

Abstract
The landscape of structural variants (SVs) in multiple myeloma remains poorly understood. Here, we performed comprehensive analysis of SVs in a large cohort of 752 multiple myeloma patients by low coverage long-insert whole genome sequencing. We identified 68 SV hotspots involving 17 new candidate driver genes, including the therapeutic targets BCMA (TNFRSF17), SLAMF and MCL1. Catastrophic complex rearrangements termed chromothripsis were present in 24% of patients and independently associated with poor clinical outcomes. Templated insertions were the second most frequent complex event (19%), mostly involved in super-enhancer hijacking and activation of oncogenes such as CCND1 and MYC. Importantly, in 31% of patients two or more seemingly independent putative driver events were caused by a single structural event, demonstrating that the complex genomic landscape of multiple myeloma can be acquired through few key events during tumor evolutionary history. Overall, this study reveals the critical role of SVs in multiple myeloma pathogenesis.
Keywords: multiple myeloma, structural variants, hotspots, chromothripsis, templated insertion, chromoplexy
Introduction
Whole genome sequencing (WGS) studies have demonstrated the importance of structural variants (SVs) in the initiation and progression of many cancers(1–8). Functional implications of SVs include gene dosage effects from gain or loss of chromosomal material, gene regulatory effects such as super-enhancer hijacking, and gene fusions(9). The basic unit of SVs are pairs of breakpoints, classified as either deletion, tandem duplication, translocation or inversion, which can manifest as simple events or form complex patterns where multiple SVs are acquired together, often involving multiple chromosomes(1–8,10).
In multiple myeloma, previous studies of SVs have had a narrow scope, usually limited to recurrent translocations involving MYC or the immunoglobulin loci (i.e. IGH, IGL and IGK)(11–17). The vast majority of established genomic drivers in multiple myeloma are single nucleotide variants (SNVs) and copy number alterations (CNAs), identified by whole exome sequencing and array-based approaches(18–22). However, important aspects of tumor biology and evolution remain poorly explained by known genomic drivers, such as progression from precursor stages to active multiple myeloma and the development of drug resistance(12,23–25).
We recently reported the first comprehensive study of SVs in multiple myeloma by WGS of sequential samples from 30 patients(21). Despite the limited sample set and the absence of gene expression data, our findings indicated that SVs are a key missing piece to understand the driver landscape of multiple myeloma. Of particular interest, we found a high prevalence of three main classes of complex SVs: chromothripsis, templated insertions and chromoplexy(21). In chromothripsis, chromosomal shattering and random rejoining results in a pattern of tens to hundreds of breakpoints with oscillating copy number across one or more chromosomes (Figure 1A-B)(26). Templated insertions are characterized by focal gains bounded by translocations, resulting in concatenation of amplified segments from two or more chromosomes into a continuous stretch of DNA, which is inserted back into any of the involved chromosomes (Figure 1C-D)(4,21). Chromoplexy similarly connects segments from multiple chromosomes, but the local footprint is characterized by copy number loss (Figure 1E-F)(27). Importantly, these complex SVs represent large-scale genomic alterations acquired by the cancer cell at a single point in time, potentially involving multiple drivers and shaping subsequent tumor evolution(2,27).
Figure 1: Complex structural variant classes in multiple myeloma.
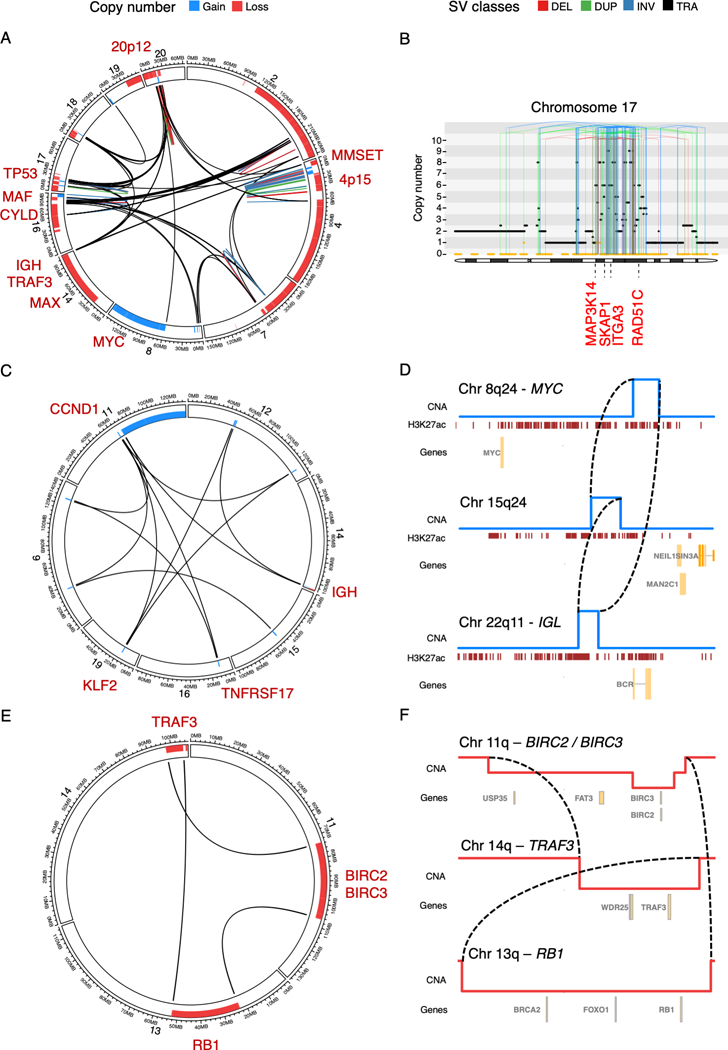
A) Chromothripsis involving IGH and 9 recurrent drivers across 10 different chromosomes (sample MMRF_1890_1_BM). B) Chromothripsis causing high-level focal gains on chromosome 17 (sample MMRF_2330_1_BM). The horizontal black line indicates total copy number; the dashed orange line minor copy number. Vertical lines represent SV breakpoints, color-coded by SV class. Selected overexpressed genes (Z-score >2) are annotated in red, including the established multiple myeloma driver gene MAP3K14, and RAD51C, an oncogene commonly amplified in breast cancer(66) (6 copies). C) Templated insertion involving 7 different chromosomes, causing a canonical IGH-CCND1 translocation and involving at least two additional drivers in the same event (i.e. KLF2 and TNFRSF17) (sample MMRF_1677_1_BM). D) Simpler templated insertion cycle (brown lines), involving IGL, MYC, and a hotspot on chromosome 15q24 (sample MMRF_1550_1_BM). Copy number profile shown in blue, with active enhancers below in brown (H3K27Ac). E) Chromoplexy involving chromosomes 11, 13, and 14, simultaneously causing deletion of key tumor suppressor genes on each chromosome (sample MMRF_2194_1_BM). F) Zooming in on the translocations and associated large deletions which make up the chromoplexy event depicted as a CIRCOS plot in C) (sample MMRF_2194_1_BM). The circos plots in panels A, C and E each show the SV breakpoints of a single complex SV (colored lines; legend above panels), with bars around the plot circumference indicating copy number changes (red = loss; blue = gain).
Here, we comprehensively characterized the role of genome-wide SVs in 752 multiple myeloma patients, revealing novel SV hotspots, rare SVs with strong impact on gene expression, and complex events simultaneously causing multiple drivers.
Results
Genome-wide landscape of structural variation in multiple myeloma
To define the landscape of simple and complex SVs in multiple myeloma, we investigated 752 newly diagnosed patients from the CoMMpass study (NCT01454297; IA13) who underwent low coverage long-insert WGS (median 4–8X) and whole exome sequencing (Table S1, Supplementary Methods). RNA sequencing was also available from 591 patients (78.6%). For each patient sample, we integrated the genome-wide somatic copy number profile with SV data and assigned each pair of SV breakpoints as either simple or part of a complex event according to the three main classes previously identified in multiple myeloma (Figure 1; Methods)(21). Templated insertions involving more than two chromosomes were considered complex. Events involving more than three breakpoint pairs which did not fulfill the criteria for a specific class of complex event were classified as unspecified “complex”(21).
Our final SV catalog was obtained by integrating the two SV calling algorithms, DELLY(10) and Manta(28), followed by a series of quality filters. First, we included all SVs called and passed by both callers. Then SVs called by a single caller were included in specific circumstances: i) SVs supporting copy number junctions; ii) reciprocal translocations; iii) translocations involving an immunoglobulin locus (i.e. IGH, IGK or IGL) (Supplementary methods). Using the final SV catalog, long-insert low-coverage WGS revealed a sensitivity of 91–92% and specificity of 97% to identify translocations involving IGH and the most common canonical drivers CCND1 and WHSC1/MMSET. Re-calculating performance metrics for canonical IGH-translocations using the same SV filtering criteria genome-wide (i.e. without the relaxed quality requirements for immunoglobulin translocations), we observed no changes in specificity, and sensitivity of 91% for IGH-CCND1 (identical as before) and 88 % for IGH-WHSC1/MMSET (down from 92 %). Overall, these performance metrics were similar to what was recently described by the PCAWG consortium using standard-coverage short-read WGS (Supplementary methods)(4,7). Furthermore, the genome-wide distribution of SV breakpoints in the low coverage WGS series corresponded with recent pan-cancer and myeloma genomes studies, showing enrichment in regions of early replication, accessible chromatin, and active enhancer regions as defined by histone H3K27 acetylation (H3K27ac) (Figure S1; Methods)(4,7,21). Taken together, this suggests that low-coverage long-insert WGS provides a representative view of the SV landscape.
We identified a median of 16 SVs per patient (interquartile range, IQR 8–32) (Figure 2A). Chromothripsis, chromoplexy and templated insertions involving >2 chromosomes were observed in 24%, 11% and 19% of patients, respectively, confirming previous observations(21); 38% of patients had an unspecified complex event. One or more complex events were identified in 63% of patients (median 1; range 0–11).
Figure 2: Distribution and clinical impact of structural variants in multiple myeloma.
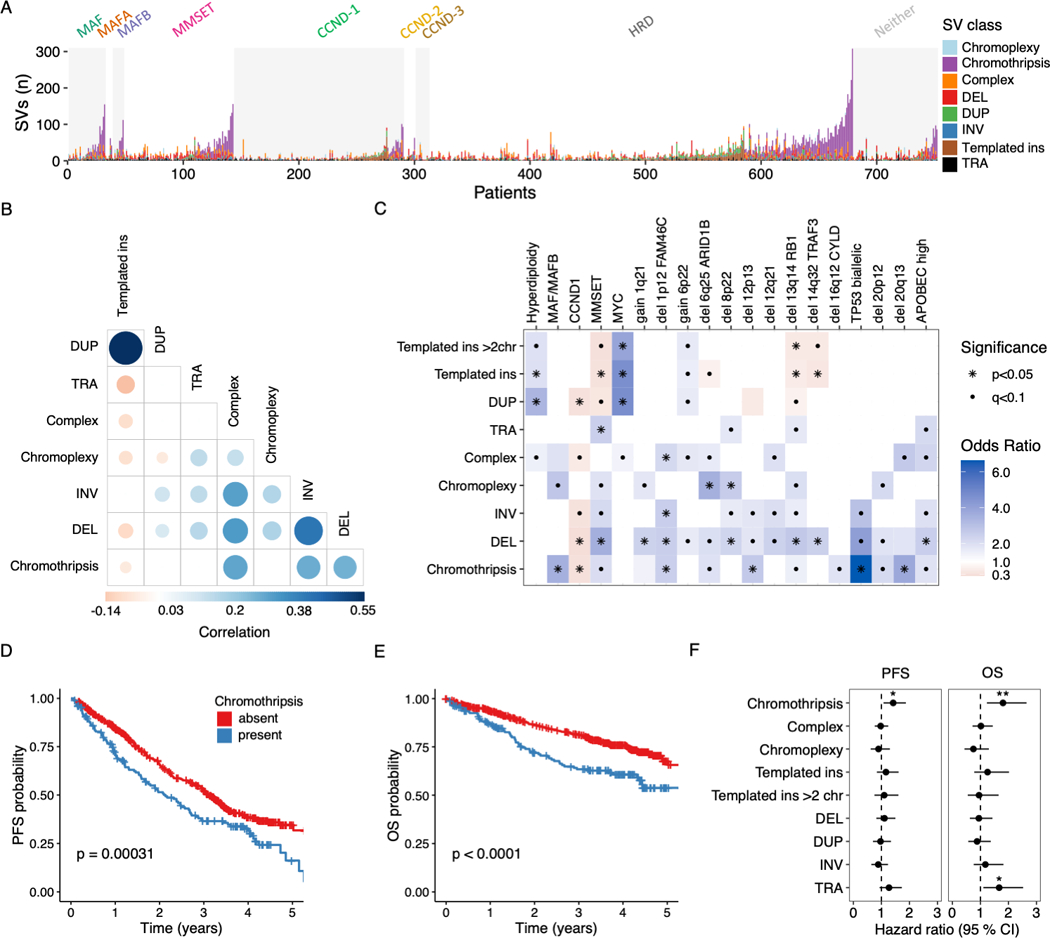
A) Stacked bars show the genome-wide burden of each structural variant (SV) class (color) in each patient (x-axis), grouped by primary molecular subgroup. B) Pairwise associations between the number of SVs of each class across patients in the CoMMpass cohort (n=752). Color and size of points are determined by the magnitude of positive (blue) and negative (red) spearman correlation coefficients, plotted only where q < 0.1. C) Association between SV classes and molecular features in the CoMMpass cohort (n = 752). Odds ratio for each pair of variables was estimated by Fisher’s exact test. Statistical significance is indicated by black dots (FDR < 0.1) and asterisks (Bonferroni-Holm adjusted p-values < 0.05). For all templated insertions, templated insertions involving >2 chromosomes, chromothripsis, chromoplexy and unspecified complex events, we compared patients with 0 versus 1 or more events. The remaining SVs were considered by their simple class (i.e. DUP, DEL, TRA and INV), comparing the 4th quartile SV burden with the lower three quartiles. D-E) Kaplan-meier plots for progression free survival (PFS) D) and overall survival (OS) E) in patients with and without chromothripsis (shown in blue and red respectively). F) Hazard ratio for PFS and OS by SV type, estimated using multivariate Cox regression. (Line indicates 95% CI from multivariate cox regression models, statistically significant features indicated by asterisks (* p<0.05; ** p < 0.01). The multivariate models included all SV variables (as defined above) as well the following clinical and molecular features: age, sex, ECOG status, ISS-stage, induction regimen, gain 1q21, del FAM46C, del TRAF3, TP53 status, del RB1, high APOBEC mutational burden, hyperdiploidy and canonical translocations involving CCND1, MMSET, MAF, MAFA, MAFB and MYC (Figure S2).
In patients with newly diagnosed multiple myeloma, different SV classes showed distinct patterns of co-occurrence, mutual exclusivity and association with recurrent molecular alterations (Figures 2A-C). Templated insertions showed a particularly striking pattern of positive correlation with single tandem duplications (spearman’s rho = 0.55, p < 2.2×10−16) and negative correlation with most other SV classes (Figure 2B). Templated insertions and single tandem duplications were both strongly enriched in patients with hyperdiploidy and MYC alterations (Figure 2C). Chromothripsis accounted for the greatest proportion of SVs among all classes (33%), and the burden of chromothripsis SVs in each patient correlated with the number of single deletions, inversions and unspecified complex events (Figure 2B). Presence of chromothripsis or a deletion burden in the 4th quartile showed striking associations with known high-risk molecular features in multiple myeloma, including primary translocations of IGH with MMSET, MAF or MAFB; high APOBEC mutational burden, and most of the recurrent aneuploidies (Figure 2C)(19,29,30). The strongest association was observed between chromothripsis and bi-allelic inactivation of TP53 (OR 6.6, 95% CI 2.7–17.15, p=4.84×10−6). Chromothripsis was previously reported as a rare event in 10 out of 764 patients with multiple myeloma (1.3%) using array-based copy number analysis, and half of these patients relapsed within 10 months(31). Despite the 18-fold higher prevalence in our WGS data, the presence of chromothripsis was associated with poor clinical outcomes and retained its significance after adjustment for established clinical and molecular risk factors, in terms of both progression free survival (PFS, adjusted HR = 1.42; 95% CI 1.08–1.87; p= 0.014) and overall survival (OS, adjusted HR = 1.81; 95% CI 1.23–2.65; p= 0.002) (Figure 2D-F and S2; Methods).
Structural basis of recurrent translocations and copy number alterations
To define the structural basis of canonical translocations in multiple myeloma, we identified all translocation-type events (single and complex) with one or more breakpoints involving the immunoglobulin loci (i.e. IGH, IGK and IGL) or canonical IGH-partners (e.g. CCND1, MMSET and MYC) (Figure 3A-B)(18). Templated insertions emerged as the cause of CCND1 and MYC translocations in 26% and 72% of cases, respectively (Figure 3A). In line with its mechanism of connecting and amplifying distant genomic segments, oncogenes and regulatory regions (e.g. super-enhancers), templated insertions of CCND1 and MYC were associated with focal amplification in 81% and 98% of cases, respectively; and involved more than two chromosomes in 42% and 44% of cases. Complex SVs involving MYC were first described in 2000(32), including insertions of the MYC gene into a partner locus or insertion of partner loci near MYC, consistent with the current definition of templated insertions(4). Although rare, we also found examples of chromothripsis and chromoplexy underlying canonical IGH translocations, resulting in overexpression of the partner gene consistent with a driver event (Figure 3B).
Figure 3: Structural variants associated with recurrent translocations, copy number changes and altered gene expression.
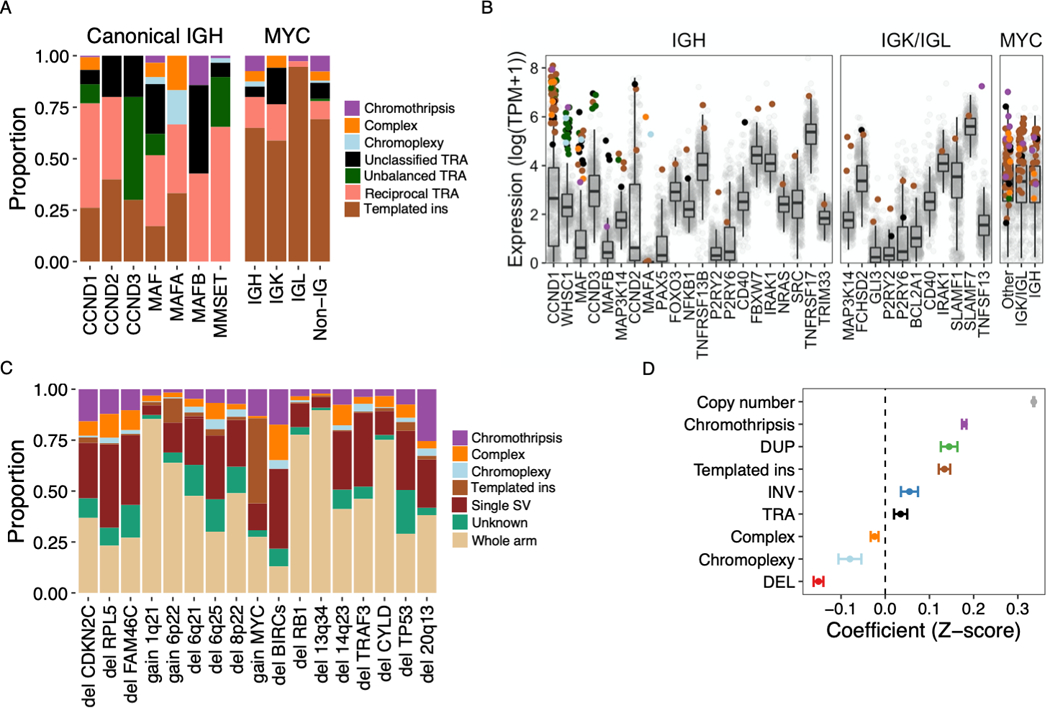
A) Relative contribution (y-axis) of simple and complex SV classes to canonical translocations (TRA) involving IGH as well as translocations of MYC with canonical and non-canonical partners (x-axis). “Non-IG” includes MYC-translocations that do not involve IGH, IGL or IGK. B) Gene expression of canonical and non-canonical partners of translocations involving IGH (left), either light chain gene locus (center) or MYC (right). Each point represents a sample, colored by the translocation class involved or absence of a translocation (gray). Boxplots shows the median and interquartile range (IQR) of expression across all patients, with whiskers extending to 1.5 * IQR. The templated insertion of IGH and MAF with low expression was part of a multi-chromosomal event involving and causing the overexpression of CCND1. C) Structural basis of established multiple myeloma CNA drivers, showing the relative contribution of whole arm events and CNAs associated with a specific SV. Intrachromosomal events without a clear causal SV were classified as “unknown” (7% of CNAs overall). D) Impact of copy number and SV involvement on normalized gene expression values (Z-scores), estimated by multivariate linear regression. Estimates with 95% CI for each parameter are shown. Pooled analysis was performed for all expressed genes on autosomes across all patients, excluding structural events involving immunoglobulin loci.
Next we went to investigate the prevalence and landscape of rare non-canonical IGH translocation partners. These events were first described in the 1990s(17), but data from a large and uniformly analyzed series has been lacking. Considering the 591 patients in our study with WGS and RNAseq, where aberrant gene expression could be confirmed, thirty-one patients (5.2%) had translocations involving at least one immunoglobulin locus (IGH = 19, IGL = 12 and IGK = 1) and a non-canonical oncogene partner, most of which were key regulators of B-cell development (e.g. PAX5 and CD40) (Figure 3B)(33,34). Non-canonical IGH translocations most commonly occurred in patients without another primary IGH translocation (15 of 19 patients, 79%), raising the possibility of non-canonical disease-initiating events. Of these, translocations involving MAP3K14 had similar prevalence (1%) as those involving CCND2 (0.8%) and MAFA (0.5%) and, which are considered among established initiating events, and showed a similar breakpoint distribution in the IGH class-switch recombination regions (Figures 3B and S3). Taken together, we show that different mechanisms of SV converge to aberrantly activate key driver genes in multiple myeloma, including rare events potentially involved in cancer initiation.
Next, we addressed the structural basis of recurrent CNAs (Table S2). Aneuploidies involving a whole chromosome arm were most common (56% of 2889 events). Among intrachromosomal CNAs, 83% could be attributed to a specific SV (Figure 3C). There was considerable variation in the proportion and class of SVs causing gains and losses between different loci, indicating the presence of distinct underlying mechanisms being active at these sites (Figure 3C). Highlighting the importance of complex SVs in shaping the multiple myeloma genome, 47% of all chromothripsis events resulted in the acquisition of at least one recurrent driver CNA (n = 116); the corresponding numbers for chromoplexy and templated insertions involving >2 chromosomes were 44% (n = 43) and 21.7% (n = 46), respectively.
SVs may exert their effect through altered gene dosage (i.e. the number of copies of a gene), or through indirect mechanisms such as the well-known super-enhancer hijacking involving the immunoglobulin loci (Figure 3B)(14,35). To quantify the effect of SVs on gene expression independently from copy number, we fit a multivariate linear regression model including all expressed genes on autosomes from all patients (Figure 3D; Supplementary Methods)(36). Structural events involving immunoglobulin loci were excluded. As expected, copy number had the strongest average effect, with an increase or decrease in expression Z-score of 0.31 for each gain or loss of a gene copy (p < 2.2×10−16). Nonetheless, all SV classes showed significant gene expression effects independent from copy number; and these effects were in the direction expected from the consequences of each SV class(36,37). Chromothripsis is associated with both gain- and loss of function(38), and the presence of high-level gains causing outlier gene expression may have skewed our model estimates. However, chromothripsis maintained a positive effect on gene expression when limiting our analysis to genes with less than 4 copies (estimate = 0.11, p < 2.2×10−16) (Figure S4). Although the specific implications of individual SVs may be difficult to predict, these data demonstrate that the average effects of SVs on gene expression are considerable.
Hotspots of structural variation
Twenty recurrently translocated regions have been previously reported in multiple myeloma, defined by a translocation prevalence of >2% within 1 Mb bins across the genome(11). These included the canonical immunoglobulin translocations, as well as MYC and recurrent partners, such as BMP6/TXNDC5, FOXO3 and FAM46C(11,14,39). We were motivated to expand the known catalogue of genomic loci where SVs play a driver role in multiple myeloma and are therefore positively selected (i.e. SV hotspots), considering all classes of single and complex SVs. To accomplish this, we applied the Piecewise Constant Fitting (PCF) algorithm, comparing the local SV breakpoint density to an empirical background model, accounting for the propensity of complex SVs to introduce large numbers of clustered breakpoints (Methods; Supplementary Methods; Data S1)(3,40). Overall, we identified 68 SV hotspots after excluding the immunoglobulin loci (i.e., IGH, IGL and IGK) and 5 known fragile sites that are prone to focal deletions (e.g. FHIT, CCSER1 and PTPRD) (Figures 4A-D, 5; Table S3). Fifty-three SV hotspots had not been previously reported in multiple myeloma. Two of the previously reported regions of recurrent translocation were not recapitulated by our hotspot analysis: 19p13.3 and the known oncogene MAFB on 20q12. This may be explained by the behavior of the PCF algorithm, which favors the identification of loci where breakpoints are tightly clustered compared with neighboring regions as well as the expected background. Indeed, SVs involving MAFB and 19p13.3, were identified in 1.5% and 8.1% of patients, but the breakpoints did not form distinct clusters (Figure S5). While MAFB is an established driver that was missed by our analysis, the implications of SVs involving 19p13.3 are unclear.
Figure 4: Genome-wide distribution of structural variation breakpoints and hotspots.
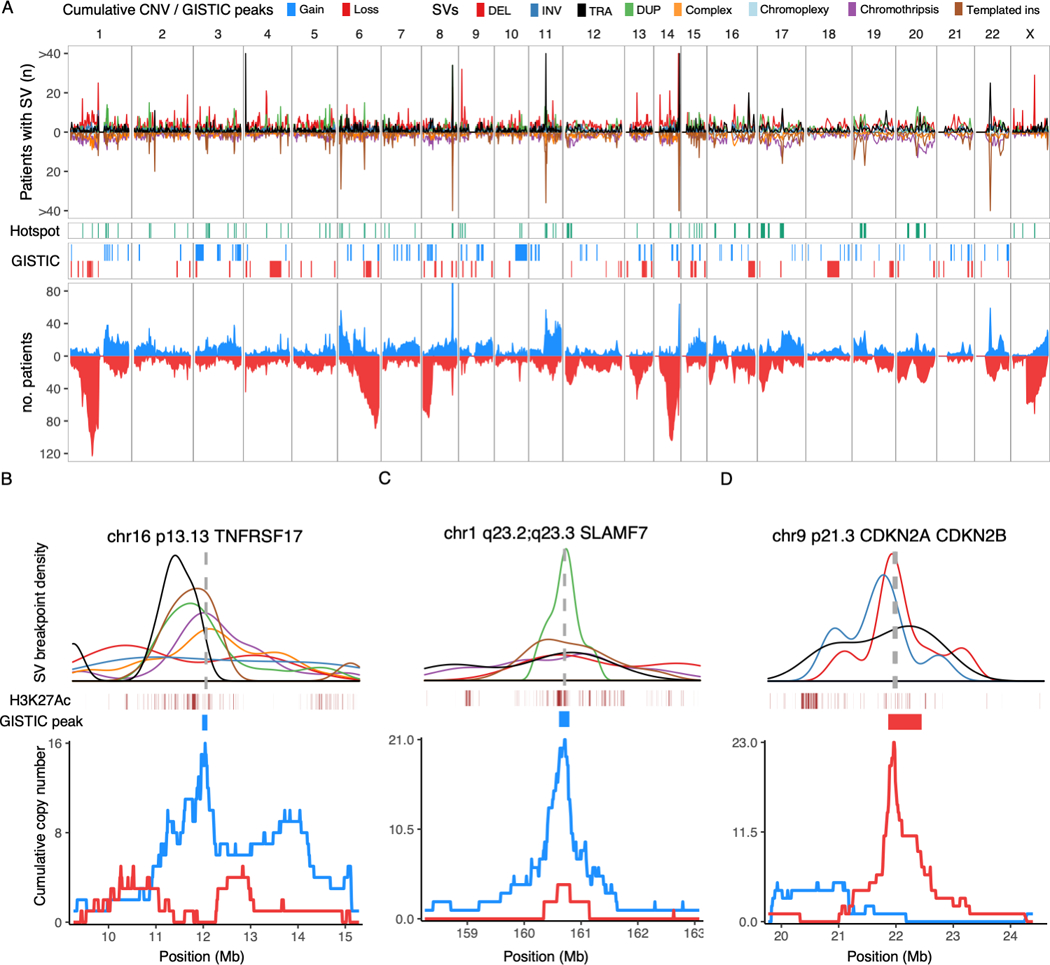
A) Top: Genome-wide density of SV breakpoints shown separately for each class (legend above figure), simple classes above the X-axis and complex classes below. Middle: Distribution of SV hotspots (green) and recurrent copy number changes (red/blue) identified by the GISTIC algorithm. Bottom: all copy number changes caused by SV breakpoints, showing cumulative plots for gains (blue) and losses (red). B-D) Zooming in on three SV hotspots, showing the breakpoint density of relevant SV classes (colors indicated in legend above A) around the hotspot; active enhancers (H3K27Ac) and supporting GISTIC peaks (middle); and cumulative copy number (bottom). The SV density plots are annotated with the location of key driver genes as vertical gray dashed lines. B) Gain-of-function hotspot centered on TNFRSF17 (BCMA), dominated by highly clustered templated insertions, associated with focal copy number gain of TNFRSF17. C) Gain-of-function hotspot involving four genes in the Signaling Lymphocyte Activation Molecule (SLAM) family of immunomodulatory receptors, including the gene encoding the monoclonal antibody target SLAMF7. D) Deletion hotspot associated with copy number loss centered on the cyclin dependent kinase inhibitors CDKN2A/CDKN2B.
Figure 5: Summary of structural variant hotspots.
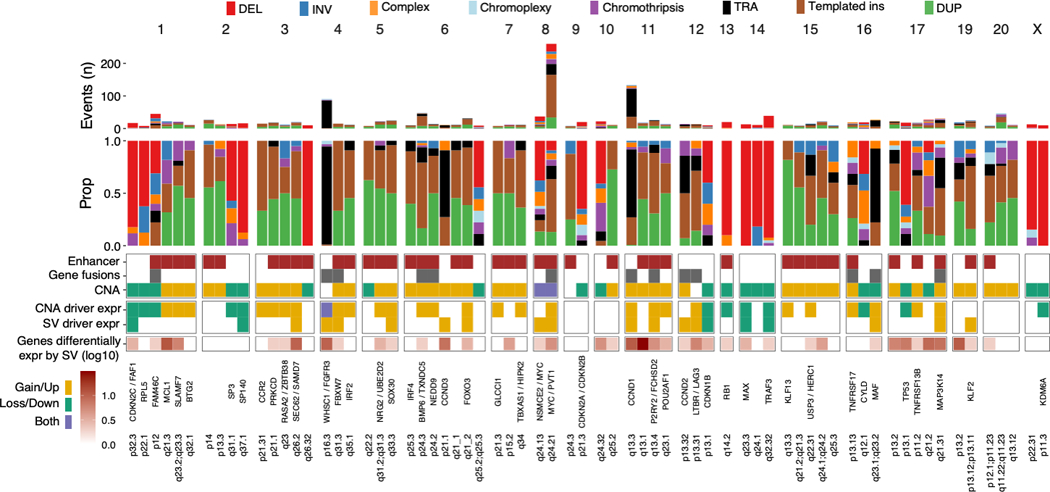
Summary of all 68 SV hotspots, showing (from the top): absolute and relative contribution of SV classes within 100 Kb of the hotspot; involvement of active enhancers in multiple myeloma, presence of putative driver gene fusions and copy number changes; differential expression of putative driver genes by copy number changes and/or SV involvement by linear regression; total number of genes in each hotspot differentially expressed by SV involvement (FDR < 0.1) after adjustment for copy number changes; known and candidate driver genes.
Given that SVs and CNAs reflect the same genomic events, we hypothesized that functionally important SV hotspots would be associated with a cluster of CNAs(4). We therefore performed independent discovery of driver CNAs using GISTIC (genomic identification of significant targets in cancer)(41). This algorithm identifies peaks of copy number gain or loss containing driver genes and/or regulatory elements based on the frequency and amplitude of observed CNAs (Figure 4A, Table S4-5). In addition, we generated cumulative copy number profiles for the patients involved by SV at each hotspot. Finally, we evaluated the impact of SV hotspots on the expression of nearby genes (Table S6), and the presence of oncogene fusion transcripts. By integrating SV, CNA, and expression data, we went on to determine the most likely consequence of each hotspot in terms of gain-of-function, loss-of-function, and potential involvement of driver genes and regulatory elements. Individual SVs within hotspots were considered as likely driver events if their functional implications corresponded to the putative driver role of that hotspot (i.e. gain- or loss-of-function); SVs with incongruous effects were removed as likely passenger events (Supplementary Methods).
Gain-of-function hotspots (n=49) were defined by clustered SVs associated with copy number gains as well as translocation-type events with or without oncogene fusions (Figures 4–5 and S6; Table S3). There was a strong tendency for templated insertions and tandem duplications to co-occur (Spearman’s rho = 0.71, p= 1.56×10−8) across hotspots, with a similar pattern being observed genome-wide (rho = 0.57, p<2.2×10−16), supporting a strong association between these events. Strikingly, gain-of-function hotspots showed 8.4-fold enrichment of super-enhancers as compared with the remaining mappable genome (2.5 vs. 0.3 super-enhancers per Mb, Poisson test p < 2.2×10−16), and 10.5-fold enrichment of transcription factors involved in key regulatory networks in multiple myeloma (Poisson test p=1.64×10−8)(42). Among gain-of-function hotspots, 16 were associated with well-defined myeloma oncogenes (e.g. WHSC1/MMSET, CCND1, IRF4 and MAP3K14)(11,18) and 17 involved a novel putative driver gene. Of particular interest, TNFRSF17 was involved by SVs in 2.5% of patients (n = 19) and encodes B-Cell Maturation Antigen (BCMA), a therapeutic target of chimeric antigen receptor T-cells (CAR-T), as well as monoclonal and bispecific antibodies (Figure 4B)(43,44). Furthermore, we report two novel SV hotspots on chromosome 1q23 involving putative driver genes with therapeutic implications: SLAMF7 (involved by SV in 2.8%, n = 21), target of the monoclonal antibody elotuzumab (Figure 4C)(43,45); and MCL1 (3%, n = 23), an apoptotic regulator implicated in resistance to the BCL2-inhibitor venetoclax(46,47) and a promising therapeutic target in its own right(48). Additional novel putative driver genes were BTG2, CCR2, PRKCD, FBXW7, IRF2, NRG2/UBE2D2, SOX30, NEDD9, GLCCI1, TBXAS1/HIPK2, POU2AF1, KLF13, USP3/HERC1, and TNFRSF13B. We also confirmed previous reports that virtually all SVs involving MYC resulted in its overexpression, including deletions and inversions acting to reposition MYC next to the super-enhancers of NSMCE2 roughly 2 Mb upstream(16).
Loss-of-function hotspots (n=19) were defined by SVs causing copy number loss, most commonly single deletions, but also inversions and complex SVs (Figures 4–5 and S6; Table S3). We identified loss of 12 known tumor suppressor genes in multiple myeloma, including CDKN2C, SP3, SP140, RPL5 and CYLD. FAM46C stood out as involved by both SVs causing copy number loss and translocation-type events which sometimes resulted in gene fusions. This is consistent with its known role as a tumor suppressor, while also serving as a target for super-enhancer hijacking(39,49).
Taken together, we identified 29 SV hotspots involving genes with established tumor suppressor or oncogene function in multiple myeloma; 17 additional hotspots, all gain-of-function, involved novel putative driver genes (Table S3; all putative driver hotspots are shown in Figures 4B-D and S6; individual patient summary in Table S7).(50)
Each patient had a median of two hotspots involved by a putative driver SV (IQR 1–3); and the number of hotspots involved was strongly associated with the overall SV burden (Spearman’s Rho = 0.46; p < 2.2×10−16). This association became even stronger when SV breakpoints associated with a single event were considered together (Spearman’s Rho = 0.51; p < 2.2×10−16). Re-analyzing published data from tandem duplication hotspots in breast cancer revealed similar results (Spearman’s Rho of 0.7 and 0.62 for rearrangement signatures 1 and 3, respectively; p<2.2×10−16; Figure S7A-C)(40). Extending this observation beyond SVs, there was a strong correlation between SNV burden in multiple myeloma and the number of SNVs in known driver genes(20,21) (Rho = 0.38, p < 2.2×10−16), which remained significant when restricting the analysis to established SNV hotspots within driver genes (rho = 0.11, p=0.001). These data indicate that genomic drivers continue to accumulate and provide selective advantage through the disease course despite multiple drivers already being present, consistent with our recent observations from re-constructing the timeline of driver acquisition in multiple myeloma(21,51,52) and multi-region WGS performed at autopsy(53).
Templated insertions and chromothripsis exemplify highly clustered versus chaotic breakpoint patterns
SVs of different classes showed different propensities to form hotspots. Templated insertion breakpoints were the most likely to be in a hotspot (logistic regression OR 4.04; 95% CI 3.65–4.49, p < 2.2×10−16), with chromothripsis breakpoints being the least likely (OR 0.48; 95% CI 0.43–0.54; p < 2.2×10−16) (Figure 6A-B). This difference remained when considering structural events instead of individual breakpoints, with 66% of 544 templated insertions involving one or more hotspot, versus 43% of 244 chromothripsis events (Fisher’s test OR 2.66; 95% CI 1.91–3.65; p=7.14×10−10), despite the vastly higher complexity of chromothripsis events as compared with templated insertions (median 17 vs 2 breakpoint pairs in each event, Wilcoxon test p < 2.2×10−16).
Figure 6: Templated insertions and chromothripsis exemplify highly clustered versus scattered breakpoint patterns.
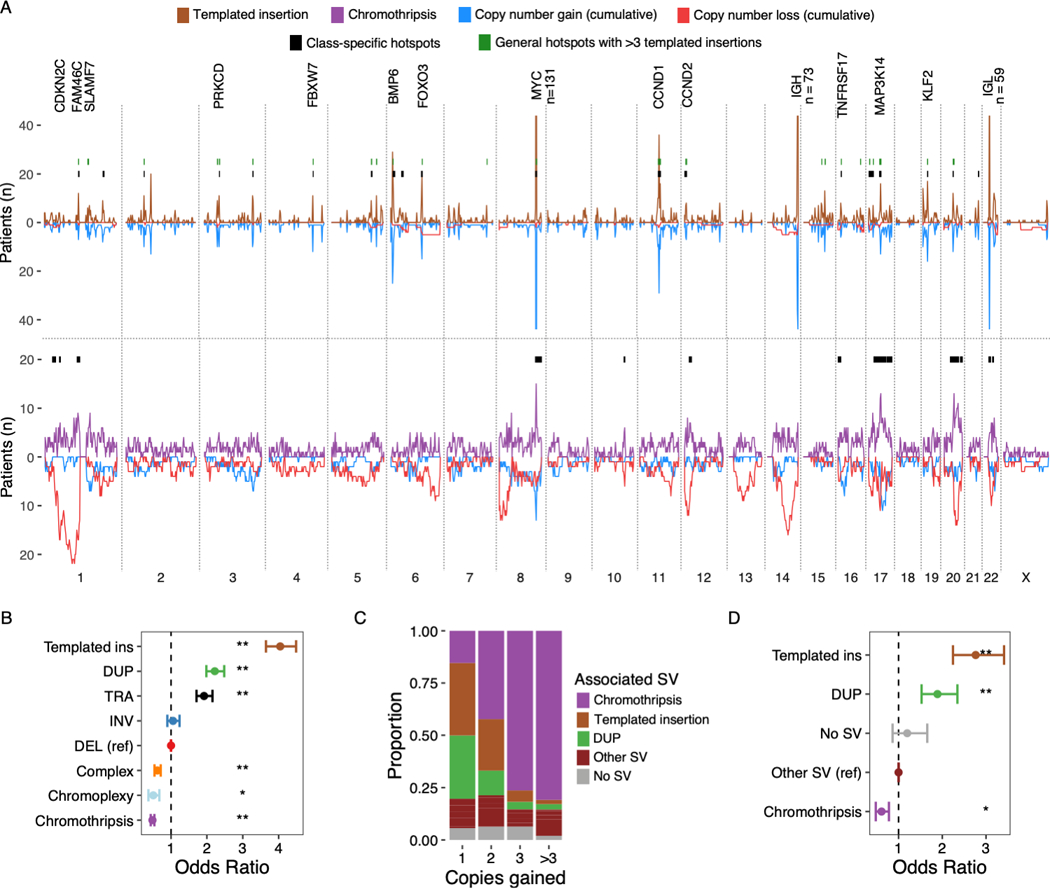
A) Distribution of templated insertions (above) and chromothripsis (below) across the genome, for each displaying SV breakpoint density above the X-axis and SV-associated cumulative copy number changes below. Results from templated insertion and chromothripsis-specific hotspot analysis drawn as black bars at y = 20. Hotspots from the main hotspot analysis which contained 6 or more templated insertions are drawn in green. Key putative driver genes involved by hotspots are annotated. Numbers are annotated where peaks extend outside of the plotting area B) The probability that a given SV breakpoint belonging to each class will fall within a hotspot region, expressed as odds ratios with 95% CI from logistic regression analysis where single deletions were used as the reference level. C) Showing the proportion of focal gains (<3 Mb) associated with each SV class, divided by the number of copies acquired relative to the baseline (x-axis). D) Shows the probability that focal gains displayed in C) contain a multiple myeloma super-enhancer, expressed as odds ratio with 95% CI from a logistic regression model adjusted for copy number. Asterisks in B and D indicate statistical significance: ** = p < 10−8; * = p<0.01.
The differences between templated insertions and chromothripsis could be clearly illustrated by the genome-wide distribution of breakpoints and association with number changes (Figure 6A). Templated insertions were associated with mainly focal copy number gain in 80.1% of cases (95% CI 78–82%); only rarely with copy number losses (5.6%; 95% CI 4.6–6.7%). Gains were almost exclusively single copy (92.3% of 1317 gains), highlighting the stability of these events. In contrast, an important feature of chromothripsis is its ability to cause both gain- and loss-of-function as part of the same event(38). Indeed, the breakpoints of chromothripsis were associated with chromosomal loss in 53.8% of cases (95% CI 52–55.6%) and gain in 37.6% (95% CI 36–39.4%). Templated insertions were predominantly associated with gain of a single copy (Fisher’s test OR 2.25 vs chromothripsis; p < 2.2×10−16), while chromothripsis dominated for gains of 2 (OR 1.7, p=7.07×10−5), 3 (OR 13.9, p<2.03×10−14) or more than 3 copies (OR 40.7, p < 2.2×10−16) (Figure 6C). The probability that focal gains involved a multiple myeloma super-enhancer was highest when associated with templated insertions (55%, logistic regression OR 2.76, p < 2.2×10−16) and lowest when associated with chromothripsis (21%, logistic regression OR 0.61, p= 7.43×10−5) (Figure 6D). In contrast to solid tumors, where chromothripsis may result acquisition of >50 copies(1,4,6,54), we observed no segments with more than 9 copies in this series (Figure 1B).
Consistent with widely different underlying mechanisms, the genome-wide distribution of templated insertion breakpoints could be predicted from genomic features such as active enhancer regions, replication time and open chromatin, but this was not the case for chromothripsis (Supplementary Methods). To test whether the clustered nature of templated insertion breakpoints can be explained solely by the local sequence context (e.g. active enhancers) or constitute real hotspots subjected to positive selection, we repeated our PCF-based hotspot analysis for templated insertions alone. Despite the considerably lower power of this analysis as compared to the combined analysis presented above, 75% of hotspots containing 6 or more templated insertions were confirmed (21 out of 28), including novel putative drivers such as FBXW7 and TNFRSF17 (BCMA) (Figure 6A; Table S8).
Since the distribution of chromothripsis breakpoints did not follow a predictable pattern, we performed separate hotspot analysis searching for regions that violated the assumption of a uniform distribution across the genome. In contrast to templated insertions, where hotspots were strongly clustered on key driver genes and regulatory regions, hotspots of chromothripsis were much larger, spanning from a few to tens of megabases (Figure 6A and Table S9). This is consistent with mechanisms where templated insertions exert gene regulatory effects disproportionate to the level of copy number gain, while the effects of chromothripsis manifest as large deletions involving recurrent regions as well as high-level amplifications and local regulatory effects (Figure 3C-D).
Multiple driver alterations caused by the same structural event
In 31% of patients (n=235), two or more seemingly independent recurrent CNAs or putative driver translocations were caused by the same SV (Figure 7A-B). The most common event classes were templated insertions causing chains of gain-of-function events in 12.7% of patients, most commonly including MYC. Chromothripsis caused two or more driver alterations in 7.2% of patients, commonly involving large deletions as well as translocation and/or amplification of oncogenes. Unbalanced translocations simultaneously causing oncogene translocations and large deletions involving tumor suppressor genes were identified in 4.4% of patients. Notably, 12 patients with canonical IGH-MMSET translocations had large deletions of 14q caused by the same unbalanced translocation, including TRAF3 (14q32) and often MAX (14q23; n=10), contributing to the known association between these events(21). Overall, SVs represents a recurrent mechanism for tumors to acquire multiple drivers simultaneously, demonstrating that the full genomic landscape of multiple myeloma can be acquired through a few key events during tumor evolutionary history(27).
Figure 7: Two or more putative driver alterations caused by a single SV.
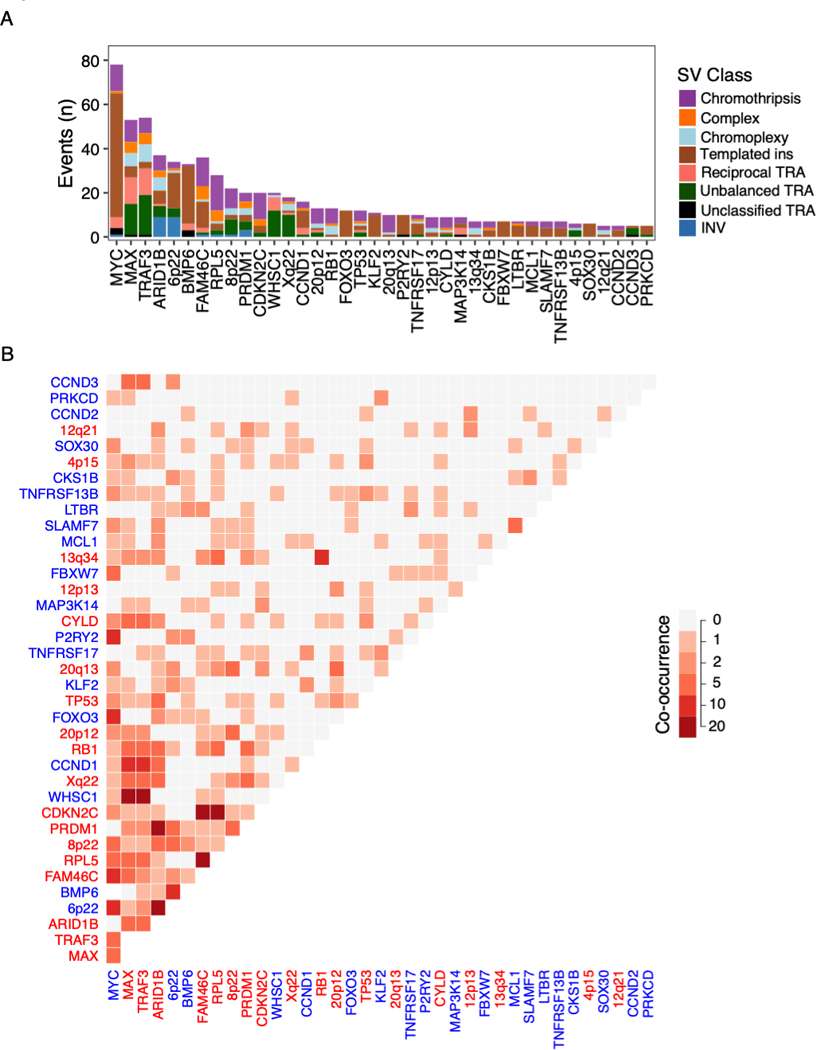
Putative driver alterations recurrently involved by multi-driver events (involved in 5 or more patients) A) Number of multi-driver events involving each gene colored by the SV class responsible. B) Heatmap showing the number of times each pair of putative drivers co-occur. Co-occurrence was defined by at least two drivers on different chromosomal copy number segments caused by the same event. Axis legends are colored according to the gain-of-function (blue) or loss-of-function (red) status of each driver.
Discussion
We describe the first comprehensive analysis of SVs in a large series of multiple myeloma patients with paired WGS and RNA sequencing. Previous studies of SVs in multiple myeloma have focused on translocations without consideration of complex events(11,15,55,56), and our previous WGS study of 30 patients lacked both the expression correlate and the power to perform comprehensive driver discovery(21). Here, applying a robust statistical approach(40), we identified 68 SV hotspots, 53 of which have not previously been reported. Integrated analysis of copy number changes, gene expression and the distribution of SV breakpoints revealed 17 new potential driver genes, including the emerging therapeutic targets TNFRSF17 (BCMA)(43,44), SLAMF7(43,45) and MCL1(48); the latter of which has also been implicated in resistance to the BCL2-inhibitor venetoclax(46). With all of these targets either currently or imminently in clinical use, it will be of great clinical importance to determine the impact of these genomic alterations as predictive biomarkers for treatment response.
From a pan-cancer perspective, the SV landscape of multiple myeloma is characterized by a lower SV burden and less genomic complexity than in many solid tumors(1,4,7). For example, we did not find any classical double minute chromosomes with tens to hundreds of amplified copies, nor did we find any of the recently proposed complex SV classes pyrgo, rigma and tyfonas(1). Nonetheless, we found that complex SVs play a crucial role in shaping the genome of multiple myeloma patients, most importantly chromothripsis, chromoplexy and templated insertions. A common feature of these SV classes is simultaneously deregulating multiple driver genes as part of a single event. Such multi-driver events are of particular importance in myeloma progression as they can provide an explanation for the rapid changes in clinical behavior that are frequently seen in the clinic(23). In myeloma precursor disease, understanding these evolutionary patterns will be crucial for early diagnosis of those patients who are on a trajectory towards progression and may benefit early intervention(23).
Of immediate translational relevance, chromothripsis emerged as a strong independent predictor for high-risk disease, detectable in 24% of newly diagnosed patients by WGS, providing a rationale for the inclusion of chromothripsis in clinical risk scores. The prevalence of chromothripsis in multiple myeloma is higher than what reported in previous studies likely for two reasons: 1) use of WGS resolution able to integrate SV and CNV data; and 2) applying the most updated criteria to define chromothripsis (4,21,57).
The use of low-coverage long insert WGS is a potential limitation of this study. We have applied extensive quality control measures to ensure specificity of our SV calls, but may have overlooked a fraction of real SVs, particularly those present at the subclonal level. Thus, the results reported in this study will be skewed towards events acquired in the early phases of tumor evolutionary history, driving tumor initiation and progression, going on to be present in the dominant tumor clone at diagnosis. Future studies using higher coverage WGS may reveal greater SV burden and additional hotspots, including subclonal events that may be selected at relapse.
Gene deregulation by SV is a major contributor to the biology of multiple myeloma constituting a hallmark feature of its genome. For decades the defining features of multiple myeloma pathogenesis and heterogeneity has been hijacking of the IGH super-enhancers to oncogenes such as CCND1 and MMSET. Our findings reveal how simple and complex SVs shape the driver landscape of multiple myeloma, with events ranging from common CNAs and canonical translocations to a large number of SV hotspots. These results focus attention on the importance of SVs in multiple myeloma and on the use of WGS analyses in order to fully understand its driver landscape and identify novel therapeutic targets.
Methods
Patients and somatic variant calling
We analyzed data from 752 patients with newly diagnosed multiple myeloma enrolled in the CoMMpass study (NCT01454297; IA13). Each sample underwent low coverage long-insert WGS (median 4–8X) and whole exome sequencing. The median physical coverage was 39 (5th percentile 28 and 95th percentile 53). The median insert size was 852bp (5th percentile = 701 and 95th percentile = 949). Paired-end reads were aligned to the human reference genome (HRCh37) using the Burrows Wheeler Aligner, BWA (v0.7.8). SV calling was performed using DELLY (v0.7.6)(10) and Manta (v.1.5.0)(28) Similarly to recent PCAWG papers, we developed a filtering process based on different criteria (see Results and Supplementary Methods)(7).
tCoNuT was used to call CNAs (https://github.com/tgen/MMRF_CoMMpass/tree/master/tCoNut_COMMPASS). To externally validate the tCoNuT workflow, we compared our results to copy number profiles generated using controlFREEC (Supplementary Methods)(20,58). The final catalogue of high-confidence SVs was obtained by integrating DELLY and Manta calls with copy number data and applying a series of quality filters (Supplementary Methods).
RNA sequencing analysis and fusion calling
RNA sequencing of 591 samples was performed to a target coverage of 100 million reads. Paired-end reads were aligned to the human reference genome (HRCh37) using STAR v2.3.1z(59). Transcript per million (TPM) gene expression values were obtained using Salmon v7.2(60). For fusion calling we employed TopHat2 v2.0.11 with the TopHat-fusion-post module(61).
Classification of structural variants
Each pair of structural variant breakpoints (i.e. deletion, tandem duplication, inversion or translocation) was classified as a single event, or as part of a complex event (i.e. chromothripsis, chromoplexy or unspecified complex), as previously described(4,21).
Translocation-type events were classified as single when involving no more than two breakpoint pairs and two chromosomes, subdivided into reciprocal translocations, unbalanced translocations, templated insertion or unspecified translocation as previously described(4,21). Templated insertions could be either simple or complex, depending on the number of breakpoints and chromosomes involved, but was always defined by translocations associated with copy number gain. Chromothripsis was defined by the presence of 10 or more interconnected SV breakpoint pairs associated with: 1) clustering of breakpoints, 2) randomness of DNA fragment joins and 3) randomness of DNA fragment order across one or more chromosomes(4,26,57). The thresholds of 10 breakpoints was imposed as a stringent criterion to avoid overestimating the prevalence of chromothripsis. Chromoplexy was defined by interconnected SV breakpoints across >2 chromosomes associated with copy number loss. Patterns of three or more interconnected breakpoint pairs that did not fall into either of the above categories were classified as unspecified “complex”(21).
Mutational signature analysis
SNV calls from whole exome sequencing were subjected to mutational signature fitting, using the previously described R package mmsig(51,52). High APOBEC mutational burden was defined by an absolute contribution of APOBEC mutations (mutational signatures 2 and 13) in the 4th quartile among patients with evidence of APOBEC activity(51).
Structural basis for recurrent CNAs in multiple myeloma
We applied the following workflow to determine the structural basis for each recurrent CNA in multiple myeloma (Table S2). First, we identified in each patient every genomic segment involved by recurrent copy number gain or loss. Gains were defined by total copy number (CN) >2; loss as a minor CN = 0. Second, whole arm events were defined when >90% of the arm had the same CN state. Third, for segments that did not involve the whole arm, we searched for SV breakpoints responsible for the CNA within 50 kb of the CN segment ends. Finally, and intrachromosomal CNAs without SV support were classified as unknown.
Gene expression effect of SV involvement
We used multivariate linear regression to determine the independent effect of SV involvement on gene expression after accounting for the effect of gene dosage (i.e. copy number). All expressed genes on autosomes were included in the analysis, defined as genes with > 0 TPM expression in >25% of patients and a median expression level of > 1. Gene expression values then underwent Z-score normalization. Genes involved by SVs were defined separately for deletion/tandem duplication type SVs and translocation/inversion types. For deletions and tandem duplications, genes were considered involved if overlapping the SV +/− 10 Kb. For translocations and inversions, genes within 1 Mb to either side of each breakpoint were considered involved. All single and complex SVs with one or more breakpoints within 1 Mb of either immunoglobulin loci were excluded, to prevent the results from being dominated by the effects of immunoglobulin enhancers. Linear regression was performed for all patients and all genes pooled together, including the total copy number of each gene as a linear feature.
Copy number changes associated with structural variant breakpoints
To determine the genome-wide footprint of copy number changes resulting from SVs, we employed an “SV-centric” workflow, as opposed to the CNA-centric workflow described above. For each SV breakpoint, we searched for a change in copy number within 50 kb. If more than one CNA was identified, we selected the shortest segment. Deletion and amplification CNAs were defined as changes from the baseline of that chromosomal arm. As a baseline, we considered the average copy number of the 2 Mb closest to the telomere and centromere, respectively. This is important because deletions are often preceded by large gains, particularly in patients with hyperdiploidy(21). In those cases, we are interested in the relative change caused by deletion, not the total CN of that segment (which may still be ≥ 2). We estimated the proportion of breakpoints associated with copy number gain or loss across patients, collapsing the data in 2 Mb bins across the genome. Confidence intervals were estimated using bootstrapping and the quantile method. For the purposes of plotting (Figures 4A and 6A), we divided the SV-associated CNAs into bins of 2 Mb. The resulting cumulative CNA plot shows the number of patients with an SV-associated deletion or amplification.
Hotspots of structural variation breakpoints
To identify regions enriched for SV breakpoints, we employed the statistical framework of piecewise constant fitting (PCF). In principle, the PCF algorithm identifies regions where SVs are positively selected, based on enrichment of breakpoints with short inter-breakpoint distance compared to the expected background and surrounding regions. We used the computational workflow previously described by Glodzik et al(40). In brief, negative binomial regression was applied to model local SV breakpoint rates under the null hypothesis (i.e. absence of selection), taking into account local features such as gene expression, replication time, non-mapping bases and histone modifications. The PCF algorithm can define hotspots without the use of binning, based on a user-defined smoothing parameter and threshold of fold-enrichment compared to the background. This allows identification of hotspots of widely different size, depending on the underlying biological processes. Moreover, there was no global threshold for the inter-breakpoint distance required to define a hotspot; instead, the genome was searched for local regions with higher than expected breakpoint density compared with local context and the background model. To avoid calling hotspots driven by highly clustered breakpoints in a few samples, we also set a minimum threshold of 8 samples involved (~ 1% of the cohort) to be considered hotspot, as previously reported (40). Despite this threshold, we found that complex SVs with tens to hundreds of breakpoints in a localized cluster (particularly chromothripsis) came to dominate the results. To account for this, we ran the PCF algorithm in two different ways: 1) considering all breakpoints of non-clustered SVs (simple classes and templated insertions); and 2) including all SV classes, but randomly downsampling the data to include only one breakpoint per 500 kb per patient. The random downsampling followed by PCF analysis was repeated 1000 times, requiring >95% reproducibility to define a hotspot. Final output from both approaches was merged for downstream analysis.
The full SV hotspot analysis workflow is attached as Data S1, drawing on generic analysis tools that we have made available on github (https://github.com/evenrus/hotspots/tree/hotornot-mm). Additional details about nomination of SV hotspots by the PCF algorithm and downstream analysis and are provided in Supplementary Methods.
Functional classification of structural variation hotspots
SV hotspots were classified based on local copy number and gene expression data as gain-of-function or loss-of-function; hotspots without clear functional implications were removed.
Copy number data was integrated from two complimentary approaches. First, we applied the GISTIC v2.0 algorithm to identify wide peaks of enrichment for chromosomal amplification or deletion (FDR < 0.1), using standard settings(41). Second, we considered the cumulative copy number profiles of each hotspot, considering only the patients with SV breakpoints within the region, looking for more subtle patterns of recurrent CNA that was not picked up in the genome-wide analysis.
To determine the effects of SV hotspot involvement on gene expression, we applied multivariate linear regression analysis for each gene within 500 Kb to either side of a hotspot(36). Genes were considered involved by SV if there was an SV breakpoint within 100 Kb to either side of the corresponding hotspot. All SV classes were considered together, and the expression level of each gene was adjusted for the total copy number of that gene in each patient. Genes differentially expressed at FDR < 0.1 were considered statistically significant.
Identification of putative driver genes involved by SV hotspots
Multiple lines of evidence were considered to identify driver genes involved by SV hotspots. Evidence of a putative driver gene included: 1) involved by driver SNVs in multiple myeloma(20,21); 2) included in the COSMIC cancer gene census (https://cancer.sanger.ac.uk/census); 3) designated as putative driver gene in The Cancer Genome Atlas(62–65); 4) enrichment of SV breakpoints in or around the gene; 5) nearby peak of SV-related copy number gain or loss; 6) SV classes and recurrent copy number changes corresponding to a known role of that gene in cancer (i.e. oncogene or tumor suppressor); and 7) differential gene expression. Having identified candidate driver genes involved by SV hotspots, we reviewed the literature for evidence of a role in multiple myeloma (Table S3).
Histone H3K27ac, super-enhancers and multiple myeloma transcription factor networks
Active enhancer (H3K27ac) and super-enhancer data from primary multiple myeloma cells, as well as key gene regulatory networks in multiple myeloma, were obtained from Jin et al (42). Enrichment of super-enhancers and key multiple myeloma transcription factors in hotspots was assessed using a Poisson test, comparing the density within 100 kb of hotspots with the remaining mappable genome.
Templated insertion hotspot analysis
We developed an empirical background model for templated insertions, which strongly out-performed a random model to predict the genome-wide distribution of templated insertion breakpoints. We then performed PCF-based hotspot analysis for templated insertions alone, searching for regions of enrichment as compared with the templated insertion background model, as described above for non-clustered SVs.
Chromothripsis hotspot analysis
Empirical background models showed very poor ability to predict the distribution of chromothripsis breakpoints, as may be expected if DNA breaks in chromothripsis tend to be random. To identify regions enriched for chromothripsis, we applied the PCF algorithm with a uniform background, only adjusting for non-mapping bases.
Enrichment of SV classes within hotspots
We used logistic regression analysis to determine the relative probability that breakpoints of different SV classes are located within 100 Kb of a hotspot. Each breakpoint was considered individually. Single deletions were considered as the reference class and results shown as OR with 95% CI.
SV classes associated with copy number gains
To determine the SV classes associated with focal copy number gains, we selected all copy number segments smaller than 3 Mb with a total copy number of >2 and a relative change of ≥1 from the baseline of that chromosome arm (as described above). Each copy number segment was assigned to the associated SV class, or as “No SV” if no breakpoints could be found within 50 Kb.
Amplification of multiple myeloma super-enhancers
To determine the relative probability of super-enhancer amplification associated with different SV classes, we applied multivariate logistic regression analysis. Focal copy number gains were assigned as associated with a super-enhancer if one was found within 100 Kb of the copy number segment. Copy number segments were grouped according to the associated SV classes: templated insertion, tandem duplication, chromothripsis, other SV or no SV. Gains associated with “Other SVs” were used as the reference level and copy number was included as a continuous variable. Results were provided as OR and 95% CI for each SV category, adjusted for the effect of copy number.
Multi-driver events
Multi-driver events were defined by the involvement of two or more independent driver copy number segments and/or SV hotspots caused by the same simple or complex SV. For example, the association between MMSET and MAX/TRAF3 deletion, was often an unbalanced translocation causing two deletion: the first involving MMSET and FGFR3 on chromosome 4p and the second involving the majority of chromosome 14q (including MAX/TRAF3). Each copy number segment was only counted once, even if more than one driver was deleted or amplified.
Data and software availability
All the raw data used in the study are already publicly available (dbGap: phs000748.v1.p1 and EGAS00001001178. Analysis was carried out in R version 3.6.1. Unless otherwise specified, we used Wilcoxon rank sum test to test for differences in continuous variables between two groups; Fisher’s exact test for 2×2 tables of categorical variables; and the Bonferroni-Holm method to adjust p-values for multiple hypothesis testing. The full analytical workflow in R to identify hotspots of structural variants is provided in Data S1. All other software tools used are publicly available.
Supplementary Material
Significance.
Previous genomic studies in multiple myeloma have largely focused on single nucleotide variants, recurrent copy number alterations and translocations. Here, we demonstrate the crucial role of structural variants and complex events in the development of multiple myeloma and highlight the importance of whole genome sequencing to decipher its genomic complexity.
Acknowledgements
This work is supported by the Memorial Sloan Kettering Cancer Center NCI Core Grant (P30 CA 008748), the Multiple Myeloma Research Foundation (MMRF), and the Perelman Family Foundation.
F.M. is supported by the American Society of Hematology, the International Myeloma Foundation and The Society of Memorial Sloan Kettering Cancer Center.
K.H.M. is supported by the Haematology Society of Australia and New Zealand New Investigator Scholarship and the Royal College of Pathologists of Australasia Mike and Carole Ralston Travelling Fellowship Award.
G.J.M is supported by The Leukemia Lymphoma Society.
Footnotes
Conflict of interest statement
No conflict of interests to declare.
References
- 1.Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Rosiene J, et al. Novel patterns of complex structural variation revealed across thousands of cancer genome graphs. bioRxiv 2019:836296 doi 10.1101/836296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018;173(3):611–23.e17 doi 10.1016/j.cell.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glodzik D, Purdie C, Rye IH, Simpson PT, Staaf J, Span PN, et al. Mutational mechanisms of amplifications revealed by analysis of clustered rearrangements in breast cancers. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2018;29(11):2223–31 doi 10.1093/annonc/mdy404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578(7793):112–21 doi 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JJ, Park S, Park H, Kim S, Lee J, Lee J, et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 2019;177(7):1842–57.e21 doi 10.1016/j.cell.2019.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature 2015;522(7555):179–84 doi 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature 2020;578(7793):82–93 doi 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015;163(7):1641–54 doi 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maciejowski J, Imielinski M. Modeling cancer rearrangement landscapes. Current Opinion in Systems Biology 2017;1:54–61 doi 10.1016/j.coisb.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28(18):i333–i9 doi 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nature communications 2019;10(1):1911 doi 10.1038/s41467-019-09555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nature communications 2018;9(1):3363 doi 10.1038/s41467-018-05058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020;34(1):322–6 doi 10.1038/s41375-019-0543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J 2014;4:e191 doi 10.1038/bcj.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergsagel PL, Kuehl WM. Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma. Immunol Rev 2003;194:96–104 doi 10.1034/j.1600-065x.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 16.Affer M, Chesi M, Chen W-DG, Keats JJ, Demchenko YN, Roschke AV, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014;28(8):1725–35 doi 10.1038/leu.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene 2001;20(40):5611–22 doi 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- 18.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 2017;14(2):100–13 doi 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 19.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019;33(1):159–70 doi 10.1038/s41375-018-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018;132(6):587–97 doi 10.1182/blood-2018-03-840132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nature communications 2019;10(1):3835 doi 10.1038/s41467-019-11680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010;116(15):e56–e65 doi 10.1182/blood-2010-04-279596. [DOI] [PubMed] [Google Scholar]
- 23.Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA oncology 2020;6(3):425–32 doi 10.1001/jamaoncol.2019.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones JR, Weinhold N, Ashby C, Walker BA, Wardell C, Pawlyn C, et al. Clonal evolution in myeloma: the impact of maintenance lenalidomide and depth of response on the genetics and subclonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019;104(7):1440–50 doi 10.3324/haematol.2018.202200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016;128(13):1735–44 doi 10.1182/blood-2016-06-723007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell 2013;152(6):1226–36 doi 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 27.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell 2013;153(3):666–77 doi 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2015;32(8):1220–2 doi 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 29.Maura F, Petljak M, Lionetti M, Cifola I, Liang W, Pinatel E, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2018;32(4):1044–8 doi 10.1038/leu.2017.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nature communications 2015;6:6997 doi 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011;118(3):675–8 doi 10.1182/blood-2011-03-344069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proceedings of the National Academy of Sciences 2000;97(1):228–33 doi 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol 2015;15(3):160–71 doi 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 34.Boothby MR, Hodges E, Thomas JW. Molecular regulation of peripheral B cells and their progeny in immunity. Genes & development 2019;33(1–2):26–48 doi 10.1101/gad.320192.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lieber MR. Mechanisms of human lymphoid chromosomal translocations. Nature reviews Cancer 2016;16(6):387–98 doi 10.1038/nrc.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Yang L, Kucherlapati M, Chen F, Hadjipanayis A, Pantazi A, et al. A Pan-Cancer Compendium of Genes Deregulated by Somatic Genomic Rearrangement across More Than 1,400 Cases. Cell Rep 2018;24(2):515–27 doi 10.1016/j.celrep.2018.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, et al. The impact of structural variation on human gene expression. Nature Genetics 2017;49:692 doi 10.1038/ng.3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nature Genetics 2020;52(3):331–41 doi 10.1038/s41588-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikulasova A, Ashby C, Tytarenko RG, Qu P, Rosenthal A, Dent JA, et al. Microhomology-mediated end joining drives complex rearrangements and over expression of MYC and PVT1 in multiple myeloma. Haematologica 2019:haematol.2019.217927 doi 10.3324/haematol.2019.217927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glodzik D, Morganella S, Davies H, Simpson PT, Li Y, Zou X, et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat Genet 2017;49(3):341–8 doi 10.1038/ng.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biology 2011;12(4):R41 doi 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin Y, Chen K, De Paepe A, Hellqvist E, Krstic AD, Metang L, et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018;131(19):2138–50 doi 10.1182/blood-2017-09-808063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho SF, Anderson KC, Tai YT. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Frontiers in immunology 2018;9:1821 doi 10.3389/fimmu.2018.01821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. The lancet oncology 2020;21(2):207–21 doi 10.1016/S1470-2045(19)30788-0. [DOI] [PubMed] [Google Scholar]
- 45.Campbell KS, Cohen AD, Pazina T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Frontiers in immunology 2018;9:2551 doi 10.3389/fimmu.2018.02551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guièze R, Liu VM, Rosebrock D, Jourdain AA, Hernández-Sánchez M, Martinez Zurita A, et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019;36(4):369–84.e13 doi 10.1016/j.ccell.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017;130(22):2401–9 doi 10.1182/blood-2017-06-788786. [DOI] [PubMed] [Google Scholar]
- 48.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538(7626):477–82 doi 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 49.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014;25(1):91–101 doi 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu G, Wei Y, Kang Y. The Multifaceted Role of MTDH/AEG-1 in Cancer Progression. Clinical Cancer Research 2009;15(18):5615 doi 10.1158/1078-0432.CCR-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nature communications 2020;11(1):1917 doi 10.1038/s41467-020-15740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maura F, Rustad EH, Yellapantula V, Luksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia 2019;34(5):1476–1480 doi 10.1038/s41375-019-0689-0. [DOI] [PubMed] [Google Scholar]
- 53.Landau HJ, Yellapantula V, Diamond BT, Rustad EH, Maclachlan KH, Gundem G, et al. Accelerated single cell seeding in relapsed multiple myeloma. Nature communications 2020;11(1):3617 doi 10.1038/s41467-020-17459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011;144(1):27–40 doi 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walker BA, Wardell CP, Johnson DC, Kaiser MF, Begum DB, Dahir NB, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood 2013;121(17):3413–9 doi 10.1182/blood-2012-12-471888. [DOI] [PubMed] [Google Scholar]
- 56.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007;12(2):131–44 doi 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nature Genetics 2020. doi 10.1038/s41588-020-0667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics (Oxford, England) 2012;28(3):423–5 doi 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods 2017;14(4):417–9 doi 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biology 2011;12(8):R72 doi 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173(2):321–37.e10 doi 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Reports 2018;23(1):282–96.e4 doi 10.1016/j.celrep.2018.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ge Z, Leighton JS, Wang Y, Peng X, Chen Z, Chen H, et al. Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Reports 2018;23(1):213–26.e3 doi 10.1016/j.celrep.2018.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Reports 2018;23(1):239–54.e6 doi 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bärlund M, Monni O, Kononen J, Cornelison R, Torhorst J, Sauter G, et al. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer research 2000;60(19):5340–4. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw data used in the study are already publicly available (dbGap: phs000748.v1.p1 and EGAS00001001178. Analysis was carried out in R version 3.6.1. Unless otherwise specified, we used Wilcoxon rank sum test to test for differences in continuous variables between two groups; Fisher’s exact test for 2×2 tables of categorical variables; and the Bonferroni-Holm method to adjust p-values for multiple hypothesis testing. The full analytical workflow in R to identify hotspots of structural variants is provided in Data S1. All other software tools used are publicly available.