

Abstract
Prostate cancer is morphologically and molecularly heterogeneous, which poses obstacles for early diagnosis and treatment. Advancements in understanding the heterogeneity of prostate cancer will help navigate through these challenges and ultimately benefit patients. In this study, we integrated single-cell sequencing for transposase-accessible chromatin and whole transcriptome in prostate cancer cell lines, aiming to decode the epigenetic plasticity upon enzalutamide (ENZ) treatment. By comparing the cell populations representing early-treatment response or resistance to the initial tumor cells, we identified seven signature gene sets; they present consistent trends of chromatin closing co-occurred with down-regulated genes during early response and chromatin opening with up-regulated genes upon maintaining drug resistance. In the molecular signatures, we found genes ZNF337, MAPK15, and ESRRG are favorable in progression-free prognosis during early response, while genes CCDC150, CCDC18, and POC1A marked poor prognosis underpinning the pre-existing drug resistance in The Cancer Genome Atlas prostate adenocarcinoma cohort. Ultimately, drug-target analyses nominated combinatory drug candidates to either enhance early-treatment response or potentially overcome ENZ resistance. Together, our integrative, single-cell multi-omics approach in pre-clinical models is effective in identifying informative signatures from complex molecular events, illustrating diverse drug responses in prostate cancer, and invoking novel combinatory drug strategies to inform clinical decision making.
Keywords: MT: Bioinformatics, single-cell RNA sequencing, single-cell ATAC sequencing, prostate cancer, enzalutamide treatment, combinatory drug candidates, tumor prognosis
Graphical abstract
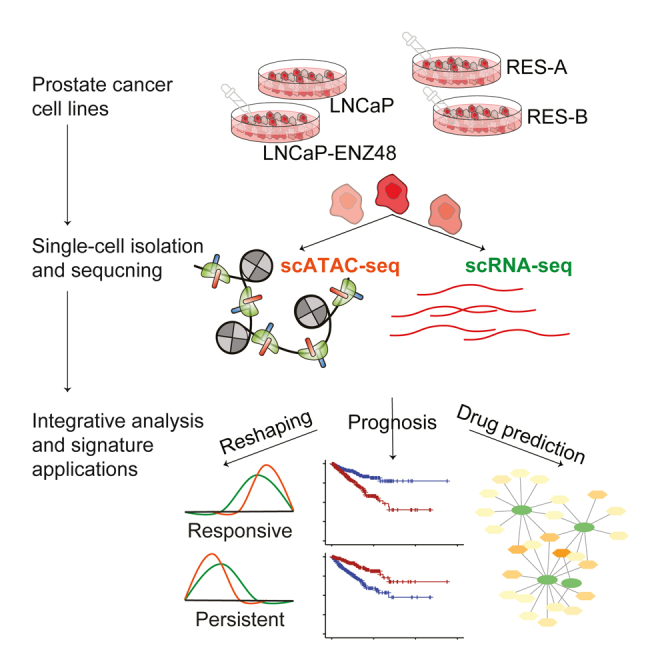
Fan et al. integrated single-cell multi-omics sequencing data in prostate cancer cell lines upon enzalutamide treatment to decode the cellular heterogeneity captured by epigenetic landscape shifting between responsive and resistant phenotypes. Seven gene signature sets were extracted with prognostic potentials and applied to nominate potential combinatory drug usage.
Introduction
Prostate cancer (PCa) is the second most frequent malignancy in men and the fifth leading cause of death worldwide.1 Androgen deprivation therapy (ADT) is the preferred standard treatment for advanced and high-risk prostate cancer, which is also known as hormone therapy2 ADT can induce remission, but its resistance brings recurrence and aggressive progression of cancer, such as castration-resistant prostate cancer (CRPC).3 Resistance to next-generation anti-androgen agents, such as enzalutamide (ENZ), accounts for a major challenge for effective treatments of advanced PCa.4,5 However, ENZ-induced molecular phenotypes in pre-clinical models remain understudied.
Agents targeting the androgen receptor (AR) signaling pathway are initially effective, but they eventually fail in the majority of CRPC patients. One of the main driving forces of treatment failure is the morphological and molecular heterogeneity within PCa.6,7 It is also the reason why patients with PCa develop ENZ resistance early and easily. Several genomic mechanisms related to ENZ resistance have been proposed, such as AR gene mutation8 and gene duplication.9,10,11 Despite the central role of AR highlighted in the development of ENZ resistance, other studies have suggested that AR-negative or AR-low PCa cells or pre-existing and genetically diverse clones in untreated primary tumors could hold the keys to therapy resistance and disease progression.4,12,13 Regardless, the majority of these genomics and/or epigenomics studies to date originate from the traditional bulk analysis of tumors, which lacks the power to capture cell population-level difference among cell types in the tumor.14,15 Advancements in single-cell sequencing techniques are thus here to help us profile the genetic and epigenetic heterogeneous landscapes of prostate cancer. Recently, a study used both single-cell RNA sequencing (scRNA-seq) and single-cell assays for transposase-accessible chromatin (scATAC-seq) to characterize a subset of pre-existing cells that were associated with tumor relapse in advanced PCa.16 Similarly, in primary PCa, pre-existing castration-resistant PCa-like cells were also observed using scRNA-seq, which promoted resistance to hormonal therapy.17 And single-cell RNA-seq analysis in localized PCa patients focused on tumor microenvironment revealed the heterogeneity of tumor-associated epithelial cell states.18,19 However, not many investigations have been particularly performed to study the effects of ENZ-induced diverse cellular phenotypes via integrating scRNA-seq and scATAC-seq data, and to understand their underlying molecular mechanisms.
Here, we integrated scRNA-seq and scATAC-seq data in PCa cell lines in response to ENZ treatment to decode the cellular heterogeneity captured by epigenetic plasticity. Seven gene signature sets were extracted by integrating differential events calculated using both scRNA-seq and scATAC-seq data. On the basis of gene set enrichment analysis, we revealed the functional difference between the ENZ-induced drug-responsive phenotype and the pre-existing drug-resistant phenotype when comparing with the initial tumor cell population. Moreover, survival analysis using the ENZ treatment-naive Cancer Genome Atlas (TCGA) prostate adenocarcinoma cohort demonstrated the prognostic values of our signature genes. Last, drug-gene network analysis predicted the combinatory use of drugs potentially to overcome ENZ resistance. Altogether, our study sheds light on the cellular heterogeneity captured by epigenetic reshaping upon drug treatment, and our signature gene sets characterized by epigenetic reshaping can be applied for novel combinatory drug discoveries and served as useful prognostic biomarkers in PCa patients.
Results
Cellular heterogeneity captured by multi-omic single-cell sequencing
Matched and publicly available single-cell RNA sequencing and single-cell assays for transposase-accessible chromatin data in four prostate cancer cell lines were used to characterize the cellular heterogeneity induced by enzalutamide treatment.16 ENZ, an oral androgen receptor inhibitor, is approved by the U.S. Food and Drug Administration (FDA) as the last line therapy to extend the survival time of castration-resistant PCa patients.20 These four cell lines included parental/control cell line LNCaP, short-term ENZ-exposed cell line LNCaP-ENZ48, and ENZ-resistant cell lines RES-A and RES-B derived from LNCaP via long-term exposure to AR inhibitor agents.21 Almost inevitably, patients develop ENZ resistance along the course of treatment. Cancer cell line models showing dynamic cellular phenotypes upon ENZ treatment will thus help us understand the molecular mechanisms underpinning ENZ-induced cellular dynamics.
Single-cell sequencing data from each PCa cell line profiled using both scRNA-seq and scATAC-seq were processed and integrated, respectively (Figures 1A, 1B, S1A, and S1B). In total, 13 cell clusters were yielded for each type of data after initial quality control procedures (Table S1; also see materials and methods) as visualized using the first two UMAP (uniform manifold approximation and projection) dimensions (Figures S1A and S1B). To define diverse cellular phenotypes in response to ENZ treatment, cellular compositions for each cell cluster were calculated and compared (Figures S1C and S1D). We then adopted the definitions for diverse phenotypes on the basis of the clustering compositions of single-cell sequencing data with similar standards from a previous study.16 From scRNA-seq clustering (Figure S1C), initial phenotype was defined as clusters of 1, 4, and 9 with their dominant compositions being cells from parental cell line LNCaP and/or LNCaP-ENZ48 with short-term treatment (see materials and methods). ENZ-induced responsive phenotype, defined as clusters most prevalent in RES-A or RES-B, contained cell clusters of 0, 5, 8, and 12. And pre-existing ENZ-resistant (i.e., persistent) phenotype, equally prevalent through all four cell lines, included cell clusters of 3, 6, 7, 10, and 11 (Figure S1C). In total, we selected 3,862, 4,549, and 4,840 cells using scRNA-seq data which represented initial, induced responsive, and drug-resistant phenotypes, separately. Meanwhile, using scATAC-seq data, 3,363, 3,191, and 2,911 cells were chosen to represent initial (cell clusters 1 and 3 included), ENZ-induced responsive (cell clusters 2 and 4 included), and drug-resistant (cell clusters 0 and 7 included) phenotypes, respectively (Figure S1D). To further examine our identified three cellular phenotypes from single-cell data, we assembled a pseudo-bulk read count table per cell cluster within each single-cell dataset. Principal components were calculated to show the membership similarity among all the cell clusters (Figures S1E and S1F). As the result, our three cellular phenotypes defined on the basis of cell line compositions totally aligned with the pseudo-bulk principal analysis. Additionally, we applied label transfer to explore whether cellular phenotypes were consistent between scRNA-seq and scATAC-seq data. Using gene activity score calculated from scATAC-seq, we could conduct label transfer from scRNA-seq to scATAC-seq data. The consensus ratio for initial and induced cell populations between scRNA-seq and scATAC-seq datasets were close to 80% (Figure S1G). As expected, the persistent cell population yielded a lower than average consensus ratio because of the vast amount of heterogeneity within this phenotype.
Figure 1.

ENZ-induced cellular phenotype shift
Dimension reduction uniform manifold approximation and projection (UMAP) plots are shown using scRNA-seq (A) and scATAC-seq data (B) in four PCa cell lines. Seurat clusters are colored according to their phenotypes as determined by their cellular compositions. Clusters with ambiguous classifications in gray are not selected for comparison analysis. Annotations for the color-coded phenotypes and shape-coded cell lines are on top of each plot. Volcano plots for comparisons between phenotypes of early response and initial and between that of persistent and initial are shown for scRNA-seq data (C and D) and for scATAC-seq data (E and F). Top differential events: differentially expressed genes (DEGs) or differentially accessible regions (DARs) are labeled on the plots.
Differential events underpinning the ENZ-induced cellular heterogeneity
Differential events including differentially expressed genes (DEGs) and differentially accessible regions (DARs) were calculated by comparing the induced to initial phenotypes and by comparing the resistant to initial phenotypes using scRNA-seq (Figures 1C and 1D; Table S2) and scATAC-seq data (Figures 1E and 1F; Table S3), respectively. In total, we identified 3,686 and 3,097 DEGs for comparison between induced and initial phenotypes, and that between resistant and initial phenotypes (Table S2; see materials and methods). Meanwhile, 3,305 and 3,887 DARs were identified using scATAC-seq data, respectively (Table S3). A reshaping in open chromatin regions was indicated upon ENZ treatment, which showed significantly higher number of open chromatin regions (∼99.9%) in cells with resistant phenotype (Figure 1F). Meanwhile, approximately 81% of the genes were up-regulated when comparing resistant to initial cell populations (Figure 1D). A clearly consistent trend between single-cell epigenetic and transcriptomic alterations was thus observed.
To demonstrate the epigenetic reshaping in response to ENZ treatment, we combined both DEGs and DARs to generate 7 different gene signature sets which represent bi-directional effects of epigenetic regulations on gene expression (Table S4; Figures 2A and 2B). Interestingly enough, a much higher number of DEGs are down-regulated while having open chromatin regions closed when comparing induced phenotype to initial phenotype (Figure 2A). On the contrary, when comparing pre-existing resistant to initial cell populations, we observed a dominated effect of open chromatin state accompanied by up-regulated genes (Figure 2B). As shown in Figure 2C, our gene signature sets are mostly exclusive to their own, with some gene sets partially overlapping with other gene sets showing the same up- or down-regulations within the two comparisons. For example, 13 down-regulated genes were shared between the two comparisons, which were coupled with open chromatin states when comparing persistent to initial phenotype but co-occurred with closed chromatin regions when comparing induced to initial phenotype. This suggests potentially different functional regulations from other layers of omics were used in ENZ-induced responsive cancer cells.
Figure 2.

Bi-directional epigenetic regulations on gene expression
(A) Gene signature sets yielded by comparing induced with initial phenotypes: closed/down (genes are down-regulated while their nearest chromatin accessibility regions are closed), closed/up, open/down, and open/up. (B) Gene signature sets generated by comparing persistent with initial phenotypes. (C) Upset plot showing the intersections among the seven gene signature sets. Heatmaps show the validation of signature genes from the comparison between induced and initial phenotype (D) and that between persistent and initial phenotype (E), using an independent bulk RNA-seq dataset. Up to top 20 differential genes are labeled.
To validate our identified gene signatures from single-cell data, we downloaded a recently released and normalized bulk RNA sequencing (RNA-seq) dataset,22 containing three similar phenotypes of initial, induced responsive, and persistent from the xenografts of prostate cancer cell line LNCaP. Despite the difference in gene expression profiling platforms and cell line culture conditions, we observed a decent proportion of our identified gene signatures consistently presented in the above independent dataset (Figures 2D and 2E). In total, we could confirm the consistency in differential regulations of 142 and 104 of the 241 and 335 up- and down-regulated signature genes, respectively, when comparing early-responsive to initial cellular phenotype (Figure 2D). For the comparison between persistent and initial phenotype, we confirmed 187 and 10 of the 377 and 66 up- and down-regulated signature genes, respectively (Figure 2E).
Functional divergence between the ENZ-induced early response and pre-existing drug resistance
Gene set enrichment analysis was carried out to demonstrate functional shift between the ENZ-induced response and pre-existing drug-resistant cell populations when comparing with the initial tumor cells (Figures 3A and 3B; Table S5). Commonly used Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology terms enriched within each comparison were curated in Table S6. Altogether, we showcased two gene signature sets with the highest number of genes from the two comparisons. Among the top 30 most enriched gene sets that formed 5 major functional clusters on the basis of semantic similarities, androgen-related gene sets were significantly down-regulated upon responding to the ENZ treatment (Figure 3A, golden branch). Aldehyde dehydrogenase 1A3 (ALDH1A3), which stands out in the gene cloud, has been implicated in the survival and proliferation of prostate cancer cells.23 ALDH1A3 protein is also a key inactivator of reactive oxygen species (ROS)-generated aldehydes, which is a perspective target for the development of new chemotherapeutic drugs thus indicating a potential combinatory use of drugs with ENZ treatment.24 On the other hand, the activation of the classic transcriptomic switch to a mitosis-phase program (Figure 3B, pink branch) as well as stemness were significantly up-regulated in tumor cells with pre-existing drug resistance.25 Interestingly, tubule cell-related gene signatures in kidneys were highly enriched in our down-regulated gene signatures (Figure 3A, pink and blue branches). Proximal tubule cell populations were reportedly associated with pathogenic expression signatures as defined by the expression quantitative trait loci (eQTL) from genome-wide association studies (GWASs).26 This down-regulation of tubule cell-regulated gene features hints at a potential back-to-normal effect in prostate cancer cells showing early response subject to ENZ treatment.
Figure 3.

Functional divergence underlying the epigenetic landscape shift
Gene set enrichment analysis using two signature gene sets from comparison between induced and initial (A) and that between persistent and initial (B). The 30 most enriched gene sets are clustered to form major functional branches on the basis of semantic similarities (on the left). Genes contained with each branch are visualized by word clouds (on the right). Genes shown in larger letters represent higher frequency.
Additionally, enrichment analysis of transcription factors (TFs) was conducted to show the most relevant regulators enriched in the differentially accessible regions identified from both the ENZ-induced responsive (Figure S2A) and pre-existing drug-resistant phenotypes (Figure S2B). Top 30 enriched TFs from each comparison are visualized in Figure S2C. In total, 20 of 40 pooled top enriched TFs were shared between comparisons. Consistently, we observed stemness-related features were enriched in persistent phenotype (Figure 3B), as illustrated by its uniquely enriched TFs, such as KLF1, KLF4, and KLF927 (Figure S2C). Meanwhile, Forkhead box (Fox) TFs, for instance FOXA1 and FOXO1, can help shape AR signaling that drives the growth and survival of prostate cancer cells (Figure S2C).28,29
In agreement with the functional shift observed, we also identified survival-related genes in both early-responsive and persistent groups of tumor cells using The Cancer Genome Atlas ENZ treatment-naive prostate adenocarcinoma cohort (Table S7; Figure 4). Particularly, we aimed to search for gene signatures responsible for better prognosis in the comparison between early-responsive and initial group (Figure 4A), and worse prognosis in the other comparison between persistent and initial group (Figure 4B). Differential directions inherited from our single-cell multi-omics analysis were also taken into consideration in order to determine whether they were prognostic genes. Among the early-responsive signature genes, we showed that lower expression of genes ZNF337 and MAPK15, and higher expression of gene ESRRG were favorable in progression-free prognosis despite different regulatory effects from chromatin accessibility on gene expression (Figure 4A). Although higher expression of genes CCDC150, CCDC18, and POC1A was indicators of poor progression-free prognosis in patients with potential drug resistance (Figure 4B). Among the six prognosis-related gene markers, MAPK15 is the only one that has been previously reported to play important roles in the pathogenesis of nasopharyngeal cancer (NPC), osteosarcoma (OS), and gastric cancer.30,31,32 Particularly, for patients with OS, inhibiting MAPK15 expression significantly decreased OS cell metastasis which remains the primary cause of death.31 Additionally, MAPK15 is also a key modulator of autophagy and, through this, of cell transformation, which might offer plenty of potential targets for future therapeutic interventions in human cancers.33
Figure 4.

Kaplan-Meier survival analysis using progression-free data in the TCGA prostate adenocarcinoma cohort
(A) Prognosis-related gene markers from ENZ-induced group. (B) Prognosis-related gene markers from ENZ-resistant group. Patients are stratified into two groups on the basis of the median gene expression of each marker gene. FDR-adjusted p values are labeled below the curves in each plot.
Identification of potential combinatory use of drugs to overcome ENZ resistance
We employed the DeSigN (Differentially Expressed Gene Signatures – Inhibitors) webserver to link our gene signatures to potential combinatory drug treatment.34 Connectivity scores were measured by DeSigN to assess the correlation of gene perturbation profile of a particular drug treatment on cancer cell lines from the Connectivity Map datasets with query gene list. In our study, the query gene lists were ranked on the basis of their gene expression fold changes upon ENZ treatment (see materials and methods). Therefore, a significant positive score would indicate a consistent trend of gene expression alterations between our query gene lists and drug-induced gene perturbation profiles. On the contrary, a significant negative score represented a nearly opposite trend between drug-induced perturbation profiles and our query gene lists. Specifically, we sought to rank drugs with positive connectivity scores in early-responsive phenotype to enhance the induced gene signatures (Table 1). Conversely, we considered negative connectivity scores in order to reverse the effects of persistent/resistant phenotype and to identify drugs that could induce an opposite perturbation profile than our query gene lists (Table 2). However, the exact drug targets, or the macromolecule which drug compounds directly bind to, may not be captured in our dataset. In such cases, “undefined target genes” were indicated. The drug-gene networks are shown in Figure 5, excluding drugs with undefined targets.
Table 1.
DeSigN prediction of drug candidates for enzalutamide-responsive phenotype
| Drug | Gene target | Connectivity score | p value |
|---|---|---|---|
| Shikonin | undefined | 1.000 | 0.000 |
| Lapatinib | EGFR, ERBB2 | 0.992 | 0.018 |
| Bleomycin | LIG1, LIG3 | 0.991 | 0.028 |
| A-443654 | undefined | 0.991 | 0.029 |
| CGP-082996 | undefined | 0.990 | 0.015 |
| Erlotinib | EGFR, NR1I2 | 0.989 | 0.021 |
| WZ-1-84 | undefined | 0.989 | 0.018 |
| CGP-60474 | undefined | 0.983 | 0.039 |
| SL-0101-1 | undefined | 0.980 | 0.046 |
| A-770041 | Undefined | 0.980 | 0.032 |
| BX-795 | CDK2, CHEK1, GSK3B, KDR, PDK1, PDPK1 | −1.000 | 0.000 |
Table 2.
DeSigN predictions of drug candidates for ENZ-resistant phenotype
| Drug | Target | Connectivity score | p value |
|---|---|---|---|
| AKT-inhibitor-VIII | undefined | 1.000 | 0.000 |
| Etoposide | TOP2A, TOP2B | 0.994 | 0.008 |
| OSU-03012 | undefined | 0.989 | 0.021 |
| Bleomycin | LIG1, LIG3 | 0.987 | 0.020 |
| PD-0332991 | CDK4, CDK6 | 0.977 | 0.031 |
| QS11 | undefined | 0.973 | 0.046 |
| AZD6482 | PIK3CA, PIK3CB, PIK3CD, PIK3CG | 0.972 | 0.040 |
| GW843682X | undefined | −0.969 | 0.043 |
| BX-795 | CDK2, CHEK1, GSK3B, KDR, PDK1, PDPK1 | −0.973 | 0.049 |
| GNF-2 | ABL1, BCR | −0.976 | 0.042 |
| Nilotinib | ABL1, KIT | −0.977 | 0.047 |
| AZ628 | undefined | −0.980 | 0.036 |
| WO2009093972 | undefined | −0.982 | 0.029 |
| Roscovitine | CDK2, CDK9 | −0.989 | 0.018 |
| AS601245 | undefined | −0.990 | 0.014 |
| Nutlin-3a | undefined | −1.000 | 0.000 |
Figure 5.

Combinatory therapeutics strategy derived using drug-gene network
Drug-gene network analysis for ENZ-induced (A) and ENZ-resistant gene signatures (B). ENZ-driven gene signatures are shown in the middle of each network, connected by dotted line. Predicted candidate drugs with potential combinatory use are shown if they target genes driven by the ENZ treatment. Predicted drugs are colored according to their connectivity score, with orange being negative and blue positive.
Particularly, we queried the DeSigN webserver using DEGs of 205 up- and 137 down-regulated genes extracted from the comparison of the early-responsive cells with initial cells (Table 1 and Figure 5A), and the DEGs of 379 up- and 66 down-regulated genes from the comparison of persistent cells with initial cells, respectively (Table 2; Figure 5B). For comparison between induced to initial phenotypes, 10 drugs in total were returned with positive connectivity scores (p < 0.05), indicating similar or enhanced responses as ENZ treatment when applied as treatments (Table 1). Among the 10 drugs identified, 6 have been already documented to be potentially beneficial in treatment for prostate cancer and/or in human cancers; these drugs included shikonin, lapatinib, erlotinib, bleomycin, A-443654, and SL0101-1.35,36,37,38,39,40 Shikonin, which ranked as the first, on the basis of connectivity score, is a traditional Chinese medicine, which has been proved to induce antitumor effects in multiple tumors, including PCa.35,41 Shikonin promotes antitumor effects by inducing apoptosis and more dominantly necroptosis in both parental and docetaxel-resistant PCa cells.35 Shikonin also inhibits the viability, proliferation, migration, and invasion of prostate cancer stem cells.42 Furthermore, it has enhanced antitumor effect of cabazitaxel, a drug indicated for advanced prostate cancer,42 and decreased the transcriptional activity of AR.43 Although shikonin has no defined target, it can inhibit the expression of ALDH3A1, ABCG2,42 MMP-2, and MMP-9.44 Lapatinib (ranked second with connectivity score) and erlotinib (ranked sixth), approved by the Food and Drug Administration, both have ErbB (EGFR/HER2/ErbB3/ErbB4) family molecules as their targets. It is expected that these molecules would be relevant to PCa, as epidermal growth factor receptor is overexpressed in a number of cancers.45,46 Lapatinib is an oral dual tyrosine kinase inhibitor that can block HER1 and HER2 tyrosine kinase activity to inhibit tumor cell growth.36 Although there is no determined evidence that lapatinib is effective for prostate cancer treatment, some research has shown that lapatinib might be a viable therapeutic option for castration-resistant prostate cancer.47 Erlotinib is used to treat non-small cell lung cancer (NSCLC) and pancreatic cancer.39 It may also be administered to patients with advanced prostate cancer and chemotherapy-naive CRPC, as it has been shown to improve clinical benefits.46 Evidence also indicates that docetaxel combined with erlotinib can be delivered safely in elderly patients with androgen-independent prostate cancer (AIPC).48 Bleomycin, which ranked third in connectivity score, is approved by FDA to treat squamous cell carcinomas, testicular cancers, and malignant lymphomas.37 A study from Ueki et al.49 showed that electroporation with bleomycin might be effective for prostate cancer, especially AIPC. Another drug of interest, A-443654, which ranked fourth in connectivity score, is a potent small-molecule inhibitor of Akt that can induce Akt Ser-473 phosphorylation in all human cancer cell lines.50 Akt kinases are central nodes in signal transduction pathways, which are important for tumor progression and overexpressed or amplified in a variety of human cancers. Furthermore, A-443654 can slow the progression of tumors as monotherapy or in combination with paclitaxel or rapamycin.51 Last, SL0101-1 (SL-0101), which ranked ninth in connectivity score, is also of interest as it inhibits proliferation of the human breast cancer cell line MCF-7.52 On the contrary, the drug BX-795 shows a connectivity score of −1, which suggests a complete opposite role in driving gene feature alterations. We would suggest avoiding the use of BX-795 on ENZ-responsive and/or ENZ-naive patients for treatment enhancement.
Meanwhile, for comparison between persistent and initial tumor cells, there were 7 drugs returned with positive connectivity scores, which demonstrate a potentially similar resistant response if given to PCa patients (Table 2). We also identified 9 drugs, nutlin-3a, AS601245, roscovitine, WO2009093972, AZ628, nilotinib, GNF-2, BX-795, and GW843682X, with p values less than 0.05. These drugs are promising to overcome ENZ resistance (Table 2; Figure 5B). Nutlin-3a, which ranked first on the basis of negative connectivity score, is a therapeutic compound with potential antineoplastic activities, which inhibits MDM2, activates wild-type p53, and induces apoptosis.53 It is suggested as a potential therapeutic agent for ovarian carcinomas expressing wild-type TP53.54 The drug AS601245, which ranked second on the basis of negative connectivity score, affects proliferation, apoptosis, and differentiation and alters the gene expression profile of human colon cancer line CaCo-2.55,56 Similarly, the drug roscovitine, which ranked third on the basis of negative connectivity score, is also of interest for treating PCa, as it has been shown to promote cell-cycle arrest as well as apoptosis induction in glioblastoma cell lines.57 Moreover, a PC-3 xenograft model in nude mouse showed a 35% reduction in tumor growth conducted with CDK inhibitor roscovitine.55,56 AZ628, another drug that ranked fifth on the basis of negative connectivity score, is a type II RAF inhibitor, which has greater effects in inhibiting cell growth than the combination of dabrafenib and trametinib. While combing AZ628 with BP-1-102, enhanced inhibition of cell proliferation and tumor growth were observed.58,59 Interestingly, nilotinib, which ranked sixth on the basis of negative connectivity score, as the tyrosine kinase inhibitor has been shown effective in targeted therapy of the Philadelphia chromosome-positive chronic myeloid leukemia (CML) after patients experienced resistance to or intolerance of imatinib.60 Another drug in our results, GNF-2, ranked seventh in negative connectivity score and is a member of novel allosteric inhibitors of the Abelson (ABL) family of tyrosine kinases. GNF-2 was also shown to overcome resistant mutations of imatinib found in CML patients.61,62 We also consider the drug BX-795, which ranked eighth in negative connectivity score, is another cell cycle inhibitor that could potentially overcome drug resistance. In oral squamous cell carcinoma, BX-795 is shown to inhibit Akt and NF-κB signaling pathways, to arrest cells in the mitotic phase, and to increase autophagy.63,64 More recently, in high-risk neuroblastoma , which is also highly drug resistant, BX-795 was tested using dual therapeutic approaches with either doxorubicin or crizotinib.65,66 Synergistic and significant inhibition of NB growth were found in contrast to either drug alone. However, note that we do not recommend using BX-795 for the ENZ-induced phenotype (Figure 5A). Last, GW843682X, which ranked ninth in negative connectivity score, is a novel selective PLK1 inhibitor, which has been shown to be effective in nasopharyngeal carcinoma, pediatric tumor cell lines, and leukemia cells.63,64,65,66,67
Discussion
Advanced prostate cancer exhibits molecular, cellular, and tissue-level heterogeneity at a significant scale, thus hampering efforts to develop effective and safe treatments. Single-cell sequencing technologies provide unprecedented resolution to accurately delineate the diverse cellular phenotypes upon drug treatment in pre-clinical models. However, a single layer of molecular profiling, such as single-cell RNA sequencing, is often difficult to resolve and to capture the comprehensive landscapes of the vast heterogeneity. It is thus crucial to integrate scRNA-seq with single-cell assays for transposase-accessible chromatin to provide a better molecular portrait of enzalutamide-induced diverse cellular phenotypes.
Using scRNA-seq, we adopted the definitions of initial, induced responsive, and pre-existing resistant cellular phenotypes from a previous study16 on the basis of cancer cell line compositions within each cell cluster. Meanwhile, similar observations were made using scATAC-seq. To study the molecular mechanisms underpinning different cellular phenotypes, we first calculated differential events (differentially expressed genes using scRNA-seq and differentially accessible regions using scATAC-seq) by comparing induced to initial phenotypes and by comparing persistent to initial phenotypes. Then, 7 signature gene sets were extracted by integrating DEGs under the influence of DARs. Among these gene sets, down-regulated DEGs coupled with open DARs or vice versa were also included as signatures. This suggests more layers of omics data, such as single-cell DNA methylation and copy number alterations, are in need to delineate the complex regulations between DARs and DEGs with opposite alteration directions. For example, mitogen-activated protein kinase 15 (MAPK15) was commonly up-regulated,68 while gene MAPK15 was identified as down-regulated in cells showing early response upon ENZ treatment but coupled with open chromatin when comparing induced to initial phenotype. This phenomenon could be partially driven by chromatin accessibility, meanwhile regulated by other stable epigenetic marks such as DNA methylation.69 Few evidence has focused on the functions of kinases in human cancers, PCa in particular. But inhibiting MAPK15 expression has been shown to significantly decrease osteosarcoma cell metastasis both in vitro and in vivo.31 Consistently, we demonstrated its favorable prognostic potential in progression-free survival using the treatment-naive TCGA prostate adenocarcinoma cohort. This is not the best practice to identify prognostic genes upon ENZ treatment among our signatures but rather to provide additional supporting evidence to show the consistency in importance among different applications of our signatures. The lack of large patient cohort treated using ENZ with well-maintained clinical information and omic data profiles is one of our limitations, which should be resolved in the near future as more relevant treatment data are expected to be generated in PCa.
Other applications of our signature gene sets include conducting a candidate drug survey that may nominate drugs to overcome potential drug resistance. Molecular biomarkers related to ENZ resistance are understudied, especially at the single-cell level. Our research highlighted 9 potential drugs on the basis of the connectivity map, which can be tested as potential candidates in terms of overcoming the resistance upon ENZ treatment. Among these candidate drugs, cell cycle inhibitor targeting agents were much common. BX-795 in particular, has been shown to be effective in multiple human cancers, such as neuroblastoma,66 oral squamous cell carcinoma,70 and bladder cancer.71 Therefore, combinatory use of BX-795 and/or cell cycle targeting agents with current therapies could be an effective and clinically tractable therapeutic approach for drug-resistant PCa.
In this study, we extracted gene signatures coupled with epigenetic alterations in PCa upon ENZ treatment. We show that these gene signatures are valuable in predicting patients’ progression-free prognoses. Moreover, we show that these gene signatures underpinning diverse cellular phenotypes in response to drug treatment can be applied for drug discovery. Even though our present research topic is centered on ENZ treatment in PCa using pre-clinical cancer cell lines, our analytical approach is portable and can be applied to other disease models with single-cell multi-omic profiles generated. The single-cell multi-omics approaches have been rapidly applied to cancer and other disease research, including molecular signatures of drug response or resistance; thus, we expect that more advanced analytic approaches will be further developed to meet the expected strong demand. Overall, we have identified and characterized gene signatures that can be used as biomarkers to present populations of ENZ-induced and ENZ-resistant cells in advanced PCa.
Materials and methods
Multi-omics single-cell sequencing data from prostate cancer cell lines
Single cell multi-omics data were collected to study biological underpinnings of prostate cancer. This multi-omics single-cell dataset included four prostate cancer cell lines: parental LNCaP cells (DMSO treated, the control), LNCaP cells treated with 10 μM enzalutamide for 48 h (LNCaP-ENZ48), and LNCaP-derived enzalutamide-resistant RES-A and RES-B cells.21 LNCaP cell lines serve as a model for PCa, as these cells originate from human prostatic adenocarcinoma.72 For single-cell RNA sequencing data, Cell Ranger-mapped (version 3.0.2) count matrices were downloaded from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE168668. Cell Ranger pre-built human genome reference (build GRCh38, version 3.0.0) was used for mapping. Matched but raw single-cell assay for transposase-accessible chromatin with sequencing data were downloaded from the Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/) under BioProject PRJNA713355.16
Processing the scRNA-seq count matrices and quality control
Cells were filtered according to a well-established quality control routine,16,72 on the basis of the number of detected genes, the total number of molecules detected, and the percentage of reads mapping to the mitochondrial genome. Detailed filtering thresholds and the total number of cells prior to and after quality control per sample are documented in Table S1. After cell filtering, scRNA-seq data from individual samples were then combined and batch-corrected using R package Harmony (version 0.1.0).73 The harmonized gene expression matrix was normalized against library size and applied for downstream analysis using R package Seurat version 4.0.6.74 The first two dimensions using the uniform manifold approximation and projection nonlinear dimensionality reduction technique after adjustment were used for visualization.
Preprocessing the raw scATAC-seq data and quality control
Raw sequencing data for scATAC-seq was preprocessed using Cell Ranger-atac (version 2.0.0)75 with default settings. To be consistent with the processed scRNA-seq data, human genome GRCh38 was used for mapping. Routine quality control16,72 was carried out on the basis of the following metrics: strength of nucleosome-binding pattern, transcription start site enrichment score (ENCODE version), total number of fragments in peaks, and the fraction of fragments in peaks. Specific thresholds per metric per sample, together with the number of cells prior to and after quality control, are included in Table S1.
Data normalization and dimensionality reduction were performed using R package Signac (version 1.6.0)76 with latent semantic indexing (LSI) embeddings, which consists of term frequency-inverse document frequency (TF-IDF) normalization and singular-value decomposition (SVD), on the basis of the top 50% most variable peaks across all cells. UMAP coordinates were calculated using the first 50 LSI components but removing the first LSI component which captures sequencing depth (i.e., technical variation) rather than biological variation in our data.
Identifying differential events in response to the treatment of enzalutamide using pseudo-bulk strategy
Cell clustering was performed separately for scRNA-seq and scATAC-seq data after sample integration. Cellular phenotypes in response to drug treatment were defined on the basis of composition of cell clusters generated in each type of data. Three cellular phenotypes were thus identified with similar standards from a previous study16: (1) the initial phenotype includes clusters dominated by cells from parental cell lines LNCaP and LNCaP-ENZ48), (2) the induced phenotype consists of clusters dominated by cells from cell lines RES-A or RES-B, and (3) the persistent phenotype (or pre-existing ENZ-resistant phenotype) consists of clusters showing no differences in cellular proportions of cells from all four different cell lines. To reduce potential bias for differential analysis, the total number of cells were balanced while merging different cell clusters into the three phenotypes. Principal-component analysis (PCA) using pseudo-bulk read count tables per cluster was conducted using R package stats with function prcomp. Additionally, we conducted a label transfer analysis to examine the consistency of the above three phenotypes between these two data types. Gene activities matrix calculated using scATAC-seq data was used as the query. We applied function FindTransferAnchors of Seurat R package to identify anchors and to annotate scATAC-seq data with scRNA-seq as reference. The fraction of cells consistently labeled as the same phenotype was used to assess the agreement between the two data types.
Differential events (i.e., differentially expressed genes and differentially accessible regions) between the induced and initial phenotype and between the persistent and initial phenotypes were calculated using R package Seurat with function FindMarkers. To enrich signal at each gene locus or open chromatin region to compute its fold change, we adopted the pseudo-bulk approach by simply summing up the total read counts from that gene or region across all the cells in a particular group. A gene was considered differentially expressed with adjusted p value of no more than 0.05 and pseudo-bulk fold change of no less than 1.2, while open chromatin regions were identified as differentially accessible if their adjusted p values were less than 1e-6 and their pseudo-bulk fold changes were higher than 2. Differentially accessible regions were then annotated with their closest gene(s) using function ClosestFeature in R package Signac.
Integrating multi-omics-based differential events to identify epigenetically regulated gene signatures and their related functions
We integrated the differential events (i.e., DEGs and DARs) to generate gene sets showing directions in epigenetic regulations. For instance, when comparing the induced phenotype with the initial phenotype, we generated four gene sets with up-regulated DEGs coupled with open (log2-transformed fold changes more than 1) and closed DARs (log2-transformed fold changes lower than −1) and down-regulated DEGs combined with open and closed DARs, respectively.
Functional enrichment analysis using hypergeometric test was carried out using all the gene sets downloaded from the MSigDB database (version 7.5.1).77 Compiled gene sets (“modules”) from a variety of resources such as KEGG and Gene Ontology in category C4 were removed before calculation because of information redundancy with categories C2 and C5. Gene sets significantly overlapped with our signatures were calculated on the basis of adjusted p values of less than 0.05 and visualized using R package clusterProfiler (version 4.2.2).78 Specifically, the hierarchical tree plots for top enriched functional terms were constructed and visualized on the basis of their pairwise similarities using Jaccard’s similarity index. Each tree plot was accompanied by gene cloud plots that were generated from using genes overlapped with our gene signatures within each functional branch. Genes with higher frequencies within a particular functional branch were shown in larger font size and visualized using R package wordcloud. Gene set enrichment analysis using KEGG pathway and Gene Ontology terms was also conducted. Significantly enriched gene sets were calculated based on p values less than 0.05 using enrichGO and enrichKEGG functions, separately, in R package clusterProfiler (version 4.2.2).
Enrichment analysis of transcription factors
The TF enrichment analysis was carried out using TF binding profiles downloaded from the JASPAR database (version 2022).79 Differentially accessible regions from each comparison were used to perform TF enrichment analysis using R package TFBSTools. Enriched TFs were then ranked on the basis of their enriched significance levels. Heatmaps were used to visualize the top 30 enriched TFs per comparison.
Validating gene signatures independently
The normalized bulk RNA sequencing data of prostate cancer cell line LNCaP xenografts comprising 54 samples spanning different treatment groups (PRE-CX [pre-castration group], POST-CX [post-castration group], CRPC [castration-resistant prostate cancer], ENZS [ENZ-sensitive], and ENZR [ENZ-resistant] groups) were downloaded from the National Center for Biotechnology Information (NCBI) GEO database under accession number GSE211856.22 Among them, groups PRE-CX (n = 9), ENZS (n = 12), and ENZR (n = 15) were used as our independent validations. However, because of data quality concerns, one sample, ATTX.ENZ.87 from group ENZR, was excluded from further analysis.
Our identified seven signature gene sets were first combined into four sets of up- and down-regulated gene sets for two comparisons. Then differential genes from bulk RNA-seq data were calculated with fold change threshold of 1.2 in both direction for each comparison. Finally, we evaluated how consistent of the differential direction from our identified signature genes and the differential genes calculated using bulk RNA-seq data. Consistent gene signatures were visualized with heatmap using bulk RNA-seq data.
Identifying potential use of combinatory drugs using drug-gene network analysis
The DeSigN webtool (https://design-v2.cancerresearch.my/query; database version CTRP) was used to identify candidate drugs using algorithm KS-IC50 with signatures of up- and down-regulated genes for each comparison as stated above.34 The webtool aims to uncover novel biomarkers either sensitive or resistant to cancer therapeutics, which curated a panel of several hundred cancer cell lines and 130 drugs in total under clinical and pre-clinical investigation.80 Visualization of the network among candidate drugs, target genes, and enzalutamide were generated using Cytoscape (version 3.9.1).81
Survival analysis
We split the cancer patients into two groups on the basis of the signature gene sets using their median gene expression. Then, we performed the Kaplan-Meier analysis using progression-free intervals with R package survival (version 3.3-1). Progression-free interval survival (PFS) was defined as the interval length of time during and after certain treatment.82 Survival curves were generated using R package survminer (version 0.4.9). A threshold of false discovery rate [FDR]-adjusted p value less than 0.05 was applied to select PFS-related prognosis genes.
All statistical analyses were performed using R Statistical Software (version 4.1.2 or higher; R Core Team 2021) and Bioconductor.
Acknowledgments
We thank the members of Bioinformatics and Systems Medicine Laboratory (BSML) for insightful discussions. This work was partially supported by National Institutes of Health grants (R01LM012806 and R01DE030122) and Cancer Prevention and Research Institute of Texas grants (CPRIT RP180734 and RP210045).
Author contributions
H.F. and Z.Z. conceived the study. H.F., J.L., and A.M.M. collected the data, performed the data analysis, and interpreted the results. H.F., J.L., A.M.M., and Z.Z. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.02.022.
Supplemental information
Data and code availability
All data and customized R scripts for data analysis are available upon request. Main processing scripts are available at https://github.com/FanBioinformatics/ProstateCancer_MTNA_2omics_SingleCell.git.
References
- 1.Rawla P. Epidemiology of prostate cancer. World J. Oncol. 2019;10:63–89. doi: 10.14740/wjon1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reiss A.B., Saeedullah U., Grossfeld D.J., Glass A.D., Pinkhasov A., Katz A.E. Prostate cancer treatment and the relationship of androgen deprivation therapy to cognitive function. Clin. Transl. Oncol. 2022;24:733–741. doi: 10.1007/s12094-021-02727-1. [DOI] [PubMed] [Google Scholar]
- 3.Ceder Y., Bjartell A., Culig Z., Rubin M.A., Tomlins S., Visakorpi T. The molecular evolution of castration-resistant prostate cancer. Eur. Urol. Focus. 2016;2:506–513. doi: 10.1016/j.euf.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Alumkal J.J., Sun D., Lu E., Beer T.M., Thomas G.V., Latour E., Aggarwal R., Cetnar J., Ryan C.J., Tabatabaei S., et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA. 2020;117:12315–12323. doi: 10.1073/pnas.1922207117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y., Chen J., Wu Z., Ding W., Gao S., Gao Y., Xu C. Mechanisms of enzalutamide resistance in castration-resistant prostate cancer and therapeutic strategies to overcome it. Br. J. Pharmacol. 2021;178:239–261. doi: 10.1111/bph.15300. [DOI] [PubMed] [Google Scholar]
- 6.Conteduca V., Ku S.Y., Fernandez L., Dago-Rodriquez A., Lee J., Jendrisak A., Slade M., Gilbertson C., Manohar J., Sigouros M., et al. Circulating tumor cell heterogeneity in neuroendocrine prostate cancer by single cell copy number analysis. NPJ Precis. Oncol. 2021;5:76. doi: 10.1038/s41698-021-00211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haffner M.C., Zwart W., Roudier M.P., True L.D., Nelson W.G., Epstein J.I., De Marzo A.M., Nelson P.S., Yegnasubramanian S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021;18:79–92. doi: 10.1038/s41585-020-00400-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joseph J.D., Lu N., Qian J., Sensintaffar J., Shao G., Brigham D., Moon M., Maneval E.C., Chen I., Darimont B., Hager J.H. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 9.Takeda D.Y., Spisák S., Seo J.H., Bell C., O'Connor E., Korthauer K., Ribli D., Csabai I., Solymosi N., Szállási Z., et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. 2018;174:422–432.e13. doi: 10.1016/j.cell.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quigley D.A., Dang H.X., Zhao S.G., Lloyd P., Aggarwal R., Alumkal J.J., Foye A., Kothari V., Perry M.D., Bailey A.M., et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. 2018;175:758–769.e9. doi: 10.1016/j.cell.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viswanathan S.R., Ha G., Hoff A.M., Wala J.A., Carrot-Zhang J., Whelan C.W., Haradhvala N.J., Freeman S.S., Reed S.C., Rhoades J., et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. 2018;174:433–447.e19. doi: 10.1016/j.cell.2018.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q., Deng Q., Chao H.P., Liu X., Lu Y., Lin K., Liu B., Tang G.W., Zhang D., Tracz A., et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018;9:3600. doi: 10.1038/s41467-018-06067-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spratt D.E., Alshalalfa M., Fishbane N., Weiner A.B., Mehra R., Mahal B.A., Lehrer J., Liu Y., Zhao S.G., Speers C., et al. Transcriptomic Heterogeneity of Androgen Receptor Activity Defines a de novo low AR-Active Subclass in Treatment Naive Primary Prostate Cancer. Clin. Cancer Res. 2019;25:6721–6730. doi: 10.1158/1078-0432.CCR-19-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shema E., Bernstein B.E., Buenrostro J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019;51:19–25. doi: 10.1038/s41588-018-0290-x. [DOI] [PubMed] [Google Scholar]
- 15.Lawson D.A., Kessenbrock K., Davis R.T., Pervolarakis N., Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018;20:1349–1360. doi: 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taavitsainen S., Engedal N., Cao S., Handle F., Erickson A., Prekovic S., Wetterskog D., Tolonen T., Vuorinen E.M., Kiviaho A., et al. Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse. Nat. Commun. 2021;12:5307. doi: 10.1038/s41467-021-25624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Q., Butler W., Zhou Y., Zhang H., Tang L., Perkinson K., Chen X., Jiang X.S., McCall S.J., Inman B.A., Huang J. Pre-existing castration-resistant prostate cancer-like cells in primary prostate cancer promote resistance to hormonal therapy. Eur. Urol. 2022;81:446–455. doi: 10.1016/j.eururo.2021.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song H., Weinstein H.N.W., Allegakoen P., Wadsworth M.H., 2nd, Xie J., Yang H., Castro E.A., Lu K.L., Stohr B.A., Feng F.Y., et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Nat. Commun. 2022;13:141. doi: 10.1038/s41467-021-27322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuong Z.K., Loudon K.W., Berry B., Richoz N., Jones J., Tan X., Nguyen Q., George A., Hori S., Field S., et al. Resolving the immune landscape of human prostate at a single-cell level in health and cancer. Cell Rep. 2021;37:110132. doi: 10.1016/j.celrep.2021.110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha Y.S., Kim I.Y. Enzalutamide: looking back at its preclinical discovery. Expet Opin. Drug Discov. 2014;9:837–845. doi: 10.1517/17460441.2014.918947. [DOI] [PubMed] [Google Scholar]
- 21.Handle F., Prekovic S., Helsen C., Van den Broeck T., Smeets E., Moris L., Eerlings R., Kharraz S.E., Urbanucci A., Mills I.G., et al. Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci. Rep. 2019;9:13786. doi: 10.1038/s41598-019-50220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chawla S., Rockstroh A., Lehman M., Ratther E., Jain A., Anand A., Gupta A., Bhattacharya N., Poonia S., Rai P., et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022;13:5680. doi: 10.1038/s41467-022-33291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S., Zhou X., Liang C., Bao M., Tian Y., Zhu J., Zhang T., Yang J., Wang Z. ALDH1A3 serves as a predictor for castration resistance in prostate cancer patients. BMC Cancer. 2020;20:387. doi: 10.1186/s12885-020-06899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zinovieva O.L., Grineva E.N., Krasnov G.S., Karpov D.S., Zheltukhin A.O., Snezhkina A.V., Kudryavtseva A.V., Mashkova T.D., Lisitsyn N.A. Treatment of cancer cells with chemotherapeutic drugs results in profound changes in expression of genes encoding aldehyde-metabolizing enzymes. J. Cancer. 2019;10:4256–4263. doi: 10.7150/jca.32608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sircar K., Huang H., Hu L., Liu Y., Dhillon J., Cogdell D., Aprikian A., Efstathiou E., Navone N., Troncoso P., Zhang W. Mitosis phase enrichment with identification of mitotic centromere-associated kinesin as a therapeutic target in castration-resistant prostate cancer. PLoS One. 2012;7:e31259. doi: 10.1371/journal.pone.0031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu C., Huang S., Park J., Park Y., Ko Y.A., Seasock M.J., Bryer J.S., Xu X.X., Song W.C., Palmer M., et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 2018;24:1721–1731. doi: 10.1038/s41591-018-0194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bialkowska A.B., Yang V.W., Mallipattu S.K. Kruppel-like factors in mammalian stem cells and development. Development. 2017;144:737–754. doi: 10.1242/dev.145441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teng M., Zhou S., Cai C., Lupien M., He H.H. Pioneer of prostate cancer: past, present and the future of FOXA1. Protein Cell. 2021;12:29–38. doi: 10.1007/s13238-020-00786-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y., Tindall D.J., Huang H. Modulation of androgen receptor by FOXA1 and FOXO1 factors in prostate cancer. Int. J. Biol. Sci. 2014;10:614–619. doi: 10.7150/ijbs.8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z., Li N., Shen L., Fu J. Quantitative proteomic analysis identifies MAPK15 as a potential regulator of radioresistance in nasopharyngeal carcinoma cells. Front. Oncol. 2018;8:548. doi: 10.3389/fonc.2018.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su Z., Yang B., Zeng Z., Zhu S., Wang C., Lei S., Jiang Y., Lin L. Metastasis-associated gene MAPK15 promotes the migration and invasion of osteosarcoma cells via the c-Jun/MMPs pathway. Oncol. Lett. 2020;20:99–112. doi: 10.3892/ol.2020.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin D.H., Lee J., Kim K.M., Kim S., Kim D.H., Park J. Overexpression of MAPK15 in gastric cancer is associated with copy number gain and contributes to the stability of c-Jun. Oncotarget. 2015;6:20190–20203. doi: 10.18632/oncotarget.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau A.T.Y., Xu Y.M. Regulation of human mitogen-activated protein kinase 15 (extracellular signal-regulated kinase 7/8) and its functions: a recent update. J. Cell. Physiol. 2018;234:75–88. doi: 10.1002/jcp.27053. [DOI] [PubMed] [Google Scholar]
- 34.Lee B.K.B., Tiong K.H., Chang J.K., Liew C.S., Abdul Rahman Z.A., Tan A.C., Khang T.F., Cheong S.C. DeSigN: connecting gene expression with therapeutics for drug repurposing and development. BMC Genom. 2017;18:934. doi: 10.1186/s12864-016-3260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markowitsch S.D., Juetter K.M., Schupp P., Hauschulte K., Vakhrusheva O., Slade K.S., Thomas A., Tsaur I., Cinatl J., Jr., Michaelis M., et al. Shikonin reduces growth of docetaxel-resistant prostate cancer cells mainly through necroptosis. Cancers. 2021;13:882. doi: 10.3390/cancers13040882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voigtlaender M., Schneider-Merck T., Trepel M. Lapatinib. Recent Results Cancer Res. 2018;211:19–44. doi: 10.1007/978-3-319-91442-8_2. [DOI] [PubMed] [Google Scholar]
- 37.Bennett J.M., Reich S.D. Bleomycin. Ann. Intern. Med. 1979;90:945–948. doi: 10.7326/0003-4819-90-6-945. [DOI] [PubMed] [Google Scholar]
- 38.Yndestad S., Austreid E., Svanberg I.R., Knappskog S., Lønning P.E., Eikesdal H.P. Activation of Akt characterizes estrogen receptor positive human breast cancers which respond to anthracyclines. Oncotarget. 2017;8:41227–41241. doi: 10.18632/oncotarget.17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdelgalil A.A., Al-Kahtani H.M., Al-Jenoobi F.I., FI Erlotinib. Profiles Drug Subst Excip Relat Methodol. 2020;45:93–117. doi: 10.1016/bs.podrm.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Duan Z., Zhang J., Ye S., Shen J., Choy E., Cote G., Harmon D., Mankin H., Hua Y., Zhang Y., et al. A-770041 reverses paclitaxel and doxorubicin resistance in osteosarcoma cells. BMC Cancer. 2014;14:681. doi: 10.1186/1471-2407-14-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang F., Yao X., Zhang Y., Tang J. Synthesis, biological function and evaluation of Shikonin in cancer therapy. Fitoterapia. 2019;134:329–339. doi: 10.1016/j.fitote.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Stadlbauer B., Lyu C., Buchner A., Pohla H. Shikonin enhances the antitumor effect of cabazitaxel in prostate cancer stem cells and reverses cabazitaxel resistance by inhibiting ABCG2 and ALDH3A1. Am. J. Cancer Res. 2020;10:3784–3800. [PMC free article] [PubMed] [Google Scholar]
- 43.Jang S.Y., Jang E.H., Jeong S.Y., Kim J.H. Shikonin inhibits the growth of human prostate cancer cells via modulation of the androgen receptor. Int. J. Oncol. 2014;44:1455–1460. doi: 10.3892/ijo.2014.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y., Zheng L., Liu J., Zhou Z., Cao X., Lv X., Chen F. Shikonin inhibits prostate cancer cells metastasis by reducing matrix metalloproteinase-2/-9 expression via AKT/mTOR and ROS/ERK1/2 pathways. Int. Immunopharmacol. 2014;21:447–455. doi: 10.1016/j.intimp.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 45.Ryan Q., Ibrahim A., Cohen M.H., Johnson J., Ko C.W., Sridhara R., Justice R., Pazdur R. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncol. 2008;13:1114–1119. doi: 10.1634/theoncologist.2008-0816. [DOI] [PubMed] [Google Scholar]
- 46.Gravis G., Bladou F., Salem N., Gonçalves A., Esterni B., Walz J., Bagattini S., Marcy M., Brunelle S., Viens P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann. Oncol. 2008;19:1624–1628. doi: 10.1093/annonc/mdn174. [DOI] [PubMed] [Google Scholar]
- 47.Whang Y.E., Armstrong A.J., Rathmell W.K., Godley P.A., Kim W.Y., Pruthi R.S., Wallen E.M., Crane J.M., Moore D.T., Grigson G., et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol. Oncol. 2013;31:82–86. doi: 10.1016/j.urolonc.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 48.Gross M., Higano C., Pantuck A., Castellanos O., Green E., Nguyen K., Agus D.B. A phase II trial of docetaxel and erlotinib as first-line therapy for elderly patients with androgen-independent prostate cancer. BMC Cancer. 2007;7:142. doi: 10.1186/1471-2407-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ueki T.i., Uemura H., Nagashima Y., Ohta S., Ishiguro H., Kubota Y. Antitumour effect of electrochemotherapy with bleomycin on human prostate cancer xenograft. BJU Int. 2008;102:1467–1471. doi: 10.1111/j.1464-410X.2008.07793.x. [DOI] [PubMed] [Google Scholar]
- 50.Han E.K.H., Leverson J.D., McGonigal T., Shah O.J., Woods K.W., Hunter T., Giranda V.L., Luo Y. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene. 2007;26:5655–5661. doi: 10.1038/sj.onc.1210343. [DOI] [PubMed] [Google Scholar]
- 51.Luo Y., Shoemaker A.R., Liu X., Woods K.W., Thomas S.A., de Jong R., Han E.K., Li T., Stoll V.S., Powlas J.A., et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol. Cancer Therapeut. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 52.Smith J.A., Poteet-Smith C.E., Xu Y., Errington T.M., Hecht S.M., Lannigan D.A. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]
- 53.Secchiero P., Bosco R., Celeghini C., Zauli G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr. Pharmaceut. Des. 2011;17:569–577. doi: 10.2174/138161211795222586. [DOI] [PubMed] [Google Scholar]
- 54.Crane E.K., Kwan S.Y., Izaguirre D.I., Tsang Y.T.M., Mullany L.K., Zu Z., Richards J.S., Gershenson D.M., Wong K.K. Nutlin-3a: a potential therapeutic opportunity for TP53 wild-type ovarian carcinomas. PLoS One. 2015;10:e0135101. doi: 10.1371/journal.pone.0135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payton M., Chung G., Yakowec P., Wong A., Powers D., Xiong L., Zhang N., Leal J., Bush T.L., Santora V., et al. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res. 2006;66:4299–4308. doi: 10.1158/0008-5472.CAN-05-2507. [DOI] [PubMed] [Google Scholar]
- 56.Cerbone A., Toaldo C., Pizzimenti S., Pettazzoni P., Dianzani C., Minelli R., Ciamporcero E., Roma G., Dianzani M.U., Canaparo R., et al. AS601245, an anti-inflammatory JNK inhibitor, and clofibrate have a synergistic effect in inducing cell responses and in affecting the gene expression profile in CaCo-2 colon cancer cells. PPAR Res. 2012;2012:269751. doi: 10.1155/2012/269751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolodziej M., Goetz C., Di Fazio P., Montalbano R., Ocker M., Strik H., Quint K. Roscovitine has anti-proliferative and pro-apoptotic effects on glioblastoma cell lines: a pilot study. Oncol. Rep. 2015;34:1549–1556. doi: 10.3892/or.2015.4105. [DOI] [PubMed] [Google Scholar]
- 58.Noeparast A., Giron P., De Brakeleer S., Eggermont C., De Ridder U., Teugels E., De Grève J. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget. 2018;9:16110–16123. doi: 10.18632/oncotarget.24576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z., Yin M., Chu P., Lou M. STAT3 inhibitor sensitized KRAS-mutant lung cancers to RAF inhibitor by activating MEK/ERK signaling pathway. Aging (Albany NY) 2019;11:7187–7196. doi: 10.18632/aging.102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ostendorf B.N., le Coutre P., Kim T.D., Quintás-Cardama A. Nilotinib. Recent Results Cancer Res. 2014;201:67–80. doi: 10.1007/978-3-642-54490-3_3. [DOI] [PubMed] [Google Scholar]
- 61.Kim H.J., Yoon H.J., Choi J.Y., Lee I.K., Kim S.Y. The tyrosine kinase inhibitor GNF-2 suppresses osteoclast formation and activity. J. Leukoc. Biol. 2014;95:337–345. doi: 10.1189/jlb.0713356. [DOI] [PubMed] [Google Scholar]
- 62.Jones J.K., Thompson E.M. Allosteric inhibition of ABL kinases: therapeutic potential in cancer. Mol. Cancer Therapeut. 2020;19:1763–1769. doi: 10.1158/1535-7163.MCT-20-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Q., Chu Y., Hu W., Peng M., Song Q. The cytotoxic effect of GW843682X on nasopharyngeal carcinoma. Anti Cancer Agents Med. Chem. 2016;16:1640–1645. doi: 10.2174/1871520616666160520112451. [DOI] [PubMed] [Google Scholar]
- 64.Bai L.Y., Chiu C.F., Kapuriya N.P., Shieh T.M., Tsai Y.C., Wu C.Y., Sargeant A.M., Weng J.R. BX795, a TBK1 inhibitor, exhibits antitumor activity in human oral squamous cell carcinoma through apoptosis induction and mitotic phase arrest. Eur. J. Pharmacol. 2015;769:287–296. doi: 10.1016/j.ejphar.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 65.Spaniol K., Boos J., Lanvers-Kaminsky C. An in-vitro evaluation of the polo-like kinase inhibitor GW843682X against paediatric malignancies. Anti Cancer Drugs. 2011;22:531–542. doi: 10.1097/CAD.0b013e3283454526. [DOI] [PubMed] [Google Scholar]
- 66.Chilamakuri R., Rouse D.C., Yu Y., Kabir A.S., Muth A., Yang J., Lipton J.M., Agarwal S. BX-795 inhibits neuroblastoma growth and enhances sensitivity towards chemotherapy. Transl. Oncol. 2022;15:101272. doi: 10.1016/j.tranon.2021.101272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikezoe T., Yang J., Nishioka C., Takezaki Y., Tasaka T., Togitani K., Koeffler H.P., Yokoyama A. A novel treatment strategy targeting polo-like kinase 1 in hematological malignancies. Leukemia. 2009;23:1564–1576. doi: 10.1038/leu.2009.94. [DOI] [PubMed] [Google Scholar]
- 68.Southekal S., Mishra N.K., Guda C. Pan-cancer analysis of human kinome gene expression and promoter DNA methylation identifies dark kinase biomarkers in multiple cancers. Cancers. 2021;13:1189. doi: 10.3390/cancers13061189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao S.G., Chen W.S., Li H., Foye A., Zhang M., Sjöström M., Aggarwal R., Playdle D., Liao A., Alumkal J.J., et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020;52:778–789. doi: 10.1038/s41588-020-0648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pai S., Yadav V.K., Kuo K.T., Pikatan N.W., Lin C.S., Chien M.H., Lee W.H., Hsiao M., Chiu S.C., Yeh C.T., Tsai J.T. PDK1 inhibitor BX795 improves cisplatin and radio-efficacy in oral squamous cell carcinoma by downregulating the PDK1/CD47/akt-mediated glycolysis signaling pathway. Int. J. Mol. Sci. 2021;22:11492. doi: 10.3390/ijms222111492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen W., Luo K., Ke Z., Kuai B., He S., Jiang W., Huang W., Cai Z. TBK1 promote bladder cancer cell proliferation and migration via Akt signaling. J. Cancer. 2017;8:1892–1899. doi: 10.7150/jca.17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan H., Jia P., Zhao Z. Cell-type-Specific profibrotic scores across multi-organ Systems predict cancer prognosis. Cancers. 2021;13:6024. doi: 10.3390/cancers13236024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castro-Mondragon J.A., Riudavets-Puig R., Rauluseviciute I., Lemma R.B., Turchi L., Blanc-Mathieu R., Lucas J., Boddie P., Khan A., Manosalva Pérez N., et al. Jaspar 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–D173. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J., et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson A.G., Booth C.M., Eisenhauer E.A. Progression-free survival as an end-point in solid tumours--perspectives from clinical trials and clinical practice. Eur. J. Cancer. 2014;50:2303–2308. doi: 10.1016/j.ejca.2014.05.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and customized R scripts for data analysis are available upon request. Main processing scripts are available at https://github.com/FanBioinformatics/ProstateCancer_MTNA_2omics_SingleCell.git.