

Abstract
Uncontrolled scarring, or fibrosis, can interfere with the normal function of virtually all tissues of the body, ultimately leading to organ failure and death. Fibrotic diseases represent a major cause of death in industrialized countries. Unfortunately, no curative treatments for these conditions are yet available, highlighting the critical need for a better fundamental understanding of molecular mechanisms that may be therapeutically tractable. The ultimate indispensable effector cells responsible for deposition of extracellular matrix proteins that comprise scars are mesenchymal cells, namely fibroblasts and myofibroblasts. In this review, we focus on the biology of these cells and the molecular mechanisms that regulate their pertinent functions. We discuss key pro-fibrotic mediators, signaling pathways, and transcription factors that dictate their activation and persistence. Because of their possible clinical and therapeutic relevance, we also consider potential brakes on mesenchymal cell activation and cellular processes that may facilitate myofibroblast clearance from fibrotic tissue - topics that have in general been understudied.
Keywords: fibroproliferative diseases, fibroblast, myofibroblast, differentiation proliferation, apoptosis, signaling pathways, de-differentiation
Introduction
Fibrosis is a process in which fibrous connective tissue is deposited in an organ or tissue. It can occur in a self-limited physiologic form in the context of wound healing, or as an excessive and progressive pathologic form which results in tissue remodeling and stiffening with eventual functional impairment of affected organs. Pathologic fibrosis can occur in virtually all organs, and such diseases are collectively termed fibrotic disorders or fibroproliferative diseases (FPDs). A startling statistic which attests to the impact of FPDs is that they account for approximately 45% of all deaths in industrialized countries [1]. Fibrosis can result from a variety of forms of acute and chronic tissue injury, and while some organ-specific differences exist, the cellular and molecular processes which drive it are largely conserved. In this review, we will briefly summarize the current understanding of cellular and molecular events involved in the initiation and evolution of FPDs. Although epithelial and bone marrow-derived cells play important facilitative roles in fibrogenesis, only mesenchymal cells, particularly fibroblasts (Fibs), are entirely indispensable. For this reason, we will focus on the regulation of resident tissue Fibs and their differentiation to myofibroblasts (MFibs) - which are most responsible for the elaboration of extracellular matrix proteins such as type I collagen (Col I) that comprise tissue scars. We will review mediators and molecular pathways important in shaping important functional phenotypes of Fibs, including proliferation, MFib differentiation, and apoptosis resistance. We will also discuss two facets that have received comparatively little attention: namely, endogenous molecular brakes on Fib activation - which are often impaired in FPDs - as well as the potential for de-differentiation of MFibs. Each of these considerations has important therapeutic implications. It should be noted that even these bodies of literature are too expansive to cover in their entirety, and we have therefore exercised selectivity in what is included in this review.
The spectrum of FPDs
FPDs comprise a large group of diverse diseases affecting virtually all organs. For most FPDs, the etiologic factors - which can be either exogenous or endogenous - are reasonably well understood. Idiopathic pulmonary fibrosis (IPF) is unusual among FPDs because, although a variety of risk factors are epidemiologically associated with disease, no direct inciting injury has been identified as responsible. Exogenous (or extrinsic) exposures to a gamut of hazardous substances are recognized to cause fibrosis of various organs. Liver injury from excessive consumption of alcohol leads to the fibrotic condition of cirrhosis [2]. Radiation therapy of malignancies can result in fibrosis of exposed organs [3]. Inhalational exposure to a variety of occupational agents elicits pulmonary fibrosis; these include asbestos (resulting in asbestosis) and silica (resulting in silicosis) [4]. The lungs are also especially sensitive to fibrotic injury to a variety of chemotherapeutic drugs, including hydroxyurea, methotrexate, cyclophosphamide, and bleomycin (the latter being the impetus for its use as a commonly employed animal model of pulmonary fibrosis). FPDs are a recognized sequella of a number of viral infections. For example, chronic infection with hepatitis virus B or C predisposes patients to cirrhosis [5]. Likewise, infections with coxsackievirus and parvovirus can lead to the development of chronic myocardial fibrosis and infection with gamma-herpesvirus to lung fibrosis [6,7]. Tissue fibrosis can also result from endogenous (or intrinsic) inflammatory insults. These include those associated with autoimmune diseases (e.g., pulmonary fibrosis in scleroderma and rheumatoid arthritis, and pancreatic fibrosis in type I diabetes) as well as those associated with ischemic injury to various organs (e.g., heart, kidney) [8–10] (see Fig.1).
Fig. 1.
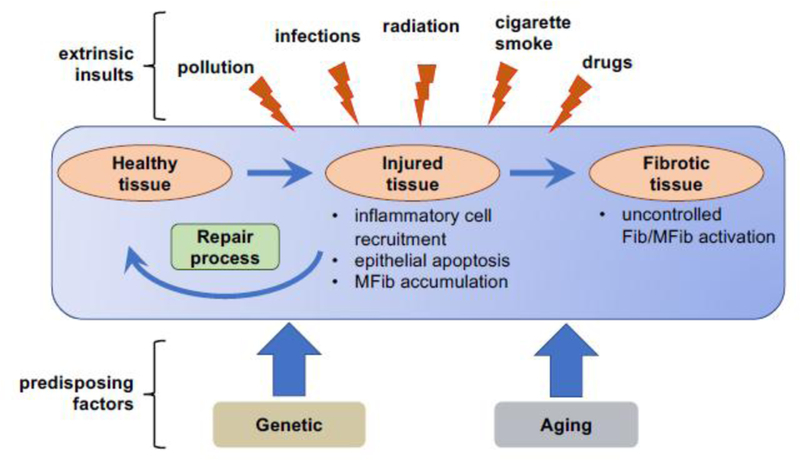
A variety of extrinsic factors can damage healthy tissue, resulting in epithelial cell death/apoptosis, local inflammation, and activation of mesenchymal cells (e.g., Fibs). Under physiological conditions, homeostatic repair processes restore healthy tissue. When repair processes fail, Fibs/MFibs become aberrantly and persistently activated, leading to deposition of excess ECM and impaired tissue function.
Physiology of wound healing and tissue repair
Wound healing is a physiologic, self-limited restorative response to breaches of tissue integrity which is intended to reduce further damage, prevent infections, and restore normal tissue functions. A brief summary of its key features is appropriate, since pathologic fibrosis (discussed below) represents an aberrant form of this homeostatic process. A typical wound involves a discrete injury to epithelial cells, and frequently extends to involve the underlying vascular endothelial cells. An early subsequent event is platelet aggregation and activation of the coagulation cascade to form fibrin clots. Platelet-derived chemokines and cytokines initiate recruitment of endothelial cells and subsequent angiogenic responses, and of macrophages which elaborate growth factors such as transforming growth factor β (TGF-β). TGF-β confers on Fibs the ability to express contractile genes such as α-smooth muscle actin (α-SMA); this hybrid myocyte/Fib is termed a MFib. Both the contractile ability of MFibs as well as their capacity to produce particularly large amounts of extracellular matrix (ECM) proteins such as Col I, fibronectin, and proteoglycans promote wound contraction and scar formation. The amount of ECM proteins deposited is further controlled by the balance of matrix metalloproteinases (MMPs) and their endogenous inhibitors, tissue inhibitors of MMPs (TIMPs). Epithelial cells from the surrounding wound margins then proliferate and migrate to re-epithelialize the denuded surface. Resolution is the last phase of the healing process and involves the loss of recruited cells as well as MFibs via apoptosis. Normal wound healing requires that each of these stages be tightly regulated and orchestrated in order to minimize any adverse impact on tissue function [11].
Pathophysiology of tissue fibrosis
While physiological wound healing is self-limited, pathological fibrotic responses are persistent and often progressive. This leads to the excessive accumulation of mesenchymal cells and ECM sufficient to disrupt normal cellular architecture and thus impair organ function. This can occur either because of repeated or persistent injury, or because of an imbalance favoring pro-fibrotic events over pro-resolution events. Events favoring fibrogenesis include impaired epithelial integrity/repair, persistent or unchecked inflammation, deregulated M1/M2 macrophage polarization, expansion of Fib/MFib numbers owing to increased proliferation and/or decreased apoptosis, and an imbalance of ECM synthesis/degradation favoring its accumulation. Each of these processes, in turn, are subject to the possible influence of genetic and epigenetic factors. We will now delve into the roles in fibrogenesis of the most critical of these cell types, providing a brief overview of epithelial cells and macrophages before shifting attention for the rest of this review to our emphasis on mesenchymal cells. Although other cells may also contribute to tissue fibrosis in a tissue- and insult-specific manner, these will not be considered here.
Epithelial cells.
Under normal conditions, the epithelium serves as a critical determinant of homeostasis and a brake on fibrogenesis. In the lung, this vital function reflects its ability to provide a physical barrier from the outside world, secrete surfactant which prevents alveolar collapse, and elaborate mediators that inhibit Fib proliferation and activation (e.g., prostaglandin E2 [PGE2], discussed below). The crucial importance of the epithelium as a curb on fibrosis is evidenced by the finding that diphtheria toxin-induced injury targeted to the alveolar epithelium was sufficient to elicit pulmonary fibrosis [12]. On the other hand, pathological fibrosis is often characterized by impaired epithelial integrity, reflecting epithelial cell dysfunction which hinders their ability to proliferate, migrate towards a site of injury, and survive. Such injury may also diminish their capacity to produce antifibrotic substances such as PGE2, allowing unchecked inflammatory and wound healing responses. Furthermore, injured epithelial cells also acquire the ability to produce TGF-β, which drives the activation of Fibs and their differentiation to MFibs. Injured epithelial cells have also been implicated as potential precursors of mesenchymal cells including MFibs in a process termed epithelial-mesenchymal transition (EMT), the significance of which will be considered below.
A diverse spectrum of exogenous forms of epithelial cell injury are recognized to cause fibrosis of various organs. Furthermore, in the lung, a variety of genetic abnormalities that impair epithelial cell integrity have also been shown to cause or to predispose to fibrosis. Although only a small proportion of patients with pulmonary fibrosis exhibit a familial pattern, this form has been linked with several mutations or SNPs in genes that predispose to epithelial damage. One class of such mutations is in genes such as TERT and TERC that result in shortening of telomeres. Because telomere shortening limits cellular replicative capacity, such abnormalities recapitulate the effects of aging - which itself has been identified as a risk factor for FPDs and in animal models involving fibrosis of the lung [13,14], heart [15], liver [16] and kidney [17]. Another class of genomic alterations identified in familial pulmonary fibrosis involves genes that encode lung surfactant proteins expressed exclusively by type II alveolar epithelial cells; accumulation of these mutant proteins leads to endoplasmic reticulum (ER) stress and induction of apoptosis in epithelial cells. Similarly, mutations in the gene encoding ATP-binding cassette protein member A3 (ABCA3, a surfactant phospholipid carrier protein specifically expressed in the alveolar epithelium) are associated with fatal neonatal interstitial pulmonary fibrosis [18]. A polymorphism in the promoter region of another epithelial gene, that encoding the mucin 5B (MUC5B) protein, has emerged from GWAS studies as the strongest genetic predisposition to sporadic IPF [19], and has also recently been reported to increase the frequency of connective tissue disease-associated pulmonary fibrosis [20].
Macrophages.
Chronic inflammation can result in fibrosis. This generally reflects the ability of inflammatory cells to secrete tissue-injurious and pro-inflammatory substances such as proteases, lipases, and reactive oxygen species, as well as pro-inflammatory cytokines, chemokines, and lipid mediators. Although many types of activated inflammatory cells can promote fibrogenesis - including neutrophils, eosinophils, lymphocytes, and mast cells - we will elaborate further only on the contributions of macrophages. Macrophages are particularly relevant in chronic FPDs because of their much longer half-lives in affected tissues than those of other inflammatory cell types.
Macrophages contribute to normal wound healing and tissue homeostasis by virtue of their well-recognized abilities to ingest and clear cell debris as well as apoptotic cells, produce MMPs, and elaborate a panoply of mediators and growth factors. The resident macrophages that populate most organs are now recognized to be largely derived from embryonic or fetal precursors, and to maintain their numbers by self-replication. When homeostasis is perturbed, bone marrow-derived monocytes are recruited from the circulation to sites of injury, thus supplementing the resident population of mononuclear phagocytic cells. These recruited cells typically manifest a more inflammatory phenotype than do resident tissue macrophages, and recent data suggest that these are particularly important in driving chronic tissue injury, inflammation, and subsequent fibrotic responses [21]. Macrophages exhibit a high degree of phenotypic plasticity, and phase-specific shifts in their phenotype during wound healing responses are also key determinants of fibrogenesis. In the early inflammatory phase, macrophages in most tissues exhibit a predominantly pro-inflammatory or M1 phenotype, characterized by a high capacity for phagocytosis and production of inflammatory cytokines and MMPs. The later phase of wound healing is dominated by a shift towards M2-like macrophages which elaborate anti-inflammatory substances as well as angiogenic and mitogenic growth factors. While these properties facilitate resolution of inflammation and restoration of homeostasis, the excessive and unchecked production by M2 cells of pro-fibrotic substances, especially TGF-β, fosters tissue fibrosis [21]. While attempts have been made to further classify M2 macrophages into several subsets, the applicability and utility of doing so remains controversial. We suggest that the subset of macrophages involved in tissue fibrosis is best reflected by their elaboration of pro-fibrotic mediators such as TGF-β, rather than by any particular classification designation.
Mesenchymal cells.
It is evident from the previous sections that epithelial cells and macrophages play important roles in the initiation and perpetuation of tissue fibrosis. However, the actions of these two cell types are ultimately directed at mesenchymal cells. By virtue of their dominant role in ECM synthesis, mesenchymal cells are the ultimate and indispensable effector cells of fibrosis. The relevant mesenchymal cell types here include Fibs and MFibs, which represent the end points of a phenotypic continuum. In the next sections we will discuss the relevant properties of and phenotypic relationship between these two related cell types. Although our lens reflects our investigative focus on pulmonary fibrosis, much of the subsequent discussion is highly applicable to FPDs of other organs as well.
Pertinent cellular properties of activated Fibs and MFibs
Fibs are well-recognized to synthesize and secrete a panoply of molecules that, in autocrine and paracrine fashion, can promote or suppress fibrotic tissue responses [22]. However, herein we will focus on a set of functional responses that are pivotal in promoting fibrosis - namely, proliferation, differentiation, migration, and persistence. These properties reflect Fib responses to a wide variety of soluble mediators as well as physical forces, discussed below.
Proliferation of Fibs
It is likely that proliferation is the predominant determinant of Fib expansion in FPDs [23]. In vitro studies with mitogens unequivocally demonstrate the proliferative capacity of Fibs and provide mechanistic understanding of the relevant signaling pathways. In vivo assessment of fibrotic tissue for proliferation specifically of Fibs can be challenging because of the lack of cellular markers that are expressed uniquely in this cell type. However, efforts utilizing serial sections of fibrotic lung tissue have demonstrated that cells staining positive for proliferation markers Ki67 or PCNA also have a characteristic spindle-shape or express Col I. Of the many growth factors listed in Table 1, fibroblast growth factor (FGF-2) and platelet-derived growth factor (PDGF) have perhaps been the most reliable in stimulating proliferation of Fibs. Numerous reports have suggested a requirement for AKT activation in Fib proliferation as well as induction of genes involved in the cell cycle, and aberrant activation of AKT has been demonstrated in fibrotic tissue of many organs [24]. A recent study identified a role for the transcription factor FOXM1 in transducing mitogen-induced AKT activation into cell cycle gene expression with subsequent proliferation of Fibs [25]. Another molecular player implicated in Fib proliferation and cell cycle activation is Hic-5, a transcriptional co-regulator [26]. Fib proliferative and activation responses have also been associated with promoter hypermethylation and transcriptional silencing of Ras protein activator like 1 (RASAL1), an endogenous brake on their activation, in renal fibrosis [21]. Molecular cross-talk among a diverse array of transcriptional regulators of Fib proliferation is therefore likely. Moreover, their relative importance may vary depending on the mitogen or the tissue.
Table 1.
Soluble factors involved in Fib activation
| Soluble drivers of fibrosis | Functions | References | |
|---|---|---|---|
| Growth factors | TGF-β | Fib differentiation | [82] |
| CTGF | [83] | ||
| ET-1 | Fib proliferation | [84,85] | |
| CTGF | [86] | ||
| PDGF | [25,87] | ||
| FGF | [25] | ||
| IGF | [88] | ||
| epidermal growth factor (EGF) | [89] | ||
| vascular endothelial growth factor (VEGF) | [90] | ||
| Angiotensin II | [91] | ||
| Cytokines | TNF-α | Fib proliferation | [92] |
| Osteopontin | [93] | ||
| IL-1β | [94] | ||
| IL-4 | [95] | ||
| IL-13 | [96] | ||
| IL-6 | [97] | ||
| Chemokine ligands (CCLs) | CCL2 | Fib proliferation | [98,99] |
| CCL11 | [100] | ||
Differentiation into MFibs
Resident tissue Fibs are considered to be quiescent until they are exposed to external activation stimuli, but different stimuli may elicit different responses. As noted previously, in vitro stimulation with TGF-β unequivocally elicits a phenotypic transition of Fibs into α-SMA-expressing MFibs. MFibs are spindle-shaped cells with phenotypic features intermediate between those of Fibs and smooth muscle cells. Like Fibs, MFibs too synthesize and secrete ECM proteins such as collagen, especially the Col I that is the predominant collagen of interstitial scar tissue. Importantly, however, the ECM protein synthetic capacity of MFibs is greater than that of Fibs. Like smooth muscle cells, MFibs also express contractile genes such as α-SMA. These unique hybrid properties of ECM generation and contractile gene expression serve to identify and mark these important cells and render them indispensable in wound contraction and tissue remodeling.
TGF-β-induced differentiation of Fibs to MFibs involves changes in expression of numerous genes besides simply α-SMA and Col I. For example, transcriptomic analysis in differentiating lung Fibs revealed that TGF-β increased expression of ~600 genes while simultaneously decreasing expression of a similar number of genes [28]. The changes in expression of many of these genes during MFib differentiation can be explained by epigenetic regulatory mechanisms [29,30]. For instance, increased expression of Mfib-specific genes including α-SMA, Col I, TGF-β itself, and TIMP1 requires methylation of histone 3, lysine 4 (H3K4). At the same time, down-regulation in skin Fibs treated with TGF-β of Fli 1, a known transcriptional repressor of the Col I gene [31], involved histone acetylation [32]. Such changes help to explain why MFibs are exuberant producers of Col I. Additionally, FN1 down-regulation has also been identified in dermal Fibs from patients with scleroderma, although the operative mechanism in this instance was promoter hypermethylation [33].
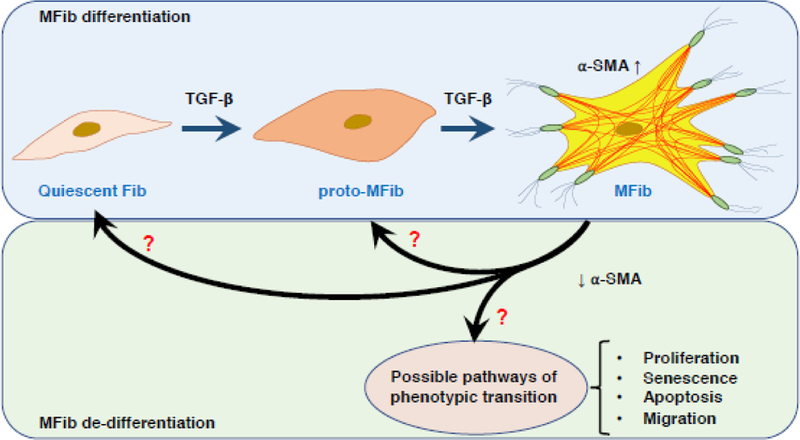
Although the differentiation of Fibs to MFibs is often framed as a dichotomous “switch,” it is far more likely to reflect a multi-step process that is better conceptualized as a transition along a continuum of discrete phenotypes. The complexity of this process may be even greater and more nuanced in vivo than it is in vitro. Evidence now suggests that prior to the process of wound contraction, activation of resident Fibs by inflammatory cytokines is necessary to elicit expression of β- and γ-cytoplasmic actins which facilitate their migration towards the wound area [34]. The morphological features of these inflammatory cytokine-activated Fibs resemble MFibs, but they fail to express α-SMA. These activated Fibs with migration capacity but lacking α-SMA have been designated “proto-MFibs [35].” Proto-MFibs synthesize and secrete two major ECM proteins, EDA-containing cellular fibronectin (EDA-FN) and Col I, which facilitate wound contraction under normal physiological conditions. Thus, proto-MFibs resemble an intermediate (activated) stage in the continuum of Fib to MFib differentiation. A number of pro-fibrotic signaling pathways (discussed below) leads to subsequent differentiation of these proto-MFibs into α-SMA-expressing MFibs.
Persistence of MFibs
As noted earlier, the contractile force generated by MFibs is necessary for physiological wound healing. However, once the tissue integrity is restored, maintenance of tissue homeostasis requires that MFibs disappear from the site of injury. The mechanisms for this loss of MFibs might theoretically include apoptosis [36], accelerated senescence [37,38], and de-differentiation (reversion to a quiescent Fib phenotype) [39]. Of these, apoptotic cell death of MFibs has been the best studied. Apoptosis is a form of programmed cell death that, unlike necrotic cell death, is compatible with tissue homeostasis. Surface expression of death receptors such as Fas, tumor necrosis factor (TNF)-receptor-l, and TNF-related apoptosis inducing ligand receptors −1 and −2 play an important role in apoptosis of MFibs. During the resolution phase of normal wound healing, surface expression of Fas receptor is necessary and sufficient for Fas ligand (FasL)-induced apoptosis of MFibs [40]. Unlike the efficient MFib apoptosis that characterizes normal wound healing, this process is very limited or absent in FPDs. This relative lack of apoptosis is an important contributor to the expansion and persistence of MFibs that characterize pathological wound healing in the context of fibrogenesis and to their uncontrolled degree of ECM deposition. Fibs from fibrotic lung have been shown to resist apoptosis elicited by FasL/Fas [41,42]. Such apoptosis resistance is also a typical characteristic of differentiated MFibs elicited by treatment with TGF-β. Apoptosis resistance in MFibs has been linked with alterations in expression of a variety of genes that mediate or regulate programmed cell death. For example, IPF Fibs that are resistant to FasL-induced apoptosis show diminished expression of Fas receptor [43], and this was subsequently attributed to epigenetic changes in histones (especially trimethylation of H3K9) [43]. Other anti-apoptotic genes whose expression is increased in MFibs include survivin [44], cellular FLICE-like inhibitory protein (c-FLIP) [42]. X-linked inhibitor of apoptosis protein (XIAP) [45], and Bcl-2 [46].
Migration of Fibs/MFibs
Although its importance (relative to proliferation and persistence) to mesenchymal cell accumulation at sites of fibrosis is not known, Fibs are also capable of migration from distal anatomic sites. Many growth factors implicated in Fib proliferation also promote their migration. As is true for proliferation, aberrant activation of AKT is crucial for Fib migration during fibrosis [47]. A property that is related to Fib migration is their invasiveness. This is positively regulated by the increased expression of cell surface receptor CD44 and hyaluronan synthase 2 [48–50]. The penetration of migrating Fibs through the interstitial matrix is facilitated by MMPs (MMP-9, −12 and −14) and opposed by TIMPs (e.g. TIMP3 and ADAM metallopeptidase with thrombospondin type 1 motif 1) [48]. Contractile proteins such as α-SMA have also been reported to contribute to the migratory capacity of Fibs. The role of contractile protein-facilitated Mfib migration in their accumulation within fibrotic foci remains uncertain.
Cellular origin of MFibs
The cellular precursors of differentiated MFibs in FPDs has been a topic of interest and some controversy. Resident tissue Fibs share mesenchymal origins with MFibs and thus are their most obvious precursors. The alternative cellular sources of MFibs that have received the most investigative attention are epithelial cells and fibrocytes. In response to pro-fibrotic factors such as TGF-β, epithelial cells can lose characteristic lineage markers (e.g., E-cadherin) and acquire mesenchymal markers in a process termed EMT [51]. Fibrocytes represent a small fraction of bone marrow-derived CD34+ circulating monocytes that express Col I [52], and which have been shown to traffic to injured tissues during fibrogenesis. Other candidate MFib precursor cells include endothelial cells [53], pericytes [54], adipocytes [55], and mesenchymal stem cells [56]. This question of MFib origin has been investigated by lineage tracing studies in various mouse fibrosis models. A number of such studies have concluded that an in vivo role for EMT in lung fibrosis is either absent or minimal [57–59]. Similar conclusions have come from studies in models of liver [60] and renal [54] fibrosis. Likewise, in a renal fibrosis model, the contribution of fibrocytes was shown to be minor [61]. In a number of these studies, the resident Fib has instead proven to be the major source of MFibs. We suggest that a variety of cell types represent potential MFib precursors, with their relative importance depending on the organ, the circumstance, and the individual; however, resident tissue Fibs are the predominant precursor cell type under most circumstances. An exception to this generalization appears to be in liver fibrosis, where lineage tracing studies have demonstrated that the dominant MFib precursor is the hepatic stellate cell [62] a cell unique to the liver with features of both pericytes and Fibs. Regardless of their origin, all MFibs within fibrotic tissue express contractile proteins such as α-SMA, produce large amounts of ECM proteins, and exhibit relative resistance to apoptosis.
Heterogeneity of Fibs and MFibs
The increasing application of single cell transcriptomic analysis is revealing that within any given tissue, cells of a given type often represent multiple heterogeneous subpopulations. Recent reports show this to be true for MFibs in lung fibrosis [63–65] and renal fibrosis [66], and this likely applies to other FPDs as well. Heterogeneity is also evident at a functional level when comparing cells from individual patients. For example, Fibs outgrown from lung tissue of different IPF patients have demonstrated variability in gene expression profiles, proliferation ability, resistance to apoptosis, and response to various growth factors [67,68]. Heterogeneity among patients certainly reflects inherent genetic variations. Heterogeneity within an individual patient likely reflects variations in the nature of the initiating injuries, the cells of origin, the mix of pertinent mediators in the local milieu, and in anatomic location that may influence determinants such as stiffness, blood flow, and oxygen tension. An example of the latter includes differences in MFibs found in the upper and lower lobes of IPF lung [69]. It is highly likely that epigenetic mechanisms mediate some of these heterogeneous responses within and among patients. Although it adds complexity, the heterogeneity of MFibs represents fertile ground for discovery of new insights into disease pathogenesis and therapeutic targeting.
Soluble drivers of tissue fibrosis
In both physiological and pathological wound healing responses, a wide variety of soluble mediators such as cytokines, chemokines, lipid mediators, and growth factors have been identified as key signals which direct the behavior of relevant cellular players in response to tissue injury. The cellular responses and phenotypes that dictate fibrogenesis ultimately reflect the net actions of pro- and anti-fibrotic mediators and signals. A large number of pro-fibrotic mediators have been identified. TGF-β has been the most extensively investigated; others that are reasonably well-studied include endothelin 1 (ET-1), connective tissue growth factor (CTGF), interleukin (IL)-13, PDGF, FGF-2, and insulin-like growth factor (IGF)-½. The actions of all of these are typically pleiotropic and often overlap with those of the others (see Table 1). We will provide a brief overview of the actions of some of these mediators. In addition to these soluble factors, mechanical forces exerted by the ECM matrix in which Fibs and MFibs reside also provide activation signals that contribute to fibrotic responses; this process will be discussed below.
TGF-β is commonly considered the master pro-fibrotic cytokine and is recognized to play a central role in FPDs involving the lung [70], heart [71,72], liver [73], and kidneys [74,75]. Macrophages and injured epithelial cells are recognized as the major cellular sources for TGF-β in fibrotic tissue [76], but numerous other cell types, including neutrophils, endothelial cells, Fibs and MFibs also produce TGF-β [77]. TGF-β exerts three critical pro-fibrotic actions, namely, its ability to promote: 1) apoptosis of epithelial cells while inhibiting apoptosis of Fibs/MFibs (termed “the apoptosis paradox”); 2) mesenchymal transition of epithelial cells, pericytes, fibrocytes, and adipocytes, and transdifferentiation of Fibs themselves, to yield MFs [61,78]; and 3) ECM protein production, most notably by Fibs and MFibs. In view of the central importance of these diverse actions of TGF-β, we will consider the mechanisms by which it signals subsequently.
Like TGF-β, ET-1, CTGF, and IL-13 have also been shown to promote Fib differentiation to a MFib phenotype with the attendant increases in α-SMA expression, Col I production, and apoptosis resistance. IL-13 is a prominent constituent of type 2 inflammatory responses and contributes to fibrotic remodeling of the airways during chronic allergic inflammation. Of note, ET-1 and CTGF are also transcriptional targets for TGF-β [79,80], implying that these mediators are likely to be co-expressed and to cooperate in many fibrogenic responses. In contrast to this set of mediators, PDGF, FGF-2, and IGF-½ have minimal capacity to induce MFib differentiation but are strong Fib mitogens. These contrasting phenotypic responses are consistent with long-held notions that cellular proliferation and differentiation may represent distinct and mutually exclusive programs [81]. In addition to these, a number of other cytokines, growth factors, and chemokines have been implicated in Fib activation (see Table 1)
Signaling pathways mediating Fib activation phenotypes
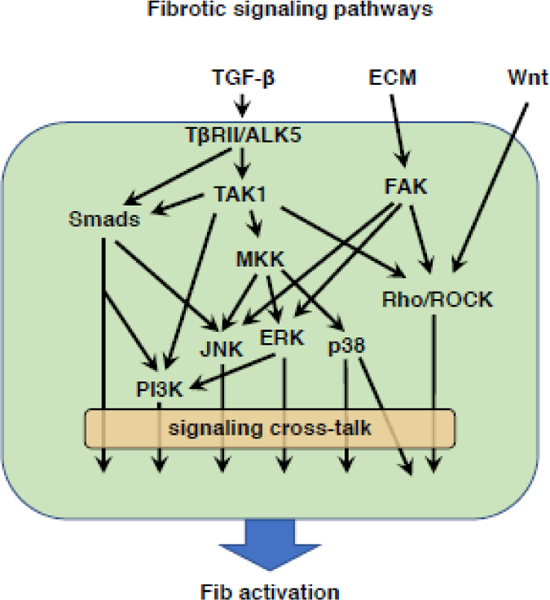
The processes of proliferation, differentiation, and survival of Fibs reflect the output of a variety of signal transduction pathways. Although certain of these pathways promote specific phenotypic endpoints, it is common for individual pathways to both interact with each other and to influence the development of more than one functional process. Because of the importance of TGF-β as a pro-fibrotic driver, we will first discuss its receptors and signaling. Subsequently, we will discuss several additional major signaling pathways mediating Fib activation and differentiation. Although these pathways are considered individually, how they interact requires much greater understanding.
TGF-β receptors and signaling.
The mechanisms by which TGF-β mediates MFib differentiation have been extensively studied. TGF-β signaling begins with its binding to, and subsequent activation to form a heteromeric complex of, its receptors - namely, two type I receptors (TβRI) and two type II receptors (TβRII). TβRI and TβRII exhibit dual serine/threonine and tyrosine kinase activity. TβRI is a ubiquitously expressed receptor also known as activin receptor-like kinase 5 (ALK5). TβRII is a constitutively active receptor and upon interaction with TGF-β, it activates TβRI through phosphorylation [82].
ALK5 initiates TGF-β signaling through Smad transcription factor-dependent (canonical) (see Transcriptional regulators below for further discussion of Smad proteins) and -independent (non-canonical) means to direct gene expression. Evidence favors the existence of cooperative interactions between these distinct signaling pathways that are crucial for TGF-β-induced phenotypic responses in Fibs and MFibs. Non-canonical TGF-β signaling involves the ALK5-mediated phosphorylation and activation of TGF-β activating kinase 1 (TAK1). This, in turn, carries out the phosphorylation and activation of a number of other downstream kinase pathways, including phosphoinositide 3-kinase (PI3K), mitogen-activated protein (MAP) kinases p38, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK) 1 and 2, as well as activation of Rho family small GTPases, such as RhoA [101].
Rho signaling.
Rho GTPases including RhoA, Rac1, and Cdc42 are important regulators of the reorganization of the actin cytoskeleton in various cellular processes including cell polarity, migration, and division. Studies of smooth muscle cell differentiation initially revealed the importance and mechanisms of Rho GTPase signaling at the molecular level. Subsequent studies of EMT and Fib differentiation into MFibs revealed that these RhoA mechanisms are conserved with respect to the regulation of TGF-β-induced expression of contractile genes such as α-SMA [102,103]. TGF-β/ALK5-mediated activation of RhoA involves the rapid exchange of bound GDP with GTP through guanine nucleotide exchange factors. The activated form of RhoA (i.e., RhoA-GTP) signals through its downstream effectors, Rho-associated protein kinase 1 and 2 (ROCK1 and 2) and mammalian homolog of Drosophila diaphanous 1 and 2 (mDial and 2). ROCK is a serine-threonine kinase of the AGC (PKA/PKG/PKC) family of protein kinases. In addition to its crucial role in regulation of actin cytoskeleton dynamics, RhoA/ROCK signaling also contributes to nucleocytoplasmic shuttling of MRTFs (discussed in Transcriptional regulators, below). Increased RhoA/ROCK signaling has been reported in fibrotic Fibs from IPF lung [104]. Knockdown of RhoA is sufficient to diminish activation characteristics in IPF Fibs, namely the expression of FN, Col I and α-SMA. Rnd3 (also known as RhoE) is an atypical Rho family protein devoid of GTP hydrolytic activity but which can antagonize RhoA signaling. Consistent with increased RhoA/ROCK activity, recent studies reported decreased expression of Rnd3 in IPF Fibs [105]. Interestingly, knockdown of Rnd3 in normal lung Fibs was sufficient to increase RhoA activity and to concomitantly enhance MFib phenotype. The parallel ability of TGF-β/ALK5 signaling to reduce the expression of Rnd3 further contributes to RhoA activation. This antagonistic function of Rnd3, however, was not identified in other tissue Fibs, and its generalized applicability thus requires further investigation [105].
PI3K signaling.
PI3K signaling via AKT has been implicated in various Fib processes including proliferation [106], migration, and apoptosis resistance [107,108]. It has also been shown in Fibs that TGF-β signaling via PI3K/AKT induces the activation of the mammalian target of rapamycin (mTOR) protein complex, particularly mTORCI, the key regulator of protein synthesis. A number of studies have implicated mTORCI in Fib activation and the development of tissue fibrosis [109–112]. Activated mTORCI has also been shown to reduce autophagy and thereby contribute to apoptotic resistance [113]. On the other hand, mTORC2 is involved in actin cytoskeleton re-organization, activation of protein kinase C alpha [114], and phosphorylation and inactivation of FOXO proteins [115] (see Endogenous negative regulators, below). In addition to activation of AKT pathways, TGF-β/PI3K signaling also leads to activation of the p21 activated kinase PAK2. Via activation of the non-receptor tyrosine kinases c-Abl and PKC5, the TGF-β/PI3K pathway also upregulates the expression of tissue transglutaminase 2, an enzyme responsible for enhanced cross-linking and stabilization of ECM proteins [116] and expression and production of Col I protein [117]. This TGF-β/PI3K/PAK2 mediated activation of c-Abl/PKCδ was observed in Fibs but not epithelial cells.
P38 signaling.
Extracellular ligands including TGF-β and ET-1 promote the production of Col I from Fibs and/or MFibs. Col I is comprised of both Col I α1 and Col I α2 chains. While the mechanisms responsible for expression of Col I are complex and still not entirely clear, p38 is important for both Col I α2 expression in response to TGF-β and Col I α1 expression in response to α2β1 integrins [118,119], p38 signaling is also implicated in TGF-β-induced α-SMA expression, reflecting the role of this kinase in the serum response factor (SRF)-mediated transcription of contractile genes including α-SMA [120] (see Transcriptional regulators, below). While it remains a matter of conjecture, some evidence suggests that the TGF-β-induced activation of p38 is the consequence of ALK5/TAK1-mediated phosphorylation and activation of MAP kinase kinase (MKK) 3 and 6 [121].
JNK signaling.
In Fibs, TGF-p/ALK5/TAK1 signaling activates the JNK pathway. JNK signaling is involved in MFib differentiation elicited by TGF-β [122], as well as by IL4- and IL13 [123]. Activation of JNK in fibrotic lung Fibs has also been reported to contribute to the persistence of MFib phenotype. Likewise, enhanced JNK activity has also been reported in liver fibrosis [124]. In human lung Fibs, JNK signaling has been implicated in TGF-β-induced expression of CTGF [125]; by contrast, CTGF expression has been shown to depend on the alternative MAP kinases p38 and ERK½ in Fibs from other tissues [126,127]. TGF-β/JNK signaling also induces the synthesis of ET-1 in lung Fibs [128]. Although Col I expression itself was not dependent on JNK signaling, ECM contraction and macromolecular assembly of collagen was. JNK has also been reported to negatively regulate the autocrine expression of TGF-β, as JNK-deficient Fibs showed increased expression of TGF-β and constitutive activation of TGF-β signaling. Interestingly, JNK-deficient Fibs have high levels of expression of a variety of TGF-β inducible pro-fibrotic genes such as procollagen type IV, plasminogen activator inhibitor and MMP9. Thus, the functional consequences of JNK signaling in Fibs are quite complex and may vary from tissue to tissue. Involvement of JNK signaling has also been described in integrin-induced differentiation into MFibs; however, in these studies, other signaling pathways were also operative and the specific role(s) of JNK in driving Fib activation is incompletely understood.
ERK 1 and 2 signaling.
TGF-β-induced phosphorylation and activation of ERK ½ has been reported in tissue resident Fibs from the skin, lung and heart [129–131]. Reports of the role of ERK½ in TGF-β-induced α-SMA expression are contradictory [132,133]. Activation of ERK½ by mitogens such as FGF-2 and PDGF has been shown to diminish TGF-β-induced α-SMA expression [134,135]. Thus, the roles of ERK½ in α-SMA expression and MFib differentiation are likely to be cell- and context-dependent. It has also been reported that in TGF-β-induced MFibs, the expression of Col I is independent of activation of ERK½. Recent findings also identified ERK½ involvement in TGF-β/Smad signaling. In addition to its direct phosphorylation by ALK5, phosphorylation of R-Smads by TGF-β/ALK5/ERK½ signaling has been reported [136,137].
Wnt signaling.
Wnt ligands comprise a large family of secreted glycoproteins that, via activation of their downstream transcriptional co-activator β-catenin, exert pleiotropic roles in organogenesis and tissue homeostasis, but also in pathologic fibrosis. Wnt ligands signal through Frizzled (Fzd) family receptors, which associate with coreceptors, lipoprotein receptor-related proteins 5 and 6 (LRP5/6). The activated Wnt/Fzd/LRP complex stabilizes β-catenin and facilitates its translocation into the nucleus, where it interacts with transcription factors, most notably T-cell factor/lymphoid-enhancing factor, to regulate gene expression. Wnt/β-catenin signaling has been shown to promote diverse activation phenotypes of Fibs, including migration, proliferation, differentiation, collagen synthesis, and apoptosis resistance [138,139]. Wnt ligands have been reported to be over-expressed in Fibs from patients with IPF [140]. Moreover, Wnt signaling is opposed by a family of decoy receptors termed secreted Fzd-related proteins, whose expression has been reported to be diminished in fibrotic lung from scleroderma patients [141] and in fibrotic Fibs from patients with skin keloids [142]. Importantly, this pathway is potentiated by TGF-β, which in Fibs can increase the expression of a number of its components, including Wnt ligands, FZDs, and β-catenin [143]. It is also noteworthy that β-catenin can similarly be activated in the absence of Wnt ligands by another pro-fibrotic stimulus, lysophosphatidic acid [144]. In considering the potential utility of targeting Wnt/β-catenin for inhibition, the concomitant role of this pathway in mediating epithelial repair could result in untowards effects.
Mechanotransduction and the YAP/TAZ pathway.
Increased stiffness is a well-recognized consequence of fibrotic tissue remodeling, and a major determinant of impaired organ function in FPDs. Such stiffness derives in large part from the mechanical forces generated by the excessive amounts of ECM proteins, including collagen, fibrin, and fibronectin, deposited within the tissue. In an organ like the lung where normal respiration requires cyclical inflation and deflation and thus a great degree of tissue compliance, tissue stiffness imposes an added burden on patients by increasing the work of breathing, leading to shortness of breath. In this circumstance, the contractile properties of MFibs themselves may further contribute to tissue stiffness. Rather than merely reflecting the consequence of tissue fibrosis, it is now appreciated from studies in which normal Fibs are cultured on substrates of varying stiffness that the degree of matrix stiffness measured in fibrotic organs actually serves as an independent stimulus that potentiates Fib activation - thus synergizing with the actions of myriad soluble pro-fibrotic mediators. This occurs because cells can sense these mechanical cues and convert them into a biochemical, intracellular response - a process called mechanotransduction. Mechanotransduction thus represents a positive feedback loop which amplifies aberrant Fib activation and fibrogenesis.
The process of mechanotransduction requires both sensory and effector arms. Integrins are the major cell surface adhesion receptors that sense mechanical cues from the ECM and transmit them to the intracellular cytoskeleton. Integrins comprise a large family of glycoproteins, with each integrin being composed of a heterodimer of α and β subunits. Distinct types of α and β subunits have been shown to influence various Fib activation phenotypes. For example, α2β1 integrins enhance proliferation of normal Fibs, yet their expression has been reported to be diminished in fibrotic Fibs from IPF patients. By contrast, α1β1 promotes MFib differentiation, while α4β1 and α5β1 are involved in MMP-1 expression. Conditioned medium elaborated by IPF Fibs as well as exogenous TGF-β itself have been reported to increase the expression of integrin a subunits [145,146]. Transduction of integrin-dependent signals proceeds by several key pathways including FAK, MAP kinases, and RhoA GTPases [147–149]. These signaling pathways activate transcriptional events that carry out the gene expression programs essential to the mechanotransductive response. YAP (Yes-associated protein) and TAZ (the transcriptional coactivator with PDZ-binding motif) are important nuclear transducers of mechanical signals. However, as YAP and TAZ themselves lack direct DNA-binding activity, they act as transcriptional co-activators by facilitating the actions of transcription factors such as TEA domain family member 1–4. Although YAP and TAZ have minimal basal expression in normal tissue, their expression increases during tissue injury and wound healing, reaching high levels in fibrotic tissue. Knockdown of both YAP and TAZ in Fibs grown on stiff matrix reduced expression of proteins associated with the MFib phenotype such as Col I and α-SMA. RhoA GTPases are crucial for the transcriptional activation of YAP/TAZ, and depletion of YAP/TAZ yields biological effects similar to those resulting from inhibition of the RhoA/ROCK pathway. Transcriptional targets of YAP/TAZ shown to be pro-fibrotic include TGF-β, CTGF, TG2, and plasminogen activator inhibitor 1 (PAI-1J. Studies from renal Fibs suggest that YAP/TAZ contributes to MFib differentiation through the actions of mTORC2 [150]. Recent studies have also shown that YAP/TAZ enhances TGF-β signaling through inhibition of the inhibitory Smad7 [151].
Interplay among discrete pro-fibrotic signaling pathways.
Although we have presented the above signaling pathways as discrete programs, (as illustrated in Fig. 2) they in fact interact or converge at a number of downstream points. Examples of this include the activation of RhoA and FAK by both TGF-β and integrin-mediated mechanical signaling. An individual soluble mediator might generate others that can in turn amplify, redirect, or limit its pro-fibrotic actions. For example, TGF-β can elicit generation of CTGF and FGF-2, and ET-1 can generate TGF-β. We have also framed ECM and soluble mediators as independent pro-fibrotic drivers, but these can physically interact with each other. For example, the ECM deposited in a fibrotic environment not only initiates mechanosensitive transduction, but it also traps and acts as a reservoir for pro-fibrotic ligands such as latent TGF-β and Wnt ligands. The fact that discrete pathways can clearly be interrelated or coexist complicates efforts to define the relative importance and contribution of individual signaling pathways in driving tissue fibrosis. Finally, tissue fibrosis is of course a result of the combinatorial effects of all the aforementioned stimuli and signaling pathways.
Fig. 2.
Schematic representation of well-characterized Fib activation signaling pathways and their crosstalk.
Transcriptional regulators of the MFib phenotype
Modulation of transcriptional programs is a critical means by which the activation of mesenchymal cells is controlled. These transcriptional regulatory mechanisms act both upstream and downstream of the signaling pathways discussed above. We will next review some of the transcription factors that are well-characterized for their role in activation and differentiation of Fibs. Subsequently, we will briefly consider the non-coding RNAs that also modulate these transcriptional programs.
Smad proteins.
Smad proteins are phosphorylation-activated transcriptional regulators of target gene expression. To date, eight different Smad proteins have been identified in mammals and based on their biological actions, they are further categorized into receptor-activated (R-Smads), common partner (Co-Smads), or inhibitory (l-Smads). R-Smads include Smad 1, 2, 3, 5 and 8; of these, Smad2 and Smad3 are well studied in the context of TGF-β signaling, whereas Smadl, 5 and 8 are activated by another TGF-β family protein, bone morphogenic protein (BMP). Smad4 is the only identified Co-Smad protein in mammals, and it partners with activated R-Smads in both TGF-β and BMP signaling. By contrast, Smad6 and Smad7 act as negative regulators of TGF-β family signaling, predominantly by blocking the activation of R-Smads and their association with Smad4 [152]. Upon TGF-β binding-induced formation of the tetrameric TβRI/TβRII complex, R-Smads Smad2/3 are phosphorylated and then complex with Smad4 and translocate into the nucleus to mediate transcription of Smad-dependent genes such as PAI-1 [153]. The Smad complex recognizes a specific GC-rich DNA sequence in target gene promoters termed the Smad binding element (SBE). However, it does so with a relatively low binding affinity [154], and its transcriptional activity is augmented by its ability to also interact with a number of other transcriptional regulators (reviewed in [155]) in the nucleus to induce expression of genes lacking a SBE. Fibs from IPF patients showed increased expression as well as nuclear accumulation of R-Smads 2 and 3 [156].
Serum response factor (SRF) and myocardin-related transcription factors (MRTFs).
Expression of contractile genes such as a-SMA is the hallmark of the MFib phenotype. The molecular regulation of TGF-β-induced a-SMA expression is well-characterized in MFibs from various tissues. Expression of α-SMA is controlled by the transcription factor serum response factor (SRF) and its co-activators, myocardin-related transcription factor (MRTF)-A or B. SRF binds to the serum response element (SRE) or CArG box in the promoter region of genes encoding contractile proteins. Although the critical role of SRF in initiating transcription of contractile protein genes was originally identified in myogenic differentiation, its ability to do so during Fib to MFib differentiation (or in epithelial cells undergoing EMT) requires this same transcriptional apparatus. Under basal conditions, the amount of SRF-MRTF complexes within the nucleus is limited by both low expression levels of nuclear SRF as well as cytoplasmic anchoring of MRTF due to their binding by monomeric G-actin. Upon TGF-β stimulation, p38 signaling increases SRF expression at the mRNA and protein levels [120]. Concurrent activation of RhoA/ROCK signaling promotes polymerization of G-actin to F-actin, facilitating the release and nuclear accumulation of MRTFs and their subsequent interaction with SRF to form the nuclear complex required to initiate α-SMA transcription [120]. In addition to its well-known role in regulating contractile gene expression and MFib differentiation, loss-of-function studies using siRNA-mediated knockdown of SRF reveal that it also is necessary for both proliferation and survival of Fibs, but the operative molecular mechanisms remain unclear. Interestingly, overexpression of SRF or its co-activators is insufficient to promote either MFib differentiation or proliferation of Fibs, suggesting a requirement for their activation by extrinsic factors [157,158]. Consistent with increased expression of SRF by pro-fibrotic mediators like TGF-β, aberrant over-expression of SRF has been reported in fibrotic Fibs from both IPF patients and animal models of lung fibrosis [120,159].
T-box (TBX) proteins.
The TBX family of transcription factors can act as either transcriptional activators or repressors in a cell- and context-dependent manner. Of these, members of the TBX2 subfamily (TBXs 2, 3, 4 and 5) play important roles in lung development. A recent study employed lineage tracing in a bleomycin-induced lung injury model and identified that TBX4-expressing progenitors were the predominant source of accumulating MFibs. In addition, ablation of TBX4-positive cells or signaling ameliorated fibrogenesis [59]. These authors also reported that TBX4 regulated the production of hyaluronan synthase 2 in fibrotic lung Fibs and facilitated their invasive activity. Other studies have noted a variety of, and sometimes discrepant, influences of TBX4 on Fib activation parameters. These include effects on Col I expression [160,161], proliferation capacity, and global gene expression [162]. Both TBX2 and TBX3 proteins have also been reported to exert anti-senescence properties in Fibs [163]. The role of TBX proteins in Fibs and in tissue fibrosis therefore requires further investigation.
Forkhead box (FOX) proteins.
FOX proteins are a large family of transcription factors that regulate expression of a variety of genes involved in cellular processes such as proliferation, differentiation, senescence, and apoptosis. Much of our current knowledge regarding the biology of FOX proteins derives from investigations in the fields of developmental biology and cancer. FOX proteins may contribute to FPDs by their abilities to promote processes involving non-mesenchymal cells such as EMT [164]. Here we will specifically focus on their role in Fibs and in MFib differentiation. FOXM1 is considered a master transcription factor for numerous cell cycle genes and controls the proliferation of a variety of cell types. As such, it has attracted particular attention as a possible therapeutic target in cancer. Considering the many parallels between tumor cells and activated Fibs, including anchorage-independent growth and apoptosis resistance [165,166], the expression and role of this transcription factor in FPDs was likewise of interest. Increased expression of FOXM1 mRNA and protein was reported in fibrotic Fibs derived from IPF patients as well as from mouse models of lung fibrosis [25]. In keeping with its known role in control of the cell cycle, FOXM1 was found to be both sufficient and necessary for growth factor (FGF-2)-induced expression of cell cycle genes and proliferation in lung Fibs. Although FOXM1 over-expression was insufficient to induce MFib differentiation, loss-of-function studies indicated that it was required for TGF-β-induced expression of genes associated with the MFib phenotype (e.g. α-SMA and Col I). In addition, FOXM1 was shown to protect Fibs (and MFibs) from FasL-induced apoptosis by modulating the expression of pro- and anti-apoptotic genes. FOXM1 has similarly been shown to protect IPF Fibs from radiation-induced cell death by increasing the expression of DNA damage response proteins (RAD51 and BRCA2) [167]. Contrary to the role of FOXM1 in activation of lung Fibs, FOXF1 inhibits their activation, and deletion of FOXF1 increased the invasiveness and collagen synthetic capacity of MFibs [168]. Although studies in mouse embryonic Fibs demonstrated that FOXF1 promoted cell migration through transcriptional upregulation of integrin β3 [169], the relevance of this finding to FPDs is unclear. Finally, one study employing RNA-seq analysis of normal and IPF lung Fibs revealed that FOXS1 was the most highly upregulated gene following treatment with TGF-β [170]. However, its potential role in MFib differentiation awaits investigation.
Runt-related (RUNX) proteins.
RUNX transcription factors control a wide range of biological process such as proliferation, differentiation and apoptosis. Three RUNX family members (RUNX1, RUNX2, and RUNX3) have been identified in mammals and their relative expression varies in a tissue-specific manner. RUNX1 has been reported to be induced by TGF-β/SMAD3 signaling and to promote EMT in a model of renal fibrosis [171]. It has also been reported to promote proliferation and the expression of genes associated with a MFib phenotype (α-SMA, tenascin-C, Fib activation protein, and Col I) in mesenchymal stem cells [172]. However, the role of RUNX proteins in Fib activation and MFib differentiation remains poorly understood. RUNX family proteins are reported to induce senescence-like growth arrest in primary human foreskin Fibs and murine Fibs [173]. In a recent study [174] expression of RUNX2 was diminished in Fibs in lungs from IPF patients and bleomycin-injured mice, while its knockdown increased Fib activation markers.
STAT6.
Signal transducer and activator of transcription (STAT) 6 is another transcription factor implicated in MFib differentiation. STAT6 is the canonical transcription factor mediating the biological actions of both IL4- and IL13, which lies downstream of their common receptor IL4Ra and which is phosphorylated and activated by Janus kinase 3. This pathway has similarly been implicated in the induction of a-SMA by these cytokines [96,175]. The precise role of STAT6 in Fib-driven fibrotic responses, versus that ascribed to other pathways elicited by IL-13 or other pro-fibrotic molecules it induces, remains uncertain.
Role of non-coding RNAs in fibrosis
Tanscriptional programs, discussed above, can also be regulated by non-coding RNAs [176]. The first subset of these is long non-coding RNAs (LncRNAs). These are >200 nucleotides in length and their expression and mechanisms of action are cell type-specific. LncRNAs regulate target gene expression through mechanisms that involve chromatin remodeling as well as transcriptional and post-transcriptional regulation. The second subset is microRNAs (miRNAs), small non-coding RNAs of 22–25 nucleotides in length that are partially complementary to mRNA molecules and which downregulate gene expression via either mRNA degradation or translational repression. RNA sequencing studies in various fibrotic tissues have revealed hundreds of differentially expressed LncRNAs and miRNAs. Although the biological significance and the mechanisms of action(s) of most of these remain unstudied, a small number of them have been examined in the context of Fib activation (Table 2 and 3). As has been suggested in recent reviews [177–179], non-coding RNAs such as these may serve as mediators and biomarkers of fibrosis as well as potential therapeutic targets.
Table 2:
LncRNAs in Fib activation and tissue fibrosis
| LncRNA | Organ | Expression | Effect on fibrosis |
References |
|---|---|---|---|---|
| H19 | Lung, Heart | ↑ | pro-fibrotic | [180,181] |
| PFAR | Lung | ↑ | pro-fibrotic | [182] |
| PFRL | Lung | ↑ | pro-fibrotic | [183] |
| PFAL | Lung | ↑ | pro-fibrotic | [184] |
| Inc-LFAR1 | Liver | ↑ | pro-fibrotic | [185] |
| MIAT | Heart | ↑ | pro-fibrotic | [186] |
| PFL | Heart | ↑ | pro-fibrotic | [187] |
| HOTAIR | Liver | ↑ | pro-fibrotic | [188] |
| GAS5 | Heart, Liver | ↓ | anti-fibrotic | [189,190] |
| MEG3 | Liver | ↓ | anti-fibrotic | [191] |
Table 3:
miRNAs in Fib activation and tissue fibrosis
| miRNA | Organ | Expression | Effect on fibrosis |
References |
|---|---|---|---|---|
| miR-21 | Lung, Liver, Heart, Kidney | ↑ | pro-fibrotic | [192–195] |
| miR-31 | Skin | ↑ | pro-fibrotic | [196] |
| miR-34a | Liver, Kidney, Lung | ↑ | pro-fibrotic | [197–199] |
| miR-96 | Lung | ↑ | pro-fibrotic | [200] |
| miR-145 | Lung | ↑ | pro-fibrotic | [201] |
| miR-154 | Lung | ↑ | pro-fibrotic | [202] |
| miR-155 | Lung, Skin, Heart | ↑ | pro-fibrotic | [203–205] |
| miR-199a-5p | Lung | ↑ | pro-fibrotic | [206] |
| miR-210 | Lung | ↑ | pro-fibrotic | [207] |
| miR-9–5p | Lung | ↓ | anti-fibrotic | [208] |
| miR-22 | Heart | ↓ | anti-fibrotic | [209] |
| miR-26a | Lung | ↓ | anti-fibrotic | [210] |
| miR-27a-3p | Lung | ↓ | anti-fibrotic | [211] |
| miR-29a,b,c | Lung | ↓ | anti-fibrotic | [212] |
| miR-101 | Lung | ↓ | anti-fibrotic | [213] |
| miR-200b,c | Lung | ↓ | anti-fibrotic | [214] |
| miR-150 & miR-194 | Liver | ↓ | anti-fibrotic | [215] |
Endogenous negative regulators of MFibs
From an evolutionary perspective, the activation of Fibs - as is true for other potentially deleterious cellular responses - must be restrained by endogenous braking mechanisms in order to maintain homeostatic organ function. The imperative of keeping fibrosis in check would predict that there be multiple molecular species of brakes, which can reinforce each other. Finally, it can be predicted that the failure of these brakes would favor the development of pathological fibrosis and FPDs. To carry this speculation even further, the development of FPDs may actually require the relative failure of these negative regulators. The amount of research on endogenous anti-fibrotic mechanisms pales in comparison to that on pro-fibrotic mechanisms, reviewed above. We will next provide a brief overview of the best understood of these anti-fibrotic molecular brakes, and their disruption in fibrosis and FPDs. Table 4 provides a list of endogenous negative regulators identified so far.
Table 4:
Negative regulators of MF differentiation
| Soluble mediators |
Key signaling pathway |
Tissue/cells | References |
|---|---|---|---|
| PGE2 | activates cAMP/PKA | Fibs | [216] |
| FGF-2 | activates PI3K & ERK1/2 | Fibs | [25] |
| Interferon (IFN)-γ | activates Jak/STAT 1 | Fibs | [217] |
| Transcriptional regulators | |||
| PPARγ | inhibits MAP kinases | MFibs | [218–220] |
| F0X03 | inhibits R-SMADs activation | MFibs | [25,221] |
| PTEN | inhibits PI3K | MFibs of lung | [47,222] |
| Smad7 | inhibits R-SMADs | MFibs of lung | [223] |
| Krupple-like factor (KLF) 15 | inhibits R-SMADs | MFibs of heart, MFibs of kidney | [224,225] |
PGE2.
Prostanoids represent a ubiquitous class of bioactive lipid mediators. PGE2 is the most abundant prostanoid produced by many cell types, including macrophages, Fibs, and epithelial cells; of these three cell types we have considered in this review, the latter have the greatest synthetic capacity on a per cell basis. The COX-2 enzyme catalyzes the conversion of membrane arachidonic acid into an unstable PGH2 endoperoxide, on which prostaglandin E synthase acts and converts it into PGE2. PGE2 plays important roles in diverse aspects of physiology and pathophysiology. Its actions are unusually pleiotropic, and sometimes even contradictory, depending on its target cell or tissue, or the biological context; this is now understood to reflect its ability to ligate and act via four distinct G protein-coupled receptors, E-type prostanoid receptors EP1–4. A feature of PGE2 actions that is critical to understanding its wide-ranging role as a brake on fibrogenesis is its opposing yet salutary effects on both epithelial cells versus Fibs. In epithelial cells, PGE2 has been shown to promote their migration [226], proliferation [227], and survival [228]. In contrast, it suppresses a number of activation phenotypes of Fibs, including proliferation [25,229], migration [47], collagen synthesis, and differentiation into MFibs [120,230], while eliciting and potentiating their apoptosis [231]. EP2 is the predominant EP receptor expressed on Fibs, and mechanistically, these inhibitory actions on Fibs are largely mediated via EP2 signaling generating cAMP and activating either protein kinase A (PKA) or guanine nucleotide exchange protein directly activated by cAMP (Epac). Interestingly, suppression of Fib proliferation by PGE2 has been attributed primarily to Epac, while suppression of collagen synthesis and MFib differentiation has been attributed primarily to PKA [216]. In-depth characterization of its inhibitory mechanisms has revealed inhibitory actions of PGE2 on a variety of genes and signaling pathways downstream from pro-fibrotic stimuli such as TGF-β and FGF-2. The importance of PGE2-EP2 signaling as an endogenous brake on fibrosis is indicated by the exaggerated pulmonary fibrosis exhibited by EP2 knockout mice [232]. Another prostanoid that signals through a G protein-coupled receptor to activate cAMP generation, prostacyclin, can engage these same signaling pathways and similarly inhibit Fib activation, MFib differentiation, and fibrogenesis [233,234]. Because phosphodiesterase inhibition prevents cAMP degradation and thus augments the signaling and actions of PGE2 and prostacyclin, it is not surprising that pharmacologic inhibitors of type IV phosphodiesterase can amplify the anti-fibrotic actions of these prostanoids [235,236]. The broad anti-fibrotic actions of PGE2 are best understood by the observations that this prostanoid can inhibit many of the pro-fibrotic mediators, signaling pathways, and transcriptional effectors discussed above. These include SRF, p38, pAKT, and FOXM1 [25,237]. Enhanced apoptosis of Fibs by PGE2 can be attributed to upregulation of the Fas receptor.
Growth factor-induced cell activation and proliferation is often accompanied by induction of COX-2; the resulting PGE2 that is generated thus serves as a built-in curb on unchecked activation elicited by these stimuli. As noted in the introductory statement above, it would be expected that this pleiotropic autocrine brake would be disrupted in FPDs. Indeed, lung Fibs from both patients with IPF and animal models of lung fibrosis exhibit diminished expression of COX-2 and decreased capacity for PGE2 synthesis. Impaired COX-2 expression in IPF Fibs has been attributed to epigenetic mechanisms [238]. A defect in COX-2 induction has also been reported in lung mesenchymal stem cells isolated from lung transplant patients exhibiting the post-transplant complication termed bronchiolitis obliterans, reflecting fibrotic remodeling of their small airways [239]. Furthermore, merely culturing normal lung Fibs on stiff matrices - which promotes their activation and differentiation - is sufficient to down-regulate COX-2 expression [240]. In addition to these examples of impaired PGE2 generation in FPDs and activated Fibs, PGE2 signaling/responsiveness is also impaired in Fibs from patients and animal models of lung fibrosis, and is attributable to epigenetic down-regulation of EP2 [241].
Peroxisome proliferator-activated receptors (PPARs).
PPARs are nuclear hormone receptors that act as ligand-inducible transcription factors. Three isoforms of PPARs have been identified - namely, PPARα, PPARγ, and PPARβ/δ (commonly identified as PPARδ). PPARs exert potent anti-fibrotic activities both in vitro and in vivo. Stimulation of Fibs with PPARδ agonists showed inhibition of proliferation. Likewise, ligands of PPARγ were shown to suppress TGF-β-induced activation of SMAD [218] as well as of p38 in Fibs [219], thereby inhibiting MFib differentiation. In animal models of fibrotic diseases of the lung, liver, kidney, and heart, it has been reported that treatment with PPARα agonists reduced collagen synthesis, and PPARα knockout mice showed worse fibrosis. An endogenous protective role for PPARγ is supported by the report of its down-regulation in fibrotic Fibs from patients with scleroderma, and the fact that its expression is diminished by Fib exposure to TGF-β [220]. Mechanistically, downregulation of PPARγ in liver MFibs has been attributed to histone methylation [242]. Of note, PPARγ has been shown to both promote and to inhibit [243] PGE2 synthetic machinery.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN).
PTEN is both a dual-specificity protein phosphatase that can dephosphorylate ser, thr and tyr residues as well as a lipid phosphatase that converts phosphatidylinositol-3,4,5-trisphosphate (PIP3) to PIP2 - thus opposing the actions of PI3K. Indeed, PI3K/AKT and PTEN represent major positive and negative regulators, respectively, of growth factor-induced signaling. A number of studies have identified inhibitory actions of PTEN on Fib proliferation and migration in response to pro-fibrotic growth factors [47]. Loss of PTEN activity results in exaggerated fibrosis in models of acute kidney, lung, and liver injury. Pro-fibrotic factors such as TGF-β repress the expression of PTEN. Diminished expression of PTEN has similarly been described in IPF Fibs. An inverse correlation between PTEN and α-SMA has been reported in IPF tissues. PTEN null Fibs likewise exhibit increased baseline expression of α-SMA in the absence of TGF-β stimulation. Taken together, these data suggest that PTEN serves as an endogenous brake on Fib activation responses that is itself diminished in fibrosis. Interestingly, PTEN has been shown both to mediate the Fib-suppressive actions of PGE2 [47] and to positively regulate EP2 expression on Fibs [222]. In addition to its fundamental ability to oppose PI3K signaling, PTEN has also been shown to interfere with other endogenous signaling pathways involved in MFib differentiation such as p38 and Rho-kinase.
Forkhead box O (FOXO) family proteins.
The FOXO group of transcription factors (F0X01, F0X03, F0X04, and F0X06) plays an important negative regulatory role in growth factor-induced signal transduction. While in the nucleus, the FOXO proteins exist in their dephosphorylated (active) form and upregulate expression of various cyclin-dependent kinase inhibitors (p21 WAF1 and p27 KIP1) while inhibiting cell cycle genes such as Cyc D1 and D2 through their direct binding to promoter elements and competition with FOXM1 for DNA binding. Pro-fibrotic factors and cytokines, via PI3K/AKT signaling, phosphorylate FOXO proteins, leading to their nuclear export and inactivation. Once in the cytoplasm, the FOXOs undergo ubiquitin-mediated proteasomal degradation and thereby favor PI3K-mediated signal transduction. FOXO proteins also promote cell death by upregulating apoptosis-associated genes such as FasL, Bim and TRAIL [244]. F0X03 expression has been shown to be diminished in IPF Fibs [245]. FOXO proteins are regulated by a number of relevant modulators. For example, mitogenic growth factors (PDGF, FGF, and IGF-I) inhibit the expression of FOXO genes. By contrast, PGE2 inhibits FGF-2-induced phosphorylation of F0X03, promoting its retention in the nucleus and its braking action on the cell cycle [25]. Similarly, PTEN can carry out the dephosphorylation and activation of FOXO proteins. In dermal Fibs, F0X01 has been shown to inhibit proliferation and to stimulate apoptosis. In both dermal and lung Fibs, down-regulation of F0X03 has been shown to accelerate their senescence [221]; although the mechanisms remain to be determined, this may further favor fibrotic activation.
FGF-2.
FGF-2 (also known as basic FGF) is a heparin-binding growth factor that possesses mitogenic activity for Fibs as well as other mesenchymal cells. FGF-2 binds to and signals through four related receptor tyrosine kinases (FGFR1, FGFR2, FGFR3 and FGFR4). FGF-2 also promotes angiogenesis under various physiological states including wound healing. As mentioned before, in parallel with its ability to stimulate proliferation of lung Fibs, FGF-2 upregulates a number of cell cycle genes including FOXM1 [25]. FGF-2 also induces the expression of pro-survival/anti-apoptotic genes such as survivin. Interestingly, however, FGF-2 fails to promote differentiation to a MFib phenotype and instead, actually suppresses TGF-β-induced expression of genes associated with a MFib phenotype (e.g., Col I, α-SMA). This ability of FGF-2 to oppose TGF-β differentiation of Fibs has been shown to proceed via ERK½ activation. Moreover, intrapulmonary administration of FGF-2 provided protection from bleomycin-induced fibrosis in a mouse model, attesting to an in vivo anti-fibrotic effect [246].
De-differentiation (reversal) of MFibs
Early fibrosis of vital organs is usually clinically silent, and by the time patients reach clinical attention with recognizable DPDs, fibrosis has typically advanced to a degree in which physiologic functions of the affected tissue are impaired. For IPF, there now exist two therapeutic agents (pirfenidone and nintedanib) that have been shown to slow the progression of fibrosis and thus, of physiologic impairment. Although these treatments represent a welcome advance, they fail to achieve the universally held therapeutic ideal of actually reversing existing fibrosis and restoring more normal organ function. Clearly, restoration of healthy tissue would require that (i) MFibs be cleared from the fibrotic tissue, (ii) ECM gets digested and removed, and (iii) tissue architecture is restored - likely requiring regenerative medicine approaches. Although removal of ECM proteins would be expected to be achievable by shifting the proteolytic balance in favor of MMPs over TIMPs, the importance of individual members of these molecular families remains uncertain. While efforts to understand and apply ECM degradation [247] and regenerative medicine capabilities [248,249] continue to progress, we will focus on this first step of clearance of MFibs from the affected tissue. Accomplishing this will likely be a requisite step in restoration of homeostasis.
One approach to clearing MFibs would involve inducing their apoptosis. This might be achieved by manipulating any of the known apoptosis-regulatory machinery. Pharmacologic agents which inhibit known anti-apoptotic molecules represent one example, and some reports employing such an approach document an improvement in tissue function in various animal models of fibrotic diseases. One important theoretical limitation of this approach is the possible promotion of apoptosis in epithelial cells as well, which could worsen fibrosis. It has been suggested that selectively targeting for inhibition anti-apoptotic proteins that are expressed to a greater degree in MFibs than in epithelial cells (e.g., XIAP) may circumvent this concern [45].
Another approach to achieving MFib clearance is to revert or reverse their differentiated phenotype back to the more quiescent Fib or proto-MFib - cells that produce less ECM per cell and are more susceptible to apoptosis than are fully differentiated MFibs. Indeed, such “de-differentiation” would be expected to render MFibs more susceptible to the pro-apoptotic strategies described above. The plausibility of a de-differentiation approach hinges on whether or not differentiation is an irreversible phenomenon. Indeed, for many years MFibs were thought to be terminally and irreversibly differentiated cells [250]. However, it is now clear that even MFibs maintain a substantial degree of phenotypic plasticity that can be exploited to achieve de-differentiation. We will next review some of the foundational research on MFib de-differentiation in response to specific mediators; most of this work has employed PGE2 or FGF-2. Possible phenotypic paths for MFib de-differentiation are summarized in Fig.3.
Fig. 3.
Schematic representation of Fib differentiation and possible phenotypic fates during the process of de-differentiation.
PGE2.
Considering its extensive ability to inhibit and prevent fibrotic Fib phenotypes, reviewed above, it was of substantial interest that PGE2 also proved capable of effecting substantial de-differentiation of established MFibs generated by in vitro treatment with TGF-β or ET-1 [251]. As is the case for its ability to prevent MFib differentiation, de-differentiation elicited by PGE2 likewise proceeded via EP2-cAMP signaling. It is thus not surprising that prostacyclin, also signaling via the second messenger cAMP, has also been reported to elicit de-differentiation [252]. The ability of cAMP-elevating prostanoids to both de-differentiate MFibs to more apoptosis-susceptible Fibs and to then directly elicit or potentiate their apoptosis provides a mechanistically attractive strategy to clear MFibs. Given that Fib to MFib transition represents a phenotypic continuum rather than a categorical duality, one can imagine that a similar continuum characterizes “de-differentiation.” Microarray transcriptomic analysis was utilized to explore the genome-wide impact of PGE2 treatment of TGF-β-differentiated MFibs beyond merely a reduction in α-SMA and Col I. PGE2 directionally reversed −55% of the genes whose expression was increased or decreased by TGF-β, indicating a broad impact on cellular programs. Clearly, however, de-differentiation was not complete, and PGE2-treated cells differed from quiescent Fibs with respect to expression of at least 412 genes [28]. These findings extend the notion of a continuum between MFib and Fib phenotypes to the process of de-differentiation. One could speculate that these partially de-differentiated cells may be similar to proto-MFibs rather than Fibs, but this question and indeed these stages require further understanding at a gene expression level. Single cell analysis of these mixed Fib populations also might prove informative. As discussed below, anti-fibrotic mediators that act independently of cAMP have also been reported to “dedifferentiate” MFibs. Whether such cells differ from those reverted by PGE2 remains to be determined, as they have not been subjected to transcriptomic analysis.
FGF-2.
FGF-2 too can effect de-differentiation of MFibs, reducing expression of α-SMA and production of Col I. Although MAP kinase activation has been implicated in its ability to prevent MFib differentiation [253], the mechanisms operative in its de-differentiation capability have not been addressed. One obvious notable difference between de-differentiated cells elicited by FGF-2 as compared to PGE2 is that the former proliferate (reflecting the mitogenic actions of FGF-2) while the latter do not (reflecting the mitogenic inhibitory actions of PGE2). The MFib de-differentiation capacity of FGF-2 - unlike that of PGE2 - then, might be understood in the context of the longstanding axiom in biology that proliferation and differentiation programs are quite distinct and possibly even mutually exclusive; further exploration of this notion requires direct investigation. It is apparent that a fibrotic milieu would be expected to contain both differentiation-causing as well as proliferation-inducing mediators. The integrated responses of Fibs to these complex mixtures of stimuli with distinctive actions have received little attention to date. It is also worth noting that the recognized mechanism of action for the FDA-approved IPF drug nintedanib involves blockade of the tyrosine kinase receptor for PDGF, FGF-2, VEGF and IGF. If FGF-2 indeed exerts certain anti-fibrotic actions, blocking its actions with nintedanib could theoretically worsen the fibrotic process in some patients, offering a possible explanation for its limited therapeutic efficacy. Additional assessment and interpretation of FGF-2 actions as well as its blockade in fibrotic diseases is needed.
PDGF.
The mitogen PDGF has also been reported to de-differentiate MFibs, as reflected by reduced expression of α-SMA. As with FGF-2, the activation of mitogenic signaling pathways (i.e., ERK½ and cyclin-dependent kinases) has been implicated in PDGF-induced de-differentiation [134]. Like FGF-2, PDGF also promotes proliferation in Fibs but it contrasts with FGF-2 in its ability to stimulate the production of Col I. Whether this reduction in α-SMA but not in Col 1 can truly be considered to reflect de-differentiation of a MFib phenotype is unclear, and underscores the potential superior value of comprehensive transcriptomic analysis in interpreting intermediate phenotypes.
Mechanistic insights into de-differentiation.
The mechanisms by which biological mediators cause de-differentiation of MFibs are largely unknown. The significance of inhibiting the molecular determinants of differentiation in effecting de-differentiation remains to be determined. However, unlike the prevention of Fib differentiation where de novo expression of contractile genes is inhibited at the transcriptional and translational levels, the process of de-differentiation requires degradation of available MFib-specific transcripts and proteins. Thus, at the molecular level, the process of dedifferentiation implies a mechanistically unique process. The current understanding of the process of fibrosis resolution has been recently reviewed [254,255].
Conclusions and Therapeutic Implications
Pathologic fibrotic remodeling of tissues resulting in impaired organ function is an important source of morbidity and mortality. Given the enormous human and economic burden of FPDs, there remains a vital unmet need for treatments capable of reversing fibrosis. It is hoped that a better understanding of the mechanisms responsible for the cellular phenotypes that promote fibrogenesis will provide the foundation for new therapeutic approaches. Although epithelial cells damage and inflammatory cell (particularly macrophage) recruitment and activation commonly underlie fibrotic responses, these processes often predate the clinical presentation of patients with FPDs. At these later clinically apparent stages of disease characterized by established fibrosis, the accumulation of mesenchymal cells and their elaboration of ECM proteins that comprise scar tissue are central pathogenic events that must be targeted if reversal of fibrosis is to be accomplished. It is for this reason that this review emphasized the proliferation of resident tissue Fibs and their differentiation into MFibs - arguably the most critical ultimate effector cell of fibrosis.
In addition to reviewing the major soluble and mechanical stimuli that drive activation of Fibs, we discussed some of the critical signaling pathways and transcription factors that mediate these responses. While it is tempting to imagine strictly linear pathways mediating particular cellular responses - i.e., a given mediator engages a single signaling pathway which activates a given transcription factor which results in a single particular functional phenotype - the realities are not that simple. More typically, a given stimulus activates a number of signaling pathways and transcriptional and phenotypic responses. Multiple pathways can converge or can act in cooperative or combinatorial ways. Moreover, because fibrotic milieus represent a mix of many soluble as well as mechanical stimuli, the ultimate complexity of responses is staggering. This complexity remains poorly understood.
Finally, fibrogenic responses depend not only on activation events, but also on the loss of endogenous suppressive mechanisms. We know far less about these endogenous anti-fibrotic brakes than we do about pro-fibrotic drivers. Therapeutic targeting to date has emphasized inhibiting the pro-fibrotic drivers. This may be a challenging approach, given the large number of such drivers, their redundancy, and their interactions. As the endogenous negative regulators typically oppose a variety of activation events in mesenchymal cells, we suggest that attempting to rescue or restore these anti-fibrotic brakes that have been lost during fibrosis may be a preferable approach. Such an approach would be especially promising if it results in MFib de-differentiation, as this may be an initial requisite step towards promoting their apoptosis. Subsequent therapeutic steps may well require the degradation of deposited ECM proteins followed by strategies to regenerate an intact epithelium.
Acknowledgments:
This work was supported by NIH grant HL094311 (to MPG) and an American Heart Association Fellowship Award (to LRP),
Funding: The authors declare no financial conflicts of interest.
References
- [1].Wynn TA (2008). Cellular and molecular mechanisms of fibrosis. J Pathol 214, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gao B. and Bataller R. (2011). Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Straub JM, New J, Hamilton CD, Lominska C, Shnayder Y. and Thomas SM (2015). Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol 141, 1985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mossman BT and Churg A. (1998). Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 157, 1666–80. [DOI] [PubMed] [Google Scholar]
- [5].Ringelhan M, McKeating JA and Protzer U. (2017). Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol Sci 372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klingel K, Sauter M, Bock CT, Szalay G, Schnorr JJ and Kandolf R. (2004). Molecular pathology of inflammatory cardiomyopathy. Med Microbiol Immunol 193, 101–7. [DOI] [PubMed] [Google Scholar]
- [7].Lok SS, Haider Y, Howell D, Stewart JP, Hasleton PS and Egan JJ (2002). Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J 20, 1228–32. [DOI] [PubMed] [Google Scholar]
- [8].Anand AS, Joseph PB and Vera-Vazquez E. (2014). A case of pulmonary fibrosis associated with rheumatoid arthritis, scleroderma sine scleroderma and ANCA associated vasculitis. Springerplus 3, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Talman V. and Ruskoaho H. (2016). Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res 365, 563–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zechner D, Knapp N, Bobrowski A, Radecke T, Genz B. and Vollmar B. (2014). Diabetes increases pancreatic fibrosis during chronic inflammation. Exp Biol Med (Maywood) 239, 670–6. [DOI] [PubMed] [Google Scholar]
- [11].Werner S. and Grose R. (2003). Regulation of wound healing by growth factors and cytokines. Physiol Rev 83, 835–70. [DOI] [PubMed] [Google Scholar]
- [12].Sisson TH et al. (2010). Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med 181, 254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Povedano JM, Martinez P, Flores JM, Mulero F. and Blasco MA (2015). Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep 12, 286–99. [DOI] [PubMed] [Google Scholar]
- [14].Thannickal VJ (2013). Mechanistic links between aging and lung fibrosis. Biogerontology 14, 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Biernacka A. and Frangogiannis NG (2011). Aging and Cardiac Fibrosis. Aging Dis 2, 158–173. [PMC free article] [PubMed] [Google Scholar]
- [16].Delire B, Lebrun V, Selvais C, Henriet P, Bertrand A, Horsmans Y. and Leclercq IA (2016). Aging enhances liver fibrotic response in mice through hampering extracellular matrix remodeling. Aging (Albany NY) 9, 98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang HC and Fogo AB (2014). Fibrosis and renal aging. Kidney Int Suppl (2011) 4, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bullard JE, Wert SE, Whitsett JA, Dean M. and Nogee LM (2005). ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med 172, 1026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stock CJ et al. (2013). Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 68, 436–41. [DOI] [PubMed] [Google Scholar]
- [20].Wang C. et al. (2014). Mucin 5B promoter polymorphism is associated with susceptibility to interstitial lung diseases in Chinese males. PLoS One 9, e104919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wynn TA and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kendall RT and Feghali-Bostwick CA (2014). Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Duffield JS, Lupher M, Thannickal VJ and Wynn TA (2013). Host responses in tissue repair and fibrosis. Annu Rev Pathol 8, 241–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xia H. et al. (2008). Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205, 1659–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Penke LR, Speth JM, Dommeti VL, White ES, Bergin IL and Peters-Golden M. (2018). FOXM1 is a critical driver of lung fibroblast activation and fibrogenesis. J Clin Invest 128, 2389–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dabiri G, Tumbarello DA, Turner CE and Van de Water L. (2008). Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-betal autocrine loop. J Invest Dermatol 128, 2518–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bechtel W. et al. (2010). Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 16, 544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wettlaufer SH, Scott JP, McEachin RC, Peters-Golden M. and Huang SK (2016). Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am J RespirCell Mol Biol 54, 114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Duong TE and Hagood JS (2018). Epigenetic Regulation of Myofibroblast Phenotypes in Fibrosis. Curr Pathobiol Rep 6, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hu B, Gharaee-Kermani M, Wu Z. and Phan SH (2010). Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am J Pathol 177, 21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK and Trojanowska M. (2001). Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem 276, 20839–48. [DOI] [PubMed] [Google Scholar]
- [32].Asano Y, Czuwara J. and Trojanowska M. (2007). Transforming growth factor-beta regulates DNA binding activity of transcription factor Fli 1 by p300/CREB-binding protein-associated factor-dependent acetylation. J Biol Chem 282, 34672–83. [DOI] [PubMed] [Google Scholar]
- [33].Wang Y, Fan PS and Kahaleh B. (2006). Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum 54, 2271–9. [DOI] [PubMed] [Google Scholar]
- [34].Li B. and Wang JH (2011). Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability 20, 108–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hinz B. (2007). Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127, 526–37. [DOI] [PubMed] [Google Scholar]
- [36].Desmouliere A, Redard M, Darby I. and Gabbiani G. (1995). Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 146, 56–66. [PMC free article] [PubMed] [Google Scholar]
- [37].Jun J.l. and Lau LF (2010). The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12, 676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jun J.l. and Lau LF (2010). Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2, 627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kisseleva T. et al. (2012). Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A 109, 9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dodi AE et al. (2018). Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli. RespirRes 19, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Buhling F, Wille A, Rocken C, Wiesner O, Baier A, Meinecke I, Welte T. and Pap T. (2005). Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir Res 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K. and Hara N. (2002). Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J 20, 359–68. [DOI] [PubMed] [Google Scholar]
- [43].Huang SK, Scruggs AM, Donaghy J, Horowitz JC, Zaslona Z, Przybranowski S, White ES and Peters-Golden M. (2013). Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis 4, e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Horowitz JC et al. (2012). Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int J Biochem Cell Biol 44, 158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ajayi IO et al. (2013). X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol 49, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ricci A. et al. (2013). Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J Cell Physiol 228, 1516–24. [DOI] [PubMed] [Google Scholar]
- [47].White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB and Peters-Golden M. (2005). Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol 32, 135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li Y. et al. (2011). Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med 208, 1459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Michael DR et al. (2011). The human hyaluronan synthase 2 (HAS2) gene and its natural antisense RNA exhibit coordinated expression in the renal proximal tubular epithelial cell. J Biol Chem 286, 19523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Patouraux S. et al. (2017). CD44 is a key player in non-alcoholic steatohepatitis. J Hepatol 67, 328–338. [DOI] [PubMed] [Google Scholar]
- [51].Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon K.l. and Tomic-Canic M. (2016). Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res 365, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bucala R, Spiegel LA, Chesney J, Hogan M. and Cerami A. (1994). Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1, 71–81. [PMC free article] [PubMed] [Google Scholar]
- [53].Piera-Velazquez S, Li Z. and Jimenez SA (2011). Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol 179, 1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Humphreys BD et al. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Marangoni RG et al. (2015). Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol 67, 1062–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Walker N. et al. (2011). Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am J Pathol 178, 2461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hung C. et al. (2013). Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 188, 820–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW and Hogan BL (2011). Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sei U S A 108, E1475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Xie T. et al. (2016). Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest 126, 3063–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA and Kisseleva T. (2010). Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139, 987–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lin SL, Kisseleva T, Brenner DA and Duffield JS (2008). Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173, 1617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP and Schwabe RF (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 4, 2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Habiel DM and Hogaboam C. (2014). Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Front Pharmacol 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Habiel DM and Hogaboam CM (2017). Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr Pathobiol Rep 5, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xie T. et al. (2018). Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22, 3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sommer M, Schaller R, Funfstuck R, Bohle A, Bohmer FD, Muller GA and Stein G. (1999). Abnormal growth and clonal proliferation of fibroblasts in an animal model of unilateral ureteral obstruction. Nephron 82, 39–50. [DOI] [PubMed] [Google Scholar]
- [67].Jordana M, Schulman J, McSharry C, Irving LB, Newhouse MT, Jordana G. and Gauldie J. (1988). Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis 137, 579–84. [DOI] [PubMed] [Google Scholar]
- [68].Raghu G, Chen YY, Rusch V. and Rabinovitch PS (1988). Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am Rev Respir Dis 138, 703–8. [DOI] [PubMed] [Google Scholar]
- [69].Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N. and Spagnolo P. (2014). Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev 23, 308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fernandez IE and Eickelberg O. (2012). The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 9, 111–6. [DOI] [PubMed] [Google Scholar]
- [71].Dobaczewski M, Chen W. and Frangogiannis NG (2011). Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol 51, 600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Leask A. (2007). TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res 74, 207–12. [DOI] [PubMed] [Google Scholar]
- [73].Fabregat I, Moreno-Caceres J, Sanchez A, Dooley S, Dewidar B, Giannelli G, Ten Dijke P. and Consortium l.-L. (2016). TGF-beta signalling and liver disease. FEBS J 283, 2219–32. [DOI] [PubMed] [Google Scholar]
- [74].Meng XM, Nikolic-Paterson DJ and Lan HY (2016). TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 12, 325–38. [DOI] [PubMed] [Google Scholar]
- [75].Meng XM, Tang PM, Li J. and Lan HY (2015). TGF-beta/Smad signaling in renal fibrosis. Front Physiol 6, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, Maeyama T. and Hara N. (2002). TGF-beta 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol 168, 6470–8. [DOI] [PubMed] [Google Scholar]
- [77].Branton MH and Kopp JB (1999). TGF-beta and fibrosis. Microbes Infect 1, 1349–65. [DOI] [PubMed] [Google Scholar]
- [78].Hong KM, Belperio JA, Keane MP, Burdick MD and Strieter RM (2007). Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J Biol Chem 282, 22910–20. [DOI] [PubMed] [Google Scholar]
- [79].Grotendorst GR (1997). Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev 8, 171–9. [DOI] [PubMed] [Google Scholar]
- [80].Rodriguez-Pascual F, Reimunde FM, Redondo-Horcajo M. and Lamas S. (2004). Transforming growth factor-beta induces endothelin-1 expression through activation of the Smad signaling pathway. J Cardiovasc Pharmacol 44 Suppl 1, S39–42. [DOI] [PubMed] [Google Scholar]
- [81].Myster DL and Duronio RJ (2000). To differentiate or not to differentiate? Curr Biol 10, R302–4. [DOI] [PubMed] [Google Scholar]
- [82].Biernacka A, Dobaczewski M. and Frangogiannis NG (2011). TGF-beta signaling in fibrosis. Growth Factors 29, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lin CH et al. (2013). Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the Rac1/MLK3/JNK/AP-1 pathway. Biochim Biophys Acta 1833, 2823–2833. [DOI] [PubMed] [Google Scholar]
- [84].Gallelli L. et al. (2005). Endothelin-1 induces proliferation of human lung fibroblasts and IL-11 secretion through an ET(A) receptor-dependent activation of MAP kinases. J Cell Biochem 96, 858–68. [DOI] [PubMed] [Google Scholar]
- [85].Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M. and Karliner JS (2000). Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol 32, 565–76. [DOI] [PubMed] [Google Scholar]
- [86].Frazier K, Williams S, Kothapalli D, Klapper H. and Grotendorst GR (1996). Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 107, 404–11. [DOI] [PubMed] [Google Scholar]
- [87].Clark JG, Madtes DK and Raghu G. (1993). Effects of platelet-derived growth factor isoforms on human lung fibroblast proliferation and procollagen gene expression. Exp Lung Res 19, 327–44. [DOI] [PubMed] [Google Scholar]
- [88].Simmons JG, Pucilowska JB, Keku TO and Lund PK (2002). IGF-I and TGF-beta1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol 283, G809–18. [DOI] [PubMed] [Google Scholar]
- [89].Laato M, Kahari VM, Niinikoski J. and Vuorio E. (1987). Epidermal growth factor increases collagen production in granulation tissue by stimulation of fibroblast proliferation and not by activation of procollagen genes. Biochem J 247, 385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hostettler KE et al. (2014). Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res 15, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Siddesha JM et al. (2013). Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J Mol Cell Cardiol 65, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Battegay EJ, Raines EW, Colbert T. and Ross R. (1995). TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J Immunol 154, 6040–7. [PubMed] [Google Scholar]
- [93].Kohan M, Breuer R. and Berkman N. (2009). Osteopontin induces airway remodeling and lung fibroblast activation in a murine model of asthma. Am J Respir Cell Mol Biol 41, 290–6. [DOI] [PubMed] [Google Scholar]
- [94].Lertchirakarn V, Birner R. and Messer HH (1998). Effects of interleukin-1 beta on human pulpal fibroblast proliferation and collagen synthesis. J Endod 24, 409–13. [DOI] [PubMed] [Google Scholar]
- [95].Monroe JG, Haidar S, Prystowsky MB and Lammie P. (1988). Lymphokine regulation of inflammatory processes: interleukin-4 stimulates fibroblast proliferation. Clin Immunol Immunopathol 49, 292–8. [DOI] [PubMed] [Google Scholar]
- [96].Saito A, Okazaki H, Sugawara I, Yamamoto K. and Takizawa H. (2003). Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol 132, 168–76. [DOI] [PubMed] [Google Scholar]
- [97].Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, Zhu S, Pugin J. and Matthay MA (2004). Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1 beta-induced IL-6 expression. J Immunol 172, 2668–77. [DOI] [PubMed] [Google Scholar]
- [98].Liao WT et al. (2010). Enhanced MCP-1 release by keloid CD14+ cells augments fibroblast proliferation: role of MCP-1 and Akt pathway in keloids. Exp Dermatol 19, e142–50. [DOI] [PubMed] [Google Scholar]
- [99].Liu X. et al. (2015). Role of human pulmonary fibroblast-derived MCP-1 in cell activation and migration in experimental silicosis. Toxicol Appl Pharmacol 288, 152–60. [DOI] [PubMed] [Google Scholar]
- [100].Puxeddu I, Bader R, Piliponsky AM, Reich R, Levi-Schaffer F. and Berkman N. (2006). The CC chemokine eotaxin/CCL11 has a selective profibrogenic effect on human lung fibroblasts. J Allergy Clin Immunol 117,103–10. [DOI] [PubMed] [Google Scholar]
- [101].Zhang YE (2017). Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb Perspect Biol 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ji H, Tang H, Lin H, Mao J, Gao L, Liu J. and Wu T. (2014). Rho/Rock cross-talks with transforming growth factor-beta/Smad pathway participates in lung fibroblast-myofibroblast differentiation. Biomed Rep 2, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lamouille S, Xu J. and Derynck R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15, 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Jiang C, Huang H, Liu J, Wang Y, Lu Z. and Xu Z. (2012). Fasudil, a Rho-kinase inhibitor, attenuates bleomycin-induced pulmonary fibrosis in mice. Int J Mol Sei 13, 8293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Monaghan-Benson E, Wittchen ES, Doerschuk CM and Burridge K. (2018). A Rnd3/p190RhoGAP pathway regulates RhoA activity in idiopathic pulmonary fibrosis fibroblasts. Mol Biol Cell 29, 2165–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Conte E, Fruciano M, Fagone E, Gili E, Caraci F, lemmolo M, Crimi N. and Vancheri C. (2011). Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS One 6, e24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z. and Thannickal VJ (2004). Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-betal in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem 279, 1359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kim G, Jun JB and Elkon KB (2002). Necessary role of phosphatidylinositol 3-kinase in transforming growth factor beta-mediated activation of Akt in normal and rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 46, 1504–11. [DOI] [PubMed] [Google Scholar]
- [109].Chen G, Chen H, Wang C, Peng Y, Sun L, Liu H. and Liu F. (2012). Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS One 7, e33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen JK, Chen J, Neilson EG and Harris RC (2005). Role of mammalian target of rapamycin signaling in compensatory renal hypertrophy. J Am Soc Nephrol 16, 1384–91. [DOI] [PubMed] [Google Scholar]
- [111].Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD, Dart A. and Du XJ (2006). Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens 24, 1663–70. [DOI] [PubMed] [Google Scholar]
- [112].Patsenker E, Schneider V, Ledermann M, Saegesser H, Dorn C, Hellerbrand C. and Stickel F. (2011). Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol 55, 388–98. [DOI] [PubMed] [Google Scholar]
- [113].Romero Y. et al. (2016). mTORCI activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 15,1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P. and Sabatini DM (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14, 1296–302. [DOI] [PubMed] [Google Scholar]
- [115].Guertin DA et al. (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11, 859–71. [DOI] [PubMed] [Google Scholar]
- [116].Jung SA et al. (2007). Upregulation of TGF-beta-induced tissue transglutaminase expression by PI3K-Akt pathway activation in human subconjunctival fibroblasts. Invest Ophthalmol Vis Sei 48, 1952–8. [DOI] [PubMed] [Google Scholar]
- [117].Wettlaufer SH, Penke LR, Okunishi K. and Peters-Golden M. (2017). Distinct PKA regulatory subunits mediate PGE2 inhibition of TGFbeta-1-stimulated collagen I translation and myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol 313, L722–L731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ivaska J, Reunanen H, Westermarck J, Koivisto L, Kahari VM and Heino J. (1999). Integrin alpha2beta1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the alpha2 cytoplasmic tail. J Cell Biol 147, 401–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Sato M, Shegogue D, Gore EA, Smith EA, McDermott PJ and Trojanowska M. (2002). Role of p38 MAPK in transforming growth factor beta stimulation of collagen production by scleroderma and healthy dermal fibroblasts. J Invest Dermatol 118, 704–11. [DOI] [PubMed] [Google Scholar]
- [120].Penke LR, Huang SK, White ES and Peters-Golden M. (2014). Prostaglandin E2 inhibits alpha-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J Biol Chem 289, 17151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang YE (2009). Non-Smad pathways in TGF-beta signaling. Cell Res 19, 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S. and Horie T. (2001). Transforming growth Factor-betal induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med 163, 152–7. [DOI] [PubMed] [Google Scholar]
- [123].Hashimoto S, Gon Y, Takeshita I, Maruoka S. and Horie T. (2001). IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase-dependent pathway. J Allergy Clin Immunol 107, 1001–8. [DOI] [PubMed] [Google Scholar]
- [124].Yoshida K. et al. (2005). Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol 166, 1029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Utsugi M, Dobashi K, Ishizuka T, Masubuchi K, Shimizu Y, Nakazawa T. and Mori M. (2003). C-Jun-NH2-terminal kinase mediates expression of connective tissue growth factor induced by transforming growth factor-betal in human lung fibroblasts. Am J Respir Cell Mol Biol 28, 754–61. [DOI] [PubMed] [Google Scholar]
- [126].Black SA Jr., Palamakumbura AH, Stan M. and Trackman PC (2007). Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase. J Biol Chem 282, 15416–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Yamanaka O, Saika S, Ohnishi Y, Kim-Mitsuyama S, Kamaraju AK and Ikeda K. (2007). Inhibition of p38MAP kinase suppresses fibrogenic reaction in conjunctiva in mice. Mol Vis 13, 1730–9. [PubMed] [Google Scholar]
- [128].Shi-Wen X. et al. (2006). Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol Cell Biol 26, 5518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Akhmetshina A. et al. (2008). Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum 58, 2553–64. [DOI] [PubMed] [Google Scholar]
- [130].Liu X, Sun SQ, Hassid A. and Ostrom RS (2006). cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase ½ and Smad signaling in cardiac fibroblasts. Mol Pharmacol 70, 1992–2003. [DOI] [PubMed] [Google Scholar]
- [131].Sun Q, Wu Y, Zhao F. and Wang J. (2017). Maresin 1 inhibits transforming growth factor-betal-induced proliferation, migration and differentiation in human lung fibroblasts. Mol Med Rep 16, 1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ju W. et al. (2012). Inhibition of alpha-SMA by the ectodomain of FGFR2c attenuates lung fibrosis. Mol Med 18, 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lai JM et al. (2016). Redox-sensitive MAPK and Notch3 regulate fibroblast differentiation and activation: a dual role of ERK½. Oncotarget 7, 43731–43745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hecker L, Jagirdar R, Jin T. and Thannickal VJ (2011). Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res 317, 1914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO and Steadman R. (2013). Transforming growth factor-betal (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem 288, 14824–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].He S, Liu X, Yang Y, Huang W, Xu S, Yang S, Zhang X. and Roberts MS (2010). Mechanisms of transforming growth factor beta(1)/Smad signalling mediated by mitogen-activated protein kinase pathways in keloid fibroblasts. Br J Dermatol 162, 538–46. [DOI] [PubMed] [Google Scholar]
- [137].Jiang Y, Wu C, Boye A, Wu J, Wang J, Yang X. and Yang Y. (2015). MAPK inhibitors modulate Smad2/¾ complex cyto-nuclear translocation in myofibroblasts via lmp7/8 mediation. Mol Cell Biochem 406, 255–62. [DOI] [PubMed] [Google Scholar]
- [138].Carthy JM, Garmaroudi FS, Luo Z. and McManus BM (2011). Wnt3a induces myofibroblast differentiation by upregulating TGF-beta signaling through SMAD2 in a beta-catenin-dependent manner. PLoS One 6, e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C. and Kaminski N. (2009). WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol 41, 583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Konigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W. and Eickelberg O. (2008). Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One 3, e2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Dees C. et al. (2014). The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis 73, 1232–9. [DOI] [PubMed] [Google Scholar]
- [142].Liu J. et al. (2018). Methylation of secreted frizzled-related protein 1 (SFRP1) promoter downregulates Wnt/beta-catenin activity in keloids. J Mol Histol 49, 185–193. [DOI] [PubMed] [Google Scholar]
- [143].Baarsma HA et al. (2011). Activation of WNT/beta-catenin signaling in pulmonary fibroblasts by TGF-beta(1) is increased in chronic obstructive pulmonary disease. PLoS One 6, e25450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cao P, Aoki Y, Badri L, Walker NM, Manning CM, Lagstein A, Fearon ER and Lama VN (2017). Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J Clin Invest 127, 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Epstein Shochet G, Brook E, Israeli-Shani L, Edelstein E. and Shitrit D. (2017). Fibroblast paracrine TNF-alpha signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir Res 18, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Margadant C. and Sonnenberg A. (2010). Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chiquet M, Renedo AS, Huber F. and Fluck M. (2003). How do fibroblasts translate mechanical signals into changes in extracellular matrix production? Matrix Biol 22, 73–80. [DOI] [PubMed] [Google Scholar]
- [148].O’Toole TE et al. (1994). Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol 124, 1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Orr AW, Helmke BP, Blackman BR and Schwartz MA (2006). Mechanisms of mechanotransduction. Dev Cell 10, 11–20. [DOI] [PubMed] [Google Scholar]
- [150].Gui Y, Li J, Lu Q, Feng Y, Wang M, He W, Yang J. and Dai C. (2018). Yap/Taz mediates mTORC2-stimulated fibroblast activation and kidney fibrosis. J Biol Chem 293, 16364–16375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Qin Z, Xia W, Fisher GJ, Voorhees JJ and Quan T. (2018). YAP/TAZ regulates TGF-beta/Smad3 signaling by induction of Smad7 via AP-1 in human skin dermal fibroblasts. Cell Commun Signal 16, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Xu F, Liu C, Zhou D. and Zhang L. (2016). TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem 64, 157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Samarakoon R. and Higgins PJ (2008). Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost 100, 976–83. [PMC free article] [PubMed] [Google Scholar]
- [154].Jonk LJ, Itoh S, Heldin CH, ten Dijke P. and Kruijer W. (1998). Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273, 21145–52. [DOI] [PubMed] [Google Scholar]
- [155].ten Dijke P, Miyazono K. and Heldin CH (2000). Signaling inputs converge on nuclear effectors in TGF-beta signaling. Trends Biochem Sci 25, 64–70. [DOI] [PubMed] [Google Scholar]
- [156].Roach KM, Wulff H, Feghali-Bostwick C, Amrani Y. and Bradding P. (2014). Increased constitutive alphaSMA and Smad2/3 expression in idiopathic pulmonary fibrosis myofibroblasts is KCa3.1-dependent. Respir Res 15, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Sandbo N, Kregel S, Taurin S, Bhorade S. and Dulin NO (2009). Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am J Respir Cell Mol Biol 41, 332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Zhang M, Fang H, Zhou J. and Herring BP (2007). A novel role of Brg1 in the regulation of SRF/MRTFA-dependent smooth muscle-specific gene expression. J Biol Chem 282, 25708–16. [DOI] [PubMed] [Google Scholar]
- [159].Plantier L, Renaud H, Respaud R, Marchand-Adam S. and Crestani B. (2016). Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways. Int J Mol Sci 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Chen J, Zhong Q, Wang J, Cameron RS, Borke JL, Isales CM and Bollag RJ (2001). Microarray analysis of Tbx2-directed gene expression: a possible role in osteogenesis. Mol Cell Endocrinol 177, 43–54. [DOI] [PubMed] [Google Scholar]
- [161].Teng H, Davis E, Abrahams A, Mowla S, Parker M.l. and Prince S. (2007). A role for Tbx2 in the regulation of the alpha2(1) collagen gene in human fibroblasts. J Cell Biochem 102, 618–25. [DOI] [PubMed] [Google Scholar]
- [162].Horie M. et al. (2018). TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 314, L177–L191. [DOI] [PubMed] [Google Scholar]
- [163].Fan W, Huang X, Chen C, Gray J. and Huang T. (2004). TBX3 and its isoform TBX3+2a are functionally distinctive in inhibition of senescence and are overexpressed in a subset of breast cancer cell lines. Cancer Res 64, 5132–9. [DOI] [PubMed] [Google Scholar]
- [164].Balli D. et al. (2013). Foxml transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J 32, 231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Paoli P, Giannoni E. and Chiarugi P. (2013). Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 1833, 3481–3498. [DOI] [PubMed] [Google Scholar]
- [166].Thannickal VJ and Horowitz JC (2006). Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. ProcAm Thorac Soc 3, 350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Im J, Lawrence J, Seelig D. and Nho RS (2018). FoxM1-dependent RAD51 and BRCA2 signaling protects idiopathic pulmonary fibrosis fibroblasts from radiation-induced cell death. Cell Death Dis 9, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Black M, Milewski D, Le T, Ren X, Xu Y, Kalinichenko VV and Kalin TV (2018). FOXF1 Inhibits Pulmonary Fibrosis by Preventing CDH2-CDH11 Cadherin Switch in Myofibroblasts. Cell Rep 23, 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Malin D. et al. (2007). Forkhead box F1 is essential for migration of mesenchymal cells and directly induces integrin-beta3 expression. Mol Cell Biol 27, 2486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Correll KA. et al. (2018). TGF beta inhibits HGF, FGF7, and FGF10 expression in normal and IPF lung fibroblasts. Physiol Rep 6, e13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zhou T, Luo M, Cai W, Zhou S, Feng D, Xu C. and Wang H. (2018). Runt-Related Transcription Factor 1 (RUNX1) Promotes TGF-beta-lnduced Renal Tubular Epithelial-to-Mesenchymal Transition (EMT) and Renal Fibrosis through the PI3K Subunit p110delta. EBioMedicine 31, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kim W, Barron DA, San Martin R, Chan KS., Tran LL, Yang F, Ressler SJ and Rowley DR (2014). RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc Natl Acad Sei U S A 111, 16389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Anderson G. et al. (2018). RUNX-mediated growth arrest and senescence are attenuated by diverse mechanisms in cells expressing RUNX1 fusion oncoproteins. J Cell Biochem 119, 2750–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Navarro V, Roig P, Nieto A, Jimenez J, Tuset C, Tuset L, Navarro R. and Juan G. (1988). A small outbreak of HIV infection among commercial plasma donors. Lancet 2, 42. [DOI] [PubMed] [Google Scholar]
- [175].Yan J, Zhang Z, Yang J, Mitch WE and Wang Y. (2015). JAK3/STAT6 Stimulates Bone Marrow-Derived Fibroblast Activation in Renal Fibrosis. J Am Soc Nephrol 26, 3060–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kaikkonen MU, Lam MT and Glass CK (2011). Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res 90, 430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Kato M. (2018). Noncoding RNAs as therapeutic targets in early stage diabetic kidney disease. Kidney Res Clin Pract 37, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Teng KY. and Ghoshal K. (2015). Role of Noncoding RNAs as Biomarker and Therapeutic Targets for Liver Fibrosis. Gene Expr 16, 155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zhang Y. et al. (2018). Critical effects of long non-coding RNA on fibrosis diseases. Exp Mol Med 50, e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Lu Q, Guo Z, Xie W, Jin W, Zhu D, Chen S. and Ren T. (2018). The IncRNA H19 Mediates Pulmonary Fibrosis by Regulating the miR-196a/COL1A1 Axis. Inflammation 41, 896–903. [DOI] [PubMed] [Google Scholar]
- [181].Tao H, Cao W, Yang JJ, Shi KH, Zhou X, Liu LP and Li J. (2016). Long noncoding RNA H19 controls DUSP5/ERK½ axis in cardiac fibroblast proliferation and fibrosis. Cardiovasc Pathol 25, 381–9. [DOI] [PubMed] [Google Scholar]
- [182].Zhao X. et al. (2018). IncRNA PFAR Promotes Lung Fibroblast Activation and Fibrosis by Targeting miR-138 to Regulate the YAP1-Twist Axis. Mol Ther 26, 2206–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Jiang H. et al. (2018). Inhibition of IncRNA PFRL prevents pulmonary fibrosis by disrupting the miR-26a/smad2 loop. Am J Physiol Lung Cell Mol Physiol 315, L563–L575. [DOI] [PubMed] [Google Scholar]
- [184].Li X. et al. (2018). IncRNA PFAL promotes lung fibrosis through CTGF by competitively binding miR-18a. FASEB J 32, 5285–5297. [DOI] [PubMed] [Google Scholar]
- [185].Zhang K. et al. (2017). The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat Commun 8, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Qu X. et al. (2017). MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep 7, 42657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Liang H. et al. (2018). LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics 8, 1180–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Bian EB et al. (2017). Hotair facilitates hepatic stellate cells activation and fibrogenesis in the liver. Biochim Biophys Acta Mol Basis Dis 1863, 674–686. [DOI] [PubMed] [Google Scholar]
- [189].Tao H, Zhang JG, Qin RH, Dai C, Shi P, Yang JJ, Deng ZY and Shi KH (2017). LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology 386, 11–18. [DOI] [PubMed] [Google Scholar]
- [190].Yu F. et al. (2015). Long Non-coding RNA Growth Arrest-specific Transcript 5 (GAS5) Inhibits Liver Fibrogenesis through a Mechanism of Competing Endogenous RNA. J Biol Chem 290, 28286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].He Y. et al. (2014). Inhibitory effects of long noncoding RNA MEG3 on hepatic stellate cells activation and liver fibrogenesis. Biochim Biophys Acta 1842, 2204–15. [DOI] [PubMed] [Google Scholar]
- [192].Dattaroy D. et al. (2015). Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 308, G298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N. and Abraham E. (2010). miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med 207, 1589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].McClelland AD et al. (2015). miR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin Sei (Lond) 129, 1237–49. [DOI] [PubMed] [Google Scholar]
- [195].Yuan J. et al. (2017). Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell Physiol Biochem 42, 2207–2219. [DOI] [PubMed] [Google Scholar]
- [196].Zhang J, Xu D, Li N, Li Y, He Y, Hu X, Lyu L. and He L. (2017). Downregulation of microRNA-31 inhibits proliferation and induces apoptosis by targeting HIF1AN in human keloid. Oncotarget 8, 74623–74634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Cui H, Ge J, Xie N, Banerjee S, Zhou Y, Antony VB, Thannickal VJ and Liu G. (2017). miR-34a Inhibits Lung Fibrosis by Inducing Lung Fibroblast Senescence. Am J Respir Cell Mol Biol 56, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Li WQ et al. (2011). The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J 278, 1522–32. [DOI] [PubMed] [Google Scholar]
- [199].Zhou Y, Xiong M, Niu J, Sun Q, Su W, Zen K, Dai C. and Yang J. (2014). Secreted fibroblast-derived miR-34a induces tubular cell apoptosis in fibrotic kidney. J Cell Sei 127, 4494–506. [DOI] [PubMed] [Google Scholar]
- [200].Nho RS, Im J, Ho YY and Hergert P. (2014). MicroRNA-96 inhibits Fox03a function in IPF fibroblasts on type I collagen matrix. Am J Physiol Lung Cell Mol Physiol 307, L632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Yang S. et al. (2013). miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J 27, 2382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Milosevic J. et al. (2012). Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol 47, 879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA and Katsikis PD (2017). Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther 19, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Pottier N. et al. (2009). Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One 4, e6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Zhang D, Cui Y, Li B, Luo X, Li B. and Tang Y. (2016). miR-155 regulates high glucose-induced cardiac fibrosis via the TGF-beta signaling pathway. Mol Biosyst 13, 215–224. [DOI] [PubMed] [Google Scholar]
- [206].Lino Cardenas CL et al. (2013). miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet 9, e1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Bodempudi V. et al. (2014). miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 307, L283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Fierro-Fernandez M. et al. (2015). miR-9–5p suppresses pro-fibrogenic transformation of fibroblasts and prevents organ fibrosis by targeting NOX4 and TGFBR2. EMBO Rep 16, 1358–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Hong Y. et al. (2016). MiR-22 may Suppress Fibrogenesis by Targeting TGFbetaR I in Cardiac Fibroblasts. Cell Physiol Biochem 40, 1345–1353. [DOI] [PubMed] [Google Scholar]
- [210].Liang H. et al. (2014). The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther 22, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Cui H, Banerjee S, Xie N, Ge J, Liu RM, Matalon S, Thannickal VJ and Liu G. (2016). MicroRNA-27a-3p Is a Negative Regulator of Lung Fibrosis by Targeting Myofibroblast Differentiation. Am J RespirCell Mol Biol 54, 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Cushing L. et al. (2011). miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol 45, 287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Huang C. et al. (2017). MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast proliferation and activation. J Biol Chem 292, 16420–16439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Yang S. et al. (2012). Participation of miR-200 in pulmonary fibrosis. Am J Pathol 180, 484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, Wu J. and Zern MA (2010). Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 298, G101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Huang SK, Wettlaufer SH, Chung J. and Peters-Golden M. (2008). Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol 39, 482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Tanaka K. et al. (2003). Inhibition of induction of myofibroblasts by interferon gamma in a human fibroblast cell line. Int Immunopharmacol 3, 1273–80. [DOI] [PubMed] [Google Scholar]
- [218].Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y. and Varga J. (2009). Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J 23, 2968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Nuwormegbe SA, Sohn JH and Kim SW (2017). A PPAR-Gamma Agonist Rosiglitazone Suppresses Fibrotic Response in Human Pterygium Fibroblasts by Modulating the p38 MAPK Pathway. Invest Ophthalmol Vis Sci 58, 5217–5226. [DOI] [PubMed] [Google Scholar]
- [220].Wei J. et al. (2010). PPARgamma downregulation by TGFss in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One 5, e13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Kyoung Kim H, Kyoung Kim Y, Song IH, Baek SH, Lee SR, Hye Kim J. and Kim JR (2005). Down-regulation of a forkhead transcription factor, F0X03a, accelerates cellular senescence in human dermal fibroblasts. J Gerontol A Biol Sci Med Sci 60, 4–9. [DOI] [PubMed] [Google Scholar]
- [222].Sagana RL et al. (2009). Phosphatase and tensin homologue on chromosome 10 (PTEN) directs prostaglandin E2-mediated fibroblast responses via regulation of E prostanoid 2 receptor expression. J Biol Chem 284, 32264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Xin Z. et al. (2018). F0X01/3: Potential suppressors of fibrosis. Ageing Res Rev 41, 42–52. [DOI] [PubMed] [Google Scholar]
- [224].Gu X, Xu D, Fu L, Wang Y, Mei C. and Gao X. (2017). KLF 15 Works as an Early Anti-Fibrotic Transcriptional Regulator in Ang ll-lnduced Renal Fibrosis via Down-Regulation of CTGF Expression. Kidney Blood Press Res 42, 999–1012. [DOI] [PubMed] [Google Scholar]
- [225].Wang B, Haidar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, Leask A. and Jain MK (2008). The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J Mol Cell Cardiol 45, 193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Blikslager AT, Roberts MC, Rhoads JM and Argenzio RA (1997). Prostaglandins 12 and E2 have a synergistic role in rescuing epithelial barrier function in porcine ileum. J Clin Invest 100, 1928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Uribe A, Alam M. and Midtvedt T. (1992). E2 prostaglandins modulate cell proliferation in the small intestinal epithelium of the rat. Digestion 52, 157–64. [DOI] [PubMed] [Google Scholar]
- [228].Nishihara H, Kizaka-Kondoh S, Insel PA and Eckmann L. (2003). Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)-2. Proc Natl Acad Sci U S A 100, 8921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Bitterman PB, Wewers MD, Rennard SI, Adelberg S. and Crystal RG (1986). Modulation of alveolar macrophage-driven fibroblast proliferation by alternative macrophage mediators. J Clin Invest 77, 700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Thomas PE, Peters-Golden M, White ES, Thannickal VJ and Moore BB (2007). PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol 293, L417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, Horowitz JC and Peters-Golden M. (2009). Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J 23, 4317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Moore BB, Ballinger MN, White ES, Green ME, Herrygers AB, Wilke CA, Toews GB and Peters-Golden M. (2005). Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J Immunol 174, 5644–9. [DOI] [PubMed] [Google Scholar]
- [233].Kamio K. et al. (2007). Prostacyclin analogs inhibit fibroblast contraction of collagen gels through the cAMP-PKA pathway. Am J Respir Cell Mol Biol 37, 113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL and Koller BH (2006). COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 291, L144–56. [DOI] [PubMed] [Google Scholar]
- [235].Sisson TH et al. (2018). Phosphodiesterase 4 inhibition reduces lung fibrosis following targeted type II alveolar epithelial cell injury. Physiol Rep 6, e13753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Udalov S. et al. (2010). Effects of phosphodiesterase 4 inhibition on bleomycin-induced pulmonary fibrosis in mice. BMC Pulm Med 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Diakov I. (1973). [Study of the dependence of oxygen consumption on the lipopolysaccharide-protein complex content in the antigenic structure of Salmonella abortus ovis]. Vet Med Nauki 10, 27–32. [PubMed] [Google Scholar]
- [238].Coward WR, Watts K, Feghali-Bostwick CA, Knox A. and Pang L. (2009). Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol 29, 4325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Walker NM, Badri LN, Wadhwa A, Wettlaufer S, Peters-Golden M. and Lama VN (2012). Prostaglandin E2 as an inhibitory modulator of fibrogenesis in human lung allografts. Am J Respir Crit Care Med 185, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM and Tschumperlin DJ (2010). Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC and Peters-Golden M. (2010). Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol 177, 2245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H. and Mann DA (2010). MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138, 705–14, 714 e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Kapoor M, Kojima F, Qian M, Yang L. and Crofford LJ (2007). Microsomal prostaglandin E synthase-1 deficiency is associated with elevated peroxisome proliferator-activated receptor gamma: regulation by prostaglandin E2 via the phosphatidylinositol 3-kinase and Akt pathway. J Biol Chem 282, 5356–66. [DOI] [PubMed] [Google Scholar]
- [244].Zhang X, Tang N, Hadden TJ and Rishi AK (2011). Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813, 1978–86. [DOI] [PubMed] [Google Scholar]
- [245].Nho RS, Peterson M, Hergert P. and Henke CA (2013). Fox03a (Forkhead Box 03a) deficiency protects Idiopathic Pulmonary Fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas. PLoS One 8, e61017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Koo HY et al. (2018). Fibroblast growth factor 2 decreases bleomycin-induced pulmonary fibrosis and inhibits fibroblast collagen production and myofibroblast differentiation. J Pathol 246, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Giannandrea M. and Parks WC (2014). Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 7, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Alvarez D, Levine M. and Rojas M. (2015). Regenerative medicine in the treatment of idiopathic pulmonary fibrosis: current position. Stem Cells Cloning 8, 61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Perrucci GL, Rurali E. and Pompilio G. (2018). Cardiac fibrosis in regenerative medicine: destroy to rebuild. J Thorac Dis 10, S2376–S2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Evans RA, Tian YC, Steadman R. and Phillips AO (2003). TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res 282, 90–100. [DOI] [PubMed] [Google Scholar]
- [251].Garrison G, Huang SK, Okunishi K, Scott JP, Kumar Penke LR, Scruggs AM and Peters-Golden M. (2013). Reversal of myofibroblast differentiation by prostaglandin E(2). Am J Respir Cell Mol Biol 48, 550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Zmajkovicova K. et al. (2018). The Antifibrotic Activity of Prostacyclin Receptor Agonism is Mediated through Inhibition ofYAP/TAZ. Am J Respir Cell Mol Biol [DOI] [PubMed] [Google Scholar]
- [253].Dolivo DM, Larson SA and Dominko T. (2017). FGF2-mediated attenuation of myofibroblast activation is modulated by distinct MAPK signaling pathways in human dermal fibroblasts. J Dermatol Sci 88, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Horowitz JC and Thannickal VJ (2019). Mechanisms for the Resolution of Organ Fibrosis. Physiology (Bethesda) 34, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Jun J.l. and Lau LF (2018). Resolution of organ fibrosis. J Clin Invest 128, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]