

Abstract
Objective
Breastfeeding is associated with a reduced risk of developing asthma in children. Using a murine model we previously demonstrated that mothers with Th1-type immunity to ovalbumin (OVA) transfer antigen-specific protection from OVA-induced allergic airway disease (AAD) to their offspring. The aim of this study was to evaluate the contribution of breastmilk and maternal B cell immunity from allergic mothers in the vertical transmission of protection from AAD.
Methods
This was investigated using an adoptive nursing strategy. Naive offspring were nursed by allergic wild-type or B cell-deficient foster mothers with histories of Th2-type immunity to OVA. Following weaning, offspring were immunized with OVA-Al(OH)3 and challenged with aerosolized OVA to induce AAD.
Results
Offspring nursed by wild-type OVA-immune foster mothers demonstrated lower levels of OVA-specific immunoglobulin E, interleukin-5, and airway eosinophilia than progeny nursed by naive control mothers. In contrast, offspring nursed by B cell-deficient OVA-immune foster mothers had similar parameters of OVA-induced AAD as progeny nursed by naive control mothers.
Conclusions
These data demonstrate the ability of breastmilk from allergic mothers to protect offspring from AAD was dependent on intact maternal B cell immunity. Nursing alone, when done by wild-type mothers with AAD, was sufficient for offspring to acquire the antigen-specific protective factor(s) from breastmilk.
Introduction
BREASTFEEDING IS HIGHLY BENEFICIAL to the development of a healthy immune system in neonates. In particular, breastmilk provides growth factors that promote cell maturation in the intestine and thus formation of this critical epithelial barrier. Overall, breastfed infants have a reduced risk of infections, especially severe infections that result in hospitalization. In addition, infants who are fed breastmilk rather than formula are generally more protected from detrimental immune reactions to innocuous environmental or self-antigens, those that can be responsible for allergic or autoimmune diseases months or years later.1-5
Mothers influence immune responsiveness in progeny via the passive transfer of antibodies,6,7 which can have beneficial or detrimental effects. For example, maternal antibodies are essential in protecting infants against a wide range of infectious diseases until their levels decline below a protective threshold.8 However, under some circumstances, preexisting maternal antibodies can inhibit infant antibody responses to active immunization.9 This is a major factor for delaying measles virus immunization until 12 months of age, when the majority of maternal antibodies have disappeared.10 Additional health concerns arise when maternal antibodies are directed against antigens expressed on fetal cells, as observed in erythroblastosis fetalis.11 It is known from animal models that maternal immunoglobulin (Ig) G can control IgE production in offspring.12,13 In rodents, the majority of maternal IgG is acquired from breastmilk,14,15 and offspring nursed by immune mothers display a selective reduction in antigen-specific IgE responses.16 These data suggest a protective effect associated with transmission of maternal IgG, but it is possible that other factors in breastmilk (e.g., IgA, cytokines, or immune cells) are also mediators of reduced IgE responses. Additional readouts of disease parameters (in addition to allergen-specific IgE levels) would advance understanding of how maternal antibodies transfer protection from allergic airway disease (AAD) to offspring. In humans, maternal IgG is acquired by children during pre- and postnatal life,17,18 and a variety of outcomes are observed as a result of this transmission. These range from increased resistance to atopy,19 likelihood of developing eczema,20 and levels of allergen-specific IgG in infancy21 and early childhood.22 Variations in genetics, routes of antibody acquisition during pre- versus postnatal life, and history of maternal allergen exposure may influence the ability of maternal antibodies to modify allergic risk in offspring. Thus, the precise contribution of transferred maternal antibodies and the circumstances in which they influence allergic susceptibility in offspring remain unclear.
We developed a model to study maternal transmission of asthma resistance using ovalbumin (OVA)-sensitized female mice as mothers subjected to secondary OVA aerosol challenge during pregnancy. Following weaning, severity of AAD (a model of human asthma) was evaluated in progeny by sensitization and aerosol challenge with OVA. We previously demonstrated that offspring exposed to the effects of maternal Th1-type (but not Th2-type) immune responses in utero and during nursing had lower levels of OVA-specific IgE in serum and airway eosinophilia than progeny of naive control mothers.23 In response to cognate OVA peptide stimulation, splenic CD4+ and CD8+ cells from mice with Th1-type immune responses produced interferon-γ (a signature cytokine of Th1-polarized cells). In contrast, splenic CD4+cells from mice with Th2-type immune responses produced interleukin (IL)-4 (a signature cytokine of Th2-polarized cells) but not interferon-γ.23 In this current study, we established conditions where offspring exposed to the effects of maternal Th2-type immune responses (allergic mothers) in utero and during nursing were more protected from AAD than progeny of naive control mothers. Furthermore, we investigated the ability of breastmilk from allergic mothers to protect offspring from AAD and determined the contribution of maternal B cell immunity to this maternally transferred protection.
Materials and Methods
Animals
Wild-type C57BL/6J (B6AAD) and B cell-deficient (Igh-6tm1Cgn) (H6AAD) mice were obtained from Jackson Laboratories (Bar Harbor, ME) or bred in our colony at the University of Connecticut Health Center (Farmington). All mice were fed sterile food and water and housed in microisolators under specific pathogen-free conditions. Their care was in accordance with institutional and Office of Laboratory Animal Welfare guidelines.
AAD in mothers and offspring
Maternal AAD was generated in 6-week-old female B6AAD or H6AAD mice, immunized at weekly intervals by intraperitoneal injection with 25 (see Figs. 1 and 2) or 8 (see Fig. 4) μg of OVA (grade V, Sigma Chemical Co., St. Louis, MO) adsorbed to 2 mg of Al(OH)3. Seven to 12 days following the second immunization, animals were exposed daily to aerosolized antigen generated from 1% OVA in normal saline with a Bioaerosol nebulizing generator (BANG, CH Technologies, Inc., Westwood, NJ). Exposures were 1 hour for 7 consecutive days delivered via a nose-only inhalation exposure chamber with space for exposing 48 mice simultaneously (In-Tox Products, Moriarty, NM). This is a standard protocol to induce AAD in C57BL/6J mice.24 Fortytwo to 55 days after the primary aerosol exposure, a time when the airway inflammatory response is resolved,25 females were bred with naive C57BL/6J males. Pregnant mice were subjected to a secondary challenge with aerosolized OVA daily, during embryonic days 11-17 of pregnancy (duration of pregnancy in C57BL/6 mice is 19-20 days).
FIG. 1.
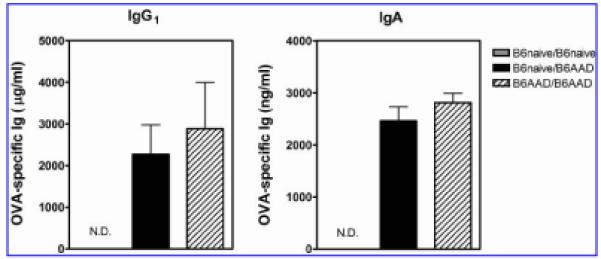
Passively acquired OVA-specific maternal antibodies in serum of 4-week-old offspring. Offspring are identified as noted in Table 1. Results are expressed as mean ± SEM values and represent five or six mice per group. N.D., not detectable.
FIG. 2.

Reduced AAD in offspring nursed by B6AAD mothers. Offspring are identified as noted in Table 1. Parameters of disease were (A) OVA-specific IgE titers and (B) airway eosinophils. Results are expressed as mean ± SEM values and represent four to six mice per group. **p ≤ 0.01, ***p ≤ 0.001 compared to B6naive/B6 naive controls.
FIG. 4.

Protection from AAD was dependent on B cell-derived factors in breastmilk. Offspring are identified as noted in Table 1. Parameters of disease were (A) OVA-specific IgE titers, (B and C) airway leukocytes, and (D) IL-5 concentrations in serum collected 24 hours after the first aerosol challenge. Results are expressed as mean ± SEM values and represent 10-15 mice per group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 compared to B6naive/B6naive controls.
Severity of OVA-induced AAD was evaluated in progeny. Following weaning, 4-week-old pups were immunized by two intraperitoneal injections with OVA (8 μg) adsorbed to 2 mg of Al(OH)3 separated by 8-10 days. Sixteen to 26 days after the second immunization, mice were subjected to challenge with aerosolized OVA daily for 4 days (1-hour exposure time). Mice were sacrificed 24 hours after the last aerosol exposure for assessment of disease as indicated by OVA-specific Ig levels in serum and distribution of leukocyte populations in the airways. In some experiments, serum was collected prior to and 24 hours after the first aerosol challenge for measurement of IL-5 levels.
Analysis of airway leukocytes
Bronchoalveolar lavage (BAL) was performed 24 hours after the last aerosol challenge, with the animals under terminal ketamine/xylazine anesthesia. Lungs from each animal were lavaged in situ with five 1-mL aliquots of sterile saline. Total leukocyte counts were performed with a Z2™ Coulter Counter® (6-20 μm; Beckman Coulter, Fullerton, CA). The differential leukocyte count in BAL was determined by analysis of cytocentrifuged slide preparations stained with Wright-Giemsa. For lung histology, tissue sections were stained with hematoxylin and eosin and examined to determine degree of allergic airway inflammation.
Flow cytometry
Monoclonal antibodies used to identify airway leukocytes were anti-CD45-fluorescein isothiocyanate (30-F11), -TCRβ-phycoerythrin (H57-597), -CD11b-PerCP-Cy5.5 (M1/70), -CD19-phycoerythrin (1D3), -CD4-PerCP (RM4-5), -CD8α-allophycocyanin (53-6.7), and -CD90.2-allophycocyanin (53-2.1) purchased from BD PharMingen (San Diego, CA). Cells (104-106) were incubated with 100 μL of appropriately diluted antibodies in phosphate-buffered saline containing 0.2% bovine serum albumin and 0.1% NaN3 for 30 minutes at 4°C and then washed with the same buffer. H-2Kb tetramer containing the OVA-derived peptide SIINFEKL was generously provided by Dr. Leo Lefranáois (University of Connecticut Health Center) and labeling of OVA-specific CD8+cells was as described.26 Relative fluorescence intensities were determined on a 4-decade log scale by flow cytometric analysis using a FACSCalibur™ (Becton Dickinson, San Jose, CA).
Determination of serum IL-5 and Ig levels
Serum IL-5 concentrations were determined by enzyme-linked immunosorbent assay (Pierce Biotechnology Inc., Rockford, IL). The assay was performed according to the manufacturer's recommendation. The minimum concentration of IL-5 detectable with this assay is 1.0 pg/mL. Serum Ig levels were measured by enzyme-linked immunosorbent assay using isotype-specific capture antibodies. Costar 3590 microtiter plates (Corning Inc., Corning, NY) were coated with rat anti-mouse IgE (R35-72) or IgG1 (A85-3) (BD PharMingen) or goat anti-mouse IgA (Southern Biotech, Birmingham, AL) at 2 μg/mL in phosphate-buffered saline for 16 hours at 4°C. After nonspecific binding was blocked, isotype-specific antibodies were captured in duplicate as three or four twofold serial dilutions of serum (within established linear ranges of the standard for each individual isotype). Detection of antigen-specific antibodies was with OVA-digoxigenin conjugates followed by anti-digoxigeninperoxidase (Roche Diagnostics, Indianapolis, IN).23,27 Development was with the 3,3′ ,5,5′ -tetramethylbenzidine microwell peroxidase substrate system (Kirkegaard & Perry Laboratories, Gaithersburg, MD), and the absorbance at 450 nm measured with a Bio-Rad (Hercules, CA) model 480 microplate reader.
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Differences between groups were determined by nonparametric Mann-Whitney test (between two groups) or one-way analysis of variance with Newman-Keuls multiple comparison post-test (multiple groups). All statistical comparisons were performed with Prism version 4 (Graph-Pad Software, San Diego). Statistical significance was defined as a value of p ≤ 0.05.
Results
Acquisition of allergen-specific maternal antibodies from breastmilk
Similar OVA-specific antibody levels and airway eosinophilia are observed in pregnant and non-pregnant C57BL/6J female mice in a discontinuous model of OVA-induced AAD (secondary challenge during embryonic days 11-17 of pregnancy).23 In this study, we modified this model to determine if breastmilk from allergic mothers can protect offspring from developing AAD. B6AAD mothers were generated as described in Materials and Methods. Breastfeeding mothers had OVA-specific IgG1 (21,323 ± 4,241 μg/mL), IgA (186,096 ± 65,856 ng/mL), and IgE (25,129 ± 4,163 ng/mL) in serum at 1 week after delivery of pups. Naive female C57BL/6J mice (B6naive) were mated, and within 14 hours of delivery one-half of the pups were adoptively nursed by B6AAD foster mothers. Thus, all fostered pups were from naive mothers to restrict the source of acquisition of allergen-specific protective factors to breastmilk, and the littermate control pups remained with their own B6naive mothers.
Sera were obtained from progeny at 4 weeks of age for measurement of passively acquired maternal antibodies. Nomenclature for offspring was denoted by birth mother's immune status followed by nursing mother's immune status (Table 1). OVA-specific antibodies were detected in serum of progeny nursed by B6AAD mothers but not by naive control mothers. In offspring born to and nursed by B6AAD mothers (B6AAD/B6AAD), acquisition of OVA-specific antibodies could have occurred via passive transfer in utero or during nursing, although previous studies have shown the majority of maternal antibodies in rodents are obtained by nursing.14,15 At 4 weeks of age, offspring adoptively nursed by B6AAD foster mothers had equivalent quantities of allergen-specific maternal IgG1 and IgA in their serum as offspring born to and nursed by B6AAD mothers (Fig. 1).
Table 1.
Identification of Offspring Based on History of Exposure to the Effects of Maternal-Derived Immune Responses
| Offspring mice | Prenatal exposure (pregnancy) | Postnatal exposure (nursing) |
|---|---|---|
| B6naive/B6naive | B6naive mother | B6naive mother |
| B6naive/B6AAD | B6naive mother | B6AAD mothera |
| B6naive/H6AAD | B6naive mother | H6AAD mothera |
| B6AAD/B6AAD | B6AAD mother | B6AAD mother |
Offspring were from B6naive or B6AAD mothers.
Offspring born to naive females were adoptively nursed by B6AAD or H6AAD foster mothers
Attenuation of disease in offspring nursed by mothers with OVA-induced AAD
Severity of OVA-induced AAD was assessed in offspring with different histories of exposure to the effects of maternal-derived immune responses. Pups were those born to and nursed by B6AAD mothers (B6AAD/B6AAD) or those born to B6naive mothers and adoptively nursed by B6AAD foster mothers (B6naive/B6AAD) shown in Figure 1. Pups born to and nursed by B6naive mothers (B6naive/B6naive) served as positive controls for AAD. Following weaning, AAD was elicited in 4-week-old pups by two immunizations of 8 μg of OVA in Al(OH)3 and four daily challenges with aerosolized OVA. Using this protocol, when compared to positive control offspring (nursed by B6naive mothers), reduced disease was evident in offspring nursed by B6AAD (allergic) mothers irrespective of their birth status (Fig. 2). Serum OVA-specific IgE in B6naive/B6AAD and B6AAD/B6AAD offspring was virtually undetectable, whereas B6naive/ B6naive positive control offspring exhibited a demonstrable response (2,390 +/- 1,068 ng/mL). Similarly, the magnitude of OVA-induced airway inflammatory response was reduced in mice nursed by B6AAD mothers. Analysis of the BAL demonstrated two- to fivefold lower numbers of eosinophils in the airways. Thus, major determinants of OVA-induced AAD were attenuated in offspring as a consequence of being nursed by B6AAD mothers.
B cell-deficient mice were competent to elicit secondary OVA-induced AAD
Previous reports using an acute model of experimentally induced AAD demonstrated that neither B cells nor antibodies (e.g., IgE) are required for T cell activation or eosinphilic airway inflammation.28-30 However, it has been shown recently that B cells are required to generate memory CD4+T cells in response to immunization with protein antigens31 or infection with Pneumocystis carinii.32 Therefore, prior to performing foster nursing studies using B cell-deficient mice, it was necessary to determine whether female B cell-deficient mice had sufficient levels of OVA-specific CD4+ T cells to initiate “recall” allergic airway inflammation when subjected to secondary OVA aerosol challenge in the discontinuous model of AAD.23 C57BL/6J wild-type (B6) or B cell-deficient (H6) mice were immunized once a week for 2 weeks with 8 μg of OVA adsorbed to 2 mg of Al(OH)3. One week after the second immunization, mice were subjected to primary aerosol challenge with OVA for 7 days. Some mice were sacrificed, whereas others were allowed to recover for a period of 6 weeks prior to secondary aerosol challenge with OVA for 7 days.
As expected, OVA-specific antibodies were present in the serum of B6AAD mice after both primary and secondary aerosol challenges but were absent from the serum of H6AAD mice (data not shown). Analysis of cell populations recovered from the BAL demonstrated virtually identical numbers of leukocytes and lymphocytes (except B cells) in the airways of B6AAD or H6AAD mice after both primary and secondary aerosol challenges (Fig. 3). Also noted were equivalent numbers of OVA-specific CD8+ cells in the airways of aerosol-challenged B6AAD mice or H6AAD mice (2-15 x 103 cells per mouse [data not shown]). There was increased severity of inflammation during the “recall” response, indicated by higher numbers of inflammatory cells recovered from the airways and a boosted serum IL-5 response in B6AAD and H6AAD mice after secondary aerosol challenge. Serum IL-5 levels in H6AAD mice were significantly greater than in B6AAD mice after secondary aerosol challenge, possibly reflecting the absence of B cells responsive to the cytokine. Histopathologic examination of lung tissue demonstrated a similar pattern of peribronchiolar eosinophilic inflammation in B6AAD mice as compared to H6AAD mice after both primary and secondary aerosol challenges. Collectively, these results indicate that in the absence of B lymphocytes and antibodies, sufficient numbers of CD4+ memory T cells were maintained to orchestrate a boosted secondary airway inflammatory response upon re-exposure to aerosolized antigen.
FIG. 3.

Similar parameters of primary and secondary AAD in wild-type and B cell-deficient mice. Parameters of disease were (A and B) airway leukocytes and (C) serum IL-5 concentrations. (D) Representative lung sections after secondary aerosol challenge. Results are expressed as mean ± SEM values and represent seven to 10 mice per group. N.D., not detectable. *p < 0.05, **p < 0.01 between groups.
Maternal B cell immunity was required for allergic mothers to transfer AAD resistance to breastfed offspring
The contribution of maternal B cell immunity in the ability of breastmilk to protect offspring from development of AAD was investigated by adoptively nursing offspring of B6naive mice by B6AAD or H6AAD foster mothers (Table 1). H6AAD mice lack B cells, so offspring nursed by H6AAD foster mothers were exposed to immune factors transmitted in breastmilk (e.g., cytokines and immune cells) except those produced by B cells, B cells themselves, and maternal antibodies. In contrast, offspring nursed by B6AAD mothers acquired the full complement of immune factors, including B cell-derived factors. Offspring that remained with their B6naive birth mothers served as positive controls for AAD. At 4 weeks of age, maternal OVA-specific antibodies were present in the serum of B6naive/B6AAD offspring but were absent in the serum of B6naive/H6AAD or B6naive/B6naive offspring (data not shown).
Following weaning, AAD was elicited in 4-week-old pups. Reduced allergic disease was evident in offspring adoptively nursed by B6AAD mothers compared to the positive control offspring nursed by B6naive mothers (Fig. 4). In contrast, essentially no protection from development of OVA-induced AAD was transmitted to offspring adoptively nursed by H6AAD mothers. After immunization and aerosol challenge of B6naive/B6AAD offspring, serum OVA-specific IgE antibodies were virtually undetectable. Analysis of the BAL also demonstrated reduced numbers of total cells, eosinophils, and lymphocytes recovered from the airways of B6naive/ B6AAD offspring as compared to B6naive/B6naive or B6niave/H6AAD offspring. In addition, serum IL-5 concentrations were notably lower in progeny nursed by B6AAD mothers as compared to B6naive/B6naive or B6niave/ H6AAD offspring.
Discussion
In this study, we investigated the contribution of maternal B cell immunity in the ability of breastmilk to protect offspring from development of AAD. Our findings demonstrated that maternal B cell immunity was required for maternal transmission of resistance to AAD from allergic mothers to nursing offspring. Offspring nursed by wild-type OVA-immune foster mothers were protected from developing severe OVA-induced AAD, whereas offspring nursed by B cell-deficient OVA-immune foster mothers were not. Thus, breastfeeding on an allergic mother protected offspring from development of AAD, but only if the mother had intact B cell immunity.
Breastmilk contains many transmissible components (e.g., antibodies, cytokines, immune cells, and fatty acids) capable of influencing immune responses in offspring. Because B cell-deficient mothers with AAD did not transfer protection from AAD to adoptively nursed offspring, we speculate that antigen-specific IgG or IgA in breastmilk is responsible for the protective effect. In rodents, the majority of maternal antibodies are transferred to offspring via the breastmilk, with absorption of IgG facilitated by a neonatal Fc receptor-dependent pathway in intestinal epithelial cells.18,33 In humans substantial amounts of maternal IgG are transmitted in utero, across the placenta.34 This process is similarly mediated by a neonatal Fc receptor localized to the synciotrophoblast of the chorionic villi.17 Thus, when considering the influence transferred maternal antibodies have on development of immune responsiveness in progeny, one must consider the possibility of species-specific effects that could result based on timing (prenatal vs. postnatal) and route (transplacental vs. intestinal absorption) of antibody acquisition.
In contrast to our previous report,23 offspring exposed to the effects of maternal-derived Th2-type immune responses were protected from developing OVA-induced AAD in the present study. B6AAD mothers and offspring were immunized with lower doses of OVA, and offspring were subjected to a shorter duration of OVA aerosol challenge as a means to enhance sensitivity of detecting protection from AAD in offspring nursed by allergic mothers. In addition, there were notably higher levels of OVA-specific IgG1 and IgA detected in offspring nursed by allergic mothers in this study, potentially contributing to increased levels of protection. Mechanistically, postnatal acquisition of allergen-specific maternal antibodies may result in the formation of immune complexes leading to clearance of the antigen, without recognition by cells of the adaptive immune system. Alternatively, a model used to examine maternal immunity on vaccine responses in progeny demonstrated that vaccineinduced T cell proliferation and cytokine production are maintained in offspring even when there is complete inhibition of antibody responses.35 Thus, in that situation at least some antigen must be available to initiate T cell responses. Another possibility is that transferred maternal IgG binds the inhibitory Fc γ receptor IIB on the surface of off-spring immune cells. It is known that simultaneous crosslinking of the Fc γ receptor IIB and the B cell receptor can result in negative regulation of B cell activation and reduced antibody production.36 The Fc γ recptor IIB is also expressed on the surface of mast cells and basophils and co-aggregation with the Fc ε receptor for IgE results in inhibition of IgE-mediated histamine release and reduced eosinophilic airway inflammation.37 These data suggest an antigen-specific mechanism by which maternal IgG may influence antibody production and airway inflammation.
To date, there are conflicting results about whether passively acquired antibodies protect against12,13 or enhance38,39 development of Th2-mediated inflammatory disease. This may be related to the nature of the IgG antibody itself and its ability to bind either activating or inhibitory Fc γ receptors.40 Our data suggest a protective effect of breastmilk, potentially associated with acquisition of maternal antibodies. Perhaps, in this model the intestinal route of maternal IgG transfer favors acquisition of IgGs that preferentially ligate inhibitory Fc γ receptors.
Although not examined in this study, it is possible that prolonging the interval from weaning to initiation of allergic challenge would result in gradual cessation of allergic protection transferred by B6AAD mothers. These results would suggest that declining concentrations of maternal protective factors in offspring results in restoration of allergic susceptibility. Irrespective of this possibility, there is great benefit to avoiding allergic airway inflammation during this critical postnatal period of immune system development and lung maturation. Even transient protection is likely to be advantageous for long-term health.
Acknowledgments
The authors thank Michelle M. Cloutier for her critical evaluation of the manuscript and Elizabeth Lingenheld, Li Zhu, and Eric Secor for their assistance. This work was funded by grants HL069083 and HL080508 from the National Institutes of Health (to L.P.) and in part by the Burr Curtis Research Endowment, Connecticut Children's Medical Center (to A.P.M.).
Footnotes
Disclosure Statement No competing financial interests exist.
References
- 1.Xanthou M. Immune protection of human milk. Biol Neonate. 1998;74:121–133. doi: 10.1159/000014018. [DOI] [PubMed] [Google Scholar]
- 2.Labbok MH, Clark D, Goldman AS. Breastfeeding: Maintaining an irreplaceable immunological resource. Nat Rev Immunol. 2004;4:565–572. doi: 10.1038/nri1393. [DOI] [PubMed] [Google Scholar]
- 3.Newton ER. Breastmilk: The gold standard. Clin Obstet Gynecol. 2004;47:632–642. doi: 10.1097/01.grf.0000136184.19927.98. [DOI] [PubMed] [Google Scholar]
- 4.Newburg DS, Walker WA. Protection of the neonate by the innate immune system of developing gut and of human milk. Pediatr Res. 2007;61:2–8. doi: 10.1203/01.pdr.0000250274.68571.18. [DOI] [PubMed] [Google Scholar]
- 5.Ip S, Chung M, Raman G, et al. AHRQ Publication No. 07-E007. Agency for Healthcare Research and Quality; Rockville, MD: 2007. Breastfeeding and maternal and infant health outcomes in developed countries. Evidence Report/Technology Assessment No. 153 (Prepared by Tufts-New England Medical Center Evidence-Based Practice Center, under Contract No. 290-02-0022) [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrlich P. “Collected Papers,” Vol. II. Z Hyg. 1892;12:31–44. [Google Scholar]
- 7.Brambell FW, Halliday R, Brierley J, et al. Transference of passive immunity from mother to young. Lancet. 1954;266:964–965. doi: 10.1016/s0140-6736(54)91571-8. [DOI] [PubMed] [Google Scholar]
- 8.Zinkernagel RM. Maternal antibodies, childhood infections, and autoimmune diseases. N Engl J Med. 2001;345:1331–1335. doi: 10.1056/NEJMra012493. [DOI] [PubMed] [Google Scholar]
- 9.Osborn JJ, Dancis J, Julia JF. Studies of the immunology of the newborn infant. II. Interference with active immunization by passive transplacental circulating antibody. Pediatrics. 1952;10:328–334. [PubMed] [Google Scholar]
- 10.Albrecht P, Ennis FA, Saltzman EJ, et al. Persistence of maternal antibody in infants beyond 12 months: Mechanism of measles vaccine failure. J Pediatr. 1977;91:715–718. doi: 10.1016/s0022-3476(77)81021-4. [DOI] [PubMed] [Google Scholar]
- 11.Murray NA, Roberts IA. Haemolytic disease of the newborn. Arch Dis Child Fetal Neonatal Ed. 2007;92:F83–F88. doi: 10.1136/adc.2005.076794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarrett EE. Stimuli for the production and control of IgE in rats. Immunol Rev. 1978;41:52–76. doi: 10.1111/j.1600-065x.1978.tb01460.x. [DOI] [PubMed] [Google Scholar]
- 13.Uthoff H, Spenner A, Reckelkamm W, et al. Critical role of preconceptional immunization for protective and nonpathological specific immunity in murine neonates. J Immunol. 2003;171:3485–3492. doi: 10.4049/jimmunol.171.7.3485. [DOI] [PubMed] [Google Scholar]
- 14.Jones EA, Waldmann TA. The mechanism of intestinal up-take and transcellular transport of IgG in the neonatal rat. J Clin Invest. 1972;51:2916–2927. doi: 10.1172/JCI107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodewald R, Kraehenbuhl JP. Receptor-mediated transport of IgG. J Cell Biol. 1984;99:159s–164s. doi: 10.1083/jcb.99.1.159s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarrett EEE, Hall E. IgE suppression by maternal IgG. Immunology. 1983;48:49–58. [PMC free article] [PubMed] [Google Scholar]
- 17.Simister NE, Story CM, Chen HL, et al. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. 1996;26:1527–1531. doi: 10.1002/eji.1830260718. [DOI] [PubMed] [Google Scholar]
- 18.Israel EJ, Taylor S, Wu Z, et al. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology. 1997;92:69–74. doi: 10.1046/j.1365-2567.1997.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vance GH, Grimshaw KE, Briggs R, et al. Serum ovalbuminspecific immunoglobulin G responses during pregnancy reflect maternal intake of dietary egg and relate to the development of allergy in early infancy. Clin Exp Allergy. 2004;34:1855–1861. doi: 10.1111/j.1365-2222.2004.02111.x. [DOI] [PubMed] [Google Scholar]
- 20.Iikura Y, Akimoto K, Odajima Y, et al. How to prevent allergic disease. I. Study of specific IgE, IgG, and IgG4 antibodies in serum of pregnant mothers, cord blood, and infants. Int Arch Allergy Appl Immunol. 1989;88:250–252. [PubMed] [Google Scholar]
- 21.Jenmalm MC, Holt PG, Bjorksten B. Maternal influence on IgG subclass antibodies to Bet v 1 during the first 18 months of life as detected with a sensitive ELISA. Int Arch Allergy Immunol. 1997;114:175–184. doi: 10.1159/000237664. [DOI] [PubMed] [Google Scholar]
- 22.Platts-Mills TA, Erwin EA, Allison AB, et al. The relevance of maternal immune responses to inhalant allergens to maternal symptoms, passive transfer to the infant, and development of antibodies in the first 2 years of life. J Allergy Clin Immunol. 2003;111:123–130. doi: 10.1067/mai.2003.10. [DOI] [PubMed] [Google Scholar]
- 23.Matson AP, Zhu L, Lingenheld EG, et al. Maternal transmission of resistance to development of allergic airway disease. J Immunol. 2007;179:1282–1291. doi: 10.4049/jimmunol.179.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schramm CM, Puddington L, Yiamouyiannis CA, et al. Proinflammatory roles of TCR γδ and TCR αβ lymphocytes in a murine model of asthma. Am J Respir Cell Mol Biol. 2000;22:218–225. doi: 10.1165/ajrcmb.22.2.3620. [DOI] [PubMed] [Google Scholar]
- 25.Schramm CM, Puddington L, Wu CA, et al. Chronic inhaled ovalbumin exposure induces antigen-dependent but not antigen-specific inhalational tolerance in a murine model of allergic airway disease. Am J Pathol. 2004;164:295–304. doi: 10.1016/S0002-9440(10)63119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masopust D, Vezys V, Marzo AL, et al. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 27.Seymour BWP, Gershwin LJ, Coffman RL. Aerosol-induced immunoglobulin (Ig)-E unresponsiveness to ovalbumin does not require CD8+ or T cell receptor (TCR)- γ/δ+ T cells or interferon (IFN)- γ in a murine model of allergen sensitization. J Exp Med. 1998;187:721–731. doi: 10.1084/jem.187.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamelmann E, Vella AT, Oshiba A, et al. Allergic airway sensitization induces T cell activation but no airway hyperresponsiveness in B cell-deficient mice. Proc Natl Acad Sci U S A. 1997;94:1350–1355. doi: 10.1073/pnas.94.4.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maclean JS, Sauty A, Luster AD, et al. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia and chemokine expression in B-cell deficient mice. Am J Respir Cell Mol Biol. 1999;20:379–387. doi: 10.1165/ajrcmb.20.3.3291. [DOI] [PubMed] [Google Scholar]
- 30.Korsgren M, Erjefält JS, Korsgren O, et al. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885–892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linton PJ, Harbertson J, Bradley LM. A critical role for B cells in the development of memory CD4 cells. J Immunol. 2000;165:5558–5565. doi: 10.4049/jimmunol.165.10.5558. [DOI] [PubMed] [Google Scholar]
- 32.Lund FE, Hollifield M, Schuer K, et al. B cells are required for generation of protective effector and memory CD4 cells in response to Pneumocystis lung infection. J Immunol. 2006;176:6147–6154. doi: 10.4049/jimmunol.176.10.6147. [DOI] [PubMed] [Google Scholar]
- 33.Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I antigens. Nature. 1989;337:184–187. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- 34.Dancis J, Lind J, Oratz M, et al. Placental transfer of proteins in human gestation. Am J Obstet Gynecol. 1961;82:167–171. doi: 10.1016/s0002-9378(16)36111-7. [DOI] [PubMed] [Google Scholar]
- 35.Siegrist CA, Barrios C, Martinez X, et al. Influence of maternal antibodies on vaccine responses: inhibition of antibody but not T cell responses allows successful early primeboost strategies in mice. Eur J Immunol. 1998;28:4138–4148. doi: 10.1002/(SICI)1521-4141(199812)28:12<4138::AID-IMMU4138>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 36.Muta T, Kurosaki T, Misulovin Z, et al. A 13 amino acid motif in the cytoplasmic domain of FcγRIIB modulates B cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 37.Zhu D, Kepley CL, Zhang K, et al. A chimeric human-cat fusion protein blocks cat-induced allergy. Nat Med. 2005;11:446–449. doi: 10.1038/nm1219. [DOI] [PubMed] [Google Scholar]
- 38.Oshiba A, Hamelmann E, Takeda K, et al. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig) E and IgG1 in mice. J Clin Invest. 1996;97:1398–1408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Garnier C, Wikstrom ME, Zosky G, et al. Allergic airways disease develops after an increase in allergen capture and processing in the airway mucosa. J Immunol. 2007;179:5748–5759. doi: 10.4049/jimmunol.179.9.5748. [DOI] [PubMed] [Google Scholar]
- 40.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]