

Abstract
Coxiella burnetii is an intracellular pathogen responsible for causing Q fever in humans, a disease with varied presentations ranging from a mild flu-like sickness to a debilitating illness that can result in endocarditis. The intracellular lifestyle of C. burnetii is unique, residing in an acidic phagolysosome-like compartment within host cells. An understanding of the core molecular biology of C. burnetii will greatly increase our understanding of C. burnetii growth, survival and pathogenesis. We used transposon-directed insertion site sequencing (TraDIS) to reveal C. burnetii Nine Mile Phase II genes fundamental for growth and in vitro survival. Screening a transposon library containing >10 000 unique transposon mutants revealed 512 predicted essential genes. Essential routes of synthesis were identified for the mevalonate pathway, as well as peptidoglycan and biotin synthesis. Some essential genes identified (e.g. predicted type IV secretion system effector genes) are typically considered to be associated with C. burnetii virulence, a caveat concerning the axenic media used in the study. Investigation into the conservation of the essential genes identified revealed that 78 % are conserved across all C. burnetii strains sequenced to date, which probably play critical functions. This is the first report of a whole genome transposon screen in C. burnetii that has been undertaken for the identification of essential genes.
Keywords: Coxiella burnetii, TraDIS, transposon sequencing
Data Summary
The DNA sequences generated in this study have been deposited in the NCBI SRA database under the BioProject PRJNA863483, sample accession number: SAMN30049071.
Impact Statement.
Coxiella burnetii causes Q-fever in animals and in humans, but we know little about the biology of the organism. We used a global mutagenesis method to identify genes which are essential for growth and in vitro survival of the bacterium. These newly identified gene products are targets for drugs to treat disease. Our work will now allow others to use our approach to identify virulence genes in C. burnetii and this will reveal additional targets for drugs to treat disease, and candidates for inclusion in vaccines.
Introduction
Coxiella burnetii is an intracellular, zoonotic pathogen responsible for causing Q fever in humans. C. burnetii is predominantly spread by farmed ruminants such as goats, sheep and cattle [1]. Human infection usually occurs via the inhalation of contaminated aerosols. In humans, Q fever is a disease with varied presentations ranging from a mild flu-like sickness to a debilitating illness that can result in endocarditis [2].
The intracellular lifestyle of C. burnetii is unique, with bacteria residing in an acidic Coxiella -containing vacuole (CCV) within host cells. CCV formation is characteristic of the normal endocytic pathway, and acidification upon lysosomal fusion – a normally bactericidal defence – stimulates the switch of C. burnetii from its small cell variant (a metabolically inactive environmental form) to a metabolically active, replicative large cell variant [3]. C. burnetii could not be cultured outside of host cells until the development of an axenic medium, ACCM-2, in 2009 [4]. As a result, little is known about the genetic factors that are responsible for the unusual lifestyle of this organism. A better understanding of essential genes will greatly contribute to the current knowledge of the critical functions of C. burnetii .
The C. burnetii Nine-mile phase I genome (NMI) reference sequence was published in 2003, revealing a 1 995 275 bp genome with a 37 393 bp QpH1 plasmid, that had been previously sequenced [5, 6]. Genome analysis identified 83 pseudogenes, suggesting ongoing genome reduction [5]. This finding also suggests that a number of genes will not be essential for the lifestyle of C. burnetii . Later, the genome sequence of the avirulent laboratory strain C. burnetii Nine-mile phase II (NMII) was published, confirming the deletion of a large (~26 kbp) chromosomal segment that results in the formation of a truncated lipopolysaccharide without O-antigen sugars [7–9].
To date, there are 83 C . burnetii sequences deposited in the NCBI database. These sequenced C. burnetii strains each fall in to one of six genomic groups (GGs), showing significant genetic diversity amongst strains which have been associated with different hosts or regions located, and show different virulence presentations in animal models [10]. GG I isolates have a global distribution, and include the NMI and NMII strains commonly used in research [11, 12]. GG II isolates are generally European, and contain strains responsible for a large Q fever outbreak in the Netherlands [13]. Recently, GG II has been divided into three subgroups (GG IIa, GG IIb and GG IIc) based on genome content and SNP profile [13, 14]. GG III isolates are associated with cattle [15], whereas GG IV isolates are associated with goats, and do not grow in axenic media [15–17]; GG IV has also been divided into three subgroups (GG IVa, GG IVb and GG X) [14]. GG V isolates are found in North America [12], and GG VI (Dugway) isolates are associated with rodents, and do not appear to cause human disease [18, 19].
Although the first genetic manipulation of C. burnetii was performed in 1996 [20], a suitable suicide plasmid system for transposon mutagenesis was not created until 2009 [21]. Since then, transposon mutagenesis has been used to identify many genes encoding proteins that play important roles in C. burnetii infection. For example, transposon insertion in ftsZ resulted in incomplete cell division, resulting in a significant growth defect in Vero cells [21], and transposon insertion into CBU_1260 (OmpA) resulted in a defect in host cell invasion [22].
In 2009, four research groups published methods combining transposon mutagenesis with high-throughput sequencing [Tn-seq, TraDIS (transposon-directed insertion site sequencing), HITs and IN-seq] [23–26]. A large population of tens, or hundreds of thousands of transposon mutants can be pooled together, put under selective conditions and simultaneously assayed to identify genes essential for growth and survival in that condition. A range of conditions can be applied to identify (for example) genes essential for infection-relevant conditions (virulence-associated genes), environmental survival and stress response. In addition, investigation of essential genes conserved in the core genome of a species allows the identification of genes which probably play critical functions.
The aim of this work was to generate a transposon library for C. burnetii to permit the identification of essential genes, thereby providing information on C. burnetii genes fundamental for the growth and survival in ACCM-2 and enabling further analysis of essential gene products as prospective drugable targets. Furthermore, conservation of essential genes in the core genome of C. burnetii has been determined. Application of this high-throughput mutagenesis technique has not been previously carried out for C. burnetii and will greatly increase our knowledge and understanding of a pathogen for which genetic manipulation techniques have only recently become available. This study provides important new information about the biology of this organism and opens opportunities to study gene fitness in a range of biologically relevant conditions. In the longer term it could lead to new pre-treatments and therapies for Q-fever.
Methods
Bacterial strains and culture conditions
C. burnetii Nine Mile Phase II (NMII) strain RSA493 clone 4 [27] was grown in laboratory-prepared ACCM-2 [4] at 37 °C in microaerobic conditions (5 % CO2, 2.5 % O2) for 7 days for liquid cultures, unless otherwise stated. To obtain colonies, bacteria were plated onto ACCM-2 plates containing agarose at a final concentration of 0.25 % (w/v) and incubated at 37 °C in microaerobic conditions (5 % CO2, 2.5 % O2) for 14 days. Escherichia coli strain DH5α was used for replication of the pKM225 plasmid (accession no. HQ386859) and grown overnight at 37 °C with agitation. Where appropriate, antibiotics were added to media as detailed in methods below.
All manipulations of C. burnetii NMII were carried out in a class I microbiological safety cabinet in a Biological Safety Level 2 (BSL2) laboratory.
Generation of C. burnetii transposon mutants
pKM225 was introduced into stationary phase C. burnetii NMII via electroporation. C. burnetii was made electrocompetent through washing with ice-cold, sterile filtered 10 % (v/v) glycerol prior to electroporation of 50 µl aliquots at 1.8 kV, 500 Ω and 25 µF using 0.1 cm cuvettes and an ECM630 electroporator (BTX Harvard Apparatus). Immediately after electroporation, 950 µl RPMI-glutamax was added to cuvettes. Five hundred microlitres of sample was used to inoculate 6 ml ACCM-2 medium in duplicate. Cultures were incubated overnight, before the addition of chloramphenicol at 3 µg ml−1, followed by a further 4 days of incubation. To obtain colonies, samples were plated onto ACCM-2 agarose plates containing chloramphenicol at 3 µg ml−1. After 14 days of incubation, colonies were washed from the plate and pooled. One hundred microlitres was used to inoculate triplicate 25 ml ACCM-2 cultures for TraDIS library preparation.
Genomic DNA extraction
Genomic DNA (gDNA) was extracted from 25 ml 7 day old cultures using the GenElute Bacterial Genomic DNA Extraction kit (Sigma-Aldrich) following the protocol for Gram-positive bacteria with slight modification. Cultures were centrifuged for 15 min at 4 000 r.p.m. and resuspended in 760 µl Gram-positive lysis solution before separating into 4×190 µl aliquots to process in four columns. The protocol was then followed with an extended 16 h step in lysis solution and addition of proteinase K. DNA was concentrated by ethanol precipitation and pooled for each 25 ml culture.
TraDIS library preparation
Unless otherwise stated, the manufacturer’s instructions were followed for all kits in this process. Five micrograms of gDNA was fragmented by ultra-sonication in a COVARIS E220 focused-ultrasonicator at a peak incident power of 140 W for 55 s. Small (<150 nt) fragments of gDNA were removed using the GeneRead Size Selection Kit (Qiagen). gDNA fragments were end-repaired using the NEBNext DNA Library Prep Master Mix Set for Illumina (New England Biolabs) and cleaned up using the QIAquick PCR purification kit (Qiagen). End-repaired samples were A-tailed using the NEBNext DNA Library Prep Master Mix Set for Illumina (New England Biolabs) followed by a further clean up using the MinElute PCR purification kit (Qiagen). Adapter ligation was performed by combining end-repaired, A-tailed gDNA with phosphorylated, annealed adapters and 5 µl ligase and incubating at room temperature for 15 min (for adapter sequences see Table S1, available in the online version of this article). Adapter ligated samples were cleaned up using the MinElute PCR purification kit (Qiagen). Parallel PCR was performed by mixing 10 µl adapter-ligated DNA with 50 µl NEBNext Q5 Hot Start HiFi PCR Master Mix (New England Biolabs), 39 µl nuclease-free water and 0.5 µl each of 100 µM primers Himar-PCR-3 and one of three custom Illumina primers MPX1–3 (for primer sequences see Table S1). Each sample was separated into 2×50 µl aliquots and PCR was performed on the following thermal profile: 2 min at 98 °C, followed by 20 cycles of 10 s at 98 °C, 30 s at 57 °C and 30 s at 65 °C, followed by 5 min at 65 °C. At the end of the reaction, aliquots were re-pooled and PCR contaminants removed with a two column clean-up using the Generead Size Selection kit (Qiagen). Size selection was performed by electrophoresis of a 2 % (w/v) agarose gel. Fragments (350–500 nt) were excised and DNA was extracted using the MinElute Gel Extraction kit (Qiagen). Samples were diluted to 2 nM and submitted for sequencing as 150 bp single-end reads on an Illumina MiSeq platform (Exeter Sequencing Service).
TraDIS data analysis
Sequencing data were analysed using the Whiteley Lab pipeline [28–30] (available at: www.github.com/WhiteleyLab/TnSeq). First, reads were screened to identify the TGTTA transposon tag, allowing 0 mismatches. The transposon tag was trimmed, and resulting reads mapped to the C. burnetii RSA493 reference genome with bowtie2 (v2.4.5) by implementing the TnSeq2.sh script. A correction for polymerase slippage was performed by taking reads that map within 1 bp of each other and collapsing them to the local maxima using slippage.sh script [29, 31]. Next, the TnSeqDESeq2Essential_mariner.R script was executed. This analysis uses estimateSizeFactors() from DESeq2 (v1.16.0) to normalize samples for sequencing depth before generating pseudodatasets containing the same number of insertion sites, and corresponding number of mapped reads as the inputted experimental dataset, randomly distributed across the genome at TA sites. One thousand pseudodatasets were created. A custom .gff file was created in Microsoft Excel to trim 10 % of the 3′ end of each gene, before insertion sites were binned to obtain gene-level insertion counts. DESeq2 (v1.16.0) was used to compare expected insertions per gene (from the pseudodatasets) to observed insertions per gene (from the experimental datasets). Genes were classified as essential if they had a P adj value of <0.05, were classified as ‘reduced’ by mClust analysis, and had an uncertainty of <0.05.
Further bioinformatic analyses
Essential genes were compared to the core genome of C. burnetii, as well as the core genomes of different C. burnetii genomic groups, using the pan-genome analysis pipeline BPGA (https://sourceforge.net/projects/bpgatool/) as described previously [14]. Clusters of orthologous groups (COGs) were assigned to essential genes using Eggnog Mapper (v2) (http://eggnog-mapper.embl.de/). Proteins encoded by C. burnetii essential genes were investigated by BlastP similarity against the Database of Essential genes (DEG) [32].
Results and discussion
Transposon mutagenesis of C. burnetii
Transposon insertion libraries were generated in C. burnetii NMII using a Himar1 Mariner transposon which inserts at TA sites in the genome, harboured on the plasmid vector pKM225. As pKM225 cannot replicate in C. burnetii, transposition results in the loss of the transposase, thus creating transposon mutants with a single, stable insertion [33]. Transposon mutagenesis yielded approximately 3000 colonies per electroporation. Mock electroporation reactions, using nuclease-free water in place of pKM225, resulted in 20 times fewer colonies. These probably represent spontaneous chloramphenicol-resistant mutants, which should not be amplified during subsequent library preparation stages. From six individual electroporation reactions, approximately 20 000 colonies were picked from ACCM-2 agarose plates and pooled to form the TraDIS library.
Identification of essential genes
The Whiteley lab pipeline is a custom Unix, Perl and R pipeline that has been implemented in previous transposon sequencing studies [28, 29, 31]. The initial step – using the TnSeq2.sh script – was to search reads for the TGTTA transposon tag, with 0 mismatches allowed. The tag is subsequently trimmed and the reads mapped to the C. burnetii RSA493 NMI reference genome using bowtie2 with one mismatch allowed. The use of the NMI reference genome as opposed to the NMII genome sequence permitted a quality check that the mapping of insertion sites was accurate, as it allowed the visualization of the absence of reads mapping to the region from CBU_0679 to CBU_0698, due to the deletion of the lipopolysaccharide (LPS) synthesis cluster in strain RSA439 (NMII) compared to RSA493 (NMI).
From a total of 22 776 700 sequencing reads, 21 358 299 (94 %) started with the transposon tag. Of these, 17 826 711 (83 %) reads were successfully mapped to the reference genome. Mapping identified 10 129 unique insertion sites, amounting to one insertion every ~200 bp. The total number of unique insertions disrupts approximately 8 % of available TA sites. The TGTTA sequence occurs naturally at 1851 positions in the C. burnetii genome, with 1846 located within coding sequences. It is possible that this could result in false identification of up to 1846 insertion sites. Another potential bias arises from the use of Himar1 which has shown reduced insertion into the sequence [CG]GNTANC[CG] [34]. As expected, Himar1 insertions into this sequence were not observed.
The C. burnetii genome contains 2134 coding sequences [5]. Up to 38 unique insertion sites per gene occurred, with a mean of four. Visualization of the insertion counts across the genome showed that 71 %(1514/2134) of genes had been disrupted (Fig. 1). These data represent the ‘observed dataset’.
Fig. 1.

Insertion site distribution across the genome as determined by the Whiteley lab pipeline. The x-axis shows gene position, and y-axis shows insertion frequency. Regions with no insertions are possible essential regions, with the exception of position CBU_0679 to CBU_0698 (purple arrow) which shows the expected LPS deletion in NMII.
Pseudodatasets were generated by arranging the ~10 000 insertions and the corresponding number of reads randomly at TA sites on the C. burnetii RSA493 NMI genome. This simulation was carried out 1000 times, although no significant difference was seen above 500 pseudodatasets. This represents the ‘expected dataset’. Finally, DESeq2 was used to determine whether insertion site abundance in the observed dataset was significantly different from the expected dataset. When plotting a histogram of the log fold change between observed insertions per gene vs expected insertions per gene (Fig. 2) a bimodal ‘line of best fit’ is fitted by the mClust R package, which also determines a log2 fold change cut-off value for separation of the two peaks, and genes with fewer insertions than expected are found in the left-hand peak, and genes with the same or more insertions than expected are found in the right-hand peak. Genes were classified as essential if they contributed to the left-hand side ‘reduced’ peak (log2 fold change <−2.3), with an uncertainty of <0.05 and an adjusted P value (Padj) <0.05. It should be noted that although a bimodal distribution has been fitted by the mClust algorithm incorporated into the pipeline, the dataset appears to show a trimodal distribution with two possible groups of genes included in the essential dataset. To determine whether the cut-off value selected by the pipeline was correct, we searched for homologues in the database of essential genes in the two groups [32]. When a more stringent cut-off of log2 fold change <−7, we found that 76.6 % (271/354) of the genes were homologues. Using the default cut-off of log2 fold change <−2.3 we found that 76.8 % (358/466) of the genes were homologues. The more stringent cut-off excluded genes known to be essential for fundamental biological processes, such as dnaA. Together, this suggests that essential genes are found at similar frequencies across both peaks. It should also be noted that there is a valley between the two peaks. Unlike other available pipelines such as BioTraDIS, the Whiteley lab pipeline does not classify any genes as ambiguous, i.e. falling close to the cut-off. This may result in the incorrect classification of genes with log2 fold changes close to the cut-off.
Fig. 2.
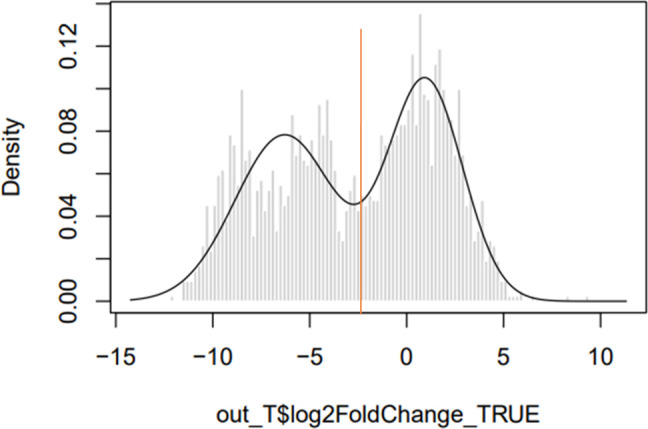
Density histogram showing the log fold change of expected vs observed insertion counts. The x-axis shows the log fold change between observed and expected datasets, and the y-axis shows the density. The left-hand peak contains genes with fewer insertions than expected, while the right-hand peak contains genes with an expected, or greater than expected number of insertion sites. The red line indicates the log2 fold change cut-off of −2.3; genes that fall on the left-hand side of this line are considered to contribute to the left-hand ‘reduced’ peak.
Based on the above criteria, the Whiteley lab pipeline revealed 505 predicted essential genes within the chromosome, and six predicted essential genes on the QpH1 plasmid. The C. burnetii essential genome contains 24 % of the total genes. This is in line with studies of gene essentiality in other microorganisms, where the essential genome can range from 5 to 35 % of the total genome content [35–39]. Notably, the list of essential genes generated with the Whiteley lab pipeine contains genes that have been shown to be essential for fundamental biological processes, such as DNA supercoiling-associated genes gyrAB and all 22 tRNA synthetases.
There have been numerous studies that isolated large numbers of C. burnetii NMII transposon mutants for individual mutant screening [22, 33, 40, 41] which obtained transposon insertions in 55 of the essential genes identified in this study. It is possible that the classification of these genes as essential could be false positives. Six of the genes where transposon insertion mutants had previously been reported in other studies had insertions in the 3′ end of the gene, potentially allowing the production of a truncated, but functional product. Single transposon mutants have been reported for the majority of the remaining 45 genes, and it is possible that the mutations could be mismapped. However, to reduce the possibility of false positives in our dataset, 45 of the genes in which others have obtained transposon insertions that are not at the extreme ends have been removed from the dataset. This left a total of 466 essential genes (Table S2).
Assignment of essential genes to clusters of orthologous groups
Essential genes were assigned to COGs [42] to give insight into the dominant essential gene functions (Fig. 3). The COG categories most highly enriched for essential genes were category J (Translation, ribosomal structure and biogenesis; 11.3 %), category H (Coenzyme transport and metabolism; 9.0 %), category C (Energy production and conservation; 8.6 %) and category L (Replication, recombination and repair; 7.7 %). Moderately enriched COG categories were category M (Cell wall and outer membrane structure and biogenesis; 6.2 %), category E (Amino acid transport and metabolism; 4.9 %) and category F (Nucleotide transport and metabolism; 4.7 %). Genes that are poorly characterized or have no COG annotation account for 22 % of the essential gene set of C. burnetii, indicating that further trends between bacterial species may emerge as genome annotations improve. These findings are in agreement with other essential gene studies. A comparison of essential gene features between 15 bacterial and one archaeal species [43] found that the COG J – Translation, ribosomal structure and biogenesis was overrepresented in all 16 species discussed, and H – Coenzyme transport and metabolism were over-represented in 87 % of species.
Fig. 3.

COG classification of essential genes. The x-axis shows the percentage of essential genes, and the y-axis shows COG categories. Ambiguously characterized groups show the percentage of essential genes that have been assigned to more than one COG category. The numbers next to the bars show the percentage of essential genes for that category.
The C. burnetii essential genome is similar to that of other bacterial species
Protein sequences of the 463 essential genes identified in this study were searched against the database of essential genes [32] by BlastP using a cut-off E value of ≤1×10−10 for hits to be considered matches. This resulted in hits for 77 % (358/463) of essential proteins against proteins within the database of essential genes. Of the proteins without hits, 47 % (51/108) were hypothetical proteins, explaining their absence in the database.
In the database of essential genes, genes encoding hypothetical proteins comprise 0–48 % of the essential genome of different species, with an average of 14 %. In the present study, 13 % (62/463) of the C. burnetii essential genes encode hypothetical proteins. Further elucidation of the structure and function of the proteins encoded by these genes may reveal important aspects related to the physiology of C. burnetii that allows it to survive and grow in the acidified endosome.
In addition, the essential gene dataset was compared to a list of 20 universally conserved essential COGs determined by Grazziotin et al. (Table 1) [44]. All of the 20 universally conserved essential COGs were represented in the list of C. burnetii essential genes.
Table 1.
A list of universally conserved essential COGs proposed by Grazziotin et al. which were also identified in this study
|
Information storage and processing |
|||||
|---|---|---|---|---|---|
|
Translation, ribosomal structure and biogenesis (J) |
CBU_2008 |
COG0018 |
Arginyl-tRNA synthetase |
||
|
CBU_0205 CBU_1488 |
COG0008 |
Glutamyl- and glutaminyl-tRNA synthetases |
|||
|
CBU_1248 |
COG0124 |
Histidyl-tRNA synthetase |
|||
|
CBU_0559 |
COG0495 |
Leucyl-tRNA synthetase |
|||
|
CBU_0081 |
COG0442 |
Prolyl-tRNA synthetase |
|||
|
CBU_1188 |
COG0172 |
Seryl-tRNA synthetase |
|||
|
CBU_0241 |
COG0090 |
Ribosomal protein L2 |
|||
|
CBU_0238 |
COG0087 |
Ribosomal protein L3 |
|||
|
CBU_0239 |
COG0088 |
Ribosomal protein L4 |
|||
|
CBU_0253 |
COG0097 |
Ribosomal protein L6P/L9E |
|||
|
CBU_1748 |
COG0102 |
Ribosomal protein L13 |
|||
|
CBU_0244 |
COG0092 |
Ribosomal protein S3 |
|||
|
CBU_0262 |
COG0522 |
Ribosomal protein S4 and related proteins |
|||
|
CBU_0255 |
COG0098 |
Ribosomal protein S5 |
|||
|
Transcription (K) |
CBU_0263 |
COG0202 |
DNA-directed RNA polymerase, alpha subunit/40 kDa subunit |
||
|
Replication, recombination and repair (L) |
CBU_0659 |
COG0592 |
DNA polymerase III sliding clamp (beta) subunit |
||
|
Cell cycle control, cell division, chromosome partitioning (D) |
CBU_0562 CBU_0770 CBU_1027 CBU_1580 |
COG0037 |
Predicted ATPase of the PP-loop superfamily implicated in cell cycle control |
||
|
Intracellular trafficking, secretion, and vesicular transport (U) |
CBU_0258 |
COG0201 |
Preprotein translocase subunit SecY |
||
|
CBU_1087 |
COG0552 |
Signal recognition particle GTPase |
|||
|
Nucleotide transport and metabolism (F) |
CBU_1830 |
COG0462 |
Phosphoribosylpyrophosphate synthetase |
||
C. burnetii essential genes are highly conserved in GG-I
Previous studies comparing different strains of C. burnetii have identified a core genome that is conserved between all strains, consisting of 1311 genes [14]. We found that only 67 % (310/463) of essential genes were conserved across all strains, highlighting the genetic diversity of C. burnetii and indicating that different genomovars may have different repertoires of essential genes. This is unsurprising as the C. burnetii core genome is derived from a range of C. burnetii strains, including those in GG IV that grow poorly (or do not grow) in ACCM-2 medium [16, 45] and the Dugway strains of GG VI, which have a much larger genome and which appear to be avirulent to humans and in immunocompetent animal models of C. burnetii infection [46–48]. Future work investigating the suitability of essential genes as novel drug targets to protect against C. burnetii infection should focus on essential genes found in the core genome, or essential genes conserved in genomic groups associated with disease in humans or animals.
Next, the conservation of essential genes between different genomovars was investigated. The core genomes of isolates assigned to GG I, IIa, IIb, III, IV, V or VI contain 1810, 1781, 1702, 1875, 1573, 1844 or 1978 genes respectively [14]. The highest number of conservation of essential genes (430/464, i.e. 92 %) was found in isolates assigned to GG I (including the NMII strain), for which the ACCM-2 culture medium used in this study was designed. In contrast, the lowest number of the essential genes (361/463, i.e. 77 %) was found in available isolates assigned to GG IV, which contains isolates shown to be unable to grow in ACCM-2 [16, 17]. The core genomes of GG IIa and GG IIb, which contain the strains associated with the Netherlands outbreak, had a similar number of conserved essential genes as GG I (426/463, i.e. 92 %; and 421/463, i.e. 91 % respectively), as did the cattle-associated isolates of GG III (423/463, i.e. 91 %). Finally, 403/463 (i.e. 87 %) of essential genes are conserved in GG V, the north American strains, and 419/463 (i.e. 90 %) of essential genes are conserved in GG VI, the strains that do not cause disease in humans.
Essential genes involved in important C. burnetii pathways
Peptidoglycan plays a key role in maintaining cell wall structure, and the majority of the genes in the peptidoglycan biosynthesis pathway were found to be essential in C. burnetii . Most genes in the mevalonate pathway were also classified as essential in this study (Fig. 4). Unlike most Gram-negative bacterial species, which source isoprenoids and isopentenyl diphosphate through the glyceraldehyde-3-phosphate pyruvate pathway [5], C. burnetii utilizes the mevalonate pathway to source isoprenoids and isopentenyl diphosphate [49, 50]. In previous work, significant upregulation of genes in the mevalonate pathway was seen during infection of Galleria mellonella, mice or Buffalo Green Monkey (BGM) cells with C. burnetii [51]. In Staphylococcus aureus, downregulation of the mevalonate pathway results in the widespread downregulation of metabolism-associated genes, and the upregulation of virulence factors and cell wall biosynthesis genes [52]. In addition, the mevalonate pathway has been shown to play a role in virulence of Listeria monocytogenes [53], Mycobacterium tuberculosis and Klebsiella pneumoniae [54–58]. Many genes in the biotin synthesis pathway and the transcriptional repressor of the biotin operon birA were identified as essential genes (Fig. 4). Biotin synthesis has been shown to be critical to the virulence of other intracellular pathogens such as Francisella tularensis [59–61] and M. tuberculosis [62, 63]. The essentiality of biotin synthesis in these species has been attributed to the absence of bioP and/or bioY in their genomes, high-affinity biotin transporters which C. burnetii is also lacking. As a result, these pathogens require high levels of exogenous biotin, compared to species which possess bioP and/or bioY [64]. The identification of genes typically associated with virulence in this essential gene screen may be due to the media used. In other bacterial species, TraDIS studies are typically carried out after growing bacteria in rich media. In contrast, ACCM-2 growth medium was developed to mimic the acidic Coxiella -containing vacuole that C. burnetii resides in within host cells [4]. Therefore, it may not be surprising that some virulence-associated genes have been identified in our study.
Fig. 4.

Essential pathways of C. burnetii. Essential routes to synthesis were identified for peptidoglycan and biotin, as well as the mevalonate pathway. Essential genes are depicted in green while non-essential genes are depicted in yellow.
Essential genes involved in axenic growth of C. burnetii
C. burnetii requires microaerobic conditions for optimal growth, as demonstrated by the expression of cytochrome bd (encoded by cydAB) [4]. Although cydB, encoding subunit II of cytochrome D, was identified as essential, cydA.1 and cydA.2 encoding subunit I were not. It may be that cydB encodes an essential subunit of cytochrome D. A similar finding was made in some strains of M. tuberculosis, where only the cydB subunit was classified as essential [65, 66]. However, although M. tuberculosis can occupy low oxygen environments [67], unlike C. burnetii it is not a strictly microaerobic bacterium. It is also possible that cydB has an essential role in protection against oxidative agents in the phagolysosome, as cydB mutants in E. coli and Brucella abortus show hypersensitivity to hydrogen peroxide [68, 69].
C. burnetii requires high levels of l-cysteine for axenic growth [4], and csdB (also known as sufS; the gene conferring a group II cysteine desulfurase) was essential in our study. Cysteine desulferases catalyse the decomposition of l-cysteine to l-alanine and sulfane sulphur, with the aid of pyridoxal 5′ phosphate as a cofactor. This process provides precursors to many biosynthetic pathways such as iron–sulphur (Fe-S) cluster assembly and the synthesis of biotin, lipocic acid, thiamine and nitrogenase [70]. Generally, csdB is co-expressed with five additional genes, sufA, sufB, sufC, sufD and sufE [71, 72]. sufBCD were classified as essential in this study. The proteins encoded by these genes form a complex for Fe-S biosynthesis [73], which has been shown to be essential under oxidative stress and iron limitation [74]. The C. burnetii genome does not appear to contain sufA (or its homologue iscA) or sufE, which is known to increase the activity of csdB 50-fold [72].
Omsland et al. previously reported that C. burnetii was able to oxidize proline under microaerobic conditions [4]. Bacteria utilize proline for protein synthesis [75, 76] and for protection against osmotic stress [77]. C. burnetii has been shown to actively transport glucose, glutamate and proline from the host cell intracellular environment in a pH-dependent manner [78, 79], and early work suggested that proline could be important for survival in the acidic CCV [78]. Later studies showed that C. burnetii uses a PmrA/B two-component system to sense the lysosomal environment and induce a transcriptional shift that mediates virulence through up-regulation of the type IV secretion system (T4SS) and associated effector proteins [80, 81], thus driving the formation of the CCV. A study by Newton et al. [82] has shown that the presence of proline (as well as phenylalanine and serine) is sensed by PmrB, and induces the expression of PmrA-regulated genes, subsequently inducing virulence of C. burnetii. Genes involved both in proline synthesis (arcB) and metabolism (putA) were essential in this study. arcB encodes ornithine cyclodeaminase, an enzyme capable of synthesizing proline in a single step through the deamination of ornithine [83–85]. arcB did not have hits against the database of essential genes, and to date, the role of arcB in C. burnetii has not been investigated. putA encodes a bi-functional enzyme with both proline dehydrogenase and Δ1-pyrroline-5-carboxylate dehydrogenase activities responsible for the metabolism of proline to glutamate. sodB and sodC encode superoxide dismutase enzymes known to protect bacteria from reactive oxygen species such as superoxide and hydrogen peroxide and have been identified as a potentially important virulence factor for C. burnetii [86], and have been identified as essential genes in Burkholderia pseudomallei [87]. The finding of these genes as essential in this study underpin the importance of superoxide dismutase enzymes in the protection of C. burnetii in the harsh intracellular environment.
Essential genes on the QpH1 plasmid
Six predicted essential genes (three hypothetical proteins of unknown function, and the CBUA0037–39 gene cluster) were identified on the QpH1 plasmid as essential (Table 2). However, a recent report demonstrated curing C. burnetii NMII of this plasmid [88]. CBUA0037, CBUA0038 and CBUA0039 confer homologues of ParA, RepB and RepA – a putative origin of replication. However, these genes were still present in the QpH1-deficient strain reported by Luo et al. because they had been cloned into the plasmid transformed into C. burnetii which then triggered the loss of QpH1 [88]. Our finding that CBUA007, 0023 and 0033 are essential whilst elimination of these genes was possible in the plasmid-cured strain suggests that there are interactions between these gene products and other plasmid gene products which would otherwise be toxic.
Table 2.
Essential genes identified in this study that are found on the QpH1 plamid
|
Locus tag |
Gene name |
Function |
|---|---|---|
|
CBUA0007 |
CBUA0007 |
Hypothetical protein |
|
CBUA0023 |
CBUA0023 |
Hypothetical protein |
|
CBUA0033 |
CBUA0033 |
Hypothetical protein |
|
CBUA0037 |
parA |
Plasmid partition protein A |
|
CBUA0038 |
repB |
DNA-binding protein |
|
CBUA0039 |
repA |
Plasmid replication initiation protein |
Alternatively, it is possible that these genes are not truly essential. This possibility might be addressed by generating additional transposon mutants to increase the size of the library or by the development of methods to prove essential genes in C. burnetii , for example using regulatable promoters to drive these genes and complementing these disrupted genes.
Some predicted T4SS effectors are essential
None of the core components of the T4SS were classified as essential, which is to be expected as this system is generally considered to be virulence associated, with transposon mutants in almost all of these genes having been made previously [22]. However, 12 predicted T4SS effector proteins were classified as essential, ten of which are designated to encode hypothetical proteins of unknown function (Table 3). A search against the Pfam database using HMMER [89] did not reveal any significant hits for the proteins encoded by these genes, and their function remains elusive. The majority of these are predicted to be effectors based on bioinformatics approaches, looking at PmrA regulatory elements, homology to Legionella pneumophilia T4SS substrates, and the presence of E-blocks. Some have experimental data in the form of CyaA translocation reporter assays, but for the majority it is possible that they are not effectors after all. Establishing the biological roles of these putative effectors should be a priority for future research. For several of the potential effector genes classified as essential, neighbouring genes have also been identified as essential (see File S2). Polar effects may be influencing the classification of these genes as essential, and further studies may be required to confirm essentiality. This could be achieved through the generation of further transposon mutants to increase saturation of the pool or by complementing the transposon mutants with wild-type genes expressed from regulatable promoters.
Table 3.
Predicted T4SS effector genes identified as essential genes in this study
|
Locus tag |
Gene name |
Function |
Reference |
|---|---|---|---|
|
CBU_0773 |
phnB |
PhnB |
[33] |
|
CBU_0794 |
CBU_0794 |
Hypothetical protein |
[90] |
|
CBU_0881 |
CBU_0881 |
Hypothetical cytosolic protein |
[90] |
|
CBU_1213 |
CBU_1213 |
Ankyrin repeat protein |
[91] |
|
CBU_1314 |
CBU_1314 |
Hypothetical cytosolic protein |
[90] |
|
CBU_1349 |
CBU_1349 |
Hypothetical cytosolic protein |
[90] |
|
CBU_1370 |
CBU_1370 |
Hypothetical membrane associated protein |
[92] |
|
CBU_1607 |
CBU_1607 |
Hypothetical protein |
[33] |
|
CBU_1614 |
CBU_1614 |
Hypothetical protein |
[81] |
|
CBU_1686 |
CBU_1686 |
Hypothetical protein |
[81] |
|
CBU_1819 |
CBU_1819 |
Hypothetical membrane associated protein |
[93] |
|
CBUA0023 |
CBUA0023 |
Hypothetical protein |
[94] |
Conclusion
We present the first report of a whole genome TraDIS study in C. burnetii . Data analysis using the Whiteley lab pipeline revealed 511 genes essential for the growth and survival of C. burnetii NMII. It was noted that some of the essential genes identified, such as predicted T4SS effectors, are typically considered to be virulence-associated. This may be a consequence of the use of ACCM-2 media to grow the bacteria, which mimics the acidic Coxiella-containing vacuole that C. burnetii would reside in, within host cells. Essential routes of synthesis were identified for the mevalonate pathway, as well as peptidoglycan and biotin synthesis. In addition, further evidence of the requirement of l-cysteine and proline for axenic growth was shown. To date, methodologies to prove essential genes in C. burnetii have not been developed. This should be considered a matter of priority. In other species, this has been achieved by putting essential genes under the control of an inducible promoter. In addition, it is important to note that C. burnetii NMII is a derogated surrogate for the fully virulent C. burnetii NMI, and therefore it would be valuable to carry out a TraDIS screen in this more clinically relevant strain. Nonetheless we have ongoing work evaluating the suitability of these essential genes as possible novel drug targets.
Supplementary Data
Funding information
This research was funded by Defence Science and Technology Laboratories (DSTL) award number DSTLX-1000123071.
Acknowledgements
We thank James Samuel for the kind gift of plasmid pKM225.
Author contribution
Conceptualization, G.M., C.H., I.N. and R.T.; methodology, G.M., C.H. and R.T.; formal analysis, G.M.; resources, I.N; writing—original draft preparation, G.M.; writing—review and editing, C.H., I.N. and R.T.; supervision, I.N. and R.T.; project administration, I.N.; funding acquisition, R.T. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Ethical statement
Not applicable.
Consent to publish
Not applicable.
Footnotes
Abbreviations: ACCM, acidified citrate cysteine medium; BGM, buffalo green monkey; CCV, Coxiella-containing vacuole; COGs, clusters of orthologous groups; gDNA, genomic DNA; GG, genomic group; LPS, lipopolysaccharide; TraDIS, transposon-directed insertion site sequencing.
All supporting data, code and protocols have been provided within the article or through supplementary data files. Two supplementary tables are available with the online version of this article.
References
- 1.Honarmand H. Q Fever: an old but still a poorly understood disease. Interdiscip Perspect Infect Dis. 2012;2012:131932. doi: 10.1155/2012/131932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angelakis E, Raoult D. Q fever. Vet Microbiol. 2010;140:297–309. doi: 10.1016/j.vetmic.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol. 2004;186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii . Proc Natl Acad Sci. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, et al. Complete genome sequence of the Q-fever pathogen Coxiella burnetii . Proc Natl Acad Sci. 2003;100:5455–5460. doi: 10.1073/pnas.0931379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiele D, Willems H, Haas M, Krauss H. Analysis of the entire nucleotide sequence of the cryptic plasmid QpH1 from Coxiella burnetti . Eur J Epidemiol. 1994;10:413–420. doi: 10.1007/BF01719665. [DOI] [PubMed] [Google Scholar]
- 7.Beare PA, Samuel JE, Howe D, Virtaneva K, Porcella SF, et al. Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J Bacteriol. 2006;188:2309–2324. doi: 10.1128/JB.188.7.2309-2324.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackstadt T, Peacock MG, Hitchcock PJ, Cole RL. Lipopolysaccharide variation in Coxiella burnetti: intrastrain heterogeneity in structure and antigenicity. Infect Immun. 1985;48:359–365. doi: 10.1128/iai.48.2.359-365.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millar JA, Beare PA, Moses AS, Martens CA, Heinzen RA, et al. Whole-genome sequence of Coxiella burnetii nine mile RSA439 (phase II, clone 4), a laboratory workhorse strain. Genome Announc. 2017;5:23. doi: 10.1128/genomeA.00471-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metters G, Norville IH, Titball RW, Hemsley CM. From cell culture to cynomolgus macaque: infection models show lineage-specific virulence potential of Coxiella burnetii . J Med Microbiol. 2019;68:1419–1430. doi: 10.1099/jmm.0.001064. [DOI] [PubMed] [Google Scholar]
- 11.Beare PA, Samuel JE, Howe D, Virtaneva K, Porcella SF, et al. Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J Bacteriol. 2006;188:2309–2324. doi: 10.1128/JB.188.7.2309-2324.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Amato F, Eldin C, Raoult D. The contribution of genomics to the study of Q fever. Future Microbiol. 2016;11:253–272. doi: 10.2217/fmb.15.137. [DOI] [PubMed] [Google Scholar]
- 13.Tilburg JJHC, Roest H-JIJ, Buffet S, Nabuurs-Franssen MH, Horrevorts AM, et al. Epidemic genotype of Coxiella burnetii among goats, sheep, and humans in the Netherlands. Emerg Infect Dis. 2012;18:887–889. doi: 10.3201/eid1805.111907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemsley CM, O’Neill PA, Essex-Lopresti A, Norville IH, Atkins TP, et al. Extensive genome analysis of Coxiella burnetii reveals limited evolution within genomic groups. BMC Genomics. 2019;20:441. doi: 10.1186/s12864-019-5833-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearson T, Hornstra HM, Hilsabeck R, Gates LT, Olivas SM, et al. High prevalence and two dominant host-specific genotypes of Coxiella burnetii in U.S. milk. BMC Microbiol. 2014;14:41. doi: 10.1186/1471-2180-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kersh GJ, Oliver LD, Self JS, Fitzpatrick KA, Massung RF. Virulence of pathogenic Coxiella burnetii strains after growth in the absence of host cells. Vector Borne Zoonotic Dis. 2011;11:1433–1438. doi: 10.1089/vbz.2011.0670. [DOI] [PubMed] [Google Scholar]
- 17.Vincent GA. Molecular Characterisation of Australian Coxiella burnetii Isolates. Murdoch, Australia: Murdoch University; 2013. [Google Scholar]
- 18.Beare PA, Jeffrey BM, Martens CA, Heinzen RA. Draft genome sequences of the avirulent Coxiella burnetii Dugway 7D77-80 and Dugway 7E65-68 strains isolated from Rodents in Dugway, Utah. Genome Announc. 2017;5:39. doi: 10.1128/genomeA.00984-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, et al. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella . Infect Immun. 2009;77:642–656. doi: 10.1128/IAI.01141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suhan ML, Chen SY, Thompson HA. Transformation of Coxiella burnetii to ampicillin resistance. J Bacteriol. 1996;178:2701–2708. doi: 10.1128/jb.178.9.2701-2708.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beare PA, Howe D, Cockrell DC, Omsland A, Hansen B, et al. Characterization of a Coxiella burnetii ftsZ mutant generated by Himar1 transposon mutagenesis. J Bacteriol. 2009;191:1369–1381. doi: 10.1128/JB.01580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez E, Cantet F, Fava L, Norville I, Bonazzi M. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog. 2014;10:e1004013. doi: 10.1371/journal.ppat.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langridge GC, Phan M-D, Turner DJ, Perkins TT, Parts L, et al. Simultaneous assay of every Salmonella typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gawronski JD, Wong SMS, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci. 2009;106:16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams JC, Peacock MG, McCaul TF. Immunological and biological characterization of Coxiella burnetii, phases I and II, separated from host components. Infect Immun. 1981;32:840–851. doi: 10.1128/iai.32.2.840-851.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanan AM, Ramsey MM, Stacy A, Whiteley M. Defining genetic fitness determinants and creating genomic resources for an oral pathogen. Appl Environ Microbiol. 2017;83:14. doi: 10.1128/AEM.00797-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stacy A, Fleming D, Lamont RJ, Rumbaugh KP, Whiteley M. A commensal bacterium promotes virulence of an opportunistic pathogen via cross-respiration. mBio. 2016;7:e00782-16. doi: 10.1128/mBio.00782-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci. 2015;112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewin GR, Stacy A, Michie KL, Lamont RJ, Whiteley M. Large-scale identification of pathogen essential genes during coinfection with sympatric and allopatric microbes. Proc Natl Acad Sci. 2019;116:19685–19694. doi: 10.1073/pnas.1907619116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang R, Ou HY, Zhang CT. DEG: a database of essential genes. Nucleic Acids Research. 2004;32:271D–272. doi: 10.1093/nar/gkh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, et al. Identification of Coxiella burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol. 2013;195:3914–3924. doi: 10.1128/JB.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio. 2017;8:e02133–16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile . mBio. 2015;6 doi: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, et al. Essential genes in the core genome of the human pathogen Streptococcus pyogenes . Sci Rep. 2015;5:9838. doi: 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz L, Bottacini F, Boinett CJ, Cain AK, O’Connell-Motherway M, et al. The essential genomic landscape of the commensal Bifidobacterium breve UCC2003. Sci Rep. 2017;7:5648. doi: 10.1038/s41598-017-05795-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barquist L, Mayho M, Cummins C, Cain AK, Boinett CJ, et al. The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics. 2016;32:1109–1111. doi: 10.1093/bioinformatics/btw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barquist L, Langridge GC, Turner DJ, Phan M-D, Turner AK, et al. A comparison of dense transposon insertion libraries in the Salmonella serovars Typhi and Typhimurium. Nucleic Acids Res. 2013;41:4549–4564. doi: 10.1093/nar/gkt148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014;10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson CL, Beare PA, Howe D, Heinzen RA. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci. 2013;110:E4770–9. doi: 10.1073/pnas.1309195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vollmer-Conna U, Piraino BF, Cameron B, Davenport T, Hickie I, et al. Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin Infect Dis. 2008;47:1418–1425. doi: 10.1086/592967. [DOI] [PubMed] [Google Scholar]
- 44.Grazziotin AL, Vidal NM, Venancio TM. Uncovering major genomic features of essential genes in Bacteria and a methanogenic Archaea. FEBS J. 2015;282:3395–3411. doi: 10.1111/febs.13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kersh GJ, Priestley RA, Hornstra HM, Self JS, Fitzpatrick KA, et al. Genotyping and axenic growth of Coxiella burnetii isolates found in the United States environment. Vector Borne Zoonotic Dis. 2016;16:588–594. doi: 10.1089/vbz.2016.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell-Lodrigue KE, Andoh M, Poels MWJ, Shive HR, Weeks BR, et al. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q fever. Infect Immun. 2009;77:5640–5650. doi: 10.1128/IAI.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.STOENNER HG, LACKMAN DB. The biologic properties of Coxiella burnetii isolated from rodents collected in Utah. Am J Hyg. 1960;71:45–51. doi: 10.1093/oxfordjournals.aje.a120088. [DOI] [PubMed] [Google Scholar]
- 48.Long CM, Beare PA, Cockrell DC, Larson CL, Heinzen RA. Comparative virulence of diverse Coxiella burnetii strains. Virulence. 2019;10:133–150. doi: 10.1080/21505594.2019.1575715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, et al. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/JB.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohdich F, Bacher A, Eisenreich W. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochem Soc Trans. 2005;33:785–791. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- 51.Kovacs-Simon A, Metters G, Norville I, Hemsley C, Titball RW. Coxiella burnetii replicates in Galleria mellonella hemocytes and transcriptome mapping reveals in vivo regulated genes. Virulence. 2020;11:1268–1278. doi: 10.1080/21505594.2020.1819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balibar CJ, Shen X, Tao J. The mevalonate pathway of Staphylococcus aureus . J Bacteriol. 2009;191:851–861. doi: 10.1128/JB.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang SX, Zhang XC, Wang SY, Shun TT, He YL. 18F-FDG PET/CT localized valvular infection in chronic Q fever endocarditis. J Nucl Cardiol. 2015;22:1320–1322. doi: 10.1007/s12350-015-0080-0. [DOI] [PubMed] [Google Scholar]
- 54.Shin SJ, Wu C, Steinberg H, Talaat AM. Identification of novel virulence determinants in Mycobacterium paratuberculosis by screening a library of insertional mutants. Infect Immun. 2006;74:3825–3833. doi: 10.1128/IAI.01742-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DiRita VJ, Lai Y-C, Peng H-L, Chang H-Y. Identification of genes induced in vivo during Klebsiella pneumoniae CG43 infection. Infect Immun. 2001;69:7140–7145. doi: 10.1128/IAI.69.11.7140-7145.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schauer K, Geginat G, Liang C, Goebel W, Dandekar T, et al. Deciphering the intracellular metabolism of Listeria monocytogenes by mutant screening and modelling. BMC Genomics. 2010;11:573. doi: 10.1186/1471-2164-11-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Begley M, Bron PA, Heuston S, Casey PG, Englert N, et al. Analysis of the isoprenoid biosynthesis pathways in Listeria monocytogenes reveals a role for the alternative 2-C-methyl-D-erythritol 4-phosphate pathway in murine infection. Infect Immun. 2008;76:5392–5401. doi: 10.1128/IAI.01376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Begley M, Gahan CGM, Kollas A-K, Hintz M, Hill C, et al. The interplay between classical and alternative isoprenoid biosynthesis controls gammadelta T cell bioactivity of Listeria monocytogenes . FEBS Lett. 2004;561:99–104. doi: 10.1016/S0014-5793(04)00131-0. [DOI] [PubMed] [Google Scholar]
- 59.Feng Y, Napier BA, Manandhar M, Henke SK, Weiss DS, et al. A Francisella virulence factor catalyses an essential reaction of biotin synthesis. Mol Microbiol. 2014;91:300–314. doi: 10.1111/mmi.12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, et al. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci. 2007;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Napier BA, Meyer L, Bina JE, Miller MA, Sjöstedt A, et al. Link between intraphagosomal biotin and rapid phagosomal escape in Francisella. Proc Natl Acad Sci. 2012;109:18084–18089. doi: 10.1073/pnas.1206411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woong Park S, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLOS Pathog. 2011;7:e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carfrae LA, MacNair CR, Brown CM, Tsai CN, Weber BS, et al. Mimicking the human environment in mice reveals that inhibiting biotin biosynthesis is effective against antibiotic-resistant pathogens. Nat Microbiol. 2020;5:93–101. doi: 10.1038/s41564-019-0595-2. [DOI] [PubMed] [Google Scholar]
- 65.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 66.Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis . PLOS Pathog. 2012;8:e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Endley S, McMurray D, Ficht TA. Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. J Bacteriol. 2001;183:2454–2462. doi: 10.1128/JB.183.8.2454-2462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goldman BS, Gabbert KK, Kranz RG. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J Bacteriol. 1996;178:6348–6351. doi: 10.1128/jb.178.21.6348-6351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mihara H, Esaki N. Bacterial cysteine desulfurases: their function and mechanisms. Appl Microbiol Biotechnol. 2002;60:12–23. doi: 10.1007/s00253-002-1107-4. [DOI] [PubMed] [Google Scholar]
- 71.Nachin L, El Hassouni M, Loiseau L, Expert D, Barras F. SoxR-dependent response to oxidative stress and virulence of Erwinia chrysanthemi: the key role of SufC, an orphan ABC ATPase. Mol Microbiol. 2001;39:960–972. doi: 10.1046/j.1365-2958.2001.02288.x. [DOI] [PubMed] [Google Scholar]
- 72.Outten FW, Wood MJ, Munoz FM, Storz G. The SufE protein and the SufBCD complex enhance SufS cysteine desulfurase activity as part of a sulfur transfer pathway for Fe-S cluster assembly in Escherichia coli . J Biol Chem. 2003;278:45713–45719. doi: 10.1074/jbc.M308004200. [DOI] [PubMed] [Google Scholar]
- 73.Hidese R, Mihara H, Esaki N. Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Appl Microbiol Biotechnol. 2011;91:47–61. doi: 10.1007/s00253-011-3336-x. [DOI] [PubMed] [Google Scholar]
- 74.Takahashi Y, Tokumoto U. A third bacterial system for the assembly of iron-sulfur clusters with homologs in archaea and plastids. J Biol Chem. 2002;277:28380–28383. doi: 10.1074/jbc.C200365200. [DOI] [PubMed] [Google Scholar]
- 75.Kumar TK, Samuel D, Jayaraman G, Srimathi T, Yu C. The role of proline in the prevention of aggregation during protein folding in vitro. Biochem Mol Biol Int. 1998;46:509–517. doi: 10.1080/15216549800204032. [DOI] [PubMed] [Google Scholar]
- 76.Pemberton TA, Still BR, Christensen EM, Singh H, Srivastava D, et al. Proline: Mother Nature’s cryoprotectant applied to protein crystallography. Acta Crystallogr D Biol Crystallogr. 2012;68:1010–1018. doi: 10.1107/S0907444912019580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Culham DE, Vernikovska Y, Tschowri N, Keates RAB, Wood JM, et al. Periplasmic loops of osmosensory transporter ProP in Escherichia coli are sensitive to osmolality. Biochemistry. 2008;47:13584–13593. doi: 10.1021/bi801576x. [DOI] [PubMed] [Google Scholar]
- 78.Hendrix L, Mallavia LP. Active transport of proline by Coxiella burnetii . J Gen Microbiol. 1984;130:2857–2863. doi: 10.1099/00221287-130-11-2857. [DOI] [PubMed] [Google Scholar]
- 79.Hackstadt T, Williams JC. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii . Proc Natl Acad Sci. 1981;78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zusman T, Aloni G, Halperin E, Kotzer H, Degtyar E, et al. The response regulator PmrA is a major regulator of the icm/dot type IV secretion system in Legionella pneumophila and Coxiella burnetii . Mol Microbiol. 2007;63:1508–1523. doi: 10.1111/j.1365-2958.2007.05604.x. [DOI] [PubMed] [Google Scholar]
- 81.Beare PA, Sandoz KM, Larson CL, Howe D, Kronmiller B, et al. Essential role for the response regulator PmrA in Coxiella burnetii type 4B secretion and colonization of mammalian host cells. J Bacteriol. 2014;196:1925–1940. doi: 10.1128/JB.01532-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newton P, Thomas DR, Reed SCO, Lau N, Xu B, et al. Lysosomal degradation products induce Coxiella burnetii virulence. Proc Natl Acad Sci. 2020;117:6801–6810. doi: 10.1073/pnas.1921344117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trovato M, Maras B, Linhares F, Costantino P. The plant oncogene rolD encodes a functional ornithine cyclodeaminase. Proc Natl Acad Sci U S A. 2001;98:13449–13453. doi: 10.1073/pnas.231320398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schindler U, Sans N, Schröder J. Ornithine cyclodeaminase from octopine Ti plasmid Ach5: identification, DNA sequence, enzyme properties, and comparison with gene and enzyme from nopaline Ti plasmid C58. J Bacteriol. 1989;171:847–854. doi: 10.1128/jb.171.2.847-854.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sharma S, Shinde S, Verslues PE. Functional characterization of an ornithine cyclodeaminase-like protein of Arabidopsis thaliana . BMC Plant Biol. 2013;13:182. doi: 10.1186/1471-2229-13-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brennan RE, Kiss K, Baalman R, Samuel JE. Cloning, expression, and characterization of a Coxiella burnetii Cu/Zn Superoxide dismutase. BMC Microbiol. 2015;15:99. doi: 10.1186/s12866-015-0430-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moule MG, Hemsley CM, Seet Q, Guerra-Assunção JA, Lim J, et al. Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. mBio. 2014;5:e00926–13. doi: 10.1128/mBio.00926-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luo S, Lu S, Fan H, Sun Z, Hu Y, et al. The Coxiella burnetii QpH1 plasmid is a virulence factor for colonizing bone marrow-derived murine macrophages. J Bacteriol. 2021;203:e00588-20. doi: 10.1128/JB.00588-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, et al. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46:W200–W204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, et al. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii . Proc Natl Acad Sci. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lifshitz Z, Burstein D, Schwartz K, Shuman HA, Pupko T, et al. Identification of novel Coxiella burnetii Icm/Dot effectors and genetic analysis of their involvement in modulating a mitogen-activated protein kinase pathway. Infect Immun. 2014;82:3740–3752. doi: 10.1128/IAI.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, et al. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun. 2015;83:661–670. doi: 10.1128/IAI.02763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, et al. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J Bacteriol. 2011;193:1493–1503. doi: 10.1128/JB.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.