

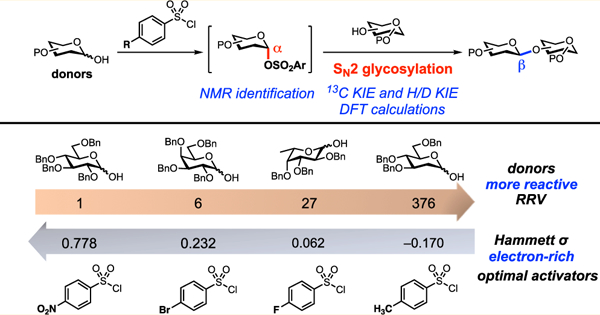
Abstract
Here we demonstrate that highly β-selective glycosylation reactions can be achieved when the electronics of a sulfonyl chloride activator and the reactivity of a glycosyl donor hemiacetal are matched. While these reactions are compatible with the acid- and base-sensitive protecting groups that are commonly used in oligosaccharide synthesis, these protecting groups are not relied upon to control selectivity. Instead, β-selectivity arises from the stereoinversion of an α-glycosyl arylsulfonate in an SN2-like mechanism. Our mechanistic proposal is supported by NMR studies, kinetic isotope effect (KIE) measurements, and DFT calculations.
Graphical Abstract
INTRODUCTION
Oligosaccharides carry out critical functions in a vast array of biochemical processes.1 However, our understanding of the molecular basis of carbohydrate function remains constrained by the challenges associated with synthesizing stereochemically pure glycosides for study. In general, the difficulty of controlling glycoside stereochemistry arises from the existence of glycosylation reactions at the border between the SN1 and SN2 mechanisms.2 Which mechanism dominates, and, consequently, what product distribution is observed, depends on complex interactions between donor, acceptor, protecting groups, and reagents.3 While many useful methods successfully leverage these interactions to generate certain glycosidic linkages in a stereoselective manner,4–25 a general solution for the construction of broad classes of linkages has yet to emerge.26 Here, we show that by matching the electronics of the leaving group to the reactivity of the glycosyl donor, it is possible to obtain SN2-type glycosylations that afford products with high β-selectivity for a range of acceptors.
Glycosylation reactions reside at the SN1–SN2 boundary because they involve nucleophilic attack on a secondary electrophile with an adjacent oxygen atom that can stabilize adjacent positive charge. In many traditional approaches to glycosylation, positive charge at C1 is generated by using a Lewis acid to promote leaving group heterolysis (Figure 1A). Although this strategy activates the donor toward nucleophilic attack, the resulting ion pair is also stereochemically labile. As a result, controlling stereoselectivity typically requires the use of specialized protecting groups to bias the approach of the nucleophile to one face of the electrophile. While the protecting group scheme can be tailored to synthesize either α- or β-glycosidic linkages, this solution greatly reduces the efficiency of oligosaccharide synthesis.
Figure 1.

(A) Classical approaches to Lewis acid-mediated glycosylation. (B) This work. LA = Lewis acid.
However, if the glycosyl donor could be activated toward nucleophilic attack without the generation of a significant amount of positive charge, then a classical SN2 process might be possible in which displacement is purely stereoinvertive (Figure 1B). In such a scenario, the product distribution would be entirely determined by the stereochemical purity of the starting material. Since SN2 reactions generally require good leaving groups, and electronegative substituents strongly favor one stereoisomer through the anomeric effect, obtaining purely α-configured starting materials is often straightforward. Thus, glycosylation reactions proceeding via an SN2 mechanism would have the potential to reduce or even eliminate the reliance on protecting groups for controlling selectivity.
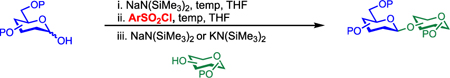
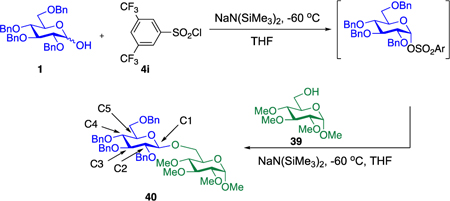
To obtain a stereoinvertive process, we examined conditions that are classically known to favor the SN2 mechanism: a donor with an excellent leaving group, an alkoxide as a strong nucleophile, and a polar aprotic solvent. We chose to use sulfonate as the leaving group based on our previous observation that 2-deoxy-pyranose α-tosylates react with acceptor alkoxides to afford β-glycosides with excellent selectivity.27–29 However, we anticipated that extending this strategy to conventional C2-substituted pyranoses might be challenging because C2-substituted sugars are more than 500 times less reactive than their 2-deoxy counterparts.30 As a result, under conditions that are sufficiently activating to render the SN2 pathway feasible, undesirable SN1 reactivity might become competitive. Indeed, when glycosyl sulfonates have previously been generated for use as donors,31–43 specialized protecting groups were required to control selectivity. For example, Srivastava and Schuerch demonstrated that a C2 sulfonate protecting group was necessary to stabilize α-mannosyl trifluoroethylsulfonates and tosylates for β-mannosylation and rhamnosylation.36,38 More recently, Crich and co-workers demonstrated the importance of the 4,6-O-benzylidene acetal in stabilizing covalent α-triflates in their β-mannosylation reaction.44–48
To balance the benefit of activation against the risk of creating stereochemical lability, we reasoned we could take advantage of the tunability of arylsufonates as leaving groups.49–51 For example, relatively unreactive donors might be expected to require more electron-poor, and presumably more activated, arylsulfonates. Conversely, for relatively reactive donors, electron-rich arylsulfonates that would be less prone to anomerization could be used. Indeed, as we show below, there is an inverse relationship between the intrinsic reactivity of the donor (as measured by Wong and coworkers’ relative reactivity values52,53) and the electronics of the optimal arylsulfonate for each glycosylation.
Furthermore, we reasoned that we could lessen the need for strong electrophilic activation of the donor by using an alkoxide as the nucleophilic acceptor.54 This prediction is consistent with previous work by the Taylor,11 Kahne,55 Yoshida,56 and Walczak19 groups, which demonstrated that activating the acceptor through either borinic acid catalysis or in situ generation of a tin ether can effect selective reactions with glycosyl sulfonate donors.
An additional advantage of our strategy is that it eliminates the need to synthesize and isolate potentially unstable species such as glycosyl halides or imidates. By generating the reactive donor in situ, through activation of the donor hemiacetal as a glycosyl sulfonate, the electrophilic donor sulfonate can react directly with the nucleophilic acceptor alkoxide. As we demonstrate below, this strategy makes it possible to engage C2-substituted sugars in SN2-like displacements, thus accessing valuable β-glycoside products (Figure 1B). Our mechanistic analysis confirms that the major products obtained in these glycosylation reactions are the result of a stereoinvertive and concerted reaction pathway.
RESULTS AND DISCUSSION
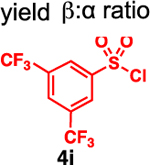
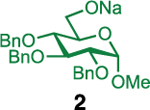
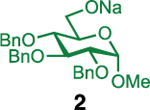
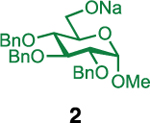
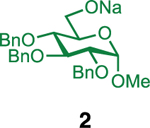
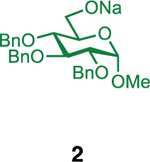
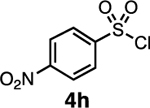
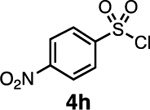
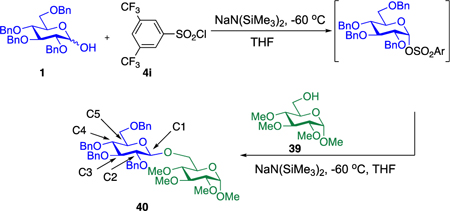
Our initial optimization efforts focused on selecting the optimal sulfonate leaving group and reaction conditions for the glycosylation between glucosyl donor 1 and acceptor 2 (Table 1). As expected, donor 1 was less reactive than its 2-deoxy-sugar analogues toward nucleophilic displacement by glucosyl acceptor 2. For example, the use of tosyl (4a) or benzenesulfonyl (4b) chloride as the promoter required an elevated reaction temperature of −15 °C (vs −78 °C for 2-deoxy-sugars).27,28 Although these conditions led to the formation of product 3 as a single β-anomer, low yields were observed (entries 1 and 2). Reasoning that the poor efficiency was due to decomposition of the putative α-sulfonate intermediate,57 we examined arenesulfonates bearing electron-donating substituents (4c–f,j–k). Once again, β-isomers were exclusively obtained, but with only somewhat improved yields (entries 3–6). Interestingly, although many arenesulfonates bearing electron-withdrawing substituents led to reduced yields (4g–h), 3,5-bis(trifluoromethyl)benzenesulfonyl chloride (4i) emerged as an effective promoter (entry 9).
Table 1.
Effect of Sulfonylating Agenta
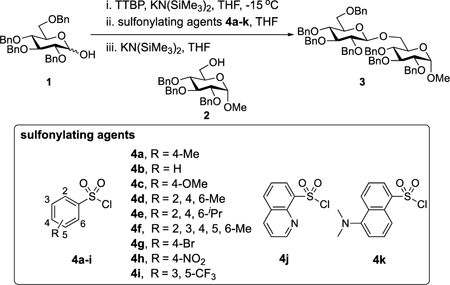 | |||
|---|---|---|---|
| entry | sulfonylating agent | yield (%)b | β/α ratioc |
| 1 | 4a | 27 | β only |
| 2 | 4b | 29 | β only |
| 3 | 4c | 29 | β only |
| 4 | 4d | 36 | β only |
| 5 | 4e | 27 | β only |
| 6 | 4f | 36 | β only |
| 7 | 4g | 29 | β only |
| 8 | 4h | 18 | β only |
| 9 | 4i | 46 | β only |
| 10 | 4j | 15 | β only |
| 11 | 4k | 17 | β only |
0.20 mmol of glucosyl donor 1, 0.13 mmol of acceptor 2, 0.20 mmol of TTBP, 0.20 mmol of sulfonylating agent, THF as the solvent, 2 h of activation time. Glycosylation [1] = 0.050 M.
Isolated yield.
All selectivities based on 1H NMR analysis of purified material (see SI). TTBP = 2,4,6-tri-tert-butylpyrimidine.
Further improvements were gained by varying the counterion, additives, and reaction conditions (Table 2). We found that sodium, rather than lithium or potassium, was the most effective alkoxide counterion (entries 1–3). Although adding the acid scavenger 2,4,6-tri-tert-butylpyrimidine (TTBP) was beneficial when the counterion was potassium, it was deleterious when the counterion was sodium (entry 1 vs entries 3 and 4). Furthermore, increasing the donor to acceptor ratio from 1.5:1 to 2:1 and decreasing the temperature to −30 °C led to the formation of the desired product 3 in 96% yield as a single β-isomer (entry 5). Under these optimized conditions, nosylate 4h was also found to be a competent promoter, affording the desired product in slightly diminished yield (85%, entry 6).
Table 2.
Optimization of Glycosylation Conditionsa
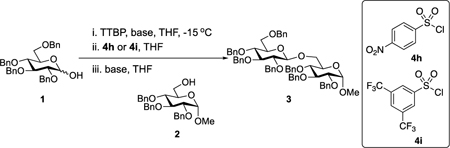 | ||||
|---|---|---|---|---|
| entry | base | sulfonylating agent | yield (%)b | β/α ratioc |
| 1 | KN(SiMe3)2 | 4i | 46 | β only |
| 2 | LiN(SiMe3)2 | 4i | NR | NR |
| 3 | NaN(SiMe3)2 | 4i | 69 | β only |
| 4d | NaN(SiMe3)2 | 4i | 81 | β only |
| 5d,e | NaN(SiMe3)2 | 4i | 96 | β only |
| 6d, e | NaN(SiMe3)2 | 4h | 85 | β only |
0.20 mmol of glucosyl donor 1, 0.13 mmol of acceptor 2, 0.20 mmol of TTBP, 0.20 mmol of sulfonylating agent, THF as the solvent, 2 h of activation time. Glycosylation was run at −15 °C. Glycosylation [1] = 0.050 M.
Isolated yield.
All selectivities based on 1H NMR analysis of purified material (see SI).
Without adding TTBP.
0.20 mmol of 1, 0.1 mmol of acceptor 2, glycosylation [1] = 0.059 M. Glycosylation was run at −30 °C. TTBP = 2,4,6-tri-tert-butylpyrimidine. NR = no reaction.
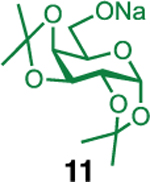
We next examined the scope of the reaction promoted by both 4h and 4i (Table 3). Although both reagents promoted reactions with acetonide-protected acceptor 11, 4h provided the desired product with higher β-selectivity (entry 2). The reaction also tolerated a variety of common protecting groups, including 2-naphthylmethyl (Nap), benzoate (Bz), acetate (Ac), and triisopropylsilyl (TIPS) ether (entries 3–7). However, the position of the benzoate ester protecting group did impact the observed yield. For example, C4-benzoates gave consistently higher yields than C3-benzoates (entries 4 vs 5), with this effect being more pronounced with 4h than 4i. Nonetheless, promoter 4i was still able to activate the C3-benzoate 7 for glycosylation to give a synthetically useful yield.
Table 3.
Scope of the Reaction between Glucosyl Donor and Acceptors to Afford β-Linked Saccharidesa
 | |||||
|---|---|---|---|---|---|
| entry | donor | acceptor | β-linked disaccharides | 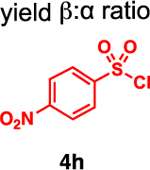 |
 |
| 1 | 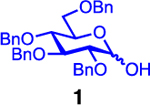 |
 |
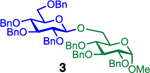 |
85%, β only | 95%, β only |
| 2 | 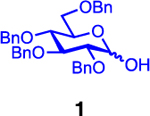 |
 |
 |
71%, 17:1b | 92%, 10:1b |
| 3 | 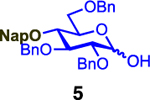 |
 |
 |
54%, β only | 69%, β only |
| 4 |  |
 |
 |
82%, β only | 90%, β only |
| 5 | 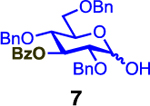 |
 |
 |
33%, β only | 65%, β only |
| 6 |  |
 |
 |
72%, β only | 83%, β only |
| 7 |  |
 |
 |
52%, β only | 57%, β only |
| 8 |  |
 |
 |
60%, 14:1b | 58%, 14:1b,c |
| 9 |  |
 |
 |
55%, 11:1b | 63%, 10:1b |
| 10 |  |
 |
 |
48%, 12:1b,d | 54%, 12:1b,c |
| 11 |  |
 |
 |
45%, β onlyb,d | 42%, β onlyb,d |
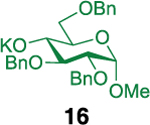
| 12 | 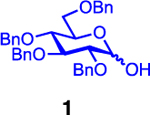 |
 |
 |
38%, 16:1b | 40%, 6:1b,c |
| 13 | 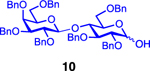 |
 |
 |
71%, β only | 90%, β only |
Reaction was run at −30 °C. The donor to acceptor ratio is 2:1. Isolated yield. All selectivities based on 1H NMR analysis of purified material (see SI).
The donor to acceptor ratio is 3:1.
Reaction was run at −40 °C.
Reaction was run at −15 °C.
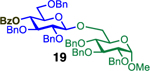
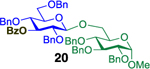
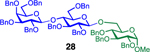
Glycosylation of the hindered secondary glycosyl acceptors 12–15 also proved to be feasible, and the corresponding disaccharides 23–26 were isolated in 42–63% yields. The selectivities ranged from 10:1 β:α to exclusively β regardless of whether 4h or 4i was used as the promoter (Table 3, entries 8–11). In contrast, reactions with fewer nucleophilic acceptors were more sensitive to the electronics of the sulfonyl chloride. For example, the union of hindered acceptor 16 with donor 1 to afford disaccharide 27 gave a much higher selectivity with 4i than 4h (entry 12). This latter result demonstrates the importance of having ready access to a collection of promoters. In addition, lactosyl donor 10 was also a competent donor in the reaction and behaved similarly to the glucosyl donors (entry 13).
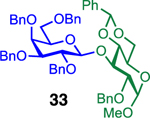
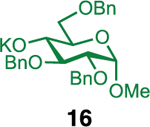
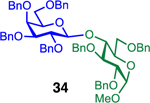
We next examined galactosyl donors, which are approximately 6 times more reactive than their glucosyl counterparts (Table 4).52,53 With both promoters 4h and 4i, the primary acceptor 2 reacted smoothly with 29 to afford disaccharide 31 in good yields and stereoselectivities. Acceptor 11, which bears acetonides, afforded product 32 in good to high yields and excellent stereoselectivities. Again, with hindered acceptors, we observed a greater dependence of selectivity on the identity of the promoter. For example, in the presence of 4i, the hindered secondary acceptor 12 reacted with 29 to afford 33 in high yield and modest selectivity. By switching to nosylate 4h as the promoter, however, we were able to obtain 33 in good yield and selectivity (73% yield, 11:1 β:α). As with the corresponding glucosyl donor, acceptor 16 reacted with galactosyl donor 29 to afford product 34, albeit with attenuated selectivity. The β-selective glycosylation of 4,6-O-benzylidene-protected galactopyranosyl donor 30 is note-worthy because this system is intrinsically biased toward α-products. For example, the triflate of 30 reacts with nucleophiles to afford products in moderate to high levels of α-selectivity.44,45 In contrast, under the current conditions, the β-anomer was the major product.
Table 4.
Examining the Scope of the Reaction between Galatosyl Donor and Acceptors to Afford β-Linked Disaccharidesa
 | ||||||
|---|---|---|---|---|---|---|
| entry | donor | acceptor | β-linked disaccharides | promoter | yield | β:α ratio |
| 1 | 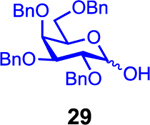 |
 |
 |
4h | 62% | 12:1 |
| 4i | 85% | 10:1 | ||||
| 2 |  |
 |
 |
4h | 58%b | β only |
| 4i | 92%c | β only | ||||
| 3 |  |
 |
 |
4h | 73% | 11:1 |
| 4i | 91%c | 5:1 | ||||
| 4 |  |
 |
 |
4h | 67% | 6:1 |
| 4i | 71%c | 5:1 | ||||
| 4g | 51%c,d | 11:1 | ||||
| 5 | 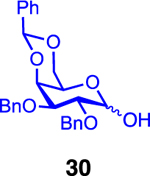 |
 |
 |
4h | 44% | 9.5:1 |
| 4i | 64% | 6:1 | ||||
Reaction was run at −30 °C. The donor to acceptor ratio is 3:1. Isolated yield. All selectivities based on 1H NMR analysis of purified material (see SI).
Reaction was run at −15 °C.
Reaction was run at −40 °C.
The donor to acceptor ratio is 4:1.
Reasoning that a better mechanistic understanding of the reaction would help guide us in improving the yields of unselective reactions, we turned to VT-NMR spectroscopy. Upon activating the glucosyl hemiacetal 1 with 4i in THF-d8 under conditions that were otherwise identical to those employed in the synthetic reaction (SI section 4.2), we observed a single anomeric doublet corresponding to the α-sulfonate (1H NMR 6.28 ppm, J = 3.3 Hz; 13C NMR 100.3 ppm).38,58 Similarly, the analogous reaction using 4h afforded a single α-linked glycosyl sulfonate (1H NMR 6.18 ppm (d, J = 3.2 Hz); 13C NMR 100.0 ppm). This selectivity is not surprising, given the established tendency of anomeric alkoxides to react with electrophiles at low temperature to form α-anomers,59 and the propensity for glycosyl sulfonates to adopt an α-configuration.34,36
While the nosylate derived from 4h is stable to room temperature, the sulfonate obtained from the reaction with 4i decomposes above 0 °C (SI section 4.2). This relative stability fits with the observation that 4h provides higher selectivity than 4i in reactions with hindered acceptors (e.g., Table 3, entry 12). This led us to consider that it should be possible to improve less selective reactions, such as the one between galactose donor 29 and acceptor 16 through the use of a less reactive sulfonate. Indeed, the selectivity of this reaction could be improved to 11:1 β:α using the 4-bromobenzenesulfonyl chloride promoter 4g (Table 4, entry 4).
To examine further the relationship between the electronics of the sulfonyl promoter and the stereoselectivity of glycosylation, we examined fucose donor 36 in detail. Because this donor is approximately 27 times more reactive than glucose,52, 53 it provides an opportunity to study a system in which the corresponding sulfonates are relatively unstable. As before, when donor 36 was reacted with primary acceptor 2, we obtained the product as a single β-isomer, regardless of which sulfonate was used. Interestingly, higher yields were obtained with less reactive sulfonates (Table 5, entries 1–3). However, when the more hindered acceptor 12 was used, only modest levels of selectivities were observed with 4g–4i, and 4l. To improve this yield, we next examined the relatively electron-rich promoter 4m (Hammett σp = 0.06). As predicted, the use of this promoter improved the selectivity of the reaction (11:1 β:α), further demonstrating the impact of sulfonate electronics on the stereochemical outcome of the reaction.
Table 5.
Scope of the Reaction between Fucosyl Donor and Acceptors to Afford β-Linked Disaccharidesa
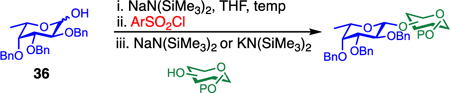 | ||||
|---|---|---|---|---|
| entry | disaccharide | promoter | yield | β:α ratio |
| 1 |  |
 |
45% | β only |
| 2 | 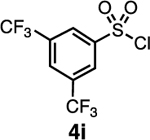 |
54% | β only | |
| 3 | 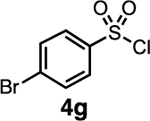 |
60% | β only | |
| 4 |  |
 |
45% | 4:1 |
| 5 | 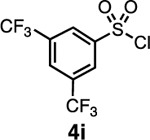 |
59% | 3:1 | |
| 6 |  |
42% | 3:1 | |
| 7 |  |
41% | 6:1 | |
| 8 |  |
34% | 11:1 | |
Isolated yield. All selectivities based on 1H NMR analysis of crude material.
Although these experiments established that these reactions proceed via the quantitative generation of a glycosyl sulfonate intermediate, further studies were required to understand the mechanism of the reaction. One possibility is that of a classical SN2 process: concerted and stereospecific inversion of the sulfonate by the acceptor alkoxide, with the development of relatively little positive charge in the transition state. Alternatively, a stereoinvertive SN1 process might occur: initial ionization of the sulfonate to form a contact ion pair, followed by highly stereoselective nucleophilic attack from the face opposite the leaving group. In this latter case, the intermediate would be a formal oxocarbenium ion, and the rate-determining transition structure would be expected to bear a significant degree of positive charge at C1.
To determine where this reaction lies along the SN2–SN1 continuum, we measured the 12C/13C kinetic isotope effects (KIEs) with respect to the donor. In an SN2 reaction, the transition state is expected to be relatively symmetric, and thus, the predicted isotope effect at C1 would be large (>1.02). In a typical SN1 reaction, where formation of the high-energy oxocarbenium ion is both rate-limiting and isotope-determining, a very late transition state is expected, and the predicted isotope effect at C1 would be small (∼1.00). This near-unity isotope effect reflects the balancing of two opposing effects: the loss of vibrational energy from leaving group heterolysis (a normal effect) and the gain of vibrational energy from hyperconjugation of neighboring σ bonds into the π* system of the oxocarbenium ion (an inverse effect).60
While the KIE at C1 reflects the degree to which the nucleophile is associated with the transition state, the KIEs at C2 and C5 reflect the degree of charge buildup in the donor ring. In an SN2 reaction, the loss of negative charge caused by leaving group departure is balanced by the gain of negative charge caused by nucleophilic attack. Accordingly, at sites removed from the reactive center (C1), the bonding is relatively unchanged, and the predicted isotope effects are small (1.007 for C2 and 1.006 for C5). In an SN1 reaction, the buildup of positive charge at C1 weakens the bonding at adjacent sites through hyperconjugation. Thus, the expected isotope effects at C2 and C5 are normal (∼1.02 at both sites). Together, the KIEs at C1, C2, and C5 are highly diagnostic of where any given glycosylation reaction lies on the SN2–SN1 mechanistic continuum (Figure 2).
Figure 2.

SN1–SN2 continuum in glycosylations. The KIE at C1 measures how early or late the transition state is, while the KIEs at C2 and C5 measure how much positive charge is present
To measure the required 12C/13C KIEs at natural abundance, we employed our previously reported DEPT methodology.61 Two glycosylation reactions using donor 1 and acceptor 39 were carried to 22% and 23% conversion at −60 °C, with 39 as the limiting reagent.62 To determine the isotopic fractionation in these partial conversion samples, the area of the peak of interest was divided by the area of a reference peak remote from the reaction center (C3). This isotopic ratio compared to the corresponding ratio in the samples fully converted to 40.
Optimizing the DEPT parameters as previously reported61 for the methines of the donor gave a tip angle of 60.25° and a magnetization transfer delay of 3.357 ms (effective 1JCH = 148.9 Hz). We also found that the addition of 0.5 mM Cr(acac)3 to the NMR samples appreciably reduced the T1 relaxation times of the methine protons, without significantly increasing T2 relaxation.63,64 This strategy allowed many more scans to be taken and increased the precision of the measurement. We recommend that concentrations of 0.5–2 mM Cr(acac)3 be used in all future applications of the DEPT methodology.
The measured KIEs are shown in Table 6. The KIE at C1 of 1.034 is relatively large for a glycosylation reaction and is consistent with an SN2 mechanism. For example, Crich et al. obtained a KIE of 1.023 in an SN2-like β-mannosylation reaction,65 while Chan, Bennet, and co-workers reported a primary 13C KIE of 1.024 for the classical SN2 reaction between a glycosyl fluoride and an azide ion.66 Similarly, the concerted enzymatic hydrolysis of methyl β-glucopyranoside gives KIEs of 1.026–1.032.67–69 The KIE at C1 is also much larger than the KIEs that are predicted for an SN1 reaction (1.00–1.01, see DFT calculations below). Conversely, the KIEs at C2 and C5 are much smaller than would be expected for an SN1 reaction (1.02 for both sites, regardless of DFT method).
Table 6.
Measured Glycosylation KIEsa
 | |||
|---|---|---|---|
| position | KIE measurement 1 | KIE measurement 2 | average KIE |
| C1 | 1.032 | 1.035 | 1.034 |
| C2 | 1.004 | 1.005 | 1.005 |
| C4 | 1.001 | 1.002 | 1.002 |
| C5 | 0.997 | 0.998 | 0.998 |
12C/13C isotopic fractionations were determined via DEPT relative to C3. The estimated standard error in these KIEs is 0.004 at all positions. 1H/2H isotopic fractionations were measured over 3 trials by 1H NMR. The 1H/2H KIE values at the anomeric position were 1.168, 1.154, and 1.160.
Further support for an SN2-like mechanism comes from secondary H/D KIE measurements at C1 of 1. Here, we measured an average secondary KIE value of 1.16 at −60 °C. This value is similar to other secondary KIE values measured for SN2-like glycosylations, such as the results reported by Crich (1.12)70,71 and Jacobsen (1.12–1.16).14 The canonical interpretation of these KIEs is that they reflect the stiffness of the out-of-plane bending mode and this stiffness is diagnostic of concertedness.
In an SN2 reaction, the hybridization at C1 remains approximately sp3 as nucleophile–C1 bonding largely replaces C1–leaving group bonding as the reaction progresses. As a result, the out-of-plane mode weakens only somewhat, and the isotope effect is expected to be small. In an SN1 reaction, C1 becomes sp2-hybridized in the oxocarbenium ion. This substantially weakens the out-of-plane mode, and the isotope effect is expected to be large.
However, glycosylation reactions naturally lie at the boundary between the SN2 and SN1 mechanisms, making clear interpretations challenging.72,73 For example, loose but concerted displacements are also expected to give large H/D isotope effects.74 Furthermore, H/D isotope effects are much more challenging to predict quantitatively through computational methods, in part due to the substantially increased role of tunneling.75 Here, the SN2 glycosylation is roughly predicted to give H/D isotope effects of 1.3–1.5 compared to 1.5–1.7 for the SN1 process (the ranges reflect conformational effects). Thus, although the experimental H/D KIE of 1.16 qualitatively supports the interpretation of an SN2 process, a definitive interpretation requires computational analysis of the 12C/13C KIEs, which are far more amenable to quantitative prediction.
Interestingly, while the 12C/13C KIE at C1 is relatively large for a glycosylation reaction, this KIE is actually much smaller than what is observed for many simple SN2 displacements at aliphatic centers (often 1.07 or larger near room temperature).76 To understand this discrepancy, and to gain atomistic-level insight into the reaction, we turned to density functional theory (DFT) calculations. To develop a realistic model of the reaction capable of capturing its many possible degrees of freedom, while maintaining good accuracy, we chose the standard method B3LYP-D3(BJ)/6–31G*/PCM. (Further analysis indicates that many other standard methods would have been acceptable; see SI section 6 for details.) Additionally, the benzyl protecting groups on the donor were simplified to methyl groups, while the sodium counterion was explicitly solvated with one dimethyl ether ligand. A comprehensive search over the conformational, donor–acceptor, and solvent degrees of freedom found 106 distinct transition states. These transition states could be clustered into three classes (Figure 3) in which the sodium counterion was (a) bound to the alkoxide nucleophile (34 structures), (b) bridging the alkoxide and the sulfonate leaving group (46 structures), or (c) bound only to the sulfonate (26 structures).
Figure 3.

Computed glycosylation transition states (B3LYP-D3(BJ)/6–31G*/PCM(THF) at −60 °C). Lowest-energy transition structures with the sodium ion: (a) bound to the alkoxide (red), (b) bridging the alkoxide and the sulfonate (green), and (c) bound to the sulfonate (blue). The sodium ion is purple. A dimethyl ether is bound to the sodium. (d) The 106 transition states found spanned a wide range of energies and geometries. (The lowest-energy transition states depicted in parts a–c are circled.) (e) Most bridging (type b) transition states gave KIE predictions at C1 that were within experimental error (highlighted), while all type a and c structures were inconsistent with experiment.
While these structures spanned a range of energies (Figure 3d) and geometries (Figure 3e), the predicted isotope effects within each class were relatively consistent. Interestingly, type a (sodium bound to alkoxide) and type c (sodium bound to sulfonate) transition states gave predicted KIEs at C1 that were too high (1.06–1.08) vs experiment (1.034). In contrast, type b (bridging) transition states gave KIEs that were very close to experiment (Table 7). Specifically, the predicted KIEs at C1 for 16 of the 24 type b structures were found to be within experimental error (highlighted points in Figure 3e).
Table 7.
Predicted vs Experimental Isotope Effectsa
 | |||||
|---|---|---|---|---|---|
| SN2 KIEs |
|||||
| SN1 EIEs | type a | type b | type c | expt | |
| C1 | 1.001 | 1.082 | 1.036 | 1.066 | 1.034 |
| C2 | 1.020 | 1.004 | 1.007 | 1.001 | 1.005 |
| C3 | 1.007 | 1.001 | 1.002 | 0.999 | 1.000b |
| C4 | 1.005 | 1.001 | 1.002 | 1.000 | 1.002 |
| C5 | 1.018 | 1.001 | 1.006 | 1.002 | 0.998 |
Only the type b SN2 transition states gave KIE predictions that were in good agreement with experiment. Predictions for the lowest-energy representative of each class are shown. These predictions include a Bell tunneling correction.
The KIE at C3 position is assumed to be 1.000.
The type b transition states were asynchronous, with a longer forming bond distance of 2.49 Å and a shorter breaking bond distance of 2.16 Å in the lowest-energy structure (Figure 3b). Type b transition states were also relatively central when compared to the type a and c transition states, with longer breaking bond distances, but similar forming bond distances. Although more central transition states might be expected to give larger isotope effects, the bridging sodium ion enforces a nonlinear nucleophile–C1–leaving group angle of 138°. Additionally, the donor oxygen–C1–nucleophile angle is 117°, and the geometry at C1 is planar. These geometric features are similar to those of nucleophilic additions to carbonyl groups, and the modest oxocarbenium character is reflected in the slightly normal predicted isotope effects at C2 and C5. Overall, it appears that the sodium may both coordinate the alkoxide and activate the sulfonate for displacement.
The large number of transition states discovered here offers a unique opportunity to examine the technique of using constrained transition states.77,78 In the unconstrained transition state approach employed above, each transition structure represents a true first-order saddle point on the potential energy surface. However, it is often the case that none of the located unconstrained structures satisfactorily reproduces the observed isotope effects. These discrepancies can be due to deficiencies in the electronic structure method, solvation protocol, or perhaps the operation of an unknown mechanism.
In the constrained transition state approach, the focus shifts from using energetic criteria to locate transition structures to finding nonstationary geometries that best reproduce experimental isotope effects. In an explicit “grid” approach, the transition structure is defined as a function of a small number of geometric parameters, such as the forming and breaking bond distances in a nucleophilic substitution. (This approach has been employed in several glycosylation studies.66,79,80) After fixing these distances at regular intervals on a predefined grid, all other geometric parameters are allowed to relax, and the isotope effects at each grid point are calculated. The “experimental” transition state can then be derived explicitly, by taking the grid point that most closely matches the experiment. Alternatively, an implicit “regression” approach might be used in which the geometric parameters of a number of known unconstrained transition states are used as features to predict the isotope effects. The experimental transition state can then be found by optimizing the parameters of the linear model.
Both approaches were tested here. To test the explicit approach, we conducted a retrospective analysis in which we imagined that only the type a transition states were known. Since none of these transition states give isotope effect predictions that are consistent with experiment, one might attempt to identify the experimental transition structure by adjusting the geometric parameters of the available structures to match the observed KIEs. To determine whether such a strategy could conceivably be successful, we assumed that the lowest-energy type b transition state is the experimental transition state. Then, we took the lowest-energy type a structure as a template and set the forming and breaking bond distances to those of the lowest-energy type b transition state. The predicted isotope effects for this constrained type a transition state did not agree with experiment (see SI section 6.3 for details). For example, the predicted KIE at C1 for the constrained type a structure is 1.065, in stark contrast to the predicted KIE of 1.036 for the unconstrained type b structure that the constrained structure is intended to mimic. The reverse process is similarly unsuccessful: constraining the lowest-energy type b structure to the forming and breaking bond distances of the lowest type a structure gives a predicted KIE at C1 of 1.059 vs the unconstrained KIE of 1.082 for unconstrained type a. Therefore, in the event that only type a (or type b) transition states were to be available, the explicitly constrained transition state would not identify the correct transition state geometry.
The finding that constrained transition states do not give the same predicted KIEs as their unconstrained counterparts, even when their key geometric parameters are the same, requires that the KIE be dependent on other factors. This inference is confirmed by a multiple linear regression analysis. The predicted isotope effect at C1 for the unconstrained type a transition states is described well by a four-parameter model that incorporates an intercept, the forming bond distance, the breaking bond distance, and the imaginary frequency (adjusted R2 = 0.95, RMS prediction error = 0.002). However, when this model was used to predict the isotope effects for the unconstrained type b structures, the KIE at C1 was significantly overpredicted by 0.01–0.02 units.
In hindsight, it is not surprising that the dependence of the KIE on geometric parameters can change significantly when the mechanism changes. The results presented here demonstrate that even subtle changes in mechanism, such as changes to the solvation sphere, are sufficient to cause the constrained transition state to give erroneous results. Therefore, constrained transition state approaches should be applied with caution in the future, particularly in glycosylation reactions.
Furthermore, this analysis highlights the power of an unconstrained transition state approach when many degrees of freedom are possible and can be explored with reasonable coverage. In addition to rationalizing the observed KIEs, the set of experimentally consistent transition states defines the precision of the experimental transition state. Specifically, the 19 highlighted points in Figure 3e define a relatively central, but asynchronous, transition state in which the forming bond is approximately 2.5 ± 0.1 Å long, the breaking bond is 2.2 ± 0.2 Å long, and the sodium ion bridges the acceptor and sulfonate oxygens.
CONCLUSION
Our study demonstrates that by matching the intrinsic reactivity of a glycosyl donor with the electronics of the sulfonate leaving group (Figure 4) it is possible to obtain highly β-selective glycosylation reactions without tailored protecting group schemes. Depending on the nature of the acceptor, achieving highly efficient β-glycosylation in this SN2 manifold can require a balance between the reactivities of the donor and arylsulfonyl activator. When relatively reactive primary acceptors are employed, the desired SN2 pathway outcompetes all others, and high levels of β-selectivity result, regardless of arylsulfonate substitution. However, when less reactive secondary acceptors are used, the selectivity between the desired SN2 and undesired SN1 pathways depends on both the reactivity and stability of the α-sulfonate intermediate. With more reactive donors, more stabilized, electron-rich arylsulfonates give more selective reactions.
Figure 4.

More reactive donors require more electron-rich sulfonyl chloride activators. RRV = relative reactivity value.52,53
Our mechanistic analysis confirms that the glycosylation reactions reported here proceed via the quantitative generation of an α-glycosyl arylsulfonate, which is then stereoinvertively displaced by a sodium alkoxide. By using a good leaving group and a strong nucleophile, an SN2-like process is favored. Kinetic isotope studies and DFT calculations indicate a concerted but asynchronous process in which only a small amount of charge develops at C1 in the transition state. Furthermore, the sodium counterion bridges the alkoxide and sulfonate oxygens in the transition state. Thus, Lewis acid activation of the sodium for displacement occurs, but only in the desired SN2 pathway, leading to a favorable trade-off between reactivity and selectivity.
This mechanistic analysis relied on the use of DEPT methodology to measure multiple KIEs simultaneously and at natural abundance as well as the use of unconstrained transition state calculations to rationalize the observed KIEs. In general, the KIE at C1 indicates whether a given glycosylation proceeds via an associative or dissociative mechanism, while the KIEs at C2 and C5 reflect the degree of positive charge development. We anticipate that this strategy will prove useful for studying other glycosylation reactions in the future.
Overall, we have shown that highly β-selective glycosylations in an SN2 manifold are feasible when the electronics of the leaving group are matched to the reactivity of the glycosyl donor. By systematically correlating the reactivity of the glycosyl donor, its leaving group, and the stereochemical outcome, we have identified conditions that favor clean and efficient stereoinversion. We hope that the insights about reactivity and selectivity gained here will prove useful for the rational design of next-generation glycosylation methodology.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the NIH for financial support (grant U01-GM120414).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07022.
Experimental details, computational analysis, and characterization data (PDF)
All computational files have been deposited at https://github.com/ekwan/bennett_glycosylation
The authors declare no competing financial interest.
REFERENCES
- (1).Essentials of Glycobiology, 3rd ed.; Varki A, Cummings RD, Esko JD, Stanley P, Hart GH, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH, Eds.; Cold Spring Harbor Laboratory Press, 2015. [PubMed] [Google Scholar]
- (2).Adero PO; Amarasekara H; Wen P; Bohé L; Crich D The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hagen B; van der Vorm S; Hansen T; van der Marel GA; Codée JDC Stereoselective Glycosylations–Additions to Oxocarbenium Ions In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett C, Ed.; Wiley-VCH: Weinheim, 2017; pp 3–26. [Google Scholar]
- (4).Mensah EA; Nguyen HM Nickel-Catalyzed Stereoselective Formation of α-2-Deoxy-2-Amino Glycosides. J. Am. Chem. Soc 2009, 131, 8778–8780. [DOI] [PubMed] [Google Scholar]
- (5).Cox DJ; Smith MD; Fairbanks AJ Glycosylation Catalyzed by a Chiral Brønsted Acid. Org. Lett 2010, 12, 1452–1455. [DOI] [PubMed] [Google Scholar]
- (6).Lu S-R; Lai Y-H; Chen J-H; Liu C-Y; Mong K-KT Dimethylformamide: An Unusual Glycosylation Modulator. Angew. Chem., Int. Ed 2011, 50, 7315–7320. [DOI] [PubMed] [Google Scholar]
- (7).Kimura T; Sekine M; Takahashi D; Toshima K Chiral Brønsted Acid Mediated Glycosylation with Recognition of Alcohol Chirality. Angew. Chem., Int. Ed 2013, 52, 12131–12134. [DOI] [PubMed] [Google Scholar]
- (8).Padungros P; Alberch L; Wei A Glycosyl Dithiocarbamates: β-Selective Couplings without Auxiliary Groups. J. Org. Chem 2014, 79, 2611–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Peng P; Schmidt RR An Alternative Reaction Course in O-Glycosidation with O-Glycosyl Trichloroacetimidates as Glycosyl Donors and Lewis Acidic Metal Salts as Catalyst: Acid-Base Catalysis with Gold Chloride-Glycosyl Acceptor Adducts. J. Am. Chem. Soc 2015, 137, 12653–12659. [DOI] [PubMed] [Google Scholar]
- (10).Kimura T; Eto T; Takahashi D; Toshima K Stereo-controlled Photoinduced Glycosylation Using an Aryl Thiourea as an Organo photoacid. Org. Lett 2016, 18, 3190–3193. [DOI] [PubMed] [Google Scholar]
- (11).D’Angelo KA; Taylor MS Borinic Acid Catalyzed Stereo-and Regioselective Couplings of Glycosyl Methanesulfonates. J. Am. Chem. Soc 2016, 138, 11058–11066. [DOI] [PubMed] [Google Scholar]
- (12).Sun L; Wu X; Xiong D-C; Ye X-S Stereoselective Koenigs-Knorr Glycosylation Catalyzed by Urea. Angew. Chem., Int. Ed 2016, 55, 8041–8044. [DOI] [PubMed] [Google Scholar]
- (13).Palo-Nieto C; Sau A; Galan MC Gold(I)-Catalyzed Direct Stereoselective Synthesis of Deoxyglycosides from Glycals. J. Am. Chem. Soc 2017, 139, 14041–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Macrocyclic bis-Thioureas Catalyze Stereospecific Glycosylation Reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lee J; Borovika A; Khomutnyk Y; Nagorny P Chiral Phosphoric Acid-Catalyzed Desymmertizative Glycosylation of 2-Deoxystreptamine and its Application to Aminoglycoside Synthesis. Chem. Commun 2017, 53, 8976–8979. [DOI] [PubMed] [Google Scholar]
- (16).Liu J-L; Zhang Y-T; Liu H-F; Zhou L; Chen J N_Heterocyclic Carbene Catalyzed Stereoselective Glycosylation of 2-Nitrogalactals. Org. Lett 2017, 19, 5272–5275. [DOI] [PubMed] [Google Scholar]
- (17).Singh Y; Wang T; Geringer SA; Stine KJ; Demchenko AV Regenerative Glycosylation. J. Org. Chem 2018, 83, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tanaka M; Nakagawa A; Nishi N; Iijima K; Sawa R; Takahashi D; Toshima K Boronic-Acid-Catalyzed Regioselective and 1,2-cis-Stereoselective Glycosylation of Unprotected Sugar Acceptors via SNi-Type Mechanism. J. Am. Chem. Soc 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- (19).Yang T; Zhu F; Walczak MA Stereoselective Oxidative Glycosylation of Anomeric Nucleophiles with Alcohols and Carboxylic Acids. Nat. Commun 2018, 9, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Levi SM; Li Q; Roötheli AR; Jacobsen EN Catalytic Activation of Glycosyl Phosphates for Stereoselective Coupling Reactions. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Meng L; Wu P; Fang J; Xiao Y; Xiao X; Tu G; Ma X; Teng S; Zeng J; Wan Q Glycosylation Enabled by Successive Rhodium(II) and Brønsted Acid Catalysis. J. Am. Chem. Soc 2019, 141, 11775. [DOI] [PubMed] [Google Scholar]
- (22).Zeng J; Wang R; Zhang S; Fang J; Liu S; Sun G; Xu B; Xiao Y; Fu D; Zhang W; Hu Y; Wan Q Hydrogen-Bonding-Assisted Exogenous Nucleophilic Reagent Effect for β-Selective Glycosylation of Rare 3-Amino Sugars. J. Am. Chem. Soc 2019, 141, 8509–8515. [DOI] [PubMed] [Google Scholar]
- (23).Hoang KM; Lees NR; Herzon SB Programmable Synthesis of 2-Deoxyglycosides. J. Am. Chem. Soc 2019, 141, 8098–8103. [DOI] [PubMed] [Google Scholar]
- (24).Hu Z; Tang Y; Yu B Glycosylation with 3,5-Dimethyl-4-(2′-phenylethynylphenyl)-phenyl (EPP) Glycosides via a Dearomative Activation Mechanism. J. Am. Chem. Soc 2019, 141, 4806–4810. [DOI] [PubMed] [Google Scholar]
- (25).Yu F; Li J; DeMent PM; Tu Y-J; Schlegel HB; Nguyen HM Phenanthroline-Catalyzed Stereoretentive Glycosylations. Angew. Chem., Int. Ed 2019, 58, 6957–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).National Research Council. Transforming Glycoscience: A Roadmap for the Future; The National Academies Press: Washington, DC, 2012; pp 86–93. [PubMed] [Google Scholar]
- (27).Issa JP; Lloyd D; Steliotes E; Bennett CS Reagent Controlled β-Specific Dehydrative Glycosylation Reactions with 2-Deoxy-Sugars. Org. Lett 2013, 15, 4170–4173. [DOI] [PubMed] [Google Scholar]
- (28).Issa JP; Bennett CS A Reagent-Controlled SN2-Glycosylation for the Direct Synthesis of β-Linked 2-Deoxy-Sugars. J. Am. Chem. Soc 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- (29).Lloyd D; Bennett CS An Improved Approach to the Direct Construction of 2-Deoxy-β-Linked Sugars: Applications to Oligosaccharide Synthesis. Chem. - Eur. J 2018, 24, 7610–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen J-H; Ruei J-H; Mong K-KT Iterative α-Glycosylation Strategy for 2-Deoxy- and 2,6-Dideoxysugars: Application to the One-Pot Synthesis of Deoxysugar-Containing Oligosaccharides. Eur. J. Org. Chem 2014, 2014, 1827–1831. [Google Scholar]
- (31).Eby R; Schuerch C The Use of 1-O-Tosyl-D-Glucopyranose Derivatives in α-D-Glucoside Synthesis. Carbohydr. Res 1974, 34, 79–90. [Google Scholar]
- (32).Koto S; Hamada Y; Zen S Direct Glucosidation of 2,3,4,6-Tetra-O-Benzyl-α-D-Glucopyranose. Chem. Lett 1975, 4, 587–588. [Google Scholar]
- (33).Lucas TJ; Schuerch C Methanolysis as a Model Reaction for Oligosaccharide Synthesis of Some 6-Substituted 2,3,4-Tri-O-Benzyl-D-Galactopyranosyl Derivatives. Carbohydr. Res 1975, 39, 39–45. [Google Scholar]
- (34).Leroux J; Perlin AS A New Synthesis of Glycosides. Reactions of Trifluoromethanesulfonic Anhydride at the Anomeric Centre. Carbohydr. Res 1976, 47, C8–C–10. [Google Scholar]
- (35).Maroussek V; Lucas TJ; Wheat PE; Schuerch C The Influence of Reactant Structure and Solvent on Galactoside Syntheses from Galactosyl Sulfonates. Carbohydr. Res 1978, 60, 85–96. [Google Scholar]
- (36).Srivastava VK; Schuerch C A Synthesis of β-D-Mannosides by Glycosidation at C-1. Carbohydr. Res 1980, 79, C13–C16. [Google Scholar]
- (37).Koto S; Sato T; Morishima N; Zen S The Glucosylation of Several Alcohols with Tetra-O-Benzyl-α-D-Glucopyranose and a Mixture of p-Nitrobenzenesulfonyl Chloride, Silver Trifluoromethanesulfonate, and Triethylamine. Bull. Chem. Soc. Jpn 1980, 53, 1761–1762. [Google Scholar]
- (38).Srivastava VK; Schuerch C Synthesis of β-D-Mannopyranosides and β-L-Rhamnopyranosides by Glycosidation at C-1. J. Org. Chem 1981, 46, 1121–1126. [Google Scholar]
- (39).Szeja W; Bogusiak J Synthesis of S-Glycosyl N,N-Diethyldithiocarbamates from Protected, Reducing Monosaccharides Under Phase-Transfer Conditions. Synthesis 1988, 1988, 224–225. [Google Scholar]
- (40).Szeja W A Convenient Synthesis of α-D-Glucopyranosides. Synthesis 1988, 1988, 223. [Google Scholar]
- (41).Crich D; Sun S Direct Formation of β-Mannopyranosides and Other Hindered Glycosides from Thioglycosides. J. Am. Chem. Soc 1998, 120, 435–436. [Google Scholar]
- (42).Crich D; Smith M 1-Benzenesulfinyl Piperidine/Trifluoromethanesulfonic Anhydride: A Potent Combination of Shelf-Stable Reagents for the Low-Temperature Conversion of Thioglycosides to Glycosyl Triflates and for the Formation of Diverse Glycosidic Linkages. J. Am. Chem. Soc 2001, 123, 9015–9020. [DOI] [PubMed] [Google Scholar]
- (43).Boebel TA; Gin DY Probing the Mechanism of Sulfoxide-Catalyzed Hemiacetal Activation in Dehydrative Glycosylation. J. Org. Chem 2005, 70, 5818–5826. [DOI] [PubMed] [Google Scholar]
- (44).Crich D; de la Mora M; Vinod AU Influence of the 4,6-O-Benzylidene, 4,6-O-Phenylboronate, and 4,6-O-Polystyrylboronate Protecting Groups on the Stereochemical Outcome of Thioglycoside-Based Glycosylations Mediated by 1-Benzenesulfinyl Piperidine/ Triflic Anhydride and N-Iodosuccinimide/Trimethylsilyl Triflate. J. Org. Chem 2003, 68, 8142–8148. [DOI] [PubMed] [Google Scholar]
- (45).Crich D; Vinogradova O On the Influence of the C2-O2 and C3-O3 Bonds in 4,6-O-Benzylidene-Directed β-Mannopyranosylation and α-Glucopyranosylation. J. Org. Chem 2006, 71, 8473–8480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Crich D; Jayalath P 2-O-Propargyl Ethers: Readily Cleavable, Minimally Intrusive Protecting Groups for β-Mannosyl Donors. Org. Lett 2005, 7, 2277–2280. [DOI] [PubMed] [Google Scholar]
- (47).Crich D; Jayalath P; Hutton TK Enhanced Diastereoselectivity in β-Mannopyranosylation through the Use of Sterically Minimal Protecting Groups. J. Org. Chem 2006, 71, 3064–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Crich D; Karatholuvhu MS Application of the 4-Trifluoromethylbenzenepropargyl Ether Group as an Unhindered, Electron Deficient Protecting Group for Stereoselective Glycosylation. J. Org. Chem 2008, 73, 5173–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Paleos CM; Varveri FS; Gregoriou GA Arenesulfonate Leaving Groups Less Reactive than the p-Toluenesulfonate Group. J. Org. Chem 1974, 39, 3594–3595. [Google Scholar]
- (50).Crossland RK; Wells WE; Shiner VJ Jr. Sulfonate Leaving Groups, Structure and Reactivity. 2,2,2-Trifluoroethanesulfonate. J. Am. Chem. Soc 1971, 93, 4217–4219. [Google Scholar]
- (51).Guthrie RD; Thang S Selective Sulfonylating Reagents. Aust. J. Chem 1987, 40, 2133–2136. [Google Scholar]
- (52).Zhang Z; Ollmann IR; Ye X-S; Wischnat R; Baasov T; Wong C-H Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc 1999, 121, 734–753. [Google Scholar]
- (53).Cheng C-W; Zhou Y; Pan W-H; Dey S; Wu C-Y; Hsu W-L; Wong C-H Hierarchical and programmable one-pot synthesis of oligosaccharides. Nat. Commun 2018, 9, 5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Phan TB; Mayr H Comparison of the Nucleophilicities of Alcohols and Alkoxides. Can. J. Chem 2005, 83, 1554–1560. [Google Scholar]
- (55).Kim SH; Augeri D; Yang D; Kahne D Concise Synthesis of the Calichemamicin Oligosaccharide Using the Sulfoxide Glycosylation Method. J. Am. Chem. Soc 1994, 116, 1766–1775. [Google Scholar]
- (56).Yamago S; Yamada T; Nishimura R; Ito H; Mino Y; Yoshida J.-i. A new Method for the Synthesis of Stannyl Ethers by Acid-Catalyzed Reaction of Alcohols with Allyltributylstannane. Chem. Lett 2002, 31, 152–152. [Google Scholar]
- (57).A reviewer suggested that elimination may be the major byproduct in the reaction; however, we were unable to detect this even upon treatment of the activated sulfonate donor with potassium t-butoxide See Supporting Information.
- (58).Resonances for the corresponding β-sulfonate, which are expected to appear at 5.5 ppm, were not observed See ref 31.
- (59).Morris WJ; Shair MD Stereoselective Synthesis of 2-Deoxy-β-glycosides Using Anomeric O-Alkylation/Arylation. Org. Lett 2009, 11, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Berti PJ; McCann JAB Toward a Detailed Understanding of Base Excision Repair Enzymes: Transition State and Mechanistic Analyses of N-Glycoside Hydrolysis and N-Glycoside Transfer. Chem. Rev 2006, 106, 506–555. [DOI] [PubMed] [Google Scholar]
- (61).Kwan EE; Park Y; Besser HA; Anderson TL; Jacobsen EN Sensitive and Accurate 13C Kinetic Isotope Effect Measurements Enabled by Polarization Transfer. J. Am. Chem. Soc 2017, 139, 43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).The stoichiometry of the donor and acceptor did not impact the stereochemical outcome of the reaction See Supporting Information.
- (63).Levy GC; Cargioli JD Spin-Lattice Relaxation in Solutions Containing Cr(III) Paramagnetic Relaxation Agents. J. Magn. Reson 1973, 10, 231–234. [Google Scholar]
- (64).Levy GC; Komoroski RA Paramagnetic Relaxation Reagents. Alternatives or Complements to Lanthanide Shift Reagents in Nuclear Magnetic Resonance Spectral Analysis. J. Am. Chem. Soc 1974, 96, 678–681. [Google Scholar]
- (65).Huang M; Garrett GE; Birlirakis N; Bohé L; Pratt DA; Crich D Dissecting the Mechanisms of a Class of Chemical Glycosylation Reactions Using Primary 13C Kinetic Isotope Effects. Nat. Chem 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chan J; Sannikova N; Tang A; Bennet AJ Transition-State Structure for the Quintessential SN2 Reaction of a Carbohydrate: Reaction of α-Glucopyranosyl Fluoride with Azide in Water. J. Am. Chem. Soc 2014, 136, 12225–12228. [DOI] [PubMed] [Google Scholar]
- (67).Zhang Y; Bommuswamy J; Sinnott ML Kinetic Isotope Effect Study of Transition States for the Hydrolysis of α- and β-Glucopyranosyl Fluorides. J. Am. Chem. Soc 1994, 116, 7557–7563. [Google Scholar]
- (68).Lee JK; Bain AD; Berti PJ Probing the Transition States of Four Glucoside Hydrolyses with 13C Kinetic Isotope Effects Measured at Natural Abundance by NMR Spectroscopy. J. Am. Chem. Soc 2004, 126, 3769–3776. [DOI] [PubMed] [Google Scholar]
- (69).Speciale G; Farren-Dai M; Shidmoossavee FE; Williams SJ; Bennet AJ C2-Oxyanion Neighboring Group Participation: Transition State Structure for the Hydroxide-Promoted Hydrolysis of 4-Nitrophenyl α-D-Mannopyranoside. J. Am. Chem. Soc 2016, 138, 14012–14019. [DOI] [PubMed] [Google Scholar]
- (70).Crich D; Chandrasekera NS Mechanism of 4,6-O-Benzylidene-Directed β-Mannosylation as Determined by α-Deuterium Kinetic Isotpe Effects. Angew. Chem., Int. Ed 2004, 43, 5386–5389. [DOI] [PubMed] [Google Scholar]
- (71).Crich and co-workers measured H/D KIE values of 1.16–1.2 at 195.15 K and corrected these to an average of 1.12 at room temperature by assuming that T ln(KH/Kd) was constant (see ref 63). Using a similar correction for our values, which were measured at 213.15 K, we can obtain an average KIE value of 1.11 at room temperature. We opted not to use this treatment as it assumes the isotope effect is purely enthalpic.
- (72).Berti PJ; Tanaka KSE Transition state analysis using multiple kinetic isotope effects: mechanisms of enzymatic and non-enzymatic glycoside hydrolysis and transfer. Adv. Phys. Org. Chem 2002, 37, 239–314. [Google Scholar]
- (73).Westaway KC Using kinetic isotope effects to determine the structure of the transition states of SN2 reactions. Adv. Phys. Org. Chem 2006, 41, 217–273. [Google Scholar]
- (74).Craze G-A; Kirby AJ; Osborne R Bimolecular substitution on an acetal. J. Chem. Soc., Perkin Trans 2 1978, 357–368. [Google Scholar]
- (75).Meyer MP; DelMonte AJ; Singleton DA Reinvestigation of the Isotope Effects for the Claisen and Aromatic Claisen Rearrangements: the Nature of the Claisen Transition States. J. Am. Chem. Soc 1999, 121, 10865–10874. [Google Scholar]
- (76).Matsson O; Dybala-Defratyka A; Rostkowski M; Paneth P; Westaway KC A theoretical investigation of a-carbon kinetic isotope effects and their relationship to the transition-state structure of SN2 reactions. J. Org. Chem 2005, 70, 4022–4027. [DOI] [PubMed] [Google Scholar]
- (77).Schramm VL Enzymatic transition states and drug design. Chem. Rev 2018, 118, 11194–11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Hirschi JS; Takeya T; Hang C; Singleton DA Transition state geometry measurements from 13C isotope effects: the experimental transition state for the epoxidation of alkenes with oxaziridines. J. Am. Chem. Soc 2009, 131, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Tanaka M; Nakagawa A; Nishi N; Iijima K; Sawa R; Takahashi D; Toshima K Boronic-acid-catalyzed regioselective and 1,2-cis-stereoselective glycosylation of unprotected sugar acceptors via SNi-type mechanism. J. Am. Chem. Soc 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- (80).Lee JK; Bain AD; Berti PJ Probing the transition states of four glucoside hydrolyses with 13C kinetic isotope effects measured at natural abundance by NMR spectroscopy. J. Am. Chem. Soc 2004, 126, 3769–3776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.