interleukin 6 (IL-6), a pleiotropic proinflammatory cytokine, is a key mediator in chronic inflammation. Classic IL6 signaling is conveyed through the IL-6/IL-6R complex and 2 molecules of the signal transducer gp130.7 Soluble IL-6R (sIL-6R), which originates from proteolytic cleavage of membrane-bound IL-6R, can form IL-6/sIL-6R complexes that convey IL-6 trans-signaling.7 Sustained activation of IL-6 signaling triggers sIL-6R release, giving rise to IL-6 trans-signaling,8,9,10,11 which is primarily responsible for chronic inflammation.
Olamkicept, a first-in-class, selective inhibitor of the sIL-6R/IL-6 complex, is a dimer formed by fusing 2 complete extracellular domains of gp130 to human IgG1 Fc12,13 that inhibits IL-6 trans-signaling by binding to and neutralizing the sIL-6R/IL-6 complexes but does not block classic IL-6 signaling. A phase 2a open-label trial (FUTURE) over 12 weeks in 16 patients with active Crohn disease or ulcerative colitis demonstrated distinct changes in mucosal pSTAT3 levels in biopsies from serial colonoscopies and was not associated with significant safety concern.7
This phase 2 clinical trial was conducted to further investigate the effect of olamkicept in active ulcerative colitis.
Methods
Study Design
This was an international, multicenter, randomized, double-blind, placebo-controlled phase 2b trial conducted between February 2018 and December 2020 at 22 clinical sites across mainland China, Taiwan, and South Korea. The trial protocol is reported in Supplement 1. All patients provided written informed consent. The trial was conducted in accordance with the Declaration of Helsinki14 and Good Clinical Practice standards, as described in the International Conference on Harmonization Guideline E6 (R2) and approved by study centers’ institutional review boards or ethics committees. A safety review committee reviewed the accumulating data regarding adverse events and provided recommendations to the sponsor on whether to continue, modify, or terminate the study.
Participants
The trial enrolled patients aged 18 to 70 years with confirmed ulcerative colitis for at least 3 months, who had active disease (total Mayo score ≥5, rectal bleeding score ≥1, and endoscopy score ≥2). The total Mayo score consists of 4 subscores: stool frequency, rectal bleeding, physician's global assessment, and endoscopic appearance. The full range for total Mayo score is 0 to 12, and each subscore ranges from 0 to 3, with a higher score indicating more severe disease. To be eligible, patients must have not responded to prior conventional nonbiological therapy and either had not received any biologic therapies or more than 8 weeks or 5 half-lives had elapsed since the last dose, whichever was longer. Conventional therapies, if still administered, were required at stable doses, with corticosteroids (≤20 mg/d prednisone or equivalent) for 2 or more weeks before randomization, 5-aminosalicylates-containing drugs (≥2 g/d 5-aminosalicylates) for at least 3 months and stable doses for 4 or more weeks before randomization, azathioprine (≥0.75 mg/kg/d) or 6-mercaptopurine (≥0.5 mg/kg/d) given for at least 6 months and stable doses for 6 or more weeks before randomization, and/or methotrexate at greater than or equal to 12.5 mg/week and stable doses for at least 12 weeks before randomization. Eligibility criteria are detailed in the full protocol (Supplement 1).
Randomization
Eligible patients were randomized 1:1:1 to receive olamkicept 600 mg or 300 mg or placebo. Central randomization was through a validated interactive web response system (Balance System) and stratified by current corticosteroid treatment (yes or no) and prior biologic treatment (yes or no). Treatment allocation was concealed so that treatment assignment was blinded from investigators, participants, and sponsors until the study was fully completed and the database was locked.
Procedures
This study included a run-in period consisting of stable conventional treatments lasting up to 6 months’ duration (if stable conventional treatment was needed), a 4-week screening period for participants eligibility, a 12-week double-blind treatment period, and a 3-week follow-up for adverse events (to day 105) as described in eFigure 1 in Supplement 2. Participants received placebo or olamkicept 600 or 300 mg by intravenous infusion at days 0, 14, 28, 42, 56, and 70. Participants were required to continue concomitant treatment with stable doses of corticosteroids, oral immunosuppressants, or 5-aminosalicylates during active treatment. Disease activity was assessed during screening, at baseline, and biweekly from week 2 to week 12 for partial Mayo score or for total Mayo score at weeks 0 and 12. All endoscopies were centrally read (1 reader at BioClinica Inc) with adjudication if results differed from the local reading.
Stool and blood samples were collected for laboratory testing, including assays for erythrocyte sedimentation rate and C-reactive protein. Other study procedures are detailed in the trial protocol in Supplement 1.
End Points
The primary efficacy end point was clinical response at week 12 using the total Mayo score and defined as a decrease of 3 or greater and of 30% or greater from baseline in total Mayo score, including a decrease of at least 1 from baseline in rectal bleeding subscore or up to 1 in rectal bleeding subscore15 with a primary comparison of olamkicept 600 mg or 300 mg vs placebo.7,13 The primary safety outcome was the frequency of adverse events and serious adverse events.
Secondary efficacy end points included clinical remission per total Mayo score (defined as a total Mayo score ≤2, no individual subscore >1, and rectal bleeding subscore = 0); remission per modified Mayo score (defined as stool frequency subscore ≤1, rectal bleeding subscore = 0, and endoscopy subscore = 0 or 1) at week 12; mucosal healing (defined as endoscopic subscore = 0 or 1); change from baseline in total Mayo score and modified Mayo score at week 12; clinical response and remission defined per 9-point partial Mayo score at weeks 4, 6, 8, 10, and 12, changes from baseline in partial Mayo scores; and Physician’s Global Assessment at weeks 4, 6, 8,10 and 12.
Immunogenicity (anti-drug antibodies,such as Anti-TJ301 antibodies), pharmacokinetics (eAppendix 3 in Supplement 2), and biomarkers (erythrocyte sedimentation rate, C-reactive protein, IL-6, s-IL6R, IL-6/sIL-6R complex, neutrophil count, platelet count and fecal calprotectin) were secondary end points.
There were 4 versions of the protocol, and the study outcomes changed between the February 2017 version and the January 2020 version of the protocol. Investigators did not review any outcome data before changing the outcome measures (Supplement 1).
Sample Size Calculation
A sample size of 27 patients per group was estimated to provide at least 70% power to detect a 30% increase in the 12-week clinical response rate between the placebo (assumed to be 30%)16 and the treatment (assumed to be 60%)7 group, with a 1-sided type I error rate of 0.05. This 30% increase in response was considered clinically meaningful given that a difference of greater than 20% has been considered as the minimal detectable difference.16,17,18 Assuming a dropout rate of 10%, 30 patients were required per group.
Statistical Analysis
The full analysis set included all randomized patients with at least 1 postbaseline 9-point partial Mayo score and was the primary analysis set for efficacy (Supplement 3). The safety set included all patients who had received at least 1 dose of the study medication and had been evaluated for adverse events and laboratory abnormalities. All patients who discontinued treatment during the 12-week treatment period advanced to the trial end assessment visit, including endoscopy if consent was not withdrawn. Participants with missing data for a dichotomized end point, such as clinical response, clinical remission, and mucosal healing were classified as a nonresponder. Missing continuous end points, such as Mayo score and Physician’s Global Assessment change from baseline, were not imputed.
The primary end point was tested at the 1-sided .05 (2-sided .1) significance level based on the P value from the logistic regression model. The 90% CI for treatment comparison, which corresponded to the 1-sided .05 significance level, was provided. The statistical significance level and 90% CI were chosen because of the exploratory nature of this proof-of-concept phase 2 trial, designed to identify preliminary signals of efficacy to inform a decision about proceeding to a larger clinical trial. This exploratory clinical trial required a small but sufficient number of participants to infer whether there was an efficacy signal from the drug. Treatment comparison on secondary end points was not adjusted for multiplicity, and nominal P values were provided. Because of the potential for type I error due to multiple comparisons, findings for analyses of secondary end points should be interpreted as exploratory.
For dichotomized end points, point estimate, and 90% CI (to correspond with the 1-sided 0.05 significance level) were presented for each treatment group using the Clopper-Pearson method. This was a phase 2 trial designed to assess whether an efficacy signal existed. Therefore, a 90% CI was selected. The point estimate, 90% CI, and P value for comparisons of treatment with placebo were calculated using a logistic regression model with treatment group, randomization stratification factors, and baseline total Mayo score as covariates. Treatment comparison was further analyzed using the Cochran-Mantel-Haenszel test adjusted by stratification factors for sensitivity.
Continuous end points were analyzed using a mixed-effects model for repeated measures, with changes from baseline as the dependent variable, and baseline score, randomization stratification factor, treatment, visit, and treatment by visit interaction as fixed effects.
Prespecified subgroup analyses by time since initial diagnosis of ulcerative colitis (<7 or ≥7 years), baseline total Mayo score (≤8 or >8), corticosteroid treatment (yes or no), and prior biologic treatment (yes or no) were performed for efficacy end points. Post-hoc analyses were performed for subgroups that included age, sex, baseline partial Mayo score (≤7 or > 7) and baseline central endoscopy score (2 or 3).
All statistical analyses were performed with SAS version 9.4 (SAS Institute, Inc).
Results
Participants
Between February 06, 2018, and December 16, 2020, there were 228 patients screened, and 91 eligible patients were randomly assigned to olamkicept 600 mg (n = 30), olamkicept 300 mg (n = 31), or placebo (n = 30) (Figure 1). The dropout rate at 12-week follow-up was 10% (n = 3, olamkicept 600 mg), 6.5% (n = 2, olamkicept 300 mg) and 23.3% (n = 7, placebo). All participants were included in the safety analysis set, and the number of participants analyzed in the full analysis set was 29 for olamkicept 600 mg, 30 for olamkicept 300 mg, and 29 for placebo (among those in each group, 4, 3, and 8 had missing total Mayo score components at week 12 with 2, 2, and 6 missing partial Mayo score). At weeks 2, 4, 6, 8, and 10 where only partial Mayo score components measured, number of missing were 0, 2, 2, 2, and 2 in the olamkicept 600 mg group, 0, 1, 1, 1, and 1 in the olamkicept 300 mg group, and 0, 2, 5, 7, and 6 in the placebo group. The 3 groups were generally comparable in demographic and baseline characteristics (Table 1).
Figure 1. Flow of Participants in the Trial of Olamkicept for Active Ulcerative Colitis.

The study had a run-in period (if stable conventional treatment was needed), a 4-week screening period, a 12-week double-blind treatment period, and a 3-week safety follow-up period (to day 105). The first patient signed the informed consent form on February 6, 2018, and the last patient completed the trial on December 16, 2020. During the double-blind treatment period, patients with active ulcerative colitis were randomized in a 1:1:1 ratio to receive olamkicept every 2 weeks at doses of 600 mg or 300 mg or placebo by intravenous infusion at days 0 (baseline), 14, 28, 42, 56, and 70. Disease activity was assessed at screening visit, baseline, and weeks 2 to 12. During screening and at week 12, assessments of disease activity included endoscopy (colonoscopy or sigmoidoscopy).
Table 1. Demographic and Baseline Characteristics.
| Variables | No. (%)a | ||
|---|---|---|---|
| Olamkiceptb | Placebo (n = 30) | ||
| 600 mg (n = 30) | 300 mg (n = 31) | ||
| Asian racec | 30 (100) | 31 (100) | 30 (100) |
| Female sex | 8 (27) | 12 (39) | 5 (17) |
| Male sex | 22 (73) | 19 (61) | 25 (83) |
| Age, mean (SD), y | 41.8 (13.8) | 40.5 (14.0) | 42.3 (12.6) |
| Weight, mean (SD), kg | 62.6 (11.4) | 63.6 (12.2) | 62.4 (10.1) |
| Duration of ulcerative colitis, median (IQR), y | 3.2 (1.4-7.4) | 4.9 (3.2-9.7) | 4.6 (2.7-7.5) |
| Mayo score, mean (SD)d | |||
| Total | 8.6 (1.4) | 8.4 (1.9) | 8.7 (1.7) |
| Partial | 6.0 (1.3) | 5.9 (1.7) | 6.2 (1.6) |
| Centrally reviewed endoscopy score | |||
| 1, Mild disease | 0 | 1 (3) | 0 |
| 2, Moderate disease | 13 (43) | 14 (45) | 15 (50) |
| 3, Severe disease | 17 (57) | 16 (52) | 15 (50) |
| IL-6, pg/mL | |||
| No. | 15 | 20 | 12 |
| Median (IQR) | 6.9 (5.0-16.8) | 6.8 (3.9-9.3) | 8.1 (4.2-11.9) |
| IL-6/IL-6 Ra complex, pg/mL | |||
| No. | 2 | 8 | 3 |
| Median (IQR) | 364.6 (347.3-382.0) | 283.7 (193.8-395.4) | 228.8 (209.5-242.5) |
| C-reactive protein, mg/L | |||
| No. | 28 | 29 | 25 |
| Median (IQR) | 3.1 (1.4-9.4) | 5.1 (2.1-14.2) | 3.2 (2.0-9.0) |
| Fecal calprotectin, mg/kg | |||
| No. | 27 | 31 | 29 |
| Median (IQR) | 3412 (1092-5169) | 3289 (1022-4690) | 3096 (728-5289) |
| Prior medications for ulcerative colitis | |||
| 5-Aminosalicylate | 28 (93) | 30 (97) | 29 (97) |
| Corticosteroids | 3 (10) | 3 (10) | 6 (20) |
| Azathioprine | 4 (13) | 1 (3) | 2 (7) |
| Biologics | 2 (7) | 2 (6) | 1 (3) |
| Methotrexate | 1 (3) | 1 (3) | 0 |
Abbreviation: sIL-6R, soluble interleukin 6 receptor.
Numeric values are reported as No. (%) unless otherwise indicated.
Administered intravenously every 2 weeks.
All patients self-reported as Asian, including 83 from 19 sites in China, 7 from 2 sites in Taiwan, and 1 from a single site in South Korea.
Total Mayo score consists of 4 subscores: stool frequency, rectal bleeding, physician's global assessment, and endoscopic appearance. The overall range for total Mayo score is 0 to 12, and each subscore ranges from 0 to 3. Partial Mayo score (total Mayo scale score excluding the endoscopic subscore) ranges from 0 to 9.
Primary End Point
In the full analysis set, significantly more patients receiving olamkicept 600 mg attained clinical response at week 12 than patients receiving placebo (58.6% [17/29] vs 34.5% [10/29]), with an adjusted difference of 26.6% (90% CI, 6.2% to 47.1%; P = .03). Thirteen patients (43.3% [13/30]) receiving olamkicept 300 mg achieved clinical response at week 12, with no significant difference from placebo (adjusted difference, 8.3% [90% CI, −12.6% to 29.1%]; P = .52) (Figure 2).
Figure 2. Primary End Point and Selected Secondary End Points in the Trial of Olamkicept for Active Ulcerative Colitis.
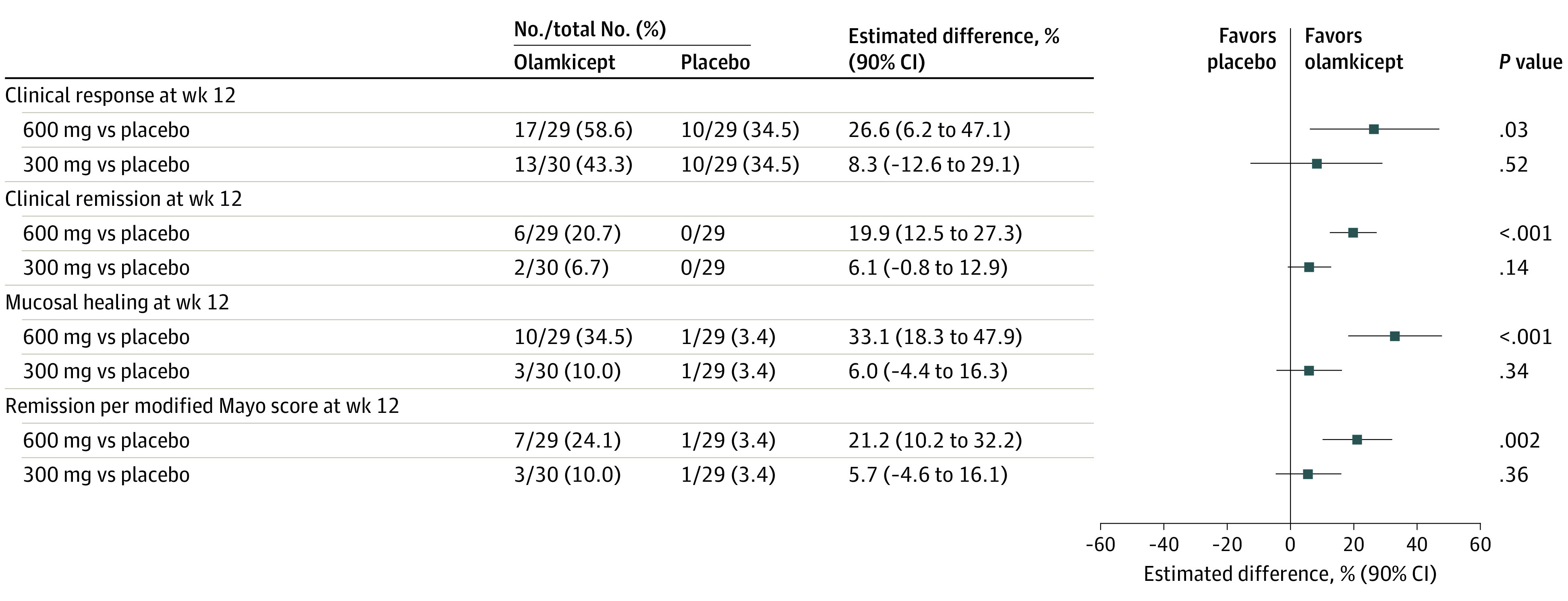
Clinical response at week 12 was defined as a decrease of 3 or greater and of 30% or greater from baseline in total Mayo score, including a decrease of 1 or greater from baseline in rectal bleeding subscore or of 1 or less1 in rectal bleeding subscore. Clinical remission at week 12 was defined as a total Mayo score of 2 or less, no individual subscore greater than 1, and a rectal bleeding subscore of 0. Mucosal healing at week 12 was defined as a Mayo endoscopic subscore of 0 or 1. Remission per modified Mayo score (ie, total Mayo score excluding Physician’s Global Assessment subscore) at week 12 was defined as a stool frequency subscore of 1 or less, a rectal bleeding subscore of 0, and an endoscopy subscore of 0 or 1. The 90% CI and P value for treatment difference were derived from a logistic regression model adjusted for treatment group, randomization stratification factors, and total Mayo score at baseline as covariates. The numbers of patients were based on the full analysis set, consisting of all randomized patients with at least 1 postbaseline 9-point partial Mayo score value, and patients with missing outcomes were imputed as nonresponders (4 patients in the olamkicept 600-mg group, 3 in the 300-mg group, and 8 patients in the placebo group).
Secondary End Points
Clinical remission based on the total Mayo score at week 12 occurred in 20.7% (6/29) of patients receiving olamkicept 600 mg and in 6.7% (2/30) of patients receiving olamkicept 300 mg but in no patients receiving placebo (Figure 2), with a significant adjusted difference between olamkicept 600 mg and placebo (19.9% [90% CI, 12.5% to 27.3%]; P < .001) and a nonstatistically significant adjusted difference between 300 mg and placebo (6.1% [90% CI, −0.8% to 12.9%]; P = .14). Significantly more patients receiving olamkicept 600 mg (34.5% [10/29]) achieved mucosal healing at week 12 than patients receiving placebo (3.4% [1/29]), with an adjusted difference of 33.1% (90% CI, 18.3% to 47.9%; P < .001) (Figure 2). Additionally, 10% [3/30] patients receiving olamkicept 300 mg achieved mucosal healing at week 12 (adjusted difference vs placebo, 6.0% [90% CI, −4.4% to 16.3%]; P = .34). Similar findings were noted in remission per modified Mayo score at week 12 (Figure 2).
Compared with placebo, a significantly greater reduction of total Mayo score occurred in the olamkicept 600-mg group between baseline and 12-week follow-up (least square mean difference between olamkicept 600 mg and placebo, −1.6 [90% CI, −2.9 to −0.4]; P = .03) (Figure 3A; eTable 1 in Supplement 2). The olamkicept 600-mg group was associated with a significant reduction in partial Mayo score compared with placebo at week 8 (least square mean difference, −1.0 [90% CI, −1.9 to 0.1]; P = .08), week 10 (least square mean difference, −1.2 [90% CI, −2.0 to −0.3]; P = .02), and week 12 (least square mean difference, −1.2 [90% CI, −2.1 to −0.2]; P = .04). The olamkicept 300-mg group had a greater reduction in partial Mayo score at each visit than the placebo group, and the difference was statistically significant at week 8 (least square mean difference, −1.3 [90% CI, −2.2 to −0.4]; P = .02) and at week 10 (least square mean difference, −1.1 [90% CI, −1.9 to −0.2]; P = .04) (Figure 3B; eTable 1 in Supplement 2). The modified Mayo score in the olamkicept 600-mg group decreased from baseline at week 12 significantly more than the placebo group, (least square mean difference between olamkicept 600 mg and placebo, −1.4 [90% CI, −2.4 to −0.4]; P = .02) (Figure 3C; eTable 1 in Supplement 2).
Figure 3. Efficacy Over Time in the Trial in the Randomized Clinical Trial of Olamkicept, Compared With Placebo, for Active Ulcerative Colitis.
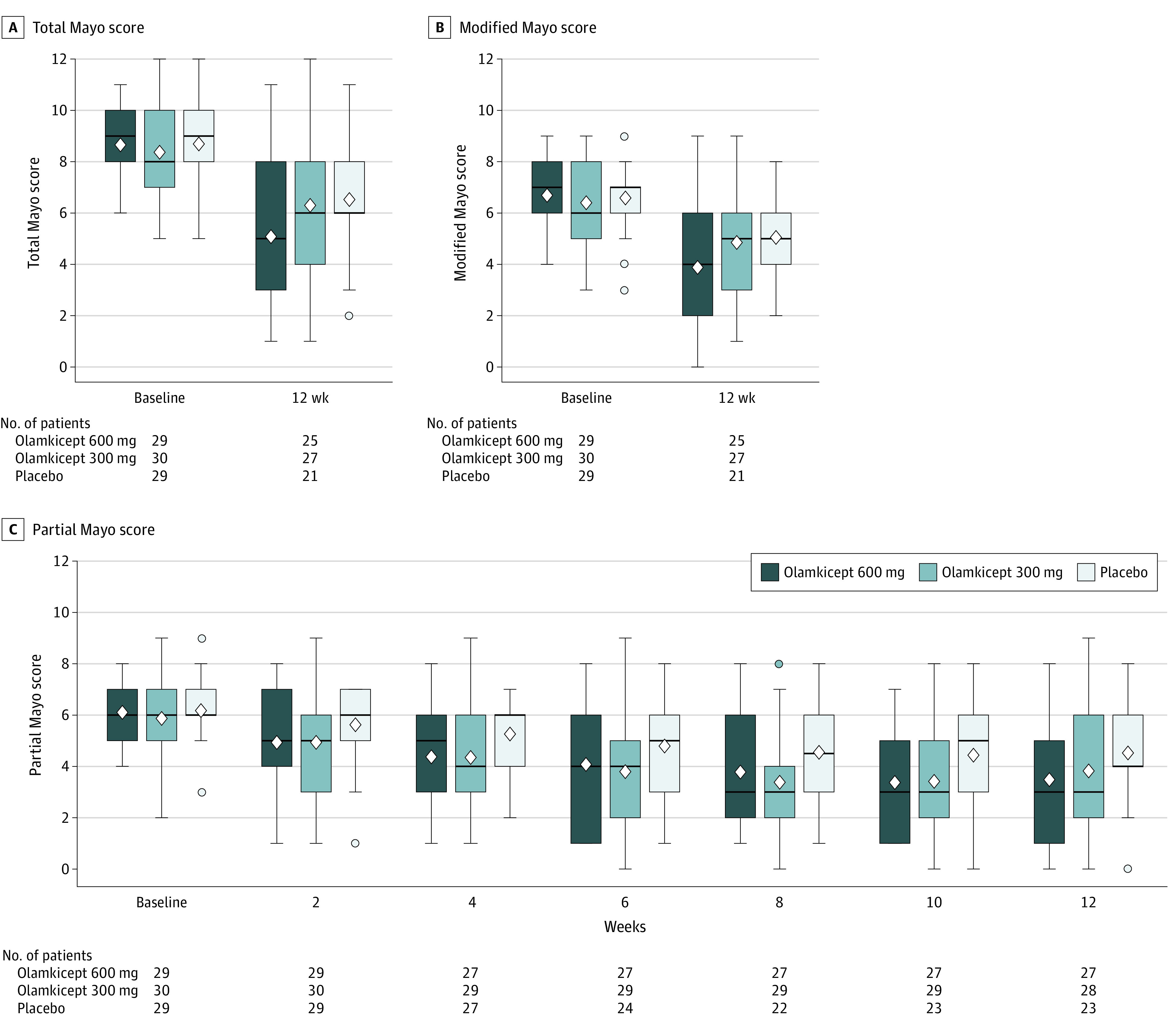
The boxplots of Mayo scores per treatment group present the median (the horizontal line in the box), mean (the diamond in the box), and IQR (25th to 75th percentiles), with whisker length equal to 1.5 times the IQR and dots indicating outliers. Summaries were based on the full analysis set, and patients with missing outcomes were not imputed. Amount of missing at each visit in each treatment group can be quantified via the No. of patients.
In the 600-mg olamkicept group, 16 of 25 secondary outcomes (specifically 8 of 9 outcomes at week 12 and 8 of 16 outcomes at weeks 4, 6, 8, and 10) were statistically significant, compared with placebo. For the 300-mg olamkicept group, 6 of 25 secondary outcomes (1 of 9 outcomes at week 12 and 5 of 16 at weeks 4, 6, 8, and 10) were statistically significant compared with placebo. (eTable 1 in Supplement 2).
Biomarkers
Fecal calprotectin significantly declined between baseline and week 12 in patients receiving olamkicept 300 mg or 600 mg, but it increased in patients receiving placebo (eTable 2 and eFigure 3A in Supplement 2). Compared with placebo, there were no significant differences in mean changes from baseline in levels of neutrophil count, platelet count, erythrocyte sedimentation rate, C-reactive protein, IL-6, sIL-6Ra, and sIL-6R/IL-6 complex at 12-week follow-up between patients receiving olamkicept 300 mg or 600 mg and those receiving placebo (eTable 2, eFigures 3B, 3C, 3D, 3E, 3F, and eFigures 4E and 4F in Supplement 2).
Subgroup Analyses
Generally consistent treatment effects were observed across subgroups, with the overall analyses for the primary end point between patients receiving olamkicept 600 mg and those receiving placebo (eFigure 2 in Supplement 2).
Adverse Events
80.0% of patients in the olamkicept 600-mg group, 87.1% in the olamkicept 300-mg group, and 66.7% in the placebo group received all 6 doses. The median drug exposure duration was 71 days in all 3 groups. One patient receiving olamkicept 600 mg and 1 patient receiving placebo withdrew due to adverse events, and treatment was temporarily discontinued due to adverse events in 1 patient receiving olamkicept 600 mg.
Adverse events occurred in 83.3% (25/30) of patients in the olamkicept 600-mg group, 93.5% (29/31) in the olamkicept 300-mg group, and 90% (27/30) in the placebo group, and serious adverse events in occurred in 3.3% (1/30) of patients in the olamkicept 600-mg group, 3.2% (1/31) in the olamkicept 300-mg group, and 6.7% (2/30) in the placebo group (Table 2). All serious adverse events resolved after treatment. Treatment-related adverse events occurred in 53.3% (16/30) of patients in the olamkicept 600-mg group, 58.1% (18/31) in the olamkicept 300-mg group, and 50% (15/30) in the placebo group. No patients died.
Table 2. Safety Outcomes.
| Variables | No. (%) | ||
|---|---|---|---|
| Olamkicepta | Placebo (n = 30) | ||
| 600 mg (n = 30) | 300 mg (n = 31) | ||
| No. of exposure days, mean (SD) | 64.8 (17.6) | 67.4 (13.7) | 61.9 (19.9) |
| Serious AEs | 1 (3) | 1 (3) | 2 (7) |
| Treatment-emergent serious AEs | 1 (3) | 1 (3) | 1 (3) |
| AEs | 25 (83) | 29 (94) | 27 (90) |
| Treatment-emergent AEs | 25 (83) | 29 (94) | 25 (83) |
| Drug-related treatment-emergent AEs | 16 (53) | 18 (58) | 15 (50) |
| Drug-related infections | 2 (7) | 3 (10) | 2 (7) |
| Most-frequent drug-related AEs (≥5% in any treatment group) | |||
| Bilirubin urine present | 2 (7) | 5 (16) | 3 (10) |
| Blood urine present | 2 (7) | 4 (13) | 3 (10) |
| Monocyte count increased | 1 (3) | 3 (10) | 3 (10) |
| Hyperuricemia | 2 (7) | 3 (10) | 1 (3) |
| Protein urine present | 2 (7) | 2 (6) | 2 (7) |
| Aspartate aminotransferase increasedb | 1 (3) | 3 (10) | 0 |
| Lymphocyte count decreased | 1 (3) | 2 (6) | 1 (3) |
| Red blood cell urine positive | 1 (3) | 2 (6) | 1 (3) |
| Blood bilirubin increased | 2 (7) | 0 | 1 (3) |
| Hypertriglyceridemia | 1 (3) | 2 (6) | 0 |
| Pyrexia | 1 (3) | 2 (6) | 0 |
| Anemia | 0 | 2 (6) | 1 (3) |
| White blood cells | |||
| Count increased | 0 | 2 (6) | 1 (3) |
| Urine positive | 0 | 2 (6) | 1 (3) |
| Alanine aminotransferase increased | 0 | 2 (6)c | 0 |
| Blood cholesterol increased | 0 | 2 (6)c | 0 |
| Blood triglycerides increased | 0 | 2 (6)c | 0 |
| C-reactive protein increased | 0 | 2 (6)c | 0 |
| Globulin increased | 0 | 2 (6)c | 0 |
| Interferon-gamma release assay positive | 0 | 2 (6)c | 0 |
| Low-density lipoprotein increased | 0 | 2 (6)c | 0 |
| AEs leading to study withdrawal | 1 (3) | 0 | 1 (3) |
| AEs leading to study treatment interruption | 0 | 1(3) | 0 |
| AEs of special interest | 2 (7) | 5 (16) | 0 |
| Interferon-gamma release assay positive | 1 (3) | 4 (13) | 0 |
| Aspartate aminotransferase increasedb | 0 | 1 (3) | 0 |
| Hypersensitivity reaction | 1 (3) | 0 | 0 |
Abbreviation: AEs, adverse events.
Administered intravenously every 2 weeks.
Listed as a subcategory beneath most-frequent drug-related AE and beneath AEs of special interest because all liver enzyme increases are not considered to be AEs of special interest. Only moderate or severe ones are considered to be AEs of special interest.
Although small in number, the AEs occurred across many participants. Numeric values do not indicate the same 1 or 2 people with multiple laboratory abnormalities.
The most common (≥5%) drug-related adverse events in the treatment group with a higher incidence rate than the placebo group were bilirubin presence in the urine (6.7% in patients receiving 600 mg olamkicept, 16.1% in patients receiving 300 mg olamkicept, and 10.0% in patients receiving placebo), hyperuricemia (6.7% in patients receiving 600 mg olamkicept, 9.7% in patients receiving 300 mg olamkicept, and 3.3% in patients receiving placebo), and increased serum aspartate aminotransferase (3.3% in patients receiving 600 mg olamkicept, 9.7% in patients receiving 300 mg olamkicept, and 0 in patients receiving placebo). No significant differences were observed in changes in platelet count and neutrophil count at weeks 4 to 12 from baseline between patients receiving olamkicept 600 mg and those receiving placebo (eFigure 4E and 4F in Supplement 2). A total of 7 patients who received olamkicept reported 7 adverse events of special interest, including a positive interferon-gamma release assay in 5 patients (8.2% [with 1 {3.3%} in the 600-mg olamkicept group and 4 {12.9%} in the 300-mg olamkicept group]), aspartate aminotransferase increased in 1 patient (3.2%) in the 300-mg olamkicept group, and hypersensitivity reaction in 1 patient (3.3%) in the 600-mg olamkicept group. No tuberculosis infection was diagnosed based on further evaluation of the positive interferon-gamma release assay test.
Pharmacokinetics and Anti-Drug Antibodies
Olamkicept plasma concentrations showed similar decline over time in both olamkicept groups (eFigure 5A and eTable 3 in Supplement 2), and the trough plasma concentrations remained stable (eFigure 5B in Supplement 2). Following the first dose, the exposure of olamkicept increased dose proportionally with a geometric mean of Cmax being 77.8 μg/mL with 300 mg olamkicept and 159.7 μg/mL with 600 mg olamkicept. Following multiple doses of olamkicept 600 mg (sixth dose), the geometric mean of half-life was 3.4 days, steady state apparent clearance was 0.126 L/hour, and apparent volume of distribution was 14.7 L.
One patient (3.3%) receiving olamkicept 600 mg and 5 patients (16.1%) receiving olamkicept 300 mg developed antidrug antibodies (eTable 4 in Supplement 2).
Discussion
Among patients with active ulcerative colitis, biweekly infusion of olamkicept 600 mg, but not 300 mg, resulted in a significantly higher rate of clinical response at 12 weeks compared with placebo. Olamkicept improved 8 of 9 secondary efficacy outcomes measured at week 12 (including clinical remission and mucosal healing) compared with placebo, using the 1-sided P value of .05. Patients with a baseline Mayo score greater than 8 or endoscopic score of 3 had lower response rates to olamkicept 600 mg compared with others who did not have these characteristics. Compared with placebo, there was a higher incidence of bilirubin in urine, hyperuricemia, and elevated aspartate aminotransferase in patients randomized to the olamkicept treatment group. Further study in a larger sample size is needed to evaluate the higher rates of these adverse events.
Serum olamkicept concentration increased dose-dependently, and olamkicept 600 mg reached a steady state after 4 weeks of biweekly administration, without obvious accumulation. Additionally, the rate of antidrug antibodies was low for olamkicept, as expected for fusion proteins like etanercept, and had no effect on efficacy and safety.19,20
Limitations
This study had several limitations. First, only 5.5% of participants had prior exposure to biologic therapies, suggesting that the participant population did not have severe ulcerative colitis. Second, the sample size was small. Third, the trial used 90% CIs, consistent with a 1-sided type I error rate of .05 in reporting given the exploratory nature of this phase 2 study. Fourth, the drop-out rate was 23.3% in the placebo group, a rate that was substantially higher than in the 600 mg (10.0%) and 300 mg (6.5%) olamkicept groups. Fifth, additional clinical trials are required to prove efficacy in induction and maintenance therapy and further evaluate potentially important adverse events.
Conclusions
Among patients with active ulcerative colitis, biweekly infusion of olamkicept 600 mg, but not 300 mg, resulted in a greater likelihood of clinical response at 12 weeks compared with placebo. Further research is needed for replication and to assess longer-term efficacy and safety.
eTable 1. Trial Protocol
Statistical Analysis Plan
eAppendix 1. List of Participating Institutions
eAppendix 2. Supplementary Methods
eAppendix 3. Pharmacokinetics, Pharmacodynamics, And Immunogenicity
eTable 1. Efficacy Outcomes in Full Analysis Set
eTable 2. Summary of Biomarker Changes from Baseline in Each Dose Group
eTable 3. Summary of Pharmacokinetic Parameters of Olamkicept in Serum (PKS)
eTable 4. Summary of Development of Anti-Drug Antibodies in Patients Receiving Olamkicept
eFigure 1. Study Schema
eFigure 2. Efficacy of Olamkicept 600 mg and Placebo by Subgroups
eFigure 3. Change from Baseline in Biomarkers Over Time
eFigure 4. Lipid Profiles, Platelet and Neutrophil Count Over Time
eFigure 5. Olamkicept Plasma Concentrations Over Time
Data Sharing Statement
References
- 1.Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769-2778. doi: 10.1016/S0140-6736(17)32448-0 [DOI] [PubMed] [Google Scholar]
- 2.Ng SC, Tang W, Ching JY, et al. ; Asia–Pacific Crohn’s and Colitis Epidemiologic Study (ACCESS) Study Group . Incidence and phenotype of inflammatory bowel disease based on results from the Asia-Pacific Crohn’s and Colitis Epidemiology Study. Gastroenterology. 2013;145(1):158-165.e2. doi: 10.1053/j.gastro.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 3.Prideaux L, Kamm MA, De Cruz PP, Chan FK, Ng SC. Inflammatory bowel disease in Asia: a systematic review. J Gastroenterol Hepatol. 2012;27(8):1266-1280. doi: 10.1111/j.1440-1746.2012.07150.x [DOI] [PubMed] [Google Scholar]
- 4.GBD 2017 Inflammatory Bowel Disease Collaborators . The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17-30. doi: 10.1016/S2468-1253(19)30333-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olfatifar M, Zali MR, Pourhoseingholi MA, et al. The emerging epidemic of inflammatory bowel disease in Asia and Iran by 2035: a modeling study. BMC Gastroenterol. 2021;21(1):204. doi: 10.1186/s12876-021-01745-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alsoud D, Verstockt B, Fiocchi C, Vermeire S. Breaking the therapeutic ceiling in drug development in ulcerative colitis. Lancet Gastroenterol Hepatol. 2021;6(7):589-595. doi: 10.1016/S2468-1253(21)00065-0 [DOI] [PubMed] [Google Scholar]
- 7.Schreiber S, Aden K, Bernardes JP, et al. Therapeutic interleukin-6 trans-signaling inhibition by olamkicept (sgp130fc) in patients with active inflammatory bowel disease. Gastroenterology. 2021;160(7):2354-2366.e11. doi: 10.1053/j.gastro.2021.02.062 [DOI] [PubMed] [Google Scholar]
- 8.Müllberg J, Oberthür W, Lottspeich F, et al. The soluble human IL-6 receptor: mutational characterization of the proteolytic cleavage site. J Immunol. 1994;152(10):4958-4968. doi: 10.4049/jimmunol.152.10.4958 [DOI] [PubMed] [Google Scholar]
- 9.Müllberg J, Schooltink H, Stoyan T, et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23(2):473-480. doi: 10.1002/eji.1830230226 [DOI] [PubMed] [Google Scholar]
- 10.Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin Ther Targets. 2012;16(2):225-236. doi: 10.1517/14728222.2012.660307 [DOI] [PubMed] [Google Scholar]
- 11.Kaur S, Bansal Y, Kumar R, Bansal G. A panoramic review of IL-6: structure, pathophysiological roles and inhibitors. Bioorg Med Chem. 2020;28(5):115327. doi: 10.1016/j.bmc.2020.115327 [DOI] [PubMed] [Google Scholar]
- 12.Waetzig GH, Chalaris A, Rosenstiel P, et al. N-linked glycosylation is essential for the stability but not the signaling function of the interleukin-6 signal transducer glycoprotein 130. J Biol Chem. 2010;285(3):1781-1789. doi: 10.1074/jbc.M109.075952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenhumberg S, Waetzig GH, Chalaris A, et al. Structure-guided optimization of the interleukin-6 trans-signaling antagonist sgp130. J Biol Chem. 2008;283(40):27200-27207. doi: 10.1074/jbc.M803694200 [DOI] [PubMed] [Google Scholar]
- 14.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 15.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-2476. doi: 10.1056/NEJMoa050516 [DOI] [PubMed] [Google Scholar]
- 16.Feagan BG, Rutgeerts P, Sands BE, et al. ; GEMINI 1 Study Group . Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699-710. doi: 10.1056/NEJMoa1215734 [DOI] [PubMed] [Google Scholar]
- 17.Sandborn WJ, Ghosh S, Panes J, et al. ; Study A3921063 Investigators . Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616-624. doi: 10.1056/NEJMoa1112168 [DOI] [PubMed] [Google Scholar]
- 18.Sandborn WJ, Su C, Sands BE, et al. ; OCTAVE Induction 1, OCTAVE Induction 2, and OCTAVE Sustain Investigators . Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723-1736. doi: 10.1056/NEJMoa1606910 [DOI] [PubMed] [Google Scholar]
- 19.Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6(5):583-588. doi: 10.1038/75068 [DOI] [PubMed] [Google Scholar]
- 20.McElvaney OJ, Curley GF, Rose-John S, McElvaney NG. Interleukin-6: obstacles to targeting a complex cytokine in critical illness. Lancet Respir Med. 2021;9(6):643-654. doi: 10.1016/S2213-2600(21)00103-X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Trial Protocol
Statistical Analysis Plan
eAppendix 1. List of Participating Institutions
eAppendix 2. Supplementary Methods
eAppendix 3. Pharmacokinetics, Pharmacodynamics, And Immunogenicity
eTable 1. Efficacy Outcomes in Full Analysis Set
eTable 2. Summary of Biomarker Changes from Baseline in Each Dose Group
eTable 3. Summary of Pharmacokinetic Parameters of Olamkicept in Serum (PKS)
eTable 4. Summary of Development of Anti-Drug Antibodies in Patients Receiving Olamkicept
eFigure 1. Study Schema
eFigure 2. Efficacy of Olamkicept 600 mg and Placebo by Subgroups
eFigure 3. Change from Baseline in Biomarkers Over Time
eFigure 4. Lipid Profiles, Platelet and Neutrophil Count Over Time
eFigure 5. Olamkicept Plasma Concentrations Over Time
Data Sharing Statement