

Abstract
Sinusoids are specialized, low pressure blood vessels in the liver, bone marrow, and spleen required for definitive hematopoiesis. Unlike other blood endothelial cells (ECs), sinusoidal ECs express high levels of VEGFR3. VEGFR3 and its ligand VEGF-C are known to support lymphatic growth, but their function in sinusoidal vessels is unknown. In this study, we define a reciprocal VEGF-C/VEGFR3-CDH5 (VE-cadherin) signaling axis that controls growth of both sinusoidal and lymphatic vessels. Loss of VEGF-C or VEGFR3 resulted in cutaneous edema, reduced fetal liver size, and bloodless bone marrow due to impaired lymphatic and sinusoidal vessel growth. Mice with membrane-retained VE-cadherin conferred identical lymphatic and sinusoidal defects, suggesting that VE-cadherin opposes VEGF-C/VEGFR3 signaling. In developing mice, loss of VE-cadherin rescued defects in sinusoidal and lymphatic growth caused by loss of VEGFR3 but not loss of VEGF-C, findings explained by potentiated VEGF-C/VEGFR2 signaling in VEGFR3-deficient lymphatic ECs. Mechanistically, VEGF-C/VEGFR3 signaling induces VE-cadherin endocytosis and loss of function via SRC-mediated phosphorylation, while VE-cadherin prevents VEGFR3 endocytosis required for optimal receptor signaling. These findings establish an essential role for VEGF-C/VEGFR3 signaling during sinusoidal vascular growth, identify VE-cadherin as a powerful negative regulator of VEGF-C signaling that acts through both VEGFR3 and VEGFR2 receptors, and suggest that negative regulation of VE-cadherin is required for effective VEGF-C/VEGFR3 signaling during growth of sinusoidal and lymphatic vessels. Manipulation of this reciprocal negative regulatory mechanism, e.g. by reducing VE-cadherin function, may be used to stimulate therapeutic sinusoidal or lymphatic vessel growth.
Keywords: sinusoid, lymphatic, VEGF-C, VEGFR3, VE-cadherin, CDH5, bone marrow, fetal liver
Introduction
The sinusoids are vessels unique to the liver, bone marrow, and spleen that are molecularly and functionally distinct from other blood vessels1. In the bone marrow and liver, the sinusoidal vasculature has been shown to play important roles in the hematopoietic stem cell niche, osteogenesis, and organ regeneration2–4. Growth and regeneration of sinusoidal vessels is essential for hematopoiesis, e.g. during embryonic development and during bone marrow transplantation3, but the signaling pathways that underlie sinusoidal vascular growth remain poorly defined. Unlike arterial and venous endothelial cells (ECs) that line other blood vessels, sinusoidal ECs (SECs) express VEGF receptor 3 (VEGFR3)5,6 a receptor for the VEGF-C and VEGF-D ligands that are known to drive lymphatic growth. The role of VEGFR3 and its ligands during the development and growth of the sinusoidal vasculature is not known.
We and others have previously investigated the role of vascular endothelial growth factor (VEGF)-C, the main ligand for VEGFR3, in developing animals. In humans, mice, and zebrafish, VEGF-C and its co-activator CCBE1 are required lymphangiogenic growth factors that enable lymph sac formation and lymphatic growth7–11. Unexpectedly, recent studies also revealed that deletion of either VEGF-C or CCBE1 resulted in decreased liver size and anemia12–14. The basis for this requirement during fetal liver hematopoiesis has been attributed to defects in erythroid maturation13,14. However, erythroid progenitors do not express VEGFR3, the primary VEGF-C receptor. Significantly, we have also found that mice with membrane-retained VE-cadherin (in which the protein is stabilized at the plasma membrane through a C-terminal fusion to α-catenin) exhibit fetal liver and lymphatic defects identical to those in animals lacking VEGF-C or CCBE1, including subtle details such as increased penetrance on a C57Bl/6 strain background15. How VEGF-C, CCBE1, VE-cadherin, and definitive hematopoiesis may be linked at the molecular and cellular levels has been unclear.
VEGF signaling negatively regulates VE-cadherin function in vivo and in vitro, and in vitro studies have suggested that the converse may also be true. VEGF-A activation of VEGFR2 signaling loosens endothelial junctions and induces tissue edema by negatively regulating VE-cadherin localization and function at the endothelial cell membrane, in part through downstream TSAD and SRC signals16 17. Conversely, in vitro studies support a reverse negative regulatory mechanism by which VE-cadherin at the cell membrane prevents activated VEGFR2 receptors from internalizing and efficiently signaling from intracellular endosomes18. In contrast to regulation of VE-cadherin by VEGFR2, the role of VE-cadherin regulation of VEGF receptors has not been demonstrated in vivo. Lymphatic deletion of VE-cadherin during development results in lymphatic hyperplasia and increased VEGFR3 phosphorylation, but how VE-cadherin might regulate VEGFR319 and whether VE-cadherin interacts in an analogous manner with VEGFR3 remains unknown.
In the present study, we elucidate the molecular and cellular connections that underlie these prior findings. We find that fetal anemia due to loss of VEGF-C function arises as a result of loss of SEC VEGFR3 signaling that is required for sinusoidal growth and establishment of the hematopoietic niche in both the fetal liver and bone marrow. Loss of VEGFR3 signaling results in loss of pERK1/2 and SEC proliferation, effects reproduced by membrane-retained VE-cadherin. Genetic haploinsufficiency of VE-cadherin rescues growth defects in the sinusoids and lymphatics in vivo following loss of VEGFR3 but not loss of VEGF-C, a finding explained by enhanced VEGF-C/VEGFR2 signaling in cultured lymphatic ECs (LECs) lacking both VEGFR3 and VE-cadherin. Finally, we find that VEGF-C/VEGFR3 signaling and VE-cadherin inhibit each other’s function through regulation of each protein’s cell membrane association. Together, these results demonstrate a reciprocal VEGF-C/VEGFR3–CDH5 signaling axis that controls both lymphatic and sinusoidal growth. The ability to rescue sinusoidal and lymphatic growth in the absence of VEGFR3 with loss of VE-cadherin function identifies VE-cadherin as a potential therapeutic target to stimulate sinusoidal and lymphatic regeneration.
Results
VEGF-C is required for sinusoidal growth in the bone marrow
Prior studies have demonstrated that VEGF-C and CCBE1 are required for fetal liver hematopoiesis12–14. Although VEGFR3 is expressed in SECs in the fetal liver, these studies excluded an angiogenic mechanism for this requirement because sinusoidal vessels were present in the small fetal liver observed in deficient mice. Since the fetal liver forms prior to the onset of its hematopoietic function in mid-gestation20, we reasoned that VEGF-C may be required specifically for the rapid expansion of sinusoidal vasculature that takes place with the initiation of definitive hematopoiesis. In mid-late gestation, definitive hematopoiesis shifts from the fetal liver to the bone marrow, coincident with the de novo growth of VEGFR3+ sinusoidal vessels in the bone marrow21, suggesting that the growth of sinusoidal vessels in the bone marrow is more tightly connected to their hematopoietic role. To test whether VEGF-C might be required to activate VEGFR3 on SECs for definitive hematopoiesis, we therefore examined the effect of VEGF-C deficiency on bone marrow sinusoidal growth and hematopoietic function.
Vegfc conditional mice were crossed onto a Rosa26-CreERT2 background to generate global inducible VEGF-C knockout mice (R26CreERT2; Vegfcfl/fl). Cre activity was induced beginning at embryonic day (E)14.5, the timepoint at which bone marrow sinusoidal angiogenesis is initiated (Figure 1a). At E18.5, control and VEGF-C knockout littermates were visually similar but qPCR revealed nearly complete loss of Vegfc mRNA in VEGF-C knockout tissues and inspection of freshly dissected femurs revealed visible blood in marrow of control embryos but not in those of VEGF-C knockout littermates (Figure 1b, c and Supplemental Figure 6d). Hematoxylin & eosin (H&E) staining of the femur confirmed a relative lack of cellularity in VEGF-C KO femurs (Figure 1d, e), while staining of Endomucin+ SECs and TER119+ erythrocytes revealed significantly reduced sinusoidal vascular area and blood-filled sinusoids in the VEGF-C KO femur (Figure 1f). Immunostaining for Endomucin, ERG, and the cell proliferation marker Ki67 demonstrated reduced SEC proliferation in the VEGF-C knockout femur vasculature (Figure 1g), while co-staining for Endomucin, ERG, and the apoptotic marker Cleaved Caspase-3 revealed slightly but significantly increased numbers of apoptotic SECs in the VEGF-C knockout femur (Figure 1h). These studies reveal that VEGF-C is required for normal sinusoidal vascular growth in the bone marrow, and that loss of VEGF-C is associated with reduced SEC proliferation and increased SEC apoptosis associated with loss of bone marrow hematopoietic function.
Figure 1. VEGF-C is required for bone marrow sinusoidal angiogenesis.
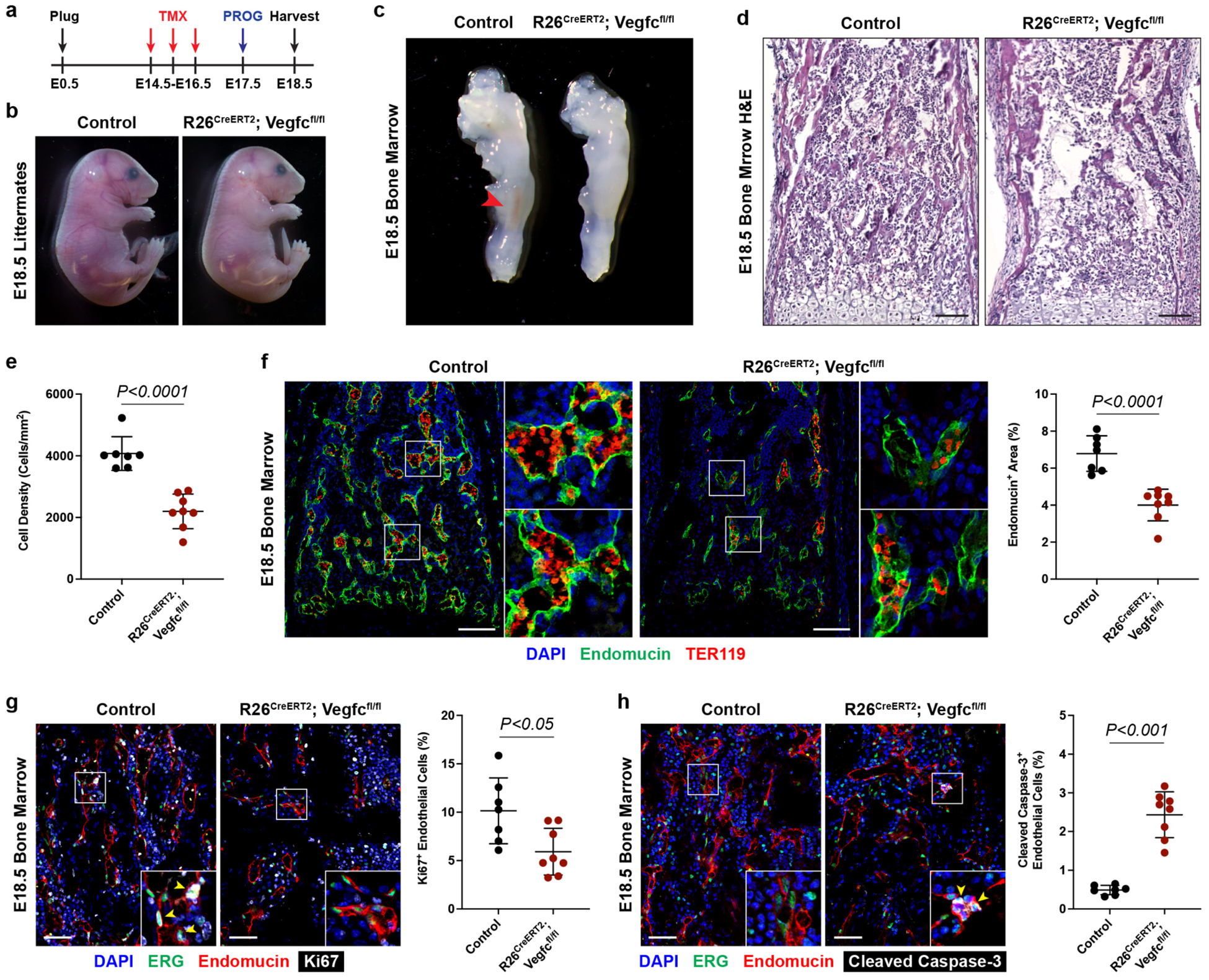
a, Tamoxifen (TMX) and medroxyprogesterone (PROG) regimen for collection of Control and R26CreERT2; Vegfcfl/fl embryonic day (E)18.5 mice. b, c, Whole embryos and femurs from Control and R26CreERT2; Vegfcfl/fl littermates. Red arrowhead indicates presence of blood in the femur marrow. d, H&E staining of femur bone marrow. Scale bars = 100μm. e, Quantification of cell density in the bone marrow. Control, n=7. R26CreERT2; Vegfcfl/fl, n=8. f, Immunofluorescence staining of bone marrow for Endomucin (green) and TER119 (red), and quantification of sinusoidal vascular area. Control, n=7. R26CreERT2; Vegfcfl/fl, n=8. Scale bars = 100μm. g, Immunofluorescence staining for ERG (green), Endomucin (red), and Ki67 (gray), and quantification of percent Ki67+ sinusoidal endothelial cells. Yellow arrowheads point to Ki67+ERG+Endomucin+ cells. Control, n=7. R26CreERT2; Vegfcfl/fl, n=8. Scale bars = 50μm. h, Immunofluorescence staining for ERG (green), Endomucin (red), and Cleaved Caspase-3 (gray), and quantification of percent Cleaved Caspase-3+ sinusoidal endothelial cells. Yellow arrowheads point to Cleaved Caspase-3+ERG+Endomucin+ cells. Control, n=7. R26CreERT2; Vegfcfl/fl, n=8. Scale bars = 50μm. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means±S.D.
Endothelial VEGFR3 is required for sinusoidal growth in the bone marrow and liver
The studies described above suggested that hematopoietic defects associated with loss of VEGF-C may be due to decreased SEC proliferation and survival as a result of reduced VEGFR3 signaling in SECs. To test this hypothesis, we next generated endothelial-specific, tamoxifen-inducible VEGFR3 knockout mice by crossing Flt4 conditional mice with Cdh5(PAC)-CreERT2 mice to generate endothelial inducible VEGFR3 knockout animals (Cdh5CreERT2; Flt4fl/fl). Following tamoxifen administration at E14.5 (Figure 2a), E18.5 Cdh5CreERT2; Flt4fl/fl mice appeared visually indistinguishable from control littermates but exhibited a loss of hematopoietic cells in in the femur, findings identical to those observed following global loss of VEGF-C at the same timepoint (Fig. 2b–e vs 1b–e). Immunofluorescence studies confirmed SEC loss of VEGFR3 protein (Supplemental Figure 1a), and decreased numbers of SECs in Cdh5CreERT2; Flt4fl/fl bone marrow (Figure 2f). As observed following loss of VEGF-C, Cdh5CreERT2; Flt4fl/fl bone marrow SECs exhibited significantly decreased proliferation (Figure 2g), however no differences in endothelial apoptosis were found (Figure 2h).
Figure 2. Endothelial VEGFR3 is required for bone marrow and liver sinusoidal angiogenesis.

a, Tamoxifen (TMX) and medroxyprogesterone (PROG) regimen for collection of Control and Cdh5CreERT2; Flt4fl/fl E18.5 mice. b, c, Whole embryos and femurs from Control and Cdh5CreERT2; Flt4fl/fl littermates. Red arrowhead indicates presence of blood in the femur marrow. d, H&E staining of femur bone marrow. Boxes in the left image denote area of magnified image on the right. Scale bars = 100μm. e, Quantification of cell density in the bone marrow. Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4. f, Immunofluorescence staining of bone marrow for Endomucin (green) and TER119 (red), and quantification of sinusoidal vascular area. Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4. Scale bars = 100μm. g, Immunofluorescence staining for ERG (green), Endomucin (red), and Ki67 (gray), and quantification of percent Ki67+ sinusoidal endothelial cells. Yellow arrowheads point to Ki67+ERG+Endomucin+ cells. Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4. Scale bars = 50μm. h, Immunofluorescence staining for ERG (green), Endomucin (red), and Cleaved Caspase-3 (gray), and quantification of percent Cleaved Caspase-3+ sinusoidal endothelial cells. Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4. Scale bars = 50μm. g, Tamoxifen (TMX) regimen for collection of Control and Cdh5CreERT2; Flt4fl/fl E14.5 mice. h, Whole embryos from Control and Cdh5CreERT2; Flt4fl/fl littermates. White dotted lines outline the liver contour, and the red arrowhead indicates severe edema. Insets show fetal livers dissected from the embryos. i, H&E staining of liver, immunofluorescence staining for LYVE1 (red) and Ki67 (green), and quantification of LYVE1 area by zone and percent Ki67+ sinusoidal endothelial cells. Control, n=7. Cdh5CreERT2; Flt4fl/fl, n=6. Scale bars = 100μm. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means±S.D.
The findings that both VEGF-C and endothelial VEGFR3 are required for bone marrow sinusoidal growth and hematopoietic activity suggested that the basis for reduced fetal liver growth and anemia in animals lacking VEGF-C or CCBE1 was likely to also be loss of VEGFR3 signaling required for SEC proliferation and growth. To test this endothelial mechanism, we induced deletion of VEGFR3 in Cdh5CreERT2; Flt4fl/fl embryos beginning at E9.5 (Figure 2i) and examined the embryos at E14.5. Immunofluorescent staining confirmed efficient loss of VEGFR3 protein in SECs and LECs (Supplemental Figure 1b, c). E14.5 Cdh5CreERT2; Flt4fl/fl embryos were severely edematous and had markedly smaller and paler fetal livers than control littermates (Figure 2j), findings identical to those observed with global loss VEGF-C or CCBE1 function or membrane-retained VE-cadherin (discussed below). H&E staining and immunofluorescent staining for the SEC marker LYVE1 revealed that Cdh5CreERT2; Flt4fl/fl fetal livers lacked RBCs and had decreased sinusoidal vasculature and proliferation in the outer, but not the central, zone (Figure 2k). These findings are consistent with Ki67 staining and prior studies demonstrating that the outer zone of the fetal liver is more angiogenic than the central zone22 (Supplemental Figure 2). These studies identify VEGFR3+ SECs as a cellular target of VEGF-C signaling required for sinusoidal vascular growth in the fetal liver and bone marrow, and support a requirement for VEGF-C/VEGFR3 signaling in SECs as the basis for the defects in embryonic hematopoiesis previously reported in mice lacking CCBE1 or VEGF-C.
Membrane-retained VE-cadherin confers sinusoidal vascular defects identical to those conferred by loss of VEGF-C/VEGFR3 function
A prior study of mice that express a mutant VE-cadherin protein in which the C-tail of the protein is fused to a-catenin to stabilize and retain VE-cadherin at the cell membrane (Cdh5a animals) revealed a defect in fetal liver hematopoiesis, as well as defects in lymphatic vascular growth15. Several lines of evidence suggested that membrane-retained/gain of function VE-cadherin phenotypes were mechanistically linked to those associated with loss of VEGF-C/VEGFR3 signaling. First, since VE-cadherin is specifically expressed in endothelial cells, both could arise due to altered endothelial cell function. Second, the fetal liver phenotypes precisely replicate each other (Supplemental Figure 3a–c), including subtleties such as a requirement for a high C57Bl/6 genetic background to observe a fully penetrant hematopoietic phenotype in CCBE1-deficient, VEGF-C-deficient, and Cdh5a mice. Third, Cdh5a animals also exhibit edema due to defects in lymphatic vascular growth, the major phenotype associated with loss of VEGF-C/VEGFR3 signaling in endothelial cells. Finally, prior studies have identified molecular crosstalk between VEGF receptors and VE-cadherin, specifically evidence that signaling by VEGFR2 negatively regulates VE-cadherin function in vitro23 and in vivo24 and that VE-cadherin may negatively regulate VEGFR2 signaling and EC proliferation in vitro18,25. These findings suggested that loss of VEGF-C/VEGFR3 signaling may result in a gain of function of VE-cadherin and/or vice-versa to restrict sinusoidal and lymphatic vascular growth.
To test this hypothesis, we first examined sinusoidal vascular growth in the bone marrow of membrane-retained VE-cadherin (Cdh5α/α) embryos. No gross differences in embryo appearance were noted (Figure 3a), but visual inspection of E18.5 Cdh5α/α and littermate femurs revealed a lack of visible blood in the Cdh5α/α femur, a finding similar to that observed in femurs from R26CreERT2; Vegfcfl/fl and Cdh5CreERT2; Flt4fl/fl mice (Figures 1c, 2c, and 3b). H&E staining and immunofluorescent staining for Endomucin+ SECs, TER119+ erythrocytes and the proliferative marker Ki67 demonstrated fewer erythrocytes, decreased sinusoidal vascular density, and attenuated SEC proliferation in Cdh5α/α bone marrow (Figure 3c–f) – findings like those seen in embryos lacking VEGF-C or endothelial cell VEGFR3 (Figures 1 and 2). Similar to loss of VEGFR3, we detected no differences in SEC apoptosis as measured by Cleaved Caspase-3 staining (Figure 3g), perhaps reflecting a slightly milder loss of growth factor signaling than that seen in SECs lacking VEGF-C. These findings demonstrate that membrane-retained VE-cadherin confers defects in bone marrow and fetal liver sinusoidal growth identical to those associated with loss of VEGF-C/VEGFR3 signaling, consistent with a shared molecular mechanism.
Figure 3. Membrane-retained VE-cadherin confers defects in bone marrow sinusoidal angiogenesis.

a, b, Whole embryos and femurs from E18.5 Control and Cdh5α/α littermates. The red arrowhead indicates presence of blood in the bone marrow. c, H&E staining of bone marrow. Boxes in the left image denote area of magnified image on the right. Scale bars = 100μm. d, Quantification of cell density in the bone marrow. Control, n=9. Cdh5α/α, n=4. e, Immunofluorescence staining of bone marrow for Endomucin (green) and TER119 (red), and quantification of sinusoidal vascular area. Control, n=9. Cdh5α/α, n=4. Scale bars = 100μm. f, Immunofluorescence staining for ERG (green), Endomucin (red), and Ki67 (gray), and quantification of percent Ki67+ sinusoidal endothelial cells. Yellow arrowheads point to Ki67+ERG+Endomucin+ cells. Control, n=6. Cdh5α/α, n=4. Scale bars = 50μm. g, Immunofluorescence staining for ERG (green), Endomucin (red), and Cleaved Caspase-3 (gray), and quantification of percent Cleaved Caspase-3+ sinusoidal endothelial cells. Control, n=6. Cdh5α/α, n=4. Scale bars = 50μm. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means±S.D.
Loss of VE-cadherin function rescues sinusoidal vascular defects conferred by VEGFR3 deficiency
The most straightforward shared molecular mechanism by which VE-cadherin and VEGF-C/VEGFR3 might impact sinusoidal and lymphatic vascular growth is through effects on VEGF-C/VEGFR3 signaling in SECs and LECs. We therefore next performed genetic experiments to test for interaction between the genes encoding these two molecular pathways and their effects on established VEGFR signaling effectors. To test for VE-cadherin-VEGFR3 interaction, we generated Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ mice in which total loss of VEGFR3 function (confirmed by immunostaining, Supplemental Figure 4a) and partial loss of VE-cadherin function was induced in endothelial cells by tamoxifen administration starting at E14.5 and littermates analyzed at E18.5. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ animals were visually indistinguishable from control littermates (Figure 4a). Examination of femurs demonstrated restoration of visible blood in the bone marrow of Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ femurs that was absent in Cdh5CreERT2; Flt4fl/fl littermates (Figure 4b). Consistent with this observation, H&E staining demonstrated restoration of normal cellularity in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ bone marrow (Figure 4c, d). Immunostaining for SECs, erythrocytes, and proliferating cells using Endomucin, TER119, and Ki67, respectively, demonstrated rescue of sinusoidal vascular growth, erythrocyte content, and SEC proliferation in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos compared with Cdh5CreERT2; Flt4fl/fl littermates (Figure 4e, f).
Figure 4. VE-cadherin haploinsufficiency rescues defects in bone marrow sinusoidal angiogenesis caused by VEGFR3 deficiency.

a, b, Whole embryos and femurs from Control, Cdh5CreERT2; Flt4fl/fl, and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ littermates. Red arrowhead indicates presence of blood in the bone marrow. c, H&E staining of bone marrow. Scale bars = 100μm. d, Quantification of cell density in the bone marrow. Control, n=4. Cdh5CreERT2; Flt4fl/fl, n=3. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. e, Immunofluorescence staining of bone marrow for Endomucin (green) and TER119 (red), and quantification of sinusoidal vascular area. Control, n=4. Cdh5CreERT2; Flt4fl/fl, n=3. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. Scale bars = 100μm. f, Immunofluorescence staining for ERG (green), Endomucin (red), and Ki67 (gray), and quantification of percent Ki67+ sinusoidal endothelial cells. Yellow arrowheads point to Ki67+ERG+Endomucin+ cells. Control, n=4. Cdh5CreERT2; Flt4fl/fl, n=3. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. Scale bars = 50μm.
To further test the genetic interaction between VEGFR3 and VE-cadherin, we next assessed mid-gestation embryos in which Cre was induced beginning at E10.5 and littermate embryos harvested at E14.5. Consistent with prior studies demonstrating a requirement for VEGFR3 during lymphatic development26–28, E14.5 Cdh5CreERT2; Flt4fl/fl embryos were severely edematous and also exhibited decreased fetal liver size (Figure 5a). VE-cadherin haploinsufficiency in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ littermates fully rescued liver size (Figure 5a), cellularity (Figure 5b), sinusoidal vascular density (Figure 5c), and SEC proliferation (Figure 5d). Immunostaining confirmed complete loss of VEGFR3 in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ fetal liver SECs with rescued sinusoidal growth (Supplemental Figure 4b). These studies reveal powerful opposing roles for VEGFR3 and VE-cadherin, such that severe defects in sinusoidal vascular growth conferred by complete loss of VEGFR3 can be fully compensated for by partial loss of VE-cadherin (discussed further below).
Figure 5. VE-cadherin haploinsufficiency rescues defects in liver sinusoidal angiogenesis caused by VEGFR3 deficiency.

a, Whole embryos from Control, Cdh5CreERT2; Flt4fl/fl, and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ littermates. White dotted lines outline the liver contour, and the red arrowhead indicates severe edema. b, H&E staining of liver. Scale bars = 100μm. c, Immunofluorescence staining and quantification of the entire liver for LYVE1 (black) in Control, Cdh5CreERT2; Flt4fl/fl, and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ E14.5 littermates. Red dotted lines demarcate the outer zone (OZ, defined as 400μm from the outer edge) and the central zone (CZ). Boxes in the top image denote areas of magnified image on the right. Control, n=8. Cdh5CreERT2; Flt4fl/fl, n=5. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. Scale bars = 500μm. d, Immunofluorescence staining of the liver for LYVE1 (red) and Ki67 (green), and quantification of percent Ki67+ sinusoidal endothelial cells. White arrowheads point to Ki67+LYVE1+ cells. Control, n=8. Cdh5CreERT2; Flt4fl/fl, n=5. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. Scale bars = 100μm. Statistical analysis was performed using one-way ANOVA with Tukey’s test for multiple comparisons. Data are shown as means±S.D.
Loss of VE-cadherin function partially rescues defects in lymphatic vascular growth conferred by VEGFR3 deficiency
Prior studies have demonstrated that both loss of VEGF-C/VEGFR3 signaling and gain of VE-cadherin function result in reduced lymphatic vascular growth12–15. Consistent with these studies15, Cdh5α/α embryos exhibited edema and a severe, 3–4 fold reduction in the density of LYVE1+PROX1+ dermal LECs compared with control littermates (Supplemental Figure 5a). A similar reduction in dermal LEC number was observed following loss of endothelial VEGFR3 in E14.5 Cdh5CreERT2; Flt4fl/fl embryos following tamoxifen induction at E10.5 (Figure 6a). Cutaneous edema due to loss of endothelial VEGFR3 was partially rescued by partial loss of VE-cadherin (Figure 5a and 6a). Haploinsufficiency of VE-cadherin conferred a highly significant rescue of skin lymphatic growth of VEGFR3-deficient Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos as shown by whole-mount immunofluorescence and tissue sections (Figure 6a, b and Supplemental Figure 5b). In contrast to fetal liver and bone marrow sinusoidal vessels in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos, however, lymphatic rescue was not complete. These studies reveal that VE-cadherin opposes VEGFR3 function during growth of both lymphatic and sinusoidal vessels. They also demonstrate that lymphatic vessels may grow in the complete absence of VEGFR3 signaling if VE-cadherin function is reduced, implicating either a VEGFR-independent mechanism by which VE-cadherin influences sinusoidal and lymphatic growth, or the use of alternative growth factor receptors and/or ligands (discussed further below).
Figure 6. VE-cadherin haploinsufficiency rescues defects in growth factor signaling and lymphatic growth associated with endothelial VEGFR3 loss.

a, Whole-mount immunofluorescence staining for PROX1 (green), LYVE1 (red), and CD31 (blue) in back skin from Control, Cdh5CreERT2; Flt4fl/fl, and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ E14.5 embryos. b, H&E and immunofluorescence staining for TER119 (green), LYVE1 (red), and PROX1 (gray) in skin sections from Control, Cdh5CreERT2; Flt4fl/fl, and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ E14.5 embryos. White arrowheads indicate LYVE1+PROX1+ lymphatic endothelial cells, and double-headed arrow with ‘E’ indicates presence of edema. Quantification of number of lymphatic endothelial cells per area. Control, n=8. Cdh5CreERT2; Flt4fl/fl, n=6. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4. Scale bars = 50μm. c, d, Immunofluorescence staining of skin (c) and bone marrow (d) for the lymphatic marker LYVE1 or sinusoidal marker Endomucin (red), TER119 (gray), and pERK1/2 (green), and quantification of percent pERK1/2+ LECs (Control, n=8. Cdh5CreERT2; Flt4fl/fl, n=6. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4) or SECs (Control, n=8. Cdh5CreERT2; Flt4fl/fl, n=5. Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+, n=4). Yellow arrowheads indicate pERK1/2+ lymphatic or sinusoidal endothelial cells. Scale bars = 25μm. e, Immunoblot analysis and quantification of human LECs treated for pERK1/2, pAKT, and pS6 expression in response to VEGF-C for 15 minutes. n=6 independent experiments per condition. Statistical analysis was performed using one-way ANOVA with Tukey’s test for multiple comparisons. Data are shown as means±S.D.
Loss of VE-cadherin restores pERK1/2 signaling in VEGFR3-deficient SECs and LECs in vivo and in vitro
VEGF-C binding to VEGF receptors induces potent growth factor signaling pathways, such as pERK1/2 and PI3K, that promote vessel growth28–30. To determine if loss of VE-cadherin rescues growth of LECs and SECs through effects on these downstream growth factor pathways, we next assessed pERK1/2 in endothelial cells in which the expression of VEGFR3 and/or VE-cadherin had been reduced. Consistent with its role during VEGFR3 signaling, pERK1/2 levels were significantly reduced in the skin LECs and bone marrow SECs of Cdh5CreERT2; Flt4fl/fl embryos (Figure 6c, d). However, pERK1/2 levels were fully restored to normal levels in bone marrow SECs and partially restored in skin LECs of Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos (Figure 6c, d). These results mirror the complete rescue of sinusoidal growth and partial rescue of lymphatic growth observed with partial loss of VE-cadherin (Figure 5a), suggesting that they are likely to explain those findings. Conversely, the levels of pERK1/2 were severely reduced in E18.5 bone marrow SECs of Cdh5α/α embryos (Supplemental Figure 5c), suggesting that loss of downstream growth factor signaling also underlies the loss of sinusoidal vascular growth with gain of VE-cadherin function.
Both VEGFR2 and VEGFR3 stimulate downstream pERK1/2 and pAKT signaling responses31. To better understand the effect of VE-cadherin on growth factor signaling pathways downstream of VEGF-C/VEGFR3, we examined primary dermal LECs that naturally express high levels of VEGFR3, allowing us to manipulate VEGFR3 and/or VE-cadherin expression in the presence or absence of VEGF-C. Treatment of LECs with VEGF-C rapidly induced pERK1/2 and pAKT with a peak response at 15 minutes (Supplemental Figure 6a), an effect reproduced by the VEGFR3-specific ligand VEGF-CC156S but not by VEGF-A (Supplemental Figure 6b). siRNA knockdown of VEGFR3 reduced pERK1/2 and pS6, but did not impact pAKT (Figure 6e and Supplemental Figure 6c). As observed in vivo, simultaneous knockdown of VE-cadherin and VEGFR3 restored pERK1/2 as well as pS6 levels to those seen with VEGF-C treatment of control LECs (Figure 6d). Notably, knockdown of VE-cadherin alone augmented the pERK1/2 response to VEGF-C to a level above that seen in control cells (Figure 6d). These in vivo and in vitro findings suggest that loss of VE-cadherin sustains sinusoidal and lymphatic vessel growth in the absence of VEGFR3 by augmenting VEGF-C induced growth factor signals such as ERK1/2 that are known to be downstream of growth factor receptors.
Loss of VE-cadherin function cannot rescue loss of lymphatic vascular growth conferred by VEGF-C deficiency: evidence for VEGF-C/VEGFR2 signaling
In vitro studies have demonstrated that VEGF-C may activate VEGFR2 receptors as well as VEGFR3 receptors18,25, albeit with lower affinity, but constitutive LEC loss of VEGFR2 receptor function has minimal impact on lymphatic vascular growth in developing mice in vivo32. The finding that loss of VE-cadherin rescues SEC and LEC proliferation and vessel growth by augmenting ERK1/2 signaling, and prior in vitro studies demonstrating that loss of VE-cadherin can augment VEGFR2 signaling in vitro18,25, suggested that loss of VE-cadherin may rescue loss of sinusoidal and lymphatic vascular growth due to VEGFR3 deficiency by augmenting VEGFR2 signals. To test this hypothesis, we next determined whether VE-cadherin haploinsufficiency could rescue sinusoidal and lymphatic vascular defects conferred by loss of VEGF-C in a manner like that observed for loss of VEGFR3. Following tamoxifen induction to delete VEGF-C at E10.5, VE-cadherin haploinsufficiency in R26CreERT2; Vegfcfl/fl; Cdh5fl/+ littermate embryos failed to rescue severe cutaneous edema, but partially rescued liver size (Figure 7a–d). Analysis of gene expression by qPCR in multiple tissues confirmed loss of Vegfc mRNA in R26CreERT2; Vegfcfl/fl; Cdh5fl/+ mice (Supplemental Figure 6d). H&E staining to assess skin edema and quantification of skin LEC numbers in skin sections demonstrated complete loss of LECs in both R26CreERT2; Vegfcfl/fl and R26CreERT2; Vegfcfl/fl; Cdh5fl/+ embryos (Figure 7b), consistent with complete lack of rescue. Quantification of liver sinusoidal vascular area and SEC proliferation showed partial rescue of the outer zone defect in R26CreERT2; Vegfcfl/fl; Cdh5fl/+ embryos (Figure 7c–e). These findings reveal (i) that loss of VE-cadherin rescues loss of VEGF-C less efficiently than loss of VEGFR3, and (ii) that in both cases rescue is more efficient in blood vascular SECs than LECs. Since VEGF-C may signal via VEGFR2 or VEGFR3, these findings do not support a VEGFR-independent mechanism of rescue but are instead consistent with augmentation of VEGFR2 signaling as the mechanism by which loss of VE-cadherin rescues VEGFR3-deficient sinusoidal and lymphatic vessel growth in vivo. To test this mechanism, we next determined whether the rescue of VEGF-C stimulated pERK1/2 in LECs lacking both VEGFR3 and VE-cadherin requires VEGFR2 in vitro. Indeed, knockdown of VEGFR2 reversed the rescue of pERK1/2 conferred by knockdown of VE-cadherin following VEGFR3 loss of function (Figure 7f).
Figure 7. VE-cadherin haploinsufficiency partially rescues sinusoidal but not lymphatic growth defects conferred by loss of VEGF-C by potentiating VEGFR2 signaling.

a, Whole embryos from Control, R26CreERT2; Vegfcfl/fl, and R26CreERT2; Vegfcfl/fl; Cdh5fl/+ littermates. White dotted lines outline the liver contour, and the red arrowheads indicate severe edema. Top and bottom rows are littermates from two different litters. b, H&E and immunofluorescence staining for TER119 (green), LYVE1 (red), and PROX1 (gray) in skin sections. White arrowheads indicate LYVE1+PROX1+ lymphatic endothelial cells, and double-headed arrow with ‘E’ indicates presence of edema. Quantification of number of lymphatic endothelial cells per area. Control, n=12. R26CreERT2; Vegfcfl/fl, n=5. R26CreERT2; Vegfcfl/fl; Cdh5fl/+, n=5. Scale bars = 50μm. c, H&E staining of liver, immunofluorescence staining for LYVE1 (red) and Ki67 (green). Scale bars = 100μm. d, Immunofluorescence staining and quantification of the entire liver for LYVE1 (black). Red dotted lines demarcate the outer zone (OZ, defined as 400μm from the outer edge) and the central zone (CZ). Boxes in the top image denote areas of magnified image on the bottom. Scale bars = 500μm. e, Quantification of LYVE1 area by zone and percent Ki67+ sinusoidal endothelial cells. Control, n=12. R26CreERT2; Vegfcfl/fl, n=5. R26CreERT2; Vegfcfl/fl; Cdh5fl/+, n=5. f, Immunoblot analysis and quantification of pERK1/2 in human LECs treated with VEGF-C with knockdown of VEGFR3, VE-cadherin, and VEGFR2. n=6 independent experiments per condition. g, Immunofluorescence staining and quantification of skin sections stained for Endomucin (green), LYVE1 (red), and VEGFR2 or pY1175 VEGFR2 (gray). White arrowheads indicate increased lymphatic VEGFR2. Yellow arrowheads indicate increased lymphatic pY1175 VEGFR2. Statistical analysis was performed using one-way ANOVA with Tukey’s test for multiple comparisons.
To further investigate whether VE-cadherin haploinsufficiency rescues loss of VEGFR3 through VEGFR2 in vivo, we stained E14.5 skin sections for VEGFR2 and pY1175 VEGFR2, a measure of VEGFR2 activation. While skin LECs in both Cdh5CreERT2; Flt4fl/fl and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos showed increased VEGFR2 protein levels relative to controls, only skin LECs from Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ mice also showed elevated levels of pY1175 VEGFR2 (Figure 7g). Similarly, VEGFR2 was also increased in bone marrow SECs of Cdh5CreERT2; Flt4fl/fl and Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos, but pY1175 VEGFR2 was only elevated in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ embryos (Supplemental Figure 7a, b). These results are consistent with prior studies showing that loss of VEGFR3 results in a compensatory increase in VEGFR2 protein level33. The lack of change in pY1175 (i.e. “activated”) VEGFR2 in Cdh5CreERT2; Flt4fl/fl embryos despite increased VEGFR2 levels may reflect inhibition by VE-cadherin that is more available for VEGFR2 inhibition in the absence of VEGFR3. Consistent with such a mechanism, augmented VE-cadherin staining observed in the bone marrow sinusoids of Cdh5CreERT2; Flt4fl/fl mice was reduced to control levels in Cdh5CreERT2; Flt4fl/fl; Cdh5fl/+ mice, a change accompanied by increased VEGFR2 pY1175 levels (Supplemental Figure 7b, c). These studies support a mechanism in which loss of VE-cadherin can rescue loss of VEGF-C/VEGFR3 signaling by augmenting VEGF-C/VEGFR2 signaling in LECs, and possibly both VEGF-C/VEGFR2 and VEGF-A/VEGFR2 signaling in SECs since we detected rescue of sinusoidal growth even in the absence of VEGF-C (Figure 7c–e and Supplemental Figure 10).
The studies above suggest that loss of VE-cadherin potentiates VEGFR2 signaling to rescue loss of VEGFR3. However, this effect seems to be dependent on the availability of VEGF-C as VE-cadherin haploinsufficiency cannot rescue lymphatic growth and only partially rescues sinusoidal growth in VEGF-C knockout mice (Figure 7a–e). One potential explanation for these observations is that LECs and SECs express different levels of VE-cadherin, VEGFR2, and VEGFR3 that may impact their sensitivity to these perturbations. Indeed, analysis of single cell RNA-sequencing (scRNA-seq) datasets of embryonic skin, liver, and bone marrow show that LECs express higher levels of VEGFR3, similar levels of VEGFR2 and lower levels of VE-cadherin compared to BECs (Supplemental Figure 8a,b). In contrast, liver and bone marrow SECs exhibit higher levels of both VEGFR2 and VEGFR3 and similar levels of VE-cadherin compared to BECs (Supplemental Figure 9a–d). Thus differential levels of VE-cadherin, VEGFR2, and VEGFR3 (summarized in Supplemental Figure 9e) may explain differences in extent of rescue between lymphatic and sinusoidal vascular beds in mice lacking VEGF-C or VEGFR3.
VEGFR3 and VE-cadherin negatively regulate each other through effects on endosomal trafficking
A large body of data has demonstrated that efficient signaling by receptor tyrosine kinases such as VEGFR3 and VEGFR2 requires receptor internalization to enable signaling from within cytoplasmic endosomes (reviewed in34). Prior studies have reported that membrane-associated VE-cadherin prevents internalization of VEGFR2 receptors in cultured endothelial cells, thereby reducing VEGF-A/VEGFR2 signaling in confluent ECs in which VE-cadherin is stabilized at cell-cell junctions18,25. Conversely, VEGF-A/VEGFR2 signaling negatively regulates VE-cadherin function by activating SRC that phosphorylates the VE-cadherin intracellular tail to drive internalization and reduce its abundance at the cell membrane17,24. These prior studies suggested that our findings in sinusoidal and lymphatic vessel growth might reflect reciprocal regulation of VEGFR and VE-cadherin trafficking from the endothelial cell membrane.
To test whether VEGF-C/VEGFR3 signaling regulates VE-cadherin trafficking and localization in a manner like that described for VEGF-A/VEGFR2 signaling, we first measured VE-cadherin phosphorylation after treatment of cultured LECs with VEGF-C and the VEGFR3-specific ligand VEGF-CC156S. Immunoblotting of LEC lysates obtained 15 minutes after treatment with both VEGF-C and VEGF-CC156S demonstrated elevated levels of both pY418 (activated) SRC and pY685 VE-cadherin, a known SRC phosphorylation site on VE-cadherin35 (Figure 8a). Consistent with a mechanism in which SRC directly phosphorylates VE-cadherin, VEGF-C stimulated strong co-localization of pY418-positive SRC and VE-cadherin at the cell membrane (Figure 8b). Immunostaining revealed reduced levels of pY418 SRC in Cdh5CreERT2; Flt4fl/fl bone marrow SECs in vivo (Figure 8c), and loss of VEGFR3 reduced the level of pY418 SRC in VEGF-C treated LECs in vitro (Figure 8d) – a finding that was not altered by concomitant loss of VE-cadherin, confirming that SRC is activated by VEGF-C/VEGFR3 signaling and not directly influenced by VE-cadherin. To test the role of SRC and its regulation of VE-cadherin on growth factor signaling, we treated LECs with the SRC inhibitor SU6656 and compared the effects to those of the MEK1/2 inhibitor U0126, which completely eliminates pERK1/2 signals. LECs treated with SU6656 prior to stimulation with VEGF-C showed blunted pERK1/2 and pY685 VE-cadherin levels (Figure 8e). In contrast, LECs treated with U0126 exhibited drastically reduced pERK1/2 levels, but no differences in pY685 VE-cadherin levels (Figure 8e). Thus VEGF-C-induced phosphorylation of VE-cadherin is dependent on SRC, and SRC signaling is in turn required for maximal activation of pERK1/2. Treatment of LECs with VEGF-C increased the number of EEA1+VE-cadherin+ endosomes, an effect that was reversed by treatment with the SRC inhibitor SU6656 (Figure 8f). Consistent with these studies in cultured ECs, immunofluorescent staining of E18.5 R26CreERT2; Vegfcfl/fl and Cdh5CreERT2; Flt4fl/fl bone marrow demonstrated stronger junctional staining for VE-cadherin in the Endomucin+ SECs of embryos lacking VEGF-C/VEGFR3 signaling (Figure 8g, h). Quantification confirmed increased fluorescence intensity normalized to endothelial area relative to controls, consistent with decreased VE-cadherin removal from the cell membrane following SEC loss of VEGF-C/VEGFR3 signaling (Figure 8g, h). These findings demonstrate that VEGF-C/VEGFR3 signaling negatively regulates VE-cadherin through SRC in a manner analogous to that described for VEGF-A/VEGFR2 signaling, and suggest that such regulation of VE-cadherin is required for optimal downstream ERK1/2 activation.
Figure 8. Reciprocal VEGFR-VE-cadherin regulation is mediated by effects on receptor trafficking.

a, Immunoblot analysis and quantification of pY685 VE-cadherin and pY418 SRC in response to treatment VEGF-C or the VEGFR3-specific VEGF-CC156S for 15 minutes. n=3 independent experiments per condition. b, Immunofluorescence staining and quantification for VE-cadherin (red) and pY418 Src (green) in human LECs after treatment with VEGF-C for 15 minutes. Bottom panels show colocalized VE-cadherin and pY418 SRC. n=3–4 independent experiments per condition. c, Immunofluorescence staining for pY418 SRC (green) and Endomucin (red) in bone marrow sections from Control and Cdh5CreERT2; Flt4fl/fl mice, and quantification of pY418 SRC FI normalized to Endomucin+ area. Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4. d, Immunoblot analysis and quantification of pY418 SRC in human LECs after knockdown of VEGFR3 and VE-cadherin and treatment with VEGF-C for 15 minutes. n=3 independent experiments per condition. e, Immunoblot analysis and quantification of pERK1/2 and pY685 VE-cadherin expression in response to VEGF-C with the SRC inhibitor SU6656 or MEK1/2 inhibitor U0126. n=3 independent experiments per condition. f, Immunofluorescence staining and quantification of human LECs that were non-treated (NT) or treated for VEGF-C for 30 minutes with or without SU6656 for the early endosome marker EEA1 (green) and VE-cadherin (red). Yellow arrows denote EEA1+VE-cadherin+ endosomes. n=16–19 fields of view from 3 independent experiments per condition. Scale bars = 25μm. g, h, Immunofluorescence staining of bone marrow from E18.5 Control and R26CreERT2; Vegfcfl/fl (g, Control, n=7. R26CreERT2; Vegfcfl/fl, n=7) or Cdh5CreERT2; Flt4fl/fl (h, Control, n=5. Cdh5CreERT2; Flt4fl/fl, n=4) littermates for Endomucin (red) and VE-cadherin (green). Boxes in the left image denote area of magnified image in gray scale below. Quantification of VE-cadherin fold change measured by VE-cadherin fluorescence intensity (FI) normalized to Endomucin+ area. Scale bars = 100μm. i, Immunofluorescence staining of human LECs that were treated with VEGF-C for 30 minutes with knockdown of VE-cadherin for EEA1 (red) and VEGFR3 (green). Yellow arrows denote EEA1+VEGFR3+ endosomes. n=50 fields of view from 4 independent experiments per condition. Scale bars = 10μm. j, Immunofluorescence staining of human LECs that were treated with VEGF-C for 30 minutes with knockdown of VEGFR3 and VE-cadherin for EEA1 (red) and VEGFR2 (green). Yellow arrows denote EEA1+VEGFR2+ endosomes. n=21 fields of view from 3 independent experiments per condition. Scale bars = 10μm. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test for a, b, c, e, g, h, and i and one-way ANOVA with Tukey’s test for multiple comparisons for d, f, and j. Data are shown as means±S.D. or median±1st/3rd quartile for violin plots.
Since VE-cadherin can negatively regulate VEGFR2 signaling by blocking receptor trafficking to signaling endosomes18,25 we next tested whether loss of VE-cadherin might act similarly on VEGFR3. siRNA knockdown of VE-cadherin in cultured LECs significantly increased the number of EEA1+VEGFR3+ endosomes after exposure to VEGF-C (Figure 8i), consistent with VE-cadherin inhibition of VEGFR3 trafficking to endosomes. The rescue of VEGFR3-deficient sinusoidal and lymphatic vessel growth and ERK1/2 signaling by loss of VE-cadherin (Figures 6b–d), and the requirement for VEGFR2 for that rescue in cultured LECs (Figure 7f), further suggested that loss of VE-cadherin might augment VEGF-C/VEGFR2 signaling by increasing VEGFR2 trafficking to endosomes. We therefore next assessed VEGFR2 internalization following VEGF-C stimulation of cultured LECs. While we found little evidence of VEGF-C-induced VEGFR2 internalization in siCTRL and siFLT4 treated LECs, simultaneous siFLT4/siCDH5 knockdown resulted in significantly increased EEA1+VEGFR2+ endosomes (Figure 8j). These findings support a mechanism in which VE-cadherin opposes growth factor signaling by both VEGFR3 and VEGFR2 via effects on endosomal trafficking of ligand-bound receptors.
Discussion
Endothelial cell growth factor signaling and adhesion are known regulators of vascular growth and development, but the molecular nature of their relationship has been challenging to define. A paradigm in which VEGF-A/VEGFR2 signaling reduces VE-cadherin function and cell-cell adhesion to facilitate cell proliferation and migration is well established16,17,23,24,36, but whether and to what extent VE-cadherin also regulates VEGFR2 or VEGFR3 signaling during vessel growth has been unclear. Our studies address these outstanding questions as a consequence of efforts to understand the similar fetal liver hematopoietic defects that arise in mice with loss of VEGF-C function or gain of VE-cadherin function. We find that VEGF-C/VEGFR3 signaling is required for the growth of sinusoidal blood vessels that support definitive hematopoiesis in the bone marrow and fetal liver of the developing embryo. VE-cadherin opposes VEGF-C/VEGFR3 signaling in sinusoidal and lymphatic endothelial cells, explaining how gain of VE-cadherin function confers hematopoietic and lymphangiogenic defects identical to those in animals lacking VEGF-C. Consistent with these observations, partial loss of VE-cadherin augments VEGF-C signaling and even enables growth of VEGFR3-deficient sinusoidal and lymphatic vessels, most likely through augmented VEGF-C/VEGFR2 signals. Our findings identify a powerful negative reciprocal regulatory mechanism in which VEGFRs and VE-cadherin balance each other’s function through regulation of receptor intracellular trafficking during sinusoidal and lymphatic vessel growth (Supplemental Figure 10). Manipulating this crosstalk, e.g. by inhibiting VE-cadherin function, may enable more efficient regeneration of sinusoidal and lymphatic vessels in patients.
The finding that VEGF-C/VEGFR3 signaling is required for the growth of sinusoidal vessels explains why sinusoidal vessels express the VEGFR3 receptor while the vast majority of blood vessels do not, and solves the mystery of why loss of CCBE1 or VEGF-C blocks fetal liver growth and hematopoiesis12,13. This phenotype was previously attributed to defective erythroid progenitor maturation, but the canonical VEGF-C receptor VEGFR3 is not expressed in those cells and we demonstrate that endothelial-specific loss of VEGFR3 confers an identical hematopoietic defect due to loss of sinusoidal angiogenesis. Why do sinusoidal vessels require VEGF-C/VEGFR3 signaling for their growth? Sinusoidal vessels are in many ways hybrid vessels that incorporate functional and molecular features of both blood and lymphatic vessels, in part to facilitate the movement of hematopoietic cells (reviewed in1). Whether VEGFR3 endows SECs with specialized, hybrid functions remains to be determined, but our work demonstrates that VEGFR2 can at least compensate for their rapid expansion during fetal liver and bone marrow development through ERK1/2 signaling.
The most significant molecular finding of this study is the discovery that the VEGF receptors VEGFR2 and VEGFR3 and VE-cadherin exhibit reciprocal, negative regulatory function in SECs and LECs, to the extent that loss of VE-cadherin enables growth of sinusoidal and lymphatic vessels in the complete absence of VEGFR3. Prior studies have established negative regulation of VE-cadherin and endothelial cell junctions by VEGF-A/VEGFR2 signaling in vivo, and we provide evidence for similar regulation by VEGF-C/VEGFR3 signaling in cultured LECs, but the converse has only been proposed on the basis of in vitro studies18,25 and is not presently part of the angiogenic paradigm. Our findings demonstrate that negative regulation of VEGFR signaling by VE-cadherin is unexpectedly powerful in vivo. The strength of this regulatory mechanism is highlighted by the observations that VE-cadherin gain of function blocks sinusoidal and lymphatic growth while VE-cadherin loss of function rescues the loss of sinusoidal and lymphatic growth conferred by loss of VEGFR3. Although the molecular mechanism(s) by which VE-cadherin regulates VEGFR signaling in sinusoidal and lymphatic vessels remains to be more fully investigated, our studies and prior work18,25 support a mechanism in which cell membrane-associated VE-cadherin opposes VEGFR internalization and efficient signal transduction from intracellular endosomes. Such a receptor-based mechanism is consistent with the observed rescue of pERK1/2 signaling in vitro and in vivo. As proposed previously18,25, such a regulatory mechanism provides an elegant molecular basis by which proliferative signals in the ECs of growing vessels can be shut down as endothelial cell-cell contacts are formed and VE-cadherin stabilized at the cell membrane. Molecularly, it is noteworthy that the endocytic mechanism of VEGFR regulation by VE-cadherin is similar to that previously reported for EphrinB229,37. Whether and to what extent EphrinB2 may participate in this regulatory process is an important future question.
It is surprising that loss of VE-cadherin function is able to rescue sinusoidal and lymphatic vascular growth despite the complete loss of VEGFR3 in LECs because the present paradigm for lymphatic vessel growth considers VEGFR3 to be required38,39. Our studies point to augmented VEGF-C/VEGFR2 signaling as the mechanism underlying this rescue in LECs because loss of VE-cadherin was completely unable to rescue loss of lymphatic growth due to VEGF-C deficiency, and rescue of VEGF-C induced ERK1/2 signaling in cultured VEGFR3-deficient LECs required VEGFR2. Biochemical and in vitro signaling studies performed more than two decades ago demonstrated that VEGF-C may bind and activate VEGFR2 (although with a KD 3-fold higher than that for VEGFR340), but these studies provide evidence that VEGFR2 may also serve as a functionally significant VEGF-C receptor in vivo. The partial rescue of sinusoidal growth in VEGF-C-deficient animals by VE-cadherin loss may be explained by the activity of other VEGFR ligands such as VEGF-A that may participate more in the growth of sinusoidal than lymphatic vessels. This explanation is also consistent with the observation that rescue of VEGFR3-deficient vessel growth is more complete in sinusoidal than lymphatic vessels, a finding that may reflect higher expression of VEGFR2 in SECs than LECs. Such a model is consistent with prior studies demonstrating that loss of VEGFR2 in lymphatics moderately attenuates growth32 but in liver and bone marrow sinusoids severely compromises sinusoidal vascular regeneration3,41.
It is noteworthy that our studies of developing sinusoidal and lymphatic vessels reveal different roles for VEGFRs and VE-cadherin than those identified by other investigators in different vascular contexts. Complete loss of VE-cadherin in the developing lymphatic system, as opposed to the partial loss utilized in this study, resulted in defective lymphangiogenesis19,42, a result likely to reflect the requirement for VE-cadherin in formation of multi-cellular vessels than its impact on VEGFR signaling. More mechanistically relevant to this study is the observation that loss of VEGFR3 in the BECs of the neonatal retina resulted in augmented VEGFR2 expression and signaling that was accompanied by reduced VE-cadherin membrane localization and vascular leak33, as opposed to augmented VE-cadherin membrane localization as observed in this study. The explanation for these opposite effects of VEGFR3 loss likely lies in the distinct ligands driving VEGFR signaling and the role of VEGFR3 in these different vascular contexts. Our genetic data demonstrate that in growing sinusoidal and lymphatic vessels VEGFR3 in SECs and LECs is responding to VEGF-C and acts as a signal-transducing receptor that opposes VE-cadherin membrane localization. In contrast, in growing blood vessels of the retina the dominant growth factor is VEGF-A that cannot bind VEGFR3; thus the role of VEGFR3 in that context is likely to be as an antagonist rather than agonist of VEGFR signaling through formation of non-signaling VEGFR2/3 heterodimers43. These observations highlight the importance of vascular context for VEGFR3 in particular and VEGFR interaction in general, a theme also recently demonstrated by genetic studies of VEGFR2 and VEGFR1 in various mouse vascular beds in vivo44.
An intriguing translational possibility raised by these studies is that manipulation of VE-cadherin could improve the growth and regeneration of sinusoidal and lymphatic vessels in mature vessels and animals. Hematopoietic recovery after bone marrow ablation and transplantation requires the regeneration of bone marrow sinusoidal vessels3. As new therapies for hematologic malignancies and hemoglobinopathies emerge, efficient bone marrow transplantation may benefit from transient blockade of VE-cadherin to augment sinusoidal angiogenesis. Similarly, the major cause of lymphedema in the first world is surgical resection of lymph nodes for staging of cancer metastasis. Lymphatic recovery after lymph node resection is minimal and simple stimulation with local or systemic VEGF-C has not yet significantly altered this outcome45,46. Transient blockade of VE-cadherin might release the lymphangiogenic activity required for such regeneration and prevention of edema. However, since the molecular basis by which VE-cadherin blocks VEGFR trafficking remains to be elucidated, future studies will be needed to determine whether pharmacologic blockade of VE-cadherin can reproduce the effect of its genetic loss on VEGFR regulation and signaling.
Materials and Methods
Generation of Mutant Mice
Rosa26-CreERT2 mice were purchased from the Jackson Laboratory (Stock No. 008463). VEGFR3 (Flt4) floxed mice47, Vegfc floxed mice47, Cdh5(PAC)-CreERT2 mice29, membrane-retained VE-cadherin (Cdh5α) mice48 and VE-cadherin (Cdh5) floxed49 were previously described. Mice were bred according to standard protocols and maintained on a C57BL/6 or mixed background. Mating pairs were set up in the afternoon and vaginal plugs checked in the morning. For E18.5 embryo collection, tamoxifen (200μl at 25mg/ml dissolved in corn oil) was administered by oral gavage to pregnant dams on E14.5, E15.5, and E16.5 and medroxyprogesterone (100μl at 15mg/ml dissolved in DMSO) was administered by subcutaneous injection on E17.5 to prevent parturition. For E14.5 embryo collection, tamoxifen (200μl at 25mg/ml dissolved in corn oil) was administered by oral gavage to pregnant dams on either E9.5, E10.5, and E11.5 (embryos in Figure 2 and Supplemental Figure 1) or E10.5, E11.5, and E12.5 (embryos in Figures 4, 5, and 6 and Supplemental Figures 4 and 5). Cre-negative and/or heterozygous littermates were used as controls. All procedures were conducted using an approved animal protocol (806811) in accordance with the University of Pennsylvania Institutional Animal Care and Use Committee.
Histology & Immunofluorescence Staining
Whole mouse embryos or dissected bones were collected and fixed in 4% paraformaldehyde (PFA) overnight at 4°C prior to dehydration in alcohol and paraffin embedding. Tissue sections underwent to dewaxing and rehydration through xylene and ethanol treatment and were then subject to hematoxylin and eosin (H&E) staining or processed for immunofluorescence. For immunodetection, 10mM citrate buffer (pH 6) was used for antigen retrieval, and sections were blocked with 10% donkey serum in 1% BSA prior to primary antibody treatment overnight at 4°C. A list of antibodies can be found below. Fluorescence-conjugated Alexa Fluor secondary antibodies were used (1:500, Invitrogen) according to the primary antibody species and counterstained with DAPI (1:1000). Sections or tissues were mounted on slides with ProLong Gold Antifade reagent. Signals were detected and images collected using a Zeiss LSM 880 confocal microscope. Percent sinusoidal coverage was measured by quantifying Endomucin- or LYVE1-positive area as a percentage of total tissue area. The ‘outer zone’ of the fetal liver was defined as <400μm from the outer edge. VE-cadherin and pY418 SRC fluorescence intensity in the bone marrow was normalized by the SEC (Endomucin+) vascular area. pERK1/2 and pY418 SRC expression in bone marrow SECs was quantified at the metaphysis, defined as <250μm from the growth plate. VE-cadherin, Ki67, and Cleaved Caspase-3 expression was quantified from the entire bone marrow (metaphysis + diaphysis). Quantification of percent positive cells was done by manual counting of double-positive cells as a proportion of single-positive cells. Images in Figures 4h, 6d, 7c, 7g, and 7h are in gray scale and have not been thresholded. All images were analyzed using ImageJ/FIJI software (NIH).
Cell Culture Experiments
Human Dermal Microvascular Endothelial Cells were purchased from Lonza (CC-2516 Batch No. 0000685447, 98% LECs as measured by CD31/Podoplanin expression) were cultured in EBM-2 Basal Medium (Lonza CC-3156) supplemented with EGM-2 MV Microvascular Endothelial Cell Growth Medium SingleQuots (Lonza CC-4147). Recombinant human VEGF-C protein was purchased from R&D (9199-VC) and used at a concentration of 100ng/ml (ED50 1.5–9ng/ml) for all experiments. Recombinant human VEGF-CC156S protein contains a cysteine-to-serine substitution and was purchased from R&D (752-VC) and used at a concentration of 50μg/ml (ED50 1–5μg/ml) for all experiments. Recombinant human VEGF-A protein was purchased from R&D (293-VE) and used at a concentration of 50ng/ml (ED50 1–6ng/ml) for all experiments. siRNAs were purchased from ThermoFisher (siFLT4 s5294, siCDH5 s223069, siKDR s535305) and transfection was done using siPORT Amine (AM4502) according to manufacturer’s instructions. For inhibition of SRC or MEK1/2, LECs were treated with 10μM SU6656 (Millipore Sigma, S9692) or 10μM U0126 (Cell Signaling Technology, 9903) for 1 hour prior to stimulation with VEGF-C. Migration assays were conducted using a 2 well culture-inserts purchased from Ibidi (Cat. No. 81176). Briefly, cells were allowed to adhere in the wells overnight, inserts were removed the next morning, and cells were permitted to migrate for 24 hours. For immunocytochemistry, cells were permeabilized in 0.05% TritonX, blocked in 10% donkey serum in 1% BSA, incubated in primary antibody overnight at 4°C, and subjected to secondary antibody treatment prior to confocal microscopy. Colocalized Pixel Map for pY418 SRC/VE-cadherin was generated using the “Colocalisation Thresholds” plugin in ImageJ using non-constant intensity for colocalized pixels. Percent colocalized pixels was calculated by dividing the area of colocalized pixels by area of VE-cadherin. All images were analyzed using ImageJ/FIJI software (NIH).
Immunoblotting
Cells were lysed in RIPA buffer (RPI, R26200) supplemented with PhosSTOP phosphatase inhibitor (Roche) and cOmplete tablet protease inhibitor (Roche). Lysates were boiled for 10 minutes prior to SDS-PAGE on Bis-Tris gels. Proteins were transferred onto nitrocellulose membranes, blocked with Odyssey blocking buffer, probed with primary antibodies overnight at 4°C, and subjected to secondary antibody treatment. Membranes were scanned and visualized using a LI-COR Odyssey imaging system. Protein levels were normalized to b-actin and phospho-proteins were divided by total level and calculated as fold change relative to control conditions. Band intensity was analyzed using ImageJ/FIJI software (NIH).
Antibodies
Antibodies for immunofluorescence (paraffin sections): Endomucin (1:400, R&D AF4666 or 1:300, Abcam ab106100), TER119 (1:300, Abcam ab91113), Ki67 (1:100, Abcam ab16667), Cleaved Caspase-3 (1:100, Millipore Sigma AB3623), mouse VEGFR3 (1:100, R&D AF743), mouse VE-cadherin (1:300, R&D AF1002), PROX1 (1:100, Abcam ab76696), pY418 SRC (1:100, Invitrogen 14–9034-82), LYVE1 (1:400, R&D AF2125 or 1:200, AngioBio 11–034), pERK1/2 (1:100, Cell Signaling Technology 4370), VEGFR2 (1:200, Cell Signaling Technology 2479), pY1175 VEGFR2 (1:200, Cell Signaling Technology 2478)
Antibodies for immunocytochemistry: human VEGFR3 (1:200, Millipore Sigma MAB3757), human VE-cadherin (1:400, R&D AF938), EEA1 (1:300, R&D MAB8047 or 1:200, R&D AF8047), VEGFR2 (1:300, Cell Signaling Technology 2479), pY418 SRC (1:100, Invitrogen 14–9034-82)
Antibodies for immunoblot: pERK1/2 (1:2000, Cell Signaling Technology 4370), total ERK (1:1000, Cell Signaling Technology 4695), pY685 VE-cadherin (1:500, Millipore Sigma ABT1760), human VE-cadherin (1:1000 R&D AF938), pAKT (1:1000, Cell Signaling Technology 4060), total AKT (1:1000, Cell Signaling Technology 4685), pS6 (1:1000, Cell Signaling Technology 4858), total S6 (1:1000, Cell Signaling Technology 2317), pY418 SRC (1:500, Abcam ab133460), total SRC (1:1000, Cell Signaling Technology 2109), human VEGFR3 (1:1000, Millipore Sigma MAB3757), β-actin (1:2000, Abcam ab8226)
Single cell RNA-sequencing analysis
Wild-type mouse embryo single cell RNA datasets derived from forelimb skin and soft tissue50, liver51, and bone marrow52, were downloaded from public repositories, and analyzed in Seurat v4.053. We tailored the normalization and CCA integration workflow, as some datasets were provided as raw counts while others were pre-normalized and even pre-clustered. Initial QC cleanup involved removal of low quality cells, batch correction, and subsetting on endothelial clusters between E12 and E17. ECs were re-clustered at higher resolution, and FindAllMarkers was used to identify gross endothelial subtypes by known marker genes54. Feature-level analysis of relevant sub-clusters, as well as result visualization, was scripted with ggplot2, Seurat, and/or ggpubr.
Statistical Analysis
All data are reported as means with n≥3 independent experiments or mice, and error bars represent standard deviation (SD). Statistical significance was determined using two-tailed, unpaired Welch’s t-test or one-way ANOVA with Tukey’s test for multiple comparisons. Differences between means were considered significant at P<0.05.
Supplementary Material
Highlights.
VEGF-C/VEGFR3 signaling is required for sinusoidal vascular growth in the fetal liver and bone marrow
CDH5 negative regulation of VEGF-C/VEGFR3 signaling restrains sinusoidal and lymphatic vessel growth
Loss of CDH5 enables growth of sinusoidal and lymphatic vessels in the absence of VEGFR3 signaling through VEGF-C/VEGFR2 signaling
Acknowledgements:
We thank the members of the Kahn lab for their thoughtful comments and advice during this work. We thank the CDB Microscopy Core for microscopy support.
Funding:
Supported by National Institute of Health grants R01 DK123528 (MLK), F30 HL158014 and T32 HL007439 (DCS), T32 HL007150 (AAR), R01 HL142905 (JPS), AHA postdoctoral fellowship No. 836238 (XC), Deutsche Forschungsgemeinschaft grants SFB1009 (A1) and SFB1348 (B1) (DV).
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability:
All data and reagents will be made available upon reasonable request. Transgenic mouse lines not available through public repositories are available from Mark Kahn under a material transfer agreement with the University of Pennsylvania.
References
- 1.Griffin CT & Gao S Building discontinuous liver sinusoidal vessels. J Clin Invest 127, 790–792, doi: 10.1172/JCI92823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itkin T et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328, doi: 10.1038/nature17624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper AT et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 4, 263–274, doi: 10.1016/j.stem.2009.01.006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramasamy SK, Kusumbe AP, Wang L & Adams RH Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507, 376–380, doi: 10.1038/nature13146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaipainen A et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A 92, 3566–3570 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Partanen TA et al. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J 14, 2087–2096, doi: 10.1096/fj.99-1049com (2000). [DOI] [PubMed] [Google Scholar]
- 7.Karkkainen MJ et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 5, 74–80 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Hogan BM et al. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet 41, 396–398, doi:ng.321 [pii] 10.1038/ng.321 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Alders M et al. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat Genet 41, 1272–1274, doi:ng.484 [pii] 10.1038/ng.484 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Le Guen L et al. Ccbe1 regulates Vegfc-mediated induction of Vegfr3 signaling during embryonic lymphangiogenesis. Development 141, 1239–1249, doi: 10.1242/dev.100495 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Saaristo A, Karkkainen MJ & Alitalo K Insights into the molecular pathogenesis and targeted treatment of lymphedema. Ann N Y Acad Sci 979, 94–110 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Fang S et al. Critical requirement of VEGF-C in transition to fetal erythropoiesis. Blood 128, 710–720, doi: 10.1182/blood-2015-12-687970 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Zou Z et al. The secreted lymphangiogenic factor CCBE1 is essential for fetal liver erythropoiesis. Blood 121, 3228–3236, doi:blood-2012–10-462689 [pii] 10.1182/blood-2012-10-462689 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bui HM et al. Proteolytic activation defines distinct lymphangiogenic mechanisms for VEGFC and VEGFD. J Clin Invest 126, 2167–2180, doi: 10.1172/JCI83967 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dartsch N, Schulte D, Hagerling R, Kiefer F & Vestweber D Fusing VE-cadherin to alpha-catenin impairs fetal liver hematopoiesis and lymph but not blood vessel formation. Mol Cell Biol 34, 1634–1648, doi: 10.1128/MCB.01526-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon EJ et al. The endothelial adaptor molecule TSAd is required for VEGF-induced angiogenic sprouting through junctional c-Src activation. Sci Signal 9, ra72, doi: 10.1126/scisignal.aad9256 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Gavard J & Gutkind JS VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8, 1223–1234, doi: 10.1038/ncb1486 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C & Dejana E Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol 174, 593–604, doi: 10.1083/jcb.200602080 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagerling R et al. Distinct roles of VE-cadherin for development and maintenance of specific lymph vessel beds. EMBO J 37, doi: 10.15252/embj.201798271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Si-Tayeb K, Lemaigre FP & Duncan SA Organogenesis and development of the liver. Dev Cell 18, 175–189, doi: 10.1016/j.devcel.2010.01.011 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Langen UH et al. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat Cell Biol 19, 189–201, doi: 10.1038/ncb3476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz L et al. Mechanosensing by beta1 integrin induces angiocrine signals for liver growth and survival. Nature 562, 128–132, doi: 10.1038/s41586-018-0522-3 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Esser S, Lampugnani MG, Corada M, Dejana E & Risau W Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 111 (Pt 13), 1853–1865 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Sun Z et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med 209, 1363–1377, doi: 10.1084/jem.20111343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grazia Lampugnani M et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol 161, 793–804, doi: 10.1083/jcb.200209019 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K & Vikkula M Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet 67, 295–301 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karkkainen MJ et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet 25, 153–159 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Shin M et al. Vegfc acts through ERK to induce sprouting and differentiation of trunk lymphatic progenitors. Development 143, 3785–3795, doi: 10.1242/dev.137901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486, doi:nature09002 [pii] 10.1038/nature09002 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Coso S, Zeng Y, Opeskin K & Williams ED Vascular endothelial growth factor receptor-3 directly interacts with phosphatidylinositol 3-kinase to regulate lymphangiogenesis. PLoS One 7, e39558, doi: 10.1371/journal.pone.0039558 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Y, Zhang X & Simons M Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol 35, 421–429, doi: 10.1161/ATVBAHA.114.304881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dellinger MT, Meadows SM, Wynne K, Cleaver O & Brekken RA Vascular endothelial growth factor receptor-2 promotes the development of the lymphatic vasculature. PLoS One 8, e74686, doi: 10.1371/journal.pone.0074686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinolainen K et al. VEGFR3 Modulates Vascular Permeability by Controlling VEGF/VEGFR2 Signaling. Circ Res 120, 1414–1425, doi: 10.1161/CIRCRESAHA.116.310477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simons M An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda) 27, 213–222, doi: 10.1152/physiol.00016.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallez Y et al. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 26, 1067–1077, doi: 10.1038/sj.onc.1209855 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Li X et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun 7, 11017, doi: 10.1038/ncomms11017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawamiphak S et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465, 487–491, doi: 10.1038/nature08995 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Zhang L et al. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res 20, 1319–1331, doi:cr2010116 [pii] 10.1038/cr.2010.116 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y et al. Heterogeneity in VEGFR3 levels drives lymphatic vessel hyperplasia through cell-autonomous and non-cell-autonomous mechanisms. Nat Commun 9, 1296, doi: 10.1038/s41467-018-03692-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joukov V et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. Embo J 16, 3898–3911, doi: 10.1093/emboj/16.13.3898 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding BS et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 468, 310–315, doi: 10.1038/nature09493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris NR et al. VE-Cadherin Is Required for Cardiac Lymphatic Maintenance and Signaling. Circ Res 130, 5–23, doi: 10.1161/CIRCRESAHA.121.318852 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tammela T et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13, 1202–1213, doi: 10.1038/ncb2331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karaman S et al. Interplay of vascular endothelial growth factor receptors in organ-specific vessel maintenance. J Exp Med 219, doi: 10.1084/jem.20210565 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunnapuu J & Jeltsch M Outside in and brakes off for lymphatic growth. Sci Signal 14, doi: 10.1126/scisignal.abj5058 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Herantis.com. Herantis Announces Inconclusive Results from Phase II Study with Lymfactin in Breast Cancer Related Lymphedema. https://herantis.com/press-releases/herantis-announces-inconclusive-results-from-phase-ii-study-with-lymfactin-in-breast-cancer-related-lymphedema/ (2021).
- 47.Keller T. C. S. t. et al. Genetic blockade of lymphangiogenesis does not impair cardiac function after myocardial infarction. J Clin Invest 131, doi: 10.1172/JCI147070 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulte D et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30, 4157–4170, doi: 10.1038/emboj.2011.304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Cha B, Motawe ZY, Srinivasan RS & Scallan JP VE-Cadherin Is Required for Lymphatic Valve Formation and Maintenance. Cell Rep 28, 2397–2412 e2394, doi: 10.1016/j.celrep.2019.07.072 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He P et al. The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature 583, 760–767, doi: 10.1038/s41586-020-2536-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X et al. Comparative analysis of cell lineage differentiation during hepatogenesis in humans and mice at the single-cell transcriptome level. Cell Res 30, 1109–1126, doi: 10.1038/s41422-020-0378-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y et al. A specialized bone marrow microenvironment for fetal haematopoiesis. Nat Commun 13, 1327, doi: 10.1038/s41467-022-28775-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529, doi: 10.1016/j.cell.2021.04.048 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalucka J et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 180, 764–779 e720, doi: 10.1016/j.cell.2020.01.015 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and reagents will be made available upon reasonable request. Transgenic mouse lines not available through public repositories are available from Mark Kahn under a material transfer agreement with the University of Pennsylvania.