

Abstract
Activation of primary afferent nociceptors produces acute, short-lived pain, and tissue or nerve injury induces long-term enhancement of nociceptive processing, manifested as hypersensitivity to thermal and mechanical stimulation. Here we used a chemical-genetic and pharmacological approach to study the contribution of the receptor tyrosine kinase (TrkB) to the generation and maintenance of injury-induced persistent pain. We performed the studies in wild type mice and transgenic (TrkBF616A) mice that express mutant, but fully functional TrkB receptors. By injecting a small molecule derivative of the protein kinase inhibitor, protein phosphatase 1 (1NM-PP1), it is possible to produce highly selective inhibition of TrkB autophosphorylation in adult mice, without interfering with the activity of other protein kinases. We report that oral administration of 1NM-PP1, at doses that blocked phosphorylation of TrkB in the spinal cord, had no effect in behavioral tests of acute heat, mechanical or chemical pain sensitivity. However, the same pretreatment with 1NM-PP1 prevented the development of tissue- or nerve injury-induced heat and mechanical hypersensitivity. Established hypersensitivity was transiently reversed by intraperitoneal injection of 1NM-PP1. Although interfering with TrkB signaling altered neither acute capsaicin nor formalin-induced pain behavior, the prolonged mechanical hypersensitivity produced by these chemical injuries was prevented by 1NM-PP1 inhibition of TrkB signaling. We conclude that TrkB signaling is not only an important contributor to the induction of heat and mechanical hypersensitivity produced by tissue or nerve injury, but also to the persistence of the pain.
Keywords: BDNF, central sensitization, Inflammation, Neuropathic pain, Neurotrophin, Spinal cord
Introduction
That brain-derived neurotrophic factor (BDNF) contributes to the processing of nociceptive messages was originally suggested by a dramatic alteration in the levels of BDNF expression in the spinal cord following tissue or nerve injury (Merighi et al, 2008b; Michael et al, 1997; Pezet et al, 2002; Cho et al, 1998; Ernfors et al, 1993; Fukuoka et al, 2001; Michael et al, 1997; Zhou et al, 1999). BDNF levels increased in peptidergic nociceptors, and there is de novo expression in cell bodies of large diameter afferents (Cho et al, 1998; Mannion et al, 1999; Michael et al, 1999). Experimental manipulation of either BDNF or TrkB, the receptor through which BDNF signals, also significantly alters nociceptive processing. For example, genetic deletion of BDNF from NaV1.8-expressing nociceptors decreased inflammation-induced thermal hyperalgesia (Zhao et al, 2006). Surprisingly, acute thermal pain responsiveness increased. In contrast, intrathecal administration of antisense directed against either BDNF or TrkB mRNA decreased both acute heat pain and inflammation-associated thermal hyperalgesia (Groth and Aanonsen, 2002), as did reduction of BDNF via intrathecal administration of anti-BDNF antibodies or by sequestration of BDNF, via intrathecal TrkB-IgG (Mannion et al, 1999; Yajima et al, 2002).
With respect to the processing of mechanical pain, some studies reported no reduction of the mechanical hypersensitivity induced by partial sciatic nerve ligation (Zhao et al, 2006), and no change in capsaicin-induced secondary mechanical hyperalgesia after sequestration of BDNF (Mannion et al, 1999). Other studies, however, found that sequestration of BDNF (Marcol et al, 2007) or siRNA-mediated reduction of microglial BDNF synthesis significantly reduced nerve injury-induced mechanical allodynia (Coull et al, 2005). In contrast, intrathecal injection of BDNF reduced the hyperalgesia produced by partial nerve injury (Lever et al, 2003). Finally, although Kerr et al (1999) found little effect of reducing BDNF levels, TrkB-IgG sequestration of BDNF decreased the hypersensitivity produced by repeated tactile stimulation in the setting of inflammation (Mannion et al, 1999).
These differences may reflect the locus and/or the magnitude of the BDNF reduction produced by the various approaches. The genetic deletion experiments (Zhao et al, 2006) indicate that peptidergic afferents are the source of BDNF that is critical to heat pain processing. By contrast, the locus of action after intrathecal administration cannot be unequivocally established. BNDF levels in primary afferents, postsynaptic neurons and/or microglia could be relevant. Most importantly, as TrkB is not the only route through which BDNF signals (Fan et al, 2008; Huang and Reichardt, 2003), studies directed at the selective contribution of TrkB signaling are required.
Here we used a transgenic mouse (TrkBF616A) that expresses a mutated TrkB (Chen et al, 2005). By administering an antagonist, 1NM-PP1, that selectively binds to the mutated receptor, it is possible to produce a profound and reversible blockade of TrkB autophosphorylation (Chen et al, 2005; Kaneko et al, 2008; Wang et al, 2003). Using this integrated chemical-genetic approach we provide evidence that TrkB signaling is critical for both the induction and maintenance of the heat and mechanical hypersensitivity produced by tissue or nerve injury.
Materials and Methods
Generation of TrkBF616A mice
TrkBF616A knock-in mice were generated as described (Chen et al, 2005). Heterozygous TrkBF616A mice were used to generate homozygous TrkBF616A mice. Wild type littermates served as controls.
Chemical synthesis of 1NM-PP1 and drug delivery
1NM-PP1 was synthesized as described previously (Wang et al, 2003) and was administered in the drinking water (5.0 μM 1NM-PP1). For systemic administration, we made an intraperitoneal injection of 5.0 μM 1NM-PP1 in saline (i.p., 10 μl/g; Wang et al, 2003).
Western immunoblots
Primary antibodies: Anti-pan Trk (C-14) polyclonal antibody (sc-139) and anti-phospho-Trk (E6) monoclonal antibody (sc-8058) were from Santa Cruz Biotechnology. Anti-TrkB monoclonal antibody was from BD Biosciences (# 610102) and anti-TrkA polyclonal from Upstate (#06-574). Secondary antibodies: Horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies were from Jackson Immunoresearch Laboratories.
Immunoprecipitation and Western blots
Spinal cords were removed quickly by hydraulic extrusion with saline containing 1.0 mM sodium orthovanadate and flash-frozen in liquid nitrogen. Cords were then homogenized in 500μl RIPA buffer (1% NP-40, 1% deoxycholic acid, 0.1% SDS, 20 mM HEPES pH7.4, 100 mM NaCl, 1mM EGTA, 1mM sodium orthovanadate, 50mM NaF, 1.0 mM PMSF, 20 mM b-glycerophosphate and Complete EDTA-free protease inhibitor cocktail from Roche) and incubated for 30 min on ice. All subsequent steps were carried out at 4 °C. Lysates were centrifuged at 16,000 × g for 15 min and equal quantities of the supernatants were incubated with TrkA antibody for 4h followed by Protein A/G agarose for 4h to pre-clear the samples of TrkA. Preliminary experiments showed that TrkA was completely removed in the first round of immunoprecipitation. The supernatant was then incubated with pan-Trk antibody overnight followed by addition of Protein A/G agarose for 4h. The resultant immunoprecipitates were washed 3× with RIPA, 1× with TBS containing 1.0 mM PMSF + 1.0 mM sodium orthovanadate and extracted by boiling in 30μl of SDS sample buffer. Proteins were resolved by SDS-PAGE, transferred to PVDF membranes and probed with anti-phospho Trk antibodies overnight. SuperSignal West-femto substrate (Pierce) was used to detect phospho-Trk by ECL. Blots were then stripped and re-probed with anti-TrkBF616A antibodies. Densitometric analysis of bands was performed using NIH Image 1.62 software. To correct for variations in loading, the data are expressed as percentage phospho-Trk of total TrkBF616A detected in each lane.
Behavior
Experiments were performed following the guidelines of the International Association for the Study of Pain and with approval of the UCSF Institutional Animal Care and Use Committee. All experiments were performed blind to the genotype of the mice. Mice of both sexes (20-35 g) were housed in a 12 hr light/dark cycle at 21 °C. Comparisons were made between TrkBF616A mice and their wild type littermates. Complete details of most of the pain models used (von Frey testing of mechanical sensitivity, Hargreaves radiant heat test of thermal sensitivity, formalin test, Complete Freund's adjuvant induced chronic inflammation and sciatic nerve injury (SNI) model of neuropathic pain) can be found in Shields et al (2007). Methods for testing post formalin mechanical hypersensitivity can be found in Zeitz et al (2004). An individual blind to genotype performed all behavioral experiments.
Capsaicin-induced licking and post-injection mechanical hyperalgesia
Mice were injected with 3.0 μg capsaicin in 10 μl (10% ethanol; 10% Tween; 80% saline) into the ankle. Thermal and mechanical sensitivity were tested as above. Post-capsaicin induced mechanical hypersensitivity was tested as for post-formalin hypersensitivity (Zeitz et al, 2004).
Statistical analyses
All data are presented as mean ± SEM. Data were analyzed using two- or one-way repeated measures ANOVA with Tukey's post hoc analysis. P<0.05 was regarded as significant.
Results
Inhibition of TrkB signaling does not alter mechanical or thermal nociceptive thresholds
Previous studies reported that PP1 derivatives, including 1NM-PP1, potently and selectively inhibited mutant TrkBF616A receptors. The IC50 for 1NM-PP1 inhibition of TrkB autophosphorylation was ∼3.0 nM. Importantly, there was no inhibition of wild type TrkA or TrkB with drug concentration as high as 10 μM and even at these high doses, there are no adverse side effects. Here we measured baseline thermal and mechanical nociceptive thresholds in wild type and TrkBF616A knock-in mice, before and after allowing the mice free access to drinking water containing 5.0 μM 1NM-PP1. We found no significant change of mechanical (von Frey test) or thermal nociceptive thresholds (Hargreaves test) in either wild type or TrkBF616A knock-in mice (data not shown). We conclude that TrkB phosphorylation is not required for acute nociceptive processing.
Inhibition of TrkB signaling prevents capsaicin-induced mechanical hyperalgesia
Although many in vitro studies established that 1NM-PP1 completely prevents TrkB phosphorylation, there is much less evidence that effective blockade could be produced in vivo. Here we validated the approach in a model of chemically-evoked persistent pain, using oral doses previously reported to block phosphorylation in vivo. In these studies, we used an algogen, capsaicin, that maximally stimulates nociceptors that express TRPV1. In the mouse, the great majority of TRPV1-expressing nociceptors express BDNF and the neuropeptides, substance P and calcitonin-gene related peptide. Capsaicin-induced activation of TRPV1-expressing nociceptors not only evokes pain behavior (shaking and licking of the injected hindpaw), but also produces a profound secondary hyperalgesia, viz., increased mechanical sensitivity outside of the area of injury (Sun et al, 2006; Walker et al, 2007). The hyperalgesia is presumed to arise from a central sensitization of spinal cord dorsal horn circuits secondary to the intense afferent discharge induced by capsaicin. By administering 1NM-PP1 in the drinking water, we could test the hypothesis that the central sensitization requires TrkB phosphorylation. To confirm that there was inhibition of TrkB phosphorylation, we collected lumbar spinal cords from a parallel group of mice treated under the same conditions. Finally, to confirm that TrkB phosphorylation occurs normally in the mice, we also studied capsaicin-treated wt and TrkBF616A knock-in mice that did not receive 1NM-PP1.
Consistent with the lack of effect of blocking TrkB signaling on acute heat or mechanical thresholds, we found no effect of 1NM-PP1 on the magnitude of the pain behavior produced by capsaicin. Figure 1A further illustrates that injection of capsaicin (3.0 μg/10μl) into the ankle of either wild type mice or transgenic mice that did not receive 1NM-PP1, induced a dramatic secondary mechanical hyperalgesia, manifested as a lowering of the mechanical pain threshold (tested on the plantar surface of the hindpaw). In addition, when we tested TrkBF616A knock-in mice treated with 5.0 μM 1NM-PP1 in the drinking water for 2-3 days prior to the injection of capsaicin, we found no change of the mechanical threshold (tested at 15, 30 or 60 minutes post capsaicin). The capsaicin treatment resulted in an expected profound increase in thermal response latencies, and this was found in both control and 1NM-PP1-treated mice (Fig 1 B). The reduced responsiveness to noxious heat presumably results either from desensitization of the TRPV1 channel, and thus cross desensitization to heat, or it may have occurred secondary to TRPV1-mediated destruction of the peripheral terminals of these afferents.
Figure 1. Inhibition of TrkB signaling by 1NM-PP1 in TrkBF616A mice prevents capsaicin-induced mechanical hyperalgesia and inhibits TrkB phosphorylation.
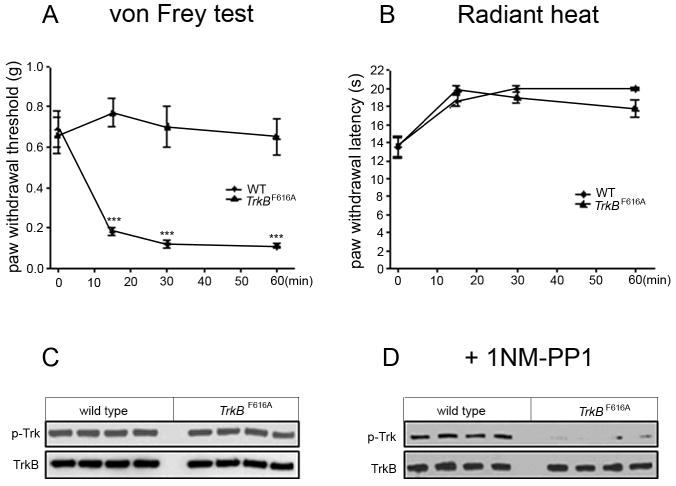
A. Changes in mechanical threshold were measured 15, 30 and 60 min after injection of capsaicin (3.0 μg/10 μl, subcutaneous) into the left ankle. Capsaicin produced a rapid drop of mechanical threshold in wt mice (n=5), but not in TrkBF616A mice, (n=5) that drank water containing 1NM-PP1 (final concentration 5.0μM); *** p<0.001.
B. Paw withdrawal latency to noxious heat did not differ between wt and TrkBF616A mice.
C, D. Western blots of lumbar spinal cords from 4 wt and 4 TrkBF616A mice show that Trk phosphorylation after capsaicin treatment in the absence of 1NM-PP1 (C) in the drinking water did not differ. Total TrkB loaded for wt and mutant mice is equivalent. By contrast, in the presence of 1NM-PP1 (D), there is a significant decrease of Trk phosphorylation in the TrkBF616A mice. (See text for quantification).
Binding of BDNF to TrkB leads to dimerization of the receptor and its auto-phosphorylation on tyrosine residues (Lindsay, 1996). To determine whether oral 1NM-PP1 inhibits TrkBF616A phosphorylation in the capsaicin model, we repeated the capsaicin experiment, in this case, under pentobarbital anesthesia. Five minutes after ankle injection of capsaicin, lumbar spinal cords were collected and flash-frozen. Trk receptors were immunoprecipitated (IP) and the relative amounts of phospho-Trk and total TrkBF616A were assessed.
Western blots showed that capsaicin-induced TrkB phosphorylation in the wild-type and TrkBF616A mice was comparable in the absence of 1NM-PP1 (Fig. 1C; PhosphoTrk/Total TrkB: 63.3% +/- 1.8 WT vs 67.0%+/-1.7 TrkBF616A). By contrast, blots of lumbar cords showed a marked increase of TrkB phosphorylation in the wild-type mice, but not in the TrkBF616A mice that received 1NM-PP1 (Figs. 1D; PhosphoTrk/Total TrkB: 47.3%+/-3.7 WT vs 5.1%+/-4.9 TrkBF616A). Importantly, the total amount of TrkB loaded into the control and 1NM-PP1 lanes was comparable (Fig 1 C, D). Based on these results, we conclude that 1NM-PP1 significantly inhibited capsaicin-induced phosphorylation of TrkB. The concurrent loss of capsaicin-induced secondary mechanical hyperalgesia suggests that TrkB signaling underlies the sensitization that is necessary for the development of the mechanical hypersensitivity.
Inhibition of TrkB signaling prevents and can reverse CFA-inflammation associated mechanical and thermal hyperalgesia
With the ability to regulate TrkB signaling in the adult, we next asked whether TrkB phosphorylation is an important contributor in other models of injury-induced persistent pain. Hindpaw injection of CFA produces a profound mechanical and thermal hyperalgesia that peaks around 3 days after its injection and persists for many days. In these studies, we allowed the mice to drink water containing 5.0 μM 1NM-PP1 for 2 days before making an intraplantar injection of CFA into the left hindpaw. Figure 2 shows that CFA induced mechanical and heat hyperalgesia were both significantly reduced (prevented) in the TrkBF616A mice treated with 1NM-PP1. Importantly, there was no effect on paw withdrawal thresholds contralateral to the CFA injection, and no effect on the hypersensitivity induced in wild type mice treated with 1NM-PP1.
Figure 2. Inhibition of TrkB signaling prevents CFA-induced mechanical and heat hyperalgesia.
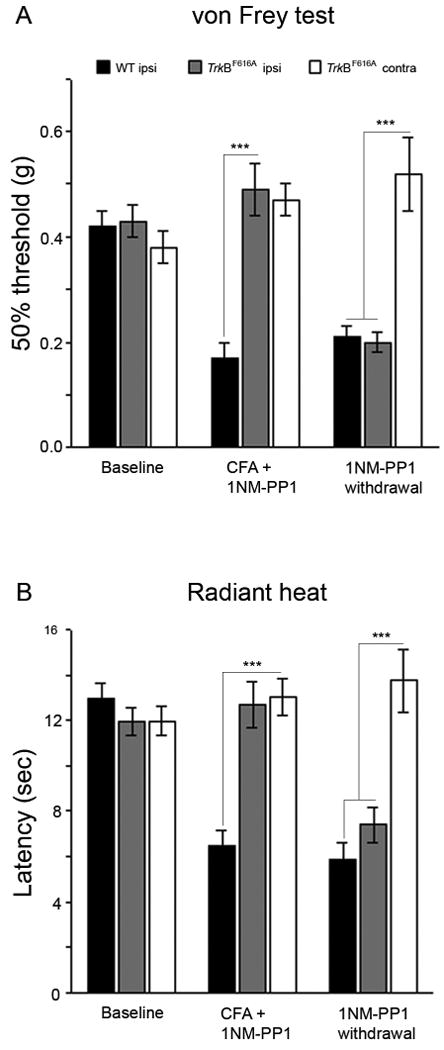
1NM-PP1 was added to the drinking water (final concentration 5.0μM) and mice had unlimited access to the water for 2-3 days before and for 2 d after injection of CFA into the hindpaw. Baseline mechanical (A) and heat (B; latency) withdrawal thresholds of the hindpaw did not differ in wt (n=5) and TrkBF616A (n=5) mice. Two days after CFA, there was a significant drop in mechanical (A) and heat thresholds (B) in wt mice (black bars), compared to the thresholds in TrkBF616A mice (gray bars) or in the hindpaw contralateral to the CFA-injected paw of TrkBF616A mice (white bars; *** p<0.001). Two days after the 1NM-PP1 was withdrawn from the drinking water, the mechanical and heat thresholds of the CFA-injected hindpaw dropped significantly, to that observed in wt mice and significantly different from the threshold of the contralateral paw of the TrkBF616A mice (*** p<0.001).
To study the reversibility of the 1NM-PP1 effects, we next asked whether switching the mice to normal drinking water would reinstate the CFA induced behavioral changes. Two days after drinking 1NM-PP1-free water, we observed that the mechanical and thermal hyperalgesia reappeared and were equivalent to the thresholds recorded in untreated TrkBF616A mice (Fig 2 A, B). These studies demonstrate that TrkB signaling contributes to the development of CFA induced hyperalgesia and suggest that there is a prolonged and possibly sustained increase of TrkB signaling as the inflammation persists.
To test the latter possibility more directly, we next asked whether blocking TrkB signaling could reverse previously established CFA-induced hyperalgesia. In these experiments we followed the protocol of Kaneko et al, (2008) who showed that an i.p injection of 1NM-PP1 transiently blocked TrkB phosphorylation. We injected CFA into the hindpaw of mice and documented the decreased heat and mechanical thresholds. Three days later we injected the mice with 1NM-PP1 (16.6ng/gm, i.p.) and measured withdrawal thresholds. Figure 3 illustrates that administration of 1NM-PP1 (i.p.) produced a significant reversal of previously established mechanical and thermal hyperalgesia. The reversal was transient; the hyperalgesia returned after 90 minutes (Fig 3 A, B). The same treatment was without effect in wild type mice. These results demonstrate that TrkB phosphorylation not only contributes to the induction of inflammation associated mechanical and thermal hyperalgesia, but also is required for its persistence. In light of this demonstration of the important contribution of TrkB signaling, it is of interest that sequestering NGF with TrkA-IgG antibodies reduced the maintenance of a non-malignant bone injury pain, but could not prevent its onset (Jimenez-Andrade et al, 2007). Taken together these results highlight the differential contribution of TrkA and TrkB signaling to the development and maintenance of inflammatory pains.
Figure 3. Inhibition of TrkB signaling can reverse established CFA-induced mechanical and heat hyperalgesia.
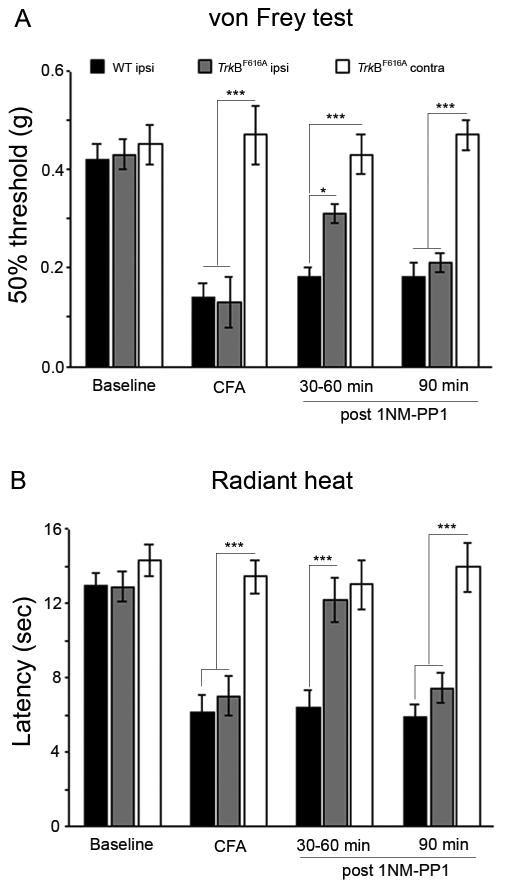
Changes in mechanical (A) and thermal threshold (B) were measured at different time points after i.p injection of 1NM-PP1 (16.6 ng/g) in CFA-treated wild type (n=5) or TrkBF616A mice (n=5). Baseline mechanical (A) and heat (B; latency) thresholds did not differ in wt and TrkBF616A mice and in both groups mechanical and heat thresholds dropped after CFA injection. However, during the first 60 min after 1NM-PP1, we observed a partial normalization of the mechanical threshold (A) and an almost complete normalization of the heat threshold B. These effects disappeared by 90m after 1NM-PP1, at which point there were no longer significant differences in mechanical or heat threshold of the CFA-injected hindpaws between wt and TrkBF616A mice, but the latter were significantly lower than the thresholds of the paw contralateral to the CFA injection in TrkBF616A mice. (*p< 0.05; *** p<0.001).
TrkB signaling is required for the induction and maintenance of peripheral nerve injury-induced mechanical hyperalgesia
Neuropathic pain represents a heightened pain sensitivity induced by peripheral nerve injury. In these conditions, normally innocuous tactile stimuli can evoke a withdrawal response, a phenomenon termed mechanical allodynia. Here we studied the effect of 1NM-PP1 on the mechanical hypersensitivity produced in the spared nerve injury (SNI) model of neuropathic pain (Shields et al, 2003). In this model, two of the three branches of the sciatic nerve are ligated, leaving the tibial nerve intact. Within 24 hours of surgery, there is a profound mechanical hypersensitivity of the partially denervated hindpaw. In these experiments, we added 1NM-PP1 to the drinking water 2 days prior to the surgical denervation and continued this treatment for 3 days after the surgery. We tested the mice 3 days after the surgery and found a profound mechanical allodynia in the control group of mice that drank the 1NM-PP1-treated water. By contrast, the magnitude of the allodynia in the TrkBF616A mice was significantly reduced. Two days after removal of the 1NM-PP1 from the drinking water, the mechanical allodynia reappeared in the TrkBF616A mice (Fig 4 A). Despite the profound mechanical allodynia observed in this model, there is limited or no change in heat thresholds. We confirmed this in the present study and further showed that 1NM-PP1 treatment did not influence the sensitivity of the nerve injury model in test of heat sensitivity (Fig 4 B).
Figure 4. Inhibition of TrkB signaling prevents nerve injury-induced mechanical hyperalgesia.
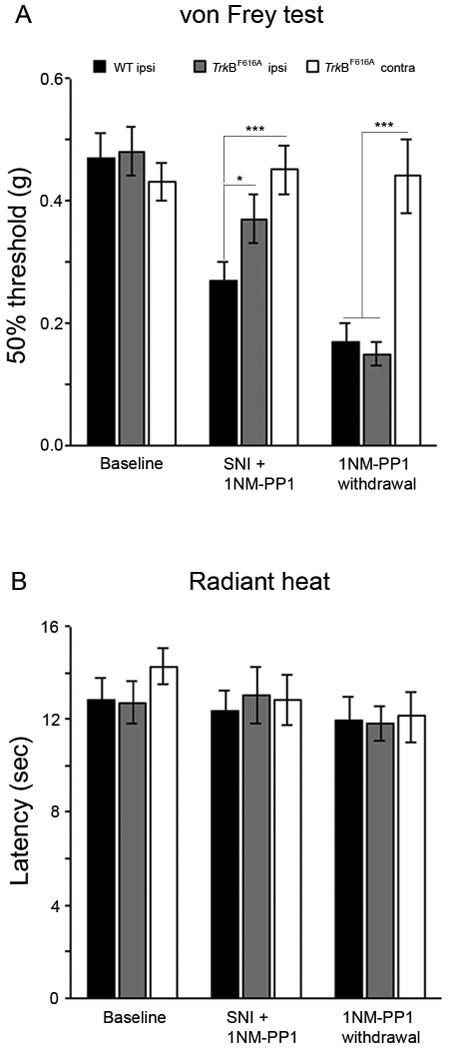
1NM-PP1 was added to the drinking water (final concentration 5.0μM) and mice had unlimited access to the water for 3 days before they underwent partial sciatic nerve injury (SNI). Baseline mechanical (A) and heat (B) thresholds prior to SNI did not differ in wt (n=5) and TrkBF616A mice (n=5). Three days after nerve injury there was a significant drop of mechanical threshold (A) in wt mice compared to TrkBF616A mice (*p< 0.05) or to the contralateral paw of the TrkBF616A mice (*** p<0.001). In neither group of mice was there a change in heat withdrawal threshold after SNI (B).
These data indicate that TrkB signaling is not only required for the induction of nerve-injury induced mechanical hypersensitivity, but also for its persistence. To provide additional evidence in support of this conclusion, we next asked whether inhibition of TrkB signaling in animals already rendered mechanically hypersensitive, could reverse the mechanical hyperalgesia. To this end, we administered 1NM-PP1 (16.6 ng/gm) 3 days after the SNI procedure and evaluated mechanical nociceptive thresholds. Figure 5A shows that post-treatment with 1NM-PP1 can partially reverse SNI-induce mechanical hyperalgesia in TrkBF616A mice. The duration of the effect of 1NM-PP1 was comparable to that observed after inflammation, i.e., the reversal of the mechanical hyperalgesia was transient (lasting no more than 90 min). As expected, we found that an i.p injection of 1NM-PP1 had no effect on heat thresholds in wild type mice or in TrkBF616A mice that did not drink water to which 1NM-PP1 was added (Fig 5 B). Taken together, these results demonstrate that TrkB phosphorylation is an important contributor, and indeed may be required, for both the induction and maintenance of nerve injury-induced mechanical hypersensitivity.
Figure 5. Inhibition of TrkB signaling can reverse already established nerve injury-induced mechanical hyperalgesia.
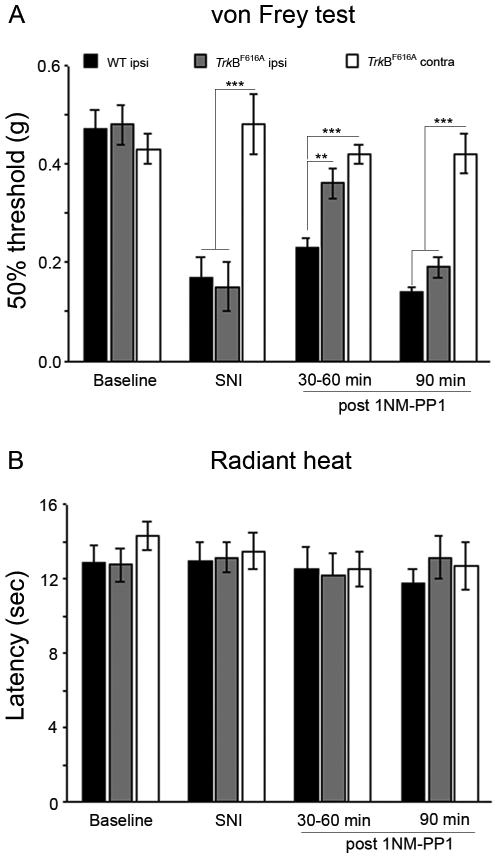
Partial sciatic nerve injury (SNI) in both wt (n=5) and TrkBF616A (n=5) produced a significant drop of the mechanical withdrawal threshold (A), compared to baseline. Injection of 1NM-PP1 (16.6 ng/gm, i.p.) 3 days after the SNI reversed the mechanical allodynia in TrkBF616A, but not in wt mice. The mechanical threshold in wt mice was significantly lower than in TrkBF616A mice after 1NM-PP1 (**p<.01) and lower than that in the uninjured contralateral paw of the TrkBF616A mice (***p<.001). The reversal lasted from 30-60, reappearing within 90 min of the 1NM-PP1 injection, at which point mechanical thresholds in wt and TrkBF616A mice did not differ and both were significantly lower than in the uninjured paw of the TrkBF616A mice, *** p<0.001. In neither group did SNI produce a change of the withdrawal threshold to noxious heat (B), and 1NM-PP1 injection was without effect in test of heat pain sensitivity.
TrkB signaling contributes to the state of hypersensitivity, but not the immediate pain behavior produced by hindpaw injection of formalin
Intraplantar injection of formalin in mice produces a stereotyped pattern of pain behavior consisting of two distinct phases (Taylor et al, 1997;Shield et al, 2007). As this model is sensitive to non-steroidal anti-inflammatory drugs and opiates, the formalin test is generally considered to be a useful model of postoperative pain. The second phase reportedly results from input processed by a spinal cord that was sensitized following the intense activity generated after the injection of formalin. Moreover, reducing BDNF levels decreased pain behavior in the second phase of the formalin test (Kerr et al, 1999; Zhao et al, 2006). Other studies questioned the importance of central sensitization to the second phase of pain behavior (Hosl et al, 2006; Taylor et al, 1997). Here we tested the effect of oral administration of 1NM-PP1 for 2 days prior to formalin injection. As illustrated in Figure 6, neither the magnitude nor the time course of the pain behavior in either phase of pain behavior in the formalin test was altered by the 1NM-PP1 pretreatment. These results suggest either that central sensitization is not required for the establishment of the second phase of pain behavior in the formalin test, or that mechanisms other than TrkB signaling are critical.
Fig 6. Inhibition of TrkB signaling does not alter formalin-evoked pain behaviors, but does prevent post-formalin mechanical hypersensitivity.
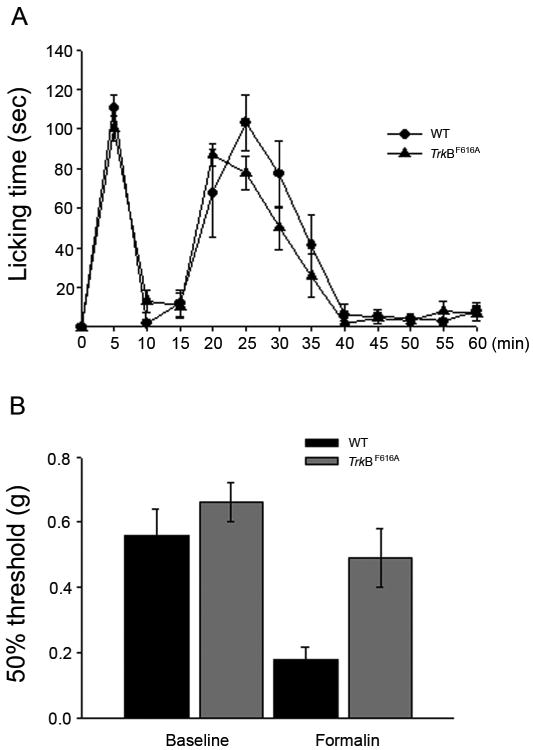
A. Wild type (n=5) and TrkBF616A (n=5) mice drank from 1NM-PP1-treated water for 2 days prior to injection of 2% formalin into the hindpaw. There was no difference in formalin-evoked licking of the hindpaw during either the first or the second phase of pain behavior. B. Baseline mechanical thresholds prior to formalin injection did not differ in wt and TrkBF616A mice. However, formalin induced a significant decrease of mechanical threshold 90 min after the formalin injection in wt, but not in TrkBF616A that were treated with 1NM-PP1, **p<0.01).
On the other hand, as we previously reported, the relatively brief duration (approximately one hour) of pain behavior produced by formalin is followed by a prolonged period of mechanical hypersensitivity. In contrast to the lack of effect of inhibiting TrkB signaling on the overt pain behavior in the formalin test, we found that oral 1NM-PP1 significantly reduced the post-formalin mechanical hypersensitivity. These results distinguish the mechanisms underlying pain behavior associated with different features of the formalin test.
Discussion
A recent comprehensive review of the contribution of BDNF to the processing of pain messages emphasized the remarkable lack of consensus in the literature (Merighi et al, 2008a). Indeed both pro and antinociceptive effects of interfering with BDNF-TrkB signaling have been reported. The disagreement likely reflects the varied methods that have been used to alter BDNF or TrkB levels, the fact that selective regulation of these molecules is difficult in the adult and the extent to which downregulation of TrkB signaling was documented. Here we took a chemical-genetic approach, one that directly targeted the phosphorylation step that is initiated when a TrkB ligand binds the receptor. We not only demonstrated profound behavioral effects of treating the TrkBF616A mice with 1NM-PP1, but also confirmed that these changes correlate with inhibition of TrkB phosphorylation. We believe that this approach provides the most direct test of the contribution of TrkB signaling to pain processing, particularly to the thermal and mechanical hypersensitivity that arises in the setting of tissue or nerve injury. Most importantly, as we could reversibly block TrkB phosphorylation in the adult, we conclude that TrkB signaling contributes both to the development of injury-induced persistent pain as well as to its maintenance.
The results were very consistent and robust. Blocking TrkB signaling in the TrkBF616A mice, by oral administration of 1NM-PP1, significantly prevented the development of tissue and nerve injury-induced persistent pain. This was true for both heat and mechanical hypersensitivity in the case of CFA induced inflammation and for the mechanical allodynia produced following partial sciatic nerve injury. By contrast, interfering with TrkB signaling did not alter acute nociceptive processing, as measured by baseline heat or mechanical thresholds, or by the pain behaviors induced by hindpaw injections of capsaicin or formalin. On the other hand, 1NM-PP1 treatment in the TrkBF616A mice prevented the mechanical hypersensitivity induced following capsaicin or formalin injection. The simplest explanation for these actions is that TrkB signaling is required for the development of the central sensitization process that underlies the development of persistent heat and mechanical hypersensitivity in the setting of tissue or injury. The fact that we found comparable effects on heat and mechanical hypersensitivity, and in both tissue and nerve injury models, suggests that a common TrkB signaling pathway contributes to the persistent pain triggered by these very different injury conditions. Our results also suggest that the contribution of BDNF to injury-induced central sensitization is via its interaction with TrkB, rather than p75, a receptor through which BDNF can also signal (Fan et al, 2008; Huang and Reichardt, 2003). Furthermore, although NT-4 also signals via TrkB, the bulk of the evidence points to BDNF, rather than NT-4, as the key TrkB ligand (Heppenstall and Lewin, 2001; Yajima et al, 2002).
Of particular interest to the clinical problem of reversing ongoing states of injury-induced persistent pain, is our finding that blocking TrkB signaling by i.p. injection of 1NM-PP1 after the hypersensitivity state was established, significantly reduced the magnitude of the hyperalgesia. The reversal was relatively brief, presumably because of the rapid metabolism of the 1NM-PP1, but the result illustrates that maintenance of the persistent pain requires long-term phosphorylation of TrkB. A similar conclusion followed from studies that implicated release of BDNF from activated microglia in the setting of peripheral nerve injury (Coull et al, 2005). Finally, we found that sustaining the blockade of CFA-induced persistent pain (hypersensitivity) also continued presence of 1NM-PP1. When we removed 1NM-PP1 from the drinking water, the mechanical and heat hypersensitivity appeared, indicating that initiation of these hyperalgesic states was delayed, and not completely prevented.
That TrkB signaling must be ongoing in order to sustain the injury induced-pain state was unexpected, but interestingly is consistent with two recent studies (Bekinschtein et al 2007; 2008) that reported that stabilization of long-term memory requires a BDNF-dependent late phase of protein synthesis. Whether the requirement for an ongoing contribution of TrkB phosphorylation to sustain persistent pain reflects continued input from the site of injury, or whether it reflects activation of a BNDF source downstream of the afferent drive (e.g. microglia, see below) remains to be determined, but it does suggest that the process of central sensitization can be reversed once established.
As we administered the 1NM-PP1 by systemic routes (either oral or i.p.) we cannot identify the particular TrkB target where phosphorylation was abrogated. We favor the view that the relevant TrkB is in the primary afferent nociceptor or in the postsynaptic neurons targeted by the nociceptor, both of which have been shown to express full-length TrkB receptor (Merighi et al, 2008a; Salio et al, 2005). It is also possible that injury-induced de novo expression of TrkB in non-nociceptive afferents (Mannion et al, 1999; Michael et al, 1999), which in turn engage pain transmission circuitry, is critical. Consistent with the view that the spinal cord or primary afferent is the locus of the relevant TrkB, the great majority of studies that implicated BNDF or TrkB in the development of persistent pain states targeted the spinal cord. Most relevant, perhaps, is that we found that capsaicin-induced secondary hyperalgesia was associated with significant TrkB phosphorylation in the lumbar spinal cord and that prevention of the mechanical hyperalgesia by 1NM-PP1 blocked the TrkB phosphorylation. Of course, systemic administration of 1NM-PP1 will concurrently interfere with the enhanced TrkB phosphorylation that occurs in the rostral ventral medulla after tissue injury, and which has also been strongly implicated in the facilitation of nociceptive processing at the level of the spinal cord (Guo et al, 2006; see also Quintao et al, 2008).
As noted above, although 1NM-PP1 blocked the mechanical hyperalgesia produced following an injection of capsaicin or formalin, the immediate pain behavior provoked by these compounds was not altered. The latter result is consistent with the conclusions that TrkB signaling is not required for acute pain responsiveness, to a variety of noxious stimuli (heat, mechanical and chemical). On the other hand, because it is generally believed that pain behavior in the second phase of the formalin test is strongly influenced by central sensitization of dorsal horn neurons following the intense burst of activity that occurs during the first phase, it was somewhat surprising that abrogation of TrkB signaling was completely without effect on pain behavior during phase 2. This result, however, is consistent with studies from our own laboratory, which argued that pain behavior during the second phase depends as much upon afferent drive that occurs during phase 2 as it does upon central sensitization produced by activity during the first phase. In our studies, we showed that selective blockade of pain behavior during phase one, with the potent, but short acting opioid agonist, remifentanil, did not reduce pain behavior in the second phase (Taylor and Basbaum, 2000; Taylor et al, 1997). The results of the present study are of particular interest in light of our finding that a mutation of CamKII completely blocked pain behavior in phase 2 of the formalin test, without influencing formalin mechanical hypersensitivity and without an effect in other persistent pain models (Zeitz et al, 2004). Taken together with the present results, we conclude that central sensitization is not a critical contributor to pain behavior in the second phase of the formalin test. The effect of CamKII modification, which can occur in nociceptors as well as in spinal cord “pain” transmission circuits, perhaps reflected disruption of afferent drive during phase 2.
In summary, using a genetic approach to selectively abrogate TrkB signaling in mice we provide evidence that signaling via TrkB is a major contributor to injury-induced heat and mechanical hypersensitivity. This is true for both tissue and nerve injury-induced persistent pain. Acute pain behavior was not altered. Although we cannot identify the particular neurons in which inhibition of TrkB phosphorylation is critical to these effects, the fact that changes produced by both tissue and nerve injury were altered argues that the locus is downstream of the primary afferent fiber. Because we found normal pain behavior in the second phase of the formalin test, but decreased post-formalin (and post-capsaicin mechanical hyperalgesia), we suggest that disruption of the central sensitization produced by these chemical activators of nociceptors, and by the different tissue and nerve injury conditions, underlies the profound antinociceptive effects that we observed. Based on these results, we suggest that the critical locus for the TrkB contribution is in the spinal cord dorsal horn. Finally, because interfering with TrkB signaling before the injury prevented development of the persistent pain state, and that interfering with TrkB signaling after the injury normalized heat and mechanical withdrawal thresholds, we conclude that TrkB signaling contributes both to the induction and maintenance of injury-induced persistent pain. Whether the same locus is involved in the induction and maintenance of the persistent pain state remains to be determined.
Acknowledgments
This work was supported by NIH grants NS48499 and 14627. The TrkBF616A mice were kindly provided by Dr. David Ginty at Johns Hopkins School of Medicine and with the permission of CGI and Princeton.
References
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH. Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron. 2007;53:261–277. doi: 10.1016/j.neuron.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci (USA) 2008;105:2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Kim JK, Park HC, Kim DS, Ha SO, Hong HS. Changes in brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia, spinal cord, and gracile nuclei following cut or crush injuries. Exp Neurol. 1998;154:224–230. doi: 10.1006/exnr.1998.6936. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Rosario CM, Merlio JP, Grant G, Aldskogius H, Persson H. Expression of mRNAs for neurotrophin receptors in the dorsal root ganglion and spinal cord during development and following peripheral or central axotomy. Brain Res Mol Brain Res. 1993;17:217–226. doi: 10.1016/0169-328x(93)90005-a. [DOI] [PubMed] [Google Scholar]
- Fan YJ, Wu LL, Li HY, Wang YJ, Zhou XF. Differential effects of pro-BDNF on sensory neurons after sciatic nerve transection in neonatal rats. Eur J Neurosci. 2008;27:2380–90. doi: 10.1111/j.1460-9568.2008.06215.x. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth R, Aanonsen L. Spinal brain-derived neurotrophic factor (BDNF) produces hyperalgesia in normal mice while antisense directed against either BDNF or TrkB, prevent inflammation-induced hyperalgesia. Pain. 2002;100:171–181. doi: 10.1016/s0304-3959(02)00264-6. [DOI] [PubMed] [Google Scholar]
- Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling:a novel mechanism for descending pain facilitation. J Neurosci. 2006;26:126–137. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppenstall PA, Lewin GR. BDNF but not NT-4 is required for normal flexion reflex plasticity and function. Proc Natl Acad Sci USA. 2001;98:8107–8112. doi: 10.1073/pnas.141015098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosl K, Reinold H, Harvey RJ, Muller U, Narumiya S, Zeilhofer HU. Spinal prostaglandin E receptors of the EP2 subtype and the glycine receptor alpha3 subunit, which mediate central inflammatory hyperalgesia, do not contribute to pain after peripheral nerve injury or formalin injection. Pain. 2006;126:46–53. doi: 10.1016/j.pain.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Martin CD, Koewler NJ, Freeman KT, Sullivan LJ, Halvorson KG, Barthold CM, Peters CM, Buus RJ, Ghilardi JR, Lewis JL, Kuskowski MA, Mantyh PW. Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain. 2007;133:183–196. doi: 10.1016/j.pain.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Hanover JL, England PM, Stryker MP. TrkB kinase is required for recovery, but not loss, of cortical responses following monocular deprivation. Nat Neurosci. 2008;11:497–504. doi: 10.1038/nn2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003;18:1169–1174. doi: 10.1046/j.1460-9568.2003.02848.x. [DOI] [PubMed] [Google Scholar]
- Lindsay RM. Role of neurotrophins and Trk receptors in the development and maintenance of sensory neurons: an overview. Philos Trans Roy Soc B. 1996;351:365–373. doi: 10.1098/rstb.1996.0030. [DOI] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcol W, Kotulska K, Larysz-Brysz M, Kowalik JL. BDNF contributes to animal model neuropathic pain after peripheral nerve transection. Neurosurg Rev. 2007;30:235–43. doi: 10.1007/s10143-007-0085-5. [DOI] [PubMed] [Google Scholar]
- Merighi A, Bardoni R, Salio C, Lossi L, Ferrini F, Prandini M, Zonta M, Gustincich S, Carmignoto G. Presynaptic functional TrkB receptors mediate the release of excitatory neurotransmitters from primary afferent terminals in lamina II (substantia gelatinosa) of postnatal rat spinal cord. Dev Neurobiol. 2008a;68:457–475. doi: 10.1002/dneu.20605. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Prog Neurobiol. 2008b;85:297–2317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Shortland PJ, Yan Q, Priestley JV. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large TrkB and TrkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur J Neurosci. 1999;11:3539–3551. doi: 10.1046/j.1460-9568.1999.00767.x. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, McMahon SB. BDNF: a neuromodulator in nociceptive pathways? Brain Res (Brain Res Rev) 2002;40:240–249. doi: 10.1016/s0165-0173(02)00206-0. [DOI] [PubMed] [Google Scholar]
- Quintao NL, Santos AR, Campos MM, Calixto JB. The role of neurotrophic factors in genesis and maintenance of mechanical hypernociception after brachial plexus avulsion in mice. Pain. 2008;136:125–133. doi: 10.1016/j.pain.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Salio C, Lossi L, Ferrini F, Merighi A. Ultrastructural evidence for a pre- and postsynaptic localization of full-length TrkB receptors in substantia gelatinosa (lamina II) of rat and mouse spinal cord. Eur J Neurosci. 2005;22:1951–1966. doi: 10.1111/j.1460-9568.2005.04392.x. [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, 3rd, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Shields SD, Mazario J, Skinner K, Basbaum AI. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain. 2007;131:8–20. doi: 10.1016/j.pain.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Yan JY, Willis WD. Activation of protein kinase B/Akt signaling pathway contributes to mechanical hypersensitivity induced by capsaicin. Pain. 2006;120:86–96. doi: 10.1016/j.pain.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Basbaum AI. Early antinociception delays edema but does not reduce the magnitude of persistent pain in the formalin test. J Pain. 2000;1:218–228. doi: 10.1054/jpai.2000.7308. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Peterson MA, Basbaum AI. Early nociceptive events influence the temporal profile, but not the magnitude, of the tonic response to subcutaneous formalin: effects with remifentanil. J Pharmacol Exp Ther. 1997;280:876–883. [PubMed] [Google Scholar]
- Walker SM, Meredith-Middleton J, Lickiss T, Moss A, Fitzgerald M. Primary and secondary hyperalgesia can be differentiated by postnatal age and ERK activation in the spinal dorsal horn of the rat pup. Pain. 2007;128:157–168. doi: 10.1016/j.pain.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Wang H, Shimizu E, Tang YP, Cho M, Kyin M, Zuo W, Robinson DA, Alaimo PJ, Zhang C, Morimoto H, Zhuo M, Feng R, Shokat KM, Tsien JZ. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc Natl Acad Sci (USA) 2003;100:4287–4292. doi: 10.1073/pnas.0636870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima Y, Narita M, Narita M, Matsumoto N, Suzuki T. Involvement of a spinal brain-derived neurotrophic factor/full-length TrkB pathway in the development of nerve injury-induced thermal hyperalgesia in mice. Brain Res. 2002;958:338–346. doi: 10.1016/s0006-8993(02)03666-1. [DOI] [PubMed] [Google Scholar]
- Zeitz KP, Giese KP, Silva AJ, Basbaum AI. The contribution of autophosphorylated alpha-calcium-calmodulin kinase II to injury-induced persistent pain. Neuroscience. 2004;128:889–898. doi: 10.1016/j.neuroscience.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, Rios M, Wood JN. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31:539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhou XF, Chie ET, Deng YS, Zhong JH, Xue Q, Rush RA, Xian CJ. Injured primary sensory neurons switch phenotype for brain-derived neurotrophic factor in the rat. Neuroscience. 1999;92:841–853. doi: 10.1016/s0306-4522(99)00027-5. [DOI] [PubMed] [Google Scholar]