

Abstract
Alternative splicing is dysregulated in cancer cells, driving the production of isoforms that allow tumor cells to survive and continuously proliferate. Part of the reactivation of telomerase involves the splicing of hTERT transcripts to produce full-length (FL) TERT. Very few splicing factors to date have been described to interact with hTERT and promote the production of FL TERT. We recently described one such splicing factor, NOVA1, that acts as an enhancer of FL hTERT splicing, increases telomerase activity, and promotes telomere maintenance in cancer cells. In normal tissues NOVA1 is involved in neurogenesis. In the present studies, we describe that polypyrimidine tract binding proteins (PTBPs), which are also typically involved in neurogenesis, are also participating in the splicing of hTERT to FL in cancer. Knockdown experiments of PTBP1 in cancer cells indicate that PTBP1 reduces hTERT FL splicing and telomerase activity. Stable knockdown of PTBP1 results in progressively shortened telomere length in H1299 and H920 lung cancer cells. RNA pulldown experiments reveal that PTBP1 interacts with hTERT pre-mRNA in a NOVA1 dependent fashion. Knockdown of PTBP1 increases the expression PTBP2 which also interacts with NOVA1, potentially preventing the association of NOVA1 with hTERT pre-mRNA. These new data highlight that splicing in cancer cells is regulated by competition for splice sites and that combinations of splicing factors interact at cis regulatory sites on pre-mRNA transcripts. By employing hTERT as a model gene, we show the coordination of the splicing factors NOVA1, PTBP1 and PTBP2 in cancer by regulating telomerase that is expressed in the vast majority of cancer cell types.
Keywords: telomerase, hTERT, alternative splicing, NOVA1, cancer
Introduction
Telomerase is a ribonucleoprotein complex that is reactivated in approximately 90% of cancers, allowing cancer cells to maintain their telomeres and providing them limitless replicative potential [1, 2] . Thus, telomerase reactivation has been characterized as a nearly universal feature of cancer [3, 4]. Telomerase activity is abundant in early fetal/embryonic development but is restricted to low levels in most adult somatic cells [1, 5]. Telomerase consists of two core components, the protein catalytic subunit human telomerase reverse transcriptase (hTERT) and the RNA template human telomerase RNA (hTERC). The limiting factor for telomerase activity in most human cancers is the expression of full-length hTERT. hTERT is a tightly regulated gene with regulatory mechanisms at all levels of gene expression [6–8]. Transcriptional regulation of hTERT has been the focus of intense study. More recently there is mounting evidence that transcriptional control is not sufficient to completely repress hTERT expression [9, 10]. We have demonstrated that TERT transcriptional regulation is controlled by telomere length through chromatin/epigenetic modifications [10]. Even though transcription of TERT mRNA increased with in vitro aging (i.e., short telomeres), neither telomerase activity or TERT transcripts containing the reverse transcriptase domain were not detected, suggesting another level regulation. Post-transcriptional regulation, for example alternative splicing, is also important for regulating telomerase activity [9, 11, 12].
The catalytic subunit of telomerase, hTERT, consists of 16 exons and is subjected to alternative splicing resulting in several variants, including the full-length (FL) reverse transcriptase (RT) competent variant. Production of the FL RT competent variant is dependent upon cell context and state. For instance, in transit amplifying stem cells and during early human fetal development, FL hTERT and several splice variants are expressed simultaneously. The major isoforms that have been extensively studied in development and in cancer involve the splicing of exons 5-9 (5 exons) which encode for part of the RT domain [9, 11, 13–16] . The FL variant of hTERT contains all 5 exons in the exon 5-9 region and is the only isoform with the potential to encode for catalytically active enzymes. The next major isoform +α−β, also known as minus beta, is the result of the skipping of exons 7 and 8 which leads to a frameshift and a premature stop codon in exon 10. Another isoform is −α+β, also known as minus alpha, lacks 36 nucleotides of exon 6 which although in frame, generates a dominant negative RT hTERT [11]. The last of the 4 isoforms involving splicing of exons 5 to 9 is −α−β. This variant is a combination of both skipping events of minus beta and minus alpha. While there is a consensus on the importance of the RT domain in telomerase activity, little is currently known about the regulation of alternative splicing that generates RT competent versus RT inactive hTERT.
Alternative splicing is regulated by a combination of cis and trans-acting factors and alternative splicing regulation is known to be aberrant in cancer [17, 18]. In the reverse transcriptase domain of hTERT three critical cis elements were discovered. Two of the elements are located in intron 6, “Block 6 repeats”, which is a variable nucleotide tandem repeat, and “direct repeat 6” were both found to be critical in the production of the minus beta (−β) containing isoforms. This study also identified a region in intron 8 that was critical in the production of FL hTERT [19]. This region called ‘direct repeat 8’ (DR8) is conserved across old world primates but not among other species such as mice and rats [19]. This is important because smaller, shorter lived mammals regulate telomerase differently from larger, longer lived mammals. Rodents have telomerase expression in somatic tissues while primates, including humans, do not [20, 21]. Thus, we hypothesized that DR8 is likely a region where splicing enhancers and repressors of FL hTERT splicing are binding. Since we are interested in factors that promote FL hTERT transcripts that can generate telomerase activity, we have focused our efforts to date on elucidating factors that bind DR8 of hTERT. We recently described a role for the splicing factor NOVA1 in the promotion of FL hTERT splicing, telomerase activity and telomere maintenance by binding to DR8 [22]. During this work, we noticed many polypyrimidine tracts located in and around the region where NOVA1 was binding to hTERT pre-mRNA in DR8. This observation led us to hypothesize that the polypyrimidine tract binding proteins (PTBPs) may also be playing a role in the regulation of hTERT splicing.
In this report, we build upon our model that NOVA1 regulates hTERT splicing by acting in conjunction with another splicing factor, PTBP1, in cancer cells to enhance the production of FL hTERT. Further, we add the discovery that NOVA1 directs PTBP1 to DR8 and promotes FL splicing to our working model of hTERT splicing. These data provide an in-depth mechanistic view of how cancer cells regulate hTERT splicing and begin to build a strong foundation for designing a cancer therapeutic approach aimed at repressing telomerase that may contribute to stable cancer remissions.
Results
Alternative splicing of hTERT as a mechanism of telomerase regulation in larger, long-lived mammals
We have previously identified cis elements in human TERT (hTERT) introns that act as splicing regulatory elements [19]. These cis elements, called direct repeat 6 and direct repeat 8 or DR6 and DR8 respectively, were shown to be conserved within old world primates. Recent work from our lab has built on this regulatory mechanism for hTERT alternative splicing by identifying the RNA binding proteins that interact with these regions, in particular DR8 [22]. To determine if the cis acting elements we previously described had a functional role in TERT splicing, we compared the splicing of TERT transcripts between humans and mice [19]. Humans and higher order primates have several intronic cis elements in the TERT gene that seem to regulate alternative splicing, however rodents and other distant organisms do not have these cis elements [19]. First, we evaluated the splicing landscape (i.e., the expressed transcript isoforms from full length cDNAs) of TERT RNAs in humans and in mice using targeted long read length RNA sequencing (long read sequencing workflow shown in Supplementary Figure 1A). As shown in Figure 1, human TERT is spliced to various transcripts (the 5 most abundant from sequencing reads are shown for the 2-16 libraries from mice and humans). In HeLa cells, full length TERT from exons 2-16 was the second most abundant transcript but only represented 40% of the transcripts in these libraries (Figure 1A). In contrast, the mouse full length Tert from exons 2-16 comprised 85% of the transcripts sequenced (Figure 1B, Supplementary Figure 1B and 1D). We prepared human libraries from exons 1-16 (HeLa cells) as well and observed that full length TERT represented about 18% of the total transcripts measured (Supplementary Figure 1C). The difference between the two library preparations among many possibilities could represent either the splicing out of exon 2 (Del2 isoform [23]) as exon 2 was skipped in the 3, 5 and, 6 most abundantly detected transcripts, or it could represent the difficulty of the reverse transcriptase and the PCR polymerases to make full length cDNAs and amplify GC rich regions, respectively. We then looked in depth at the reverse transcriptase domain of human TERT and mouse Tert (exons 5-9 specifically), since we previously observed cis elements in the human intron 8 that appear to be critical for RNA binding proteins (trans-factors) that influence splicing of exons 5-9.
Figure 1. Alternative splicing of hTERT as a mechanism of telomerase regulation in long-lived larger mammals.
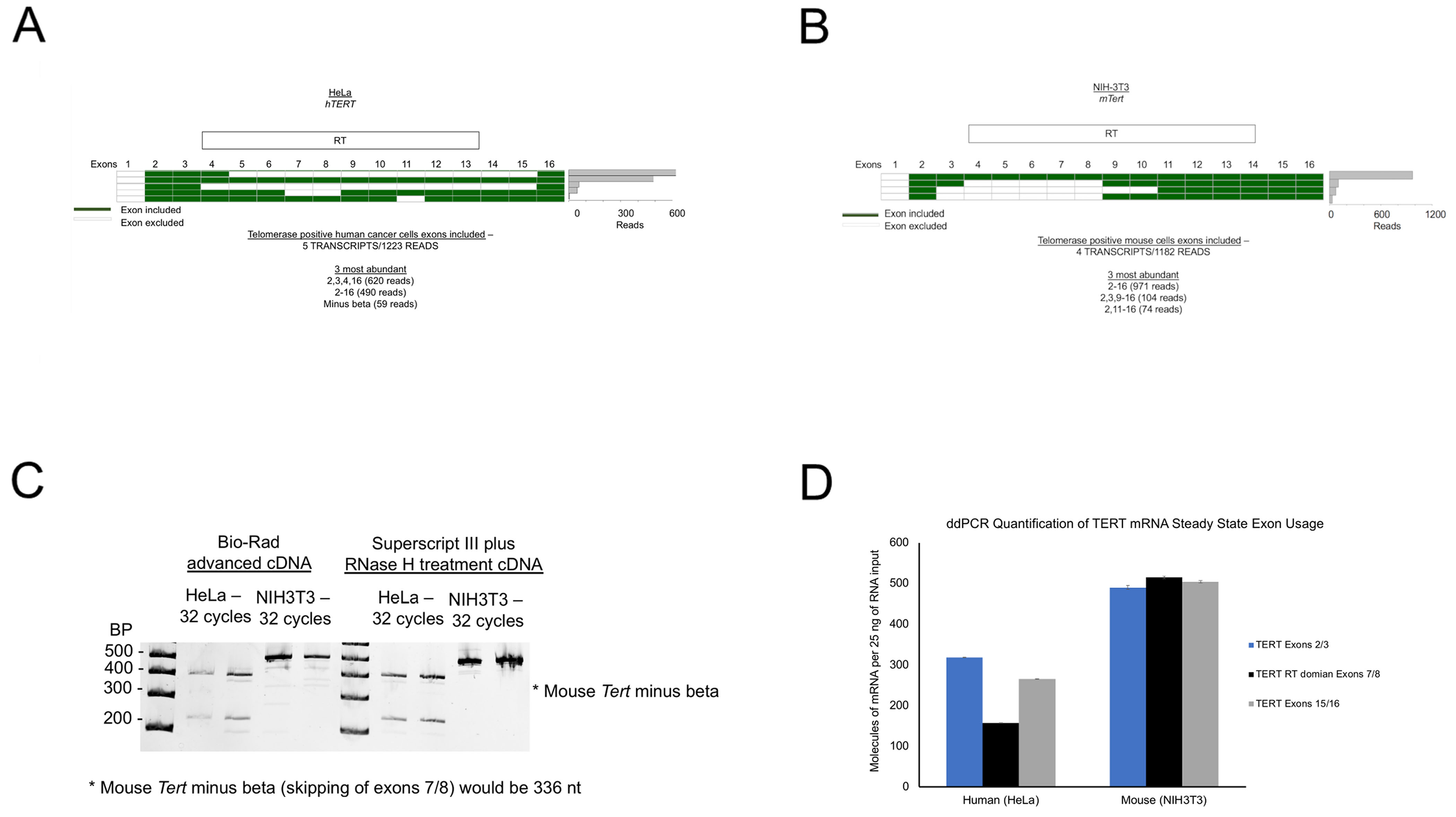
Long-read sequencing of TERT specific libraries was performed to analyze the splicing isoforms of TERT between humans and mice. (A) Human TERT libraries were generated with primers in exon 2 and exon 16 and sequenced by long-read sequencing (Pacific Biosciences). A heatmap of included versus excluded exons is shown. A frequency plot displays the number of reads each isoform that was observed. (B) Mouse Tert libraries were generated with primers in exon 2 and exon 16 and sequenced by long-read sequencing (Pacific Biosciences). A heatmap of included versus excluded exons is shown. A frequency plot displays the number of reads each isoform that was observed. (C) Mouse (NIH3T3) and human (HeLa) cDNAs were amplified with PCR primers spanning TERT exons 5 through 9 of the TERT reverse transcriptase domain (species specific primers were used). N = 3 per species, representative image shown. (D) Species specific TERT quantification across the gene body at three different regions of TERT using droplet digital RT-PCR. N = at least 6 replicates per primer pair.
Inspection of the mouse Tert intron 8 revealed that this species lacks a similar intronic element as compared to human TERT. Mouse Tert also lacks elements in intron 6 that have been characterized as important for splicing in human TERT as well [19]. We used RT-PCR to test for splicing in the region of exons 5-9 and observed that humans generate two prominent splice products (potential full length TERT at 426 bp, Figure 1C) and minus beta (skipping of exons 7 and 8, 246bp; Figure 1C), as expected. When we designed similar primers for mouse Tert exons 5-9, we did not observe robust splicing in this region of the reverse transcriptase domain (Figure 1C). In order to quantify the difference in transcripts containing different combinations of exons across the mouse Tert and human TERT we designed primers to three different regions. Primers were designed to exons 2 and 3 (5-TERT), 7 and 8 (RT domain) and exons 15 and 16 (3-TERT) for both mouse and human. Using ddPCR, we observed about a 2-fold difference in total transcripts between human cancer cells (HeLa) and mouse immortal fibroblasts (NIH3T3; Figure 1D; Average of transcripts measured at all three regions – average ± standard error - human 247.5 ± 47.6 versus mouse 503.2 ± 7.3 molecules of mRNA per 25 ng of RNA input; Student’s t –test, p < 0.01). These data confirmed that the RT domain of human TERT is spliced out of the majority of the transcripts, while in mouse the majority of the transcripts contain the RT domain. All together, these data provide strong evidence that the intronic cis elements found in humans and other old-world primates act as splicing regulatory regions where trans-acting factors likely bind to either promote or repress full length TERT production.
Basal levels of splicing factors, telomerase activity and hTERT splicing in a cell panel
We previously provided evidence that DR8 was a potential binding site for RNA binding proteins [19, 24]. A mini-gene screen targeting RNA binding proteins led to the identification and validation of NOVA1 as a target for regulating hTERT FL splicing [22]. NOVA1’s influence on hTERT splicing and activity was mediated through its binding of YCAY (Y = C or T) elements within DR8 (Figure 2A, red text). Upon further examination of DR8’s sequence, we identified an enrichment of polypyrimidine tracts (PPTs, purple text underlined) (Figure 2A). The PPTs were located in close proximity to NOVA1’s YCAY motifs and even overlapped in one case. Polypyrimidine-tract binding proteins, (PTBs) bind PPTs and regulate mRNA metabolism. PTBs are well known, ubiquitously expressed splicing factors involved in alternative splicing, mRNA stability and translation initiation through its interaction with the ribosome [25]. Humans express 3 homologues (70-80% homology) in a tissue specific manner. The most abundant homologue is PTBP1 that is ubiquitously expressed. The second homologue PTBP2, also known as neuronal PTB (nPTB), is expressed in adult brain, muscle and testis [25]. The third homologue PTBP3, also known as ROD1, is restricted to hematopoietic stem cells [25]. PTBs are known to both activate or repress splicing in a context specific manner [25]. We then tested if PTBs, in conjunction with NOVA1, bind to DR8 in hTERT mRNA and promote the FL splicing of hTERT in cancer (Figure 2A). This model would provide another layer of potential regulation to hTERT alternative splicing to further dissect the larger complexity of telomerase regulation.
Figure 2: Basal levels of splicing factors, telomerase activity and telomerase splicing in a cell panel.

(A) Cartoon model depicting NOVA1 binding to cis element DR8 in intron 8. PTBP1 and NOVA1 binding motifs in intron 8 depicted in purple and red font respectively. (B) Western blots for splicing factors NOVA1, PTBP1 and PTBP2 in cell panel. Cell panel consists of normal human bronchial epithelial cells, HBEC, indicated by asterisk as well as a variety of cancer cells. Histone H3 was employed as an internal protein loading control. Repeated 3 times, representative images shown. (C) Droplet digital TRAP assay detecting telomerase activity using whole cell lysates from the cancer cells in the cell panel (n=3). NTC (no template control) which is used as a negative control. (D) Droplet digital PCR assay quantification of hTERT splicing in cancer cells from cell panel. Percentage of FL (exons 7/8) hTERT mRNA to -beta (exons 6/9 splicing) hTERT mRNA. FL bars are indicated in black and minus beta in blue as indicated in figure legend (n=3). Data are expressed as means and standard error of the mean where applicable.
We first analyzed baseline levels of NOVA1, PTBP1 and PTBP2 in a panel of human tissue culture cell lines consisting of lung epithelial cells (HBEC), non-small cell lung cancer cells (H920, A549, H1299, H2887, H2882 and Calu6), small cell lung cancer (SHP-77 and H82), colon cancer (HCT-116), cervical cancer (HeLa) and transformed embryonic kidney (293T) cells. We observed heterogeneity in the levels of NOVA1 across the cancer lines (7 of 11 tumor cell lines express NOVA1; HBECs did not express NOVA1), with some lines such as Calu6 expressing no NOVA1 and others such as SHP-77 expressing high levels (Figure 2B). More importantly, no NOVA1 was detected in the human bronchial epithelial cells (HBEC), a model of normal somatic lung cells. This is important because HBECs are non-transformed telomerase negative cells and NOVA1 may be acquired during transformation in certain lung cancers providing them with greater telomere maintenance programs (i.e., more hTERT FL). PTBP1 was more readily detected across lines and elevated levels of PTBP1 were observed in the vast majority of cancer lines compared to the normal HBECs. Finally, we observed little to no PTBP2 protein in the panel of cells. The lack of PTBP2 along with elevated levels of PTBP1 in cancer led us to focus our efforts on PTBP1 [26].
We employed droplet digital TRAP and PCR (ddTRAP and ddPCR) to assess levels of telomerase activity and hTERT splicing in our cell panel [27]. As expected, we observed heterogeneity in the both telomerase activity and hTERT splicing in cancer cells (Figure 2C and D).
Stable and transient knockdown of PTBP1 reduces telomerase activity
Since PTBP1 is abundant in cancer cells we moved forward with studies focused on PTBP1 over PTBP2. Knockdown (KD) experiments were performed to determine the role of PTBP1 in telomerase regulation and hTERT alternative splicing. If PTBP1 was promoting FL splicing of hTERT then knockdown of PTBP1 would be expected to result in a decrease in telomerase activity due to the decrease in FL splicing of hTERT. We started with transient knockdown of PTBP1 in a panel of non-small cell lung cancer cell lines: H1299, H2882, H920, and HCC1359. After cells were incubated with siRNAs targeting PTBP1, we assayed telomerase activity [27]. We observed a significant 50% reduction in telomerase activity in all of the lines (Student’s t test; p<0.05 and p<0.005 as indicated; Figure 3A). In order to prevent changes in proliferation rates, we used a lower dose of siRNA (10 nm).
Figure 3: Transient siRNA and stable shRNA knockdown of PTBP1 in non-small cell lung cancer cells, NSCLC.
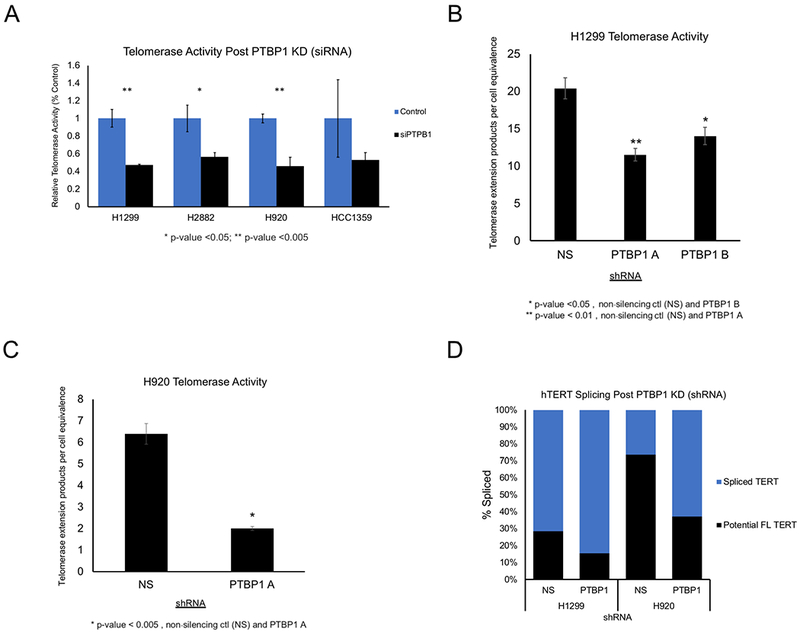
(A) Droplet digital TRAP assay detecting telomerase activity post siRNA knockdown of PTBP1 in H1299, H2882, H920 and HCC1359 (50 cell equivalents, n = 3, Student’s t test set at p < 0.05 for significance, *, and p < 0.005, **). (B) and (C) shRNA’s targeting PTBP1 were employed to generate stable knockdowns in H1299 and H920. Telomerase activity was detected using ddTRAP assay and plotted for H1299 in panel B and H920 in panel C (50 cell equivalents, n = 3, Student’s t test, p < 0.05 for significance, *). (D) Splicing of hTERT mRNA was detected and quantified using ddPCR. Spliced TERT, indicated in blue, represents all spliced variants for hTERT. Potential FL TERT represents the total steady state hTERT mRNAs minus the spliced variants. NS represents non-silencing control shRNAs (n = 3). Data are expressed as means and standard error of the mean where applicable.
Next, we generated stable cell knockdown of PTBP1 in both H1299 and H920cells. We focused on H1299 and H920 lung cancer cells because both lines have robust NOVA1 and PTBP1 expression and showed changes in telomerase activity following transient knockdown of PTBP1 (Figure 3A). In H1299 cells we introduced two shRNAs targeting PTBP1 (shRNA A and B). Following PTBP1 knockdown we measured telomerase activity and hTERT splicing. Stable KD of PTBP1 significantly reduced telomerase activity in H1299 cells compared to control shRNAs (Figure 3B), with shRNA A being more potent in reducing telomerase activity. For this reason, we continued our studies focusing only on shRNA A targeting PTBP1. To control for cell line specific phenotypes of PTBP1 targeting shRNAs we introduced shRNA A or control shRNAs into a second NSCLC cell line H920. Following selection, we observed reduced telomerase activity compared to controls in H920 cells as well (Figure 3C). Consistent with our telomerase enzyme activity results, upon PTBP1 KD, we found a decrease in the percentage of FL hTERT splicing in both H1299 and H920 cells compared to control cells (Figure 3D; 46% in H1299 cells and 50% in H920 cells).
Long-term PTBP1 KD leads to an increase in critically short telomeres in lung adenocarcinoma cells
In order to determine the impact of PTBP1 on telomere length maintenance, we employed the stable shRNA PTBP1 KD and control shRNA cell lines generated with H1299 and H920. Telomere Shortest Length Assay, also known as TeSLA, was used to assess the length and percentage of the shortest telomeres [28]. We compared control shRNA cells to PTBP1 KD cells in two different cells lines at similar population doublings post selection. The complete inhibition of telomerase results in the loss of approximately 50 to 100 nucleotides per cell division. In our experiments we were able to inhibit telomerase by 40-60% in both lines. Given the amount of telomerase inhibition we achieved, we observed the expected progressive telomere shortening overtime in culture. For example, if we assume 100 nucleotide loss per population doubling (PD) in H1299 cells we would have observed 2000 nucleotides loss of telomere DNA (100 nucleotides × 20 PD). Applying the 40% telomerase activity inhibition due to PTBP1 knockdown (Figure 3B) to this assumption we would have expected to observe an 800 nucleotides loss of telomere DNA. We observed a 780 nucleotides loss according to the TeSLA quantification putting us close to our expectations. Upon PTBP1 KD, the length of the shortest 20% of telomeres in both H1299 and H920 decreased compared to earlier passaged cells (H1299 PTBP1 KD change of 0.76 kb, Figure 4A; H920 PTBP1 KD change of 0.14 kb, Figure 4B). Furthermore, an increase in the percentage of short telomeres, both below 1.6 kb and 0.8 kb was observed. Although modest, PTBP1 KD in both lung cancer lines increased the critically short telomeres. These data are consistent with the model that PTBP1 KD decreases FL hTERT splicing, thus leading to a decrease in telomerase activity, and an increase in the load of short telomeres due to improper telomere maintenance.
Figure 4: Telomere length analysis in stable knockdown PTBP1 cancer lines.
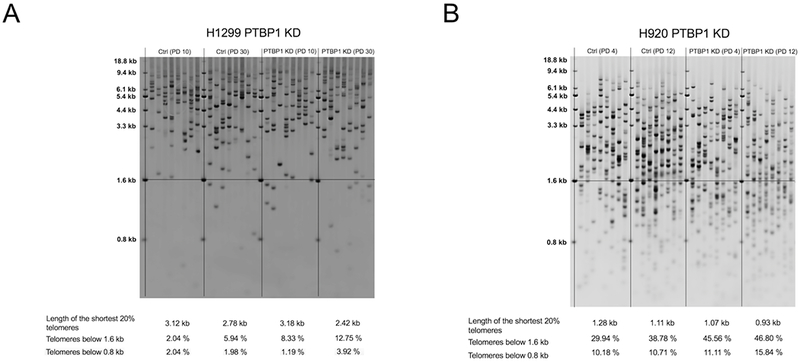
Telomere Shortest Length Assay, TeSLA, was performed to quantitatively assess long term effects of PTBP1 KD in the cancer lines H1299 and H920. (A) and (B) Stable shRNA H1299 and H920 along with control cells were analyzed with TeSLA. Telomere lengths for both control and PTBP1 KD H1299 cells were measured at population doubling 10 and 30 (PD10 and PD30). Telomere lengths for both control and PTBP1 KD H920 cells were measured at PD4 and PD12. Data are indicated in the tables below blots. Telomere bands were automatically detected and quantified with accompanying TeSLA software. Each lane represents a separate PCR replicate. In total 8 lanes/replicates per sample.
PTBP1 binds hTERT pre-mRNA at DR8 via polypyrimidine tract motifs
To understand the mechanism by which PTBP1 impacts hTERT FL splicing and telomerase activity, we designed a series of RNA baits that mapped in and around DR8 in intron 8 (Figure 5A). Performing an RNA-IP would allow us to identify the region of intron 8 that PTBP1 binds. Since hTERT is a very low abundant pre-mRNA typical cross-linking RNA-IP experiments are difficult to perform compared to analyzing more abundant target pre-mRNAs. To overcome this abundance issue, we turned to a common in vitro RNA-IP pulldown using synthesized RNA baits and cell lysates to probe for proteins that bind to specific RNA sequences. To perform the RNA-IP experiments we transiently overexpressed NOVA1 in 293T cells. NOVA1 binds in oligos 3, 4, and 5, with the strongest binding in oligo 3, as previously shown [22]. Here we show that PTBP1 predominately binds in oligo 4 and to a lesser extent in oligo 3 and oligo 5 (Figure 5B). Oligo 4 encompasses the last 125 nts of the DR8 region in intron 8. This region did not include the large polypyrimidine tract that we had expected PTBP1 to have a greater affinity for (the PPT is in oligo 2, Figure 2A). Interestingly, PTBP1’s binding profile was in close proximity to and to a certain extent overlapped with the binding profile of NOVA1, suggesting the potential for crosstalk or interaction between the two splicing factors.
Figure 5: Analysis of PTBP1 and NOVA1 binding to DR8 using RNA baits.

(A) Cartoon schematic displaying hTERT mRNA. A large polypyrimidine tract located upstream of DR8 was included in our analysis. Five oligo baits (oligo 1-5) were designed to encompass ~ 1 kb of hTERT mRNA. Baits were then in vitro transcribed into RNA and biotinylated in order to tether on to streptavidin beads. (B) Whole cell lysates from H1299 and Calu6 were used to test PTBP1 binding to the 5 baits. Eluted samples from the RNA-IP were probed via western blot for PTBP1. (C) and (D) Oligo 4 was mutated by altering all T residues to A. PTBP1 was then RNA-IP and compared with wild type (WT) bait 4. Western blot probing of PTBP1 showed a reduction in PTBP1 binding to the mutant bait. Quantification of western blot band intensity determined a significant reduction in PTBP1 binding (n = 3, Student’s t test set at p < 0.05 for significance, *). (E) and (F) Mutant bait was designed for oligo 3 converting all YCAY NOVA1 binding motifs to YAAY. NOVA1 in the elution was probed via western blot and quantified. (n = 3, Student’s t test set at p < 0.05 for significance, *).
Next, we performed RNA-IP using mutated baits to further validate the binding of PTBP1 as well as NOVA1 to their respective baits (bait 4 for PTBP1 and bait 3 for NOVA1). For PTBP1 we used the mutant bait 4 in which we replaced all the thymidine bases (T) to adenine (A) disrupting the polypyrimidine tracts. As for NOVA1, we mutated the YCAY binding motif to YAAY in bait 3 to generate mutant bait 3, which is known to abolish binding (where Y is C or T) [29]. An RNA-IP comparing the sets of control and mutant baits was performed in 293T cells with NOVA1 transiently overexpressed. When we exposed a 293T lysate to a mutant bait 4 (PTBP1 mutant bait 4), we observed a 65% reduction in binding for PTBP1 (Figure 5C and D). Similarly, when we used a mutant bait 3 (NOVA1 mutant bait 3) we observed a 50% reduction in binding of NOVA1.
NOVA1 is both necessary and sufficient for PTBP1 binding to DR8 in hTERT pre-mRNA
Using quantitative assays for telomerase activity and FL hTERT splicing pointed out significant heterogeneity in our cancer cell line panel. We leveraged this heterogeneity by comparing cell lines at opposite ends of the spectrum such as Calu6, a low telomerase activity and low hTERT FL expressing line, and H1299, a high telomerase activity and high hTERT FL expressing line (Figure 2C and 2D). Since these two lines display opposite phenotypes with regards to activity and splicing of telomerase, we predicted that they likely have different mechanisms of alternative splicing and potentially employ different sets of trans-factors. To test this idea, we designed a 1 kb RNA bait that again encompassed DR8 of hTERT pre-mRNA. This bait was then incubated with lysates from Calu6 and H1299 cells (Figure 6A). We probed for PTBP1, and found PTBP1 was able to bind the hTERT pre-mRNA bait in H1299, as seen in the elution, but not in Calu6 (Figure 6B and 6C). Since both lines expressed PTBP1 (H1299 slightly more than Calu6) this suggests the presence or absence of a limiting factor in the lysate of Calu6 prevents or allows PTBP1 to bind hTERT pre-mRNA. One possibility is that the missing factor may be NOVA1. NOVA1 protein is present in H1299 cells and not in Calu6 cells (Figure 2B). Secondly, NOVA1’s binding to hTERT mRNA was in close proximity and overlapped with PTBP1’s binding (baits 3 and 4). Finally, previous evidence from a yeast two-hybrid screen documented that NOVA1 and PTBP2 proteins interact but it was not known if PTBP1 and NOVA1 interact [30].
Figure 6: NOVA1 is sufficient and necessary for directing PTBP1 to DR8 in hTERT mRNA.

(A) A 1 kb bait was generated to pulldown and detect splicing factors a NOVA1 expressing cell line (H1299). Elution from RNA-IP was probed for PTBP1 to determine PTBP1 binding to bait. (B) The same 1 kb bait was employed in cell line that does not express NOVA1 (Calu6). PTBP1 was probed for to determine binding. (C) Calu6 lines were manipulated to either overexpress NOVA1, silence PTBP1 (siRNA) or both. NOVA1, PTBP1 and PTBP2 levels were detected via western blot from whole cell lysates. (D) Calu6 (scrambled short interfering RNA control, siCTL), Calu6 + NOVA1, Calu6 + siPTBP1 and Calu6 + NOVA1 + siPTBP1 whole cell lysates were employed for RNA-IP with RNA bait oligo 4. PTBP1 and PTBP2 levels were probed in input and elution samples for each cell line. Red arrows indicate PTBP1 and PTBP2 binding in elution. (E) Droplet digital TRAP assay was performed on whole cell lysates to determine effects on telomerase activity. (n = 3, Student’s t test set at p < 0.05 for significance, *, and p < 0.005 ,**). (F) Droplet digital PCR assay to determine FL (exons 7/8) and minus beta (exons 6/9) levels in Calu6 manipulated cells. (n = 3) (G) and (H) Quantification and western blot image displaying PTBP1 binding to NOVA1 mutated oligo 3 (YCAY to YAAY) in H1299 whole cell lysates. Western blot shows elution samples from WT and mutant bait. (n = 4, Student’s t test set at p < 0.05 for significance, *). (I) Working model cartoon for NOVA1, PTBP1 and PTBP2 action at hTERT DR8.
Since Calu6 cell normally do not express NOVA1, we expressed NOVA1 in Calu6 cells, Calu6+NOVA1, to generate a set of cells lines similar besides the expression of NOVA1. To test if NOVA1 must be present in order for PTBP1 to bind the hTERT bait, we transiently knocked down PTBP1 in the Calu6 cells in the presence or absence of NOVA1. We then probed for NOVA1, PTBP1 and PTBP2 levels in the Calu6 cell series (Figure 6C). NOVA1 was successfully overexpressed and unaffected by siRNA treatments. Furthermore, siRNAs targeting PTBP1 successfully knocked down PTBP1. Previous studies have shown cells upregulate PTBP2 in the absence or depletion of PTBP1 [31]. We observed the same phenomena in the Calu6 lines. PTBP2 protein levels significantly increased upon PTBP1 KD (Figure 6C).
Calu6 and Calu6+NOVA1 were treated with control or PTBP1 siRNAs and RNA IP lysates of each cell line were probed using oligo/bait 4. Oligo 4 was the primary RNA bait bound by PTBP1. As previously shown, PTBP1 from Calu6 siCTL was unable to bind the bait (Figure 6D). However, after introduction of NOVA1 (Calu6+NOVA1 siCTL) PTBP1 was now able to bind to hTERT pre-mRNA bait (indicated by red arrow, Figure 6D). This suggests that the presence of NOVA1 is sufficient to direct PTBP1 to hTERT pre-mRNA. As expected, the Calu6 siPTBP1 sample displayed little to no binding of the RNA bait (oligo 4). Interestingly, in the Calu6+NOVA1 siPTBP1 sample, PTBP2 was upregulated and now was able to bind (indicated by red arrow). This was not the case for PTBP2 in the Calu6 siPTBP1 lysate. The presence of NOVA1 was shown to not only direct PTBP1 but also PTBP2 to hTERT pre-mRNA bait.
Next, we assessed telomerase activity following transient siRNA mediated knockdown (KD) of PTBP1(Figure 6E). NOVA1 was able to increase telomerase activity by increasing the levels of FL hTERT mRNA [22]. However, KD of PTBP1 in Calu6 cell line had no impact on telomerase activity compared to siRNA control (Figure 6E). In the Calu6+NOVA1 line, PTBP1 KD abolished the increase in telomerase activity compared to Calu6+NOVA1 siRNA control (Figure 6E). The decrease in activity can be attributed to an increase in PTBP2 levels. PTBP2 has been previously shown to interact with NOVA1 in mouse neurons [30]. We have also confirmed this interaction via Co-IP in humans (Supplementary Figure 2). Interestingly, we were not able to determine whether or not an interaction between PTBP1 and NOVA1 exists. Taken together, this suggests a potential role for PTBP2 as a repressor complex of FL hTERT splicing via direct interaction with NOVA1 at DR8. Percent FL hTERT mRNA was reduced by PTBP1 knockdown only in Calu6 cells with NOVA1 expression, similar to the telomerase activity results (Figure 6F).
The experiments performed above demonstrate that NOVA1 was sufficient to direct PTBP1 to hTERT pre-mRNA. In order to determine whether NOVA1 was necessary for PTBP1 binding to hTERT pre-mRNA, we employed the mutated oligo 3 in an RNA-IP. Oligo 3 was one of the baits that provided an overlap of binding between the two proteins. By mutating the baits YCAY motifs to YAAY we were able to reduce NOVA1 binding significantly (Figure 5D and 5F). If NOVA1 was necessary for PTBP1 binding, then the reduction of NOVA1’s binding to mutant oligo/bait 3 would be predicted to also abolish PTBP1 binding regardless of the polypyrimidine tracts present. RNA-IP using NOVA1 mutant oligo 3 determined that PTBP1 binding was reduced by 50% (Figure 6G and H). These data provide evidence that NOVA1 is both necessary and sufficient for PTBP1 binding to hTERT pre-mRNA at DR8.
Discussion
Understanding the regulatory mechanisms that govern telomerase activation and activity is critical towards a better understanding of both aging and cancer. Recent evidence has also implicated telomerase in a spectrum of genetic diseases known as “telomeropathies” in which the underlying mechanism is improper maintenance of stem cell telomeres [32], thereby further cementing its importance in biomedical research. In the current study, we focused on the alternative splicing of hTERT, a critical yet less understood mechanism of telomerase regulation. We previously reported important cis elements, specifically direct repeat 8 in intron 8, that acts as an alternative splicing enhancer of full length (FL) hTERT pre-mRNA splicing [19, 24]. More recently we have identified a role for the splicing factor NOVA1 in binding to DR8 and promoting FL telomerase splicing and activity in non-small cell lung cancer cells [22]. While this has shed new light on telomerase splicing, the process of splicing and the trans-acting RNA binding proteins that dictate splicing outcomes is relatively understudied due to the low abundance of hTERT. In this report, we build upon the importance of the cis element DR8 in intron 8 and the splicing factor, NOVA1 in telomerase regulation with the goal of eventually finding a means to target hTERT splicing to promote durable cancer remissions.
The long-read sequencing data of TERT in mouse and human cells reveals a remarkable evolutionary trade-off between telomerase regulation and growth/cancer. The major transcript, approximately 85%, for Tert in mice is FL which is capable of producing TERT protein and active telomerase enzyme. In humans however, most TERT transcripts are alternatively spliced to inactive variants. Larger, long-lived mammals, such as humans, trade strict regulation on TERT through alternative splicing (as well as other mechanisms of regulation) for future reproduction or survival while smaller short-lived mammals, such as mice, forgo the strict regulations on TERT for current reproduction or growth. TERT splicing potentially poses as a “current-future” life history trade-off [33]. These types of regulations on TERT through alternative splicing also are consistent with Peto’s paradox [34]. The investment into regulatory mechanisms of critical genes, such as TERT, ensures somatic maintenance and allows for larger long-lived mammals to have lower cancer incidence even though they are made up of more cells that divide over longer periods of time than their smaller short-lived counterparts [33, 35]. The cis element DR8 plays a critical role in this regulation. The long-read sequencing data reveals that mTert alternative splicing is inherently different than that of human TERT. The minus beta isoform in humans, lacking the RT domains exons 7 and 8 necessary for telomerase activity, is not detected in mice. The lack of intronic cis elements (both DR6 and DR8) in mTert pre-mRNA appears to prevent the splicing of mTert in the RT domain. DR8 and other cis elements are conserved in old world primate’s TERT pre-mRNA potentially allowing TERT to be regulated by alternative splicing via trans-acting factors binding to them in those species. The evidence presented provides insights into the importance of DR8 and rationale to further understand the trans-factors that bind hTERT cis elements in order to determine if manipulation of hTERT splicing is a tractable cancer therapeutic approach.
The challenges associated with TERT mRNA abundance and the assays used to quantify them have been a significant obstacle. Despite these obstacles, we have reported a novel role for a well characterized and studied neuronal splicing factor, NOVA1 [22]. This led us to believe that there may be other splicing factors, whether they be common or rare, involved in this process and building on the role of DR8 we can now piece together a more complete picture of hTERT splicing. The polypyrimidine tracts in and around DR8 represent an ideal motif for PTBP1 and PTBP2. Using a panel of cells, we confirmed that PTBP2 protein is not present outside of neurons (cancerous and non-cancerous) and thus PTBP1 became our prime target. Upon transient and stable KD of PTBP1, both telomerase activity and hTERT FL splicing were reduced. Furthermore, using the TeSLA assay [28] we were able to report a change in the percentage of short telomeres upon KD of PTBP1 in H1299 and H920 cells, as well as a change in average telomere lengths. We provided evidence that PTBP1 and NOVA1 are indeed binding hTERT pre-mRNA sequences. Together, NOVA1 and PTBP1 play a role in driving the FL splicing of hTERT in cancer. Interestingly, the data we present here display a master regulator role for NOVA1 as both sufficient and necessary in directing PTBP1 to DR8 in hTERT mRNA to promote FL splicing (Figure 6I). In the absence of PTBP1 (siPTBP1), NOVA1 was able to recruit PTBP2 to hTERT mRNA baits. This resulted in a decrease in telomerase activity and FL hTERT splicing (Figure 6F and 6G). We propose a working model in which the absence of PTBP1 leads NOVA1 to recruit PTBP2 to DR8 and form a repressor complex to prevent FL splicing of hTERT (Figure 6I). This model may have greater implications in cells with elevated levels of PTBP2, such as neurons, where hTERT is repressed. Taken together, these data provide significant insights into splicing factor interactions at DR8 in hTERT pre-mRNA that may be targeted as a form of therapy in order to disrupt the formation of active telomerase molecules in cancer cells.
Materials and Methods
Plasmids
GFP pGIPZ shRNA plasmids for control (non-targeting), NOVA1 (Openbiosystems, NOVA 1 – 5’- TTGGACTTAGACAGCTTGA), and PTBP1 (Openbiosystems, PTBP1 – 5’-TCTGGAAGAACTTGAATCC) were obtained. Lentivirus was made by co-transfecting 5 μg of proviral shRNA plasmids and 2 μg of packaging plasmids pMD2.G and psPAX2 using Polyjet transfection reagent (SignaGen Laboratories) into 293T cells. CCSB-Broad lentiviral human NOVA1 full length cDNA with a C-terminal V5 tag and blasticidin selection in mammalian cells (accession: BC075038, clone ID: ccsbBroad304_01104) was purchased and sequence verified (GE, Dharmacon). Viral particles were produced as above.
Cell culture and cell lines
All cancer cell lines (H1299, H920, Calu6, HeLa, A549, HCT116, H2887, H82, SHP77, 293T and H2882) were cultured at 37ºC in 5% CO2 in 4:1 DMEM:Medium 199 containing 10% calf serum (HyClone, Logan, UT). Briefly, HBECs were maintained in low oxygen conditions in serum free media containing supplements from the Keratinocyte-SFM media (Invitrogen/Gibco catalog # 17005-42) on a collagen/gelatin coated tissue culture dish [36]. All cell lines were obtained from ATCC, or as a kind gift from Drs. John Minna and Adi Gazdar. All cell lines were verified by STR profiling and were tested for mycoplasma contamination.
Long read length mRNA/cDNA sequencing –
Human 2-16 hTERT libraries –
RNA was extracted from approximately 100 million HeLa cells using the RNeasy plus kit (Qiagen). cDNA was synthesized using two different reverse transcriptases (Superscript III and Bio-Rad Advanced iScript) following manufactures instructions. We use a gene specific priming strategy (5- GTACAGGGCACACCTTTGGT) to generate hTERT specific cDNAs with 1000ng of RNA input. Following cDNA synthesis, fifteen individual cDNAs were pooled and prepared for pulldown using a biotinylated hTERT exon 1 oligo (Biotin-5’-AATAATAAT AGCGCTGCGTCCTGCTGCGCACGTGGGAAG). To pulldown hTERT cDNAs, the pooled reactions were heated in the presence of 25 μM biotinylated hTERT exon 1 oligo to 95ºC for 10 min in a heat block. The heat block was then removed from the heating coil and the block placed on the bench and allowed to cool to room temperature. The cDNAs were then collected with magnetic streptavidin beads (5 μL, Dyna beads). To prepare the beads for binding they were first washed twice in binding buffer (with 0.1% Tween 20 added). Following the beads were blocked in MS2 RNA (0.08 μg/mL MS2 RNA in binding buffer with 0.1% Tween 20) for 10 minutes at room temperature with rotation. The beads were then collected with the magnet and the blocking solution removed and then added 140 μL of 2 × binding buffer. Next, 140 μL of the annealed cDNAs were added to the beads and mixed via a rotator for a minimum of 60 minutes at room temperature. After the beads were collected by the magnet and washed twice in 1 × binding buffer with 0.05% Tween 20. Following the washes captured cDNAs were eluted off the beads by adding 20 μL of 10 mM Tris (pH. 8.0) and heated to 85ºC for 5 minutes and collected immediately on the magnet and the supernatant (cDNAs) collected and placed in a new tube. Following, the exon 1 captured cDNAs were then diluted 1:4 prior to PCR with exon 2 (5’-AAGCATGCCAAGCTCTCG) and exon 16 hTERT (5’-AACAATGGCGAATCTGGGGATGGACTATTCCTAT) primers. Following PCR with Emerald AMP GC HS PCR master mix (Takara/Clonetech), 8 reactions were pooled and the DNA purified by phenol:chloroform followed by AMPure beads (Agencourt AMPure XP, Beckman Coulter) to remove primers. An agarose gel was then run and fragments from 3 kb to 1.5 kb and 1.5 kb to 0.5 kb were excised from the gel and purified. The samples were then barcoded via PCR to be able to distinguish them following pooled sequencing. Using this strategy, we can assume that all reads we observed contained exon 1. We used this exon 2 strategy because exon 1 of hTERT is extremely GC rich and difficult to PCR.
Human 1-16 hTERT libraries –
To generate the human 1-16 hTERT libraries the following procedures were performed. We generated gene specific cDNAs as described above with the following modifications. We made 8 cDNAs from HeLa RNA (1000 ng input). The pooled cDNAs were then diluted 1:4 with water. We then performed a first round of PCRs with primers in exon 1 (5’-AGCGCTGCGTCCTGCTGCGCACGTGGGAAG) and exon 16 (5’-AACAATGGCGAATCTGGGGATGGACTATTCCTAT) for 25 cycles using Emerald Amp GC high specificity master mix (8 reactions). The 8 reactions were then pooled and purified with AMPure beads (Agencourt AMPure XP, Beckman Coulter). Following the entire first round of PCR was amplified a second time for 35 cycles with barcoded primers, gel purified into two size bins of 4 kb to 2.5 kb and 2.5kb to 1.5 kb. The size selected barcoded hTERT PCR products were then sent to Pacific Biosciences for long read sequencing on the Isoseq platform.
Mouse 2-16 mTert libraries –
We generated mouse Tert exon 2-16 libraries. We observed that exon 1 and exon 2 of mouse Tert were similarly expressed and that our focus was on the splicing of the reverse transcriptase domain (exons 4-13) thus we only generated 2-16 libraries for the mouse experiments (Supplementary Figure 1B). To generate the libraries, we isolated RNA from NIH 3T3 cells and prepared cDNA using two priming strategies. cDNAs were prepared by inputting 1000 ng of total RNA into the Superscript III (Invitrogen) priming reaction with either a 1:1 ratio of oligo dT:random hexamers (total cDNA) or a gene specific reverse (GSP) primer in exon 16 of mouse Tert (5’-TCCGGCACAGCAGTTTTT). Following cDNA synthesis (using manufactures instructions at 55ºC) cDNAs were diluted 1:4 in water. We then prepared 8 PCR reactions with barcoded primers and thermocycled for 40 cycles using Emerald Amp HS PCR mix (Takara). The PCR products were analyzed for size on an agarose gel (5 μL of each PCR). Once PCR size and success was confirmed the remaining PCRs were pooled and cleaned up with AMPure beads ((Agencourt AMPure XP, Beckman Coulter) to remove excess primers. Samples were then sent for sequencing. Data are deposited and publicly available (accession number coming soon).
Sequence analysis
Following sequence acquisition, data were processed first through quality control and demultiplexing (Pacific Biosciences SMRT analysis software (v1.3.3)). Circular consensus reads (CCS) were counted as reads that had the DNA polymerase pass at least once. SAM files were obtained by aligning FASTQ (of files of the CCS reads) sequences to the genome (either mouse (mm10) or human (hg19)) using GMAP (version 2013-08-19) [37]. Samtools (version 0.1.19) was used to generate BAM and BED files for use in IGV [38]. Data were viewed in IGV and manually counted as primer to primer reads. The plots were generated in R.
Transient siRNA experiments
For transient knockdown experiments, cells were plated in 6-well plates (150,000 cells per well) and were transfected with non-silencing controls (Santa Cruz Biotechnology, sc-37007) or a pool of three siRNAs targeting PTBP1 (Santa Cruz Biotechnology, sc-38280: sense RNA sequences – 1. 5’-CCAAGAACUUCCAGAACAUtt, 2. 5’-CUUGUGGUAUUACCUUGUAtt, 3. 5’-GCAAUUCCAGGCUCAGUAUtt). Cells were plated 24hrs prior to transfections. On the day of transfection, media was switched to 2% serum and transfection complexes were prepared with 50nM of siRNAs using MEM (Gibco, Invitrogen) and RNAi max (Invitrogen) following manufactures procedures. Following 72hrs of exposure to siRNAs, cells were washed, trypsinized, counted and pelleted for downstream assays. For transient transfection assays 3 biological replicates were completed in technical duplicate resulting in 6 measures of each condition. These assays were repeated twice in the laboratory.
Western blot analysis
Total protein lysates were extracted from tissue culture cells using Laemmli buffer, boiled and the protein concentration determined (BCA protein assay, Pierce). Thirty micrograms of protein was resolved on SDS-PAGE gels, transferred to PVDF membranes and detected with a rabbit monoclonal antibody for NOVA1 (Abcam, EPR13847, ab183024, 1:1000 dilution in 5% NFDM), PTBP1 (Abcam, EPR9048B, ab133734, 1:10,000 dilution in 5% NFDM), or PTBP2 (Abcam, EPR9891, ab154853, 1:1000 dilution in 5% NFDM). Protein loading was determined with antibodies against with histone H3 (Anti-Histone H3 antibody produced in rabbit, H0164; Sigma). Blots were imaged with Bio-Rad Chemidoc XRS+ Molecular Imager and quantified with Bio-Rad Image Lab software. Analysis shown in Figure 2B was one cell lysate with the blot repeated twice. Blots shown in Figure 5B were repeated twice from two separate pulldowns. Figure 5D and F were performed twice in the laboratory with 3 biological replicates. Figure 6A and B blots were from 1 biological replicated repeated twice in the laboratory. Figure 6C and H are from biological triplicates repeated twice in the laboratory.
Reverse transcriptase-droplet digital PCR
All cDNAs were diluted 1:4 before use and stored at −80ºC. For hTERT splicing analyses we used iScript Advanced (Bio-Rad) to generate cDNAs, diluted 1:4 and used within 48 hours of production in ddPCR measures. Primer sequences and methods for calculating percent spliced TERT transcripts for TERT are from [22]. Briefly, minus beta, minus alpha, INS3, INS4, and 3’ hTERT (exons 15/16) were measured. Total hTERT was represented by the summation of INS4, and 3’ hTERT (exons 15/16), percentages of the specific splice products calculated, and the remainder assumed to be full length. Individual splice isoform percentages were added together with the percentage of full length TERT and were graphed as percent spliced (Figure 3D; 3 biological replicates and 2 technical replicates). Samples analyzed in Figure 2 represent 3 biological replicates of each cell line. Samples analyzed in Figure 6F are from 3 biological replicates repeated twice in the laboratory.
Droplet digital TRAP assay (telomerase activity)
Quantitation of telomerase enzyme activity was performed using a modified telomeric repeat amplification protocol. Briefly, cells were lysed, diluted and added to the telomerase extension reaction for 40 minutes followed by heat inactivation of telomerase. An aliquot of the extension products was amplified in a droplet digital PCR for 40 cycles and fluorescence measured and droplets read and counted on the droplet reader (QX200, Bio-Rad). Following, data was processed and telomerase extension products per cell equivalents determined. Each droplet digital TRAP assay was repeated in biological triplicate and technical duplicate.
Shortest telomere length measurement
Genomic DNA was extracted using the Gentra Pure DNA extraction kit (Qiagen) according to the manufacturer’s instructions. Each DNA sample was quantified on a Nanodrop (Thermo Scientific) for concentration and purity, and integrity of DNA was determined as previously indicated [28].
The Telomere Shortest Length Analysis (TeSLA) method was performed exactly as described [28]. Briefly, 50 ng of genomic DNA was added to a final volume of 20 μl of ligation buffer containing 1000 units of T4 DNA ligase (New England Biolabs), 1X Cut Smart Buffer (New England Biolabs), 1 mM ATP and 1 nM of TeSLA telorettes (TeSLA Telo 1–6) and incubated at 35°C for 16 hours followed by heat inactivation at 65°C for 10 min. After ligation, genomic DNA was digested using a set of restriction enzymes (2 Units each of CviAII, BfaI, NdeI, and MseI, New England Biolabs) and then treated with 1 unit of Shrimp Alkaline Phosphatase (rSAP, New England Biolabs) at 37°C for 60 min in a final volume of 50 μl. This mixture was subsequently heat inactivated at 80°C for 20 min and 10 μl of sample were added to 10 μl of adapter ligation mix (1 μM AT adapter, 1 μM TA adapter, 1mM ATP, 1X Cut Smart Buffer and 2000 units of T4 DNA Ligase) and incubated at 16°C for 16 hours. After adapter ligation, the sample was heat inactivated at 65°C for 10 min and subsequently diluted to a concentration of 15 pg DNA/ul (1:25 dilution). For each sample analyzed we performed eight independent PCR reactions (94°C for 2 min followed by 26 cycles of 94°C for 15 sec, 60°C for 30 sec and 72°C for 15 min) using a total of 25 μl mix containing 30 pg DNA, 2.5 units of FailSafe enzyme (Epicenter), 1X FailSafe buffer H (Epicenter) and 250 nM primers (adapter and TeSLA TP). PCR products were run on a 0.85 % agarose gel (1.5 V/cm for 19 hours) followed by Southern blot analysis to detect amplified telomeres as previously described [28]. Southern blot images were analyzed by using MATLAB-based software to automatically and accurately detect and size annotate the telomere bands including the percentage of shortest telomeres and average telomere lengths [28]. These southern blot gels were repeated twice in the laboratory.
RNA pulldown with biotinylated RNA baits
A plasmid was generated (TOPO TA) via PCR from a BAC containing hTERT (RP11-990A6, CHORI) using primers that contianed a 1 kb fragment of hTERT intron 8 including DR8. Following integration into the TOPO TA vector, in vitro transcription was performed using the T7 promoter (Ampliscribe T7 kit, Ambion, Life technologies) following the manufacturer instructions including a 45 min DNase step prior to RNA precipitation. RNA was isolated and biotinylated at the 3’ end (Pierce RNA 3’ end biotinylation kit). Biotinylated RNA was purified with streptavidin beads. Cell lysates were prepared following the kit instructions (Peirce Magnetic RNA-protein pull-down kit). Protein-RNA complexes were immunoblotted for either NOVA1, PTBP1 or PTBP2 following pulldown. To produce the smaller RNA baits, T7 promoter sequences were incorporated into the 5’end of the forward primers of each region of interest in and surrounding hTERT DR8. The same procedure was followed as previously described for the 1 kb baits to generate the smaller RNA baits. Furthermore, we generated mutant baits with the same procedure. Due to the low abundance of endogenous NOVA1 in most cancer cells, to perform the NOVA1 pulldowns 293T cells were transfected with V-5-tagged NOVA1 cDNA construct using lipofectamine 2000. After 48 hours, triplicate samples of 10 × 106 cell were washed, trypisinized, counted, pelleted and frozen at −80ºC until analysis. For PTBP1 pulldowns endogenous PTBP1 was able to be probed because it is very abundant. Sample sizes for each pulldown are reported in the western blot section with each figure represented. Sequences for primers used to generate baits (1 kb, wild type and mutant) are as follows:
| T7 Tagged Forward Primers for Oligos 1-5 (PTBP1) | Sequence (5’-3’) |
| PT_1 T7 F LY | TAATACGACTCACTATAGGGCGTATCTGCTTGCGTTGAC |
| PT_2 T7 F LY | TAATACGACTCACTATAGGCACCAGCAAGGAAAGCCTC |
| PT_3 T7 F LY | TAATACGACTCACTATAGCGATCTCAACTCACTGCAACCTC |
| PT_4 T7 F LY | TAATACGACTCACTATAGAGGCTGGTCTCGAACTCCTG |
| PT_5 T7 F LY | TAATACGACTCACTATAGGCAAGCGTCTCTTAGCAACAGG |
| Revers Primers for Oligo 1-5 (PTBP1) | Sequence (5’-3’) |
| PT_1 R LY | GCTTTCCTTGCTGGTGCAGA |
| PT_2 R LY | CGGAGGTTGCAGTGAGTTGAG |
| PT_3 R LY | TGGGTGGATCACCTGAGGTC |
| PT_4 R LY | AAGAGACGCTTGCAGCCTAC |
| PT_5 R LY | GACAAACAGTGAGAGCAGAATAGC |
| Primers Used for Mutant Construct (PTBP1) | |
| Forward Primers | Sequences (5’−3’) |
| T7 Mut PTBP1 Oligo 3 F | TAATACGACTCACTATAGCGATCTCAACTCACTGCAACCTC |
| T7 Mut PTBP1 Oligo 4 F | TAATACGACTCACTATAGAGGCTGGTCTCGAACTCCT |
| T7 Mut NOVA1 Oligo 3 F | TAATACGACTCACTATAGCGATCTCAACTaACTGCAACCTC |
| T7 Mut NOVA1 Oligo 4 F | TAATACGACTCACTATAGAGGCTGGTCTCGAACTCCT |
| Reverse Primers | Sequences (5’-3’) |
| Mut PTBP1 Oligo 3 R | TGGGTGGATCACCTGAGGTC |
| Mut PTBP1 Oligo 4 R | TTGTGTCGCTTGCAGCC |
| Mut NOVA1 Oligo 3 R | TtGGTtGATCACCTGAGGTC |
| Mut NOVA1 Oligo 4 R | AAGAGACGCTTGCAGCC |
Statistics
Unless otherwise noted in methods section, figure legend, or in the results section, pairwise Student’s t tests (2-sided) were used to determine statistically significant differences between group means. For detailed information please see the methods subsections above and/or the figure legends for exact sample sizes and replicates of each experiment. Unless otherwise noted the ‘error’ bars represent standard error of the mean (s.e.m.). Significant differences were accepted at a p value less than 0.05.
Supplementary Material
Supplementary Figure 1: Long-read length sequencing work flow. (A) Work flow cartoon showing sources of the human RNA (HeLa cells) and how the gene specific cDNA was made. We used two reverse transcriptases to make cDNA. We then constructed PCR libraries using either an exon 1 or exon 2 primer with an exon 16 primer. Following PCR and barcoding, libraries were size selected into specific pools and sent for sequencing. For sequencing, hairpin adapters were added to PCR products prior to sequencing. Data were then processed, de-multiplexed and aligned to the genomes (human or mouse). We visualized the reads with IGV and counted primer to primer full length reads (circular consensus reads-CCS reads) and recorded the isoforms observed. (B) Agarose gel image of mouse TERT amplified with exon 1 and 16 primers and exon 2 and 16 primers. (C). Splicing landscape of hTERT following sequencing and data analysis of exon 1 to 16 libraries.
Supplementary Figure 2: PTBP2 Co-IP with NOVA1 in 293T + NOVA1 cells. Western blot probing for PTBP2 post crosslinking with NOVA1 in 293T cells that overexpress NOVA1.
Acknowledgements
We thank Drs. John Minna and Adi Gazdar for cell lines. We acknowledge Henrietta Lacks and her family members for the important contributions “HeLa” cells have made to this work. This work was performed in laboratories constructed with support from NIH grant C06 RR30414. This work was supported by AG01228 from the National Institute on Aging (WEW and JWS) and by a T32 Training Grant CA124334 (JWS). A Pathway to Independence award to ATL (K99/R00 CA197672-01A1, National Cancer Institute (NCI)). We also acknowledge the Harold Simmons NCI Designated Comprehensive Cancer Center Support Grant (CA142543), the NCI SPORE (CA070907), and the Southland Financial Corporation Distinguished Chair in Geriatric Research (J.W.S. and W.E.W.).
Footnotes
Conflict of interest
The authors declare no financial or other conflicts of interest related to this manuscript.
Data has been deposited in a public repository https://www.ncbi.nlm.nih.gov/sra/PRJNA506254
Supplementary information is available online at the ONCOGENE website
References
- 1.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266: 2011–2015. [DOI] [PubMed] [Google Scholar]
- 2.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer 1997; 33: 787–791. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 5.Wright WE, Brasiskyte D, Piatyszek MA, Shay JW. Experimental elongation of telomeres extends the lifespan of immortal × normal cell hybrids. EMBO J 1996; 15: 1734–1741. [PMC free article] [PubMed] [Google Scholar]
- 6.Avilion AA, Piatyszek MA, Gupta J, Shay JW, Bacchetti S, Greider CW. Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res 1996; 56: 645–650. [PubMed] [Google Scholar]
- 7.Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997; 90: 785–795. [DOI] [PubMed] [Google Scholar]
- 8.Yi X, Shay JW, Wright WE. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic acids research 2001; 29: 4818–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hrdlickova R, Nehyba J, Bose HR Jr. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol Cell Biol 2012; 32: 4283–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim W, Ludlow AT, Min J, Robin JD, Stadler G, Mender I et al. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol 2016; 14: e2000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi X, White DM, Aisner DL, Baur JA, Wright WE, Shay JW. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2000; 2: 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res 1998; 58: 4168–4172. [PubMed] [Google Scholar]
- 13.Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK et al. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 1997; 6: 2011–2019. [DOI] [PubMed] [Google Scholar]
- 14.Ulaner GA, Hu JF, Vu TH, Oruganti H, Giudice LC, Hoffman AR. Regulation of telomerase by alternate splicing of human telomerase reverse transcriptase (hTERT) in normal and neoplastic ovary, endometrium and myometrium. International journal of cancer 2000; 85: 330–335. [PubMed] [Google Scholar]
- 15.Yi X, Shay JW, Wright WE. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res 2001; 29: 4818–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong MS, Wright WE, Shay JW. Alternative splicing regulation of telomerase: a new paradigm? Trends Genet 2014; 30: 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene 2014; 33: 5311–5318. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 2013; 3: 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong MS, Chen L, Foster C, Kainthla R, Shay JW, Wright WE. Regulation of telomerase alternative splicing: a target for chemotherapy. Cell Rep 2013; 3: 1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomes NM, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR et al. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 2011; Accepted Article; Doi: 10.1111/j.1474-9726.2011.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seluanov A, Chen Z, Hine C, Sasahara TH, Ribeiro AA, Catania KC et al. Telomerase activity coevolves with body mass not lifespan. Aging Cell 2007; 6: 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ludlow AT, Wong MS, Robin JD, Batten K, Yuan L, Lai T-P, Dahlson N, Zhang L, Mender I, Tedone E, Sayed ME, Wright WE, Shay JW NOVA1 regulates hTERT splicing and cell growth in non-small cell lung cancer. Nature communications 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Withers JB, Ashvetiya T, Beemon KL. Exclusion of exon 2 is a common mRNA splice variant of primate telomerase reverse transcriptases. PLoS One 2012; 7: e48016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong MS, Shay JW, Wright WE. Regulation of human telomerase splicing by RNA:RNA pairing. Nature communications 2014; 5: 3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romanelli MG, Diani E, Lievens PM. New insights into functional roles of the polypyrimidine tract-binding protein. Int J Mol Sci 2013; 14: 22906–22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis 2014; 3: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludlow AT, Robin JD, Sayed M, Litterst CM, Shelton DN, Shay JW et al. Quantitative telomerase enzyme activity determination using droplet digital PCR with single cell resolution. Nucleic Acids Res 2014; 42: e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai TP, Zhang N, Noh J, Mender I, Tedone E, Huang E et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nature communications 2017; 8: 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito Y, Miranda-Rottmann S, Ruggiu M, Park CY, Fak JJ, Zhong R et al. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. Elife 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polydorides AD, Okano HJ, Yang YY, Stefani G, Darnell RB. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc Natl Acad Sci U S A 2000; 97: 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linares AJ, Lin CH, Damianov A, Adams KL, Novitch BG, Black DL. The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife 2015; 4: e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holohan B, Wright WE, Shay JW. Cell biology of disease: Telomeropathies: an emerging spectrum disorder. J Cell Biol 2014; 205: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young AJ. The role of telomeres in the mechanisms and evolution of life-history trade-offs and ageing. Philos Trans R Soc Lond B Biol Sci 2018; 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aviv A, Anderson JJ, Shay JW. Mutations, Cancer and the Telomere Length Paradox. Trends Cancer 2017; 3: 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monaghan P, Eisenberg DTA, Harrington L, Nussey D. Understanding diversity in telomere dynamics. Philos Trans R Soc Lond B Biol Sci 2018; 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 2004; 64: 9027–9034. [DOI] [PubMed] [Google Scholar]
- 37.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005; 21: 1859–1875. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Long-read length sequencing work flow. (A) Work flow cartoon showing sources of the human RNA (HeLa cells) and how the gene specific cDNA was made. We used two reverse transcriptases to make cDNA. We then constructed PCR libraries using either an exon 1 or exon 2 primer with an exon 16 primer. Following PCR and barcoding, libraries were size selected into specific pools and sent for sequencing. For sequencing, hairpin adapters were added to PCR products prior to sequencing. Data were then processed, de-multiplexed and aligned to the genomes (human or mouse). We visualized the reads with IGV and counted primer to primer full length reads (circular consensus reads-CCS reads) and recorded the isoforms observed. (B) Agarose gel image of mouse TERT amplified with exon 1 and 16 primers and exon 2 and 16 primers. (C). Splicing landscape of hTERT following sequencing and data analysis of exon 1 to 16 libraries.
Supplementary Figure 2: PTBP2 Co-IP with NOVA1 in 293T + NOVA1 cells. Western blot probing for PTBP2 post crosslinking with NOVA1 in 293T cells that overexpress NOVA1.