

Summary
Underutilized crops are, by definition, under‐researched compared to staple crops yet come with traits that may be especially important given climate change and the need to feed a globally increasing population. These crops are often stress‐tolerant, and this combined with unique and beneficial nutritional profiles. Whilst progress is being made by generating reference genome sequences, in this Tansley Review, we show how this is only the very first step. We advocate that going ‘beyond a reference genome’ should be a priority, as it is only at this stage one can identify the specific genes and the adaptive alleles that underpin the valuable traits. We sum up how population genomic and pangenomic approaches have led to the identification of stress‐ and disease‐tolerant alleles in staple crops and compare this to the small number of examples from underutilized crops. We also demonstrate how previously underutilized crops have benefitted from genomic advances and that many breeding targets in underutilized crops are often well studied in staple crops. This cross‐crop population‐level resequencing could lead to an understanding of the genetic basis of adaptive traits in underutilized crops. This level of investment may be crucial for fully understanding the value of these crops before they are lost.
Keywords: crop improvement, food security, genomics, pangenome, population genomics, reference genome, underutilized crops
| Contents | ||
|---|---|---|
| Summary | 1583 | |
| I. | Introduction | 1583 |
| II. | Genomics of underutilized crops to‐date | 1585 |
| III. | The way forward | 1589 |
| Acknowledgements | 1592 | |
| References | 1592 |
I. Introduction
In 2020, at least 720 million people (≥ 9.9% of the world’s population) faced hunger, an increase on previous years, and the greatest percentage of the total population since 2010 (FAO et al., 2021). With ongoing climate change, the increasing global population and the COVID19 pandemic, the number of people facing hunger is expected to rise significantly. It is increasingly recognized that achieving Sustainable Development Goal (SDG) 2 (‘Zero Hunger’) by 2030 is in doubt (Global Panel on Agriculture & Food Systems for Nutrition, 2020). To overcome this challenge, one of the most favourable approaches is to breed new germplasm to face these stresses. Crop breeders have identified germplasm (either local varieties or crop wild relatives (CWRs)) with beneficial traits, and introgressed the adaptive alleles into elite varieties. Although substantial efforts and many successes have been made to breed climate‐resilient crops, yield has started to plateau because of negative effects of extreme weather events in at least some years and in some parts of the world (Zhao et al., 2017). Homogenization of diets and the increasing consumption of calorie‐rich yet nutrition‐deficient crops has also led to an increase in hidden hunger, a significant factor threatening human health (Khoury et al., 2014; Dawson et al., 2019). A greater understanding of the effect of environmental stresses on crop quality, for example nutrient content and composition, is also needed.
Staple crops are limited in their tolerance of a changing climate, forcing researchers and breeders to start to investigate new pathways to ensure future food security. Underutilized crops (sometimes known as semidomesticated, orphan and/or forgotten crops) are locally important crops grown in limited regions with low‐input conditions. They are currently impossible or inefficient to be produced at large scale due to low yield, antinutrient content, consumer preferences, harvestability or other properties. Because of the diverse nutrient and local adaptations, these crops are often locally important or vital, and represent a broad gene pool for future crop improvement. Examples include high iron, zinc and calcium content in tepary bean and bambara groundnut (Chibarabada et al., 2017), tolerance of drought stress in lablab, horsegram and cowpea (Muchow, 1985; Ewansiha & Singh, 2006), and tolerance of heat in amaranth and cassava (Kuo et al., 1992). Thus, underutilized crops have significant potential to aid food security (Massawe et al., 2016; Mabhaudhi et al., 2017; X. Li et al., 2020; Siddique et al., 2021). However, some underutilized crops are becoming abandoned due to globalization, migration and other economic forces and therefore are at risk of loss of diversity or complete loss of the crop, or the loss of associated indigenous knowledge (Mabhaudhi et al., 2017).
This review will summarize advances made in major crops, and how genome sequencing has brought about a step‐change in agricultural research. We also discuss some crops which could have been described as underutilized only c. 30 yr ago but have become more mainstream, and the genomics‐assisted research that has taken place. Beyond this, we discuss the benefits of venturing ‘beyond a reference genome’ (i.e. resequencing, population genomics and pangenomics) and what advantages this can bring. We encourage the research community to incorporate underutilized crops into wider research and to envisage the benefits that come from this increased investment.
1. Genomics and its contribution to agricultural improvements
For staple crops, notable successes have been made using genomic‐related approaches to uncover the genes responsible for important phenotypes.
Genome‐wide association study
Genome‐wide association study (GWAS) relies on genome‐scale data (dense single nucleotide polymorphisms (SNPs) or whole genome sequences) to associate genomic variation with trait variation (Liu & Yan, 2019). Typically, the analysis of hundreds of varieties/lines is necessary to generate sufficient power to resolve quantitative trait loci (QTL), as well as to ensure rare causative variants are included (e.g. Xing et al., 2015). This breadth of variation encompasses orders of magnitude more recombination than QTL mapping and therefore can resolve even small‐effect QTL and with typically greater resolution. Thousands of GWAS have been reported and do not just cover the staple cereals, such as cucumber, peach, sesame and lettuce (Liu & Yan, 2019), but GWAS of crops that fit the definition of underutilized are in their infancy (see the Section ‘Population genomics in underutilized crops’).
Nested‐association mapping
Nested‐association mapping (NAM) involves the development of multiple mapping populations sharing one parent, circumventing in part the issues related to traditional QTL mapping approaches: only covering the variation present in two parents and the low power to tightly define QTL due to limited recombination events (McMullen et al., 2009). With this increased power (Bouchet et al., 2017), resources are being developed for a range of crops, and have provided candidate genes for a range of agronomic traits, for example flowering time in maize (Buckler et al., 2009), barley (Maurer et al., 2015) and sorghum (Bouchet et al., 2017), and plant architectural traits in maize (Brown et al., 2011) and durum wheat (Kidane et al., 2019). Many of these studies have confirmed previous work as well as identified novel QTL for follow up, but are currently restricted to only the main crops.
Pangenomes
It has become increasingly clear that across varieties of a crop, presence/absence variation is common; this means that any one reference genome contains only a subset of the species’ genome, potentially lacking causative loci (Della Coletta et al., 2021). Sequencing and assembling multiple varieties and adding novel genomic regions to the reference results in the pangenome. Genes present in all accessions are defined as ‘core’ and others as ‘dispensable’. In some cases, half or less of some crop genomes are core (Gordon et al., 2017; Wang et al., 2018; Haberer et al., 2020), although these values are highly dependent on the number of accessions sequenced, and whether wild relatives are examined. This rare variation can underlie loci of agronomic importance, for example a flavour‐related gene in tomato (Gao et al., 2019). Pangenomics has also revealed some important evolutionary genomic insight; core maize genes exhibit much greater expression than dispensable genes (Haberer et al., 2020), and dispensable regions tend to have higher transposable element (TE) content than core regions (e.g. Gao et al., 2019).
Most of the above studies have focused on agronomic traits such as yield and its associated components (flowering time, plant stature and size), but because of ensuing climate change, a concerted effort to increase the study of climate change resilience phenotypes, for example root traits and stress response, would be prudent. Whilst some studies have investigated this, for example using NAM to explore drought‐adaptive phenotypes in barley (Pham et al., 2019) and maize (Li et al., 2016), these are comparatively rare.
2. Genome‐assisted advances in staple crops
Here we focus on rice and maize as two of the most widely grown staple crops, collectively grown on 360 million ha in 2019 (http://faostat.fao.org; accessed August 2021), to highlight how genomic advances have led to significant crop improvement for hundreds of millions of consumers.
Genomic advances in rice
Rice is one of the most important crops providing staple food for more than half of the world’s population. Due to the relatively small genome size (430 Mb), colinearity with other cereals, a highly efficienct genetic transformation system and abundant genetic resources, rice has become a model grass species equivalent to the eudicot Arabidopsis. Significant efforts have been made to assemble and annotate the rice genome, for the japonica subspecies (cv Nipponbare; Goff et al., 2002) and the indica subspecies (93‐11; Yu et al., 2002). Compared to Arabidopsis, rice exhibits a gradient in GC content, which means a large proportion of rice genes have no obvious homologue in Arabidopsis (Yu et al., 2002).
Rice is an excellent candidate for population genomic analysis because of its extremely strong population structure and the large extent of linkage disequilibrium (LD) owing to self‐pollination. Resequencing of 527 rice landraces clearly separated the two cultivated subspecies and further divided these by latitudinal photoperiod and temperature clines (Huang et al., 2010). Genome analysis of 446 wild rice (Oryza rufipogon) accessions and 1083 cultivated varieties identified a population of O. rufipogon in southern China where it appears that the domestication into the japonica subspecies took place; from there, hybridization with local wild rice formed indica rice, which spread into Southeast and South Asia (Huang et al., 2012). By combining genomic and phenotypic data through GWAS, a variety of loci associated with agronomic traits in rice have been identified (Supporting Information Table S1).
Whilst mapping short sequence reads of other varieties onto one of the reference genomes can allow these types of analyses, this means that genomic regions absent from the reference will be ignored. Therefore, more recently the genomes of other varieties with beneficial agronomic traits (Zhao et al., 2018; Choi et al., 2020), weedy rice (Sun et al., 2019) and other species of Oryza (W. Li et al., 2020) have been sequenced and assembled, forming a potential rice pangenome resource. A rice pangenome initiative, involving the comparison of 16 de novo assembled genomes from the main population genomic groups (sequenced and assembled using long reads and optical mapping, and hence are described as ‘platinum’ genomes), showed that an average of 33.7 Mb of genome was absent among all pairwise comparisons (Zhou et al., 2020). This highlights how using one reference will only ever describe variation in a subset of the entire crop’s (pan)genome. It is worth noting that the majority (nearly 90%) of the presence/absence fraction of the rice pangenome is made up of TEs, indicating these are more evolutionary labile, but the remaining 10% contains potentially protein‐coding loci. These genome resources will promote evolutionary studies and the identification of adaptive genetic variation in rice.
Genomic advances in maize
Maize was domesticated from teosinte in southwestern Mexico c. 10 000 yr ago. From its wild progenitor, maize has evolved a strikingly different morphology, forming an unbranched plant with large cobs and sweet, naked kernels (Doebley, 1990; Matsuoka et al., 2002). Since then, maize has been continuously improved, and an array of hybrid lines suitable for modern agricultural practice have been developed. In the past century, maize yield has increased eight‐fold due to the increase in yield per plant and plant density adapted by harnessing heterosis. The ancestor genome of maize experienced a tetraploid intermediate stage (n = 20), and then a series of chromosome fusions led to diploidization and recovery of the chromosome number n = 10 (Schnable, 2015). Thus, the maize genome is very large (Gaut & Doebley, 1997) and is especially known for its array of TEs; indeed it is from maize that McClintock (1950) first hypothesized that some genetic elements could be mobile.
Using bacterial artificial chromosome and fosmid clones, the genome of maize variety B73 was assembled, and revealed that long‐terminal repeat retrotransposons (LTRs) account for 74% of the genome. Proliferation of the LTRs was the primary reason why the maize genome is so expanded relative to other grasses (Schnable et al., 2009). Resequencing of 17 wild relatives, 23 traditional landraces and 35 improved maize lines and mapping back to the B73 reference genome suggested that introgression from wild relatives could be responsible for diversity recovery in maize following domestication and identified genes with diverse biological functions having been under selection during domestication (Hufford et al., 2012).
Although significant advances have been made in maize genome sequencing and population genomics, GWAS in maize is a challenge because LD decays within 2 kb. The development of the large NAM panel (McMullen et al., 2009) has significantly increased the efficiency of GWAS in maize, and loci associated with multiple agronomic traits have been identified (Table S2).
Using single‐molecule real‐time sequencing and high‐resolution optical mapping, an improved B73 genome was more recently assembled (Jiao et al., 2017). Due to significant structural variation among inbred lines, the genome of B73 alone is not sufficient to fully explain the variation among other inbred lines. Thus, the genomes of other inbred lines, including PH207 (Hirsch et al., 2016), W22 (Springer et al., 2018), Mo17 (Sun et al., 2018), HuangZaoSi (Li et al., 2019), small‐kernel (Yang et al., 2019) and B73‐Ab10, a variant of B73 containing Abnormal chromosome 10 (J. Liu et al., 2020), have been assembled. This and other ongoing work will facilitate increased understanding of maize genome diversity, as well as the breeding and improvement of maize.
II. Genomics of underutilized crops to‐date
The genetic improvement of underutilized crops is, in part, constrained by limited genome resources. Recent developments in genome technology and the reduction of sequencing costs means genome‐scale research is no longer limited to major food crops (Table 1).
Table 1.
Underutilized crops for which reference genomes are available; crops are organized into families (subfamilies) and the genome statistics are given.
| Family | Species | Common name | Chromosome no. | Estimated genome size | Sequencing method 1 | Scaffold N50 | Number of contigs | Assembled scaffolds size | References |
|---|---|---|---|---|---|---|---|---|---|
| Poaceae (Pooideae) | Secale cereale | Rye | 2n = 2x = 14 | 7.92 Gb | H2000 | 9.4 kb | 1581 707 | 2.80 Gb | Bauer et al. (2017) |
| Aegilops tauschii | Wheat (D‐genome) | 2n = 2x = 14 | 4.36 Gb | H2000 | 58.0 kb | 179 145 | 4.23 Gb | Jia et al. (2013) | |
| Poaceae (Chloridoideae) | Eleusine coracana | Finger millet | 2n = 4x = 36 | 1.45 Gb | H4000, N500 | 23.7 kb | 525 759 | 1.20 Gb | Hittalmani et al. (2017) |
| 1.5 Gb | NS500, PB | 905.3 kb | 2812 919 | 1.20 Gb | Hatakeyama et al. (2018) | ||||
| Eragrostis tef | Tef | 2n = 4x = 40 | 772 Mb | H2000, 454 | 85.0 kb | 672 Mb | Cannarozzi et al. (2014) | ||
| 622 Mb | PB, H4000, Hi‐C | 1.6 Mb | 1344 | 576 Mb | VanBuren et al. (2020) | ||||
| Poaceae (Panicoideae) | Setaria italica | Foxtail millet | 2n = 2x = 18 | 510 Mb | GAII | 12.3 Mb | 397 Mb | Bennetzen et al. (2012) | |
| 490 Mb | GAII, H2000 | 1.0 Mb | 16 903 | 423 Mb | Zhang et al. (2012) | ||||
| Setaria viridis | Green millet | 2n = 2x = 18 | 500 Mb | PB, H2000 | 11.2 Mb | 75 | 395 Mb | Mamidi et al. (2020) | |
| 401 Mb | ONT, NS500 | 19.5 Mb | 44 | 397 Mb | Thielen et al. (2020) | ||||
| Cenchrus americanus | Pearl millet | 2n = 2x = 14 | 1.79 Gb | H2000, PB | 885.0 kb | 175 708 | 1.79 Gb | Varshney et al. (2017b) | |
| Panicum miliaceum | Broomcorn/Proso millet | 2n = 4x = 36 | 887 Mb | PB, X‐ten, BN, Hi‐C | 8.24 Mb | 1308 | 848 Mb | Shi et al. (2019) | |
| 1 Gb | X‐ten | 89 kb | 171 982 | 823 Mb | Ott et al. (2018) | ||||
| 923 Mb | PB, H2500 | 46.7 Mb | 5541 | 855 Mb | Zou et al. (2019) | ||||
| Echinochloa crus‐galli | Barnyard millet | 2n = 6x = 54 | 1.38 Gb | H2000, PB | 1.8Mb | 1.27 Gb | Guo et al. (2017) | ||
| PB, H4000 | 4.1 Mb | 1.34 Gb | Ye et al. (2020) | ||||||
| Digitaria exilis | Fonio millet | 2n = 4x = 36 | 893Mb | H2500, No6000, Hi‐C, BN | 10.7 Mb | 29 155 | 716 Mb | Abrouk et al. (2020) | |
| PB, H3000, H4000 | 1.73 Mb | 3329 | 761Mb | X. Wang et al. (2021) | |||||
| Coix lacryma‐jobi | Adlay/Job's tears | 2n = 2x = 20 | 1.68 Gb | H2000 | Cai et al. (2014) | ||||
| PB, H2500 | 2.24 Mb | 4519 | 1.62 Gb | Guo et al. (2020) | |||||
| PB, H2500 | 594.3 kb | 13 691 | 1.28 Gb | Kang et al. (2020) | |||||
| PB, H2000, BN, Hi‐C | 14.0 Mb | 3321 | 1.73 Gb | H. Liu et al. (2020) | |||||
| Sorghum bicolor | Sorghum | 2n = 2x = 20 | 730 Mb | BAC | 35 Mb | 12 873 | 679 Mb | Paterson et al. (2009) | |
| BAC, H2500 | 68.7 Mb | 2688 | 655 Mb | McCormick et al. (2018) | |||||
| Fabaceae | Cajanus cajan | Pigeonpea | 2n = 2x = 22 | 833 Mb | BAC, GAII, H2000 | 516.1 kb | 173 708 | 606 Mb | Varshney et al. (2012) |
| BAC, 454 | 14.0 kb | 332 766 | 511 Mb | Singh et al. (2012) | |||||
| Cicer arietinum | Chickpea | 2n = 2x = 16 | 738 Mb | BAC, GAII, 454 | 77.3 kb | 520 Mb | Jain et al. (2013) | ||
| BAC, H2000, BAC | 40.0 Mb | 62 619 | 532 Mb | Varshney et al. (2013) | |||||
| 454, GAII | 39.9 Mb | 182 734 | 510 Mb | Parween et al. (2015) | |||||
| Vigna radiata | Mungbean/green gram | 2n = 2x = 22 | 543 Mb | H2000, 454 | 1.52 Mb | 180 372 | 431 Mb | Kang et al. (2014) | |
| Vigna. angularis | Adzuki bean | 2n = 2x = 22 | 542 Mb | H2000 | 1.3 Mb | 25 426 | 467 Mb | Yang et al. (2015) | |
| 612 Mb | H2000, 454 | 703 kb | 36 516 | 443 Mb | Kang et al. (2015) | ||||
| Vigna unguiculata | Cowpea | 2n = 2x = 22 | 640 Mb | H2000, BAC, PB | 16.4 Mb | 765 | 519 Mb | Lonardi et al. (2019) | |
| Lupins albus | White lupin | 2n = 2x = 50 | 584 Mb | PB, H2000, Hi‐C | 18.7 Mb | 3171 | 474 Mb | Xu et al. (2020) | |
| PB, H3000, BN | 17.4 Mb | 451 Mb | Hufnagel et al. (2020) | ||||||
| Lupinus angustifolius | Narrow leafed lupin | 2n = 2x = 40 | H2500 | 703.2 kb | 14 379 | 609 Mb | Hane et al. (2017) | ||
| PB, H2000 | 30.8 Mb | 123 | 616 Mb | P. Wang et al. (2021) | |||||
| Vigna mungo | Blackgram | 2n = 2x = 22 | 574 Mb | X‐ten and ONT | 1.4 Mb | 475 Mb | Jegadeesan et al. (2021) | ||
| Trifolium subterraneum | Subterranean clover | 2n = 2x = 16 | 540 Mb | H2000, M, 454 | 287.6 kb | 101 010 | 472 Mb | Hirakawa et al. (2016) | |
| BN | 1.4 Mb | 264 | 512 Mb | Kaur et al. (2017) | |||||
| Polygonaceae | Fagopyrum esculentum | Common buckwheat | 2n = 2x = 16 | 1.2 Gb | H2000 | 25.1 Mb | 1.18 Gb | Yasui et al. (2016b) | |
| Fagopyrum tataricum | Tartary buckwheat | 2n = 2x = 16 | 540 Mb | H2000, H2500, M, PB, GBS | 550.7 kb | 8778 | 489.3 Mb | Zhang et al. (2017) | |
| Amaranthaceae | Chenopodium quinoa | Quinoa | 2n = 4x = 36 | 1.5 Gb | H2500, PB | 86.9 kb | 110 092 | 1.1 Gb | Yasui et al. (2016a) |
| PB, BN | 3.8 Mb | 4232 | 1.18 Gb | Jarvis et al. (2017) | |||||
| H2500, PB | 1.2 Mb | 10 795 | 1.3 Gb | Zou et al. (2017) | |||||
| Euphorbiaceae | Manihot esculentum | Cassava | 2n = 2x = 36 | 746 Mb | H2000, 454, BAC | 67 kb | 432 Mb | Wang et al. (2014) | |
| Ricinus communis | Castor bean | 2n = 2x = 20 | 320 Mb | Plasmid, Sanger | 561.4 kb | 25 800 | 325 Mb | Chan et al. (2010) | |
| Convolvulaceae | Ipomoea batatas | Sweet potato | 2n = 6x = 90 | 4.4 Gb | H2500, N500, H4000, 454 | 200.7 kb | 35 919 | 836 Mb | Yang et al. (2017) |
| Dioscoreaceae | Dioscorea dumetorum | Trifoliate yam | 2n = 2x = 36 | 322 Mb | H1500, ONT | 3.2 Mb | 1172 | 485 Mb | Siadjeu et al. (2020) |
| Dioscorea rotundata | White Guinea yam | 2n = 2x = 40 | 579 Mb | H2500, BAC | 2.1 Mb | 4723 | 594 Mb | Tamiru et al. (2017) |
Sequencing approaches are abbreviated as follows: 454, Roche 454; BAC, Bacterial Artificial Chromosome sequencing; BN, BioNano; GAII, Illumina Genome Analyzer II; H, Illumina Hiseq; Hi‐C, high‐throughput chromosome conformation capture; M, Illumina Miseq; N500, NextSeq 500; No6000, Novaseq6000; ONT, Oxford Nanopore Technologies; PB, PacBio; X‐ten, Illumina X‐ten.
1. Reference genome sequences for underutilized crops and cross‐crop comparisons
The Poaceae (grasses) is the second largest plant family, with c. 12 000 species. Besides the staple crops rice, maize, wheat and sugarcane, and some previously common cereals such as barley, oats and rye, Poaceae also contain many underutilized crops, including sorghum, foxtail millet, finger millet and broomcorn millet that all use C4 photosynthesis, in which photorespiratory losses induced by hot and arid environments are reduced. The conversion of C3 rice and wheat towards C4 photosynthesis is a long‐standing biotechnological goal. Comparative genomics has revealed that genes involved in C4 carbon fixation are all present in C3 plants (Zhang et al., 2012), and therefore the C4 pathway might have evolved from ancestral C3 isoforms. The panicoid grasses maize and sorghum show greater conservation of these genes compared to the pooid grasses rice and Brachypodium (Bennetzen et al., 2012). Furthermore, a tandem duplication of the carbonic anhydrase Caβ subfamily, which hydrates atmospheric CO2 to bicarbonate in the mesophyll, was found in C4 plants, potentially vital for C4 evolution (Zhang et al., 2012). Genome comparisons between underutilized C4 crops and the staple C3 crops in the Poaceae will provide new suggestions for the evolution of C4 photosynthesis, with the potential to improve the photorespiration efficiency and subsequent drought tolerance of other underutilized and staple crops.
The Fabaceae is the third‐largest plant family, including many agronomically important grain and forage species. Legumes can improve soil fertility through the fixing of atmospheric nitrogen via root nodule‐specific bacteria. The discovery of many genes involved in nitrogen fixing has been aided through the study of underutilized legume genomes (Jain et al., 2013; Lu et al., 2018; Zhuang et al., 2019). In addition, the legumes also contain species with unique nutritional features; for example, adzuki bean, widely cultivated in Asia, is referred to as the ‘weight loss bean’ due to its sweet taste but low caloric and fat content. Genomic comparisons with other legumes found that adzuki bean has fewer starch and fatty acid biosynthesis genes, which could play a role in its unique nutritional profile (Yang et al., 2015).
Several underutilized crops are advocated as worthy of investment because of their extreme stress resilience, often greater than staple crops (Massawe et al., 2015; Cullis & Kunert, 2017). Further comparative genomics in the Poaceae has identified numerous gene family expansions associated with stress tolerance in underutilized crops, and these might explain the high stress resistance in underutilized crops. Drought‐tolerant foxtail millet and sorghum (compared to drought‐susceptible rice and maize) contain expansions of stress response gene families, including those encoding cytochrome P450 proteins, expansins, lipid transfer proteins and several others, as well as miRNA169 targeting drought stress‐associated transcription factor nuclear factor‐YB (Paterson et al., 2009; The International Brachypodium Initiative, 2010). Tef, a drought‐tolerant cereal mainly distributed in Ethiopia, contains a tandem duplication of the nucleotidase/phosphatase SAL1, a gene family involved in drought tolerance, relative to other grasses investigated (Cannarozzi et al., 2014). Pearl millet possesses more members of cutin, suberin, wax biosynthetic and metabolite transporter genes, which might be responsible for the heat and drought tolerance in this underutilized crop (Varshney et al., 2017b). The number of BTB ubiquitin E3 ligases is greater in grasses than in Arabidopsis, and one subgroup, the BTB‐BACK subgroup, was only expanded in the underutilized cereal broomcorn millet (Zou et al., 2019), which may contribute to its excellent stress tolerance. Clearly, genome comparison of these underutilized crops will provide a new pool of stress‐targeted genes for well‐studied main crops. Similar results were found in the genus Dioscorea, in which draft genomes have been assembled for two yam species (Tamiru et al., 2017; Siadjeu et al., 2020), and phylogenetic analyses show that Dioscorea has more bulb‐type lectin genes than the Poaceae and Arabidopsis, with potential roles in the insecticidal properties of Guinea yam (Tamiru et al., 2017).
In summary, reference genomes of underutilized crops can help resolve the genetic basis of agronomic traits, especially as a (sometimes unique) resource for improving the photorespiratory efficiency, nutritional value and stress tolerance of related major food crops currently challenged by climate change.
2. Population genomics in underutilized crops
Population genomics of underutilized crops can help researchers to understand population structure and domestication history, as well as aid in identifying candidate genes modulating key agronomic traits through GWAS and to develop molecular markers for marker‐assisted breeding (MAB; Fig. 1).
Fig. 1.
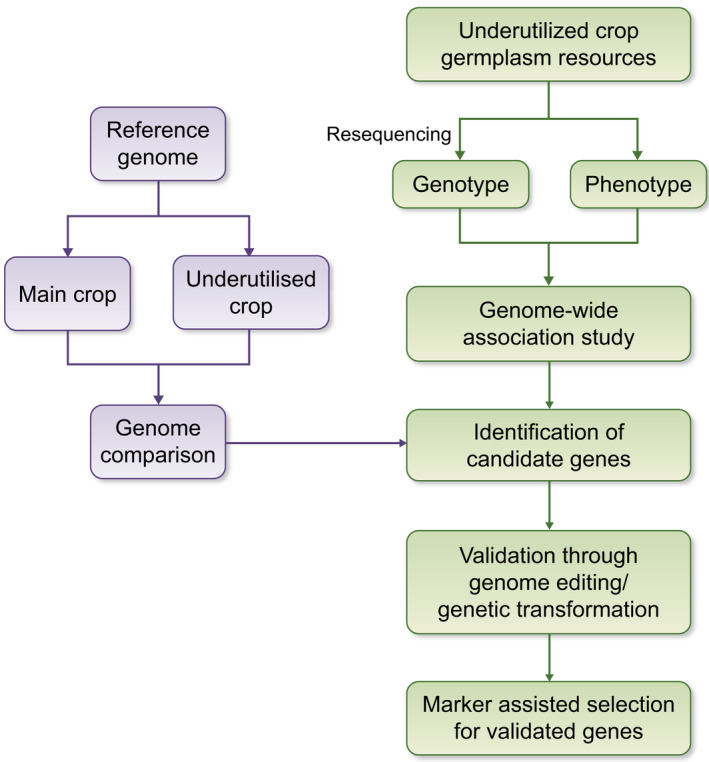
Schematic diagram highlighting the main sequencing and breeding approaches discussed in this article. First (left), by comparing the reference genomes of underutilized crops and staple/main crops, genome variants responsible for superior agronomic traits (such as specific resistance and nutrient quality or quantity) of underutilized crops could be explored. In addition (right), the combination of genome resequencing and phenotyping through genome‐wide association study could help to identify candidate genes responsible for agronomic traits of underutilized crops. Through genetic transformation or genome editing, the function of these candidate genes could be verified. Finally, by associating genomic data to phenotypic information of different accessions, germplasm resources can be effectively screened and bred by means of molecular marker‐assisted breeding and crossing, to improve the resistance and nutritional value of underutilized crops in addition to staple crops.
Identifying crop wild relatives and untapped resources
The combination of genome resequencing and phylogenetic analysis enables us to better understand the population structure and identify wild relatives. For example, resequencing of 994 pearl millet lines identified four main clusters and pinpointed a West African origin for the crop (Varshney et al., 2017b). Resequencing of 166 fonio millet accessions and 17 proposed progenitors found a significant separation between cultivated and wild accessions, and the cultivated accessions were genetically closest to wild accessions from Southern Togo and West Guinea (Abrouk et al., 2020). Furthermore, diversity of the grain size gene GS5 was significantly reduced in fonio millet cultivars (with larger grains than the wild progenitor), suggesting human selection on this locus during domestication. Resequencing of 510 accessions of Tartary buckwheat from the entire global distribution identified three major clades and indicated two geographically distinct domestication events (Zhang et al., 2021). Similar investigations in other underutilized crops have identified wild relatives and genetic subpopulations of the domesticates, for example adzuki bean (Yang et al., 2015), lupin (Hufnagel et al., 2020; P. Wang et al., 2021) and coix (H. Liu et al., 2020). Resequencing of Guinea yam and its potential wild relatives resolved a hybrid origin from a cross between the wild rainforest species Dioscorea praehensilis and the savanna species D. abyssinica (Sugihara et al., 2020).
GWAS and selection analyses
The high‐density SNP data obtained from population‐scale genome resequencing can be used to identify the genetic basis of adaptive traits through GWAS and selection analyses. From this, genetic markers can be designed and used for MAB. Beyond breeding, genetic modification‐type approaches can be used to insert the candidate genes from a stress‐tolerant or otherwise novel underutilized crop into a susceptible or trait‐lacking staple crop.
Seed size and weight are some of the most important traits of many crops, affecting price and milling qualities. GWAS of 368 cowpea accessions found candidate genes involved in endosperm development, embryo development and cell elongation associated with seed size, some of which also play a role in common bean (Lo et al., 2019). Similar analyses in castor bean identified candidate genes for seed traits that differentiate the wild and cultivated types (Xu et al., 2021). Recently, GWAS of Tartary buckwheat identified a mutation in the GCC cis‐element of an AP2 transcription factor associated with grain weight (Zhang et al., 2021).
Underutilized crops can possess multiple agronomic traits that are not present in staple crops, and therefore present untapped resources for traits such as abiotic tolerances and novel nutrients. GWAS has been used to identify genes involved in tuber quality in cassava (S. Zhang et al., 2018) and anthocyanin content in mungbean (Noble et al., 2018). Regarding stress tolerance, genes involved in lateral root development, stress tolerance and phosphorus use efficiency of mungbean have been identified (Reddy et al., 2020).
Comparing wild and domesticated cassava genomes has identified selective sweeps in genes involved in photosynthesis, starch accumulation and stress response (Wang et al., 2014). Extending this to other crops will facilitate the development of markers associated with domestication‐related traits. Candidate genes identified in underutilized crops through GWAS can help improve the quality and stress tolerance not only of the underutilised crops but also of related staple crops, to better suit our needs in a changing climate.
Extended population genomics, for example using reduced representation technologies, can be used to link genetic markers to agronomic traits, without knowing the precise genetic basis of the trait. Markers linked to several diverse agronomic traits in foxtail millet have been identified using these approaches, for example coloration, leaf size and shape, grain yield and weight, and flowering time (Upadhyaya et al., 2015; Jaiswal et al., 2019). Similarly, QTL mapping approaches at sufficient density can provide marker–trait associations, for example markers associated with yield and flowering in pea (Annicchiarico et al., 2017) and dormancy in groundnut (Kumar et al., 2020).
Genomics‐assisted breeding of underutilized crops has been limited due to the lack of molecular markers linked to traits of interest, but the recent recognition of the importance of underutilized crops and the development of genome technology have clearly started to remedy this. The use of these markers to accelerate breeding (i.e. genomic selection) has been shown for a handful of underutilized crops (Ye & Fan, 2020).
3. Genetic transformation and gene editing in underutilized crops
In recent years, genetic engineering has been widely used to elucidate gene function and for crop improvement. Compared with traditional hybridization and crossing of varieties, genetic engineering could deliver agronomically useful traits into plants faster and in a more targeted manner. Agrobacterium‐based transformation systems are widely used for genetic transformation in plants, facilitating the integration of foreign gene copies into the host plant’s genome. Although Agrobacterium transformation has been successfully used for transformation in several major crops, the inherent limitations associated with resistance to Agrobacterium infection and their recalcitrance to in vitro regeneration limit the transformation of many orphan crops. At present, Agrobacterium‐mediated transformation has only been successful in shoot apex explants of finger millet (Ceasar & Ignacimuthu, 2011) and foxtail millet (Ceasar et al., 2017), callus derived from mature seeds of finger millet (Hema et al., 2014), green millet (Martins et al., 2015a; Nguyen et al., 2020), sorghum (Zhao et al., 2000; Belide et al., 2017) and foxtail millet (Santos et al., 2020), embryonic axis explants of pigeonpea (Ghosh et al., 2017), germinated seedlings of chickpea (Senthil et al., 2004), and hairy roots of chickpea (Aggarwal et al., 2018) and buckwheat (Mi et al., 2020). However, the recalcitrant tissue culture efficiency and occasional and unpredictable chimerism lower the efficiency of these tissue culture‐based methods.
Recently, using the floral‐dip Agrobacterium‐mediated transformation method, the wild ancestor of foxtail millet, green millet, was successfully transformed (Martins et al., 2015b). This is a significant advance because millets are model C4 grasses, and green millet it diploid, with a rapid life cycle, small genome size, simple growth requirements and high transformation efficiency.
Despite ongoing challenges of carrying out gene editing in even the best studied crops (Yang, 2020), CRISPR/Cas9‐based gene editing has been conducted in underutilized crops with relatively high tissue culture efficiency, including green millet (Weiss et al., 2020) and sorghum (Jiang et al., 2013; Che et al., 2018). These approaches will provide the necessary technical support for improving the efficiency in confirming the function of unique genes and the development of advantageous varieties of underutilized crops.
III. The way forward
1. The successful transition from underutilized to mainstream
In the past 20 yr, a few previously underutilized crops, such as quinoa, chickpea and pigeonpea, have seen a significant boost in research and recognition. For these crops we have seen a parallel 20–500% increase in the area grown worldwide between the 1960s and 2010s (http://faostat.fao.org; accessed August 2021). Chickpea and pigeonpea were among the first underutilized crops to have their genomes sequenced (Varshney et al., 2012, 2013) with the (tetraploid) quinoa genome being made available more recently (Jarvis et al., 2017). Clearly the availability of genome sequence was a major stepping‐stone in resolving the genetic basis of adaptive and agronomic phenotypes in these crops.
Using pigeonpea as an exemplar, this crop was recognized as worthy of significant investment in the 1970s, with the Pigeonpea Genomics Initiative (PGI) established in 2006 (Varshney et al., 2010). After genome sequencing (Varshney et al., 2012), significant advances have been made in identifying genomic regions underlying adaptive traits that could be crossed between varieties using MAB (Varshney et al., 2017a), for example markers associated with sterility mosaic disease (Saxena et al., 2017a) and fusarium wilt (Saxena et al., 2017b). Genomic analysis has revealed fewer genes involved in lipid biosynthesis in pigeonpea than in soybean, and more cellulose synthesis genes, which together might underlie the biochemical and morphological differences between pigeonpea and other legumes (Singh et al., 2012). In addition, a pigeonpea gene involved in disease resistance was cloned and transferred to soybean, conferring resistance to Asian soybean rust (Kawashima et al., 2016), which would have been impossible without using the pigeonpea genome sequence.
More recent GWAS of nearly 300 pigeonpea accessions (Varshney et al., 2017a) identified dozens of associations and provided significant resources for MAB (Bohra et al., 2020). Pigeonpea is probably the only underutilized crop for which a pangenome has been sequenced (Zhao et al., 2020); this has 55 512 genes, compared to the reference genome (Varshney et al., 2012), which has only 53 612 (when annotated in exactly the same way as the pangenome). Using this pangenome, novel GWAS associations have been identified (Zhao et al., 2020), which were absent using the single reference genome (Varshney et al., 2017a). This further highlights the additional insights that can be made when a pangenome is made available.
Chickpea is grown and consumed worldwide, but 30 yr ago could have been considered underutilized. Although productivity has steadily increased, the development of accessions with greater yield, improved nutrition and stress resistance is essential to meet increasing demands. Comparative genomics of legumes has identified a lack of some resistance and nodulation genes, potential reasons for the low stress resistance (Jain et al., 2013; Varshney et al., 2013). Resequencing panels have identified genetic groups of cultivars (primarily the desi and kabuli types), identified the origin of the crop, and uncovered genes involved in drought tolerance and heat stress response through GWAS (Varshney et al., 2013, 2019).
Several other previously underutilized crops are seeing a revolution in their investigation, suggesting they are on the path to escaping some of the reasons they were previously underutilized. The following examples are case studies of crop species early on this trajectory and provide ideas to circumvent issues such as large genomes and examples of crops with unique attributes which have received investment.
For species with polyploid genomes, investigations of related diploids can shed light on agronomic traits. Oat is a nutritional crop containing abundant calcium, dietary fibre (especially β‐glucan) and unsaturated fatty acids (Joyce et al., 2019). Due to the cholesterol‐lowering properties and the antidiabetic effect of β‐glucan, oat has been widely used in adjuvant treatment of diabetes and cardiovascular disorders. The rotation of oat with other crops can improve soil structure and reduce diseases in other crops. This disease resistance has been attributed to the production of avenacins, specialized antifungal metabolites. Oat is allohexaploid, with a relatively large, highly repetitive and rearranged genome, and thus brings challenges for genome assembly. Current sequencing has mainly focused on wild diploid oats. For example, through genome assembly of the diploid extant progenitors, candidate genes regulating flowering time and disease resistance were identified (Maughan et al., 2019). Genome assembly of other diploid accessions identified a 12‐gene cluster responsible for avenacin biosynthesis, and this cluster was located in a subtelomeric region which may have formed since oat diverged from other crops (Li et al., 2021). These results shed light on the evolution of oat and will help in breeding oat varieties with modified and improved health benefits.
For other underutilized crops, they bring qualities and traits which are lacking in mainstream staple crops, and as such significant investment has begun to start their escape from being underutilized. Quinoa is one example, a crop of the Chenopodiaceae, which has been cultivated for c. 7000 yr. Its diverse environmental adaptability means it is grown from the sea level of Chile to altitudes above 4500 m in Bolivia (Suárez‐Estrella et al., 2018). Due to its extraordinary balance of essential amino acids, and abundant vitamins, minerals, dietary fibre and unsaturated fatty acids, it was recognized as a complete food and has attracted the attention of the scientific community (Filho et al., 2017). However, quinoa contains bitter and astringent antinutritional factors such as saponins. Although these substances are health‐promoting, their bitter taste has limited the utilization of quinoa (Suárez‐Estrella et al., 2018). Thus, selection of genotypes with low saponin content is one of the most important quinoa breeding objectives for the future. In addition, due to its outcrossing nature, genome assembly of quinoa was not trivial, requiring repeated self‐pollination to reduce heterozygosity (Yasui et al., 2016a). Analysis of the subsequently assembled quinoa genome (Yasui et al., 2016a) identified expansions of gene families involved in lysine, vitamins, polyphenol and betalain synthesis, as well as abscisic acid (ABA) signalling, which together may relate to the unique profile of nutritional and antinutritional factors and abiotic stress tolerance in quinoa (Yasui et al., 2016a; Zou et al., 2017).
Another example of an underutilized crop with novel attributes is buckwheat, a pseudocereal originating from and domesticated in China > 4000 yr ago (Zhang et al., 2021). This crop possesses an outstanding nutritional profile (especially flavonoids) and an excellent ability to grow under adverse climatic and soil conditions. The main cultivated species are common buckwheat and Tartary buckwheat. Similar to quinoa, being outcrossing makes genome sequencing of common buckwheat more challenging and requires repeated self‐pollination to reduce heterozygosity (Yasui et al., 2016b). In contrast to common buckwheat, the sequencing of Tartary buckwheat was relatively simple because of a smaller genome and because its is predominantly a selfer. Comparative genomics using the chromosome‐scale Tartary buckwheat genome revealed a whole‐genome duplication event after buckwheat divergence from sugar beet, with some evidence that this might play a role in buckwheat tolerance of extremely harsh environments (Zhang et al., 2017). Genome resequencing of Tartary buckwheat identified two independent domestication events, in southwestern and in northern China, which has resulted in the diversity of modern Tartary buckwheat varieties (Zhang et al., 2021). Candidate genes responsible for flavonoid biosynthesis were also identified and will help breeding of buckwheat with improved health and medical benefits.
Given these findings from the significant investment and the ongoing work in representative orphan crops (Roorkiwal et al., 2020), we feel encouraged that the resources and investment needed for these crops to be elevated to the national stage are in place. However, these are only the tip of the underutilized crop iceberg; dozens of underutilized crops have a single reference genome, and in some cases small resequencing panels (Table 1), but significant population and GWAS resources or pangenomes are absent for the vast majority.
2. What do we need and why? The advantages of going beyond a reference genome
To efficiently breed improved varieties of underutilized crops we need to have reliable linkage between genetic markers and traits of interest. Markers identified in a single QTL mapping experiment may not be reliable given that many QTL are only expressed in some environments (genotype × environment interaction) and do not always tightly define the genomic region (therefore the QTL spans dozens or hundreds of genes). More precision can be gleaned from LD mapping approaches (Thornsberry et al., 2001), including GWAS, which requires extensive panels of germplasm and high marker density. A reference genome is an asset to begin to understand important and adaptive phenotypes in underutilized crops, yet it is becoming clear that significant advances in breeding improved varieties are only possible when the genomic variants are identified, thus requiring a population of genomes and potentially a pangenome.
Quality trait‐marker linkage
One main advantage to having population‐level sequencing is to tie this to trait data using GWAS‐type approaches. This is an efficient way to start to narrow down the genetic basis of quantitative traits such as yield, seed and organ size, plant stature, etc., all traits which need to be optimized to ensure a crop is cost‐effective to be grown at scale (Fig. 2). A reference genome is an asset, but without the resequencing (or high‐density SNP genotyping), GWAS cannot be done. Examples of GWAS‐style analyses in the underutilized crops cowpea, castor bean, Tartary buckwheat, cassava and mungbean are given above.
Fig. 2.
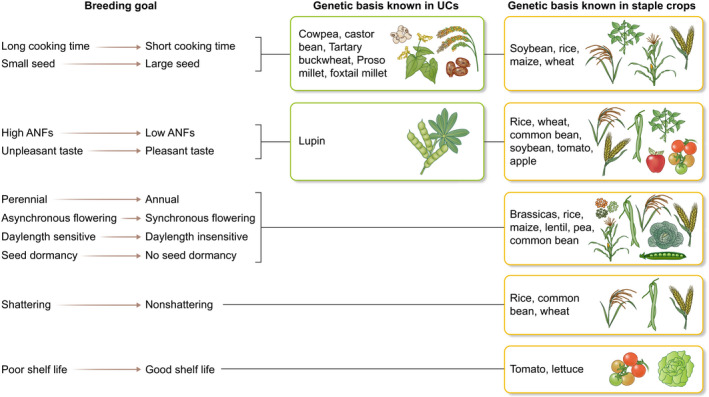
Schematic diagram highlighting common breeding targets for any crop, and whether the genetic basis has been investigated in underutilized crops (UCs) and/or staple crops. ANF, antinutrient factor.
Using reduced‐representation technology (e.g. genotyping‐by‐sequencing, or restriction site‐associated DNA sequencing) is an efficient way to screen large numbers of genetic loci throughout the genome at lower cost than resequencing. The downside is that unless LD extends for very long distances, most markers will be so widely spaced that they will be too far from the underlying causative loci to be associated. Nevertheless, these approaches have yielded marker‐trait associations (MTAs) in some underutilized crops and pave the way for more in‐depth analyses. For example, in Proso millet (Panicum milaceum L.) 13 MTAs for seven traits were resolved, but no MTAs were found for another nine traits (Boukail et al., 2021). In foxtail millet, 81 MTAs for 10 traits were resolved, but most did not pass false discovery rate correction (Jaiswal et al., 2019), and in Kersting’s groundnut, 10 MTAs for five traits were resolved (Akohoue et al., 2020). It is important to bear in mind that because of the marker spacing, partly due to the location of cut sites in the genome, and partly because of uneven sequencing coverage across loci (Beissinger et al., 2013), these studies are likely to report only a subset of genomic loci involved in the traits of interest.
Cross‐crop analyses
Whilst underutilized crops may lack some desirable traits (fast growth, ease of harvest, high harvest index), or have additional phenotypes that are unwanted (antinutrients, perenniality), significant advances have been made in understanding the genetic basis of many of these traits in other crops (Fig. 2). Thus, where these traits have been studied in detail, there may be candidate genes that can be followed up if population resequencing or pangenomic data are available in the underutilized crop.
Several underutilized crops would benefit from having more predictable flowering/fruiting or being adapted to novel environments where the daylength is different. The legume lablab is typically a short‐day plant, and therefore expansion outside its native tropical latitudes is unlikely to be successful (Sennhenn et al., 2017). Bambara groundnut, another tropical underutilized legume, is typically short‐day although a few semiimproved varieties can be grown further from the equator, but it is acknowledged that this is still a barrier to more widespread adoption (Mayes et al., 2019). Daylength response (and therefore flowering time) is relatively well studied in staple crops, including rice, maize and the typically long‐day legumes, lentil and pea (Hung et al., 2012; Weller et al., 2012; Itoh & Izawa, 2013), offering candidate genes for the development of underutilized cereal and legume varieties for adaptation to nonnative latitudes. Candidate genes, or genomic regions, underlying annual vs perennial growth have been identified in Brassicaceae species (Heidel et al., 2016; Kiefer et al., 2017); this is another trait which might help the adoption of underutilized crops.
An often‐cited reason for the poor adoption, or decline in use, of underutilized crops is their antinutrient content. Antinutrient factors (ANFs) inhibit the uptake of beneficial minerals and vitamins, so a high‐nutrient crop with high ANF content will have low nutrient bioavailability. This is especially the case in legumes where several ANFs have been identified that affect iron, zinc and protein uptake. Whilst cooking and fermentation can reduce the presence of these compounds (e.g. Samtiya et al., 2020), these take time or energy (e.g. fuel for cooking). However, ANFs are usually vital for crop disease resistance, and therefore breeding for high ANF during the growth period coupled with low ANF in the maturation period would clearly be advantageous. Progress has been made in understanding the genetic basis of these traits (Campion et al., 2013; Sparvoli & Cominelli, 2015), opening the door for understanding the genetic basis of these traits in underutilized crops.
There are other traits which make underutilized crops less attractive as a choice for a farmer or the consumer, for example poor shelf‐life, unpleasant taste or lengthy cooking times (and an increase cost for fuel). Genes involved in shelf‐life in tomato have been elucidated (Casals et al., 2012; L. Zhang et al., 2018), along with QTL for alkaloid content in lupin (Rychel & Książkiewicz, 2019) and for seed hardness, and therefore cooking time, in legumes (Sandhu et al., 2018; Diaz et al., 2021).
Relatedly, many underutilized crops are known for their extreme resilience phenotypes. Any analysis of the genetic basis of drought or heat tolerance in any underutilized crops, probably requiring population sequencing for GWAS, for example, will be of significant value to other more mainstream crops. This could identify novel alleles or even undercover novel genes and pathways involved in these climate‐change‐relevant tolerances. The sequencing of one reference genome of an underutilized crop cannot offer this.
Population‐level resequencing mapped to one reference will not be able to examine the fraction of the genome that is only present in some accessions (presence–absence variation lacking from the sequenced reference, which would only be identified in a pangenome). This problem could be underestimated for underutilized crops where variation in genome size might not be recognized; for example, the underutilized legume lablab was probably domesticated twice (Robotham & Chapman, 2015; Maass et al., 2017), and the two gene pools differ in genome size by c. 20% (MAC, unpublished).
Next steps
We propose that efforts should be made not only to generate a reference genome but also to carry out population‐level sequencing and pangenomics. In parallel we encourage the continued collection and long‐term archiving of seed resources, and addressing the challenges associated with archiving the required indigenous knowledge associated with these under‐investigated species (Mabhaudhi et al., 2017; Kamenya et al., 2021). Researchers should make data free to use, and collaborations between institutes worldwide should be encouraged to expedite the production of results and limiting unnecessary overlap and wasted resources.
Whilst the cost and time implications of multiple reference genomes, resequencing and collecting global germplasm are not trivial, we believe that, given the climate crisis and the need to fast‐track the development of mainstream and novel crops, this is the most reliable way to ensure that underutilized crops are investigated to the depth at which reliable and meaningful data can be used. It is likely that some underutilized crops hold vital genetic variants to help the human population combat food insecurity in the next few decades; this genetic erosion is under‐investigated even in staple crops (Khoury et al., 2022). Without fully investigating underutilized crop genomes, we do not know where these variants lie, and if we delay too long, we may lose alleles, varieties and crops entirely.
Author contributions
MAC, YH and MZ researched the topic and wrote the manuscript.
Supporting information
Table S1 List of important traits dissected by GWAS in rice.
Table S2 List of important traits dissected by GWAS in maize.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This research was supported by grants from the National Key R&D Program of China (2019YFD1001300/2019YFD1001302) and National Natural Science Foundation of China (31871536 and 32161143005) to MZ and from the Natural Environment Research Council and Biotechnology and Biological Sciences Research Council (NE/S002022/1) to MAC. We thank our lab groups for ongoing discussions into these crops as well as three anonymous reviewers who gave insightful comments and suggestions on a previous version of this review.
Contributor Information
Mark A. Chapman, Email: m.chapman@soton.ac.uk.
Meiliang Zhou, Email: zhoumeiliang@caas.cn.
References
- Abrouk M, Ahmed HI, Cubry P, Šimoníková D, Cauet S, Pailles Y, Bettgenhaeuser J, Gapa L, Scarcelli N, Couderc M et al. 2020. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nature Communications 11: 4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal PR, Nag P, Choudhary P, Chakraborty N, Chakraborty S. 2018. Genotype‐independent Agrobacterium rhizogenes‐mediated root transformation of chickpea: a rapid and efficient method for reverse genetics studies. Plant Methods 14: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akohoue F, Achigan‐Dako EG, Sneller C, Van Deynze A, Sibiya J. 2020. Genetic diversity, SNP‐trait associations and genomic selection accuracy in a west African collection of Kersting’s groundnut [Macrotyloma geocarpum (Harms) Maréchal & Baudet]. PLoS ONE 15: e0234769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annicchiarico P, Nazzicari N, Pecetti L, Romani M, Ferrari B, Wei Y, Brummer EC. 2017. GBS‐based genomic selection for pea grain yield under severe terminal drought. Plant Genome 10: 1–13. [DOI] [PubMed] [Google Scholar]
- Bauer E, Schmutzer T, Barilar I, Mascher M, Gundlach H, Martis MM, Twardziok SO, Hackauf B, Gordillo A, Wilde P et al. 2017. Towards a whole‐genome sequence for rye (Secale cereale L.). The Plant Journal 89: 853–869. [DOI] [PubMed] [Google Scholar]
- Beissinger TM, Hirsch CN, Sekhon RS, Foerster JM, Johnson JM, Muttoni G, Vaillancourt B, Buell CR, Kaeppler SM, de Leon N. 2013. Marker density and read depth for genotyping populations using genotyping‐by‐sequencing. Genetics 193: 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belide S, Vanhercke T, Petrie JR, Singh SP. 2017. Robust genetic transformation of sorghum (Sorghum bicolor L.) using differentiating embryogenic callus induced from immature embryos. Plant Methods 13: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, Estep M, Feng L, Vaughn JN, Grimwood J et al. 2012. Reference genome sequence of the model plant Setaria. Nature Biotechnology 30: 555–561. [DOI] [PubMed] [Google Scholar]
- Bohra A, Saxena KB, Varshney RK, Saxena RK. 2020. Genomics‐assisted breeding for pigeonpea improvement. Theoretical and Applied Genetics 133: 1721–1737. [DOI] [PubMed] [Google Scholar]
- Bouchet S, Olatoye MO, Marla SR, Perumal R, Tesso T, Yu J, Tuinstra M, Morris GP. 2017. Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 206: 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukail S, Macharia M, Miculan M, Masoni A, Calamai A, Palchetti E, Dell’Acqua M. 2021. Genome wide association study of agronomic and seed traits in a world collection of proso millet (Panicum miliaceum L.). BMC Plant Biology 21: 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PJ, Upadyayula N, Mahone GS, Tian F, Bradbury PJ, Myles S, Holland JB, Flint‐Garcia S, McMullen MD, Buckler ES et al. 2011. Distinct genetic architectures for male and female inflorescence traits of maize. PLoS Genetics 7: e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint‐Garcia S, Garcia A, Glaubitz JC et al. 2009. The genetic architecture of maize flowering time. Science 325: 714–718. [DOI] [PubMed] [Google Scholar]
- Cai Z, Liu H, He Q, Pu M, Chen J, Lai J, Li X, Jin W. 2014. Differential genome evolution and speciation of Coix lacryma‐jobi L. and Coix aquatica Roxb. hybrid guangxi revealed by repetitive sequence analysis and fine karyotyping. BMC Genomics 15: 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion B, Glahn RP, Tava A, Perrone D, Doria E, Sparvoli F, Cecotti R, Dani V, Nielsen E. 2013. Genetic reduction of antinutrients in common bean (Phaseolus vulgaris L.) seed, increases nutrients and in vitro iron bioavailability without depressing main agronomic traits. Field Crops Research 141: 27–37. [Google Scholar]
- Cannarozzi G, Plaza‐Wüthrich S, Esfeld K, Larti S, Wilson YS, Girma D, de Castro E, Chanyalew S, Blösch R, Farinelli L et al. 2014. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genomics 15: 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals J, Pascual L, Cañizares J, Cebolla‐Cornejo J, Casañas F, Nuez F. 2012. Genetic basis of long shelf life and variability into Penjar tomato. Genetic Resources and Crop Evolution 59: 219–229. [Google Scholar]
- Ceasar SA, Baker A, Ignacimuthu S. 2017. Functional characterization of the PHT1 family transporters of foxtail millet with development of a novel Agrobacterium‐mediated transformation procedure. Scientific Reports 7: 14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceasar SA, Ignacimuthu S. 2011. Agrobacterium‐mediated transformation of finger millet (Eleusine coracana (L.) Gaertn.) using shoot apex explants. Plant Cell Reports 30: 1759–1770. [DOI] [PubMed] [Google Scholar]
- Chan AP, Crabtree J, Zhao QI, Lorenzi H, Orvis J, Puiu D, Melake‐Berhan A, Jones KM, Redman J, Chen G et al. 2010. Draft genome sequence of the oilseed species Ricinus communis . Nature Biotechnology 28: 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che P, Anand A, Wu E, Sander JD, Simon MK, Zhu W, Sigmund AL, Zastrow‐Hayes G, Miller M, Liu D et al. 2018. Developing a flexible, high‐efficiency Agrobacterium‐mediated sorghum transformation system with broad application. Plant Biotechnology Journal 16: 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibarabada TP, Modi AT, Mabhaudhi T. 2017. Expounding the value of grain legumes in the semi‐ and arid tropics. Sustainability 9: 60. [Google Scholar]
- Choi JY, Lye ZN, Groen SC, Dai X, Rughani P, Zaaijer S, Harrington ED, Juul S, Purugganan MD. 2020. Nanopore sequencing‐based genome assembly and evolutionary genomics of circum‐basmati rice. Genome Biology 21: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis C, Kunert KJ. 2017. Unlocking the potential of orphan legumes. Journal of Experimental Botany 68: 1895–1903. [DOI] [PubMed] [Google Scholar]
- Dawson IK, Powell W, Hendre P, Bančič J, Hickey JM, Kindt R, Hoad S, Hale I, Jamnadass R. 2019. The role of genetics in mainstreaming the production of new and orphan crops to diversify food systems and support human nutrition. New Phytologist 224: 37–54. [DOI] [PubMed] [Google Scholar]
- Della Coletta R, Qiu Y, Ou S, Hufford MB, Hirsch CN. 2021. How the pan‐genome is changing crop genomics and improvement. Genome Biology 22: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz S, Ariza‐Suarez D, Ramdeen R, Aparicio J, Arunachalam N, Hernandez C, Diaz H, Ruiz H, Piepho H‐P, Raatz B. 2021. Genetic architecture and genomic prediction of cooking time in common bean (Phaseolus vulgaris L.). Frontiers in Plant Science 11: 622213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley J. 1990. Molecular evidence and the origin of maize. Economic Botany 44: 6–27. [Google Scholar]
- Ewansiha SU, Singh BB. 2006. Relative drought tolerance of important herbaceous legumes and cereals in the moist and semi‐arid regions of West Africa. Journal of Food Agriculture and Environment 4: 188–190. [Google Scholar]
- FAO, IFAD, UNICEF, WFP, WHO . 2021. The State of Food Security and Nutrition in the World 2021: transforming food systems for food security, improved nutrition and affordable healthy diets for all. Rome, Italy: FAO. [Google Scholar]
- Filho AM, Pirozi MR, Borges JT, Pinheiro Sant'Ana HM, Chaves JB, Coimbra JS. 2017. Quinoa: nutritional, functional, and antinutritional aspects. Critical Reviews in Food Science and Nutrition 57: 1618–1630. [DOI] [PubMed] [Google Scholar]
- Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM, Burzynski‐Chang EA, Fish TL, Stromberg KA, Sacks GL et al. 2019. The tomato pan‐genome uncovers new genes and a rare allele regulating fruit flavor. Nature Genetics 51: 1044–1051. [DOI] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF. 1997. DNA sequence evidence for the segmental allopolyploid origin of maize. Proceedings of the National Academy of Sciences, USA 94: 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, Ganguly S, Purohit A, Chaudhuri RK, Das S, Chakraborti D. 2017. Transgenic pigeonpea events expressing Cry1Ac and Cry2Aa exhibit resistance to Helicoverpa armigera. Plant Cell Reports 36: 1037–1051. [DOI] [PubMed] [Google Scholar]
- Global Panel on Agriculture and Food Systems for Nutrition . 2020. Future food systems: for people, our planet, and prosperity. London, UK: Global Panel on Agriculture and Food Systems for Nutrition. [Google Scholar]
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100. [DOI] [PubMed] [Google Scholar]
- Gordon SP, Contreras‐Moreira B, Woods DP, Des Marais DL, Burgess D, Shu S, Stritt C, Roulin AC, Schackwitz W, Tyler L et al. 2017. Extensive gene content variation in the Brachypodium distachyon pan‐genome correlates with population structure. Nature Communications 8: 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Wang Y, Yang A, He J, Xiao C, Lv S, Han F, Yuan Y, Yuan Y, Dong X et al. 2020. The Coix genome provides insights into Panicoideae evolution and papery hull domestication. Molecular Plant 13: 309–320. [DOI] [PubMed] [Google Scholar]
- Guo L, Qiu J, Ye C, Jin G, Mao L, Zhang H, Yang X, Peng Q, Wang Y, Jia L et al. 2017. Echinochloa crus‐galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nature Communications 8: 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberer G, Kamal N, Bauer E, Gundlach H, Fischer I, Seidel MA, Spannagl M, Marcon C, Ruban A, Urbany C et al. 2020. European maize genomes highlight intraspecies variation in repeat and gene content. Nature Genetics 52: 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hane JK, Ming Y, Kamphuis LG, Nelson MN, Garg G, Atkins CA, Bayer PE, Bravo A, Bringans S, Cannon S et al. 2017. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant‐microbe interactions and legume evolution. Plant Biotechnology Journal 15: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M, Aluri S, Balachadran MT, Sivarajan SR, Patrignani A, Grüter S, Poveda L, Shimizu‐Inatsugi R, Baeten J, Francoijs K‐J et al. 2018. Multiple hybrid de novo genome assembly of finger millet, an orphan allotetraploid crop. DNA Research 25: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel AJ, Kiefer C, Coupland G, Rose LE. 2016. Pinpointing genes underlying annual/perennial transitions with comparative genomics. BMC Genomics 17: 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hema R, Vemanna RS, Sreeramulu S, Reddy CP, Senthil‐Kumar M, Udayakumar M. 2014. Stable expression of mtlD gene imparts multiple stress tolerance in finger millet. PLoS ONE 9: e99110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa H, Kaur P, Shirasawa K, Nichols P, Nagano S, Appels R, Erskine W, Isobe SN. 2016. Draft genome sequence of subterranean clover, a reference for genus Trifolium . Scientific Reports 6: 30358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch CN, Hirsch CD, Brohammer AB, Bowman MJ, Soifer I, Barad O, Shem‐Tov D, Baruch K, Lu F, Hernandez AG et al. 2016. Draft assembly of elite inbred line PH207 provides insights into genomic and transcriptome diversity in maize. Plant Cell 28: 2700–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittalmani S, Mahesh HB, Shirke MD, Biradar H, Uday G, Aruna YR, Lohithaswa HC, Mohanrao A. 2017. Genome and Transcriptome sequence of Finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genomics 18: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Kurata N, Wei X, Wang Z‐X, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W et al. 2012. A map of rice genome variation reveals the origin of cultivated rice. Nature 490: 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wei X, Sang T, Zhao Q, Feng QI, Zhao Y, Li C, Zhu C, Lu T, Zhang Z et al. 2010. Genome‐wide association studies of 14 agronomic traits in rice landraces. Nature Genetics 42: 961–U976. [DOI] [PubMed] [Google Scholar]
- Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia J‐M, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM et al. 2012. Comparative population genomics of maize domestication and improvement. Nature Genetics 44: 808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel B, Marques A, Soriano A, Marquès L, Divol F, Doumas P, Sallet E, Mancinotti D, Carrere S, Marande W et al. 2020. High‐quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nature Communications 11: 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung H‐Y, Shannon LM, Tian F, Bradbury PJ, Chen C, Flint‐Garcia SA, McMullen MD, Ware D, Buckler ES, Doebley JF et al. 2012. ZmCCT and the genetic basis of day‐length adaptation underlying the postdomestication spread of maize. Proceedings of the National Academy of Sciences, USA 109: E1913–E1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H, Izawa T. 2013. The coincidence of critical day length recognition for florigen gene expression and floral transition under long‐day conditions in rice. Molecular Plant 6: 635–649. [DOI] [PubMed] [Google Scholar]
- Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G et al. 2013. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). The Plant Journal 74: 715–729. [DOI] [PubMed] [Google Scholar]
- Jaiswal V, Gupta S, Gahlaut V, Muthamilarasan M, Bandyopadhyay T, Ramchiary N, Prasad M. 2019. Genome‐wide association study of major agronomic traits in foxtail millet (Setaria italica L.) using ddRAD sequencing. Scientific Reports 9: 5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis DE, Ho YS, Lightfoot DJ, Schmöckel SM, Li BO, Borm TJA, Ohyanagi H, Mineta K, Michell CT, Saber N et al. 2017. The genome of Chenopodium quinoa . Nature 542: 307–312. [DOI] [PubMed] [Google Scholar]
- Jegadeesan S, Raizada A, Dhanasekar P, Suprasanna P. 2021. Draft genome sequence of the pulse crop blackgram [Vigna mungo (L.) Hepper] reveals potential R‐genes. Scientific Reports 11: 11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X et al. 2013. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496: 91–95. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP. 2013. Demonstration of CRISPR/Cas9/sgRNA‐mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Research 41: e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang BO, Campbell MS, Stein JC, Wei X, Chin C‐S et al. 2017. Improved maize reference genome with single‐molecule technologies. Nature 546: 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce SA, Kamil A, Fleige L, Gahan CGM. 2019. The cholesterol‐lowering effect of oats and oat beta glucan: modes of action and potential role of bile acids and the microbiome. Frontiers in Nutrition 6: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenya SN, Mikwa EO, Song B, Odeny DA. 2021. Genetics and breeding for climate change in Orphan crops. Theoretical and Applied Genetics 134: 1787–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Kim B, Choi BS, Lee HO, Kim NH, Lee SJ, Kim HS, Shin MJ, Kim HW, Nam K et al. 2020. Genome assembly and annotation of soft‐shelled adlay (Coix lacryma‐jobi Variety ma‐yuen), a cereal and medicinal crop in the Poaceae Family. Frontiers in Plant Science 11: 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Kim SK, Kim MY, Lestari P, Kim KH, Ha B‐K, Jun TH, Hwang WJ, Lee T, Lee J et al. 2014. Genome sequence of mungbean and insights into evolution within Vigna species. Nature Communications 5: 5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Satyawan D, Shim S, Lee T, Lee J, Hwang WJ, Kim SK, Lestari P, Laosatit K, Kim KH et al. 2015. Draft genome sequence of adzuki bean, Vigna angularis . Science Reports 5: 8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Bayer PE, Milec Z, Vrána J, Yuan Y, Appels R, Edwards D, Batley J, Nichols P, Erskine W et al. 2017. An advanced reference genome of Trifolium subterraneum L. reveals genes related to agronomic performance. Plant Biotechnology Journal 15: 1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima CG, Guimarães GA, Nogueira SR, MacLean D, Cook DR, Steuernagel B, Baek J, Bouyioukos C, Melo Bdo V, Tristão G et al. 2016. A pigeonpea gene confers resistance to Asian soybean rust in soybean. Nature Biotechnology 34: 661–665. [DOI] [PubMed] [Google Scholar]
- Khoury CK, Bjorkman AD, Dempewolf H, Ramirez‐Villegas J, Guarino L, Jarvis A, Rieseberg LH, Struik PC. 2014. Increasing homogeneity in global food supplies and the implications for food security. Proceedings of the National Academy of Sciences, USA 111: 4001–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury CK, Brush S, Costich DE, Curry HA, Haan S, Engels JMM, Guarino L, Hoban S, Mercer KL, Miller AJ et al. 2022. Crop genetic erosion: understanding and responding to loss of crop diversity. New Phytologist 233: 84–118. [DOI] [PubMed] [Google Scholar]
- Kidane YG, Gesesse CA, Hailemariam BN, Desta EA, Mengistu DK, Fadda C, Pè ME, Dell'Acqua M. 2019. A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnology Journal 17: 1380–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer C, Severing E, Karl R, Bergonzi S, Koch M, Tresch A, Coupland G. 2017. Divergence of annual and perennial species in the Brassicaceae and the contribution of cis‐acting variation at FLC orthologues. Molecular Ecology 26: 3437–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Janila P, Vishwakarma MK, Khan AW, Manohar SS, Gangurde SS, Variath MT, Shasidhar Y, Pandey MK, Varshney RK. 2020. Whole‐genome resequencing‐based QTL‐seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnology Journal 18: 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CG, Chen HM, Sun HC. 1992. Membrane thermostability and heat tolerance of vegetable leaves. In: Kuo CG, ed. Adaptation of food crops to temperature and water stress. Tainan, Taiwan: AVRDC ‐ The World Vegetable Center, 160–168. [Google Scholar]
- Li C, Song W, Luo Y, Gao S, Zhang R, Shi ZI, Wang X, Wang R, Wang F, Wang J et al. 2019. The HuangZaoSi maize genome provides insights into genomic variation and improvement history of maize. Molecular Plant 12: 402–409. [DOI] [PubMed] [Google Scholar]
- Li C, Sun B, Li Y, Liu C, Wu X, Zhang D, Shi Y, Song Y, Buckler ES, Zhang Z et al. 2016. Numerous genetic loci identified for drought tolerance in the maize nested association mapping populations. BMC Genomics 17: 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li K, Zhang Q‐J, Zhu T, Zhang Y, Shi C, Liu Y‐L, Xia E‐H, Jiang J‐J, Shi C et al. 2020. Improved hybrid de novo genome assembly and annotation of African wild rice, Oryza longistaminata, from Illumina and PacBio sequencing reads. The Plant Genome 13: e20001. [DOI] [PubMed] [Google Scholar]
- Li X, Yadav R, Siddique KHM. 2020. Neglected and underutilized crop species: the key to improving dietary diversity and fighting hunger and malnutrition in Asia and the Pacific. Frontiers in Nutrition 7: 593711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Leveau A, Zhao Q, Feng QI, Lu H, Miao J, Xue Z, Martin AC, Wegel E, Wang J et al. 2021. Subtelomeric assembly of a multi‐gene pathway for antimicrobial defense compounds in cereals. Nature Communications 12: 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Shi J, Cai Z, Huang Y, Lv M, Du H, Gao Q, Zuo YI, Dong Z, Huang W et al. 2020. Evolution and domestication footprints uncovered from the genomes of Coix . Molecular Plant 13: 295–308. [DOI] [PubMed] [Google Scholar]
- Liu HJ, Yan J. 2019. Crop genome‐wide association study: a harvest of biological relevance. The Plant Journal 97: 8–18. [DOI] [PubMed] [Google Scholar]
- Liu J, Seetharam AS, Chougule K, Ou S, Swentowsky KW, Gent JI, Llaca V, Woodhouse MR, Manchanda N, Presting GG et al. 2020. Gapless assembly of maize chromosomes using long‐read technologies. Genome Biology 21: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S, Muñoz‐Amatriaín M, Hokin SA, Cisse N, Roberts PA, Farmer AD, Xu S, Close TJ. 2019. A genome‐wide association and meta‐analysis reveal regions associated with seed size in cowpea [Vigna unguiculata (L.) Walp]. Theoretical and Applied Genetics 132: 3079–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonardi S, Muñoz‐Amatriaín M, Liang Q, Shu S, Wanamaker SI, Lo S, Tanskanen J, Schulman AH, Zhu T, Luo MC et al. 2019. The genome of cowpea (Vigna unguiculata [L.] Walp.). The Plant Journal 98: 767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Li H, Hong Y, Zhang G, Wen S, Li X, Zhou G, Li S, Liu H, Liu H et al. 2018. Genome sequencing and analysis of the peanut B‐genome progenitor (Arachis ipaensis). Frontiers in Plant Science 9: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass BL, Robotham O, Chapman MA. 2017. Evidence for two domestication events of hyacinth bean (Lablab purpureus (L.) Sweet): a comparative analysis of population genetic data. Genetic Resources and Crop Evolution 64: 1221–1230. [Google Scholar]
- Mabhaudhi T, Chimonyo VGP, Chibarabada TP, Modi AT. 2017. Developing a roadmap for improving neglected and underutilized crops: a case study of South Africa. Frontiers in Plant Science 8: 2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidi S, Healey A, Huang PU, Grimwood J, Jenkins J, Barry K, Sreedasyam A, Shu S, Lovell JT, Feldman M et al. 2020. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nature Biotechnology 38: 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins PK, Nakayama TJ, Ribeiro AP, Cunha B, Nepomuceno AL, Harmon FG, Kobayashi AK, Molinari HBC. 2015a. Setaria viridis floral‐dip: a simple and rapid Agrobacterium‐mediated transformation method. Biotechnology Reports 6: 61–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins PK, Ribeiro AP, Cunha B, Kobayashi AK, Molinari HBC. 2015b. A simple and highly efficient Agrobacterium‐mediated transformation protocol for Setaria viridis. Biotechnology Reports 6: 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massawe FJ, Mayes S, Cheng A, Chai HH, Cleasby P, Symonds R, Ho WK, Siise A, Wong QN, Kendabie P et al. 2015. The potential for underutilised crops to improve food security in the face of climate change. Procedia Environmental Sciences 29: 140–141. [Google Scholar]
- Massawe F, Mayes S, Cheng A. 2016. Crop diversity: an unexploited treasure trove for food security. Trends in Plant Science 21: 365–368. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez GJ, Buckler E, Doebley J. 2002. A single domestication for maize is shown by multilocus microsatellite genotyping. Proceedings of the National Academy of Sciences, USA 99: 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maughan PJ, Lee R, Walstead R, Vickerstaff RJ, Fogarty MC, Brouwer CR, Reid RR, Jay JJ, Bekele WA, Jackson EW et al. 2019. Genomic insights from the first chromosome‐scale assemblies of oat (Avena spp.) diploid species. BMC Biology 17: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer A, Draba V, Jiang Y, Schnaithmann F, Sharma R, Schumann E, Kilian B, Reif JC, Pillen K. 2015. Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genomics 16: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes S, Ho WK, Chai HH, Gao X, Kundy AC, Mateva KI, Zahrulakmal M, Hahiree MKIM, Kendabie P, Licea LCS et al. 2019. Bambara groundnut: an exemplar underutilised legume for resilience under climate change. Planta 250: 803–820. [DOI] [PubMed] [Google Scholar]
- McClintock B. 1950. The origin and behavior of mutable loci in maize. Proceedings of the National Academy of Sciences, USA 36: 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick RF, Truong SK, Sreedasyam A, Jenkins J, Shu S, Sims D, Kennedy M, Amirebrahimi M, Weers BD, McKinley B et al. 2018. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. The Plant Journal 93: 338–354. [DOI] [PubMed] [Google Scholar]
- McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun QI, Flint‐Garcia S, Thornsberry J, Acharya C, Bottoms C et al. 2009. Genetic properties of the maize nested association mapping population. Science 325: 737–740. [DOI] [PubMed] [Google Scholar]
- Mi Y, Zhu Z, Qian G, Li Y, Meng X, Xue J, Chen Q, Sun W, Shi Y. 2020. Inducing hairy roots by agrobacterium rhizogenes‐mediated transformation in tartary buckwheat (Fagopyrum tataricum). Journal of Visualized Experiments 157: e60828. [DOI] [PubMed] [Google Scholar]
- Muchow RC. 1985. Phenology, seed yield and water use of grain legumes grown under different soil water regimes in a semi‐arid tropical environment. Field Crops Research 11: 81–97. [Google Scholar]
- Nguyen DQ, Van Eck J, Eamens AL, Grof CPL. 2020. Robust and reproducible Agrobacterium‐mediated transformation system of the C(4) genetic model species Setaria viridis . Frontiers in Plant Science 11: 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble TJ, Tao Y, Mace ES, Williams B, Jordan DR, Douglas CA, Mundree SG. 2018. Characterization of linkage disequilibrium and population structure in a mungbean diversity panel. Frontiers in Plant Science 8: 2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Schnable JC, Yeh CT, Wu L, Liu C, Hu HC, Dalgard CL, Sarkar S, Schnable PS. 2018. Linked read technology for assembling large complex and polyploid genomes. BMC Genomics 19: 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parween S, Nawaz K, Roy R, Pole AK, Venkata Suresh B, Misra G, Jain M, Yadav G, Parida SK, Tyagi AK et al. 2015. An advanced draft genome assembly of a desi type chickpea (Cicer arietinum L.). Scientific Reports 5: 12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A et al. 2009. The Sorghum bicolor genome and the diversification of grasses. Nature 457: 551–556. [DOI] [PubMed] [Google Scholar]
- Pham A‐T, Maurer A, Pillen K, Brien C, Dowling K, Berger B, Eglinton JK, March TJ. 2019. Genome‐wide association of barley plant growth under drought stress using a nested association mapping population. BMC Plant Biology 19: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VRP, Das S, Dikshit HK, Mishra GP, Aski M, Meena SK, Singh A, Pandey R, Singh MP, Tripathi K et al. 2020. Genome‐wide association analysis for phosphorus use efficiency traits in mungbean (Vigna radiata L. Wilczek) using genotyping by sequencing approach. Frontiers in Plant Science 11: 537766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robotham O, Chapman MA. 2015. Population genetic analysis of hyacinth bean (Lablab purpureus (L.) Sweet, Leguminosae) indicates an East African origin and variation in drought tolerance. Genetic Resources and Crop Evolution 64: 139–148. [Google Scholar]
- Roorkiwal M, Bharadwaj C, Barmukh R, Dixit GP, Thudi M, Gaur PM, Chaturvedi SK, Fikre A, Hamwieh A, Kumar S et al. 2020. Integrating genomics for chickpea improvement: achievements and opportunities. Theoretical and Applied Genetics 133: 1703–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychel S, Książkiewicz M. 2019. Development of gene‐based molecular markers tagging low alkaloid pauper locus in white lupin (Lupinus albus L.). Journal of Applied Genetics 60: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samtiya M, Aluko RE, Dhewa T. 2020. Plant food anti‐nutritional factors and their reduction strategies: an overview. Food Production, Processing and Nutrition 2: 6. [Google Scholar]
- Sandhu KS, You FM, Conner RL, Balasubramanian PM, Hou A. 2018. Genetic analysis and QTL mapping of the seed hardness trait in a black common bean (Phaseolus vulgaris) recombinant inbred line (RIL) population. Molecular Breeding 38: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CM, Romeiro D, Silva JP, Basso MF, Molinari HBC, Centeno DC. 2020. An improved protocol for efficient transformation and regeneration of Setaria italica . Plant Cell Reports 39: 501–510. [DOI] [PubMed] [Google Scholar]
- Saxena RK, Kale SM, Kumar V, Parupali S, Joshi S, Singh V, Garg V, Das RR, Sharma M, Yamini KN et al. 2017a. Genotyping‐by‐sequencing of three mapping populations for identification of candidate genomic regions for resistance to sterility mosaic disease in pigeonpea. Scientific Reports 7: 1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena RK, Singh VK, Kale SM, Tathineni R, Parupalli S, Kumar V, Garg V, Das RR, Sharma M, Yamini KN et al. 2017b. Construction of genotyping‐by‐sequencing based high‐density genetic maps and QTL mapping for fusarium wilt resistance in pigeonpea. Scientific Reports 7: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable JC. 2015. Genome evolution in maize: from genomes back to genes. Annual Review of Plant Biology 66: 329–343. [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. [DOI] [PubMed] [Google Scholar]
- Sennhenn A, Odhiambo JJO, Maass BL, Whitbread AM. 2017. Considering effects of temperature and photoperiod on growth and development of Lablab purpureus (L.) Sweet in the search of short‐season accessions for smallholder farming systems. Experimental Agriculture 53: 375–395. [Google Scholar]
- Senthil G, Williamson B, Dinkins RD, Ramsay G. 2004. An efficient transformation system for chickpea (Cicer arietinum L.). Plant Cell Reports 23: 297–303. [DOI] [PubMed] [Google Scholar]
- Shi J, Ma X, Zhang J, Zhou Y, Liu M, Huang L, Sun S, Zhang X, Gao X, Zhan W et al. 2019. Chromosome conformation capture resolved near complete genome assembly of broomcorn millet. Nature Communications 10: 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siadjeu C, Pucker B, Viehöver P, Albach DC, Weisshaar B. 2020. High contiguity de novo genome sequence assembly of trifoliate yam (Dioscorea dumetorum) using long read sequencing. Genes 11: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique KHM, Li X, Gruber K. 2021. Rediscovering Asia's forgotten crops to fight chronic and hidden hunger. Nature Plants 7: 116–122. [DOI] [PubMed] [Google Scholar]
- Singh NK, Gupta DK, Jayaswal PK, Mahato AK, Dutta S, Singh S, Bhutani S, Dogra V, Singh BP, Kumawat G et al. 2012. The first draft of the pigeonpea genome sequence. Journal of Plant Biochemistry and Biotechnology 21: 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparvoli F, Cominelli E. 2015. Seed biofortification and phytic acid reduction: a conflict of interest for the plant? Plants 4: 728–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben‐Zvi G et al. 2018. The maize W22 genome provides a foundation for functional genomics and transposon biology. Nature Genetics 50: 1282–1288. [DOI] [PubMed] [Google Scholar]
- Suárez‐Estrella D, Torri L, Pagani MA, Marti A. 2018. Quinoa bitterness: causes and solutions for improving product acceptability. Journal of the Science of Food and Agriculture 98: 4033–4041. [DOI] [PubMed] [Google Scholar]
- Sugihara Y, Darkwa K, Yaegashi H, Natsume S, Shimizu M, Abe A, Hirabuchi A, Ito K, Oikawa K, Tamiru‐Oli M et al. 2020. Genome analyses reveal the hybrid origin of the staple crop white Guinea yam (Dioscorea rotundata). Proceedings of the National Academy of Sciences, USA 117: 31987–31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ma D, Tang L, Zhao M, Zhang G, Wang W, Song J, Li X, Liu Z, Zhang W et al. 2019. Population genomic analysis and de novo assembly reveal the origin of weedy rice as an evolutionary game. Molecular Plant 12: 632–647. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, Song W, Zhang M, Cui Y, Dong X et al. 2018. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nature Genetics 50: 1289–1295. [DOI] [PubMed] [Google Scholar]
- Tamiru M, Natsume S, Takagi H, White B, Yaegashi H, Shimizu M, Yoshida K, Uemura A, Oikawa K, Abe A et al. 2017. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biology 15: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Brachypodium Initiative . 2010. Genome sequencing and analysis of the model grass Brachypodium distachyon . Nature 463: 763–768. [DOI] [PubMed] [Google Scholar]
- Thielen PM, Pendleton AL, Player RA, Bowden KV, Lawton TJ, Wisecaver JH. 2020. Reference genome for the highly transformable Setaria viridis ME034V. G3 (Bethesda) 10: 3467–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES IV. 2001. Dwarf8 polymorphisms associate with variation in flowering time. Nature Genetics 28: 286–289. [DOI] [PubMed] [Google Scholar]
- Upadhyaya HD, Vetriventhan M, Deshpande SP, Sivasubramani S, Wallace JG, Buckler ES, Hash CT, Ramu P. 2015. Population genetics and structure of a global foxtail millet germplasm collection. Plant Genome 8: eplantgenome2015.07.0054. [DOI] [PubMed] [Google Scholar]
- VanBuren R, Man Wai C, Wang X, Pardo J, Yocca AE, Wang H, Chaluvadi SR, Han G, Bryant D, Edger PP et al. 2020. Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nature Communications 11: 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MTA, Azam S, Fan G, Whaley AM et al. 2012. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource‐poor farmers. Nature Biotechnology 30: 83–89. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Penmetsa RV, Dutta S, Kulwal PL, Saxena RK, Datta S, Sharma TR, Rosen B, Carrasquilla‐Garcia N, Farmer AD et al. 2010. Pigeonpea genomics initiative (PGI): an international effort to improve crop productivity of pigeonpea (Cajanus cajan L.). Molecular Breeding 26: 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Saxena RK, Upadhyaya HD, Khan AW, Yu Y, Kim C, Rathore A, Kim D, Kim J, An S et al. 2017a. Whole‐genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nature Genetics 49: 1082–1088. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Shi C, Thudi M, Mariac C, Wallace J, Qi P, Zhang H, Zhao Y, Wang X, Rathore A et al. 2017b. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nature Biotechnology 35: 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar'an B et al. 2013. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nature Biotechnology 31: 240–246. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Thudi M, Roorkiwal M, He W, Upadhyaya HD, Yang W, Bajaj P, Cubry P, Rathore A, Jian J et al. 2019. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nature Genetics 51: 857–864. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhou G, Jian J, Yang H, Renshaw D, Aubert MK, Clements J, He T, Sweetingham M, Li C. 2021. Whole‐genome assembly and resequencing reveal genomic imprint and key genes of rapid domestication in narrow‐leafed lupin. The Plant Journal 105: 1192–1210. [DOI] [PubMed] [Google Scholar]
- Wang W, Feng B, Xiao J, Xia Z, Zhou X, Li P, Zhang W, Wang Y, Møller BL, Zhang P et al. 2014. Cassava genome from a wild ancestor to cultivated varieties. Nature Communications 5: 5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T, Fuentes RR, Zhang F et al. 2018. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen S, Ma X, Yssel AEJ, Chaluvadi SR, Johnson MS, Gangashetty P, Hamidou F, Sanogo MD, Zwaenepoel A et al. 2021. Genome sequence and genetic diversity analysis of an under‐domesticated orphan crop, white fonio (Digitaria exilis). GigaScience 10: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Wang C, Kang X, Zhao H, Elena Gamo M, Starker CG, Crisp PA, Zhou P, Springer NM, Voytas DF et al. 2020. Optimization of multiplexed CRISPR/Cas9 system for highly efficient genome editing in Setaria viridis . The Plant Journal 104: 828–838. [DOI] [PubMed] [Google Scholar]
- Weller JL, Liew LC, Hecht VFG, Rajandran V, Laurie RE, Ridge S, Wenden B, Vander Schoor JK, Jaminon O, Blassiau C et al. 2012. A conserved molecular basis for photoperiod adaptation in two temperate legumes. Proceedings of the National Academy of Sciences, USA 109: 21158–21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing A, Gao Y, Ye L, Zhang W, Cai L, Ching A, Llaca V, Johnson B, Liu L, Yang X et al. 2015. A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize. Journal of Experimental Botany 66: 3791–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wu DI, Yang T, Sun C, Wang Z, Han B, Wu S, Yu A, Chapman MA, Muraguri S et al. 2021. Genomic insights into the origin, domestication and genetic basis of agronomic traits of castor bean. Genome Biology 22: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Zhang Q, Yuan W, Xu F, Muhammad Aslam M, Miao R, Li Y, Wang Q, Li X, Zhang X et al. 2020. The genome evolution and low‐phosphorus adaptation in white lupin. Nature Communications 11: 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B. 2020. Grand challenges in genome editing in plants. Frontiers in Genome Editing 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Moeinzadeh M‐H, Kuhl H, Helmuth J, Xiao P, Haas S, Liu G, Zheng J, Sun Z, Fan W et al. 2017. Haplotype‐resolved sweet potato genome traces back its hexaploidization history. Nature Plants 3: 696–703. [DOI] [PubMed] [Google Scholar]
- Yang K, Tian Z, Chen C, Luo L, Zhao B, Wang Z, Yu L, Li Y, Sun Y, Li W et al. 2015. Genome sequencing of adzuki bean (Vigna angularis) provides insight into high starch and low fat accumulation and domestication. Proceedings of National Academy of Sciences, USA 112: 13213–13218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Liu J, Gao Q, Gui S, Chen LU, Yang L, Huang J, Deng T, Luo J, He L et al. 2019. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nature Genetics 51: 1052–1059. [DOI] [PubMed] [Google Scholar]
- Yasui Y, Hirakawa H, Oikawa T, Toyoshima M, Matsuzaki C, Ueno M, Mizuno N, Nagatoshi Y, Imamura T, Miyago M et al. 2016a. Draft genome sequence of an inbred line of Chenopodium quinoa, an allotetraploid crop with great environmental adaptability and outstanding nutritional properties. DNA Research 23: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Hirakawa H, Ueno M, Matsui K, Katsube‐Tanaka T, Yang SJ, Aii J, Sato S, Mori M. 2016b. Assembly of the draft genome of buckwheat and its applications in identifying agronomically useful genes. DNA Research 23: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Fan L. 2020. Orphan crops and their wild relatives in the genomic era. Molecular Plant 14: 27–39. [DOI] [PubMed] [Google Scholar]
- Ye C‐Y, Wu D, Mao L, Jia L, Qiu J, Lao S, Chen M, Jiang B, Tang W, Peng Q et al. 2020. The genomes of the allohexaploid Echinochloa crus‐galli and its progenitors provide insights into polyploidization‐driven adaptation. Molecular Plant 13: 1298–1310. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–92. [DOI] [PubMed] [Google Scholar]
- Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, Xie M, Zeng P, Yue Z, Wang W et al. 2012. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nature Biotechnology 30: 549–554. [DOI] [PubMed] [Google Scholar]
- Zhang K, He M, Fan YU, Zhao H, Gao B, Yang K, Li F, Tang YU, Gao Q, Lin T et al. 2021. Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biology 22: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li X, Ma B, Gao Q, Du H, Han Y, Li Y, Cao Y, Qi M, Zhu Y et al. 2017. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Molecular Plant 10: 1224–1237. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhu M, Ren L, Li A, Chen G, Hu Z. 2018. The SlFSR gene controls fruit shelf‐life in tomato. Journal of Experimental Botany 69: 2897–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen X, Lu C, Ye J, Zou M, Lu K, Feng S, Pei J, Liu C, Zhou X et al. 2018. Genome‐wide association studies of 11 agronomic traits in Cassava (Manihot esculenta Crantz). Frontiers in Plant Science 9: 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Liu B, Piao S, Wang X, Lobell DB, Huang Y, Huang M, Yao Y, Bassu S, Ciais P et al. 2017. Temperature increase reduces global yields of major crops in four independent estimates. Proceedings of the National Academy of Sciences, USA 114: 9326–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bayer PE, Ruperao P, Saxena RK, Khan AW, Golicz AA, Nguyen HT, Batley J, Edwards D, Varshney RK. 2020. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnology Journal 18: 1946–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Feng QI, Lu H, Li Y, Wang A, Tian Q, Zhan Q, Lu Y, Zhang L, Huang T et al. 2018. Pan‐genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nature Genetics 50: 278–284. [DOI] [PubMed] [Google Scholar]
- Zhao ZY, Cai T, Tagliani L, Miller M, Wang N, Pang H, Rudert M, Schroeder S, Hondred D, Seltzer J et al. 2000. Agrobacterium‐mediated sorghum transformation. Plant Molecular Biology 44: 789–798. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Chebotarov D, Kudrna D, Llaca V, Lee S, Rajasekar S, Mohammed N, Al‐Bader N, Sobel‐Sorenson C, Parakkal P et al. 2020. A platinum standard pan‐genome resource that represents the population structure of Asian rice. Scientific Data 7: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang W, Chen H, Yang M, Wang J, Pandey MK, Zhang C, Chang W‐C, Zhang L, Zhang X, Tang R et al. 2019. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nature Genetics 51: 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Chen A, Xiao L, Muller HM, Ache P, Haberer G, Zhang M, Jia W, Deng P, Huang RU et al. 2017. A high‐quality genome assembly of quinoa provides insights into the molecular basis of salt bladder‐based salinity tolerance and the exceptional nutritional value. Cell Research 27: 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Li L, Miki D, Li D, Tang Q, Xiao L, Rajput S, Deng P, Peng LI, Jia W et al. 2019. The genome of broomcorn millet. Nature Communications 10: 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 List of important traits dissected by GWAS in rice.
Table S2 List of important traits dissected by GWAS in maize.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.