

Abstract
Purpose
Endocrine therapy resistance (ETR) remains the greatest challenge in treating patients with hormone receptor-positive breast cancer. We set out to identify molecular mechanisms underlying ETR through in-depth genomic analysis of breast tumors.
Experimental Design
We collected pre-treatment and sequential on-treatment tumor samples from 35 patients with estrogen receptor-positive breast cancer treated with neoadjuvant then adjuvant ET; 3 had intrinsic resistance, 19 acquired resistance, and 13 remained sensitive. Response was determined by changes in tumor volume neoadjuvantly and by monitoring for adjuvant recurrence. Twelve patients received 2 or more lines of ET, with subsequent treatment lines being initiated at the time of development of resistance to the previous ET. DNA whole-exome sequencing and RNA-sequencing were performed on all samples, totalling 169 unique specimens. DNA mutations, copy number alterations and gene expression data were analyzed through unsupervised and supervised analyses to identify molecular features related to ETR.
Results
Mutations enriched in ETR included ESR1 and GATA3. The known ESR1 D538G variant conferring ETR was identified, as was a rarer E380Q variant that confers endocrine hypersensitivity. Resistant tumors which acquired resistance had distinct gene expression profiles compared to paired sensitive tumors, showing elevated pathways including ER, HER2, GATA3, AKT, RAS and p63 signaling. Integrated analysis in individual patients highlighted the diversity of ETR mechanisms.
Conclusions
The mechanisms underlying ETR are multiple and characterized by diverse changes in both somatic genetic and transcriptomic profiles; to overcome resistance will require an individualized approach utilizing genomic and genetic biomarkers and drugs tailored to each patient.
Introduction
Breast cancer is one of the leading causes of mortality in women. Over 75% of breast cancers are estrogen receptor positive (ER+) and approximately 65% of these are also progesterone receptor positive (PR+). ER+ and/or PR+ breast cancer is also known as hormone receptor positive (HR+), and has the best prognosis among all breast cancers, with five-year relative survival rates estimated to be around 90% (1). Good outcomes are largely attributed to success in therapeutically targeting hormone receptors. However, not all HR+ patients respond to endocrine therapy due to a variety of reasons including molecular heterogeneity (15-20%: intrinsic resistance) as well as cellular phenotypes that develop and evolve during treatment (30-40%: acquired resistance) (2). Breast cancers exhibit unique somatic mutations, copy number alterations (CNA) and transcriptomic profiles, which are particularly frequent among the HR+ subgroup (3–5) and can contribute to endocrine resistance.
Commonly used endocrine agents in clinical practice that target the estrogen receptor include the selective estrogen receptor modulator tamoxifen and the selective estrogen receptor degrader fulvestrant (6). An alternative option is to reduce levels of circulating estrogen using the aromatase inhibitors (AI: letrozole, anastrozole and exemestane); these work by inhibiting aromatase, and thus blocking estrogen production in postmenopausal women. They can be used in premenopausal women when combined with ovarian suppression or ablation. Despite there being a variety of agents with different modes of action, resistance can develop to each of these treatments, and is inevitable in the metastatic setting.
Extensive studies have investigated resistance mechanisms to endocrine therapies (7–9). These fall mainly into the following categories: changes in the estrogen signaling pathway, activation of growth factor signaling pathways and cell cycle dysregulation. Changes in the estrogen signaling pathway represent the most common resistance mechanisms where genetic and/or epigenetic alterations in ER or ER-associated transcription factors and co-activators have been identified (10–13). Specifically, somatic mutations in ESR1 following AI treatment alter ER-signaling and result in endocrine insensitivity due to constitutive activation (14). Growth factor signaling pathway activation including EGFR/HER2, FGFR, MAPK, and PI3K has also been associated with resistance (15–18). Cell cycle regulator changes including amplification of Cyclins, CDKs, MYC, and loss of RB can uncouple the cell cycle from estrogen-mediated entry into G1/S and represent a further resistance mechanism (19–21).
Breast cancer is a highly heterogeneous disease and the full range of mechanisms underlying endocrine resistance has not as yet been fully characterized. To best identify and characterize known and postulated resistance mechanisms, studies using patient derived materials utilizing comprehensive DNA and RNA sequencing can provide optimal insight, however, there have been few comprehensive studies addressing this using a multi-omics approach. Here, we present an integrated analysis of both DNA and RNA sequencing of a cohort of 35 ER+ breast cancer patients treated with primary endocrine therapy, who had tumor biopsies performed before and during treatment with endocrine therapy, and at the time of recurrence in those with progressive disease. Clinical response was defined according to institutional standards by monitoring changes in tumor volume by 3D ultrasound in the neoadjuvant setting and conventional monitoring for recurrence in the adjuvant setting. Twelve patients received two or more lines of endocrine therapy agents, with second-line agents being initiated at the time of development of resistance to first-line agents.
The aim of this study was to delineate the mechanisms of endocrine resistance in these patients. We have examined mutation and copy number landscapes, and transcriptional profiles, and found genetic and transcriptomic changes specific to resistant tumors. These results highlight the genetic diversity of the resistance process.
Materials and Methods
Patients, consent and tissue processing
Tumor tissue was obtained from 35 patients with estrogen receptor positive breast cancer who consented to participate in this research study at the Edinburgh Breast Unit and University of Edinburgh. Written informed consent was obtained from all patients. Ethical approval for the study was granted under the Lothian NRS BioResource approval number 20/ES/0061 and the study was conducted in accordance with Scottish Common Law. Patient characteristics are presented in Table 1 and Supplementary Table S1. All patients received neoadjuvant or primary endocrine therapy, which was continued in the adjuvant setting for those patients who eventually underwent surgery. In total, 23 patients were treated with a single endocrine agent only, while 12 patients received 2 or more lines, with second-line agents being initiated at the time of resistance to first-line agents in those patients who did not wish surgery or were deemed unfit for surgery at that time. Neoadjuvant treatment in this cohort ranged from 3.6 months to 61.2 months with a median duration of 7.8 months. Of 34 patients who underwent surgery, 23 had a wide local excision and 11 had mastectomies. Six patients had surgery at the time of cancer recurrence while receiving adjuvant endocrine therapy with samples at surgery being obtained for analysis (Figure 1).
Table 1. Clinical characteristics of the study population (n = 35). Pathologic characteristics are from institutional clinical pathologic assessment without central review.
| Age at diagnosis | 75 yr (mean) - 43-95 yrs (range) |
|---|---|
| ER Status | |
| ER positive (Allred 7) | 7 |
| ER positive (Allred 8) | 28 |
| HER2 Status | |
| HER2 positive | 3 |
| HER2 negative | 32 |
| Histological Grade | |
| 1 | 2 |
| 2 | 22 |
| 3 | 11 |
| Lymph Node Status | |
| Positive | 14 |
| Negative | 20 |
| Unknown (no surgery) | 1 |
| Menopausal Status | |
| Pre | 1 |
| Post | 34 |
| Surgery | |
| Wide local excision | 23 |
| Mastectomy | 11 |
| No surgery | 1 |
| Adjuvant Therapy | |
| Endocrine Therapy | 35 |
| Chemotherapy | 6 |
| Radiotherapy | 17 |
Figure 1. Patient scheme.

Swimmerplot indicating timing of endocrine therapy treatment and tumor tissue sampling for each patient stratified by patient response to endocrine therapy. For each tumor sample, response to treatment defined by assessment of tumor volume change and number of biological replicates were indicated. Clinically HER2+ patients with positive immunohistochemistry (IHC) staining were highlighted in pink box.
Neoadjuvant clinical response was assessed by monitoring dynamic changes in 3D tumor volume, determined by repeat ultrasound measurements recorded during the neoadjuvant treatment window and performed by a single sonographer (JMD). Clinical response classification was determined using RECIST 1.1 criteria. All patients had long-term follow-up and for those who had surgery (n=34), adjuvant response was determined by monitoring for cancer recurrence. Of the 35 patients, 13 patients remained endocrine therapy responsive throughout treatment, 3 patients had intrinsic resistance to neoadjuvant therapy and 19 patients acquired resistance: 13 during neoadjuvant therapy and 6 developed a recurrence on adjuvant endocrine therapy following previous neoadjuvant therapy. Of the 6 who developed a recurrence, 3 went on to develop further recurrences on subsequent adjuvant endocrine agents - each had surgery followed by a new adjuvant endocrine therapy agent at the time of each recurrence. Overall, 13 patients remained sensitive to the first endocrine agent (Supplementary Table S1). All patients received clinical follow-up for 10 years from diagnosis or until death. No responsive patients developed a recurrence in that time, and none died from breast cancer. For each patient, tumor core biopsies were taken during the neoadjuvant treatment window using ultrasound guided 14-guage needle core biopsy. On average 3 sequential tumor tissue samples were taken from each patient (range 2-8 samples). Biopsies were taken at diagnosis (pre-treatment), at 2-4 weeks on-treatment, and obtained at progression or every 3-6 months in those patients who declined or were not deemed fit enough for surgery at the time and were instead managed by longer-term primary endocrine therapy. A relative increase in tumor volume of 20% or greater prompted surgery or re-biopsy and initiation of a second-line or third-line endocrine agent. Tumor tissue was collected at surgery and when recurrence developed. All surgeries were performed to clear margins by a single surgeon (JMD). Wherever feasible, multiple tumor samples were taken from each recurrence. In total, sufficient RNA and DNA was successfully extracted from 32 primary tumors and 137 tumor specimens during treatment and was used for DNA whole exome sequencing and RNA-seq.
For 17 patients, fresh frozen tissue was available. Core biopsy and surgical tissues were snap frozen in liquid nitrogen immediately after the procedure. For the remaining 18 patients, FFPE tissues were analyzed. For fresh frozen tissues, DNA and RNA were isolated using QIAGEN RNAeasy and DNAeasy kits, respectively, according to the manufacturer’s protocols. For FFPE tissues, RNA and DNA were isolated with QIAGEN AllPrep FFPE Kit, according to the manufacturer’s protocols. RNA quality was assessed with an Agilent BioAnalyzer RNA 6000 Nano Kit.
DNA whole exome sequencing
DNA was prepared for sequencing using the Agilent Technologies SureSelect XT library protocol. Fresh-frozen tumors were processed according to the manufacturer’s protocol for 3 μg input, while FFPE tumors were processed with the low-volume input according to the manufacturer’s protocol for 200 ng input. DNA libraries were captured and amplified with Agilent Technologies SureSelect Human All Exon, version 5 for fresh frozen tissues or version 6 for FFPE tissues, according to the manufacturer’s protocol. The quality of both the DNA libraries and DNA exome capture and concentration were quantified with Agilent TapeStation DNA 1000 and High Sensitivity D1000, respectively. Paired-end sequence data (2 x 100 bp) were generated using the Illumina HiSeq 2500 for each tumor or normal sample, with 3 samples per lane. Illumina reads were mapped to the hg19 reference sequence with BWA 0.7.9a (22), realigned with ABRA, version 0.96 (23), and processed by biobambam2 (24). Viral alignments were counted with Samtools (25) and BEDTools-Version- 2.15.0. Picard 1.92 (26) was used to calculate sequencing metrics. ISAAC (27) and Freebayes were used to call germline mutations with quality scores above 30. SnpSIFT, version 1.3.4, band SnpEFF (28) was used to annotate alterations with population-level frequencies. CADABRA SomaticLocusCaller was used for further filtration. Somatic variants were called with STRELKA (29) using strelka_config_bwa_default.ini.
Minor allele frequencies of highly variable SNPs called by Freebayes in the general population were used to for sample identity. All tumor normal pairs had an expected 89%–100% identity from the same patient.
DNA Copy number alterations were identified with SynthEx (30) using 50,000-bp–sized bins and K nearest neighbors = 4 from the pool of 31 available normal tissues. Briefly, the ratio of on-target and off-target exome reads of the tumor were compared with a normal tissue selected from the data set by the highest degree of similarity by K-nearest neighbor (KNN) based on library size and fold enrichment. Segment-level ratios were calculated and log2 transformed. Copy number levels greater than 0.25 were considered gains, and levels below –0.2 were considered losses. Using Ensemble hg19 gene annotations, genes were mapped to segments in each sample and a gene by sample copy number matrix was constructed. Specifically, a gene totally falls into a segment was assigned the copy number of that segment.
RNA sequencing
Fresh-frozen RNA ribo-zero libraries were prepared with rRNA removed from total RNA using Epicentre’s Ribo-Zero rRNA Removal kit (Cat# RZH11042). 30-100 ng Ribo-Zero RNA was used for the construction of the library using the Illumina TruSeq™ RNA Sample Prep Kit (Cat# RS-122-2001) and followed the manufacturer’s instruction, except for omitting the purification step before fragmentation. FFPE RNA was prepared with the Illumina TruSeq FFPE RiboZero Gold protocol according to the manufacturer’s instructions. RNA libraries were sequenced as 2 x 50 bp paired end reads with 2 samples per lane on Illumina HiSeq 2500 sequencers. Reads were aligned with STAR2.4.2a (31), and gene values were quantitated with Salmon (32). Raw gene counts were upper quartile normalized, filtered to genes that present in over 70% of samples and log2 transformed. To correct the batch effect between fresh frozen and FFPE samples, log2 transformed gene expression values for fresh frozen and FFPE sample sets were separately median centered and column standardized, then merged to a uniform data set for downstream analysis (33,34).
PAM50 subtyping was implemented as described in Fernandez-Martinez et al. (35). Briefly, batch-corrected gene expression data were normalized to ER+ samples in the PAM50 training set (36). Correlation to each subtype centroids were calculated, and subtypes were called according to the nearest centroid.
Computational and statistical analysis
Gene expression signatures
A panel of 654 previously published gene expression signatures were used to fully characterize cancer expression phenotypes (Supplementary Table S2). These 654 signatures were obtained from multiple publications or GSEA (37) and were summarized in Xia et al. (26). Signature scores were calculated in a way consistent to how they were derived, noting that the large majority were median expression of a predetermined set of genes (38).
Linear mixed model of CNA, gene expression and gene signatures
To identify differential Copy Number Alterations (CNA), gene expressions and gene signatures, biological replicates in CNA data, batch corrected gene expression data or gene signatures data were first collapsed into pseudo-samples by taking the mean value of multiple biological replicates (For example, sample 020-4-1A, 020-4-1B, 020-4-1C, 020-4-1D and 020-4-2A were collapsed into 020-4 by averaging each CNA/gene/gene signature values). Patients were then filtered to those with documented acquired resistance and those that had at least one sensitive tumor and at least one resistant tumor, resulting in N=13 patients with 38 samples for CNA analysis and N=17 with 53 samples for gene expression and gene signature analysis. For gene expression, genes were further filtered to those with Entrez ID. Each CNA, gene or signature was then tested for differential expression in the resistant tumors versus sensitive tumors, with the patient taken into account as a confounding variable using lme4 package in R (39): lmer(value ~ sensitive/resistant + (1|patient)). Permutation based false discovery rate (FDR) was calculated by permuting the tumor sensitive/resistant labels 100 times.
Hierarchical clustering of gene expression and gene signatures
For gene expression, hierarchical clustering was done using correlation distance metric and complete linkage. Clustering for gene signatures was done using Euclidean distance metric and complete linkage. All clustering was done using R package pheatmap.
Computational re-interrogation of somatic mutations in related tumors
Low read coverage or low tumor cell purity can cause the rigorous somatic mutation caller to miss mutations (23,40). Thus, all of the high-confidence somatic mutations from every tumor taken from 1 patient were re-interrogated within the same tumors from that same patient. First, all of the somatic mutations from the tumors within a patient were collapsed into one file, excluding any guanine-to-adenine or cytosine-to-thymine mutations from FFPE samples. For each mutation from a single patient, we then counted the mutant and reference alleles at that position from the original BAM file of each tumor from that patient. Variant allele frequencies (VAF, alternate counts/total read counts) were recalculated from the new calls. All mutations from the data set were interrogated in the normal sequence for all tumors in this data set to account for false-positives. Mutations with VAFs of greater than 20% in at least 2 normal tissues from unrelated patients were excluded from future analyses.
Count table for mutations
We filtered patients to those with acquired resistance and 12 patients had at least 1 sensitive and 1 resistant tumor sample with successful DNA sequencing. For each gene in the Pan-Cancer 299 significantly mutated genes, we divided the 12 patients into the following four categories according to mutation status of the gene: (i) patients with mutation present in both sensitive and resistant tumors, (ii) patients with mutation present in resistant tumors only, (iii) patients with mutation present in sensitive tumors only, and (iv) patients with mutation present in neither sensitive or resistant tumors. All sensitive tumors and all resistant tumors from each patient were considered as a whole.
Mutational signatures
Mutational signatures were calculated using R package DeconstructSigs v1.8.0 to determine the weights of known mutational processes (COSMIC mutational signatures v2 – March 2015: https://cancer.sanger.ac.uk/signatures/signatures_v2/) in each tumor sample. We then focused on 4 signatures that are known to be important in breast cancer including Signature1 – Age related; Signature 2 and Signature 13 - Apobec related; and Signature3 – Homology directed repair (HDR) related.
R version
All statistical analyses were performed using R v. 3.5.2 in RStudio.
Results
Patient characteristics
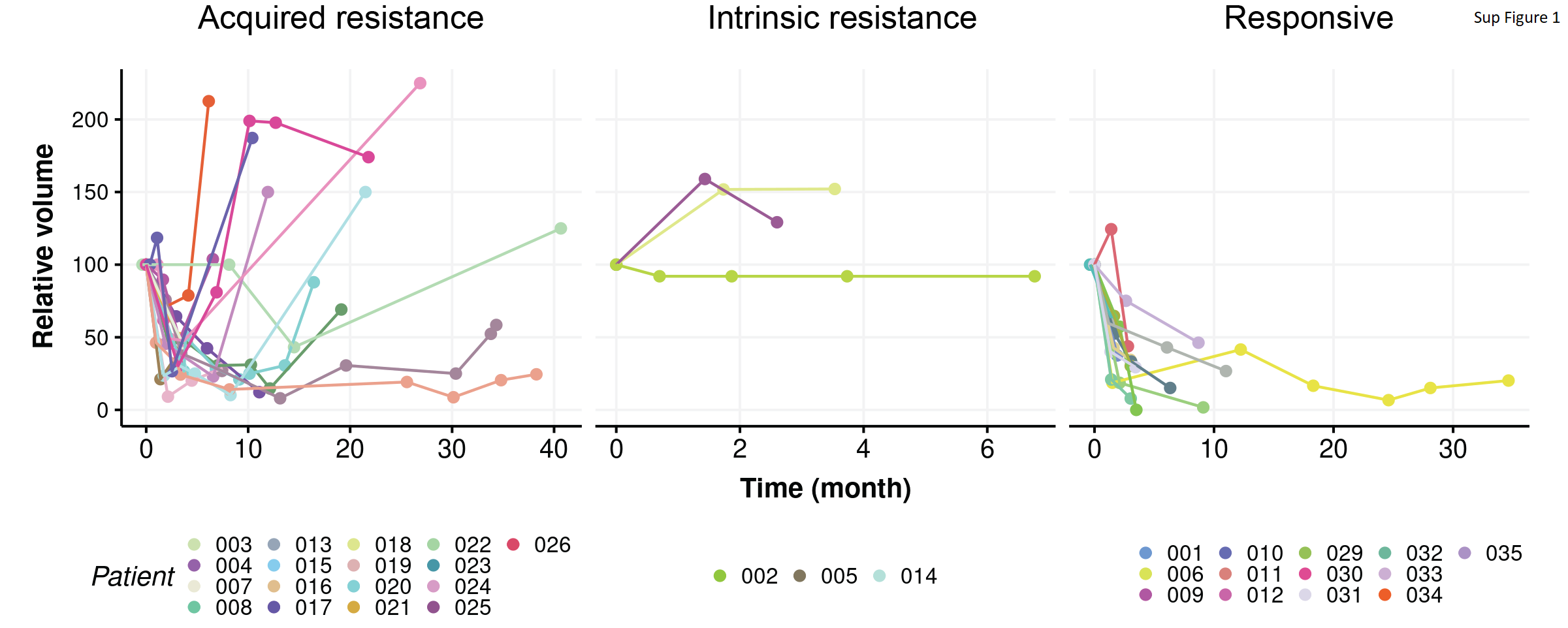
We examined the clinical features and molecular subtypes of all 169 samples, representing 35 patients (Supplementary Table S3). This cohort had a median age of 75 years at the diagnosis of breast cancer and a median follow-up of 4 years (Table 1). Responses to endocrine therapy were categorized as intrinsic resistance (non-response, n=3), acquired resistance (initial response followed by tumor regrowth, n=19), or ongoing sensitivity (initial response that was maintained or no evidence of recurrence, n=13) (Fig. 1, Supplementary Fig. 1). 33 patients received letrozole as initial neoadjuvant therapy, 1 received anastrozole and 1 received 2-weeks of fulvestrant prior to surgery and subsequent adjuvant tamoxifen, as part of a clinical study. Of the 33 who received primary letrozole, 3 were changed to alternative endocrine therapies (2 received tamoxifen and 1 received anastrozole) due to side-effects with letrozole. All patients had ER+ tumors (Allred score 7-8, Table 1) by clinical pathologic report and 3 had HER2+ tumors by immunohistochemistry (IHC 3+) (Figure. 1). We applied the PAM50 subtype predictor to determine their intrinsic molecular subtype (35,36). Of 32 primary tumors with successful RNA-seq, 14 were Luminal A (LumA); 14 were Luminal B (LumB), 3 were Her2-enriched and 1 was Normal-like. Among the three HER2-enriched primary tumors, 2 were HER2+ by IHC and 1 was HER2- by IHC. Of note, the three intrinsically resistant primary tumors had subtypes of LumA, LumB and Normal-like. This Normal-like primary tumor had very close correlation to LumA centroid and to the Normal-like centroid, which is not uncommon for many low cellularity LumA tumors (4). Of all 167 tumor specimens subtyped including the primary tumors, 79 were LumA, 50 were LumB, 17 were Her2-enriched, 1 was Basal-like and 20 were Normal-like. Only 8 patients had consistent subtypes across all samples from that patient, and there were cases when even two specimens of the same tumor showed different subtypes, indicating that spatial and temporal tumor heterogeneity does occur.
Genomic landscape of endocrine resistance
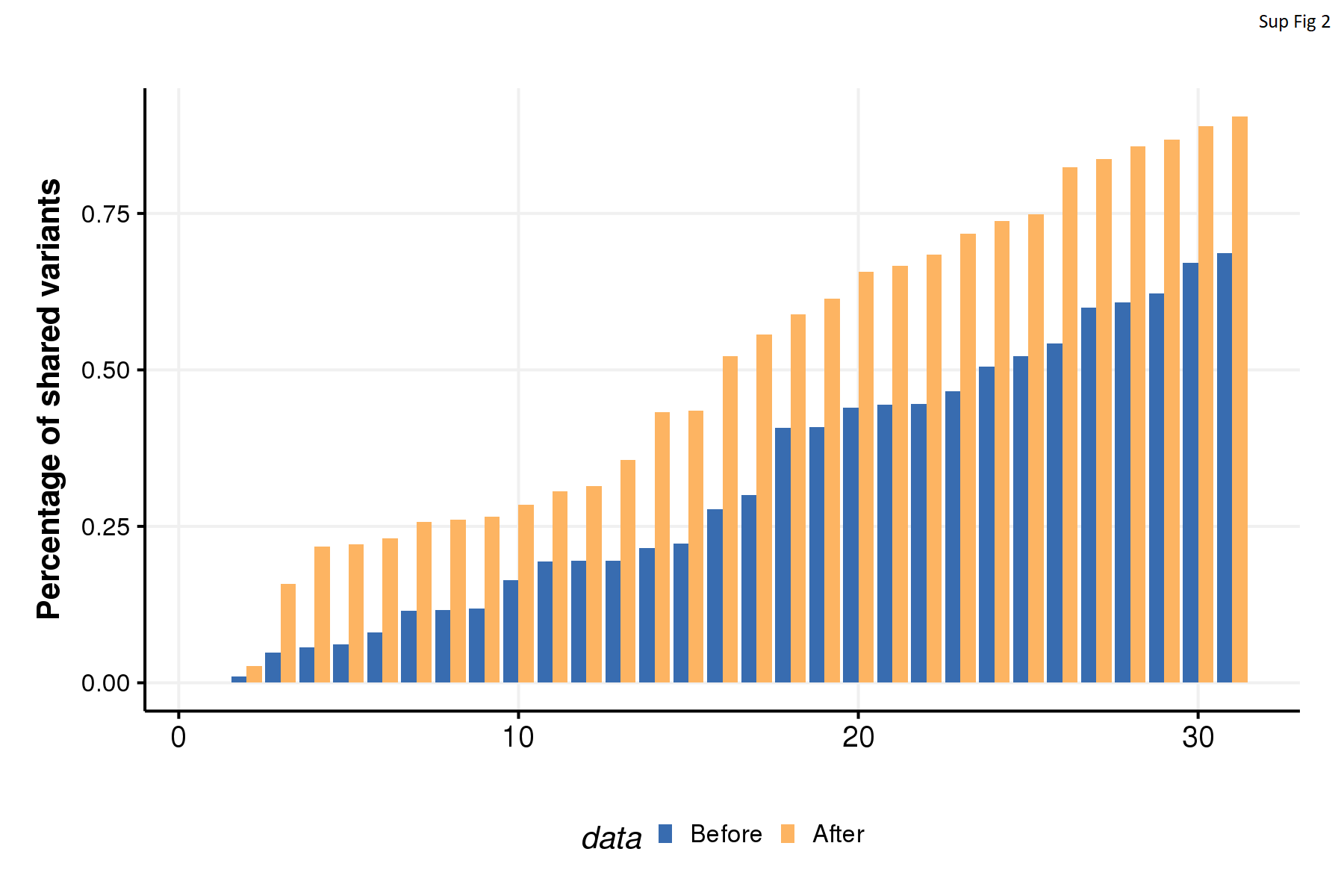
Whole exome sequencing revealed many somatic mutations and especially many copy number alterations (CNA). Previous work has shown that low-frequency subclones present at 1% to 5% in the primary tumor can be enriched to greater than 40% in related metastases (41). Despite this some have used cutoffs of variant allele of frequency (VAF) to exclude low frequency variants (42,43). To minimize false positives while maintaining high sensitivity, we followed a computational re-interrogation approach described by our group previously (34). Briefly, high-quality somatic variants called from the multiple samples of the same patient were combined into a single file per patient. The combined file containing all variants from the same patient were then re-interrogated across all samples from that patient. This method greatly improved detection of variants of low VAF and increased the percentage of shared variants across samples from each patient (Supplementary Fig. 2). Using the re-interrogated variants set, we explored the somatic mutation landscapes (Supplementary Table S4).
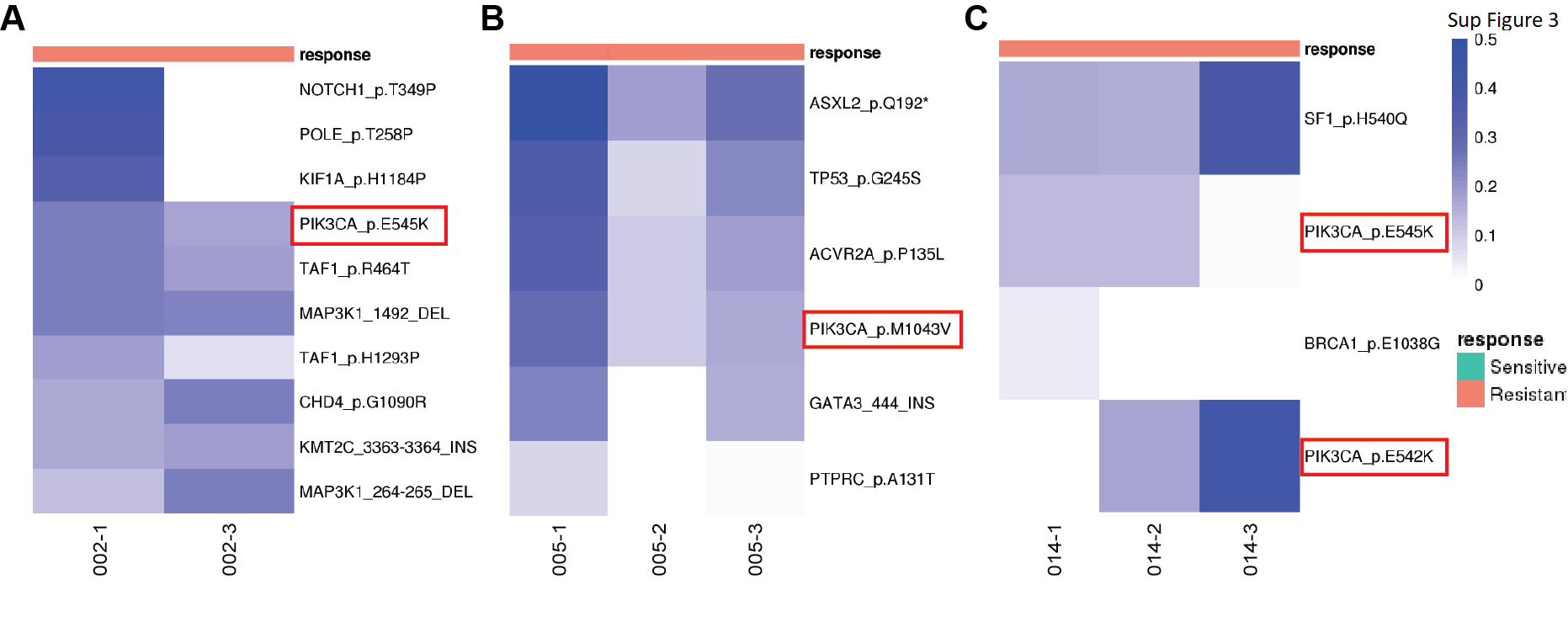
Genes previously shown to be recurrently altered in breast cancers were prevalent in this cohort. PIK3CA had the highest mutation rate with occurrence in 15 out of 35 patients. The other frequently mutated genes in our set included GATA3, CDH1, TP53 and ESR1, with mutation occurrences over 7 patients. The frequency in gene alterations reflected the multiple timepoints for a patient and/or multiple biopsies at a given timepoint (Fig. 2A). Of note, PIK3CA hotspot mutations were observed recurrently in all 3 patients that had de novo endocrine resistance, providing supporting evidence for previous findings that demonstrate hyperactivation of the PI3K pathway can promote endocrine therapy resistance (Supplementary Fig. 3) (44). Mutations in the ESR1 gene have been extensively linked to endocrine therapy resistance with the missense mutation D538G being the most prevalent (45,46); this mutation confers ligand-independent constitutive activation. Consistent with previous findings, ESR1 D538G had the highest frequency in our cohort, showing a pattern of enrichment in resistant tumors (Fig. 2B). It was observed in none of the intrinsic resistant patients and in 5 acquired resistance patients during treatment with aromatase inhibition. Of these, 1 patient had the variant in the primary tumor with a very low variant allele frequency which increased in frequency in the subsequent endocrine-resistant tumor; 1 patient did not have the variant in primary tumor and acquired the variant in the subsequent resistant tumor; and the remaining 3 patients had the variant in resistant tumors with unknown status in primary tumors (DNA sequencing not available). Interestingly, we also found an ESR1 E380Q variant present at diagnosis in one responsive patient with ongoing response to letrozole. The VAFs gradually decreased in subsequent samples, in line with this mutants’ estradiol hypersensitivity nature (47). GATA3 was the second highest mutated gene; with a range of detected variants (Fig. 2C). GATA3 is an ESR1-cooperating transcription factor with frame-shift mutations being most commonly reported, however, there have been few studies of its potential role in endocrine resistance (48,49). Recently Takaku et al. demonstrated that a specific type of GATA3 mutations in the second zinc-finger region reprogramed progesterone receptor signaling (50). Accordingly, we saw mostly frame-shift mutations in our cohort, with 4 variants residing in the second zinc finger region and 2 acquired in resistant tumors (Fig. 2C).
Figure 2. Genomic characteristics.

A, The pattern, frequency and type of genomic alterations of key breast cancer genes across patients. Samples were ordered by patient and response to endocrine therapy treatment. B-C, The type of variants detected shown in lollipop plot with labeling color indicating variants observed in sensitive tumors only (cyan), that observed in resistant tumors only (orange and boxed) and that observed in both sensitive and resistant tumors (orange) for ESR1 (N=8 patients with 23 samples) (B) and GATA3 (N=13 patient with 31 sampels) (C). D, Copy number frequency landscape plots showing copy number altered genes in sensitive and resistant tumors respectively. For each patient, copy number values were averaged among all sensitive samples and resistant samples from that patient, resulting in 23 sensitive tumors and 21 resistant tumors. Copy number gains are plotted above the x axis in red and copy number losses are plotted below the x axis in green. The frequency of alterations is indicated on the y axis from 0 to 100%.
To identify genes enriched in resistant tumors genome wide, we focused on the patients with DNA-seq matched pre- and on-treatment samples who acquired resistance in the neoadjuvant setting (N=12) and performed a matched mutation analysis (i.e. primary vs. matched later resistant samples per patient). We constructed count tables for each gene summarizing its mutation status in any of the sensitive and resistant tumors for each patient. Using a previously identified Pan-Cancer significantly mutated gene list (51), we found a variety of driver genes that were acquired in resistance in at least 1 patient (Supplementary Table 2). ESR1 appeared at the top of the list with 2 patients having the mutation acquired in resistant tumors. We identified 38 such genes including GATA3.
DNA copy number alterations including chromosome arm and subarm level changes were called using SynthEx (Supplementary Table S5) (30). The copy number gain and loss frequencies were comparable to the TCGA ER+ cohort (4). For example, we saw frequent 1q gain and 16q loss, noting that 16q loss is specific to luminal/ER+ breast cancers (Fig. 2D). Across the whole data set, there were some differences between copy number landscapes of sensitive and resistant tumors, however, using a linear mixed model comparing copy number values between sensitive (N=18 from 13 patients) and resistant tumors (N=20 from 13 patients, see Methods section for detail), and accounting for patients in the acquired resistant patient group, no significant regions of loss or gain were identified. This may be due to our small sample size.
We calculated the weights of known DNA mutational signatures (COSMIC v2) for each sample using DeconstructSigs. We then investigated both known DNA copy number changes that confer endocrine resistance, i.e. ERBB2 and FGFR1, and mutational signatures that are important in breast cancer, i.e. age, apobec and homologous directed repair (HDR) related signatures in each patient that acquired endocrine resistance (Supplementary Fig. 4). Results demonstrated that few patients showed potential resistance mechanisms related to investigated CNA and mutational signatures. For example, patient 020 showed acquired ERBB2 amplification in resistant samples and patient 022 showed acquired FGFR1 amplification in one of the resistant samples. These selected genetic features also showed the vast genetic heterogeneity among patients and spatial biological replicates.
Differential transcriptional program in endocrine resistant tumors compared to paired sensitive tumors
To identify genes differentially expressed in resistant versus sensitive tumors, we used linear mixed models to compare matched sensitive tumors to resistant tumors accounting for patients using the RNA-seq data. Briefly, biological replicates of the same tissue were first collapsed to construct a pseudo-sample by taking mean expression values across samples for each gene. We used RNA-seq data from 19 patients with acquired resistance and did matched comparisons of the untreated primary and the same tumor after acquisition of resistance. The t-statistic derived from the linear mixed model defines how consistently a gene is altered comparing resistant and sensitive tumors from each patient. The labels indicating treatment response were randomized 100 times to calculate false discovery rates (FDR).
We found 316 up-regulated genes and 123 down-regulated genes in resistant tumors that have FDR < 5% and fold change > 4 (for up-regulated genes) or < 0.25 (for down-regulated genes) (Supplementary Table S6). Hierarchical clustering of these significantly expressed genes across the whole dataset encompassing all patients and samples largely separated sensitive and resistant tumors (Fig. 3A, Supplementary Table S7). Primary tumors and biopsy samples from tumors that retained endocrine sensitivity clustered together and resistant tumors also clustered together. Gene ontology (GO) analysis revealed that the up-regulated genes in acquired resistant tumors were associated with cell proliferation, with GO terms chromatin assembly, nucleosome assembly and DNA packaging (Fig. 3B). We then checked a known proliferation score and it was higher in resistant tumors among the 19 patients (Fig. 3C) (52).
Figure 3. Differential gene expression in resistant tumors.

A, Hierarchical clustering of batch corrected RNA gene expression of significantly differentially expressed genes in resistant tumors (N=24 from 17 patients) compared to matched sensitive tumors (N=29 from 17 patients) using linear mixed models. B, Gene ontology terms associated with up-regulated genes in resistant tumors. C, Boxplot indicating median score and inter-quartile range of proliferation scores in resistant and sensitive tumors.
To fully characterize tumor transcriptional portraits, we used a panel of 654 previously published gene expression signatures measuring a variety of tumor phenotypes including amplicon signatures, oncogenic pathways, proliferation and the tumor microenvironment (Supplementary Table S2) (26). We calculated gene expression signature scores for each tumor and performed similar differential expression analysis using these signatures features. Among the acquired resistance tumors compared with their matched untreated primary, we found 37-upregulated gene signatures with FDR <0.01 and fold change >1.15, and 3 down-regulated gene signatures with FDR <0.05 and fold change <1 (Supplementary Table S8). Hierarchical clustering of differentially expressed signature scores again separated resistant and sensitive tumors (Fig. 4A, Supplementary Table S9). In particular, PAM50 subtype correlation to LumA centroids signature was one of the 3 down-regulated signatures, concordant with the fact that resistant tumors were enriched with HER2-enriched and LumB subtypes. Among the up-regulated signatures include 2 amplicon signatures, i.e. 16q23 and 8q, hypoxia, and several oncogenic pathway signatures; both estrogen and HER2 signaling pathways were also up-regulated. A signature composed of GATA3 induced genes was also up-regulated in resistant samples. In addition, AKT, RAS and p63 pathways showed higher expression in resistant tumors, consistent with previous findings that growth factor signaling pathways play important roles in endocrine resistance (Fig. 4B) (53). These results demonstrate resistant tumors harbor many transcriptional program changes compared to sensitive tumors.
Figure 4. Differential expression of gene signatures in resistant tumors.

A, Hierarchical clustering of significantly differentially expressed gene signatures in resistant tumors (N=24 from 17 patients) compared with matched sensitive tumors (N=29 from 17 patients) identified through linear mixed models. B, Boxplots indicating median score and inter-quartile range of significant signatures showing elevated signaling pathways in resistant tumors.
Molecular Portraits of selected patients
Lastly, we sought to integrate our DNA and RNA analysis for each patient to identify the mechanisms of resistance in individual patients. Patient 006 was responsive to letrozole treatment, and the tumor volume changes clearly showed a good response to treatment (Supplementary Fig. 5A). VAF plots showed a decreased number of variants and lower VAF likely due to low tumor purity, although cancer cell cellularity remained high as assessed by a pathologist (Supplementary Fig. 5B). This patient had ESR1 E380Q hypersensitivity variant in the primary tumor and the following two samples had a lower VAF in later tumors specimens, suggesting selective death of these endocrine sensitive cells (Supplementary Fig. 5C). E380Q is characterized as hypersensitive to estrogen ligand, which likely contributed to this tumor’s responsiveness. Accordingly, proliferation rate was initially high in the primary tumor, which subtyped as LumB, and decreased in subsequent LumA tumor specimens from this patient (Supplementary Fig. 5D).
Patient 020 acquired resistance and had only received letrozole treatment (Fig. 5A). The primary tumor and the second specimen were taken while the patient was responding, while the third and the fourth specimens were collected when the cancer was growing and thus was resistant. A number of somatic mutations were detected (Fig. 5B), and there was a clear clonal shift from the second sensitive specimen to the fourth resistant specimen, from which there were five biological replicates (Fig. 5C). This patient gained both ESR1 and GATA3 mutations along with many other mutations including CHD4, CDH1, PIK3R1. Gene expression showed a decreased proliferation rate in the second sensitive tumor that increased in the subsequent resistant tumors. Molecular subtype switched from primary tumor of LumB to LumA and back to LumB (Fig. 5D). Copy number landscapes showed increased genome instabilities in resistant specimens as well (Fig. 5E).
Figure 5. Molecular portraits of a luminal patient with acquired endocrine resistance.

A, Line plot showing tumor volume change indicating resistance to endocrine therapy acquired for the last two samples. Red dots indicate timing for tissue sampling. B, Box plots indicating median score and inter-quartile range of VAF of detected variants stratified by synonymous and nonsynonymous variants. C, Heatmap showing VAF of nonsynonymous variants of Pan-Cancer drivers. D, Heatmap showing gene expression of hormone receptors and key signaling pathways. E, Heatmap showing copy number landscapes of all tumors from the patient.
Lastly, patient 028 had high ERBB2 mRNA expression and had a HER2-enriched gene expression subtype but lacked clinical HER2 status. This patient had multiple lines of endocrine therapy including fulvestrant, tamoxifen and letrozole. Detected somatic variants showed clear clonal shifts (Fig. 6A, B), and acquired mutations included FGFR3 and GABRA6. There was also expansion of an existing clone in primary tumor containing mutations of PTEN, PIK3R1 and TP53, which are all recurrent variants. In terms of gene expression, all samples were classified as HER2-enriched subtype, with high ERBB2 and FGFR4 mRNA expression. Recently Garcia et al. showed that high FGFR4 expression plays an important role in HER2-enriched breast cancers and was associated with disease progression (54). Proliferation was lowest in the primary tumor and increased in later resistant tumors (Fig. 6C). The resistant tumor samples showed higher levels of copy number gains and losses (Fig. 6D).
Figure 6. Molecular portraits of a HER2-enriched patient with acquired endocrine resistance.

A, Box plots indicating median score and inter-quartile range of VAF of detected variants stratified by synonymous and nonsynonymous variants. B, Heatmap showing VAF of nonsynonymous variants of Pan-Cancer drivers. C, Heatmap showing gene expression of hormone receptors and key signaling pathways. D, Heatmap showing copy number landscapes of all tumors from the patient.
Discussion
Endocrine therapy resistance remains the biggest barrier to prolonged survival and cure for patients with ER+ breast cancer. There have been many studies investigating mechanisms of resistance and several have been defined, including acquired ESR1 mutations (14,45,53). Here we sought to understand more about the development of endocrine resistance in a series of ER+ patients during hormonal therapy treatment utilizing integrated DNA and RNA sequencing. We identified mutations that were enriched in resistant tumors, and we identified transcriptional profiles specific to tumor resistance, thus providing a molecular mechanism of resistance for some patients. This study is unique in its ability, through repeated and serial tissue acquisition during different phases of endocrine sensitivity within individual patients, to examine real-time alterations contributing to endocrine resistance.
Our work confirmed previous findings related to endocrine resistance mechanisms. Mutations enriched in resistant tumors include ESR1, GATA3 and FGFR3. Multiple signaling pathways were activated in resistant tumors including HER2, AKT, and RAS signaling, leading to increased proliferation rates within resistant specimens. Due to our sample size, larger cohort studies are needed to confirm mutations we identified that only were acquired by one patient, and increased power may allow detection of recurrent copy number changes.
Studies of sensitive breast cancers showed that clones that are sensitive to endocrine therapy can disappear under selective drug pressure. This was clearly seen in the patient with a sensitizing ESR1 E380Q variant that was present in the primary tumor and gradually disappeared in later samples. The genomic complexity seemed to reduce in sensitive cancers during treatment with a possible explanation being reduced cancer cellularity although the samples analyzed contained significant amounts of tumor on histological assessment. It is well known that multiple clones can exist within a primary cancer. Our observations show some clones are eliminated or reduced while others present at a low VAF at diagnosis increase and new clones with new mutations not detectable evolve possibly under the pressure of drug treatment.
A powerful aspect of this study is our unique series of patients that included patients who remained sensitive during treatment, and others with acquired resistance, with serial sampling over time from the same tumor in vivo. This study shows very clearly that there is no one mechanism that explains resistance in even a majority of patients. Each patient was unique in genetic and transcriptomic profiles. The most common ESR1 mutation only accounts for the resistance mechanism for 2 out of the 19 patients with acquired resistance by paired sensitive/resistant tumor analysis, and suggests that, although an important mechanism of resistance, it accounted for only a small number of resistant tumors in this study.
We collected multiple biological replicate samples from the same resistant tissue to assess tumor spatial heterogeneity. We observed clear heterogeneity between biological replicates in genetic and transcriptomic features. The biological replicates had both shared and unique mutations (Fig. 5C, Fig. 6C). As we used computational re-interrogation to recover low-frequency variants in related specimens, the unique mutations specific to each replicate are likely truly unique clones. Overall, gene expression profiles were highly correlated, and hierarchical clustering showed that replicates tended to cluster together (Fig. 3A). Subtle differences in gene expression were observed (Fig. 5D), with these findings suggesting that spatial heterogeneity (i.e. intra-tumor heterogeneity) could play a role in resistance to endocrine therapy. This study has confirmed some known mechanisms of resistance and identified some novel mechanisms. Most importantly it has highlighted the heterogeneity that underlies breast tumor biology and shows that endocrine resistance is complex and that more studies of patient-derived breast cancers are required. Taken together, it is clear that multiple molecular mechanisms underlying endocrine therapy resistance exist and there is a need to understand the mechanism in individual patients to effectively combat resistance in each patient.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational Relevance.
Many patients with estrogen receptor positive breast cancer develop drug resistance during endocrine therapy treatment. The molecular mechanisms underlying endocrine therapy resistance are not fully understood. We sought to investigate this by utilizing comprehensive and integrated DNA and RNA analysis of samples of breast cancers from 35 patients treated over time with endocrine therapy, whose cancers were responsive, had intrinsic resistance or developed acquired resistance. We identified known and novel mutations enriched in resistant tumors, as well as unique transcriptomic profiles specific to resistant tumors leading to increased activation of multiple oncogenic signaling pathways. Each patient displayed distinct molecular profiles, showing a variety of mechanisms of resistance, corroborating the highly heterogeneous nature of endocrine resistance. Improving the treatment of resistant tumors will require a personalized approach tailored to patient-specific causes of resistance.
Acknowledgements
We would like to acknowledge the contribution and support of our friend and colleague Dr Andrew Sims, University of Edinburgh, who sadly passed away prior to submission of this journal article. We are deeply grateful to the patients who allowed us to sample and use their tumor samples for this study. Thanks to Breast Cancer Now who funded the Translational Oncology Research Group at the Edinburgh Cancer Research Centre (JMD, AT). Funds from a generous grant from the Lyda Henderson Fund held at the Edinburgh and Lothians Health Foundation also supported this research. Funds from a generous grant from the Chewning Family to LAC also supported this research. This work was also supported by Breast Cancer Research Foundation (CMP, LAC). Samples were collated and identified with help from the Edinburgh Breast Unit team and Cancer Research UK who fund the Edinburgh Tissue Group.
Footnotes
Authors’ Contributions
J Michael Dixon, Lisa A. Carey, Charles M. Perou and Arran Turnbull conceived and designed the study. J Michael Dixon and Lorna Renshaw collected all the samples and JMD monitored response. Lorna Renshaw, Andrew Sims, Carlos Martinez Perez, Charlene Kay, Mark Gray, James Meehan worked with patient samples. Xiaping He performed library preparation. Youli Xia, Arran Turnbull and Joel S. Parker performed analyses. Youli Xia, J Michael Dixon, Charles M. Perou and Arran Turnbull wrote the paper. All authors have reviewed and approved the final paper.
Disclosure of Potential Conflicts of Interest
C.M.P is an equity stock holder and consultant of BioClassifier LLC. C.M.P and J.S.P are listed as an inventor on patent applications on the Breast PAM50 assay. The other authors declare no competing interests.
Data availability
DNA whole exome sequencing data are available at the EMBL-EBI ArrayExpress database under the accession number E-MTAB-10080. RNA sequencing data are available at the EMBL-EBI ArrayExpress database under the accession number E-MTAB-9917.
References
- 1.American Cancer Society. Cancer Facts & Figures 2020. Available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-andstatistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf.
- 2.Lei JT, Anurag M, Haricharan S, Gou X, Ellis MJ. Endocrine therapy resistance: new insights. Breast. 2019;48(Suppl 1):S26–s30. doi: 10.1016/S0960-9776(19)31118-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atals Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163(2):506–19. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlson RW. The history and mechanism of action of fulvestrant. Clin Breast Cancer. 2005;6(Suppl 1):S5–8. doi: 10.3816/cbc.2005.s.008. [DOI] [PubMed] [Google Scholar]
- 7.Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12(4):721–47. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 8.Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15(5):261–75. doi: 10.1038/nrc3920. [DOI] [PubMed] [Google Scholar]
- 9.Rani A, Stebbing J, Giamas G, Murphy J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front Endocrinol (Lausanne) 2019;10:245. doi: 10.3389/fendo.2019.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22(47):7316–39. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 11.Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27(21):3423–9. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95(5):353–61. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 13.Redmond AM, Bane FT, Stafford AT, McIlroy M, Dillon MF, Crotty TB, et al. Coassociation of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clin Cancer Res. 2009;15(6):2098–106. doi: 10.1158/1078-0432.CCR-08-1649. [DOI] [PubMed] [Google Scholar]
- 14.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–30. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, et al. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004;10(17):5670–6. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 16.Bergqvist J, Elmberger G, Ohd J, Linderholm B, Bjohle J, Hellborg H, et al. Activated ERK1/2 and phosphorylated oestrogen receptor alpha are associated with improved breast cancer survival in women treated with tamoxifen. Eur J Cancer. 2006;42(8):1104–12. doi: 10.1016/j.ejca.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 17.Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, et al. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13(2):137–44. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 18.Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005;18(2):250–9. doi: 10.1038/modpathol.3800296. [DOI] [PubMed] [Google Scholar]
- 19.Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer. 2005;12(Suppl 1):S47–59. doi: 10.1677/erc.1.00993. [DOI] [PubMed] [Google Scholar]
- 20.Perez-Tenorio G, Berglund F, Esguerra Merca A, Nordenskjold B, Rutqvist LE, Skoog L, et al. Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response to tamoxifen in breast cancer. Int J Oncol. 2006;28(5):1031–42. [PubMed] [Google Scholar]
- 21.Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117(1):218–28. doi: 10.1172/JCI28803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30(19):2813–5. doi: 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tischler German, L S. biobambam: tools for read pair collation based algorithms on BAM files. SOURCE CODE FOR BIOLOGY AND MEDICINE. 2014 [Google Scholar]
- 25.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Y, Fan C, Hoadley KA, Parker JS, Perou CM. Genetic determinants of the molecular portraits of epithelial cancers. Nat Commun. 2019;10(1):5666. doi: 10.1038/s41467-019-13588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baier H, Schultz J. ISAAC - InterSpecies Analysing Application using Containers. BMC Bioinformatics. 2014;15:18. doi: 10.1186/1471-2105-15-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 30.Silva GO, Siegel MB, Mose LE, Parker JS, Sun W, Perou CM, et al. SynthEx: a synthetic-normal-based DNA sequencing tool for copy number alteration detection and tumor heterogeneity profiling. Genome Biol. 2017;18(1):66. doi: 10.1186/s13059-017-1193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–9. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics. 2014;15(1):419. doi: 10.1186/1471-2164-15-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siegel MB, He X, Hoadley KA, Hoyle A, Pearce JB, Garrett AL, et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J Clin Invest. 2018;128(4):1371–83. doi: 10.1172/JCI96153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Martinez AA-O, Krop IA-O, Hillman DA-O, Polley MA-O, Parker JS, Huebner LA-O, et al. Survival, Pathologic Response, and Genomics in CALGB 40601 (Alliance), a Neoadjuvant Phase III Trial of Paclitaxel-Trastuzumab With or Without Lapatinib in HER2-Positive Breast Cancer. 2020:1527–7755. doi: 10.1200/JCO.20.01276. (Electronic)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan C, Prat A, Parker JS, Liu Y, Carey LA, Troester MA, et al. Building prognostic models for breast cancer patients using clinical variables and hundreds of gene expression signatures. BMC Med Genomics. 2011;4:3. doi: 10.1186/1755-8794-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bates D, Machler M, Bolker B, Walker S. Fitting Linear Mixed-Effects Models Using lme4. Journal of statistical Software. 2015;67(1):48. [Google Scholar]
- 40.Wilkerson MD, Cabanski CR, Sun W, Hoadley KA, Walter V, Mose LE, et al. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 2014;42(13):e107. doi: 10.1093/nar/gku489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoadley KA, Siegel MB, Kanchi KL, Miller CA, Ding L, Zhao W, et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016;13(12):e1002174. doi: 10.1371/journal.pmed.1002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015;5(11):1164–77. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savas P, Teo ZL, Lefevre C, Flensburg C, Caramia F, Alsop K, et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program "CASCADE". PLoS Med. 2016;13(12):e1002204. doi: 10.1371/journal.pmed.1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–61. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reinert T, Saad ED, Barrios CH, Bines J. Clinical Implications of ESR1 Mutations in Hormone Receptor-Positive Advanced Breast Cancer. Front Oncol. 2017;7:26. doi: 10.3389/fonc.2017.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73(23):6856–64. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 47.Pakdel F, Reese JC, Katzenellenbogen BS. Identification of charged residues in an N-terminal portion of the hormone-binding domain of the human estrogen receptor important in transcriptional activity of the receptor. Mol Endocrinol. 1993;7(11):1408–17. doi: 10.1210/mend.7.11.8114756. [DOI] [PubMed] [Google Scholar]
- 48.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23(1):12–22. doi: 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cottu P, Bieche I, Assayag F, El Botty R, Chateau-Joubert S, Thuleau A, et al. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin Cancer Res. 2014;20(16):4314–25. doi: 10.1158/1078-0432.CCR-13-3230. [DOI] [PubMed] [Google Scholar]
- 50.Takaku M, Grimm SA, Roberts JD, Chrysovergis K, Bennett BD, Myers P, et al. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat Commun. 2018;9(1):1059. doi: 10.1038/s41467-018-03478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173(2):371–85.:e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics. 2015;8:54. doi: 10.1186/s12920-015-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Recio S, Thennavan A, East MP, Parker JS, Cejalvo JM, Garay JP, et al. FGFR4 regulates tumor subtype differentiation in luminal breast cancer and metastatic disease. J Clin Invest. 2020 doi: 10.1172/JCI130323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA whole exome sequencing data are available at the EMBL-EBI ArrayExpress database under the accession number E-MTAB-10080. RNA sequencing data are available at the EMBL-EBI ArrayExpress database under the accession number E-MTAB-9917.