

Abstract
Rationale
Patients with severe asthma (SA) are less responsive to the beneficial effects of corticosteroid (CS) therapy and relative CS insensitivity has been shown in airway smooth muscle cells (ASMC) from SA patients.
Objectives
We investigated whether there was a defect in the actions of the glucocorticoid receptor (GR) underlying the ability of CS to suppress the inflammatory response in ASMC of patients with SA. ASMC from healthy subjects (n=10), severe (n=8) and non-severe asthma (N-SA; n=8) were cultured from endobronchial biopsies.
Measurements and Main Results
GR expression in ASMC from SA and N-SA was reduced compared to that from healthy subjects by 49% (p<0.01). While baseline levels of nuclear GR were similar, GR nuclear translocation induced by dexamethasone (10−7 M) in SA was 60% of that measured in either healthy or N-SA. TNF-α induced greater NF-κB (p65) mRNA expression in ASMC from SA (5.6 vs 2.0-fold; p<0.01), whereas baseline and TNFα-induced nuclear translocation and dexamethasone-mediated suppression of p65 expression were similar between groups. Dexamethasone, while not modulating TNFα-induced p65 nuclear translocation, attenuated p65 recruitment to the CCL11 promoter in the healthy and N-SA group, but this suppressive effect was impaired in SA.
Conclusions
Decreased GR expression with impaired nuclear translocation in ASMC, associated with reduced dexamethasone-mediated attenuation of p65 recruitment to NFκB-dependent gene promoters, may underlie CS insensitivity of severe asthma.
Keywords: airway smooth muscle, asthma, corticosteroid insensitivity, glucocorticoid receptor, nuclear translocation, CCL11
INTRODUCTION
Asthma is a chronic disease characterised by airway inflammation, hyper-responsiveness and remodelling(1). A proportion of asthma patients do not achieve adequate asthma control, despite taking oral corticosteroids (CS) and β-adrenergic agonists leading to frequent hospital admissions and use of emergency services. These patients, referred to as having severe or refractory asthma, are relatively insensitive to the therapeutic benefits of CS as demonstrated in lung macrophages(2) and airway smooth muscle cells(3) (ASMC) by the lesser suppressive effect of dexamethasone on induced release of proinflammatory cytokines and on induced proliferation of ASMCs compared to cells from non-severe asthma or healthy subjects. In patients with severe asthma, ASM mass is increased in the airways(4, 5) contributing to increased thickening and narrowing of the airways(4) and bronchial hyper-responsiveness(6). ASMCs can synthesise cytokines and growth factors and express cell-surface molecules that allow them to interact with the extracellular matrix and inflammatory cells, and may play a central role in orchestrating the inflammatory response within the bronchial wall(7, 8).
The anti-inflammatory effects of corticosteroids are mediated through the glucocorticoid receptor (GR), a ligand-activated transcription factor that modulates both inflammatory and anti-inflammatory gene expression. Following corticosteroid binding to GR, GR dissociates from chaperone proteins and rapidly translocates into the nucleus, where GR either binds to specific glucocorticoid-responsive elements (GRE) on DNA to enhance transcription of anti-inflammatory genes, or represses transcription of pro-inflammatory genes by interaction with inflammatory transcription factors, such as nuclear factor kappa-B (NF-κB) and activated protein-1 (AP-1). Changes in the phosphorylation pattern of GR as a consequence of its activation leads to many alterations of its function(9). Six serine residues have been identified as phosphorylation targets in the human GR(10) and phosphorylation of GR at serine 211 (Ser211) has been associated with ligand binding, nuclear translocation and transcriptional activition(9).
NF-κB is of paramount importance in asthmatic inflammation(11). Upon cellular stimulation, NF-κB translocates to the nucleus and mediates gene transcription. NF-κB consists of hetero- or homodimers of the DNA-binding Rel family of proteins of which the p65 subunit is ubiquitously expressed and confers transcriptional regulation(12). The NF-κB binding sites in the promoters of the pro-inflammatory genes such as CCL11 (eotaxin) and CXCL8 have been identified(13) and recruitment of the p65 subunit of NF-κB to the CCL11 promoter in lung epithelial cells is attenuated by dexamethasone(14). Moreover, we have shown that the suppressive effect of dexamethasone on TNFα-induced CCL11 and CXCL8 release is impaired in ASMC of severe asthma compared to non-severe asthma(3).
Insensitivity to CS may be attributed to a reduced ability of GR to bind to DNA or to an increase in the expression of pro-inflammatory transcription factors, such as NF-κB and AP-1(15). CS insensitivity of ASMC from severe asthma patients is associated with a greater p38 mitogen activated protein kinase (MAPK) activation induced by TNFα(3), similar to findings in alveolar macrophages from severe asthma where p38 MAPK inhibition reversed CS insensitivity(2);(16). In this study, we hypothesised that the regulation of GR and NF-κB (p65), in terms of their expression, nuclear translocation and recruitment to gene promoters, is perturbed in ASMC of patients with severe asthma compared those of patients with non-severe and healthy subjects. Some of the results of these studies have been previously reported in the form of an abstract (Po- Jui Chang et al. Eur Respir J 2012; 40: Suppl. 56, 615s)
MATERIALS AND METHODS
More details are provided in the Supplementary Material.
Subject characteristics (Table 1)
Table 1.
Subject characteristics
| Healthy Controls | Non-severe Asthma | Severe Asthma | |
|---|---|---|---|
| Number | 10 | 9 | 9 |
| Age, years | 39.0 ±11.3 | 31.8 ± 16 | 41.4 ± 12 |
| Gender, F/M | 5/5 | 4/5 | 5/4 |
| Duration of asthma, years | N/A | 18.2 ± 3.8 | 25.7 ± 9.7 * |
| Inhaled corticosteroid dose, μg BDP equivalent | N/A | 622 ± 290 | 2160±466 * |
| Atopy (n)$ | 3 | 6 | 5 |
| Receiving oral prednisolone (n) | N/A | N/A | 6 |
| FEV1, L | 3.15 ± 1.860 | 2.96 ± 0.46 | 2.0 ± 0.47 ** |
| FEV1,% predicted | 91.0 ±15.6 | 81.5 ± 8.3 | 61.59 ± 4.9 *** |
| FEV1/FVC, % | 77.2 ± 3.9 | 75.0 ± 3.1 | 69.0 ± 5.9 * |
| β-agonist reversibility#, % | N/A | 24.0 ± 3.5 | 31.0 ± 4.11* |
| PC20, mg/mL | > 16 | 2.7 ± 0.104 | 0.331 ± 0.13 (4/9) |
BDP, beclomethasone dipropionate; FEV1, forced expiratory volume in 1s; FVC, forced vital capacity; PC20, provocative concentration of metacholine causing a 20% fall in FEV1; N/A: not applicable.
Defined as positive skin prick tests to one or more common aeroallergens.
Measured as percent increase in FEV1 after 400 μg salbutamol.
p<0.05
p<0.001 vs non-severe asthma.
Severe asthma patients were on high-dose inhaled corticosteroids and sometimes on additional daily oral corticosteroid therapy to achieve a level of mild-to-moderate persistent asthma (17, 18). Patients with non-severe asthma used inhaled beclomethasone (0-1000 μg/day or equivalent) with good control of asthma. Current and ex-smokers of > 5 pack-years were excluded. All patients gave informed consent to participate in this study approved by the local Ethics Committee. They underwent fiberoptic bronchoscopy during which endobronchial biopsies were obtained.
ASMC isolation, culture and activation
Bronchial biopsies were cut into small pieces (<1 mm2), and transferred to 6-well culture plates. At confluence, cells were harvested and split into larger flasks at each passage. ASMC were identified by the characteristic “hill and valley” morphology, and by their expression of calponin, smooth muscle α-actin and myosin heavy chain in more than 95% of the cells(19). Cells were plated in 6-well culture plates for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) or in 75 cm2-flasks for Western Blot and chromatin immunoprecipitation (ChIP assay). At 90% confluence, cells were serum-starved for 24 hours, and stimulated with dexamethasone and/or TNF-α for times as indicated. Cells were used at passage 4 or 5.
Determination of mRNA expression
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Crawley, UK) and reverse-transcribed with random primers and AMV reverse transcriptase (Promega, Southampton, UK) using manufacturer’s instructions. cDNA was quantified by RT-qPCR (Rotor Gene 3000; Corbett Research, UK) using SYBR Green PCR Master Mix Reagent (Qiagen) and gene-specific primer sets for CCL11, p65 and 18S (Qiagen).
Chromatin immunoprecipitation (ChIP) assay
Cells were fixed in 1% formaldehyde for 10 minutes and DNA fragmented by sonication (5 × 10 second pulses). After adding ChIP dilution buffer, 4 μg of antibody was added to pre-cleared chromatin solution overnight. Antibody/DNA complexes were captured, washed, eluted, and reverse cross-linked. Both the DNA and input fractions were purified by phenol/chloroform wash and ethanol precipitation. The precipitated DNA was resuspended and quantitative PCR was performed using primers for the CXCL11 promoter region.
Western Blotting
40 μg of protein from each sample was loaded for electrophoresis. For protein from whole cell lysates, the transfer membrane was incubated with a rabbit anti-phospho-GR antibody, followed by anti-rabbit-HRP antibody. Antibody-bound proteins were visualized by ECL or ECL plus (GE Healthcare, CT, USA). The membrane was then re-probed with a rabbit anti-total GR antibody or with mouse anti-β-actin monoclonal antibody to control for protein loading. For proteins from cytoplasmic or nuclear extracts, the membrane was incubated with rabbit anti-GR p65 ant, and with α-tubulin or TBP (to control for protein loading. Relevant band intensities were quantified by scanning densitometric analysis.
Statistical analysis
Repeated measures analysis of variance (ANOVA) with Dunnet multiple comparison test was used for intra-group analysis. Mann-Whitney test or Kruskal-Wallis test with Dunn’s multiple comparison was used to compare results between the groups. p<0.05 was taken as significant.
RESULTS
Participant characteristics
Severe asthmatics had a longer duration of asthma, used higher doses of ICS, and had a lower FEV1 (% predicted) and increased obstruction (as determined by a reduced FEV1/FVC% ratio) compared to non-severe asthmatics. Six out of eight patients with severe asthma were on a daily dose of oral prednisolone (Table 1).
Expression and nuclear translocation of GR
Baseline GR expression, measured by Western blot, was lower in ASMC of severe astma (Kruskal-Wallis, post hoc test: p < 0.01) and non-severe asthma (Kruskal-Wallis, post hoc test: p < 0.05) compared to healthy subjects, and there was no difference between non-severe and severe asthma (Fig. 1A). Dexamethasone-induced nuclear translocation of GR in ASMC was confirmed by Western blot (Supplemental Fig. E1) and dexamethasone did not influence GR expression up to four hours post-stimulation (Fig. 1B). To compare nuclear translocation induced by dexamethasone in ASMC, cells were stimulated with dexamethasone (10−7 M) for 30 minutes to 4 hours. The quantity of nuclear GR at baseline was similar among ASMC of healthy subjects and non-severe and severe asthma (Fig. 2A). In ASMC of healthy subjects and non-severe asthma, dexamethasone induced a 3-fold increase in nuclear GR abundance at 30 minutes, which was maintained at one hour post-stimulation, followed by a gradual decrease to 50% of induced levels at the 4 hour time point. In contrast, in ASMC of patients with severe asthma, nuclear GR was induced by less than 2-fold which was significantly less than that observed in healthy subjects and patients with non-severe asthma (Fig. 2B).
Figure 1. Comparison of GR expression at baseline and effect of dexamethasone on GR expression.

(A) Baseline GR expression in ASMC of healthy subjects and asthma patients. After serum starvation for 24 hours, GR and β-actin protein in whole cell lysates from ASMC of healthy subjects and non-severe and severe asthma patients were measured in duplicate along with a standard (STD) protein by Western Blot followed by densitometric analysis. Representative blots are shown. Further analysis shows the comparison of baseline GR expression between the non-severe and severe asthma patients. Horizontal lines represent median. Kruskal-Wallis, post hoc test: **p<0.01, *p<0.05. (B) Effect of dexamethasone on GR expression. ASMC were treated with dexamethasone (10−7 M) for times indicated. GR and β-actin protein in the whole cell lysates were measured by Western Blot followed by densitometric analysis. Bars represent mean ± SEM from 3 ASMC of healthy subjects. ** p<0.01 vs unstimulated (US).
Figure 2. Impaired nuclear translocation of GR in ASMC of severe asthma.

(A) Nuclear GR expression at baseline. After serum starvation for 24 hours, GR and TBP protein in nuclear extracts from ASMC of healthy subjects and non-severe and severe asthma patients were measured in duplicate along with a standard (STD) protein by Western Blot followed by densitometric analysis; representative blot is shown. Horizontal lines represent median. (B) Dexamethasone-induced nuclear translocation of GR. ASMC of healthy subjects (●, n=8) and non-severe (■, n=8) and severe asthma (▲, n=8) patients were treated with dexamethasone (10−7 M) for times indicated, GR and TATA-box binding protein (TBP) in the nuclear extracts, as well as a standard (STD) protein, was assessed by Western blot followed by densitometric analysis. Points represent mean ± SEM. * p<0.05 vs healthy subjects. # p<0.05, ## p<0.01 vs non-severe asthma.
In order these translate our in vitro ASMC culture findings to that in the lung we examined GR expression ex vivo, in a limited number of biopsies from healthy subjects, and non-severe and severe asthmatics, using immuno-histochemistry. We analysed the relative percentage of GR positive nuclei in airway smooth muscle bundles (Figure E2A-C). We show that, in support of our in vitro data, that there are fewer GR positive nuclei in severe asthmatic biopsies compared to non-severe asthmatics (Kruskal-Wallis; post hoc test: p<0.05; Fig.E2D) and normal subjects (Kruskal-Wallis; post hoc test: p<0.05).
Phosphorylation of GR at serine 211 by dexamethasone
To investigate the effect of dexamethasone on phosphorylation of GR at the serine residue 211 (Ser211), ASMC of healthy subjects were stimulated with dexamethasone (10−6 M) for 5 minutes to 4 hours. GR phosphorylation was induced by dexamethasone at 30 minutes (p<0.01), which further increased (5.2-fold) at one hour and was maintained at 4 hours (Fig. 3A; p<0.001). To compare dexamethasone-induced phosphorylation of GR at Ser211 between groups, cells were stimulated with dexamethasone (10−6 M) for 2 hours. In ASMC of healthy subjects, dexamethasone induced a 6-fold increase in GR phosphorylation at Ser211, and this induction was similar in ASMC from patients with severe and non-severe asthma (Fig. 3B-C).
Figure 3. Comparison of dexamethasone-induced GR phosphorylation at serine 211.

(A) Time-dependent, dexamethasone-induced, phosphorylation of GR Ser211. After serum starvation for 24 hours, ASMC of healthy subjects were treated with dexamethasone (10−6 M) for times indicated. A representative blot is shown. Bars represent mean ± SEM from 3 ASMC of healthy subjects. ** p<0.01, *** p<0.001 vs unstimulated (US). (B) Comparison of GR Ser211 phosphorylation. ASMC of healthy subjects and non-severe and severe asthma patients were treated with dexamethasone (10−6 M) for 2 hours. Phosphorylated (Ser211) and total GR protein in whole cell lysates were measured by Western blot followed by densitometric analysis. Horizontal lines represent median.
Nuclear translocation of p65 and effect of dexamethasone
At 24 hours, TNF-α induced a ~2-fold increase in p65 mRNA expression in ASMC of healthy subjects and non-severe asthmatics compared to 5.6-fold in severe asthma, (Fig. E3A; p<0.01). TNF-α-induced nuclear translocation of p65 was also examined (Fig. E3B). The total quantity of nuclear p65 (Fig. E3C) and the time-course of TNFα-induced p65 nuclear abundance in ASMC of patients with non-severe and severe asthma and of healthy subjects (Fig. E3D) was the same. In ASMC of healthy subjects, dexamethasone suppressed TNFα-induced p65 protein expression by 21.3% at 24 hours (p<0.05; Fig. E4A) and similar effect was observed in ASMC of non-severe asthma (p<0.001) and severe asthma (p<0.01). TNFα-induced p65 mRNA was inhibited in a concentration-dependent manner in all three groups (Fig. E4B), with maximal suppression of ~50% by dexamethasone at 10−7 and 10−6 M (p<0.01, respectively). We also investigated the effect of dexamethasone on nuclear translocation of p65 induced by TNF-α in ASMC of healthy subjects (Fig. E4C) and non-severe asthma (Fig. E4D). TNF-α increased the nuclear abundance of p65, but this was not affected by dexamethasone.
Effect of dexamethasone on p65 recruitment to the CCL11 promoter
Having previously demonstrated that dexamethasone attenuates the recruitment of p65 to the fractalkine gene promoter in lung epithelial cells(20), we investigated the effect of dexamethasone on induced p65 recruitment to the gene promoter of CCL11 in ASMC. Cells were pretreated with dexamethasone (10−7 M) for 2 hours and then stimulated with TNF-α (10 ng/mL) for one hour. In ASMC of healthy subjects, TNFα-induced an 11.4-fold increase in the recruitment of p65 (p<0.001), which was suppressed by dexamethasone by 52.7% (Fig. 4A; p<0.05). This effect was similar in ASMC of patients with non-severe asthma, where dexamethasone suppressed TNFα-induced p65 recruitment by 51.4% (Fig. 4B). In contrast, the induced p65 recruitment to the gene promoter was not attenuated by dexamethasone in ASMC of severe asthma (Fig. 4C). We also compared the effect of dexamethasone on p65 recruitment to the CCL11 (eotaxin) gene promoter in ASM cells from patients with SA and NSA. Our results show a significant difference in the percentage reduction in p65 recruitment mediated by dexamethasone between NSA and SA, (52 ±12% vs 13 ± 8%, respectively; p<0.05).
Figure 4. Impaired dexamethasone mediated attenuation of TNFα induced p65 recruitment to the CCL11 promoter in ASMC of severe asthma.

After serum starvation for 24 hours, ASMC were pre-treated with dexamethasone (10−7 M) for 2 hours and stimulated with TNF-α (10 ng/mL) for 1 hour. p65 recruitment to the CCL11 promoter in ASMC of (A) healthy subjects and (B) non-severe and (C) severe asthma patients was measured by ChIP assay. Bars represents mean ± SEM. US= unstimulated, Dex=dexamethasone and NS=not significant. * p<0.05, *** p<0.001.
DISCUSSION
In the light of our recent observation of corticosteroid insensitivity in ASM of patients with severe asthma(3), we report that GR expression in severe and non-severe asthma is reduced compared to that in the healthy subjects. Furthermore, dexamethasone-induced GR nuclear translocation is attenuated in severe asthma compared of that in either the healthy or non-severe asthma. Baseline levels of nuclear GR were similar in the three groups. TNFα-induced greater p65 mRNA expression in severe asthmatics whereas baseline and TNFα-induced nuclear translocation and dexamethasone-mediated suppression of p65 expression were similar in all groups. Dexamethasone, while not modulating TNFα-induced p65 nuclear translocation, attenuated p65 recruitment to the CCL11 promoter in the healthy and non-severe asthma. However, this suppressive effect was impaired in severe asthma. Thus, decreased GR expression with impaired nuclear translocation in ASMC, associated with reduced dexamethasone-mediated attenuation of p65 recruitment to gene promoters, underlies the mechanism of corticosteroid insensitivity in severe asthma and this may contribute to the chronic inflammation observed in this disease (see Figure 5).
Figure 5. Model of corticosteroid insensitivity in severe asthma.
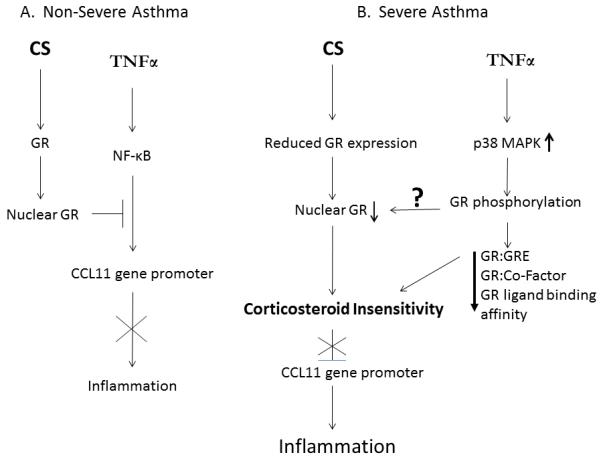
In non-severe asthma (A) TNFα-induced inflammation is suppressed by CS through GR-mediated attenuation of NF-κB recruitment to the CCL11 promoter. In severe asthma (B) reduced GR expression and impaired nuclear translocation results in corticosteroid insensitivity and an inability to inhibit inflammation. p38 MAPK-mediated phosphorylation of GR may also affect GR nuclear translocation
A reduction in GR may account for the failure of corticosteroid suppression of serum-induced ASMC proliferation in asthma(3, 21). In contrast, equivalent GR expression in ASMC of non-severe and severe asthma is supported by reports of similar levels of mRNA expression of either GRα or GRβ in peripheral lymphocytes of severe or moderate asthma(22) and similar protein and mRNA expression of both GRα and GRβ in peripheral blood mononuclear cells (PBMC) of steroid-dependent well-controlled asthma(23). The abundance of GR protein is reduced by dexamethasone at 24 hours post-stimulation, indicative of a down-regulatory effect of glucocorticoids on GR expression. GR mRNA is negatively regulated by glucocorticoids(24), which could be attributed to their inhibitory effects at the GRE, AP-1, NF-κB and cAMP response element-binding (CREB) regulatory motifs located in the promoter of GR(25). Alternatively, glucocorticoids may post-transcriptionally regulate the expression of GR, perhaps via destabilization of the GR mRNA(26). Ligand-activated GR protein could also be removed upon prolonged exposure to glucocorticoids by the proteasome-ubiquitin degradation pathway(27).
Nuclear translocation of GR is the critical step in corticosteroid-mediated anti-inflammatory effects. Based on our observation that total GR expression is not influenced by dexamethasone, we surmised that the increase in nuclear GR abundance is a direct consequence of GR nuclear translocation. However, the reduced dexamethasone-induced nuclear translocation of GR in ASMC of severe asthma, which extends the observation from peripheral blood mononuclear cells(28, 29) to airway resident cells, could underlie the mechanism of corticosteroid insensitivity in severe asthma.
Our study shows a reduction in GR expression in ASMC of both severe and non-severe asthma and yet corticosteroid insensitivity is only observed in the former. CS insensitivity is influenced by many factors, not only the number of GR but also by the affinity of GR and by interference with the glucocorticoid signalling pathways(30). The low affinity state of GR is achieved through binding of HSP70, HSP40 and HSP70–HSP90 organizing protein (HOP)(31), while a switch to a high affinity state occurs on binding of HSP90 and p23. A reduced HSP90:GR ratio has been reported in PBMC from patients with glucocorticoid-resistant asthma(32). Increased expression of the co-chaperone FKB51, reported in PBMC of severe asthma, could also reduce ligand affinity of GR(33). Transcriptional activity of GR can also be impaired via direct interaction with cytokine-induced proinflammatory transcriptional factors such as NF-κB, which prevents GR:GRE binding, which is increased in severe asthma.
The defective GR nuclear translocation in corticosteroid-insensitive severe asthma may be partly attributed to hyper-phosphorylation of GR(29), and this can also contribute to impaired effect of corticosteroids as a result of either decreased GR-GRE binding(34) or failure to suppress histone acetyltransferase activity induced by inflammatory stimuli(28). GR phosphorylation can be mediated by mitogen-activated protein kinases (MAPK) p38, c-Jun N-terminal kinase (JNK), or extracellular signal-regulated kinase (ERK)(30). We have reported heightened p38 MAPK activity in alveolar macrophages(35) and ASMCs(3) of severe asthma, and in addition, we have shown that inhibition of p38 MAPK leads to reversal of corticosteroid insensitivity in ASM and alveolar macrophages from patients with severe asthma(3, 16). This raises the possibility that p38 inhibition may improve corticosteroid responsiveness in severe asthma by reversal of defective nuclear translocation of GR, as demonstrated in an IL2- and IL4-induced corticosteroid-resistant T-cells(36). Phosphorylation of GR at serine 211 has been proposed as a marker for the transcriptional potential of GR(37). Absence of differential degrees of phosphorylation, at Ser211 between the asthmatic groups, indicates that this residue may not directly contribute to corticosteroid insensitivity in severe asthma.
TNFα-induced expression of the p65 and p50 components of NF-κB is suppressed by dexamethasone in ASMC of healthy subjects at both protein and mRNA levels(38), which we now demonstrate in ASMC of asthmatic patients. However, whereas corticosteroid insensitivity is displayed in ASMC of severe asthma, in terms of impaired suppression of the NFκB-dependent genes such as CCL11 and CXCL8(3), the suppressive effect of dexamethasone on p65 expression is similar between the normal and asthma groups. This suggests that reduced response to corticosteroids in severe asthma does not extend to impaired suppression of total NF-κB expression.
The inability of dexamethasone to inhibit induced p65 nuclear translocation is consistent with report in the literature in both ASMC(14) and in human lung epithelial cells(20). It is also reported that dexamethasone does not attenuate either TNFα-induced NF-κB DNA binding or NFκB-mediated reporter activity in ASMC(39). However, TNFα-induced p65 recruitment to the CCL11 promoter in AMSC from healthy subjects, is attenuated by dexamethasone. This is associated with reduced acetylation of histone H4, resulting in condensation of chromatin and subsequent hindered access of p65 to DNA(14). However, this effect of dexamethasone is impaired in ASMC from severe asthma, and this suggests a possible mechanism by which corticosteroids fail to suppress NFκB-medicated inflammatory genes, such as CCL11 and CXCL8, in ASMC from severe asthma(3). These results provide a better mechanistic understanding of corticosteroid insensitivity observed in airway smooth muscle cells obtained from patients with severe asthma. We have shown an attenuation of the suppression of p65 recruitment by dexamethasone associated with a reduced translocation of GR from the cytoplasm to the nucleus in airway smooth muscle cells from severe asthmatics compared to non-severe asthmatics. This observation is likely to explain the poor therapeutic effects of corticosteroid therapy in patients with severe asthma, where the magnitude of the reduction in p65 recruitment to pro-inflammatory gene promoters caused by corticosteroids determines their effects in controlling asthmatic inflammation.
Decreased GR expression with impaired nuclear translocation and subsequent inability to suppress p65 recruitment to the gene promoters contribute to the defective corticosteroid suppression of NF-κB-mediated chemokine expression in ASMC of severe asthma. These mechanisms may underlie CS insensitivity in severe asthma.
Supplementary Material
Scientific Knowledge on the Subject.
Expression of the glucocorticoid receptor (GR) in airway smooth muscle cells (ASMC) of patients with severe asthma and non-severe asthma is reduced compared to that of healthy subjects. GR nuclear translocation induced by dexamethasone in ASMC of severe asthma is 40% lower than that in non-severe asthma or healthy subjects. Dexamethasone attenuates NF-κB recruitment to the CCL11 promoter in ASMC of healthy subjects and non-severe asthma but this effect is impaired in severe asthma.
What This Study Adds to the Field
Reduced expression of the glucocorticoid receptor with impaired nuclear translocation and subsequent inability to suppress recruitment of pro-inflammatory transcription factor to gene promoters contribute to corticosteroid insensitivity in airway smooth muscle of severe asthma.
ACKNOWLEDGEMENTS
We thank Florence Chow and Sally Meah for the recruitment of patients. This study was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
This study was supported by project grants from Wellcome Trust (085935), Asthma UK (08/041), Chang Gung Memorial Hospital and College of Medicine (CMRPG371781) and by Respiratory Disease Biomedical Research Unit at the Royal Brompton NHS Foundation Trust and Imperial College London. KFC is a Senior Investigator of NIHR, UK.
Footnotes
This article has an online data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org.
Reference List
- 1.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From Bronchoconstriction to Airways Inflammation and Remodeling. American Journal of Respiratory and Critical Care Medicine. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 2.Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, Chung KF. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008;63:784–790. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- 3.Chang PJ, Bhavsar PK, Michaeloudes C, Khorasani N, Chung KF. Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma. Journal of Allergy and Clinical Immunology. 2012;130:877–885. doi: 10.1016/j.jaci.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway Structural Alterations Selectively Associated with Severe Asthma. American Journal of Respiratory and Critical Care Medicine. 2003;167:1360–1368. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- 5.Macedo P, Hew M, Torrego A, Jouneau S, Oates T, Durham A, Chung KF. Inflammatory biomarkers in airways of patients with severe asthma compared with non-severe asthma. Clin Exp Allergy. 2009;39:1668–1676. doi: 10.1111/j.1365-2222.2009.03319.x. [DOI] [PubMed] [Google Scholar]
- 6.Begueret H, Berger P, Vernejoux JM, Dubuisson L, Marthan R, Tunon-de-Lara JM. Inflammation of bronchial smooth muscle in allergic asthma. Thorax. 2007;62:8–15. doi: 10.1136/thx.2006.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirota N, Martin JG. MEchanisms of airway remodeling. CHEST Journal. 2013;144:1026–1032. doi: 10.1378/chest.12-3073. [DOI] [PubMed] [Google Scholar]
- 9.Weigel NL, Moore NL. Steroid Receptor Phosphorylation: A Key Modulator of Multiple Receptor Functions. Molecular Endocrinology. 2007;21:2311–2319. doi: 10.1210/me.2007-0101. [DOI] [PubMed] [Google Scholar]
- 10.Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Molecular and Cellular Biology. 2011;31:4663–4675. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adcock IM, Ito K, Barnes PJ. Glucocorticoids: Effects on Gene Transcription. Proceedings of the American Thoracic Society. 2004;1:247–254. doi: 10.1513/pats.200402-001MS. [DOI] [PubMed] [Google Scholar]
- 12.Hayden MS, Ghosh S. Shared Principles in NF-κB Signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, Casolaro V, Schleimer RP. Activation of Eotaxin Gene Transcription by NF-κB and STAT6 in Human Airway Epithelial Cells. The Journal of Immunology. 1999;163:6876–6883. [PubMed] [Google Scholar]
- 14.Nie M, Knox AJ, Pang L. β2-Adrenoceptor Agonists, Like Glucocorticoids, Repress Eotaxin Gene Transcription by Selective Inhibition of Histone H4 Acetylation. The Journal of Immunology. 2005;175:478–486. doi: 10.4049/jimmunol.175.1.478. [DOI] [PubMed] [Google Scholar]
- 15.Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy ClinImmunol. 2006;117:522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Bhavsar P, Khorasani N, Hew M, Johnson M, Chung KF. Effect of p38 MAPK inhibition on corticosteroid suppression of cytokine release in severe asthma. European Respiratory Journal. 2010;35:750–756. doi: 10.1183/09031936.00071309. [DOI] [PubMed] [Google Scholar]
- 17.Salmon M, Liu YC, Mak JCW, Rousell J, Huang TJ, Hisada T, Nicklin PL, Fan Chung K. Contribution of Upregulated Airway Endothelin-1 Expression to Airway Smooth Muscle and Epithelial Cell DNA Synthesis after Repeated Allergen Exposure of Sensitized Brown-Norway Rats. American Journal of Respiratory Cell and Molecular Biology. 2000;23:618–625. doi: 10.1165/ajrcmb.23.5.3909. [DOI] [PubMed] [Google Scholar]
- 18.Proceedings of the ATS Workshop on Refractory Asthma. Current Understanding, Recommendations, and Unanswered Questions. Am J Respir Crit Care Med. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- 19.Oltmanns U, Walters M, Sukkar M, Xie S, Issa R, Mitchell J, Johnson M, Chung KF. Fluticasone, but not salmeterol, reduces cigarette smoke-induced production of interleukin-8 in human airway smooth muscle. Pulmonary Pharmacology & Therapeutics. 2008;21:292–297. doi: 10.1016/j.pupt.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Bhavsar PK, Sukkar MB, Khorasani N, Lee KY, Chung KF. Glucocorticoid suppression of CX3CL1 (fractalkine) by reduced gene promoter recruitment of NF-{kappa}B. The FASEB Journal. 2008;22:1807–1816. doi: 10.1096/fj.07-094235. [DOI] [PubMed] [Google Scholar]
- 21.Roth M, Johnson P, Borger P, Bihl MP, Rudiger JJ, King GG, Ge Q, Hostettler K, Burgess JK, BLACK JL, Tamm M. Dysfunctional Interaction of C/EBP{alpha} and the Glucocorticoid Receptor in Asthmatic Bronchial Smooth-Muscle Cells. The New England Journal of Medicine. 2004;351:560–574. doi: 10.1056/NEJMoa021660. [DOI] [PubMed] [Google Scholar]
- 22.Jakiela B, Bochenek G, Sanak M. Glucocorticoid receptor isoforms in steroid-dependent asthma. Pol Arch Med Wewn. 2010:214–222. [PubMed] [Google Scholar]
- 23.Gagliardo R, Chanez P, Vignola AM, Bousquet J, Vachier I, Godard P, Bonsignore G, Demoly P, Mathieu M. Glucocorticoid receptor alpha and beta in glucocorticoid dependent asthma. AmJ Respir Crit Care Med. 2000;162:7–13. doi: 10.1164/ajrccm.162.1.9911032. [DOI] [PubMed] [Google Scholar]
- 24.Shimojo M, Hiroi N, Yakushiji F, Ueshiba H, Yamaguchi N. Differences in Down-Regulation of Glucocorticoid Receptor mRNA by Cortisol Prednisolone and Dexamethasone in HeLa Cells. Endocrine Journal. 1995;42:629–636. doi: 10.1507/endocrj.42.629. A. aM, Y. [DOI] [PubMed] [Google Scholar]
- 25.Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. The Journal of Steroid Biochemistry and Molecular Biology. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Vedeckis WV, Ali M, Allen HR. Regulation of Glucocorticoid Receptor Protein and mRNA Levels. Cancer Research. 1989;49:2295s–2302s. [PubMed] [Google Scholar]
- 27.Alarid ET. Lives and Times of Nuclear Receptors. Molecular Endocrinology. 2006;20:1972–1981. doi: 10.1210/me.2005-0481. [DOI] [PubMed] [Google Scholar]
- 28.Matthews JG, Ito K, Barnes PJ, Adcock IM. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy ClinImmunol. 2004;113:1100–1108. doi: 10.1016/j.jaci.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Mercado N, To Y, Kobayashi Y, Adcock IM, Barnes PJ, Ito K. p38 Mitogen-Activated Protein Kinase-γ Inhibition by Long-Acting β2 Adrenergic Agonists Reversed Steroid Insensitivity in Severe Asthma. Molecular Pharmacology. 2011;80:1128–1135. doi: 10.1124/mol.111.071993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. The Journal of Steroid Biochemistry and Molecular Biology. 2010;120:76–85. doi: 10.1016/j.jsbmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. MolCell Endocrinol. 2007:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Qian X, Zhu Y, Xu W, Lin Y. Glucocorticoid receptor and heat shock protein 90 in peripheral blood mononuclear cells from asthmatics. Chin Med J (Engl) 2001:1051–1054. [PubMed] [Google Scholar]
- 33.Chun E, Lee HS, Bang BR, Kim TW, Lee SH, Kim JH, Cho SH, Min KU, Kim YY, Park HW. Dexamethasone-Induced FKBP51 Expression in Peripheral Blood Mononuclear Cells Could Play a Role in Predicting the Response of Asthmatics to Treatment with Corticosteroids. J Clin Immunol. 2011;31:122–127. doi: 10.1007/s10875-010-9463-9. [DOI] [PubMed] [Google Scholar]
- 34.Adcock IM, Lane SJ, Brown CR, Peters MJ, Lee TH, Barnes PJ. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. The Journal of Immunology. 1995;154:3500–3505. [PubMed] [Google Scholar]
- 35.Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, Chung KF. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008;63:784–790. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- 36.Goleva E, Li Lb, Leung DYM. IFN-+| Reverses IL-2GÇô and IL-4GÇôMediated T-Cell Steroid Resistance. American Journal of Respiratory Cell and Molecular Biology. 2009;40:223–230. doi: 10.1165/rcmb.2007-0327OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beck IME, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocrine Reviews. 2009;30:830–882. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF, Kannan MS. Transcriptional regulation of CD38 expression by tumor necrosis factor-+| in human airway smooth muscle cells: role of NF-κ|B and sensitivity to glucocorticoids. The FASEB Journal. 2006;20:1000–1002. doi: 10.1096/fj.05-4585fje. [DOI] [PubMed] [Google Scholar]
- 39.Amrani Y, Lazaar AL, Panettieri RA. Up-Regulation of ICAM-1 by Cytokines in Human Tracheal Smooth Muscle Cells Involves an NF-κB-Dependent Signaling Pathway That Is Only Partially Sensitive to Dexamethasone. The Journal of Immunology. 1999;163:2128–2134. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.