

Abstract
Recent findings regarding the cellular biology and immunology of BST-2 (also known as tetherin) indicate that its function could be exploited as a universal replication inhibitor of enveloped respiratory viruses (e.g., influenza, respiratory syncytial virus, etc.). BST-2 inhibits viral replication by preventing virus budding from the plasma membrane and by inducing an antiviral state in cells adjacent to infection via unique inflammatory signaling mechanisms. This review presents the first comprehensive summary of what is currently known about BST-2 antiviral function against respiratory viruses, how these viruses construct countermeasures to antagonize BST-2, and how BST-2 function might be targeted to develop therapies to treat respiratory virus infections. We address the current gaps in knowledge, including the need for mechanistic understanding of BST-2 antagonism by respiratory viruses, that should be bridged to achieve that goal.
Keywords: respiratory virus, antiviral, interferon, innate immunity, influenza, respiratory infection
1. Introduction
Respiratory viruses represent an enormous worldwide health burden. They are a common cause of respiratory tract infections, which are the third leading cause of death worldwide (and the most deadly communicable diseases). Current options to prevent or clear respiratory virus infections are limited and have notable shortcomings. For example, the effectiveness of vaccination against seasonal influenza A virus (IAV) usually hovers around 50–60%,[1] and there is often a six-month delay between strain identification and vaccine production.[2] There is currently no vaccine for respiratory syncytial virus (RSV). Few antiviral drugs are presently approved for treating respiratory virus infections and most are IAV neuraminidase inhibitors.[3] For individuals suffering from the chronic airway diseases asthma and chronic obstructive pulmonary disease (COPD), respiratory virus infections can cause debilitating, and potentially deadly, exacerbations. In addition to acute disease, frequent and severe respiratory viral infections in youth have been implicated in detrimental airway remodeling and in the development of chronic diseases asthma and COPD in adulthood.[4] Thus, there is a current unmet need to develop new antivirals to treat respiratory virus infections.
The majority of respiratory viruses are RNA viruses,[5] which initially infect epithelial cells of nasal passages and airways. These viruses primarily infect with a minimal infectious dose, then utilize host cell machinery to replicate and produce new virions that then infect other cells, eventually causing epithelial cell death and airway remodeling. Efficient amplification, including virus release and infection of new cells, is obligatory for virus survival and disease pathogenesis. To subvert this process, host cells encode several interferon (IFN)-inducible antiviral factors that constitute a potent battery of innate immune defense. These antiviral factors interfere with various steps in the viral replication pathway to aid in clearance.[6] A comprehensive mechanistic understanding of how host antiviral factors function could potentially be exploited to develop new treatments to limit disease caused by respiratory viruses.
This essay will focus on the function of one particularly prolific host antiviral factor called bone marrow stromal antigen 2 (BST-2, also known as tetherin or CD317). We will examine whether BST-2 could be targeted to inhibit the replication of respiratory viruses and aid in their clearance. BST-2 has demonstrated broad-spectrum antiviral activity against numerous enveloped viruses,[7] and in response, most viruses have evolved functions to antagonize BST-2. BST-2 has not been shown to restrict non-enveloped viruses. Thus, our discussion will focus on common enveloped respiratory viruses that cause disease in humans [5, 8] (e.g., IAV; RSV; parainfluenza virus, PIV; metapneumovirus, MPV; coronaviruses, CoV) and mouse models (Sendai virus, SeV) but we will not discuss common non-enveloped respiratory viruses (e.g., rhinovirus, RV; enterovirus 68, EV68). We will review what is known regarding the regulation of BST-2 expression, the structure and function of BST-2, the mechanism of BST-2 antiviral function, modes of BST-2 antagonism by viral proteins, BST-2-mediated and -modulated signaling, and other immunological and cellular functions of BST-2, paying special attention to the settings of airway epithelial cells and respiratory viruses.
2. Is BST-2 Expressed in Airway Epithelia Following Viral Infection?
In order to most optimally inhibit respiratory virus replication BST-2 would need to be expressed in epithelial cells of the airway and mucosal surfaces. A number of signaling pathways have been demonstrated to induce expression of BST-2. The promoter region contains consensus sites for the transcription factors STAT3, IRF1, and ISGF1.[9] Accordingly, interferons (IFNs) from all three classes (i.e., IFN-α, -β, -γ, -τ, -λ3, and -ω) induce the expression of BST-2 in numerous cell types.[10, 11] In naïve mice, BST-2 expression appears to be limited to plasmacytoid dendritc cells, but is upregulated on most cell types by interferons that are induced and secreted in response to viral infection.[10] However, upregulation is not limited to IFN signaling, as a number of IFN-independent stimulants have also been reported. For example IL-27 is a potent inducer,[12] and TLR3 and TLR8 stimulation induce BST-2 expression in the absence of IFN.[13] In contrast, despite the apparent presence of an IRF1 binding site in the BST-2 promoter region, TNFα has little impact on BST-2 expression levels.[10, 14] Expression is downmodulated by TGFβ.[15]
Because the response to IFN-I is generally conserved in all cells,[16] BST-2 expression should increase in the airway as part of the antiviral response. However, there have been differing observations as to whether or not BST-2 expression is induced in airway epithelial cells. In cultured primary mouse airway epithelial cells and mouse airway cell lines (LA-4), BST-2 expression is induced by IFN-α, but only modestly increases 24 hours after IAV infection.[17] In contrast, we have observed increased BST-2 expression in vivo in mouse airway epithelial cells following both SeV and IAV infection (unpublished data), and BST-2 expression is upregulated in human airway cell lines (A549, NCI-H196, NCI-H358) following viral infection [18–20] and IFN-I.production.[19, 21] Overall, these observations suggest BST-2 is expressed in airway and mucosal epithelia in response to viral infection.
3. BST-2 Restricts Cellular Virion Release for a Broad Range of Enveloped Viruses
The antiviral function of BST-2 was first reported in 2008, when it was demonstrated that it inhibited the release of HIV-1 viral particles that were deficient in the viral membrane protein Vpu.[22] BST-2 inhibits viral replication by preventing release of viral progeny from infected cells, leading to their subsequent internalization and degradation.[23] Since then, BST-2 has been shown to potently inhibit several enveloped viruses that bud from the plasma membrane.[7] Due to this pressure, most viruses have developed antagonist strategies to block BST-2 function. Because of this, the BST-2 viral tethering function has typically been demonstrated in assays using mutant viruses deficient in BST-2-antagonizing proteins (such as Vpu-deficient HIV-1) or virus-like particles (VLPs). These types of studies have shown that BST-2 restricts the budding of VLPs or mutant viruses from the following families: retroviruses (alpha-, beta-, delta-, lenti-, and spuma-);[24, 25] arenaviruses (Lassa and Machupo);[26, 27] herpesviruses (KSHV);[28] filoviruses (Ebola and Marburg);[24, 27] rhabdoviruses (vesicular stomatitis);[29] paramyxoviruses (Nipah);[26] and flaviviruses (Hepatitis C).[30] BST-2 has been shown to effectively inhibit budding of some intact infectious viruses, including Lassa and Machupo arenaviruses,[26] and vesicular stomatitis rhabdovirus,[29] but does not restrict other infectious viruses such as Ebola, Marburg, cowpox, or Rift Valley fever bunyavirus.[26]
These observations demonstrate that BST-2 inhibits the budding of a broad spectrum of enveloped viruses and suggest that it might inhibit release of enveloped respiratory viruses from infected epithelial cells. More recent studies have investigated the potential of BST-2 to restrict budding of respiratory viruses (Table 1). For IAV, results appear to be dependent on virus strain and cell type used. Some groups have reported that BST-2 does not restrict IAV release,[17, 19, 31] while others report that BST-2 does inhibit release of IAV VLPs [18, 32, 33] and partially restricts infectious virus.[20, 21, 34] These observations appear to be dependent on IAV-encoded BST-2 antagonists, the identity and effectiveness of which may be strain dependent (discussed below). BST-2 also partially restricts the human pathogens PIV2,[35] hCoV-229E,[36] and SARS,[37] and this restriction is enhanced by deletion of viral antagonists. It also restricts animal model respiratory virus SeV,[38] but interestingly does not seem to inhibit pRRSV.[39] Thus, BST-2 appears to restrict a number of enveloped respiratory viruses in vitro, and this ability is hindered by encoded viral antagonists (discussed below).
Table 1 –
Summary of experimental observations for respiratory virus release inhibited by BST-2
| Virus | Family | Experimental Observations |
|---|---|---|
| Influenza A | Orthomyxovirus | Observations have been strain-specific. Some report BST-2 does not inhibit IAV release.[17, 19, 31] Others report BST-2 inhibits IAV VLP release; dependent on neuraminidase sequence.[18, 20, 21, 32, 33] Others report BST-2 inhibits infectious IAV and IAV VLPs, enhanced by loss of IAV M2.[34] NS1, which broadly targets ISG expression at the RNA level, may partially inhibit BST-2 by lowering expression. [31, 32] |
| PIV2 | Paramyxovirus | BST-2 inhibits PIV2 release.[35] |
| Sendai (SeV) | Paramyxovirus | BST2 inhibits SeV release from PM.[38] |
| SARS CoV | Coronavirus | BST-2 inhibits SARS release from PM; more strongly inhibits SARS-ORF7abΔ release from PM.[37] |
| Human CoV 229E (hCoV-229E) | Coronavirus | BST-2 inhibits hCoV-229E release from the PM.[36] |
| Porcine reproductive and respiratory syndrome virus (pRRSV) | Arterivirus | pRRSV E protein interacts with BST-2 by yeast two-hybrid and mislocalizes it from surface; BST-2 doesn’t inhibit pRRSV.[39] |
Key: SARS = severe acute respiratory syndrome; CoV = coronavirus; PIV = parainfluenza virus; PM = plasma membrane; SARS-ORF7abΔ = SARS with ORF7a and ORF7b deleted.
4. Structural Features of BST-2 Optimally Facilitate Tethering of Viruses
The structural organization of BST-2 is highly unique among mammalian proteins. It is a type II transmembrane protein consisting of a short N-terminal cytoplasmic tail (CT), a single transmembrane region (TM), an ectodomain (ED), and a second membrane anchor- a C-terminal glycosylphosphotidylinositol (GPI) (Fig. 1A). Sequence analysis suggests this dual-anchor topology is only found in one other protein in the human genome, a special form of the prion protein.[40] These structural features are important for the antiviral function of BST-2. Deletion of either one of the membrane anchors (TM or GPI) ablates the ability of BST-2 to restrict viral budding.[22, 41, 42] In addition to being a membrane anchor required for viral tethering, the GPI-anchor is required for cellular trafficking as BST-2 is sequestered in the ER of cells that have defects in the GPI-biosynthesis pathway.[42] The GPI-anchor enriches BST-2 to lipid rafts to enhance its antiviral activity, since this is a preferred site of budding for many enveloped viruses.[40] Mature BST-2 recycles between the plasma membrane, endosomes, and trans-Golgi network (TGN).[40, 43] Despite localization to lipid rafts, BST-2 is internalized via clathrin-dependent endocytosis, mediated by an evolutionarily conserved YxY motif in the CT, which recruits the adaptor AP-2.[43]
Figure 1.
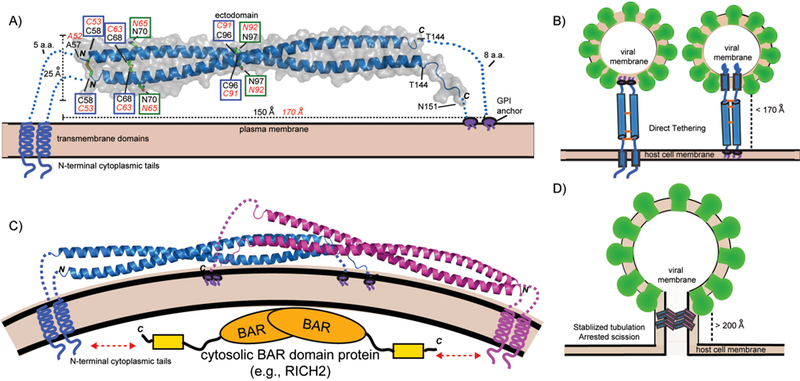
Structure of BST-2 and mechanisms of viral tethering. A: Crystal structure of mouse BST-2 ectodomain.[44] Structural features are highlighted: N-linked glycosylation sites = green boxes; disulfide bonds = blue boxes. Transmembrane domains, GPI anchors, and N-terminal cytoplasmic tails are shown as cartoons. Black labels correspond to mouse BST-2 sequence and features while the corresponding residues and values for human BST-2 are shown in red italics. B: The direct tethering models of BST-2 mediated inhibition of viral budding. Parallel BST-2 dimers incorporate one set of membrane anchors each into the viral membrane and host membrane. There is a 3–5 fold preference for incorporation of the GPI-anchor into the viral membrane (left) versus the other orientation (right).[51] C: A dimeric assembly created by crystallographic symmetry in the mouse BST-2 crystal structure. The dimer has a similar shape to BAR domains that bind membranes to generate or stabilize curvature. The N-terminal tail of BST-2 could bind cytoplasmic BAR domain proteins (e.g., RICH2)[55] to enhance mechanical stability in the stabilized tabulation model. (D): Stabilized tubulation and prevention of scission model of BST-2 mediated inhibition of viral budding. BAR-like BST-2 oligomers stabilize the tubulating membrane and prevent membrane scission. These tubules could be further stabilized by cytosolic BAR domain proteins that bind the cytoplasmic tail of BST-2 (e.g., RICH2)[55]. This hypothetical model is based on the observation of a BAR-like dimer in the mouse BST-2 ectodomain crystal structure [44] and the observation of long tube-like structures longer than the ectodomain of BST-2 (>200 Å) tethering HIV-1 VLPs to the plasma membrane by cryo-EM.[47]
The BST-2 ectodomain has numerous structural features required for optimal antiviral function (Fig. 1A). The ectodomain spans the two membrane anchors, forming a coiled-coil parallel dimer containing 3 inter-dimer disulfides and resembles a molecular “rope” spanning roughly 150 to 170 Å.[44–46] The ectodomain coiled-coil is unique in that it contains conserved patterns of residues at the dimer interface that destabilize the packing, causing the ectodomain to act less like a rigid rod and more like a flexible rope in solution, which likely aids its ability to adjust to the dynamics of virus assembly at the plasma membrane.[44, 45, 47] The ectodomain also contains two conserved N-linked glycosylation sites (Fig. 1A), which appear to be required for proper vesicular transport and possibly folding of BST-2, but are not required for antiviral activity.[42] Although isolated BST-2 ectodomains form parallel homodimers, the transmembrane regions of BST-2 also form parallel dimers in lipid environments and likely aid the formation of the parallel dimer.[48] Collectively, these structural features serve to inhibit viral budding from the plasma membrane.
5. What are the Mechanisms By Which BST-2 Restricts Viral Infection?
The bulk of evidence in the literature suggests that BST-2 restricts enveloped virus release by directly bridging the host and viral membranes through simultaneous inclusion of the two opposing membrane anchors (Fig. 1B).[42, 49] Cryo-EM and cryo-ET studies have shown budding viruses and VLPs anchored to cells by short tethers containing BST-2.[22, 47, 49, 50] Two orientations for viral tethering are possible (Fig. 1B). However insertion of the GPI-anchor into the viral membrane is favored 3–5 fold.[51] This orientation would optimize the ability of the N-terminal CT to interact with the cytoskeleton, which likely aids in BST-2 clustering at the host membrane. BST-2 accumulates at HIV budding sites, where it clusters at around 4 to 7 molecules per site.[52] Mutations to the BST-2 ectodomain prevent clustering and render it unable to restrict the release of VLPs.[53] This orientation and clustering also likely enhances the ability of BST-2 to form signaling complexes, which will be discussed in section 7.1.
Crystallographic and cryo-EM studies suggest a second potential mechanism for BST-2-mediated inhibition of viral budding. Analysis of the crystal structure of mouse BST-2 revealed a dimer-of dimers assembly that resembled those formed by BAR domains (Fig. 1C).[44] BAR domains are α-helical bundles that dimerize to form crescent-shaped structures that can bind to and stabilize curved membranes that form during membrane trafficking and budding events.[54] Clustering BST-2 could form such assemblies, which could be stabilized on the cytosolic side by the BAR-domain protein RICH2, which binds BST-2 CT using a C-terminal domain.[55] Such assemblies could stabilize tubulating membranes and prevent viral scission (Fig. 1D). Interestingly, cryo-EM studies of HIV-infected cells have observed some tethers that span at least 500 Å, much longer than a single BST-2 ectodomain,[47] hence lending further support to this potential assembly.
6. Viruses Encode Antagonists That Inhibit BST-2 Antiviral Function Via Diverse Mechanisms
BST-2 is highly efficient at trapping a wide variety of enveloped viruses. Consequently, most viruses have developed countermeasures to evade BST-2 antiviral function. These viral antagonists impair BST-2 by a number of different mechanisms, including removal from or prevention of trafficking to the cell surface, and targeting of BST-2 for proteosomal degradation.[7, 56] For example, HIV-1 encodes Vpu, a small membrane protein that interacts with BST-2 via transmembrane regions.[41, 57, 58] Vpu then facilitates recruitment of ubiquitin ligases and AP-1 to enable targeting of BST-2 for endolysosomal degradation.[59, 60] KSHV uses a similar strategy, encoding the protein K5 that is itself an E3 RING ubiquitin ligase that recognizes the BST-2 CT, thus enabling ubiquitination of BST-2 and subsequent degradation[28, 61] SIV Nef protein directs removal from the cell surface by binding the BST-2 CT and linking it to the endocytic adaptor AP-2 to remove BST-2 from the plasma membrane.[62] HIV-2 Env protein interacts with the BST-2 ectodomain and likewise facilitates BST-2 internalization by (again via AP-2) via a GYXXΦ internalization motif in the HIV-2 Env cytoplasmic tail.[63] Ebola envelope glycoprotein (GP) also interacts with the BST-2 ectodomain. However, this interaction appears to be independent of protein sequence, also requires the GP transmembrane domain, and does not lead to significant surface removal of BST-2.[64] Thus, viruses have developed a myriad of methods to inhibit BST-2 antiviral function and are facilitated by interactions involving various structural features of the protein.
The theme of viruses encoding BST-2 antagonists extends to respiratory viruses (Fig. 2). As mentioned earlier, IAV release appears to be partially restricted by BST-2 in a strain-dependent manner.[20] These differences appear to be mostly due to variations in the sequence of the encoded neuraminidase.[20, 21, 32, 33] Neuraminidase has been the most widely reported IAV-encoded BST-2 antagonist, but little known about its mechanism of action. Despite this lack of information, differences of a single amino acid have been reported to switch a restricted IAV VLP to one not inhibited by BST-2.[18] Interactions appear to be mediated between the ectodomains of the two proteins. However, this interaction does not appear to promote surface removal of BST-2. A more recent study suggests the IAV M2 channel protein is a BST-2 antagonist.[34] In contrast to neuraminidase, this interaction promotes surface removal of BST-2, ending in proteasomal degradation. In addition, IAV encodes the general IFN-I antagonist NS1, which can broadly inhibit expression of ISGs. NS1 may partially inhibit BST-2 via this broadly targeted strategy.[31, 32] PIV2 encodes the V protein, which targets BST-2 for surface removal, but does not appear to catalyze proteasomal destruction of BST-2.[35] PIV2 V may also prevent viral-mediated induction of BST-2.[65] SARS-CoV encodes ORF7a, which binds BST-2 via interaction of their ectodomains.[37] This interaction appears to inhibit maturation of BST-2 glycosylation, which prevents surface expression. Infection of cells with hCoV-229E promotes surface removal of BST-2, but the mechanism and viral protein involved are unknown.[36] SeV infection promotes proteasomal degradation of BST-2. This degradation appears to be mediated by the fusion (F) and hemagluttanin-neuraminidase (HN) proteins in concert, although further mechanistic details are unknown.[38] In summary, respiratory viruses have also evolved countermeasures to inhibit BST-2 viral tethering demonstrating evolutionary pressure to evade this potent viral restriction factor.
Figure 2.
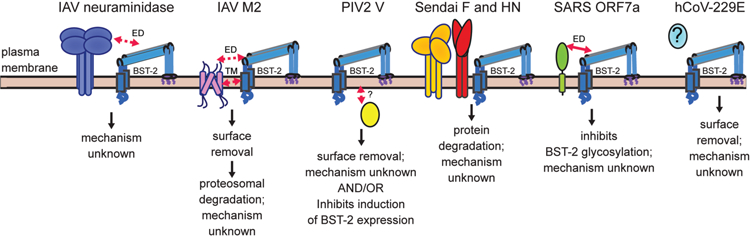
Respiratory virus antagonism of BST-2 and mode of action. Schematic summarizes what is currently understood about how various respiratory viruses antagonize BST-2. Structural domains demonstrated to mediate direct interactions are indicated by solid red arrows. Implied interactions are denoted by dashed red arrows. Antagonism of BST-2 by IAV is strain-dependent. In some strains, the neuraminidase protein partially inhibits IAV release.[18, 21, 33] This antagonism is thought to be mediated by the ectodomains (ED) of neuraminidase and BST-2. Another study suggests IAV antagonizes BST-2 using hemagglutinin, but the effect requires neuraminidase.[20] A more recent study suggests that the IAV M2 channel protein antagonizes BST-2 targeting interactions with the ectodomain and/or transmembrane (TM), causing surface removal.[34] The cytoplasmic V protein of PIV2 may antagonize BST-2 by two different mechanisms. It may bind and target BST-2 for surface removal [35, 80] and it may also inhibit induction of BST-2 expression.[65] How interaction is mediated is unknown. SeV F and HN proteins are required to degrade BST-2. However, it is unknown how interaction is mediated or the mechanism.[38] SARS CoV ORF7a ectodomain mediates an interaction with BST-2, which leads to inhibited glycosylation of BST-2 and its antagonism.[37] Human CoV-229E triggers surface removal of BST-2. The identity of the antagonist protein(s) is unknown.[36]
7. BST-2 Mediates and Modulates Immune Signaling
7.1. Pro-inflammatory signaling
BST-2 is known to have roles in inflammatory signaling. In addition to its function as a viral tethering protein, BST-2 also acts as an innate immune viral sensor that signals and triggers activation of NF-κB to elicit production of inflammatory cytokines that alert neighboring cells, extending its protection abilities to adjacent cells by inducing production of antiviral proteins. Clustering of BST-2 captured in budding virions initiates assembly of a signaling pathway that causes NF-κB activation.[66, 67] This clustering creates the formation of a specialized signaling motif called a HemITAM including residues Y6 and Y8 in the BST-2 CT, which recruit Src-family kinases and become phosphorylated (Fig. 3). The phosphorylation specifically recruits the kinase Syk, which then assembles a signaling complex composed of adaptors TRAF2 and TRAF6, as well as TAK1, and culminates in downstream activation of NF-κB.[68] BST-2 is coupled to the actin cytoskeleton via interaction with the BAR and Rho-GAP containing protein RICH2, which binds the BST-2 CT. Mutations that inhibit this interaction prevent or reduce phosphorylation of BST-2 and subsequent downstream NF-κB activation.[68, 69] Thus, it appears that pre-concentration of BST-2 on the cell surface is required for efficient clustering by viruses. This signaling can induce expression of inflammatory and antiviral signaling cytokines IFN-β, CXCL10, and IL-6.[68] Virus-encoded BST-2 antagonists have varying effects on BST-2-mediated activation of NF-κB, which appears to be linked to the ability to direct degradation of BST-2. HIV-1 Vpu, which targets BST-2 for surface removal and destruction, also prevents BST-2-mediated activation of NF-κB.[67, 70] However, Ebola GP and HIV2 Env, which engage BST-2 to inhibit its viral tethering ability but do not direct its degradation, do not inhibit this signaling ability of BST-2.[71] The impact of antagonists encoded by respiratory viruses on BST-2-mediated NF-κB activation has not been investigated.
Figure 3.
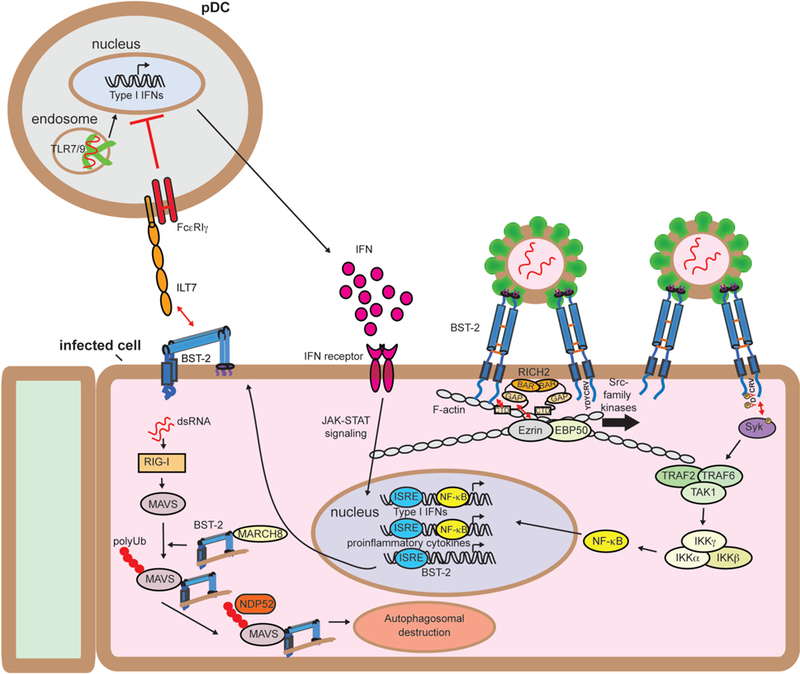
Immune signaling functions of BST-2. IFN produced in response to viral infection stimulates a cellular antiviral state by inducing the expression of hundreds of genes, including BST-2. IFN-induced BST-2 inhibits budding of newly-forming virions, creating BST-2 clusters at the plasma membrane. Clustered BST-2 is linked to the cortical actin cytoskeleton via RICH2, a BAR domain containing protein. This leads to phosphorylation of tyrosines in the YDYCRV motif in the cytoplasmic tail by Src-family kinases followed by the recruitment and activation of Syk. Syk activation recruits TRAF2, TRAF6, and TAK1 into a complex that leads to the activation of NF-kB and the induction of proinflammatory cytokines.[68] The upregulated BST-2 expressed on the surface of infected cells engages the plasmacytoid dendritic cell (pDC) receptor ILT7, resulting in inhibitory signaling that reduces the expression of IFNs by pDCs.[72, 73] BST-2 also negatively regulates RIG-I-like receptor innate immune signaling. RIG-I recognizes uncapped viral dsRNA and induces MAVS, resulting in IFN-I expression and subsequently BST-2 expression. BST-2 then recruits the ubiquitin E3-ligase MARCH8 to catalyze addition K27-linked polyUb chains to MAVS at K7. This chain is then recognized by NDP52, which recruits BST-2/MAVS to autophagosomes for destruction, thereby dampening IFN-I production.[74]
7.2. Anti-inflammatory signaling
BST-2 has also been implicated in inhibitory signaling in plasmacytoid dendritic cells (pDCs), thereby dampening inflammatory signaling. In this capacity, it serves as a ligand for the inhibitory receptor ILT-7, which is exclusively expressed on pDCs (Fig. 3). Circulating pDCs produce IFN-I in response to virus-derived TLR7/9 stimulation, which then initiates the antiviral state in infected and surrounding cells, including upregulation of BST-2 expression. The BST-2 expressed on infected and neighboring cells can then engage ILT-7 on the surface of pDCs, which signals through FcRγI to shut down production of IFN-I in a negative-feedback manner to prevent overstimulation.[72] HIV-1 Vpu appears to exploit this mechanism by specifically targeting BST-2 clustered to viral budding sites for destruction, and enriching distribution of BST-2 outside of the viral budding site, thus enhancing the probability of BST-2/ILT7 interaction to suppress pDC antiviral response.[73] This interaction has also been suggested to occur in cis between ILT7 and BST-2 both expressed on the surface of pDCs, possibly to prevent premature maturation.[14]
BST-2 can also suppress virus-induced interferon production by altering innate immune signaling. It was recently reported that BST-2 can direct the degradation of the RIG-I-like receptor MAVS to dampen IFN-I production.[74] In this scenario, BST-2 engages MAVS and recruits the ubiquitin E3 ligase MARCH8, which directs K27-linked poly-Ub chains to K7 of MAVS. This polyubiquitination then engages NDP52, leading to autophagic degradation (Fig. 3). This function highlights the need for tight regulation of IFN-I production to initiate proper antiviral immune response while avoiding development of autoimmune disorders.
7.3. Other signaling roles:
In addition to these roles in signaling pathways, BST-2 may also play a role in mediating intercellular signaling by tethering exosomes.[75] Exosomes are small, secreted extracellular vesicles that carry cargo such as growth factor cytokines and nutrients between cells. BST-2 can trap exosomes on the surface of parent cells, thereby controlling whether exosomes are involved in short-range or long-range communication by cells.
8. Strategies to Harness BST-2 Antiviral Function to Accelerate Clearance of Respiratory Viruses.
BST-2 was first identified as a potent HIV-1 antiviral factor in 2008.[22] In the decade since, there has been much research activity on the antiviral and immunological functions of BST-2: these investigations have demonstrated that it can restrict a wide variety of enveloped viruses. A number of recent studies indicate that BST-2 can also restrict the budding of respiratory viruses. This is further exemplified by the fact that these viruses have developed BST-2 countermeasures against this significant pressure. So how could BST-2 function be exploited to clear respiratory virus infections and what knowledge is needed to design such treatments? In this section we will discuss how this could be achieved given the current state of knowledge and examine what gaps need to be addressed.
8.1a. Pharmacological inhibition of viral BST-2 antagonists.
Nearly all enveloped viruses investigated to date encode an antagonist that at least partially blocks BST-2 ability to prevent virus release. In theory, inhibiting these antagonists (most specifically by disrupting interaction with BST-2) would allow BST-2 to exert its function at the cell surface and trap virion release. This concept has been actualized, at least experimentally, for HIV-1. BST-2 antiviral function against HIV-1 and how it is antagonized by Vpu has been investigated comprehensively. This understanding has allowed the design of unique membrane-targeted peptides[76] as well as high-throughput assays, which have identified compounds that prevent Vpu-mediated surface down-regulation of BST-2.[77] These small molecules inhibit HIV-1 release and suppress replication.[78] Similar pursuits could be engaged for respiratory viruses. However, a similar level of comprehensive knowledge would need to be obtained.
8.1b. Gaps in knowledge and how to address them.
The identity of BST-2 antagonists encoded by various respiratory viruses as well as their mechanism of action is poorly understood. The action of BST-2 against some established (e.g., RSV) and emerging respiratory pathogens (e.g., metapneumovirus, bocavirus) has not been investigated. Additionally, it is unknown whether BST-2 exerts inhibitory effects toward any non-enveloped viruses. Within this class, rhinovirus and enterovirus-68 are prominent respiratory pathogens, causing severe acute disease and triggering asthma exacerbations. For others that have been studied (Table 1), few details are known about how the putative antagonists engage BST-2 or their mechanism of action. In fact, the only virus for which structural details of antagonist action have been elucidated is HIV-1 Vpu.[58, 59] Detailed mechanistic and structural studies of BST-2 antagonist action and their interactions with BST-2 are required to design inhibitors or develop screens to discover them. In addition, mechanistic studies should be carried out in airway epithelial cell lines and verified in primary airway epithelial cells and animal models. Treatments would be virus-specific, as current knowledge indicates that each respiratory virus employs a unique antagonist mechanism.
8.2a. Pharmacological tuning of BST-2 signaling.
It is now understood that BST-2 plays a role in innate immune and downstream inflammatory signaling. Viral clustering of BST-2 initiates a signaling cascade that results in NF-κB activation and production of cytokines involved in inflammatory and antiviral responses (Fig. 3). Additionally, BST-2 serves as a ligand for ILT-7 that dampens production of antiviral IFN-I. Third, BST-2 can dampen innate immune signaling through MAVS. Inhibition of some of these aspects may be needed to create the optimal tuning of antiviral versus anti-inflammatory response in the airway.
8.2b. Gaps in knowledge and how to address them.
Although some mechanistic details are known for each of the three pathways, detailed structural and biophysical information regarding BST-2 interactions in these events have not been reported (e.g., BST-2 engagement of RICH2, which is central to NF-κB activation; BST-2 binding ILT-7 to signal for downregulation of IFN production). Such details are required to develop methods to block or enhance these interactions. In addition, it is currently unknown whether BST-2 antagonists encoded by respiratory viruses alter any of these signaling pathways.
8.3a. Pharmacological enhancement of BST-2 cell surface expression.
BST-2 needs to be on the cell surface in order to restrict virus release and suppress further infection. Its expression is mostly controlled by interferon signaling. However, induction of BST-2 has also been reported in the absence of IFN.[13] This suggests that multiple transcription factors maybe be able to regulate BST-2 expression and implies that pharmacological induction, independent of the pleiotropic effects of IFN, is likely possible.
8.3b. Gaps in knowledge and how to address them.
Long-term BST-2 overexpression has been implicated in the growth and progression of some cancers,[79] so methods to enhance BST-2 surface expression should be approached with caution. However, it is unclear whether enhanced surface expression over a short term would be a significant risk.
9. Conclusions and prospects.
In this essay, we present the first comprehensive review of BST-2 antiviral function and signaling with respect to respiratory viruses. Emerging evidence indicates that BST-2 can restrict budding and replication of important respiratory viruses, which is counteracted by encoded viral antagonists. Detailed structural, biophysical, and cellular immunologic studies utilizing airway cells are now needed to develop a mechanistic understanding of how respiratory viruses antagonize BST-2 antiviral function and, potentially, immunologic signaling. This understanding could lead to the development of novel therapies that exploit BST-2 function to enhance recovery from respiratory virus infections.
Acknowledgements
This work was supported by NIH R01-HL119813 (to T.J.B.), American Heart Association SDG-196051 (to T.J.B.), NIH F30-HL140783 (to K.N.B), NIH T32-HL007317 (to K.N.B.), T32-GM007200 (to K.N.B.), American Heart Association Predoctoral Fellowships 15PRE22110004 and 17PRE32780001 (to D.L.K.), and NIH T32-GM7067 (to D.L.K.).
Abbreviations:
- COPD
chronic obstructive pulmonary disease
- IFN
interferon
- IFN-I
type-I interferon
- IAV
influenza A virus
- ISG
interferon stimulated gene
- RSV
respiratory syncytial virus
- VLP
virus-like particle
- cryo-EM
cryo-electron microscopy
- cryo-ET
cryo-electron tomography
- KSHV
Kaposi-Sarcoma Herpesvirus
- BST-2
bone marrow stromal antigen 2
- ERGIC
ER-Golgi intermediate compartment
- PM
plasma membrane
- PIV2
parainfluenzavirus 2
- SARS CoV
severe acute respiratory syndrome coronavirus
- SeV
Sendai virus
- hCoV-229E
human coronavirus 229E
- pRRSV
porcine reproductive and respiratory syndrome virus
Footnotes
The authors declare no conflicts of interest.
References
- [1].Jackson ML, Chung JR, Jackson LA, Phillips CH, Benoit J, Monto AS, Martin ET, Belongia EA, McLean HQ, Gaglani M, Murthy K, Zimmerman R, Nowalk MP, Fry AM, Flannery B, N Engl J Med 2017, 377, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Koszalka P, Tilmanis D, Hurt AC, Influenza Other Respir Viruses 2017, 11, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ison MG, Clin Chest Med 2017, 38, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, Hartert TV, J Allergy Clin Immunol 2009, 123, 1055; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, Printz MC, Lee WM, Shult PA, Reisdorf E, Carlson-Dakes KT, Salazar LP, DaSilva DF, Tisler CJ, Gern JE, Lemanske RF Jr., Am J Respir Crit Care Med 2008, 178, 667; [DOI] [PMC free article] [PubMed] [Google Scholar]; Edwards MR, Strong K, Cameron A, Walton RP, Jackson DJ, Johnston SL, J Allergy Clin Immunol 2017, 140, 909; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jartti T, Gern JE, J Allergy Clin Immunol 2017, 140, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hodinka RL, Microbiol Spectr 2016, 4. [DOI] [PubMed]
- [6].Villalon-Letelier F, Brooks AG, Saunders PM, Londrigan SL, Reading PC, Viruses 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swiecki M, Omattage NS, Brett TJ, Mol Immunol 2013, 54, 132. [DOI] [PubMed] [Google Scholar]
- [8].Pawelczyk M, Kowalski ML, Curr Allergy Asthma Rep 2017, 17, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Hirano T, Tsuchiya M, Biochem Biophys Res Commun 1999, 258, 583. [DOI] [PubMed] [Google Scholar]
- [10].Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M, J Immunol 2006, 177, 3260. [DOI] [PubMed] [Google Scholar]
- [11].Arnaud F, Black SG, Murphy L, Griffiths DJ, Neil SJ, Spencer TE, Palmarini M, J Virol 2010, 84, 4415; [DOI] [PMC free article] [PubMed] [Google Scholar]; Dietrich I, McMonagle EL, Petit SJ, Vijayakrishnan S, Logan N, Chan CN, Towers GJ, Hosie MJ, Willett BJ, J Virol 2011, 85, 5840; [DOI] [PMC free article] [PubMed] [Google Scholar]; Amet T, Byrd D, Hu N, Sun Q, Li F, Zhao Y, Hu S, Grantham A, Yu Q, Curr Mol Med 2014, 14, 349; [DOI] [PubMed] [Google Scholar]; Cobos Jimenez V, Booiman T, de Taeye SW, van Dort KA, Rits MA, Hamann J, Kootstra NA, Sci Rep 2012, 2, 763; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu MQ, Zhou DJ, Wang X, Zhou W, Ye L, Li JL, Wang YZ, Ho WZ, PLoS One 2012, 7, e35902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guzzo C, Jung M, Graveline A, Banfield BW, Gee K, Sci Rep 2012, 2, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bego MG, Mercier J, Cohen EA, J Virol 2012, 86, 3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tavano B, Galao RP, Graham DR, Neil SJ, Aquino VN, Fuchs D, Boasso A, J Immunol 2013, 190, 2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sayeed A, Luciani-Torres G, Meng Z, Bennington JL, Moore DH, Dairkee SH, PLoS One 2013, 8, e67191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schreiber G, J Biol Chem 2017, 292, 7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Londrigan SL, Tate MD, Job ER, Moffat JM, Wakim LM, Gonelli CA, Purcell DF, Brooks AG, Villadangos JA, Reading PC, Mintern JD, PLoS One 2015, 10, e0142925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Leyva-Grado VH, Hai R, Fernandes F, Belicha-Villanueva A, Carter C, Yondola MA, J Mol Biol 2014, 426, 1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Winkler M, Bertram S, Gnirss K, Nehlmeier I, Gawanbacht A, Kirchhoff F, Ehrhardt C, Ludwig S, Kiene M, Moldenhauer AS, Goedecke U, Karsten CB, Kuhl A, Pohlmann S, PLoS One 2012, 7, e43337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gnirss K, Zmora P, Blazejewska P, Winkler M, Lins A, Nehlmeier I, Gartner S, Moldenhauer AS, Hofmann-Winkler H, Wolff T, Schindler M, Pohlmann S, J Virol 2015, 89, 9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, Kaiser L, Piguet V, J Biol Chem 2012, 287, 22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neil SJ, Zang T, Bieniasz PD, Nature 2008, 451, 425; [DOI] [PubMed] [Google Scholar]; Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J, Cell Host Microbe 2008, 3, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sauter D, Specht A, Kirchhoff F, Cell 2010, 141, 392; [DOI] [PubMed] [Google Scholar]; Evans DT, Serra-Moreno R, Singh RK, Guatelli JC, Trends Microbiol 2010, 18, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD, J Virol 2009, 83, 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jones PH, Maric M, Madison MN, Maury W, Roller RJ, Okeoma CM, Virology 2013, 438, 37; [DOI] [PMC free article] [PubMed] [Google Scholar]; Mahauad-Fernandez WD, Jones PH, Okeoma CM, J Gen Virol 2014, 95, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SP, Bavari S, J Virol 2010, 84, 10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J, J Virol 2009, 83, 2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K, J Virol 2009, 83, 9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT, J Virol 2010, 84, 12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dafa-Berger A, Kuzmina A, Fassler M, Yitzhak-Asraf H, Shemer-Avni Y, Taube R, Virology 2012, 428, 98. [DOI] [PubMed] [Google Scholar]
- [31].Bruce EA, Abbink TE, Wise HM, Rollason R, Galao RP, Banting G, Neil SJ, Digard P, J Gen Virol 2012, 93, 963. [DOI] [PubMed] [Google Scholar]
- [32].Watanabe R, Leser GP, Lamb RA, Virology 2011, 417, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yondola MA, Fernandes F, Belicha-Villanueva A, Uccelini M, Gao Q, Carter C, Palese P, J Virol 2011, 85, 2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hu S, Yin L, Mei S, Li J, Xu F, Sun H, Liu X, Cen S, Liang C, Li A, Guo F, Biochem J 2017, 474, 715. [DOI] [PubMed] [Google Scholar]
- [35].Ohta K, Goto H, Yumine N, Nishio M, J Gen Virol 2016, 97, 561. [DOI] [PubMed] [Google Scholar]
- [36].Wang SM, Huang KJ, Wang CT, Virology 2014, 449, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Taylor JK, Coleman CM, Postel S, Sisk JM, Bernbaum JG, Venkataraman T, Sundberg E, Frieman MB, J Virol 2015, 89, 11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bampi C, Rasga L, Roux L, J Gen Virol 2013, 94, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang X, Li C, Zhou L, Zhang N, Wang X, Ge X, Guo X, Yang H, Virus Res 2014, 191, 92. [DOI] [PubMed] [Google Scholar]
- [40].Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G, Traffic 2003, 4, 694. [DOI] [PubMed] [Google Scholar]
- [41].Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, Sata T, Tokunaga K, J Biol Chem 2009, 284, 35060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD, Cell 2009, 139, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Masuyama N, Kuronita T, Tanaka R, Muto T, Hirota Y, Takigawa A, Fujita H, Aso Y, Amano J, Tanaka Y, J Biol Chem 2009, 284, 15927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Swiecki M, Scheaffer SM, Allaire M, Fremont DH, Colonna M, Brett TJ, J Biol Chem 2011, 286, 2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hinz A, Miguet N, Natrajan G, Usami Y, Yamanaka H, Renesto P, Hartlieb B, McCarthy AA, Simorre JP, Gottlinger H, Weissenhorn W, Cell Host Microbe 2010, 7, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schubert HL, Zhai Q, Sandrin V, Eckert DM, Garcia-Maya M, Saul L, Sundquist WI, Steiner RA, Hill CP, Proc Natl Acad Sci U S A 2010, 107, 17951; [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang H, Wang J, Jia X, McNatt MW, Zang T, Pan B, Meng W, Wang HW, Bieniasz PD, Xiong Y, Proc Natl Acad Sci U S A 2010, 107, 18428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hammonds J, Wang JJ, Yi H, Spearman P, PLoS Pathog 2010, 6, e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cole G, Simonetti K, Ademi I, Sharpe S, Biochemistry 2012, 51, 5033. [DOI] [PubMed] [Google Scholar]
- [49].Fitzpatrick K, Skasko M, Deerinck TJ, Crum J, Ellisman MH, Guatelli J, PLoS Pathog 2010, 6, e1000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Strauss JD, Hammonds JE, Yi H, Ding L, Spearman P, Wright ER, J Virol 2016, 90, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Venkatesh S, Bieniasz PD, PLoS Pathog 2013, 9, e1003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Habermann A, Krijnse-Locker J, Oberwinkler H, Eckhardt M, Homann S, Andrew A,Strebel K, Krausslich HG, J Virol 2010, 84, 4646; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lehmann M, Rocha S, Mangeat B, Blanchet F, Uji IH, Hofkens J, Piguet V, PLoS Pathog 2011, 7, e1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hammonds J, Ding L, Chu H, Geller K, Robbins A, Wang JJ, Yi H, Spearman P, J Virol 2012, 86, 2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Frost A, Unger VM, De Camilli P, Cell 2009, 137, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rollason R, Korolchuk V, Hamilton C, Jepson M, Banting G, J Cell Biol 2009, 184, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sauter D, Front Microbiol 2014, 5, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dube M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA, PLoS Pathog 2010, 6, e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J, J Biol Chem 2012, 287, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, Suarez M, Guatelli J, Xiong Y, Elife 2014, 3, e02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV, J Virol 2009, 83, 7931; [DOI] [PMC free article] [PubMed] [Google Scholar]; Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC, PLoS Pathog 2009, 5, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pardieu C, Vigan R, Wilson SJ, Calvi A, Zang T, Bieniasz P, Kellam P, Towers GJ, Neil SJ, PLoS Pathog 2010, 6, e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT, PLoS Pathog 2009, 5, e1000429; [DOI] [PMC free article] [PubMed] [Google Scholar]; Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F, Cell Host Microbe 2009, 6, 409; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T, Cell Host Microbe 2009, 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM, Retrovirology 2010, 7, 51; [DOI] [PMC free article] [PubMed] [Google Scholar]; Le Tortorec A, Neil SJ, J Virol 2009, 83, 11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lopez LA, Yang SJ, Hauser H, Exline CM, Haworth KG, Oldenburg J, Cannon PM, J Virol 2010, 84, 7243; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P, Proc Natl Acad Sci U S A 2009, 106, 2886; [DOI] [PMC free article] [PubMed] [Google Scholar]; Vande Burgt NH, Kaletsky RL, Bates P, Viruses 2015, 7, 5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ohta K, Matsumoto Y, Yumine N, Nishio M, Med Microbiol Immunol 2017. [DOI] [PubMed]
- [66].Cocka LJ, Bates P, PLoS Pathog 2012, 8, e1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Galao RP, Le Tortorec A, Pickering S, Kueck T, Neil SJ, Cell Host Microbe 2012, 12, 633; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J, J Virol 2013, 87, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Galao RP, Pickering S, Curnock R, Neil SJ, Cell Host Microbe 2014, 16, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sauter D, Hotter D, Engelhart S, Giehler F, Kieser A, Kubisch C, Kirchhoff F, Retrovirology 2013, 10, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Manganaro L, de Castro E, Maestre AM, Olivieri K, Garcia-Sastre A, Fernandez-Sesma A, Simon V, J Virol 2015, 89, 9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dufrasne FE, Lucchetti M, Martin A, Andre E, Dessilly G, Kabamba B, Goubau P, Ruelle J, Virology 2018, 513, 11; [DOI] [PubMed] [Google Scholar]; Rizk MG, Basler CF, Guatelli J, J Virol 2017, 91. [DOI] [PMC free article] [PubMed]
- [72].Cao W, Bover L, Cho M, Wen X, Hanabuchi S, Bao M, Rosen DB, Wang YH, Shaw JL, Du Q, Li C, Arai N, Yao Z, Lanier LL, Liu YJ, J Exp Med 2009, 206, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bego MG, Cote E, Aschman N, Mercier J, Weissenhorn W, Cohen EA, PLoS Pathog 2015, 11, e1005024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, Wu Y, Cui J, Mol Cell 2017, 68, 308. [DOI] [PubMed] [Google Scholar]
- [75].Edgar JR, Manna PT, Nishimura S, Banting G, Robinson MS, Elife 2016, 5. [DOI] [PMC free article] [PubMed]
- [76].Mi Z, Wang X, He Y, Li X, Ding J, Liu H, Zhou J, Cen S, Biopolymers 2014, 102, 280. [DOI] [PubMed] [Google Scholar]
- [77].Zhang Q, Liu Z, Mi Z, Li X, Jia P, Zhou J, Yin X, You X, Yu L, Guo F, Ma J, Liang C, Cen S, Antiviral Res 2011, 91, 321; [DOI] [PubMed] [Google Scholar]; Zhang Q, Mi Z, Huang Y, Ma L, Ding J, Wang J, Zhang Y, Chen Y, Zhou J, Guo F, Li X, Cen S, Retrovirology 2016, 13, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mi Z, Ding J, Zhang Q, Zhao J, Ma L, Yu H, Liu Z, Shan G, Li X, Zhou J, Wei T, Zhang L, Guo F, Liang C, Cen S, Sci Rep 2015, 5, 18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mahauad-Fernandez WD, Okeoma CM, Immun Inflamm Dis 2016, 4, 4; [DOI] [PMC free article] [PubMed] [Google Scholar]; Mahauad-Fernandez WD, Okeoma CM, Sci Rep 2018, 8, 4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ohta K, Matsumoto Y, Ito M, Nishio M, Med Microbiol Immunol 2017. [DOI] [PubMed]